

Suppressing the genes encoding napin and cruciferin leads to an increase in oleosin accumulation and a reorganization of the intracellular architecture in the developing embryo of oilseed rape.
Abstract
The tradeoff between protein and oil storage in oilseed crops has been tested here in oilseed rape (Brassica napus) by analyzing the effect of suppressing key genes encoding protein storage products (napin and cruciferin). The phenotypic outcomes were assessed using NMR and mass spectrometry imaging, microscopy, transcriptomics, proteomics, metabolomics, lipidomics, immunological assays, and flux balance analysis. Surprisingly, the profile of storage products was only moderately changed in RNA interference transgenics. However, embryonic cells had undergone remarkable architectural rearrangements. The suppression of storage proteins led to the elaboration of membrane stacks enriched with oleosin (sixfold higher protein abundance) and novel endoplasmic reticulum morphology. Protein rebalancing and amino acid metabolism were focal points of the metabolic adjustments to maintain embryonic carbon/nitrogen homeostasis. Flux balance analysis indicated a rather minor additional demand for cofactors (ATP and NADPH). Thus, cellular plasticity in seeds protects against perturbations to its storage capabilities and, hence, contributes materially to homeostasis. This study provides mechanistic insights into the intriguing link between lipid and protein storage, which have implications for biotechnological strategies directed at improving oilseed crops.
INTRODUCTION
Cellular architecture depends inter alia on the size, number, and disposition of the various subcellular organelles (Rafelski and Marshall, 2008). In the seed of oilseed rape/canola (Brassica napus), storage parenchyma cells are responsible for the synthesis of triacylglycerols (TAGs), deposited in the form of lipid droplets (Chapman et al., 2012), while the major seed storage proteins cruciferin and napin are deposited in large storage vacuoles (Borisjuk et al., 2013). Cruciferin and napin account for 60 and 20%, respectively, of the seed’s overall protein content (Crouch and Sussex, 1981), representing around one-quarter of a seed’s dry mass. Lipid content represents about one-half of the seed’s dry mass and is largely in the form of TAGs. Therefore, the metabolism of developing seeds is dominated by a repertoire of enzymes and transporters involved in the synthesis of both TAGs and storage proteins (Troncoso-Ponce et al., 2011; Schwender et al., 2015; Chao et al., 2017).

Amino acids are the building blocks for the elaboration of seed storage proteins, while sucrose is converted into both fatty acids and TAGs. Both glycerolipids and storage proteins are assembled in the endoplasmic reticulum (ER) simultaneously during seed development and may thus compete for limited resources, including precursors and cofactors (Schwender, 2008; Schwender and Hay, 2012). Additionally, the site of accumulation within the embryo overlaps extensively (Borisjuk et al., 2013), suggesting that perturbations in the synthesis of proteins may affect lipid synthesis, or vice versa. The existence of crosstalk between lipid and protein metabolism has also been established by showing that certain gene loci influence both the protein and lipid contents of the B. napus seed, but in the opposite direction (Chao et al., 2017). The outcome, as has been noted by breeders, is that the lipid content of mature seed tends to be negatively associated with that of protein (Chao et al., 2017; Patil et al., 2018). It has been suggested that this competition might be transgenically manipulated by redirecting substrate from protein to lipid synthesis (or vice versa), but little success has been achieved in implementing this strategy. Indeed, in B. napus, the repression of genes encoding certain storage proteins was found to favor the compensatory synthesis of other storage proteins (a process named protein rebalancing) rather than to boost the accumulation of lipids (Kohno-Murase et al., 1994, 1995). A similar effect has been noted in soybean (Glycine max; Schmidt et al., 2011), in barley (Hordeum vulgare; Hansen et al., 2007), in maize (Zea mays; Wu and Messing, 2014), and in wheat (Triticum aestivum; Barro et al., 2016). Shifts in carbon partitioning and central metabolism remain extremely difficult to engineer (Sweetlove et al., 2017), and the mechanistic link between oil and protein storage in seeds is far from being understood.
B. napus is one of the most important and widely cultivated temperate zone oilseed crops. Increasing its seed lipid content remains a major aim of a number of breeding programs (Abbadi and Leckband, 2011). Here, the dependency between protein and lipid accumulation was analyzed by transgenic suppression of genes encoding the two dominant storage protein classes (napin and cruciferin) using RNA interference (RNAi), along with phosphoenolpyruvate carboxylase (PEPC), an enzyme that channels glycolytic intermediates toward organic and amino acid synthesis (Rolletschek et al., 2004). The developing seeds set by these transgenic plants were characterized at the transcriptomic, proteomic, metabolomic, and lipidomic levels, and the visible consequences of RNAi suppression were explored using microscopy, NMR, and mass spectrometry (MS) imaging. Metabolic fluxes and energy cofactor demands were predicted by flux balance analysis. The identification of the numerous changes at the ultrastructural, molecular, and metabolic levels has led to a number of mechanistic insights, highlighting cellular and metabolic plasticity to ensure a sufficient level of homeostasis to support the maturation and viability of the seed.
RESULTS
The Effectiveness of RNAi in Suppressing Napin and Cruciferin Synthesis
The pool of storage proteins in B. napus is encoded by numerous genes (Gacek et al., 2018); thus, BAC library screening results and in silico analysis were applied to identify all genes of interest. For cruciferin, we identified 8 genes encoding the P1, P2, and P3 subfamilies of cruciferin, which were all targeted by the hairpin (Supplemental Data Set 1). For napin, 15 (out of 17 identified) genes were targeted, and for PEPC, 8 (out of 11 identified) were targeted. The effectiveness with which the targeted genes were downregulated in the developing embryo by RNAi was evaluated by RT-qPCR of three independent transgenic lines (nos. 576, 578, and 580) and compared with those of their respective wild-type segregant (WTS) lines (nos. 577, 579, and 581; Figures 1A to 1C). The abundance of transcripts generated by different genes encoding various cruciferins was reduced by up to 32-fold (−5 at a log2 scale; Figure 1A). The extent of the suppression was even higher for napins, shown for three napin-encoding genes in Figure 1B. The suppression of other targeted napin- and cruciferin-encoding genes became evident in the RNA-seq data sets (see further below). A time series analysis showed that the extent of downregulation was most pronounced in the later stages of seed filling (Figure 1D). The level of suppression of PEPC was marginal at the levels of both transcript abundance and enzyme activity (Figures 1C and 1E). Thus, the PEPC RNAi construct was ineffective.
Figure 1.

The Effects of Suppressing Cruciferin and Napin Achieved by Introducing an RNAi Construct.
(A) to (C) Transcript abundances in the developing embryo (30 DAF) of genes encoding cruciferin (Cru; [A]), napin (Nap; [B]), and PEPC (C). Abundances are given in the form of log2 fold changes in the transgenic embryos (lines #576, #578, and #580) versus the respective WTS embryos (lines #577, #579, and #581). Values are shown as individual data points; n = 3 biological repeats, each comprising pooled RNA extracted from 10 embryos.
(D) Log2 fold changes in transcript abundances over the course of embryo development of genes encoding napin and cruciferin in transgenic line #580 (versus WTS line 581). n = 3 biological repeats with nine embryos each.
The expression levels in (A) to (D) were normalized to that of BnUBC9.
(E) Maximum catalytic activity of PEPC in embryo extracts. n = 6 replicates with three embryos each)
(F) to (H) Contents of lipids (F), fiber (G), and protein (H) in mature seeds.
Values in (E) to (H) are shown in the form mean ± sd (error bars); n = 12 replicates each comprising ∼5 seeds in (F), and n = 6 replicates each comprising ∼20 seeds in (G) and (H). Mature seeds from three independent batches were analyzed regarding parameters (F) to (H) with similar results. DW, dry weight; FW, fresh weight.
Asterisks indicate that means differed significantly from WTS values (P < 0.05, one-way ANOVA); ns, not statistically significant.
The RNAi Approach Alters the Composition but Not the Carbon-to-Nitrogen Ratio of Mature Seeds
The visible phenotype of the transgenic plants was essentially identical to those displayed by WTS plants. Measurements of seed content showed that mature transgenic seeds contained 10 to 15% less protein and lipid and 30 to 35% more fiber than WTS seeds (Figures 1F to 1H). Further compositional analysis revealed that the total nitrogen content and the carbon-to-nitrogen ratio were unchanged, while the water content was statistically lower by ∼9% in the transgenic seeds (Supplemental Figure 1). Starch content (only transiently accumulated in B. napus seeds) was below 1% of dry weight without any consistent changes in the transgenic versus wild-type lines (Supplemental Figure 1).
Suppressing the Production of Napin and Cruciferin Lowers the Embryo’s Portion of Mature Seeds
At maturity, the transgenic seeds had a lower weight than WTS seeds despite little difference in seed size (Figure 2B). To determine what changes may have resulted internally due to RNAi suppression of cruciferin and napin proteins, magnetic resonance imaging (MRI) was used to visualize the seed’s internal structure noninvasively (Supplemental Movie 1). MRI revealed that the cotyledons of mature transgenic seeds were irregularly shaped and left a number of air-filled cavities (void spaces), as exemplified in Figure 2A. The volume of void spaces was significantly greater by 62 to 73% than in WTS seeds (n = 8 to 12 seeds; Figure 2E). In WTS seeds, the radicle is usually covered by cotyledons and only rarely protrudes beyond the cotyledons (8%), whereas the radicle tip protruded in 87% of the transgenic seed models (white arrows in Figure 2A; Supplemental Movie 2).
Figure 2.

The Effects on Seed Phenotype of Suppressing Cruciferin and Napin via the Introduction of an RNAi Construct.
(A) Three-dimensional seed models for two transgenic lines (#578 and #580) and their corresponding WTS lines (#579 and #581) acquired by MRI. Red arrows indicate air-filled cavities (void space), and white arrows indicate radicle tips.
(B) Weight of mature seeds. n = 6 with ∼20 seeds each.
(C) to (F) Volume of embryo (C), seed coat (D), and void space (E) and the percentage of each component to total seed volume (F).
Asterisks in (B) to (E) indicate that means differed significantly from WTS values (P < 0.05, one-way ANOVA); ns, not statistically significant. Data in (C) to (F) were derived from the MRI-based analysis of eight individual seeds per genotype. See also Supplemental Movies 1 to 3. em, embryo; ic, inner cotyledon; oc, outer cotyledon; ra, radicle; sc, seed coat; vs, void space.
When assayed using MRI, the transgenic embryos were 7 to 9% smaller than WTS embryos (Figure 2C), and the volume of the seed coat was 5 to 8% greater (Figure 2D). This led to a lower ratio of embryo (rich in oils and protein) to seed coat (rich in fiber) in transgenic seeds and thus to an altered seed composition (Figure 2F). Despite these changes, there was no loss in the ultimate viability of transgenic seeds, except a delay in the timing of germination (Supplemental Figure 2).
The Intracellular Architecture Is Markedly Changed by the Loss of Napin and Cruciferin
Substantial differences in the subcellular organization were observed in the embryos of transgenic and WTS plants. In the early stages of development, parenchyma cells in WTS embryos already featured numerous protein bodies, which expanded over the course of seed development (Figures 3A, 3C, and 3E; Supplemental Figure 3). By contrast, in the transgenic embryos, protein bodies were detected only later in seed development, remained substantially smaller in size, and exhibited a crystalline structure (Figures 3B, 3D, and 3F; Supplemental Figure 3). Perhaps the most striking subcellular change was the accumulation of large membrane stacks evident throughout storage parenchyma cells of the transgenic embryos (Figure 3G).
Figure 3.

Fine Structure of the Seed Embryo’s Storage Tissues.
(A) and (B) Protein bodies, visualized using Ponceau staining, in early storage stage embryos formed by WTS line no. 581 (A) and transgenic line #580 (B).
(C) and (D) Electron micrographs of protein bodies in early storage stage embryos formed by WTS line #581 (C) and transgenic line #580 (D).
(E) and (F) Electron micrographs of protein bodies in mature storage stage embryos formed by WTS line #581 (E) and transgenic line #580 (F).
(G) The transgenic embryos formed distinct large oleosin-containing membrane stacks (marked with arrows).
The protein bodies formed in WTS embryos were surrounded by densely packed lipid droplets (Figure 4A). Immunolabeling confirmed that both napin and cruciferin were associated with the WTS protein bodies (Figures 4C and 4D), while oleosins (structural proteins of oil body membranes; Huang, 2018) were associated with lipid droplets (Figure 4B). In the cells of transgenic embryos, the abundance of cruciferin was greatly reduced, and what was present was scattered throughout the protein bodies (Figure 4H); napin was barely detectable (Figure 4G). On the other hand, oleosin was well represented in the cells of transgenic embryos, both within lipid droplets and throughout the membrane stacks (Figure 4F). All principal changes in intracellular architecture described above for transgenic line no. 580 versus WTS line no. 581 were also evident for other transgenic lines, as exemplified in Supplemental Figure 4.
Figure 4.

The Ultrastructure of the Embryo Storage Tissues.
(A) and (E) Protein bodies (pb) surrounded by lipid bodies (lb) in the embryos of WTS line #581 (A) and transgenic line #580 (E). The oleosin-containing membrane stacks present in embryos formed by the transgenic line are arrowed.
(B) to (D) and (F) to (H) Immunolabeling used to highlight the presence of oleosin ([B] and [F]), napin ([C] and [G]), and cruciferin ([D] and [H]) in the embryos of WTS lines ([B] to [D]) and transgenic lines ([F] to [H]).
Both the Storage and Membrane Lipids Are Altered in Transgenic Seeds
Analysis of lipids from intact, mature seeds using MRI showed reduction in total lipid content by ∼20% in both transgenic lines analyzed despite similar seed sizes to WTS lines (Supplemental Movie 3). Regarding lipid distribution, the cotyledons accumulated a higher proportion of lipids relative to the radicle (Supplemental Movie 2), confirming earlier analyses (Borisjuk et al., 2013). This was apparent in all genotypes.
MS analysis of developing seeds (∼42 days after flowering [DAF]) revealed that both the quantity and composition of TAGs and the common membrane lipid, phosphatidylcholine (PC), were affected by suppression of napin and cruciferin production (Figure 5A). In plants, PC serves as both a structural component of membranes as well as an important metabolic precursor for the synthesis of TAGs. The quantity of PC was nearly fourfold greater in the seeds from transgenic line no. 580 than in those from WTS line no. 581, whereas the quantity of TAGs was ∼20% lower in the transgenic seeds. For both classes of lipids, there was a marked shift in their saturation levels. Polyunsaturated fatty acids were more abundant in both PC and TAG from lipid extracts of transgenic seeds compared with WTS seeds (Figure 5B).
Figure 5.

The Level and Distribution of Storage and Membrane Lipids in the Seed.
(A) Mean contents of PC and TAGs as measured using electrospray ionization (ESI)-MS. Data are given in nmol/mg seed.
(B) Mean contents of individual molecular species of PC/TAGs as measured using ESI-MS. Data are given in mol%.
Data in both (A) and (B) are plotted for each of the three replicates with the sd lines centered around the mean.
(C) MS-acquired images showing the spatial distribution of PC and TAGs. The abundance of each compound type is shown as mol% on a colored scale from yellow (low) to green (high). Images captured by light microscopy of the sections used for MS imaging are shown to the left of the MS-acquired ones. See also Supplemental Figure 5 for biological replicates. Bars = 1 mm.
MS-based imaging of seed sections confirmed the marked reduction in saturation in the transgenic seeds with respect to TAGs and PC and also emphasized the heterogeneous distribution of lipid molecular species in different tissues (Figure 5C). The extent of the reduced saturation was more variable seed to seed in transgenic seeds than in WTS seeds (Supplemental Figure 5).
Analysis of lipid extracts of dissected seed tissues confirmed the MS imaging results and demonstrated that the quantitative shift in saturation for both PC and TAG to more unsaturated species occurred in all parts of the transgenic embryos (Supplemental Figure 6). In addition, membrane lipids not involved in TAG accumulation also showed a similar shift to more unsaturated molecular species in the different tissues of the embryo, as shown for phosphatidylethanolamine (PE) and phosphatidylinositol (PI; Supplemental Figure 7). This may suggest that the membrane stacks observed in cells of the embryo (Figures 3 and 4) were associated with alterations in lipid composition, favoring membranes (and storage lipids) with a greater degree of unsaturation. Particularly, marked differences were observed in the region of the embryonic axis, where the composition of PCs shifted from 34:1 to 34:2 and 34:3 (Figure 5B), that of PEs from 34:1 to 34:2 and from 36:3 to 36:4 (Supplemental Figure 7), and that of PI from 34:1 to 34:2 and 34:3 (Supplemental Figure 7). However, all parts of the transgenic embryos exhibited shifts in unsaturation in both membrane and storage lipids (Figure 5C; Supplemental Figure 6).
Comparative Transcriptome Analysis Indicates a Major Shift in Global Gene Expression
RNA sequencing (RNA-seq) profiling detected transcripts from 67,885 protein-encoding genes (Supplemental Data Sets 2A to 2J). A comparison of transcript abundances between the transgenic (line no. 580) and its WTS (line no. 581) embryos showed that 94 genes were downregulated in the transgenic embryos, while 97 were upregulated (Supplemental Data Set 2C). Not surprisingly, the most strikingly affected genes were those encoding storage proteins (Supplemental Data Set 2D). The transcript abundance from 12 genes predicted to encode napin and 7 to encode cruciferin were significantly lower in line no. 580 (transgenic) than in line no. 581 (WTS) embryos (Figure 6A). In most cases, downregulation was >10-fold, but it was as high as 111-fold in at least one case. In the WTS embryos, ∼50% of the reads were generated from a set of 25 napin- or cruciferin-encoding genes, but this was the case for just 4% of the reads in the transgenic embryos. This represents a large quantitative shift in gene expression. A number of genes encoding proteins associated with sulfur assimilation were downregulated in the transgenic embryos (see also Supplemental Data Set 2F): the Arabidopsis (Arabidopsis thaliana) orthologs (At1g04770, At5g48850, and At1g36370) of nine of these genes are known to be induced by sulfur deficiency (Maruyama-Nakashita et al., 2005). The transcript abundance of several genes involved in the reduction of sulfate to H2S also was reduced; their Arabidopsis putative orthologs encode a sulfate transporter of the chloroplast envelope (AT5G13550), a chloroplastidic isoform of ATP sulfurylase (AT3G22890), 5′-adenylylsulfate reductase (AT1G62180), and a sulfite reductase (AT5G04590). The set of downregulated genes also included two putative orthologs of the Arabidopsis genes encoding adenylyl sulfate kinase (AT2G14750), an enzyme that provides activated sulfate used in the synthesis of secondary metabolites, as well as a gene encoding Ser acetyltransferase (AT2G17640), an enzyme active in Cys synthesis.
Figure 6.

The Impact of the RNAi Transgene on the Transcriptome.
Read counts of B. napus genes were ranked according to their expression levels in WTS line #581 (A) or transgenic line #580 (B); only the 30 highest ranking genes are shown. The genes associated with the highest read counts in the WTS line were mostly identified as encoding storage protein (napin or cruciferin). These represented ∼50% of the counts in WTS line #581 but only 4% in transgenic line #580. In the transgenic line, 50% of the read counts are contained in 614 of the highest ranking genes. For gene identifiers and further information, see Supplemental Data Set 2J. n = 3 biological repeats, each comprising pooled RNA extracted from 10 embryos. The significance threshold was set as 5% for the adjusted P value and a more than fourfold change in transcript abundance. Asterisks indicate that means differed significantly between the WTS and transgenic lines.
Among the upregulated genes, those encoding oleosin tended to be expressed at higher levels in the transgenic line than in the WTS line (Figure 6B), although none of the individual 19 oleosin-encoding genes were statistically significantly different (Supplemental Data Set 2D). Several members of a gene family encoding nonspecific lipid transfer proteins were upregulated in the transgenic line no. 580 (Supplemental Data Set 2D), the most abundant being BnaC04g30640D (Arabidopsis orthologs At5g38180 and At5g38195). The B. napus putative orthologs of At3g59970, an Arabidopsis gene encoding methylenetetrahydrofolate reductase (C1 metabolism), were upregulated in the transgenic embryos, and their products were noted as being more abundant at the proteomic level.
Gene Ontology term enrichment analysis indicated that among the downregulated genes in the transgenic line, the biological processes “sulfate assimilation,” “serine family amino acid metabolic process,” and “sulfur compound biosynthetic process” were the most significantly overrepresented terms, while among the upregulated genes, “lipid transfer” and “lipid localization” were the most overrepresented (Supplemental Data Set 2I).
The Rebalancing of the Proteome Induced by the Loss of Napin and Cruciferin Results in the Hyperaccumulation of Oleosin
Two-dimensional gel electrophoresis-based proteome profiling enabled the identification of 1124 distinct proteins in B. napus embryos (Supplemental Figure 8). According to an MS-based analysis, 49 proteins were shown to have increased in abundance and 165 to have decreased in abundance in transgenic embryos relative to WTS embryos (Table 1; for a full list of proteins, see Supplemental Data Set 3). As expected, the list of proteins with lower abundance was dominated by the vacuolar storage proteins napin2, cruciferin1, and cruciferin4, each of which was reduced in abundance by between fivefold and 10-fold. The set of reduced-abundance proteins also included several enzymes involved in the tricarboxylic acid cycle, glycolysis, the synthesis of amino acids and polyamines, and sulfur and cofactor (C1) metabolism. Cruciferin1 was recovered from several spots, pointing to multiple isoforms (which confirms earlier observations by Nietzel et al. [2013]). There was an indication that the α P1 and β P2 cruciferin chains were reduced in abundance, while the quantities of the α P2, α P3, and β P1 + P3 chains were largely unaffected in the transgenic seeds. Among the upregulated proteins, the most prominent were oleosin2 (6.7-fold increase in abundance), biotin carboxyl carrier protein2 (a subunit of the acetyl-CoA carboxylase complex), and plastidic pyruvate kinase, all of which are involved in the synthesis of fatty acids and/or storage of lipids (Supplemental Data Set 3). Notable among the other upregulated proteins were two related to translation (elongation factor 1-α1 [EF1-α] and the 60S ribosomal protein L10), the ER stress-related protein disulfide isomerase-like1-2, and other proteins involved in either cell wall synthesis or amino acid metabolism. Among the latter group was nitrogen regulatory protein PII, coordinating carbon/nitrogen metabolism (Chellamuthu et al., 2014), and part of the acetyl-CoA carboxylase complex (Feria Bourrellier et al., 2010). We did not observe an increase of other vacuolar storage proteins.
Table 1. Proteins Classified as Differentially Abundant between Transgenic and WTS Embryos.
| Spot Volume Difference | Up-Regulated Proteins in Transgenic Line no. 580 | ATG Code |
|---|---|---|
| 6.75 | Oleosin S2-2 | At3g27660 |
| 4.71 | Biotin carboxyl carrier protein of acetyl-CoA carboxylase2, chloroplastic (BCCP2) | At5g15530 |
| 3.73 | Elongation factor 1-α-1 | At1g07940 |
| 3.32 | 60S ribosomal protein L10 | At1g26910 |
| 3.32 | Osmotin-like protein | At4g11650 |
| 3.18 | MLP-like protein31 | At1g70840 |
| 2.74 | Oleosin S2-2 O | At3g27660 |
| 2.72 | Oleosin 21.2 kD | At5g40420 |
| 2.66 | Glutathione S-transferase U5 | At2g29450 |
| 2.56 | Nitrogen regulatory protein PII homolog | At4g01900 |
| 2.35 | β-Galactosidase3 | At4g36360 |
| 2.35 | Probable Ser protease EDA2 | At2g18080 |
| 2.32 | Isopentenyl-diphosphate Δ-isomerase II | At3g02780 |
| 2.25 | α-Galactosidase | At5g08370 |
| 2.25 | UDP-sulfoquinovose synthase, chloroplast precursor | At4g33030 |
| 2.19 | Importin subunit α-1 | At3g06720 |
| 2.19 | Protein disulfide isomerase-like1-2 | At1g77510 |
| 2.19 | Pyruvate kinase isozyme A, chloroplastic | At3g22960 |
| Down-Regulated Proteins in Transgenic Line no. 580 | ||
| 10.17 | Cruciferin CRU1 | AT4G28520 |
| 10.17 | Malate dehydrogenase, mitochondrial | AT1G53240 |
| 5.94 | 12S seed storage protein CRU1 | AT5G44120 |
| 5.94 | 12S seed storage protein CRU4 | At1g03890 |
| 5.63 | Napin-2 | AT4G27150 |
| 4.89 | Elongation factor 1-γ1 | AT1G09640 |
| 4.89 | Elongation factor Tu, chloroplastic | AT4g20360 |
| 4.89 | Monodehydroascorbate reductase, cytoplasmic isoform 3 | AT3G09940 |
| 4.89 | S-Adenosylmethionine synthase | AT1G02500 |
| 4.89 | S-Adenosylmethionine synthase 2 | AT4G01850 |
| 4.89 | S-Adenosylmethionine synthase 3 | AT2G36880 |
| 4.53 | Methylenetetrahydrofolate reductase1 | AT3G59970 |
| 4.35 | Succinyl-CoA ligase [GDP-forming] subunit α-1 | At5g08300 |
| 4.22 | Biotin synthase | At2g43360 |
| 4.22 | Fructose-bisphosphate aldolase, cytoplasmic isozyme | At4g26520 |
| 4.08 | Branched chain amino acid aminotransferase-like protein2 | At5g27410 |
| 3.96 | Eukaryotic translation initiation factor 3 subunit H | At1g10840 |
| 3.77 | Phosphopantothenate-Cys ligase2 | At5g02080 |
| 3.51 | Cruciferin | AT4G28520 |
| 3.51 | Napin | At4g27140 |
| 3.51 | Napin-3 | At4g27160 |
| 3.45 | Probable E3 ubiquitin-protein ligase ARI12 | At1g05880 |
Based on two-dimensional gel electrophoresis profiling followed by their identification by MS. A full list is given in Supplemental Data Set 3.
Amino Acid Metabolism Is a Focal Point of the Metabolic Adjustment to the Loss of Napin and Cruciferin
A set of 79 free metabolites were quantified using a liquid chromatography/MS platform. Soluble sugars (especially sucrose) were by far the most abundant metabolite category, followed by amino acids (especially Arg, Asp, Gln, and Glu) and organic acids (especially citrate and malate; Figure 7A; Supplemental Data Set 4). Notably, the amount of amino acids and polyamines was almost doubled in the transgenic versus WTS embryos. There were statistically significant increases in the levels of Gln (+163%), Arg (+118%), Val (+38%), His (+136%), and Trp (+ 34%) and, although not statistically significant, strong increases for some other amino acids (Asn, +192%; Asp, +55%; Orn, +184%; and citrulline, +75%). Oxaloacetate (a product of the PEPC reaction) was significantly increased by 24% in the transgenic versus WTS embryos. Apart from these variations, the loss of napin and cruciferin induced minor changes in the levels of other intermediates of cellular metabolism (Figure 7B).
Figure 7.
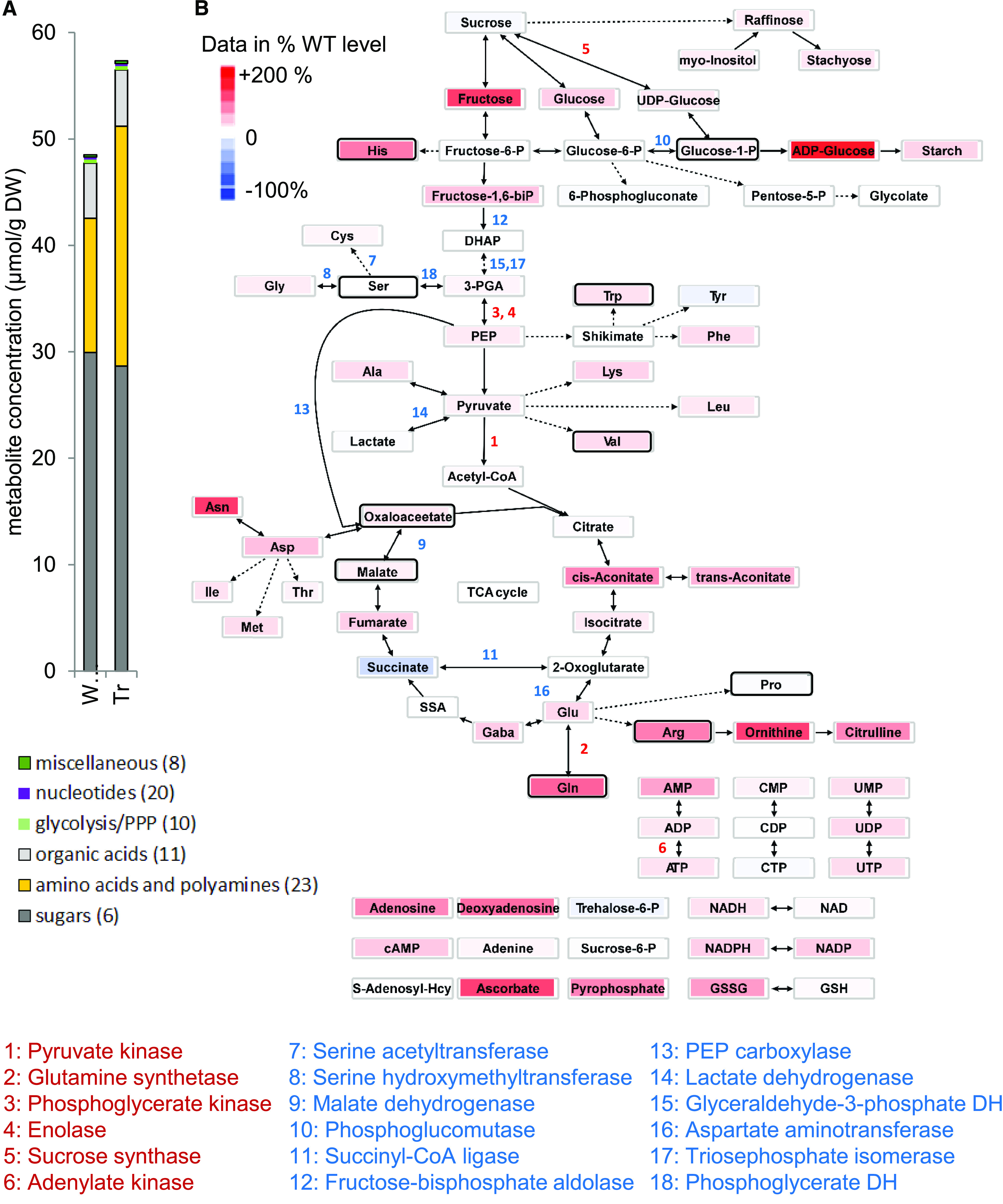
Steady State Profile of Metabolites in the Developing B. napus Embryo and Related Changes in Gene Expression.
(A) Metabolite abundance (ordered by categories; numbers in parentheses indicate quantified compounds) present in the embryos of transgenic line #580 and WTS line #581. DW, dry weight.
(B) Central metabolism network view. The heat map of metabolites indicates mean values relative to that obtained in the WTS line: increased (decreased) abundance of compounds in red (blue) color. Three biological replicates with each sample consisting of ∼10 embryos were analyzed. Numbers refer to enzymes for which differential gene expression was evident at the level of either the transcriptome or the proteome. Black frames refer to statistical significance (one-way ANOVA; P < 0.05).
Neither the Respiration nor the Photosynthetic Activity of the Embryo Is Affected by the Loss of Napin and Cruciferin
The operational efficiency of PSII in embryos was assessed by pulse-amplitude-modulated (PAM) fluorescence, while photosynthetic energy transfer occurring within the seed was measured as the linear electron transport rate (ETR). Both transgenic and WTS embryos exhibited a similar ETR without the formation of any distinct gradient across the embryo (Supplemental Figure 9A). A comparison of rapid light response curves (Supplemental Figure 9B) showed that both genotypes exhibited comparable levels of photosynthetic activity when illuminated by normal light levels, equivalent to a maximum of 100 µmol m−2 s−1 PAR (Borisjuk et al., 2013), but at higher light levels, transgenic embryos had lower activity than WTS embryos. There were no apparent differences in the respiratory activity (measured as the rate of total oxygen consumption) between the transgenic and WTS embryos (Supplemental Figure 9C).
Modeling the Metabolic Flux and the Energy Cofactor Demands for Biosynthesis of Lipids and Proteins
Since the transgenic interventions were found to change seed composition and metabolic profiles (Figures 1, 5, and 7), there should be associated changes in metabolic flux. To compare central metabolism flux between the genotypes, we first determined the growth rate and the biomass composition of the developing embryo at ∼30 DAF for the WTS and transgenic lines nos. 580 and 581 (Supplemental Data Sets 5A and 5B). In transgenic embryos, there was a statistically significant 0.75-fold reduction in protein level relative to WTS (P = 10−4), while free metabolites were 1.8-fold higher (P = 5 × 10−5; Supplemental Data Set 5B). The decrease in protein in the growing embryos parallels the measurements in dry mature seeds of transgenics (Figure 1H). The theoretical amino acid requirements for the expressed proteins as determined from the RNA-seq data of expressed genes suggests that transgenic embryos would use 52% less Gln and 31% less Cys (Supplemental Data Set 5C), and the measured sulfur content was ∼15% lower in transgenic embryos than in the WTS embryos (4.3 versus 5.1 mg/g), which together is consistent with the proposed shift in Cys demand and sulfur metabolism.
The embryo dry weight composition data were used to parametrize a metabolic model of the B. napus developing embryo (Hay et al., 2014). The accumulation of free sugars, amino acids, and other polar metabolites was defined in the model according to the free metabolite dry weight fraction (Supplemental Data Set 5B) and compositional data (Figure 7). Metabolic fluxes were predicted for the two genotypes by flux balance analysis (Supplemental Data Set 5D). Based on the biosynthetic pathway fluxes, energy cofactor demands for the biosynthesis of lipids and proteins were derived (Figure 8A; Supplemental Data Set 5E). In the transgenic line, the model revealed an overall reduction in biosynthetic ATP demand (Figure 8A). ATP demand for protein synthesis was diminished while ATP demand for lipid synthesis increased by a lesser extent (Figure 8A). With regard to the balance of reducing cofactors, the total redox demand for the transgenic line increased but the redox changes were overall less pronounced than the changes in ATP demand (Figure 8A). Since it was observed that lipids of transgenic embryos were overall higher in unsaturation than those of WTS embryos, this may suggest a higher demand for reducing equivalents for fatty acid desaturation and an overall impact on total redox balance. The balance of redox for desaturation of fatty acids, shown in Figure 8A, is increased by 1.2-fold in transgenic embryos. However, this increase, amounting to 0.5 mmol H2 equivalents/g dry weight, is only 2% of the total redox demand for lipid biosynthesis in the WTS embryos (26.1 mmol/g dry weight). Therefore, the overall requirements for reducing equivalents were only slightly impacted by the change in lipid saturation.
Figure 8.

Modeling of the Impact of the RNAi Transgene on the Embryonic Cells of B. napus.
(A) Demands in energy cofactors (ATP and reducing equivalents) for the biosynthesis of proteins and lipids. The reducing equivalents for fatty acid desaturation are shown separately of the balance for lipids. Details are given in Supplemental Data Set 5E. DW, dry weight.
(B) Allocation of nitrogen among different biomass fractions (based on biomass and elemental composition in Supplemental Data Set 5C).
(C) Increase of surface area in response to ellipsoid parameters. Increasing one of the axes of a sphere while keeping its volume constant will result in an increase of the surface area of the ellipsoid over the sphere. Calculation was based on the formula in (D).
(D) Gain of surface by reshaping lipid bodies (LB). The left panel shows distinctly shaped lipid bodies in an electron micrograph. The right panel exemplifies how the transition from sphere to ellipsoid (at constant volume) causes tremendous gain in surface area (SA). Transgenic embryonic cells apply this strategy to incorporate the excess membrane protein oleosin; the stacks are partially a consequence of the excess oleosin and the preponderance of phospholipid at the partial expense of TAGs.
In addition to the balances of energy cofactors, we examined the total nitrogen balance of the embryo (Figure 8B). In particular, since in the transgenic embryo PC was found to be increased almost fivefold (Figure 5A), it was of interest to know if the associated increase in PC-bound nitrogen had a substantial impact on the total nitrogen balance. Overall, the total nitrogen content per gram of dry weight in transgenic embryos was reduced relative to the WTS embryos (Figure 8B). The reduction in the protein nitrogen level was in part compensated by an increase in nitrogen in the free metabolite fraction (Figure 8B). Although PC-bound nitrogen was increased in the transgenic embryos by almost fivefold, this increase accounted only for ∼16% of the decrease found in protein nitrogen. This would suggest that changes in PC had only minor impacts on overall nitrogen balance.
DISCUSSION
The RNAi-mediated suppression of the synthesis of cruciferin and napin was balanced by a remarkable level of plasticity at the ultrastructural, molecular, and metabolic levels. The formation of large, oleosin-containing membrane stacks replaced the normal protein-filled vacuoles. Although the precise mechanistic basis for these subcellular changes remains to be determined, several factors revealed here suggest insights into a homeostatic organellar mechanism in seeds that maintains the appropriate storage and compartmentalization of carbon and nitrogen reserves for seed viability.
Reprogramming of Protein Storage Is Accompanied by Intracellular Rearrangement
Despite the transgene-induced reprogramming of the storage process, the seeds appeared to develop normally, were viable, and hardly showed any outwardly visible aberration (although their germination was somewhat slower than that of WTS seeds). The most prominent effect of suppressing the synthesis of cruciferin and napin was to alter the intracellular architecture of the embryo’s storage tissues. Along with the expected reduction in the size of protein bodies, the cytoplasm developed numerous small oil bodies and large, oleosin-rich membrane stacks. This latter phenomenon may well be a consequence of the somewhat unusual features of oleosin (Hsieh and Huang, 2004), which possesses a central, uninterrupted hydrophobic domain required for its association with membranes and its deposition into oil bodies (Hope et al., 2002). Oleosins are synthesized on the ER membrane and targeted to intracellular lipid droplets. As neutral lipids are generated in the same compartment, the formation of lipid droplets parallels the incorporation of oleosins into membranes (Wahlroos et al., 2003) together with other lipid droplet-related proteins (Chapman et al., 2012; Huang, 2018). Immunolabeling showed that oleosin was attached to the lipid droplets in storage cells (Figure 4), while electron microscopy imaging confirmed a marked increase in the number of small lipid droplets (Figure 3; Supplemental Figure S3). An in vitro study performed by Vance and Huang (1987) established that many small lipid droplets would contain greater amounts of oleosin than fewer larger lipid droplets due to increased surface area. The formation of small, oleosin-enriched lipid droplets and membrane stacks is thus in accord with the severalfold increase in oleosin content found in the transgenic seed. During the process of lipid droplet formation and oleosin synthesis, a gradient of enrichment with respect to oleosins and TAG is generated, and, as a result, the quantity of TAG produced has an impact on the final shape of the lipid droplet. As TAG content of transgenic embryos was lower than that of WTS embryos (Figure 5), the lipid droplets may have been less efficiently filled, resulting in the creation of smaller and irregularly shaped lipid droplets. A similar phenotype was observed in maize kernel (Ting et al., 1996), where a reduction in the proportion of oleosin to lipid was shown to result in the formation of large lipid droplets (Siloto et al., 2006; Shimada et al., 2008). The mechanistic basis of lipid droplet formation remains obscure (Herman, 2008; Gao and Goodman, 2015), but it seems reasonable to conclude that both a reduced size and aberrant/ellipsoid shape (Figure 3; Supplemental Figure 3) in the transgenic embryos were influenced, possibly even determined, by the surfeit of oleosin. The loading of membranes with oleosin and the formation of many small lipid droplets provide the means for the embryo to accommodate an excess of oleosin protein. The geometric shift from spherical to ellipsoid lipid droplets (and in its extreme to membrane stacks) also involves a preponderance of phospholipid (Figure 5). Thereby, the transgenics can maximize the surface area of lipid storage while maintaining their volume. The mathematical description (formula in Figure 8D; Michon, 2019) reveals that a fivefold increase of one of the axes of a sphere (while keeping its volume constant) will cause the surface area to increase by ∼75%. In other words, the intracellular plasticity allows the plant to easily store excessively abundant membrane proteins (oleosin) without large compromises on overall oil storage capacity (lipid droplet volumes).
The consequences of the intracellular rearrangements could be widespread. Lipid droplets are involved in intracellular communication due to their contact with most of the subcellular organelles (Valm et al., 2017), and they are associated with several functions relating to intracellular homeostasis (Gao and Goodman, 2015; Huang, 2018; Shimada et al., 2018). The observed upregulation of the gene encoding protein disulfide isomerase-like1-2 (At1g77510) in our transgenic plants is possibly a response to ER stress (Lu and Christopher, 2008). A further suggested effect on ER functionality relates to the increased presence of both EF1-α and a 60S ribosomal protein, implying that translation may be compromised in the transgenic embryo. Finally, there were numerous changes in the contributions of PC, PE, and PI to the membrane lipid content (Figure 5; Supplemental Figures 5 to 7), all of which are assembled in the ER. Note that the composition of membrane lipids (and membrane proteins) can affect both the fluidity and curvature of the membrane (van Meer et al., 2008), so part of the homeostatic control here may be to synthesize additional membrane lipids with increased numbers of double bonds to accompany changes in membrane architecture required to store adequate amounts of protein and lipid for seed viability.
The Metabolic Adjustments Associated with the Process of Intracellular Rearrangement
Although the introduction of the RNAi construct delivered a major reduction in the abundance of transcript encoding cruciferin and napin (Figure 6A), the overall reduction in total seed protein was only moderate (Figure 1H). The shift in protein synthesis had the effect of lowering the embryos’ demand for amino acids, most notably for Gln, which most likely accounted for a marked increase in the steady state levels of both Gln and other amino acids (Figure 7). In the transgenic lines, processes controlling the supply and demand of nitrogenous precursors are likely perturbed. Part of the decrease of nitrogen in storage proteins is offset by an increase of nitrogen in the fraction of free metabolites and, to a lesser extent, in the nitrogen-containing membrane lipid PC (Figure 8B). In soybean, proteome rebalancing involves only minor transcriptional changes, with posttranscriptional/translational regulation playing a greater role (Herman, 2014).
An unexpected (but nevertheless clear) result was that the transgenic seeds accumulated less storage lipid than WTS seeds. The following scenarios can be invoked to explain this observation: first, the portion of seed coat versus embryo slightly increases in the transgenic plants (Figure 2); second, the increased abundance of PII protein (Table 1) may imply a strengthened inhibition over the activity of ACCase, responsible for initiating the synthesis of fatty acids in the plastids (Feria Bourrellier et al., 2010); third, the reduced abundance of several enzymes involved in glycolysis (Supplemental Data Set 3) may lower the flux into fatty acids/TAGs; fourth, the increased level of PC (Figure 5A) may either indicate a failure to incorporate fatty acids into TAGs and/or reflect an increased demand for PC to synthesize the observed membrane stacks; and fifth, the size and/or spatial distribution of lipid droplets (Figure 4; Supplemental Figure 3) may affect the dynamics of lipid accumulation (Miquel et al., 2014).
Another unexpected finding was that the degree of fatty acid unsaturation was elevated in transgenic embryos (Figure 5). While the increased unsaturation observed was too small stoichiometrically to make a significant contribution to rebalancing redox demand (outcome of flux balance analysis modeling; Figure 8A), the question remains why a reduction in the seed storage proteins cruciferin and napin would increase the degree of unsaturation in membrane and storage lipids. Plausible explanations are (1) greater resident time of PC in the ER prolonging the availability for fatty acid desaturase enzymes to act, thereby supporting the increase in polyunsaturated fatty acids; (2) a concerted ER subdomain configuration change that favors membrane lipid assembly versus TAG; and (3) inefficient packaging of newly synthesized TAGs that leads to a buildup of improperly assembled cytoplasmic lipid droplets.
Implications for Seed Improvement
The proteome rebalance in seeds of B. napus seems to be ineffective as compared with soybean and maize (Schmidt et al., 2011; Herman, 2014; Wu and Messing, 2014). Knowing that the rules can be different in different crops is critical for breeding and genetic intervention to improve agriculture.
A standout conclusion from this research is that suppressing the production of one class of storage compound (in this case napin/cruciferin) does not necessarily lead to the redirection of assimilate to an alternative class (TAGs/seed oil). Rather, the growth of the embryo is slowed as a result of a presumed feedback between protein and lipid storage. The conclusion is that attempting to raise lipid or seed oil yields by repressing the accumulation of storage proteins is unlikely to succeed. Instead, a more promising approach would be to focus on increasing the size of the seed, for example by stimulating the supply of assimilate and its uptake into the seed. It has been suggested that overexpressing the genes encoding oleosin could boost the production of storage lipids (Huang, 2018), a strategy that appears to be effective in tissues characterized by a rather minor lipid content, such as the rice (Oryza sativa) grain (Liu et al., 2013) or the potato (Solanum tuberosum) tuber (Liu et al., 2017); however, as yet, there is no support in the literature for this approach working in an oilseed crop. Our data imply that raising the production of oleosin alone will possibly fail to have a positive effect on the seed lipid content. This conclusion would need to be tested more stringently by engineering the upregulation of oleosin-encoding gene(s) in a background where the production of cruciferin and napin was normal. Upregulation of oleosin-encoding genes combined with increases in the transcriptional activator WRINKLED1 (Li et al., 2015), diacylglycerol acyltransferase (Weselake et al., 2008), and other lipid assembly genes offers better chances of actually driving higher oil production.
Overall, the outcome of our experiments supports the quantal synthesis model proposed for animal embryos (Rafelski and Marshall, 2008). That model predicts that the determination of an organelle’s size depends on the amount of precursor synthesized. In our B. napus transgenic plants, a surplus of oleosins and PC had the effect of inducing the formation of large, oleosin-containing membrane stacks, which replaced the normal protein-filled vacuoles but were not filled with storage lipids. The remarkable plasticity of the seed’s cellular architecture prevents aberrations in the overall seed phenotype and thereby ensures the homeostasis required for sustaining seed viability and growth.
METHODS
Plant Transformation
To simultaneously downregulate the genes encoding PEPC1, PEPC2, napin, and cruciferin, protoplasts recovered from hypocotyls of 2-d-old seedlings of Brassica napus var PPS02-144B were isolated following Glimelius (1984) and then cocultivated for 3 d with Agrobacterium tumefaciens strain C58C1Rif (pGV4000) harboring the binary vector pTJCL032. The pTJCL032 T-DNA contained a chimeric hairpin fragment consisting of a 120-nucleotide segment of PEPC2 (BnaA05g34260D-1), a 103-nucleotide segment of PEPC1 (BnaA08g01200D-1) driven by the Arabidopsis (Arabidopsis thaliana) oleosin promoter (sequence description of promoter in Thomas and Li, 1998), as well as a 179-nucleotide segment of napin (BnaA01g16230D-1) and a chimeric 200-nucleotide segment comprising portions of the genes encoding the P1, P2, and P3 subfamiles of cruciferin, both driven by the B. napus napinA promoter (Stålberg et al., 1993). The relevant sequences are given in Supplemental Table 1. The fragment was isolated from B. napus accession PPS02-144B. In addition, the vector harbored the gene bar to allow for the phosphinothricin-based selection of transformed cells. Following 14 to 20 d in culture, colonies were plated onto K3 medium (Nagy and Maliga, 1976) containing 0.6% (w/v) Seaplaque agarose, 0.1 M Suc, 5 mg/L AgNO3, 250 mg/L carbenicillin, 250 mg/L ticarcillin, 0.5 g/L MES, 20 mg/L phosphinothricin, 0.25 mg/L 2,4-D, 0.025 mg/L naphthaleneacetic acid, and 0.025 mg/L 6-benzylaminopurine. To induce shoots, colonies of diameter 2 to 3 mm were transferred onto Murashige and Skoog medium solidified with 0.5% (w/v) agarose (Invitrogen) and supplemented with 2% (w/v) Suc, 5 mg/L AgNO3, 250 mg/L carbenicillin, 250 mg/L ticarcillin, 0.5 g/L MES, 10 mg/L phosphinotricin, 1 mg/L 6-benzylaminopurine, and 0.5 mg/L naphthaleneacetic acid. Regenerants were potted into soil and raised in a greenhouse. Based on DNA gel blot analysis, only transgenic plants with single inserts were selected for further analysis (Supplemental Figure 10). Three independent transgenic lines were isolated from each primary transformant and were paired with a WTS line. All WTS lines were checked for the absence of hairpin sequences using TaqMan assays (Supplemental Figure 11) according to the procedure described by Martin et al., 2005.
DNA Gel Blot Analysis
Genomic DNA was isolated from four leaf discs according to the cetyl-trimethyl-ammonium bromide protocol (Doyle and Doyle, 1987). Ten milligrams of genomic DNA samples was digested overnight with either EcoRI or EcoRV restriction enzyme (New England Biolabs), and two hybridization membranes were prepared for the transformation events, one for hybridization with the marker gene-specific probe (bar) and the other for hybridization with the target DNA-specific probe (pdk-intron). The digested DNA was separated on a 1% (w/v) Tris-acetate EDTA agarose gel (12 h at 25 V) and transferred to a Hybond-XL membrane (Amersham). For DNA probes, template DNA was PCR amplified by Taq polymerase (Promega), purified using the Qiagen gel purification kit, and radioactively labeled with either [α-32P]dCTP (bar probe) or [α-32P]dTTP (pdk-intron probe) using the High Prime DNA Labeling Kit (Roche). Hybridizations were performed using a standard hybridization protocol (Sambrook and Russell, 2001). After washing, the hybridization membranes were exposed to a Kodak Hyperfilm for 5 d, after which the films were developed.
Plant Growth and Sampling
Plants were grown in a chamber set to deliver a constant temperature of 18°C, a 16-h photoperiod (400 µmol quanta m−2 s−1), and a relative humidity of 60%. The same embryo materials were used for the transcriptomic, proteomic, and metabolomic analyses: developing embryos, sampled 30 to 45 DAF, were harvested, snap-frozen in liquid nitrogen, and stored at −80°C until required.
Assays for PEPC Activity and Respiration
The maximum catalytic activity of PEPC was measured in embryo extracts using an indirect spectrophotometric assay (Rolletschek et al., 2004). Embryonic respiration was deduced from the rate of oxygen consumption: intact embryos harvested at ∼35 DAF were incubated in air-tight flasks (5 embryos per flask, 10 replicates per genotype) following Munz et al. (2017).
Chlorophyll Fluorescence Imaging
The chlorophyll fluorescence generated by freshly sectioned embryos harvested at ∼35 DAF was quantified using an Imaging-PAM chlorophyll fluorometer (Borisjuk et al., 2013). The resulting data relate to the effective quantum yield of PSII (measured at 37 µmol quanta m−2 s−1), used to represent the ETR across the embryo section.
Metabolite, Starch, and Sulfur Analysis of the Immature Embryo
Metabolic intermediates were extracted from frozen immature embryo samples in a chloroform/methanol/water mixture and analyzed using liquid chromatography coupled to MS, as detailed by Schwender et al. (2015). Each sample was measured three times (technical replicates), and each genotype was replicated three times (biological replicates with ∼10 embryos each). The data were quantified using authentic standards and external calibration. Starch contents in the immature embryo were assessed by subjecting the insoluble residue to spectrophotometry, following Borisjuk et al. (2013). Total sulfur was measured using an iCAP 6500 inductively coupled plasma optical emission spectroscope (Thermo Fisher Scientific) combined with the CETAC ASXPRESS PLUS rapid sample introduction system and a CETAC autosampler (CETAC Technologies). A 50-mg sample of freeze-dried, ground embryos was digested in 2 mL of 69% (v/v) HNO3 (Bernd Kraft) using an UltraClave IV microwave device (MLS). Digested samples were made up to 15 mL with deionized water. Element standards (Bernd Kraft) were used as external standards, and yttrium (ICP Standard Certipur, Merck) was used as an internal standard for matrix correction. For each genotype, three replicate samples were measured pooled from ∼50 seeds from three different plants each.
Proteomics
For proteomics, three replicate samples each with ∼50 embryos (35 DAF) were used. Embryo proteomes were profiled by two-dimensional isoelectric focusing/SDS gel electrophoresis following methods provided by Schwender et al. (2015). Visible spots were detected and numbered using Delta2D software. Spots classed as differentially abundant were excised from the gel, and their protein constitution was analyzed using MS. The threshold applied for differential expression was a 1.5-fold difference in the relative spot volume.
Mature Seed Phenotyping
Seeds were harvested at ∼65 DAF and stored at 4°C until required. The lipid content in ∼50-mg seed samples was measured using an MQ60 time domain NMR device (Bruker) following Borisjuk et al. (2013). The seeds’ contents of fiber, starch, and moisture were measured in the same samples using a near-infrared spectroscope (MPA; Bruker), calibrated according to the supplier’s protocol. Total nitrogen and total carbon contents were measured in pulverized samples of mature seeds using an elemental analyzer (vario EL cube; Elementar Analysensysteme) according to the manufacturer’s instructions with acetanilide as an elemental standard.
Histological Procedures
Sections of embryos were stained with Ponceau. For staining, 0.1% (w/v) Ponceau S was dissolved in 5% (w/v) acetic acid. The microscope slides were placed on a heating plate (60°C) and covered with the staining solution. After 2 min, the slides were carefully rinsed with distilled water and dried. Immunostaining based on affinity-purified anti-cruciferin, anti-oleosin, and anti-napin polyclonal antibodies used in 1:100 to 1:500 dilution (Tiedemann et al., 2008) and Alexa488 anti-rabbit antibodies (1:500 to 1:1000) for secondary labeling (catalog no. A-11008; Invitrogen/Thermo Fisher Scientific) was performed following Borisjuk et al. (2013). Transmission electron microscopy including tissue preparation was performed exactly as described previously (Verboven et al., 2013).
Germination Assay
Mature seeds were spread on filter paper moistened with 1 mL of sterile water in a sealed Petri dish and held at 23°C under a 14-h photoperiod (100 µE m−2 s−1). Each dish contained 25 seeds, and each genotype was represented by three dishes. The formation of a visible radicle protrusion was taken as a successful germination.
MRI
MRI was used to generate three-dimensional models of seeds and to map the distribution of lipids. The analysis relied on an Avance III HD 400 MHz NMR spectrometer (Bruker) based on the procedures detailed by Borisjuk et al. (2013). NMR data processing was performed using MATLAB (MathWorks). Segmentation and seed modeling was performed using AMIRA software (FEI Visualization Sciences Group). For details, see Munz et al. (2017).
ESI-MS
Lipids were extracted from both whole seeds (20 to 30 mg) and dissected seed tissues and then subjected to direct-infusion ESI-MS analysis as described by Sturtevant et al. (2017, 2019). Hand-dissected tissue samples were prepared from 20 to 30 mg of seed imbibed under vacuum. The internal standards PC-14:0/14:0 (1,2-dimyristoyl-sn-glycero-3-phosphocholine) and TAG-17:0/17:0/17:0 (glyceryl triheptadecanoate) were added to the samples to quantify PC and TAGs.
MALDI-MS Imaging of Seed Sections
Seeds (40 to 45 DAF) were embedded in 10% (w/v) gelatin held at 40°C. The solidified gelatin block encapsulating a single seed was trimmed and frozen at −80°C and then held at −20°C for 3 d prior to cryosectioning. Seed sections (30 μm thick) were prepared using a CM1950 cryomicrotome (Leica Biosystems) and thaw-mounted on Superfrost Plus microscope glass slides (Fisherbrand, 12-550-15; Waltham). The sections were lyophilized for 2.5 to 3 h and then held under vacuum until required for MS imaging. Sections used to image PC and TAGs were coated with 2,5-dihydroxybenzoic acid, while those used to image PE and PI were coated with 1,5-diaminonaphthalene; the sublimation coating procedure followed Hankin et al. (2007). The sections were then subjected to MALDI-MS imaging using a MALDI-LTQ-Orbitrap-XL mass spectrometer (Thermo Fisher Scientific). The 2,5-dihydroxybenzoic acid-coated sections were analyzed in positive ionization mode and the 1,5-diaminonaphthalene-coated sections in negative ionization mode. The MALDI ionization conditions were as follows: 12 μJ/pulse, 10 laser shots per step, and raster step size of 40 μm. The device was set to a resolution of 60,000 and an m/z scan range of 600 to 1200. Raw data were processed as described by Horn and Chapman (2014) using Metabolite Imager software and plotted as mol%: low values are shown in green and high values in red. The PC images were derived from the [M+H]+ adduct, those for TAGs from the sum of the [M+Na]+ and [M+K]+ adducts, and those for PE and PI from the [M−H]− adduct.
RNA Extraction and RT-qPCR
Total RNA was extracted from developing embryos (harvested at different stages from 22 to 45 DAF), following the protocol supplied with a Spectrum Plant Total RNA kit (Sigma-Aldrich), and then treated with RNase-free DNaseI (New England Biolabs) to remove any contaminating DNA. A 1-μg aliquot of total RNA was converted to single-strand cDNA using a RevertAid First Strand cDNA Synthesis kit (Thermo Fisher Scientific). A 100-ng template of the single-strand cDNA provided the template for RT-qPCR formulated with SYBR Green PCR Master Mix (Invitrogen). The necessary primers, designed using Primer3 software (primer3.ut.ee) to generate an amplicon of size 150 to 200 bp, targeted the 3′-untranslated region sequence of the targeted genes (Supplemental Table 2). The specificity and amplification efficiency of each primer pair were checked by a melting curve analysis. Each reaction was subjected to a 95°C/60-s denaturation, followed by 40 cycles of 95°C/30 s, 55°C/30 s, and 72°C/30 s, and the reactions were completed with a 72°C/10-min final extension. Estimates of transcript abundances, based on three biological replicates, each represented by three technical replicates, were derived by applying the 2−ΔΔCt method (Livak and Schmittgen, 2001). The reference sequence was UBC9 (UBIQUITIN-CONJUGATING ENZYME9; accession No. XM_013800933).
Library Construction and RNA-Seq
RNA was extracted and processed from three samples of embryos (30 DAF) harvested from the transgenic line #580 and the WTS line #581 using the TruSeq RNA Kit (Illumina) following the manufacturer’s instructions. The resulting libraries were sequenced using an Illumina HiSeq 2500 device.
Reference Genomes and Data Analysis
The version 5 genome assembly of B. napus ‘Darmor-bzh’ (Chalhoub et al., 2014) was retrieved from www.genoscope.cns.fr/brassicanapus/data/. This assembly comprises a set of 101,040 protein-encoding gene models. Gene annotation data for the various gene identifiers were obtained from the Universal Protein Resource (www.uniprot.org/). Since only one transcript per locus is predicted to be generated, the analysis did not consider alternative transcripts. As an additional reference, The Arabidopsis Information Resource (TAIR10) genome release (Lamesch et al., 2012) was accessed to retrieve protein sequences (representative gene models; ftp://ftp.arabidopsis.org/home/tair/Sequences/blast_data sets/TAIR10_blastsets/TAIR10_pep_20110103_representative_gene_model_updated.fa), genomic coordinates (ftp://ftp.arabidopsis.org/home/tair/Genes/TAIR10_genome_release/TAIR10_gff3/TAIR10_GFF3_genes.gff), and functional descriptions (ftp://ftp.arabidopsis.org/home/tair/Genes/TAIR10_genome_release/TAIR10_functional_descriptions_20140331.txt). The SynOrths v1.0 package (Cheng et al., 2012; brassicadb.org/brad/tools/SynOrths/) was used to link the protein-encoding genes in the B. napus and Arabidopsis genomes. RNA-seq reads were aligned using bowtie2 v2.2.3 software (Langmead and Salzberg, 2012; www.bioconductor.org), and differential transcription was deduced using the DEseq v1.20.0 package (Anders and Huber, 2010; www.bioconductor.org). The BLAST algorithm v2.2.27+ (Altschul et al., 1997) was used to derive sequence alignments.
Gene Annotation and Read Quantification
Our analysis of RNA-seq data emphasizes higher level aggregation of gene expression over detailed quantification of single gene products. This is meaningful if, for example, the combined transcript abundance originating from different syntelog gene copies is considered to represent the totality of transcript encoding an active enzyme. In addition, as inferring transcript abundances at the gene level is likely a more reliable method than attempting the quantification of individual transcript isoforms (Soneson et al., 2015), it has been assumed that gene expression aggregated onto a higher level might be more robust. These factors have particular relevance to B. napus given its tetraploid status (Chalhoub et al., 2014). The strategy adopted was therefore to first establish orthologies between B. napus and Arabidopsis genes, taking advantage of the SynOrths v1.0 package with B. napus set as the query genome and Arabidopsis as the reference genome; the resulting B. napus RNA-seq read counts were thereby transferred to their Arabidopsis gene orthologs. Inconsistencies arising in the SynOrths output were manually resolved based on BLAST alignments. For this integration of orthology relations, it needs to be considered that SynOrths simplifies the complexity resulting from tandem array duplications in the reference genome by reporting only one gene as a representative for each group of genes in a tandem gene array. Accordingly, we applied the tandem array mapping as obtained from the SynOrths output to the Chalhoub et al. (2014) synteny information. Finally, for those B. napus genes for which no Arabidopsis ortholog was found, nonsyntenic orthology was deduced based on a BLASTP alignment against the Arabidopsis proteome. Orthology was assumed for the best alignment between a B. napus and an Arabidopsis sequence, provided that the e value was <10, the alignment included >40% identity, and the aligned length represented at least 60% of the length of the query sequence. The whole procedure resulted in the association of 95,197 B. napus genes with 21,282 Arabidopsis genes (Supplemental Data Set 2H), with 5843 B. napus genes (5.8%) remaining without any Arabidopsis ortholog.
Alignment of RNA-Seq Reads to Transcripts
High-throughput paired end sequencing reads were aligned against predicted B. napus transcripts using bowtie2 v2.2.3 software, counting only paired end alignments. According to the SAM-format output (“mapping quality”), for 87% of the alignments there was a <1% probability of no correspondence with the read’s true point of origin in the genome. The alignment to 101,040 predicted B. napus transcripts resulted in the detection of 67,885 (67%) of the B. napus protein-encoding genes. All but 1591 B. napus reads (equivalent to 99.5%) were associated with 19,062 Arabidopsis genes.
Differential Transcription
Differential transcription between transgenic and WTS samples was detected using the DEseq package (Anders and Huber, 2010), applying default parameter settings. The read counts were step normalized in order to adjust for differences in sequencing depth between samples (Anders and Huber, 2010). To guard against the possibility that the presence of a small number of highly abundant transcripts introduced a bias following the normalization procedure, the impact of omitting the 22 most highly represented genes from the data set was tested. (For the WTS line no. 581, ∼50% of the reads were derived from genes encoding ∼20 putative 2S and 12S seed storage proteins, while in the transgenic line no. 580, this was the case for only ∼5% of the reads.) The test resulted in only one change in the call of differential transcription (gene BnaC06g21090D). To limit the number of false positives, the Benjamini-Hochberg multiple testing adjustment was included (adjusted P values). As recommended by Schurch et al. (2016), the significance threshold was set as 5% for the adjusted P value and a more than fourfold change in transcript abundance (equivalent to adjusted P < 0.01 and log2 fold change > 2; Supplemental Data Set 2C).
Metabolic Seed Model
Simulations of metabolic flux distributions in developing WTS (line 581) and transgenic (line 580) embryos by flux balance analysis were performed based on bna572 (Schwender and Hay, 2012; Hay et al., 2014), a metabolic model describing growth and storage accumulation in B. napus developing embryos. In the study by Borisjuk et al. (2013), this model was parametrized for in planta-relevant conditions at 30 DAF, specifically into three tissue-specific embryo submodels representing the inner cotyledon, outer cotyledon, and radicle, respectively. In this study, the same in planta developmental stage (30 DAF) is to be modeled, and therefore some essential model constraints were adapted for the former study by essentially superimposing the three submodels according to biomass proportions. This way estimates were derived for the intensity of photosynthetic active light reaching the embryo within the developing silique (photon flux) as well as for the ATP-drain flux, a generic reaction summarizing the impact of non-growth-associated ATP consumption on metabolism. As further model inputs, embryo-specific growth rates were determined from embryo dry weight determinations between 17 and 37 DAF (Supplemental Data Set 5A). The biomass composition of embryo material was determined at 30 DAF (Supplemental Data Set 5B). Organic solvent extraction and liquid:liquid fractionation into chloroform-soluble (lipid), methanol/water-soluble (polar), and insoluble cell polymer fractions as well as elemental analysis were performed as reported by Lonien and Schwender (2009). To integrate lipidomic data into the model (Supplemental Data Set 5C), the lipid metabolism subnetwork of bna572 was expanded for explicit simulation of lipid molecule species. Flux simulations were performed using the COBRA toolbox 2.0.5 (Schellenberger et al., 2011) with TOMLAB CPLEX solver package (version 12.3) implemented in MATLAB (version R2013a; MathWorks). Simulations were done by minimization of substrate uptakes (Suc and Gln) and optimization of the models with function optimizeCBmodel and option allowLoops set to false. The SBML standard-compliant XML model is given in Supplemental Data Set 6.
Calculations and Statistics
Mathematical calculations were performed using Excel 2010 (Microsoft) and statistical analyses using software MATLAB (version R2019b; MathWorks). The significance of differences between mean abundances was tested using one-way ANOVA with α = 0.05. All test results are given in Supplemental Data Set 7.
Accession Numbers
Accession numbers are as follows: Cru1, BnaC07g48660D; Cru2, BnaA02g22500D; Cru3, BnaA01g08350D; Nap2, BnaA01g16180D; Nap3, BnaC01g43250D; Nap4, BnaC01g19300D; PEPC1, BnaA08g01200D; and PEPC2, BnaC03g69540D. Primary transcriptome data are available at the EMBL-EBN database (https://www.ebi.ac.uk/ena) under accession number PRJEB36839. Proteome raw data are available via ProteomeXchange with identifier PXD018919.
Supplemental Data
Supplemental Figure 1. Compositional analysis of mature transgenic seeds (lines 576, 578, 580) versus the respective WTS (lines 577, 579, 581).
Supplemental Figure 2. Germination of transgenic seed carrying an RNAi construct designed to repress genes encoding PEPC, napin and cruciferin.
Supplemental Figure 3. The intracellular structure of early storage stage cells.
Supplemental Figure 4. Intracellular architecture in transgenic line #578 versus its WTS line #579.
Supplemental Figure 5. Distribution of lipid molecular species.
Supplemental Figure 6. The content of phosphatidylcholine (PC) and triacylglycerols (TAGs) of dissected parts of the whole embryo.
Supplemental Figure 7. The level and distribution of membrane lipids in the seed.
Supplemental Figure 8. Proteomic profiling of the embryo.
Supplemental Figure 9. Photosynthetic and respiratory activity in the seed.
Supplemental Figure 10. Southern-blot analysis of transgenic lines.
Supplemental Figure 11. Outcome of TaqMan assays and glufosinate treatment.
Supplemental Table 1. Hairpin sequence information.
Supplemental Table 2. Primers used for quantitative real-time PCR analysis of PEPC, napins, cruciferins and the reference gene UBC9.
Supplemental Movie 1. Representative, three-dimensional models of mature B. napus seeds of transgenic line 580 versus the WTS line 581, based on magnetic resonance imaging.
Supplemental Movie 2. Three-dimensional models of lipid distribution in mature B. napus seeds based on magnetic resonance imaging.
Supplemental Movie 3. Comparison of mature B. napus seeds of transgenic lines 578/580 versus the respective WTS lines 579/581, based on magnetic resonance imaging.
Supplemental Data Set 1. Identification of hairpin targets for the knock-down of cruciferin, napin and PEPC.
Supplemental Data Set 2. Transcriptome data set.
Supplemental Data Set 3. Proteome data set; MS-based identification of differentially expressed proteins.
Supplemental Data Set 4. Levels of metabolic intermediates measured using MS and chromatography.
Supplemental Data Set 5. Flux balance analysis modeling results of the biomass composition, metabolic fluxes and bioenergy cofactors for lipid and protein biosynthesis of embryos (30 DAF).
Supplemental Data Set 6. Model files (SBML and spreadsheet formats) and code for simulation of metabolic flux distributions.
Supplemental Data Set 7. Statistical test results.
DIVE Curated Terms
The following phenotypic, genotypic, and functional terms are of significance to the work described in this paper:
Acknowledgments
We thank Julie Claeys (BASF) for the production of the plant transformation vectors, Volodymyr Radchuk (Leibniz Institute of Plant Genetics and Crop Plant Research [IPK]) and Anne Fiebig (IPK) for support in bioinformatics, Yudelsy A.T. Moya (IPK) for performing the inductively coupled plasma optical emission spectroscopy experiments, Henning Tschiersch (IPK) for help with the PAM imaging, and Steffen Wagner (IPK) and Sabine Herrmann (IPK) for technical support. This work was supported by the U.S. Department of Energy (grant DE-SC0012704 to J.S., H.S., and S.M. and grant DE-SC0016536 to K.D.C.) and by the Deutsche Forschungsgemeinschaft (grants 223207907 and 397750294 to H.R. and L.B.).
AUTHOR CONTRIBUTIONS
L.B. and H.R. designed and carried out the research, analyzed the data with the help of H.-P.B. and P.D., and wrote the article; C.L. and C.K. were responsible for the generation of the proteomic, respiration, and PAM imaging data and sampling; J.S., S.M., and H.S. were responsible for the transcriptomic data and flux balance analysis modeling; A.G. was responsible for near-infrared spectroscopy, biomass fractionation, and statistics; E.M. was responsible for the MRI data; Tr.R. and K.D.C. were responsible for the lipidomic and MALDI imaging data; N.H. was responsible for the metabolomic data; and S.O., Tw.R., and T.B. were responsible for the histological data and structural modeling; K.V.A., S.B., and M.V.A. constructed the transgenics.
Footnotes
Articles can be viewed without a subscription.
References
- Abbadi A., Leckband G.(2011). Rapeseed breeding for oil content, quality, and sustainability. Eur. J. Lipid Sci. Technol. 113: 1198–1206. [Google Scholar]
- Altschul S.F., Madden T.L., Schäffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J.(1997). Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S., Huber W.(2010). Differential expression analysis for sequence count data. Genome Biol. 11: R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barro F., Iehisa J.C.M., Giménez M.J., García-Molina M.D., Ozuna C.V., Comino I., Sousa C., Gil-Humanes J.(2016). Targeting of prolamins by RNAi in bread wheat: Effectiveness of seven silencing-fragment combinations for obtaining lines devoid of coeliac disease epitopes from highly immunogenic gliadins. Plant Biotechnol. J. 14: 986–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borisjuk L., et al. (2013). Seed architecture shapes embryo metabolism in oilseed rape. Plant Cell 25: 1625–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalhoub B., et al. (2014). Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 345: 950–953. [DOI] [PubMed] [Google Scholar]
- Chao H., Wang H., Wang X., Guo L., Gu J., Zhao W., Li B., Chen D., Raboanatahiry N., Li M.(2017). Genetic dissection of seed oil and protein content and identification of networks associated with oil content in Brassica napus. Sci. Rep. 7: 46295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman K.D., Dyer J.M., Mullen R.T.(2012). Biogenesis and functions of lipid droplets in plants. Thematic Review Series: Lipid Droplet Synthesis and Metabolism: From Yeast to Man. J. Lipid Res. 53: 215–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chellamuthu V.R., Ermilova E., Lapina T., Lüddecke J., Minaeva E., Herrmann C., Hartmann M.D., Forchhammer K.(2014). A widespread glutamine-sensing mechanism in the plant kingdom. Cell 159: 1188–1199. [DOI] [PubMed] [Google Scholar]
- Cheng F., Wu J., Fang L., Wang X.(2012). Syntenic gene analysis between Brassica rapa and other Brassicaceae species. Front. Plant Sci. 3: 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crouch M.L., Sussex I.M.(1981). Development and storage-protein synthesis in Brassica napus L. embryos in vivo and in vitro. Planta 153: 64–74. [DOI] [PubMed] [Google Scholar]
- Doyle J.J., Doyle J.L.(1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19: 11–15. [Google Scholar]
- Feria Bourrellier A.B., Valot B., Guillot A., Ambard-Bretteville F., Vidal J., Hodges M.(2010). Chloroplast acetyl-CoA carboxylase activity is 2-oxoglutarate-regulated by interaction of PII with the biotin carboxyl carrier subunit. Proc. Natl. Acad. Sci. USA 107: 502–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gacek K., Bartkowiak-Broda I., Batley J.(2018). Genetic and molecular regulation of seed storage proteins (SSPs) to improve protein nutritional value of oilseed rape (Brassica napus L.) seeds. Front. Plant Sci. 9: 890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Q., Goodman J.M.(2015). The lipid droplet: A well-connected organelle. Front. Cell Dev. Biol. 3: 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glimelius K.(1984). High growth rate and regeneration capacity of hypocotyl protoplasts of some Brassicaceae. Physiol. Plant. 61: 38–44. [Google Scholar]
- Hankin J.A., Barkley R.M., Murphy R.C.(2007). Sublimation as a method of matrix application for mass spectrometric imaging. J. Am. Soc. Mass Spectrom. 18: 1646–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen M., Lange M., Friis C., Dionisio G., Holm P.B., Vincze E.(2007). Antisense-mediated suppression of C-hordein biosynthesis in the barley grain results in correlated changes in the transcriptome, protein profile, and amino acid composition. J. Exp. Bot. 58: 3987–3995. [DOI] [PubMed] [Google Scholar]
- Hay J.O., Shi H., Heinzel N., Hebbelmann I., Rolletschek H., Schwender J.(2014). Integration of a constraint-based metabolic model of Brassica napus developing seeds with 13C-metabolic flux analysis. Front. Plant Sci. 5: 724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman E.M.(2008). Endoplasmic reticulum bodies: Solving the insoluble. Curr. Opin. Plant Biol. 11: 672–679. [DOI] [PubMed] [Google Scholar]
- Herman E.M.(2014). Soybean seed proteome rebalancing. Front. Plant Sci. 5: 437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hope R.G., Murphy D.J., McLauchlan J.(2002). The domains required to direct core proteins of hepatitis C virus and GB virus-B to lipid droplets share common features with plant oleosin proteins. J. Biol. Chem. 277: 4261–4270. [DOI] [PubMed] [Google Scholar]
- Horn P.J., Chapman K.D.(2014). Metabolite Imager: Customized spatial analysis of metabolite distributions in mass spectrometry imaging. Metabolomics 10: 337–348. [Google Scholar]
- Hsieh K., Huang A.H.C.(2004). Endoplasmic reticulum, oleosins, and oils in seeds and tapetum cells. Plant Physiol. 136: 3427–3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang A.H.C.(2018). Plant lipid droplets and their associated proteins: Potential for rapid advances. Plant Physiol. 176: 1894–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohno-Murase J., Murase M., Ichikawa H., Imamura J.(1994). Effects of an antisense napin gene on seed storage compounds in transgenic Brassica napus seeds. Plant Mol. Biol. 26: 1115–1124. [DOI] [PubMed] [Google Scholar]
- Kohno-Murase J., Murase M., Ichikawa H., Imamura J.(1995). Improvement in the quality of seed storage protein by transformation of Brassica napus with an antisense gene for cruciferin. Theor. Appl. Genet. 91: 627–631. [DOI] [PubMed] [Google Scholar]
- Lamesch P., et al. (2012). The Arabidopsis Information Resource (TAIR): Improved gene annotation and new tools. Nucleic Acids Res. 40: D1202–D1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L.(2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q., Shao J., Tang S., Shen Q., Wang T., Chen W., Hong Y.(2015). Wrinkled1 accelerates flowering and regulates lipid homeostasis between oil accumulation and membrane lipid anabolism in Brassica napus. Front. Plant Sci. 6: 1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q., et al. (2017). Genetic enhancement of oil content in potato tuber (Solanum tuberosum L.) through an integrated metabolic engineering strategy. Plant Biotechnol. J. 15: 56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W.X., Liu H.L., Qu Q.(2013). Embryo-specific expression of soybean oleosin altered oil body morphogenesis and increased lipid content in transgenic rice seeds. Theor. Appl. Genet. 126: 2289–2297. [DOI] [PubMed] [Google Scholar]
- Livak K.J., Schmittgen T.D.(2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25: 402–408. [DOI] [PubMed] [Google Scholar]
- Lonien J., Schwender J.(2009). Analysis of metabolic flux phenotypes for two Arabidopsis mutants with severe impairment in seed storage lipid synthesis. Plant Physiol. 151: 1617–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D.P., Christopher D.A.(2008). Endoplasmic reticulum stress activates the expression of a sub-group of protein disulfide isomerase genes and AtbZIP60 modulates the response in Arabidopsis thaliana. Mol. Genet. Genomics 280: 199–210. [DOI] [PubMed] [Google Scholar]
- Martin C., Sheils O., O’Leary J.(2005). Real-time PCR gene analysis In The Science of Laboratory Diagnosis, Crocker J., and Burnett D., eds (Hoboken, New Jersey: Wiley; ), pp. 495–504. [Google Scholar]
- Maruyama-Nakashita A., Nakamura Y., Watanabe-Takahashi A., Inoue E., Yamaya T., Takahashi H.(2005). Identification of a novel cis-acting element conferring sulfur deficiency response in Arabidopsis roots. Plant J. 42: 305–314. [DOI] [PubMed] [Google Scholar]
- Michon G.P. (2019). Spheroids & scalene ellipsoids.http://www.numericana.com/answer/ellipsoid.htm#spheroid.
- Miquel M., Trigui G., d’Andréa S., Kelemen Z., Baud S., Berger A., Deruyffelaere C., Trubuil A., Lepiniec L., Dubreucq B.(2014). Specialization of oleosins in oil body dynamics during seed development in Arabidopsis seeds. Plant Physiol. 164: 1866–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munz E., Rolletschek H., Oeltze-Jafra S., Fuchs J., Guendel A., Neuberger T., Ortleb S., Jakob P.M., Borisjuk L.(2017). A functional imaging study of germinating oilseed rape seed. New Phytol. 216: 1181–1190. [DOI] [PubMed] [Google Scholar]
- Nagy J.J., Maliga P.(1976). Callus induction and plant regeneration from mesophyll protoplasts of Nicotiana sylvestris. Z. Pflanzenphysiol. 78: 453–455. [Google Scholar]
- Nietzel T., Dudkina N.V., Haase C., Denolf P., Semchonok D.A., Boekema E.J., Braun H.P., Sunderhaus S.(2013). The native structure and composition of the cruciferin complex in Brassica napus. J. Biol. Chem. 288: 2238–2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil G., et al. (2018). Dissecting genomic hotspots underlying seed protein, oil, and sucrose content in an interspecific mapping population of soybean using high-density linkage mapping. Plant Biotechnol. J. 16: 1939–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafelski S.M., Marshall W.F.(2008). Building the cell: Design principles of cellular architecture. Nat. Rev. Mol. Cell Biol. 9: 593–602. [DOI] [PubMed] [Google Scholar]
- Rolletschek H., Borisjuk L., Radchuk R., Miranda M., Heim U., Wobus U., Weber H.(2004). Seed-specific expression of a bacterial phosphoenolpyruvate carboxylase in Vicia narbonensis increases protein content and improves carbon economy. Plant Biotechnol. J. 2: 211–219. [DOI] [PubMed] [Google Scholar]
- Sambrook J., Russell D.(2001). Molecular Cloning: A Laboratory Manual.. (Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; ). [Google Scholar]
- Schellenberger J., et al. (2011). Quantitative prediction of cellular metabolism with constraint-based models: The COBRA Toolbox v2.0. Nat. Protoc. 6: 1290–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M.A., Barbazuk W.B., Sandford M., May G., Song Z., Zhou W., Nikolau B.J., Herman E.M.(2011). Silencing of soybean seed storage proteins results in a rebalanced protein composition preserving seed protein content without major collateral changes in the metabolome and transcriptome. Plant Physiol. 156: 330–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurch N.J., Schofield P., Gierliński M., Cole C., Sherstnev A., Singh V., Wrobel N., Gharbi K., Simpson G.G., Owen-Hughes T., Blaxter M., Barton G.J.(2016). How many biological replicates are needed in an RNA-seq experiment and which differential expression tool should you use? RNA 22: 839–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwender J.(2008). Metabolic flux analysis as a tool in metabolic engineering of plants. Curr. Opin. Biotechnol. 19: 131–137. [DOI] [PubMed] [Google Scholar]
- Schwender J., Hay J.O.(2012). Predictive modeling of biomass component tradeoffs in Brassica napus developing oilseeds based on in silico manipulation of storage metabolism. Plant Physiol. 160: 1218–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwender J., Hebbelmann I., Heinzel N., Hildebrandt T., Rogers A., Naik D., Klapperstück M., Braun H.-P., Schreiber F., Denolf P., Borisjuk L., Rolletschek H.(2015). Quantitative multilevel analysis of central metabolism in developing oilseeds of oilseed rape during in vitro culture. Plant Physiol. 168: 828–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada T.L., Hayashi M., Hara-Nishimura I.(2018). Membrane dynamics and multiple functions of oil bodies in seeds and leaves. Plant Physiol. 176: 199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada T.L., Shimada T., Takahashi H., Fukao Y., Hara-Nishimura I.(2008). A novel role for oleosins in freezing tolerance of oilseeds in Arabidopsis thaliana. Plant J. 55: 798–809. [DOI] [PubMed] [Google Scholar]
- Siloto R.M.P., Findlay K., Lopez-Villalobos A., Yeung E.C., Nykiforuk C.L., Moloney M.M.(2006). The accumulation of oleosins determines the size of seed oilbodies in Arabidopsis. Plant Cell 18: 1961–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soneson C., Love M.I., Robinson M.D.(2015). Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000 Res. 4: 1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stålberg K., Ellerström M., Josefsson L.G., Rask L.(1993). Deletion analysis of a 2S seed storage protein promoter of Brassica napus in transgenic tobacco. Plant Mol. Biol. 23: 671–683. [DOI] [PubMed] [Google Scholar]
- Sturtevant D., Dueñas M.E., Lee Y.J., Chapman K.D.(2017). Three-dimensional visualization of membrane phospholipid distributions in Arabidopsis thaliana seeds: A spatial perspective of molecular heterogeneity. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1862: 268–281. [DOI] [PubMed] [Google Scholar]
- Sturtevant D., Romsdahl T.B., Yu X.H., Burks D.J., Azad R.K., Shanklin J., Chapman K.D.(2019). Tissue-specific differences in metabolites and transcripts contribute to the heterogeneity of ricinoleic acid accumulation in Ricinus communis L. (castor) seeds. Metabolomics 15: 6. [DOI] [PubMed] [Google Scholar]
- Sweetlove L.J., Nielsen J., Fernie A.R.(2017). Engineering central metabolism: A grand challenge for plant biologists. Plant J. 90: 749–763. [DOI] [PubMed] [Google Scholar]
- Thomas T., Li Z. (1998). An oleosin 5′ regulatory region for the modification of plant seed lipid composition. https://patents.google.com/patent/WO1998045461A1/en11.
- Tiedemann J., Rutten T., Mönke G., Vorwieger A., Rolletschek H., Meissner D., Milkowski C., Petereck S., Mock H.P., Zank T., Bäumlein H.(2008). Dissection of a complex seed phenotype: Novel insights of FUSCA3 regulated developmental processes. Dev. Biol. 317: 1–12. [DOI] [PubMed] [Google Scholar]
- Ting J.T.L., Lee K., Ratnayake C., Platt K.A., Balsamo R.A., Huang A.H.C.(1996). Oleosin genes in maize kernels having diverse oil contents are constitutively expressed independent of oil contents: Size and shape of intracellular oil bodies are determined by the oleosins/oils ratio. Planta 199: 158–165. [DOI] [PubMed] [Google Scholar]
- Troncoso-Ponce M.A., Kilaru A., Cao X., Durrett T.P., Fan J., Jensen J.K., Thrower N.A., Pauly M., Wilkerson C., Ohlrogge J.B.(2011). Comparative deep transcriptional profiling of four developing oilseeds. Plant J. 68: 1014–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valm A.M., Cohen S., Legant W.R., Melunis J., Hershberg U., Wait E., Cohen A.R., Davidson M.W., Betzig E., Lippincott-Schwartz J.(2017). Applying systems-level spectral imaging and analysis to reveal the organelle interactome. Nature 546: 162–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance V.B., Huang A.H.(1987). The major protein from lipid bodies of maize: Characterization and structure based on cDNA cloning. J. Biol. Chem. 262: 11275–11279. [PubMed] [Google Scholar]
- van Meer G., Voelker D.R., Feigenson G.W.(2008). Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 9: 112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verboven P., Herremans E., Borisjuk L., Helfen L., Ho Q.T., Tschiersch H., Fuchs J., Nicolaï B.M., Rolletschek H.(2013). Void space inside the developing seed of Brassica napus and the modelling of its function. New Phytol. 199: 936–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlroos T., Soukka J., Denesyuk A., Wahlroos R., Korpela T., Kilby N.J.(2003). Oleosin expression and trafficking during oil body biogenesis in tobacco leaf cells. Genesis 35: 125–132. [DOI] [PubMed] [Google Scholar]
- Weselake R.J., et al. (2008). Metabolic control analysis is helpful for informed genetic manipulation of oilseed rape (Brassica napus) to increase seed oil content. J. Exp. Bot. 59: 3543–3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y., Messing J.(2014). Proteome balancing of the maize seed for higher nutritional value. Front. Plant Sci. 5: 240. [DOI] [PMC free article] [PubMed] [Google Scholar]