

Abstract
Stochasticity between independent LC–MS/MS runs is a challenging problem in the field of proteomics, resulting in significant missing values (i.e., abundance measurements) among observed peptides. To address this issue, several approaches have been developed including computational methods such as MaxQuant’s match-between-runs (MBR) algorithm. Often dozens of runs are all considered at once by MBR, transferring identifications from any one run to any of the others. To evaluate the error associated with these transfer events, we created a two-sample/two-proteome approach. In this way, samples containing no yeast lysate (n = 20) were assessed for false identification transfers from samples containing yeast (n = 20). While MBR increased the total number of spectral identifications by ~40%, we also found that 44% of all identified yeast proteins had identifications transferred to at least one sample without yeast. However, of these only 2.7% remained in the final data set after applying the MaxQuant LFQ algorithm. We conclude that false transfers by MBR are plentiful,0020but few are retained in the final data set.
Keywords: match-between-runs, label-free quantitation, two-proteome mixture, false transfer rate
Graphical Abstract
INTRODUCTION
Stochasticity is an important issue for quantitative multirun data-dependent acquisition (DDA) LC–MS/MS experiments as lack of observable evidence does not prove absence.1 Attempts to address this issue have been both chemical and computational.2–4 Chemically, various labeling methods have been developed to analyze multiple MS experiments simultaneously and address missing values.5 Isobaric labeling techniques like tandem mass tags (TMT) and isobaric tags for relative and absolute quantitation (iTRAQ) provide peptide level modification and simplify downstream analysis while increasing sample throughput.5–7 These chemical labels, however, are limited by the number of samples that can be analyzed simultaneously, currently 11 for TMT and 8 for iTRAQ. Attempts to expand the multiplexing capabilities of these compounds often requires computational manipulation and experimental rearrangement (e.g., use of a bridge channel) or the complete redesign of the chemical compound.8,9
Label-free quantitation inherently does not require chemical or metabolic labeling, but is susceptible to stochasticity.2,10 As such, attempting to quantify across independent, label-free LC–MS/MS analyses can result in inaccuracies and missing data. To combat this, several computational methods have been applied.3,10 However, most of these strategies employ imputation to fill in the missing values.
An alternative method to statistical imputation is to perform an identification transfer by leveraging chromatographic and mass-to-charge information. The most popular variation of this technique is the match-between-runs (MBR) algorithm, which is included within the MaxQuant software suite.11,12 Briefly, the MBR algorithm assesses each identified peak in an MS1 spectrum from an LC–MS/MS run and compares its retention time to unidentified peaks in another. An identification is transferred if an unidentified peak with the same properties (e.g., m/z and charge state) is found within a specified retention time window. As retention time is critical for the algorithm to function, the MBR algorithm first realigns compared chromatograms (by default, up to 20 min deviations) before attempting to transfer identifications. Thereby, identifications through peptide-spectrum matches (PSMs) from one run can be transferred to peaks having no tandem MS information in another run. However, this strength also presents the primary difficulty in assessing the accuracy of MBR.
With no tandem MS information, the authenticity of an identification transfer is not guaranteed. The need to validate identification transfers becomes more important as research groups begin to utilize the MBR algorithm for experiments with many runs. For example, a recent publication comparing the proteomes of 29 different healthy human tissues utilized MBR with a total of over 1800 MS/MS RAW files analyzed during the study.13 Previously, work to assess the quality of identification transfers by MBR has utilized alignment of “ID-pairs” to investigate false transfer rate.14 Simply put, peptide identifications in MBR compared LC–MS/MS runs should align closely after MBR chromatogram recalibration; transfers in regions where “ID-pairs” do not align well are assumed to be incorrect. By this method, MBR was found to have between 2% (LC–MS/MS runs analyzed on the same day) and 74% (LC–MS/MS a month apart) false transfers regardless of realignment and recalibration of sample chromatograms.14
Here, we present a novel method utilizing a human-only Sample (H) compared with a human + 10% yeast spike-in Sample (HY) to assess the false transfer rate via the MBR algorithm. To eliminate issues of carry over and to reduce the previously identified effects of column consistency on the MBR analysis, we first analyzed Sample H 20 consecutive times immediately followed by 20 consecutive analyses of the Sample HY. By leveraging Sample H as ground truth due to its single proteome composition, we are able to measure how often a false transfer occurs by counting the number of yeast protein identifications found in Sample H after MBR transfers compared to a standard analysis.
Through this study, we find that while the use of MBR greatly improves the missing values problem, false transfers from the MBR algorithm do occur at a measurable rate. When observing identifications across the experiment, we find that 44% of yeast proteins in the two-proteome sample were incorrectly transferred at least once to the human-only sample. Additionally, most of these incorrect transfers result in one-hit-wonder identifications in line with the belief that false transfers are spurious. However, by processing the MBR data with LFQ enabled in MaxQuant, these spurious transfers were frequently assigned zero or near zero quantification values, thereby preventing incorrect quantitation.
MATERIALS AND METHODS
Human Cell Culture
HCT116 cells were obtained from ATCC and cultured as described previously.15 Briefly, cells were cultured in DMEM supplemented with 10% fetal calf serum and 5% penicillin/streptomycin. Cells were kept at 37 °C with 5% CO2 until harvest. Harvesting occurred after cells reached 80% confluency by visual inspection. After ice-cold PBS wash, cells were lysed on-plate with 1 mL of an 8 M urea lysis solution containing 200 mM EPPS, pH 8.5, and protease inhibitors. Lysate was homogenized by trituration through a 21-gauge needle followed by sedimentation by centrifugation at 21 000g for 15 min. Clarified lysate was flash frozen and stored at −80 °C.
Yeast Samples
Saccharomyces cerevisiae was acquired from Dharmacon and cultured in standard yeast-peptone-dextrose (YPD) media as described previously.16,17 Briefly, when the culture reached mid log phase (measured by optical density of 0.6/mL), the culture was pelleted by centrifugation and resuspended in an 8 M urea lysis solution containing 200 mM EPPS, pH 8.5, and complete-mini, EDTA-free protease inhibitors (Roche). The resuspension was lysed via bead beating in microcentrifuge tubes. Lysate was clarified by centrifugation, flash frozen, and stored at −80 °C.
Sample Preparations
Human cell lysate and yeast cell lysate were prepared for label-free LC–MS/MS analysis following protocols previously described.2 Briefly, a protein level bicinchoninic acid protein assay (Pierce) was performed on the clarified lysates to determine protein concentration. Lysates were then reduced at room temperature in the dark with incubation of 5 mM tris(2-carboxyethly)-phosphine (TCEP), followed by alkylation with 10 mM iodoacetamide to covalently block reactive cysteine groups. The reaction was quenched with the addition of 15 mM dithiotreitol. Blocked lysates were then chloroform– methanol precipitated.
Precipitated proteins were resuspended in 200 mM EPPS pH 8.5 and placed in an orbital shaker at room temperature for overnight digestion with Lys-C at a 1:100 protease:protein ratio (Wako). Sequencing grade trypsin (Promega) was added to the Lys-C digest and incubated for 6 h on an orbital shaker at 37 °C. A Quantitative Colorometric Peptide assay (Pierce) was then performed to measure the concentration of digested peptides present.
At this point, the human sample was split into 2 aliquots of 30 μg each. Three μg of yeast peptides were added to one of the human samples, while an equal volume of HPLC grade water was added to the other sample. Both samples were independently desalted using stop-and-go-extraction tips containing a C18 solid phase.18 Desalted samples were then dried in a vacuum centrifuge and resuspended in a 5% acetonitrile, 5% formic acid buffer for LC–MS/MS analysis.
Liquid Chromatography and Tandem Mass Spectrometry (LC–MS/MS)
Both unfractionated samples were each analyzed 20 times consecutively on an Orbitrap Fusion Lumos mass spectrometer operated in positive-mode with a Proxeon EASY-nLC 1200 liquid chromatograph (Thermo Fisher Scientific) as described previously.2 Peptide fractionation was performed on a 100 μm inner diameter microcapillary column packed with 35 cm of Accucore C18 resin (2.6 μm, 150 Å, Thermo Fisher Scientific). Approximately 1 μg of peptide was loaded onto the column for LC–MS/MS analysis.
Separation occurred across a 90 min gradient from 4% to 35% acetonitrile in 0.125% formic acid. The flow rate was set to 525 nL/min over the gradient. To prevent carry over, the 20 analyses of the human only sample (H) were queued first followed by the 20 analyses of the human + 10% yeast spike-in sample (HY).
A data dependent Top Speed (3 s) method was used to collect spectra: high resolution MS1 spectra (Orbitrap resolution: 120 000; mass range: 350–1400 Th; and automatic gain control (AGC) target: 4 × 105; maximum injection time 50 ms) and high resolution MS2 spectra (Quadrupole isolation window: 1.6 Th; Orbitrap resolution: 7500; HCD energy: 30%; AGC target: 5 × 104; maximum injection time: 22 ms). Dynamic exclusion was enabled with a duration time of 120 s.
Data Analysis
Spectra collected from our 40 LC–MS/MS analyses were analyzed with the MaxQuant software package (Version 1.6.3.4).2,11 Spectra were searched against a concatenated human (Uniprot ID: UP000005640, Downloaded November 8, 2018) and yeast (Uniprot ID: UP000002311, Downloaded November 8, 2018) database. Database reversal for false discovery rate determination using the target-decoy method was performed by MaxQuant.19 To prevent bias, MaxQuant default parameters were used. All 40 LC–MS/MS runs were analyzed simultaneously and given a unique Experiment tag in the “Raw data” upload section of MaxQuant. LFQ was enabled with default settings.
LC–MS/MS runs were analyzed once without and once with MBR enabled. Proteins containing even a single shared peptide between human and yeast databases were removed to avoid artificially increasing the amount of yeast identifications due to common peptides between species (n = 77). To account for leucine/isoleucine isomers, a regular expression search was conducted for peptides containing those amino acids, allowing them to match sequences containing either form of the isomers from both databases. MBR was enabled through the “Identification” subtab in the “Global Parameters” tab of MaxQuant. The default settings for MBR were used (0.7 min match window and 20 min alignment time).
Extracted ion chromatograms (XICs) of peptides were viewed using the Skyline client.20 A peptide list was imported into the software and the “msms.txt” file generated from the MaxQuant software suite was supplied along with the raw files used in this study. Default Skyline settings were used. For Skyline analysis, raw files were aligned and calibrated within Skyline. All graphs generated by Skyline for a given XIC were synchronized such that the x- and y-axes were identical.
RESULTS
Assessing Experimental Stochasticity
We devised a two-sample, two-proteome system consisting of a pure-sample and a mixed-sample to measure how frequently the MBR algorithm incorrectly transfers an identification. The pure sample, Sample H, consisted of a human-only peptide mixture from a digested HCT116 cellular lysate. Sample HY, the mixed sample, was an identical aliquot of Sample H with an additional spike-in of yeast (S. cerevisiae) at 10% of the human lysate mass (Figure 1A). Forty, 90 min, back-to-back LC–MS/MS analyses were performed such that the first 20 analyses were conducted on Sample H (single-proteome sample) and the last 20 analyses were on Sample HY (two-proteome sample) to avoid sample carryover. As such, Sample H serves as ground truth as it is devoid of yeast proteins. The raw data were analyzed twice by MaxQuant, once with MBR off and again with MBR on to assess how many yeast proteins would be identified by matching in the single-proteome sample (Figure 1B).
Figure 1.
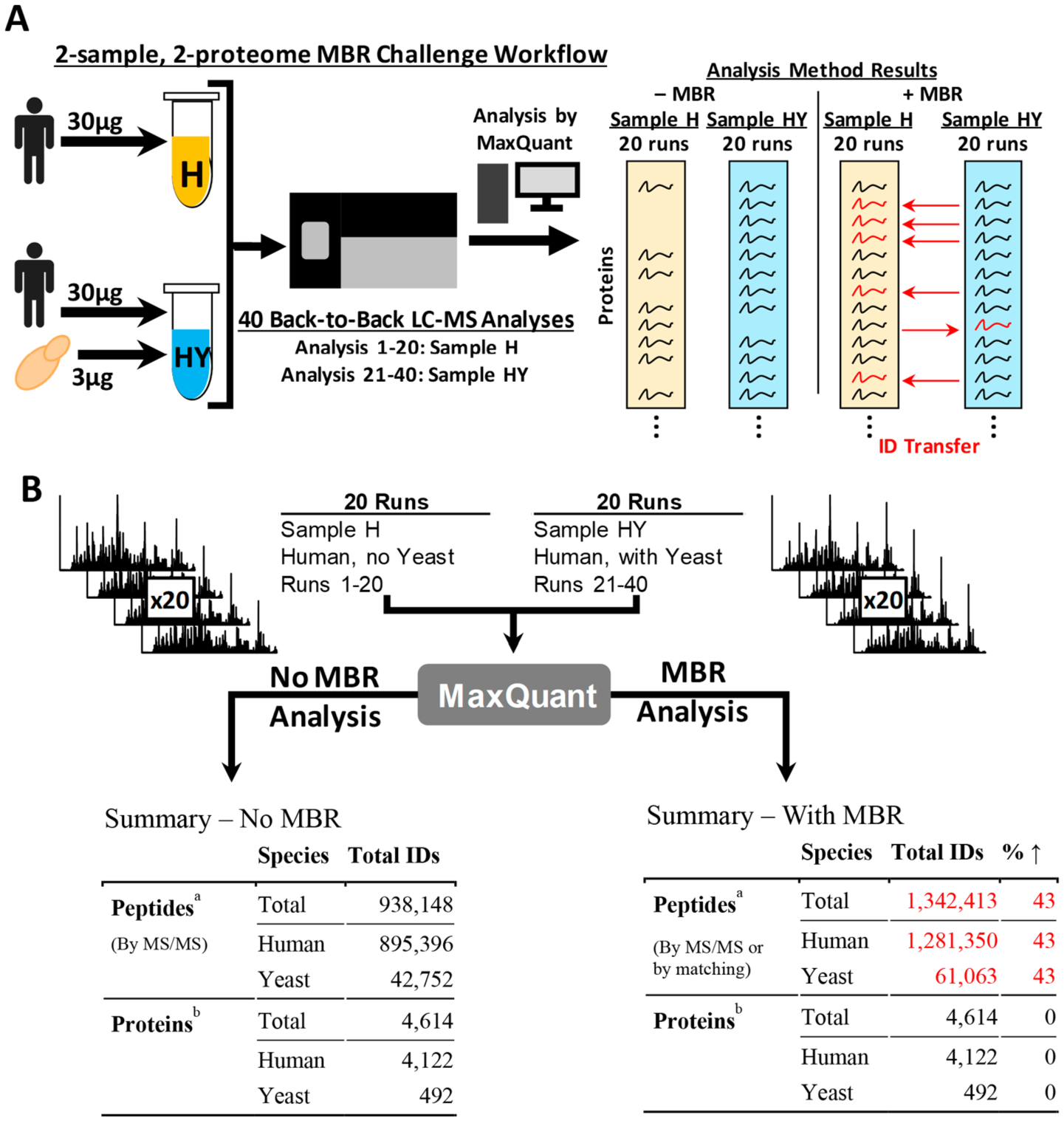
Experiment setup and overview. (A) Diagram of experimental workflow. Sample H (yellow) and HY (blue) were generated from the same human lysate. Sample HY was only differentiated from Sample H by a spike-in of yeast whole cell lysate at 10% of the human lysate mass. Both samples were analyzed 20 times each by LC–MS/MS back-to-back on the same column with analysis of Sample H performed first to prevent carry over. MaxQuant was used to process the LC–MS/MS data with and without MBR. (B) Diagram summarizing the results of the 40 MS/MS runs by MaxQuant analysis type. aPeptides that overlapped the human and yeast databases were marked as human. Peptides were blasted against the human database and allowed to match either leucine and isoleucine isomers. bProteins containing peptides that overlapped the human and yeast databases were excluded from analysis to remove confounding data. Protein groups that included human proteins were marked as human.
To summarize the complete data set and assess stochasticity, we examined the number of total and unique peptide identifications over all MS/MS analyses performed during this study (Figure 1B and Supporting Table S1). Without MBR enabled, 938 148 peptides were identified over the 40 MS/MS analyses split between 895 396 human identifications compared to 42 752 yeast identifications. This translated into 4122 human and 492 yeast proteins. Note that any yeast protein which shared one or more peptides with a human protein was removed. By enabling MBR, peptide identifications were increased by 43%, equally split between both species. Furthermore, no protein identifications were added as MBR does not increase total protein identifications within the data set.
Match-between-Runs Aids in the Completion of Data Sets
To evaluate the effect of MBR on multiple MS/MS experiments, we first looked to establish a baseline identification rate by assessing the number of unique peptides and proteins identified, by species, in each of the 40 MS/MS analyses (Figure 1B, Table 1, Supporting Figure S1). Without MBR, only 57% and 56% of all observed human peptides were detected in each MS/MS analysis of Sample H and HY, respectively (Supporting Figure S1). A similar per run identification rate of 59% was observed for yeast peptides in the 20 MS/MS analyses of Sample HY. By enabling MBR, the peptide level identification rate for human peptides improved to an average of 81% across all 40 analyses, while the average identification rate of yeast peptides in the 20 analyses of Sample HY increased to 83%.
Table 1.
Effect of Match-between-Runs (MBR) on Protein Identifications Per Run
| sample | n | species | no MBR | with MBR | |||
|---|---|---|---|---|---|---|---|
| protein IDs | % totala | protein IDs | % totala | fold ↑b | |||
| all runs | 40 | totalc | 3738 ± 190 | 81 ± 4 | 4290 ± 195 | 93 ± 4.2 | 1.15 ± 0.01 |
| human | 3521 ± 47 | 85 ± 1 | 4023 ± 18_ | 98 ± 0.4 | 1.14 ± 0.01 | ||
| yeast | 217 ± 212 | 44 ± 43. | 267 ± 209 | 54 ± 42. | 4.49 ± 3.61 | ||
| H (human only) | 20 | totalc | 3554 ± 40 | 77 ± 1 | 4097 ± 10 | 89 ± 0.2 | 1.15 ± 0.01 |
| human | 3546 ± 40 | 86 ± 1 | 4037 ± 9 | 98 ± 0.2 | 1.14 ± 0.01 | ||
| yeast | 8±2 | 1.6 ± 0.4 | 60 ± 5 | 12 ± 0.9 | 7.87 ± 1.64 | ||
| HY (human with yeast) | 20 | totalc | 3922 ± 43 | 85 ± 1 | 4482 ± 14 | 97 ± 0.3 | 1.14 ± 0.01 |
| human | 3496 ± 40 | 85 ± 1 | 4009 ± 14 | 97 ± 0.3 | 1.15 ± 0.01 | ||
| yeast | 426 ± 6 | 87 ± 1 | 473 ± 3 | 93 ± 0.7 | 1.11 ± 0.01 | ||
Percent total is calculated with respect to protein identification counts in Figure 1.
Fold increase is calculated per run as the ratio of protein identifications with MBR to that without MBR.
Total protein is the sum of human and yeast proteins (4122 + 492).
A more holistic analysis was conducted at the protein level (Table 1). On average, 3738 of the 4614 proteins (81%), across both species, were identified in each MS/MS run. By species, an average of 85% of the 4122 human proteins were identified per run. This trend was preserved when analyzing Sample H and HY separately. An equivalent average identification rate was observed in Sample HY for yeast proteins (87% of the 494 were identified) while only 1.6% of the yeast proteins were identified in Sample H.
Assessing False Transfer Rates Using a Two-Proteome Model
With the baseline case established, we repeated the analysis with MBR enabled to measure the increase in average identification rate (Table 1). Globally, an average of 4290 proteins (or 93%) were identified per run with 98% of all human proteins being identified per run (up from 85% without MBR) showing an increase in completeness of the data set. Between samples H and HY, this identification rate was preserved for the human proteins −98% and 97%, respectively.
To measure the false transfer rate, we next looked at the yeast proteins transferred between samples H and HY. For Sample HY, the same completeness trend observed for human proteins was found for yeast proteins with an average of 93% of the 492 yeast proteins identified in each analysis. However, the number of yeast proteins identified on average in Sample H increases to 60 proteins out of 492, or 12%. This is an average 7.87-fold increase of yeast protein identifications in a human-only sample.
Furthermore, we utilized the Skyline suite to assess the differences between the XICs of yeast peptide identifications transferred to Sample H by the MBR algorithm. By comparing the yeast peptide XICs from the 20 LC–MS/MS analyses of Sample H to the 20 from Sample HY, we were able to observe that while some incorrectly transferred peptides had little to no signal in the calibrated retention time window in the 20 LC– MS/MS, others contained various incorrect peaks with the appropriate m/z and charge state. For example, yeast peptide IIDDDVPTILQGAK contained no signal in Sample H while peptide FGPIVSASLEK contained several peaks within the identified retention time window (Supporting Figure S2 and S3).
Requiring Identifications in Multiple Runs Reduces Spurious Identifications
Next, we assessed how often any given yeast peptide or protein was identified in the 20 LC–MS/MS runs of Sample H (Figure 2A). Without MBR, only 57 yeast peptides corresponding to 37 proteins were identified in the 20 analyses. When requiring identification in at least 5 runs, this number reduced to 10 proteins. However, analysis with MBR resulted in the identification of 215 yeast proteins in the 20 samples containing only human proteins. Furthermore, when requiring identification in at least 5 runs, 76 yeast proteins were still identified. However, of the 215 yeast proteins identified in at least one MBR enabled analysis of Sample H, 167 transferred identifications were subsequently reported as having 0 intensity by the LFQ algorithm, 36 proteins had been identified previously without MBR, and 12 yeast proteins were transferred and quantified by LFQ as having a non-0 intensity (Figure 2B). As such, while 44% of all yeast proteins were identified in at least one run with MBR enabled, only 2.4% received an intensity value greater than 0 from the downstream LFQ algorithm. This result is due to the default setting of the LFQ algorithm requiring a minimum of 2 peptides per protein for quantitation.
Figure 2.
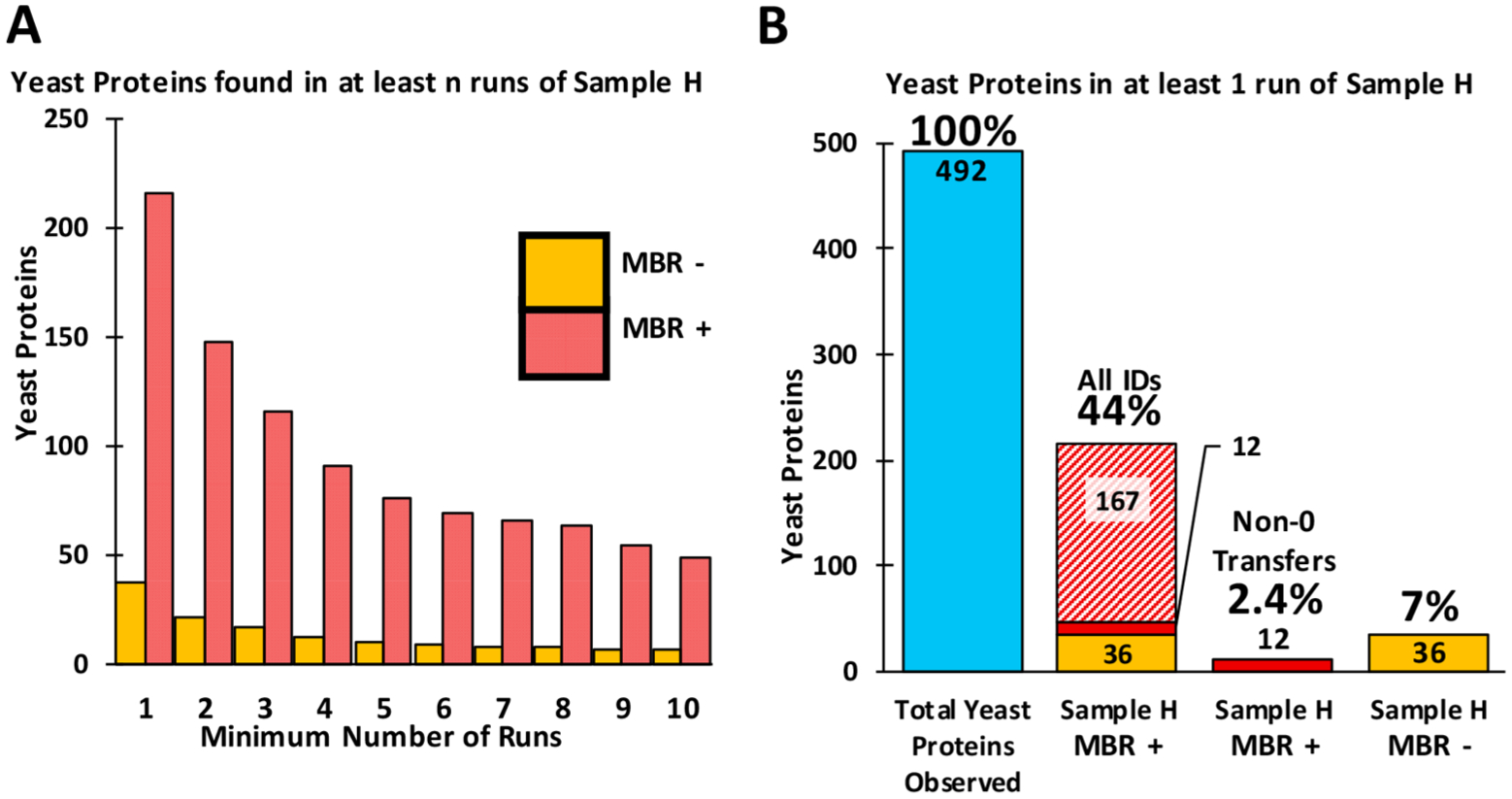
Analysis of yeast protein identifications occurring in sample H (no yeast). (A) Bar plot showing the number of yeast proteins found in n or more runs. (B) Stacked bar plots showing the type of yeast protein identifications occurring in at least one run from sample H. Blue bar shows the number of yeast proteins observed across the 40 MS/MS runs. Yellow bars represent yeast proteins identified constantly irrespective of MBR. Diagonally dashed bar depicts the number of identifications transferred by MBR, but then removed by the downstream LFQ algorithm. Solid red bars show transfers remaining after MBR and downstream LFQ analysis.
Incorrect Identifications by Match-between-Runs Are Quantified Near-0 When LFQ Is Implemented
To evaluate the effect MBR has on label-free quantitation, we assessed further how the MaxQuant LFQ algorithm handled the 12 yeast protein identifications receiving LFQ intensity values greater than 0 when transferred to Sample H. These proteins were not identified by MS/MS during any of the Sample H analyses, but each identification was transferred, on average, to 12 analyses of Sample H (Table 2). Conversely, these 12 yeast proteins were identified by MS/MS in all 20 analyses performed on Sample HY.
Table 2.
Overview of 12 Yeast Proteins Remaining after MBR and LFQ Analysis in Sample H
| protein name | sample H | sample HY | average LFQ ratio H:HY | |||
|---|---|---|---|---|---|---|
| MS/MS ID | matching ID | LFQ quantifieda | MS/MS ID | LFQ quantifieda | all transfers for protein | |
| PABP | 0 | 19 | 6 | 20 | 0.21 | 0.064 |
| HXKA | 0 | 18 | 2 | 20 | 0.09 | 0.009 |
| RL6B | 0 | 19 | 1 | 20 | 0.18 | 0.009 |
| FAS1 | 0 | 11 | 1 | 20 | 0.03 | 0.002 |
| DHE4 | 0 | 15 | 3 | 20 | 0.32 | 0.048 |
| G6PI | 0 | 11 | 1 | 20 | 0.08 | 0.004 |
| ALF | 0 | 4 | 1 | 20 | 0.01 | 0.000 |
| EF3A | 0 | 5 | 1 | 20 | 0.01 | 0.000 |
| PFKA2 | 0 | 11 | 1 | 20 | 0.14 | 0.007 |
| FAS2 | 0 | 14 | 2 | 20 | 0.27 | 0.027 |
| UGPA1 | 0 | 8 | 2 | 20 | 0.36 | 0.036 |
| PURA | 0 | 4 | 1 | 20 | 0.50 | 0.025 |
| average | 0 | 12 | 2 | 20 | 0.18 | 0.019 |
LFQ quantified is defined as a protein assigned an intensity greater than 0 during LFQ analysis.
Despite many Sample H analyses receiving an identification of the 12 proteins by matching, only an average of 2 Sample H analyses per yeast protein contained a transfer that would be quantified as non-0 during LFQ analysis. When analyzing the average LFQ ratio of these quantifiable transfers, an average H:HY ratio was 0.18—indicating that on average, these non-0 assignments at the LFQ level resulted in a quantitation ratio near 1:5. The largest average ratio was 1:2 and observed in the protein PURA, transcriptional activator protein Pur-alpha (Table 2). Meanwhile elongation factor 3A, EF3A, and the alpha-tubulin folding protein had the smallest ratio of 1:100. However, when including transfers that were quantified with an intensity of 0 by LFQ, the average H:HY LFQ ratio of the 12 proteins is near 2:100, indicating a negligible identification.
DISCUSSION
Match-between-runs is a popular approach which partially addresses the problem of stochastic identifications in LC–MS/MS by leveraging chromatographic data. No standardized method, however, is available to assess the false transfer rate when utilizing the MBR algorithm in MaxQuant. Here we present a novel method that utilizes single-proteome and dual-proteome samples to quantitatively measure false transfers. While our data set is biased by primarily focusing on assessing the presence or absence of low abundant proteins, it does mimic the exclusivity of rare proteins when comparing between treatments or tissues.
Our findings suggest that, on average, an ~8-fold increase in incorrect identifications can occur at the protein level when allowing the algorithm to perform matches between 40 independent MS/MS runs −20 of a single proteome sample and 20 of a dual-proteome sample. These spurious identifications are often “one-hit-wonders” and are a result of nonsystematic transfers of peptide identifications.
Despite these shortcomings, MBR is not without its merits. We found that in identical samples, MBR increased the number of peptides identified by an average of 43%. This increase was reduced to 15% at the protein level due to the assignment of multiple peptides to a single protein. As such, 98% of all detected human proteins were observed in all 40 MS/MS runs, up from the initial 86% identification rate without MBR. The result of enabling MBR in identical samples is near-complete identification of all observed proteins in the data set, which alleviates the missing-value problem. Furthermore, enabling LFQ in the MaxQuant algorithm resolved most spurious matches due to false transfer. Although a small subset remained after LFQ analysis, on average these represent a 1:50 ratio between the human-only (H) and human + yeast sample (HY). Until further developments to the MBR algorithm to measure, quantify, and control the false transfer rates are added, it is recommended to utilize MBR in conjunction with some form of postprocessing software that can address false transfers (i.e., LFQ).
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank members of the Gygi Lab at Harvard Medical School for their insightful and encouraging discussions. We specifically would like to thank Brandon Gassaway for lending his expertise with the Skyline Windows Client. This work was funded in part by an NIH/NIDDK grant GM132129 (J.A.P.) and GM67945 (S.P.G).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteome.9b00492.
Table S1: Formatted tables of MaxQuant “peptide.txt” and “proteinGroups.txt” output files separated by species (yeast and human) (XLSX)
Figure S1: Analysis of MBR false transfers as a percentage of peptide identifications; Figure S2: Extracted ion chromatograms for peptide IIDDDVPTILQGAK across 40 runs; Figure S3: Extracted ion chromatograms for yeast peptide FGPIVSASLEK across 40 runs (PDF)
The authors declare no competing financial interest.
RAW files and data for LC–MS/MS experiments have been uploaded to the ProteomeXchange Consortium via the PRIDE partner repository.21 The data set identifier is PXD014415.
REFERENCES
- (1).Stead DA; et al. Information quality in proteomics. Briefings Bioinf. 2007, 9, 174–188. [DOI] [PubMed] [Google Scholar]
- (2).O’Connell JD; Paulo JA; O’Brien JJ; Gygi SP Proteome-Wide Evaluation of Two Common Protein Quantification Methods. J. Proteome Res 2018, 17, 1934–1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Lazar C; Gatto L; Ferro M; Bruley C; Burger T Accounting for the Multiple Natures of Missing Values in Label-Free Quantitative Proteomics Data Sets to Compare Imputation Strategies. J. Proteome Res 2016, 15, 1116–1125. [DOI] [PubMed] [Google Scholar]
- (4).O’Brien JJ; et al. The effects of nonignorable missing data on label-free mass spectrometry proteomics experiments. Ann. Appl. Stat 2018, 12, 2075–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Bantscheff M; Lemeer S; Savitski MM; Kuster B Quantitative mass spectrometry in proteomics: Critical review update from 2007 to the present. Anal. Bioanal. Chem 2012, 404, 939–965. [DOI] [PubMed] [Google Scholar]
- (6).Thompson A; et al. Tandem mass tags: a novel quantificaiton strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem 2003, 75, 1895–1904. [DOI] [PubMed] [Google Scholar]
- (7).McAlister GC; et al. Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. Anal. Chem 2012, 84, 7469–7478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Dephoure N; Gygi SP Hyperplexing: A Method for Higher-Order Multiplexed Quantitative Proteomics Provides a Map of the Dynamic Response to Rapamycin in Yeast. Sci. Signaling 2012, 5, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Braun CR; et al. Generation of Multiple Reporter Ions from a Single Isobaric Reagent Increases Multiplexing Capacity for Quantitative Proteomics. Anal. Chem 2015, 87, 9855–9863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Wiberg HK; et al. Review, Evaluation, and Discussion of the Challenges of Missing Value Imputation for Mass Spectrometry-Based Label-Free Global Proteomics. J. Proteome Res 2015, 14, 1993–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Cox J; et al. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res 2011, 10, 1794–1805. [DOI] [PubMed] [Google Scholar]
- (12).Cox J; et al. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics 2014, 13, 2513–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Wang D; et al. A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol. Syst. Biol 2019, 15, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Bielow C; Mastrobuoni G; Kempa S Proteomics Quality Control: Quality Control Software for MaxQuant Results. J. Proteome Res 2016, 15, 777–787. [DOI] [PubMed] [Google Scholar]
- (15).Lim MY; O’Brien J; Paulo JA; Gygi SP Improved Method for Determining Absolute Phosphorylation Stoichiometry Using Bayesian Statistics and Isobaric Labeling. J. Proteome Res 2017, 16, 4217–4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Paulo JA; O’Connell JD; Gygi SP A Triple Knockout (TKO) Proteomics Standard for Diagnosing Ion Interference in Isobaric Labeling Experiments. J. Am. Soc. Mass Spectrom 2016, 27, 1620–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Paulo JA; Gygi SP A comprehensive proteomic and phosphoproteomic analysis of yeast deletion mutants of 14-3-3 orthologs and associated effects of rapamycin. Proteomics 2015, 15, 474–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Ishihama Y; Rappsilber J; Mann M Modular Stop and Go Extraction Tips with Stacked Disks for Parallel and Multidimensional Peptide Fractionation in Proteomics. J. Proteome Res 2006, 5, 988–994. [DOI] [PubMed] [Google Scholar]
- (19).Elias JE; Gygi SP Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 2007, 4, 207–214. [DOI] [PubMed] [Google Scholar]
- (20).MacLean B; et al. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Perez-Riverol Y; et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019, 47, 442–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.