

Abstract
Unlike many epithelial tissues, the corneal epithelium is insulin insensitive, meaning it does not require insulin for glucose uptake. In this study, we show that insulin differentially regulates mitochondrial respiration in two human mucosal epithelial cell types: insulin-insensitive corneal epithelial cells and insulin-sensitive bronchial epithelial cells. In both cell types, insulin blocks glycogen synthase kinase beta (GSK3β) activity. In the corneal epithelium however, insulin selectively regulates PTEN-induced kinase 1 (PINK-1)-mediated mitophagy and mitochondrial accumulation of insulin receptor (INSR). While insulin blocked basal levels of PINK-1-mediated mitophagy in bronchial epithelial cells, mitochondrial trafficking of INSR was not detectable. We further show that in corneal epithelia, INSR interacts with the voltage-dependent anion channel-1 (VDAC1) in mitochondria and that INSR knockdown triggers robust mitochondrial fragmentation, alterations in mitochondrial polarization, and blocks the induction of PINK-1-mediated mitophagy. Collectively, these data demonstrate that INSR interacts with VDAC1 to mediate mitochondrial stability. We also demonstrate unique interactions between VDAC1 and other receptor tyrosine kinases, indicating a novel role for this family of receptors in mitochondria.
Keywords: bronchial epithelial cells, corneal epithelial cells, insulin, mitochondria, receptor tyrosine kinases, VDAC1
1 ∣. INTRODUCTION
Insulin and insulin receptor (INSR) play a critical role in regulating cell growth through the activation of key signaling pathways involved in cell metabolism, proliferation, and survival.1 The corneal epithelium, a mucosal epithelia that covers the front surface of the eye, has been reported to be insulin insensitive, with insulin sensitivity defined as the need for insulin to promote glucose uptake.2,3 This is due to the presence of a constitutively active glucose uptake transporter (GLUT1). Insulin is present in the precorneal tear film however, and is readily secreted by the lacrimal gland.4,5 While insulin has been shown to regulate proliferation and cell migration in cultured corneal epithelial cells and, at suprathreshold concentrations to promote wound healing in the diabetic cornea, the role of insulin in the homeostasis of this stratified mucosal epithelial surface remains largely undefined.6,7 Our prior work has shown that insulin is required for mitochondrial respiration in corneal epithelial cells, suggesting a new role for insulin and INSR in the regulation of mitochondrial homeostasis.8,9
Mitochondria are respiratory organelles that produce the majority of adenosine triphosphate (ATP) in the cell through aerobic respiration, while a much smaller amount of ATP is produced by anaerobic glycolysis.10 In addition to functioning as a primary energy source, mitochondria produce fatty acids, function as a storage depot for calcium ions, generate heat, and are also known modulators of cell death and survival through the regulation of apoptosis.11,12 Mitochondrial dysfunction contributes to the development of pathological complications during aging and in many diseases, including type 1 and type 2 diabetes mellitus. To protect against disease, mitochondria have developed multiple quality control mechanisms, including fission/fusion and mitophagy. Mitophagy, a form of selective autophagy, functions to remove damaged mitochondria through degradation by autophagolysosomes.13
The glycogen synthase kinase, GSK-3β, is a serine/threonine kinase that has also been associated with diabetes.14-16 Regulation of GSK-3β is phosphorylation dependent. Akt-induced phosphorylation of GSK-3β at Ser9 is known to inhibit GSK3β activity, whereas desphosphorylation of GSK-3β at Ser9, when cells are unstimulated, is associated with cytochrome c release and the induction of apoptosis.17 The voltage-dependent anion channel-1 (VDAC1) localizes to the mitochondrial outer membrane where it regulates protein and metabolite trafficking in and out of the mitochondria.18 Phosphorylation of VDAC1 by GSK-3β modulates mitochondrial activities by interfering with VDAC1 binding to Hexokinase (HK). Disruption of the interaction between HK and VDAC1 frees VDAC1 to bind to pro- or anti-apoptotic B-cell lymphoma 2 (BCL2) family members to trigger or inhibit mitochondrial-mediated apoptosis.18-21
We previously showed that insulin regulates nuclear accumulation of the INSR/insulin-like growth factor type 1 receptor (IGF-1R) hybrid in corneal epithelial cells.8,9,22 In the present study, we now show that insulin also mediates mitochondrial accumulation of INSR, in addition to other tyrosine kinase family receptors. These include IGF-1R, the epidermal growth factor receptor (EGFR), and hepatocyte growth factor receptor (HGFR). We further compare the role of insulin in mediating metabolic activity, mitochondrial stability, and mitophagy, in two mucosal epithelial cell types: human corneal epithelial (hTCEpi) cells and human bronchial epithelial cells (HBECs). Unlike human corneal epithelial cells, human bronchial epithelial cells are considered to be insulin sensitive, meaning that insulin stimulates glucose uptake through the glucose transporter GLUT4.23 In the current study, we show that insulin differentially regulates mitochondrial respiration and homeostasis in corneal and bronchial epithelium. We further identify a novel, direct interaction between INSR and VDAC1 in the mitochondria that is critical to maintaining stability and function of this organelle in corneal epithelia.
2. ∣. MATERIALS AND METHODS
2.1 ∣. Cell lines and primary cultures
The human telomerized corneal epithelial (hTCEpi) cell line used in this study was previously developed and characterized by our laboratory.24 The human telomerized bronchial epithelial cell line (HBEC) were a generous gift from Dr Jerry Shay in the Department of Cell Biology at UT Southwestern Medical Center.25 hTCEpi cells and HBECs were cultured in serum-free keratinocyte basal media containing 0.15 mM calcium and 6.0 mM glucose with supplements at 37°C and 5% CO2 (KGM2, Promocell, Germany). Primary cultures of human corneal epithelial cells (HCECs) were derived from cadaver donors. Donor corneas were obtained from our on campus eye bank (Transplant Services Center, UT Southwestern Medical Center, Dallas, TX) and cultures were generated as previously reported.24 Briefly, corneas were digested in 5 U/mL of dispase (Invitrogen, Carlsbad, CA) overnight at 4°C. Intact epithelial cell sheets were carefully removed and subject to a second digestion in 5 U/mL dispase for 2 hours at 37°C. Cells were then separated by gentle pipetting and seeded onto plastic tissue culture dishes coated with Type IV collagen (Biocoat, BD Biosciences, San Jose, CA). HCECs were cultured in CnT20 cell culture media enriched for progenitor cell culture for 10-15 days (Zen Bio, Research Triangle Park, NC). After their first passage, HCECs were then transitioned to serum-free keratinocyte basal media containing 0.15 mM calcium with supplements at 37°C and 5% CO2 (KGM2, Promocell, Germany). To induce growth factor deprivation, all cell types were cultured in keratinocyte basal media containing 0.15 mM calcium and 6.0 mM glucose without additional supplements (KBM).
2.2 ∣. Real-time metabolic studies
Real-time measurement of cellular oxygen consumption rate (OCR) and intracellular acidification rate (ECAR) was performed using a Seahorse Metabolic Analyzer XFp (Agilent Technologies, Santa Clara, CA). hTCEpi cells and HBECs were seeded onto Seahorse XFp mini-plates at 30 000 cells per well and allowed to adhere overnight. Cells were then cultured in KGM or KBM with or without 5 μg/mL of human recombinant insulin (Sigma, St. Louis, MO) at 37°C and 5% CO2 for 24 and/or 48 hours. Prior to initiating measurements, cells were first incubated for 1 hour at 37°C in Seahorse XF Dulbecco's modified Eagle's medium (DMEM) Base Medium, containing 5 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES, pH 7.4) and supplemented with 1 mM pyruvate, 2 mM glutamine, and 10 mM glucose in a non-CO2 incubator. OCR and ECAR were analyzed using Seahorse XFp Cell Energy Phenotype and XFp Cell Mito Stress Test kits (Agilent Technologies, Santa Clara, CA). All assays were performed according to manufacturer instructions. For the XFp Cell Energy Phenotype Test assay, 10 μM oligomycin was added to inhibit ATP synthase at 18 minutes followed by three separate injections of 1.25, 2.5, and 5 μM carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) at 20-minute intervals to alter mitochondrial membrane potential and allow for maximal oxygen consumption due to uninhibited electron flow through the electron transport chain. Measurements were obtained every 5-7 minutes for a total of 94 minutes. For the XFp Cell Mito Stress assay, the first injection was 10 μM oligomycin followed by one injection of 5 μM FCCP and a final injection of 5 μM of a rotenone/antimycin A mix (R/A). FCCP and R/A are inhibitors of complex I and III, respectively, and function to inhibit mitochondrial respiration. This allows for measurement of non-mitochondrial respiration. For both assays, mean OCR and ECAR values were calculated from the basal measurement at the third time point, prior to the addition of any test compounds. The ratio for OCR/ECAR was also calculated for each experiment. Total pmol per minute of ATP produced were measured using the manufacturer provided Wave software, version 2.6.0 (Agilent Technologies, Santa Clara, CA). All data were normalized per cell number using a Celigo Imaging Cytometer (Nexcelom Bioscience, Lawrence, MA). All assays were performed in hexplicate and repeated a minimum of two additional times.
2.3 ∣. siRNA knockdown
For siRNA experiments, hTCEpi cells and HBECs were seeded at 50%-60% confluence in a 6-well tissue culture plate and grown overnight. Cells were transfected with double-stranded inhibitory RNA oligonucleotides (INSR #GS3643 FlexiTube, Qiagen, Germantown, MD) using Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA) in antibiotic-free basal media. These commercially available oligonucleotides have been tested and validated to confirm the absence of off target effects. Twelve pmol of siRNA oligonucleotides were added to 100 μL antibiotic-free KBM and incubated for 5 minutes at room temperature. siRNAs were then mixed with 2 μL Lipofectamine and allowed to incubate for an additional 20 minutes. After that, the transfection mix was added directly to hTCEpi cells containing 1 mL of KBM and incubated for 24 hours. Following the incubation period, media was removed and cells were then cultured in KGM or KBM for another 24 hours as indicated, with or without 5 μg/mL of human recombinant insulin (Sigma, St. Louis, MO). The AllStars negative control siRNA was used as a non-targeting control (#1027280, Qiagen, Germantown, MD) for all experiments. For metabolic analysis following knockdown, transfected cells were detached with trypsin-EDTA (Gibco, Thermo Fisher, Rockford, IL) and seeded in Seahorse mini-plates (Agilent Technologies, Santa Clara, CA) as described.
2.4 ∣. Plasmid transfection
For GFP-LC3 experiments, hTCEpi cells were seeded at 50%-60% confluence onto 35-mm glass bottom dishes (MatTek Corporation, Ashland, MA) and allowed to adhere overnight. Cells were transfected with a plasmid encoding GFP-LC3 (a generous gift from Dr Ciro Isidoro, UNIPO, Novara, Italy) using Lipofectamine 3000 (Invitrogen, Carlsbad, CA) in antibiotic-free KBM and cultured with or without 5 μg of human recombinant insulin for 24 hours. Five μg of GFP-LC3 plasmid and 10 μL of P3000 reagent were added to 125 μL of Opti-MEM medium (Invitrogen, Carlsbad, CA). Five μL of Lipofectamine 3000 (Invitrogen, Carlsbad, CA) was then diluted in 125 μL of Opti-MEM medium and added to the DNA mix. The resultant mixture was allowed to incubate for 15 minutes, after which the transfection mix was added directly to hTCEpi cells containing 1 mL of KBM and incubated for 24 hours. The media was then removed and cells were cultured in KGM or KBM for another 24 hours as indicated, with or without 5 μg of human recombinant insulin for 24 hours.
2.5 ∣. Mitochondrial fractionation and immunoblotting
To confirm the mitochondrial localization of INSR, hTCEpi cells, HCECs, and HBECs were subjected to mitochondrial and cytoplasmic fractionation. Confluent cells were collected using trypsin-EDTA and washed with phosphate-buffered saline (PBS). A reagent-based mitochondrial fractionation kit was used to isolate the two subcellular fractions (Thermo Fisher, Rockford, IL). Protein concentration was measured using a Qubit 3.0 Fluorometer (Thermo Fisher, Rockford, IL). The cytosolic lysate and mitochondrial pellet were boiled and then electrophoresed through a 4%-15% linear gradient precast polyacrylamide gel (BioRad, Hercules, CA) and immunoblotted as described below.
2.6 ∣. SDS PAGE and immunoblotting
For experiments involving whole cell lysates, hTCEpi cells and HBECs were lysed in 6-well culture plates using lysis buffer containing 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, and a protease and phosphatase inhibitor cocktail (Thermo Fisher, Rockford, IL) on ice for 10 minutes. Lysates were then centrifuged for 10 minutes at 10 000g at 4°C in a microcentrifuge (BioRad, Hercules, CA). Supernatants were removed and boiled for 5 minutes in 2× sample buffer (pH 6.8), containing 65.8 mM Tris-HCL, 26.3% (w/v) glycerol, 2.1% SDS, 5.0% β-mercaptoethanol and 0.01% bromophenol blue (Bio-rad, Hercules, CA). Samples were resolved on a 4%-15% precast linear gradient polyacrylamide gel (Bio-rad, Hercules, CA) and transferred to a polyvinyl difluoride (PVDF) membrane (Bio-rad, Hercules, CA). Membranes were blocked in 5% non-fat milk in TBS 0.1% Tween 20 (Bio-rad, Hercules, CA) for 1 hour at room temperature and incubated in primary antibody overnight at 4°C. The following primary antibodies were used: COX IV #4850, SOD1 #4266, SOD2 # #13141, GSK-3β #9832, pGSK-3β #5558, p-IGF-1Rβ (Tyr1131)/InsulinRβ (Tyr1146) #3021, VDAC1 #4661, PDI #3501, EGFR #4267, HGFR #8198, IGF-1R #3027 (Cell Signaling, Danvers, MA); Insulin Rβ #sc-57342, #sc-711, GAPDH #sc-66163, β-actin # sc-47778, VDAC1 #sc-390996, Na+/K+-ATPase α1 (C464.6) # sc-21712 (Santa Cruz, CA); VDAC/Porin #ab15895; p-GSK3β (Tyr279/Tyr216) #05413 (Millipore, Temecula, CA); LC3B #L7543 (Sigma, St. Louis, MO); and, PINK-1 #BC100-494, p62 #H00008878 (Novusbio, Littleton, CO). Membranes were washed and incubated for 1 hour with an anti-mouse or anti-rabbit secondary antibody (Santa Cruz, CA). Proteins were visualized using ECL Prime Detection Reagent (Thermo Fisher, Rockford, IL) and imaged on an Amersham Imager 600 (Amersham Biosciences, Piscataway, NJ). β-actin was used as a loading control for whole cell lysates. For the fractionation blots, VDAC/Porin and protein disulfide-isomerase (PDI) were used as loading controls for the mitochondrial and cytosolic fractions, respectively. Na+/K+-ATPase α1 was used to confirm the absence of any plasma membrane contamination.
Immunoblots were quantified using ImageQuant TL Toolbox v8.1 software (Amersham Bioscience, Piscataway, NJ). Images were analyzed using area and profile-based tools. The bands of all proteins were normalized using actin. In the case of phosphorylated proteins, the respective total protein was used for normalization.
2.7 ∣. Immunoprecipitation
To test for the presence of INSR and interactions between INSR and VDAC1 in hTCEpi cells, mitochondrial fractions were immunoprecipitated with anti-INSRα (specific for the homo-tetrameric insulin receptor) and anti-VDAC1 monoclonal antibodies (VDAC1 #sc-390996, Insulin Rα #sc-57344, Santa Cruz, CA). A mouse IgG was used as a control (mouse IgG #sc-2025, Santa Cruz, CA). Two micrograms of antibody were incubated with 500 μg of proteins from mitochondrial fractions overnight at 4°C with continuous rocking in a refrigerated environmental chamber (Thermo Fisher, Rockville, IL). Antibody-protein complexes were added to 25 μL immobilized protein A/G plus magnetic agarose beads (Thermo Fisher, Rockville, IL) prewashed with 0.05% Tween-20 in TBS and incubated for 1 hour at room temperature with rocking. Beads were separated from buffer using a DynaMag-2 magnet (Thermo Fisher, Rockville, IL). Precipitates were washed twice in TBS and once in purified water. Next, 60 μL of 4× sample buffer (pH 6.8) containing 0.25 M Tris, 8% lauryl sulfate, 40% glycerol, 20% mercaptoethanol, and 0.04% bromophenol blue were added to the eluate from the immunoprecipitated beads. The supernatant was then collected using a DynaMag-2 magnet (Thermo Fisher, Rockville, IL) and boiled for 5 minutes. The supernatant was subjected to SDS PAGE and immunoblotted as described above. Membranes were blotted for INSR, IGF-1R, EGFR, HGFR, PDI, and VDAC1.
2.8 ∣. Immunofluorescence
For immunofluorescent studies, hTCEpi cells and HBECs were seeded onto 35-mm glass bottom dishes (MatTek Corporation, Ashland, MA) and allowed to adhere overnight. Cells were cultured in KBM with or without 5 μg of human recombinant insulin for 48 hours or 10 minutes in carbonyl cyanide 3-chlorophenylhydrazone (CCCP, Sigma, St. Louis, MO). CCCP was used as a control for mitochondrial depolarization and mitophagy. For INSR siRNA knockdown, cells were transfected with double-stranded inhibitory RNA oligonucleotides as described and cultured for 24 hours in KGM or KBM. After treatment, cells were rinsed with cold PBS, fixed in 1% paraformaldehyde (Electron Microscopy Sciences, Fort Washington, PA) in PBS for 7 minutes, and washed three times with PBS. Next, cells were permeabilized in 0.1% Triton X-100 in PBS for 10 minutes and blocked using 0.5% bovine serum albumin (Sigma, St. Louis, MO) in PBS for 30 minutes. Samples were then incubated in primary antibodies against SOD1, SOD2, INSRβ, COX IV, and cytochrome c at 4°C. Following an overnight incubation, samples were washed in PBS and stained with Alexa Fluor 488 and 555 (Cell Signaling, Danvers, MA) secondary antibodies for 1 hour at room temperature. All samples were mounted on slides using Prolong gold anti-fade reagent containing 4′,6-diamidino-2-phenylindole (DAPI) to label epithelial cell nuclei (Vector Laboratories Inc, Burlingame, CA). Cells were imaged on a Leica SP8 laser scanning confocal microscope (Leica Microsystems, Heidelberg, Germany) using a 63× oil objective. All images were sequentially scanned to avoid spectral crosstalk between channels.
2.9 ∣. Transmission electron microscopy (TEM)
hTCEpi cells and HBECs were seeded onto 35-mm glass bottom dishes (MatTek Corporation, Ashland, MA) and allowed to adhere overnight. Cells were cultured in KGM or KBM with or without human recombinant insulin for 48 hours or transfected as described. Cells were then fixed with 2.5% glutaraldehyde/0.1 M cacodylate buffer pH 7.4 for 15 minutes at room temperature. After three rinses in 0.1 M sodium cacodylate buffer, cells were post-fixed in 1% osmium tetroxide and 0.8% K3[Fe(CN6)] in 0.1 M sodium cacodylate buffer for 1 hour at room temperature. Cells were rinsed with water and stained en bloc with 2% aqueous uranyl acetate overnight. Cells were again rinsed three times with water, samples were dehydrated with increasing concentration of ethanol, infiltrated with Embed-812 resin, and polymerized in a 60°C oven overnight. Blocks were sectioned with a diamond knife (Diatome) on a Leica Ultracut UCT7 ultramicrotome (Leica Microsystems) and collected onto copper grids, post-stained with 2% Uranyl acetate in water and lead citrate. Images were acquired on a JEOL 1400 Plus (JEOL) equipped with a LaB6 source using a voltage of 120 kV.
2.10 ∣. Mitochondrial and lysosomal probes
hTCEpi cells and HBECs were seeded onto 35-mm glass bottom dishes (MatTek Corporation, Ashland, MA) and allowed to adhere overnight. Cells were cultured in KGM or KBM with or without human recombinant insulin for 48 hours or transfected as described. As a depolarization control, cells were incubated with 50 μM CCCP in dimethyl sulfoxide (DMSO) for 10 minutes. Ten minutes prior to the end of the 48-hour treatment period, cells were incubated at 37°C with 10 μg/mL of tetraethylbenzimidazolyl-carbocyanine iodide (JC-1) dye, 200 nM of MitoTracker deep red, 100 nM of MitoTraker green (MTG, Invitrogen, Carlsbad, CA), or and 50 nM of tetramethylrhodamine, ethyl ester (TMRE) (Abcam, Cambridge, MA). The TMRE probe detects the loss of mitochondrial membrane potential. A 50 nM concentration of LysoTracker Red (Invitrogen, Carlsbad, CA) dye was used to stain lysosomes. After incubation with the respective dyes, cells were washed twice in PBS and imaged on a Leica SP8 laser scanning confocal microscope (Leica Microsystems, Heidelberg, Germany) enclosed within an environmental chamber that maintained cells at 5% CO2 and 37°C. A 63× oil objective was used. J-aggregates (multimers) were scanned using a 488 nm excitation laser. The J-monomers, indicating mitochondrial polarization, were scanned using a 568 nm laser. MTG staining was scanned using a 488 nm excitation laser, and TMRE was imaged using a 561 nm excitation laser. For MitoTracker deep red, a 633 nm excitation laser was used. All images were sequentially scanned to avoid spectral crosstalk between channels.
2.11 ∣. Statistical analysis
All data are expressed as mean ± standard deviation. For comparisons between two groups, a two-tailed t test was used. For comparison between three groups, a one-way ANOVA was used with an appropriate post-hoc multiple comparison test. Statistical significance was set at P < 0.05.
3 ∣. RESULTS
3.1 ∣. Insulin regulates mitochondrial respiration in hTCEpi cells and HBECs
Here we compare mitochondrial activity in hTCEpi cells and HBECs using a Seahorse Mito-Stress assay. Both cell lines were cultured in defined keratinocyte growth media (KGM) or in basal media (KBM) with or without 5 μg/mL of insulin for 48 hours. Metabolic activity was characterized by measuring the oxygen consumption rate (OCR) to monitor mitochondrial respiration and extracellular acidification rate (ECAR) to monitor glycolysis. Using this assay, we observed a significant difference in OCR among treatment groups compared to the KGM control for both hTCEpi cells and HBECs (Figure 1A,B). After 48 hours of culture, mitochondrial respiration was significantly reduced in hTCEpi cells cultured in KBM compared to KGM. The addition of insulin to KBM prevented this decrease. A similar decrease in mitochondrial respiration in KBM was also seen in HBECs. Co-treatment with insulin, however, had a much greater effect on HBECs and resulted in a significant increase in mitochondrial respiration compared to the KGM control.
FIGURE 1.
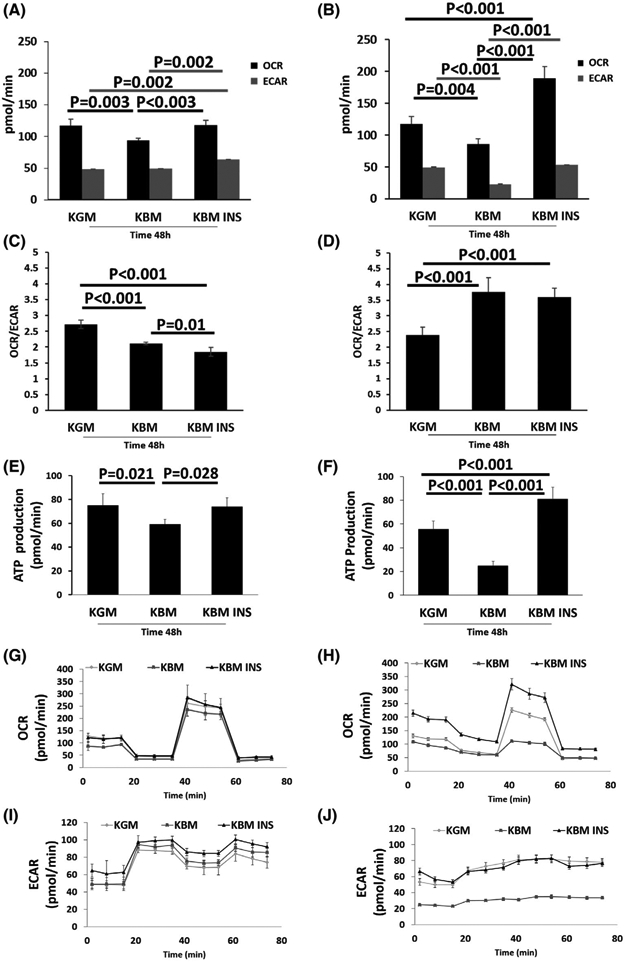
Insulin regulates respiration in human mucosal epithelial cells. hTCEpi cells and HBECs were cultured in growth media (KGM), basal media (KBM), and KBM containing 5 μg/mL of human recombinant insulin (KBM INS) for 48 hours. A,B, OCR and ECAR measurements in hTCEpi cells and HBECs. A, In hTCEpi cells, culture in KBM reduced cellular oxygen uptake (OCR). This decrease was blocked by co-treatment with insulin. Insulin also increased the extracellular acidification rate (ECAR). B, In HBECs, OCR and ECAR both decreased in KBM and were increased with insulin. C,D, The ratio of OCR to ECAR characterizes the metabolic phenotype. C, hTCEpi cells exhibit a more glycolytic phenotype in KBM, which was further increased in the presence of insulin. D, HBECs demonstrated a more respiratory phenotype in KBM that was unchanged by insulin. E,F, ATP production per min for hTCEpi cells and HBECs. ATP levels paralleled respiration for both cell types. G-J, OCR and ECAR in hTCEpi cells (G, I) and HBECs (H, J) plotted as a function of time. Data expressed as mean ± standard deviation from one representative experiment. One-way ANOVA, Holm-Sidak multiple comparisons test. n = 5
The effects of KBM on glycolysis also varied between cell types. In hTCEpi cells, basal levels of ECAR were unchanged between KGM and KBM, but were significantly increased in the presence of insulin (Figure 1A,B). In HBECs however, ECAR was decreased in KBM compared to the KGM control. Co-treatment with insulin blocked this effect. Analysis of the ratio between OCR and ECAR showed a decrease in hTCEpi cells after 48 hours of culture in KBM. The ratio was further decreased in the presence of insulin. This reduction in the OCR/ECAR ratio in hTCEpi cells indicates a metabolic shift toward a higher glycolytic phenotype (Figure 1C). In contrast, HBECs cultured in KBM shifted toward a more respiratory phenotype, evident by the increase in the OCR/ ECAR ratio. The ratio was unchanged by insulin (Figure 1D). We next looked at the effects of these metabolic phenotypes on the molar amount of ATP produced per minute during the assay. Not surprisingly, we found that ATP levels paralleled the OCR data, indicating that the majority of ATP was produced by mitochondrial respiration (Figure 1E,F) in both cell types.
OCR and ECAR were again measured after adding oligomycin to block ATP production through the inhibition of ATP synthase activity, followed by FCCP to stimulate OCR. These measurements were then used to calculate respiratory capacity. Finally, a mix of rotenone and antimycin was added to inhibit mitochondrial complexes I and III, respectively, shutting down mitochondrial respiration. As shown in Figure 1G,H, the maximal respiratory capacity was higher in KGM than KBM in both hTCEpi cells and HBECs. While maximal respiration in hTCEpi cells treated with insulin paralleled those cultured in KGM, HBECs were significantly more responsive to insulin and showed a much greater increase in maximal respiration. Evaluation of ECAR over time further demonstrated that culture in KBM did not alter glycolysis in hTCEpi cells, but glycolysis was increased when co-treated with insulin (Figure 1I). In HBECs, the shape of the ECAR curve was unchanged by any of the injected compounds. ECAR measurements in HBECs cultured in KBM remained flat and reduced compared to the KGM control and the insulin-treated group (Figure 1J).
3.2 ∣. Insulin restores mitochondrial polarization in hTCEpi cells but not in HBECs
To confirm that regulation of metabolic activity by insulin is correlated to a change in mitochondrial structure and function, we analyzed mitochondrial morphology and membrane polarization using two dyes: MitoTracker green (MTG) and TMRE (red). MTG is a mitochondrial marker that can be used to assess morphology. TMRE is used to stain polarized mitochondria. We found that mitochondria in hTCEpi cells were depolarized after 48 hours in KBM (Figure 2A). In the presence of insulin, not only was there no depolarization, but TMRE staining was much brighter, suggesting that mitochondria were hyperpolarized. Hyperpolarization was also associated with mitochondrial elongation (Figure 2A, white arrows), which suggests an increase in mitochondrial respiration. These findings were confirmed using JC-1, a second probe that measures mitochondrial polarization. JC-1 monomers (green) serve as a mitochondrial marker, while JC-1 aggregates (red) indicate mitochondrial polarization. In agreement with our MTG-TMRE data, JC-1 staining also showed a loss of polarization in KBM. Co-treatment with insulin again blocked depolarization and promoted elongation (Supplementary data 1a). In stark contrast to hTCEpi cells, HBECs did not exhibit any changes in mitochondrial morphology or polarization in any of the conditions tested (Figure 2A, Supplementary data 1b). Together, these data highlight disparate roles for insulin in regulating mitochondrial activity in these two cell lines. CCCP was used as control for mitochondrial depolarization and fragmentation for both mitochondrial dyes and cell lines.
FIGURE 2.

Insulin selectively mediates mitochondrial polarization and mitochondrial protein distribution in hTCEpi cells. hTCEpi cells and HBECs were cultured in growth media (KGM), basal media (KBM), and KBM containing 5 μg/mL of human recombinant insulin (KBM INS) for 48 hours. A, MTG (green) and TMRE (red) were used to analyze mitochondrial morphology and polarization. In hTCEpi cells, polarization was decreased in KBM. Co-treatment with insulin blocked depolarization and triggered hyperpolarization, when compared to culture in growth conditions. Elongated mitochondria (white arrows) were visible. HBECs did not display any change in mitochondrial polarization or morphology in any of the treatments tested. CCCP was used as control for depolarization. Scale bar: 10 μm; zoomed images: 5 μm. B, TEM was used to analyze changes in mitochondrial morphology in hTCEpi cells (top row) and HBECs (bottom row). White arrows indicate mitochondria; black arrows indicate autophagic structures). Scale bar: 500 nm. n = 3
To further analyze mitochondrial ultrastructure, we imaged hTCEpi cells and HBECs using transmission electron microscopy (TEM, Figure 2B). In KGM, consistent with our MTG staining, hTCEpi cells displayed healthy, elongated mitochondria suggestive of actively respiring cells, whereas hTCEpi cells cultured in KBM had small, fragmented mitochondria. Double membrane autophagosomal structures were also observed. The presence of insulin in KBM blocked mitochondrial fragmentation and cells appeared elongated, similar to the KGM control (white arrows: mitochondria, black arrows autophagosomal structures). In HBECs, TEM images showed mitochondrial elongation in all culture conditions analyzed; however, despite elongation, in KBM the mitochondria were much thinner. This shift in morphology may explain the decrease in respiration and ATP production in this condition (Figure 1B,F). No autophagosomal structures were seen in HBECs in KBM. When co-treated with insulin, mitochondria exhibited normal thickness and morphology, similar to their appearance in KGM (white arrow: mitochondria).
3.3 ∣. Insulin regulates INSR and cellular distribution of mitochondrial enzymes
We have shown that insulin regulates expression and nuclear localization of IGF-1R and INSR in hTCEpi cells.8,9 Consistent with this, using immunofluorescence, we found the same increase and nuclear (DAPI, blue) accumulation of INSR (green) in hTCEpi cells cultured for 48 hours in KBM (Supplementary data 2a). The presence of insulin in KBM blunted translocation of INSR to the nucleus. We also found that SOD1 (red) shifted to the nucleus with INSR (Supplementary data 2a). Immunofluorescence for SOD2 (green) demonstrated robust nuclear localization (DAPI, blue) with some cytoplasmic staining in hTCEpi cells cultured in KGM. Nuclear SOD2 shifted from the nucleus to the cytoplasm after 48 hours in KBM (Supplementary data 2b). The addition of insulin to KBM triggered the accumulation of SOD2 near the cell border and only partially blocked the shift from the nucleus to other compartments (Supplementary data 2b). In contrast to hTCEpi cells, HBECs did not display any changes in the localization of INSR (green) or SOD1 (red, Supplementary data 2c). However, SOD2 (green) appeared to accumulate in the nucleus in KBM with or without insulin (Supplementary data 2d).
3.4 ∣. Insulin differentially regulates mitochondrial protein expression in hTCEpi cells and HBECs
To elucidate the pathways involved in regulating mitochondrial activity in hTCEpi cells and HBECs, cells were cultured in KBM and the expression and localization of key mitochondrial markers including VDAC, COX IV, SOD1, and SOD2 were analyzed by immunoblotting (Figure 3A,C, Supplementary data 3a,b). Localization of COX IV and cytochrome c was further evaluated using immunofluorescence (Figure 3B,D). In hTCEpi cells, we found a decrease in expression of COX IV and pan-VDAC in KBM. The decrease in these two mitochondrial proteins indicates a reduction in total mitochondria. Treatment with insulin led to the accumulation of both proteins, suggesting that insulin blocked the loss of mitochondria in KBM. Treatment with insulin showed no significant changes in SOD1 and SOD2 following the addition of insulin (Figure 3A, Supplementary data 3a). While immunofluorescent staining for COX IV (green) and cytochrome c (red, Figure 3B) also were decreased in KBM, there was a corresponding change in the distribution of both markers. This suggests a change in mitochondrial morphology and initiation of mitochondrial fragmentation (indicated by the white arrows, Figure 3B).
FIGURE 3.

Insulin regulates mitochondrial protein expression. hTCEpi cells and HBECs were cultured in growth media (KGM), basal media (KBM), and KBM containing 5 μg/mL of human recombinant insulin (KBM INS) for 48 hours. A, Immunoblotting was used to measure the expression of mitochondrial markers COX IV, pan-VDAC, SOD1, and SOD2 in hTCEpi cells. Actin was used as loading control. COX IV and VDAC were decreased in basal media, but accumulated in the presence of insulin. SOD1 was slightly decreased in KBM, while SOD2 was unchanged. While SOD1 was increased with insulin, SOD2 was decreased. n = 3. B, Immunofluorescence was used to analyze expression and localization of the mitochondrial markers COX IV (green) and cytochrome c (red) in hTCEpi cells. COX IV and cytochrome c were decreased in KBM and showed a shift in distribution, indicating mitochondrial damage (white arrow). Cytochrome c was not localized to the cytosol. Co-treatment with insulin blocked the decrease in COX IV and the change in cytochrome c expression and localization. Scale bar: 10 μm. n = 3. C, Immunoblotting for mitochondrial markers in HBECs. In contrast to hTCEpi cells, COX IV, VDAC, SOD1, and SOD2 were increased in KBM and further increased by insulin. n = 3. D, Assessment of HBECs using immunofluorescence showed no changes in COX IV or cytochrome c expression and distribution. Scale bar: 10 μm. n = 3. E,F, Immunoblotting was used to measure the expression of INSR, GSK3β, p-Ser9-GSK3β, and p-Tyr 216-GSK3β. Actin was used as loading control. E, In hTCEpi cells, expression of INSR was increased in KBM but decreased with insulin. GSK3β was less phosphorylated at Ser9 in KBM. Phosphorylation at Ser9 was increased in the presence of insulin. Tyr216 phosphorylation was also decreased with insulin. F, In HBECs, there was a drop in phosphorylation of GSK3β at Ser9 and Tyr216 in KBM. Phosphorylation of Ser9 and Tyr216 were both increased with insulin. n = 3
Collectively, the decrease in mitochondrial respiration measured using the Seahorse assay (Figure 1A), the loss of mitochondrial polarization (Figure 2A), and the decrease in key mitochondrial proteins suggest that there is significant mitochondrial damage in hTCEpi cells after 48 hours of culture in KBM. The addition of insulin is critical to block mitochondrial damage, maintain mitochondrial marker expression, and maintain mitochondrial function similar to cells cultured in growth media (Figure 3A,B). In contrast to this, there was no change in mitochondrial polarization of HBECs after 48 hours in KBM (Figure 2A). Instead, immunoblotting for the same mitochondrial markers showed only an increase in SOD2 after KBM treatment that was further increased by insulin. The mitochondrial marker VDAC was also increased with insulin (Figure 3C, Supplementary data 3b). This may explain the higher metabolic response to insulin in HBECs (Figure 1B). Immunofluorescence for COX IV and cytochrome c showed that expression and localization of these two proteins were unchanged (Figure 3D).
3.5 ∣. Insulin regulates GSK3β activity in hTCEpi cells and HBECs
We previously reported that insulin regulates Akt phosphorylation through IGF-1R in hTCEpi cells. GSK3β is a known substrate for Akt.9 Here we show, by immunoblotting, that phosphorylation of GSK3β at Ser9 is decreased in KBM (Figure 3E, Supplementary data 3c). Ser9 is an inhibitory phosphorylation site for GSK3β. Instead, GSK3β remains phosphorylated at Tyr216, a known activation phosphorylation site (Figure 3E, Supplementary data 3c). In HBECs, as expected from our metabolic data and mitochondrial protein expression, GSK3β activity was also different than what we observed in hTCEpi cells. In KBM, phosphorylation of GSK3β at Ser9 was unchanged, while there was an increase in Tyr216 phosphorylation (Figure 3F, Supplementary data 3d). In both cell lines, the addition of insulin to KBM inhibited GSK3β activity by increasing phosphorylation at Ser9 (Figure 3E,F, Supplementary data 3c,d).
3.6 ∣. Mitochondrial respiration compensates for the reduction in glycolysis during the first 24 hours of growth factor withdrawal in hTCEpi cells, but not in HBECs
Our data indicate that the reduction in respiration seen in hTCEpi cells after 48 hours in KBM is due to mitochondrial damage. To better understand the differences in the metabolic response between hTCEpi cells and HBECs, we analyzed the metabolic activity of these two cell lines at an earlier time point. To accomplish this, cells were cultured as previously described and their metabolic phenotype was measured at 24 hours using a Seahorse Metabolic Analysis Cell Characterization assay. In hTCEpi cells, mitochondrial respiration and glycolysis were different at 24 hours compared with 48 hours. In fact, after 24 hours of culture in KBM, mitochondrial respiration was increased in hTCEpi cells compared to the KGM control. The increase in respiration coincided with a decrease in glycolysis. Interestingly, co-treatment with insulin did not further increase respiration but did increase glycolysis (Figure 4A,C,D). In HBECs however, the metabolic response at 24 hours mirrored the 48-hour response with a decrease in both mitochondrial respiration and glycolysis in KBM. This decrease was not evident following co-treatment with insulin (Figure 4E,G,H). Together, these data suggest that in hTCEpi cells, there is a fuel switch during prolonged culture in KBM that mediates the balance between respiration and glycolysis. This is illustrated by the initial reduction in glycolysis that is seen during the first 24 hours (Figure 4A), followed by recovery at 48 hours to compensate for the reduction in respiration at that latter time point (Figure 1A). This fuel switch was not evident in HBECs.
FIGURE 4.
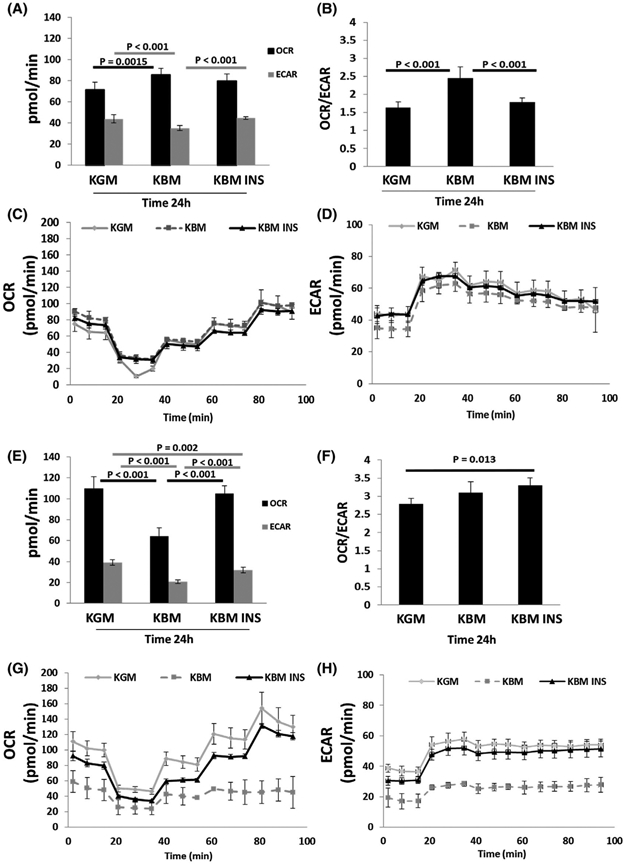
hTCEpi cells increase their respiratory phenotype in the first 24 hours in basal media. hTCEpi cells and HBECs were cultured in growth media (KGM), basal media (KBM), and KBM with 5 μg/mL of human recombinant insulin (KBM INS) for 24 hours. A, Basal OCR and ECAR in hTCEpi cells. Culture in KBM decreased ECAR. This was accompanied by a compensatory increase in OCR. Insulin blocked the changes in KBM. B, The OCR/ECAR ratio in hTCEpi cells was increased in KBM, indicating a shift toward a higher respiratory phenotype. This was blocked by insulin. C, OCR plotted as a function of time in hTCEpi cells. D, ECAR plotted as a function of time in hTCEpi cells. E, Basal OCR and ECAR in HBECs. OCR and ECAR were both decreased in KBM and increased with insulin. F, In HBECs, there was no change in the metabolic phenotype in KBM, but there was a significant increase toward a respiratory phenotype when treated with insulin. G, OCR plotted as a function of time in HBECs. H, ECAR plotted as a function of time in HBECs. Data shown as mean ± standard deviation from one representative experiment. One-way ANOVA, Holm-Sidak multiple comparisons test. n = 3
The combination of an increase in respiration and a corresponding drop in glycolysis at 24 hours shifted the OCR/ ECAR ratio in hTCEpi cells toward a more respiratory phenotype. This phenotype shifted back toward glycolytic when cultured with insulin (Figure 4B). In HBECs, unlike the 48-hour time point (Figure 1D), 24 hours of culture in KBM did not shift cells toward a more respiratory phenotype, but required insulin to increase the OCR/ECAR ratio (Figure 4F).
OCR and ECAR were again measured after the addition of oligomycin to block ATP production by inhibiting ATP synthase activity. FCCP was then added to stimulate OCR and calculate the maximal respiratory capacity. As shown in the OCR line graph in Figure 4C, the basal respiratory capacity in hTCEpi cells was higher in KBM than the KGM control and unchanged by insulin in the first 24 hours. In HBECs on the other hand, the basal OCR decreased in the first 24 hours of culture in KBM. This drop in OCR was blocked by treatment with insulin (Figure 4G). In contrast to OCR, there was a decrease in ECAR in KBM for both hTCEpi cells and HBECs. Again, this drop in glycolysis was blocked by co-treatment with insulin for both cell types (Figure 4D,H).
We have shown significant mitochondrial damage and dysfunction in hTCEpi cells after 48 hours in KBM, while mitochondria appeared healthy and functional at the earlier time point. To investigate the effects of culture in KBM on hTCEpi cells at 24 hours and evaluate mitochondrial quality control, we immunoblotted for key mitophagy markers: LC3-II/LC3-I and p62. We also measured changes in the mitochondrial proteins COX IV and VDAC (Figure 5A-D, Supplementary data 4). After 24 hours in KBM, both hTCEpi cells and HBECs showed an increase in the transport protein p62 and in the autophagosome marker LC3-II, suggesting an increase in autophagy. To further investigate the potential increase in autophagic flux, we used Bafilomicin-1 (Baf-1). Baf-1 blocks autophagic fusion and autophagolysosomal degradation. After blocking with Baf-1, we measured total accumulation of autophagosomes in each culture condition. We found that by blocking autophagic fusion, we inhibited p62 degradation and increased the accumulation of autophagosomes in KBM, confirming an increase in autophagy in both cell lines. As shown in Figure 5A,C and Supplementary data 4a,b, neither p62 nor LC3-II were proportionally increased following co-treatment with insulin, compared to the other treatments. These data indicate that autophagy is activated in response to growth factor deprivation (as seen in KBM) and inhibited by insulin.
FIGURE 5.

Induction of mitophagy regulates mitochondrial function in hTCEpi cells. hTCEpi cells and HBECs were cultured in growth media (KGM), basal media (KBM) and KBM with 5 μg/mL of human recombinant insulin (KBM INS) for 24 hours with or without 10 nM of bafilomycin (Baf-1). A-D, Immunoblotting was used to measure the expression of p62, LC3-I/LC3-II, PINK-1, COX IV, and VDAC. Actin was used as loading control. A, In hTCEpi cells, p62 and LC3-II were increased in KBM with and without insulin. Blocking fusion of the autophagosome with Baf-1 confirmed the increase in autophagic flux in KBM. Autophagic flux was blocked by insulin. B, In hTCEpi cells, PINK-1 was increased in KBM and decreased with insulin, whereas COX IV accumulated in KBM with and without insulin. VDAC was unchanged. C, Similar to hTCEpi cells, p62 and LC3-II were increased in HBECs in KBM independent of insulin. D, In HBECs, PINK-1 was unchanged in KBM and decreased with insulin. COX IV and VDAC accumulated in KBM with and without insulin. n = 3 E, In hTCEpi cells, MitoTracker (blue) was used to stain mitochondria and TMRE (red) was used to stain polarized mitochondria in GFP-LC3-transfected cells. In KBM, there was an increase in the number of GFP-LC3-positive puncta in hTCEpi cells and circles (white arrows) and mitochondria were still polarized. Co-treatment with insulin blocked this process. A GFP empty vector (GFP-θ vector) was used as a control. Scale bar: 10 μm. n = 3
To determine whether the increase in autophagy was due to an increase in mitophagy and not canonical macroautophagy, we next measured levels of PINK-1, an outer mitochondrial membrane protein that is stabilized by mitochondrial depolarization (Figure 5B,D, Supplementary data 4c,d). In hTCEpi cells, PINK-1 was increased in KBM (Figure 5B, Supplementary data 4c). Together, these data suggest that PINK1-dependent mitophagy is activated in the first 24 hours of culture in KBM. As shown in Figure 5B, insulin blocked the increase in PINK-1 leading to an accumulation of COX IV and VDAC (however, the accumulation is not statistically significant). This increase in PINK-1 was not seen in HBECs cultured in KBM (Figure 5D). Consistent with hTCEpi cells, the presence of insulin decreased PINK-1 and increased COX IV and VDAC, indicating a block in PINK-1-dependent mitophagy (Figure 5D, Supplementary data 4d).
To confirm activation of mitophagy in hTCEpi cells, we transfected cells with a GFP-LC3 expression plasmid (green) and stained mitochondria with MitoTracker (blue) and TMRE (red) (Figure 5E). In KGM, GFP-LC3 localized diffusely throughout the cytoplasm, indicating the presence of GFP-LC3-I. In KBM, GFP-LC3 was localized in discrete puncta. These puncta indicate the presence of GFP-LC3-II. Thus, the observed increase in GFP-LC3-II puncta at 24 hours of culture in KBM indicates an increase in autophagosomes. In contrast to puncta, the formation of GFP-LC3 circles, indicated by white arrows, has been previously associated with mitophagy induction in hepatocytes during nutrient deprivation (Figure 5E).26 Consistent with our PINK-1 findings, the number of puncta were decreased when cells were co-treated with insulin. These data confirm that insulin is blocking mitophagy in hTCEpi cells.
3.7 ∣. INSR regulates mitochondrial function in hTCEpi cells, but not in HBECs
To verify whether insulin is regulating mitochondrial activity through INSR, we transfected cells with siRNA oligonucleotides targeting INSR or non-targeting control siRNA oligonucleotides. Following knockdown, cells were then cultured in KGM or KBM for 24 hours and mitochondrial morphology and polarization was visualized using confocal imaging for MTG and TMRE (Figure 6). In hTCEpi cells, MTG-TMRE staining showed a slight decrease in polarization after 24 hours in the KBM control, but there were no obvious changes in mitochondrial morphology (Figure 6A). INSR knockdown in KGM induced mild depolarization and a slight shift in morphology (Figure 6A), whereas INSR knockdown in KBM induced robust depolarization and ring/donut shaped mitochondrial fragmentation in a large proportion of cells. In remaining non-fragmented cells, mitochondria were hyperpolarized, indicated by the strong yellow signal (Figure 6A). In HBECs, knockdown of INSR in KBM failed to alter membrane polarization or mitochondrial morphology (Figure 6B).
FIGURE 6.

INSR regulates mitochondrial polarization in hTCEpi cells. hTCEpi cells and HBECs were treated with siRNA oligonucleotides targeting INSR in KBM for 24 hours. A,B, Mitochondria were stained with MTG (green) and TMRE (red) to measure morphology and polarization, respectively. A, After INSR knockdown in hTCEpi cells cultured in KBM, mitochondrial morphology took on a ring/donut shape (mitochondrial fragmentation). Some hyperpolarized mitochondria were also detectable. 2X zoomed image of hTCEpi cells after INSR knockdown also shown. B, In HBECs, there were no changes in mitochondrial morphology and/or polarization with or without INSR. Scale bar: 10 μm. n = 3
To determine whether the change in mitochondrial morphology observed in KBM was associated with changes in mitochondrial respiration, we next used a Seahorse Mito-Stress assay to measure metabolic activity. OCR and ECAR were analyzed in cells transfected with INSR or non-targeting control siRNA oligonucleotides for 24 hours. In hTCEpi cells, OCR and ECAR were proportionally decreased in response to INSR knockdown (Figure 7A). The OCR/ECAR ratio was unchanged (Figure Supplementary data 5c). These data suggest that INSR modulates both glycolysis and respiration in corneal epithelial cells. Consistent with the decreases in glycolysis and respiration, ATP levels (Figure 7B), maximal respiratory capacity (Figure 7C,D), ECAR, and the spare respiratory capacity (Supplementary data 5a,b) were also decreased. To confirm ultrastructural changes in mitochondrial morphology, we imaged mitochondria using TEM (Figure 7E,F). Consistent with the immunofluorescent findings, mitochondria were smaller and showed signs of degradation following INSR knockdown (Figure 7E,F).
FIGURE 7.
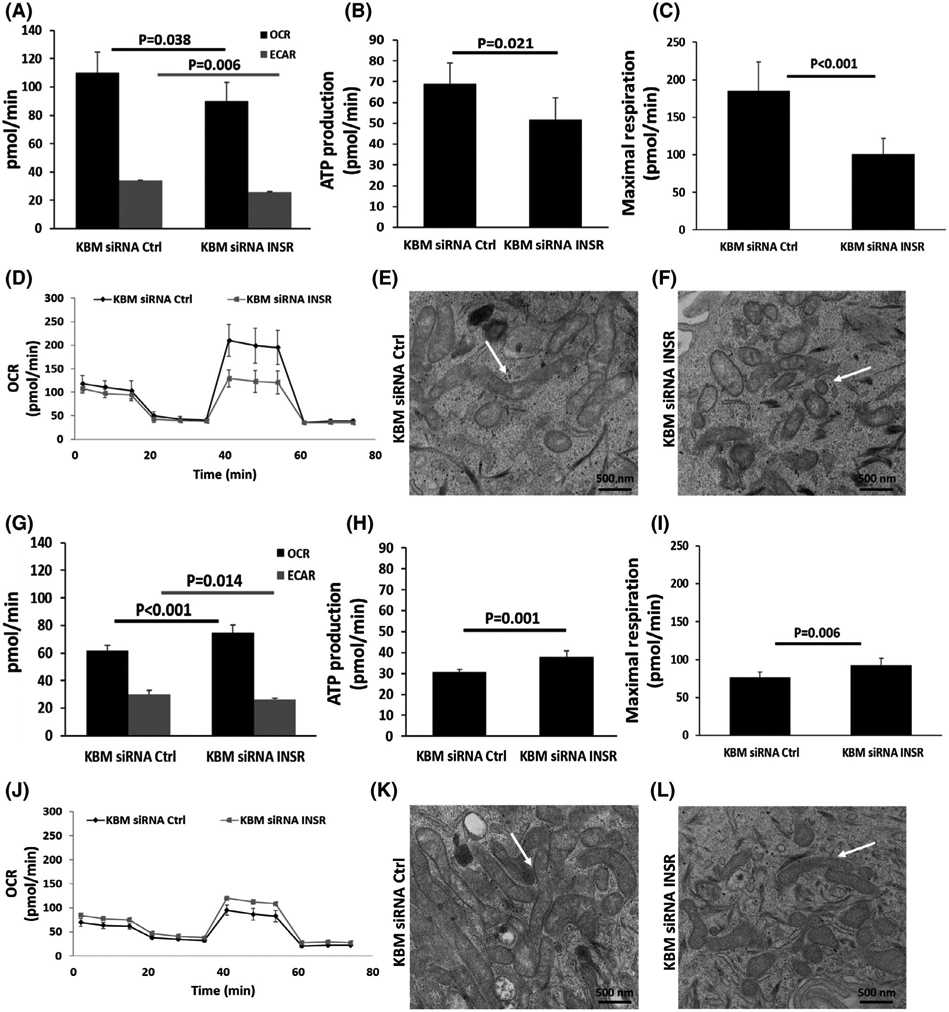
INSR regulates mitochondrial function in hTCEpi cells. hTCEpi cells and HBECs were treated with siRNA oligonucleotides targeting INSR in basal media for 24 hours. A-D, In hTCEpi cells, (A) OCR and ECAR were decreased in KBM after INSR knockdown. B, ATP production per minute was decreased, and (C) the maximal respiratory capacity was also decreased. D, Representative line graph of OCR over time. E,F, TEM showing changes in mitochondrial morphology after INSR knockdown in hTCEpi cells cultured in KBM. E, Healthy mitochondria are visible in controls cells transfected with siRNA non-targeting oligonucleotides (white arrow); F, altered mitochondrial morphology in cells after knockdown of INSR. Scale bar: 500 nm. G-J, In HBECs, (G) OCR increased and ECAR decreased in KBM after INSR knockdown. H, ATP production per minute was increased, as was the (I) maximal respiratory capacity. J, Representative line plot of OCR as a function of time. K,L, TEM showing mitochondrial structure in HBECs. K, Healthy mitochondria were visible after transfection with the control siRNA oligonucleotide. L, Morphological changes were evident in mitochondria after INSR knockdown. Despite obvious mitochondrial thinning, mitochondria remained elongated and functional. Data represented as mean ± standard deviation from one representative experiment, t-test. n = 3
In contrast to hTCEpi cells, in HBECs, there was an increase in OCR after INSR knockdown, while ECAR was significantly reduced (Figure 7G). This shifted the OCR/ECAR ratio toward a more respiratory phenotype (Supplementary data 5f). As expected, there was an increase in ATP due to the increase in OCR (Figure 7H). The OCR/ECAR line graphs also showed a consistent increase in basal respiration and maximal respiratory capacity (Figure 7I,J). While respiration was increased in HBECs after INSR knockdown, there was a corresponding decrease in glycolysis and no detectable change in spare respiration capacity (Supplementary data 5d,e). TEM analysis of HBECs also showed some ultrastructural differences. Specifically, some mitochondria appeared elongated, whereas others were smaller after INSR knockdown compared to controls (Figure 7K,L). Despite these changes, mitochondria remained polarized, suggesting that the mitochondria were still functional.
3.8 ∣. INSR regulates mitochondrial proteins expression and distribution in hTCEpi cells
To further demonstrate that INSR mediates mitochondrial function in hTCEpi cells and not HBECs, we knocked down INSR in KGM and KBM for 24 hours and analyzed the expression of mitochondrial markers by immunoblotting and immunofluorescence (Figure 8, Supplementary data 6). Immunoblotting for INSR was used to confirm knockdown (Figure 8A,C, Supplementary data 6). We found that mitochondrial marker expression was unchanged in the siRNA control in both KGM and KBM after 24 hours (Figure 8A, Supplementary data 6a). INSR knockdown in KBM, however, led to a decrease in the mitochondrial markers, pan-VDAC, COX IV, SOD 1, and SOD 2, by immunoblotting (Figure 8A, Supplementary data 6a). Immunofluorescence also showed a corresponding change in the distribution of COX IV and cytochrome c, suggesting differences in mitochondrial morphology (Figure 8B). No cytochrome c release into the cytosol was noted. Unlike hTCEpi cells, in HBECs, the loss of INSR did not alter expression or distribution of mitochondrial proteins in KBM (Figure 8C,D, Supplementary data 6b).
FIGURE 8.

INSR regulates mitochondrial protein expression and localization. hTCEpi cells and HBECs were treated with siRNA oligonucleotides targeting INSR in KBM for 24 hours. A, To determine the expression of mitochondrial markers in hTCEpi cells, we immunoblotted for VDAC, COX IV, SOD1, and SOD2. Immunoblotting for INSR was used to confirm knockdown. Actin was used as loading control. Immunoblotting showed a decrease in mitochondrial proteins cultured in KBM following INSR knockdown. B, Immunofluorescence was used to analyze expression and localization of the mitochondrial markers COX IV (green) and cytochrome c (Cyto C, red). In hTCEpi cells, COX IV and cytochrome c were decreased in KBM after INSR knockdown. Mitochondrial morphology was also altered, indicating mitochondrial damage. Scale bar: 10 μm. C, Analysis of mitochondrial markers in HBECs did not show a similar effect in KBM. D, Using immunofluorescence, there were no changes in COX IV or cytochrome c expression or localization in HBECs. Scale bar: 10 μm. n = 3
We next investigated GSK3β. In hTCEpi cells, activation of GSK3β was unaltered following INSR knockdown, as there were no changes in the phosphorylation status of either Ser9 or Tyr216 (Supplementary data 7a). In contrast to this, in HBECs, INSR knockdown decreased phosphorylation of both the activation and inhibitory sites (Supplementary data 7b). These findings suggest that phosphorylation of GSK3β in HBECs is mediated by INSR, while in hTCEpi cells, GSK3β is regulated by non-INSR pathways.
We next examined the impact of INSR on mitophagy. In hTCEpi cells, there was an increase in mitophagy after 24 hours in KBM (Figure 5). To determine whether INSR was affecting mitophagy, we knocked down INSR for 24 hours in KBM with or without Baf-1 and measured expression of autophagy markers p62, LC3-II, and PINK-1 (Figure 9A, Supplementary data 8). We found an increase in p62, LC3-II, and PINK-1 in KBM that was confirmed by blocking autophagosome degradation with Baf-1. Thus, Baf-1 treatment demonstrated a high level of autophagy in KBM that was completely blocked by INSR knockdown (Figure 9A, Supplementary data 8). We also found that INSR was necessary for mitophagy in hTCEpi cells using the mitochondrial marker MTG (green) and co-labeling with the lysosomal marker LysoTracker (red). This confirmed fusion of the autophagosome containing mitochondria with the lysosome (Figure 9B) in our transfection control (white arrows, Figure 9B). After INSR knockdown, as expected, mitochondria were fragmented, there were fewer lysosomes, and of the lysosomes that were present, we were unable to detect any co-localization between lysosomes and mitochondria (Figure 9B). Using TEM, we further confirmed the presence of autophagolysosomes (Figure 9C black arrow) in the KBM control. After siRNA knockdown of ISNR however, there were distinct morphological changes in mitochondria. We failed to detect any autophagolysosomes in these samples. Since mitophagy is an essential quality control mechanism, the absence of autophagolysosomes suggests that there is an accumulation of damaged mitochondria that are not being effectively cleared from corneal epithelial cells following loss of INSR (Figure 9D).
FIGURE 9.

INSR regulates mitophagy and accumulates in mitochondria in hTCEpi cells. hTCEpi cells were treated with siRNA oligonucleotides targeting INSR in KBM for 24 hours with or without 10 nM of bafilomycin (Baf-1). A, Immunoblotting for PINK-1, p62 and LC3 was used to show autophagic flux. Actin was used as loading control and blotting for INSR was used to confirm knockdown. A siRNA non-targeting oligonucleotide was used as transfection control. INSR knockdown increased LC3-II and p62 but decreased PINK-1. Baf-1 blocked autophagic flux and showed accumulation of LC3-II, p62 and PINK-1. Accumulation was completely blocked by INSR knockdown. n = 3. B, MTG (green) was used to stain mitochondria and LysoTracker (red) to stain lysosomes. In KBM, there was co-localization of MTG and LysoTracker (white arrows). INSR knockdown blocked co-localization between MTG and LysoTracker, decreased the number of lysosomes, and induced mitochondrial fragmentation. Scale bar: 10 μm. n = 3. C,D, TEM was used to show the absence of autophagolysosomes containing mitochondria in hTCEpi cells after INSR knockdown in KBM. C, Autophagolysosomes were visible in the siRNA non-targeting control (white arrows: mitochondria; black arrows: autophagolysosome). D, Autophagosomes and autophagolysosomes were absent following siRNA for INSR. Scale bar: 300 nm. hTCEpi cells, HCECs, and HBECs were cultured in growth (KGM) and basal media (KBM) for 48 hours and separated into cytosolic (C) and mitochondrial fractions (M). E, Immunoblotting for INSR in hTCEpi cells showed accumulation of the receptor in mitochondria in KBM. P-INSR immunoblotting also showed a decrease in phosphorylation of INSR in the mitochondrial fraction. E, The presence of INSR in the mitochondrial fraction was confirmed in HCECs. F, In HBECs, INSR was undetectable in the mitochondria. VDAC was used as a loading control for the mitochondrial fraction and PDI for the cytosolic fraction. G, INSR was also found in the mitochondrial fraction in HCECs. H, hTCEpi cells were cultured in growth (KGM) and basal media (KBM) for 48 hours and subject to mitochondrial fractionation (M). Samples were immunoprecipitated with an antibody specific for the homo-tetrameric INSR, VDAC1, and an IgG control. Subsequent immunoblotting for INSR and pan-VDAC confirmed pull down and indicated the presence of an interaction between INSR and VDAC. I, Immunoblotting for IGF-1R, EGFR, and HGFR confirmed the presence of these receptors in the mitochondria. J, Mitochondrial fractions isolated from hTCEpi cells were immunoprecipitated using antibodies for IgG and VDAC1. Immunoblotting confirmed mitochondrial localization and interactions with IGF-1R, EGFR, and HGFR. n = 3
3.9 ∣. INSR binds mitochondrial VDAC1 to maintain mitochondrial stability and function in hTCEpi cells
Our previous studies show that INSR and IGF-1R undergo nuclear translocation in hTCEpi cells in KBM.8,9 To determine whether INSR was regulating mitochondrial function and stability through accumulation in mitochondria, we performed a subcellular fractionation to isolate cytosolic and mitochondrial fractions. Indeed, INSR was present in the mitochondrial fraction of hTCEpi cells and that the amount of mitochondrial-localized INSR was increased after 48 hours in KBM. Not surprisingly, there was also a reduction in the phosphorylated form of INSR in KBM compared to KGM (Figure 9E). In HBECs, we were unable to detect INSR in the mitochondrial fraction (Figure 9F). To further confirm these findings and show that mitochondrial localization of INSR was not an artifact of the cell line, we used primary cultures of human corneal epithelial cells (HCECs). As shown in Figure 9G, INSR localized to the mitochondrial fraction and increased after 48 hours of culture in KBM. To exclude the presence of plasma membrane contamination in the mitochondrial fractions, we immunoblotted for the plasma membrane marker Na+/K+-ATPase α1 in hTCEpi cells, HBECs, and HCECs. We confirmed the absence of contamination (Supplementary data 9).
VDAC1 has been shown to form complexes with certain receptors and proteins in the mitochondria to regulate apoptosis and/or survival.18,27,28 To test for an interaction between INSR and VDAC1 in hTCEpi cells, we isolated the mitochondrial fraction and performed reciprocal immunoprecipitations for INSRβ and VDAC1 (Figure 9H). An irrelevant IgG was used as a control. Immunoblotting demonstrated that INSRβ was binding VDAC1 in the mitochondria. We next interrogated hTCEpi cells to determine whether other receptor tyrosine kinases were also playing a role in mitochondrial function. To accomplish this, we performed a subcellular fractionation in hTCEpi cells cultured in KGM or KBM for 48 hours and immunoblotted for IGF-1R, EGFR and HGFR. Our results showed that all three of the receptors tested were present in the mitochondria (Figure 9I). To determine whether these receptors were also binding VDAC1, we performed an additional immunoprecipitation for VDAC1 using only the mitochondrial fraction and immunoblotted for IGF-1R, EGFR, and HGFR (Figure 9J). Our data confirmed that all three of the receptors were binding VDAC1 in mitochondria in all culture conditions tested. Interestingly, immunoblotting for IGF-1R demonstrated the presence of a higher molecular weight receptor (~140 kDa instead 95 kDa) in the pull-down blot compared with the input lysate. Based on this data, we speculate that IGF-1R is complexed with VDAC1 and other proteins in the mitochondria. Together, our findings indicate that tyrosine kinase receptors have a previously unrecognized role in regulating mitochondrial activity via VDAC1. A summary of our findings is detailed in Figure 10.
FIGURE 10.
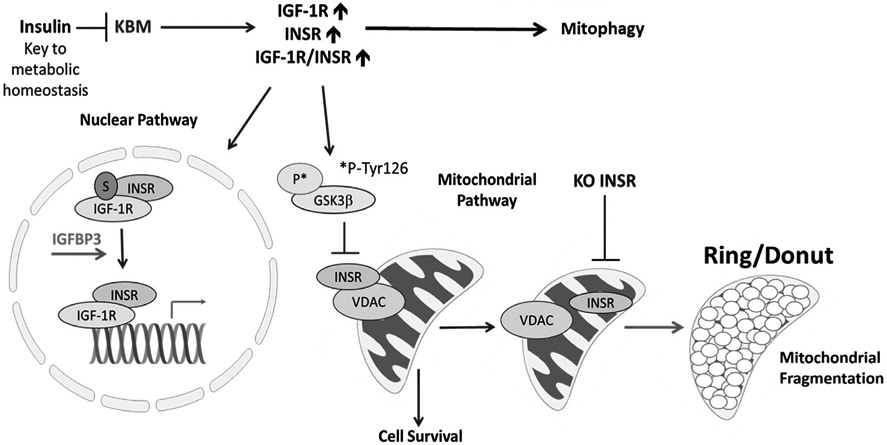
Schematic summary of the role of insulin in regulating nuclear and mitochondrial pathways
4 ∣. DISCUSSION
Our first novel finding in this paper is the identification of insulin as an important regulatory molecule for mitochondrial health and cellular metabolism in corneal epithelial cells. It is well established that INSR is a well-known regulator of metabolism. This is due to the ability of insulin to activate INSR and the subsequent stimulation of glucose uptake. However, in corneal epithelial cells, insulin is not required for glucose uptake, due to the presence of a constitutively active GLUT1.2,3 Metabolic analysis of corneal epithelial cells cultured in basal media without insulin demonstrate a temporal bimodal response. In short-term culture, corneal epithelial cells initially exhibit a drop in glycolysis and a corresponding increase in mitochondrial respiration. Accompanying this is an increase in PINK-1-mediated mitophagy to recycle damaged mitochondria and maintain metabolic activity. PINK-1 is normally degraded during active cell growth and requires a loss of membrane polarization for stabilization and the subsequent recruitment of Parkin. Thus, despite the increase in mitochondrial respiration that is evident after 24 hours, the stabilization of PINK-1 indicates early perturbations in the mitochondrial membrane. After prolonged (48 hour) culture, robust mitochondrial depolarization drives a decrease in respiration and ATP production. This overrides the ability of mitophagy to sustain mitochondrial quality control. Glycolysis is then upregulated to sustain the cell. This represents the presence of a critical fuel switch that mediates the change from respiration to glycolysis during chronic stress that is essential for corneal epithelial cell survival.
In contrast to corneal epithelial cells that need insulin to maintain mitochondrial membrane polarization, bronchial epithelial cells do not depolarize after long-term culture in basal media devoid of insulin. Glycolysis and respiration are decreased proportionally in both short- and long-term culture, indicating the absence of a fuel switch. This is associated with an increase in canonical autophagy and not PINK-1-mediated mitophagy. Despite the persistent drop in respiration, the mitochondria maintain their elongated morphology. While they do appear much thinner, their membrane remains polarized, and the cells maintain a respiratory metabolic phenotype. The ability of bronchial epithelial cells to maintain their polarization under basal conditions means that they are not dependent on insulin to remain polarized. Instead, this likely accounts for the exaggerated metabolic response after re-stimulation with insulin. The maintenance of mitochondrial polarization also explains the absence of an increase in PINK-1-mediated mitophagy in basal media, since mitochondrial depolarization is required for PINK-1 accumulation in the mitochondrial membrane. Since some cell types are able to upregulate the necessary growth factors required for cellular homeostasis when cultured in basal conditions, we speculate that this may explain the differences seen between bronchial and corneal epithelia.
Our second, and most important key finding is the demonstration of the novel interaction between INSR and VDAC1 in mitochondria in corneal epithelial cells. Importantly, the disruption of this interaction through knockdown of INSR appeared to play a key role in mediating mitochondrial stability and function, since INSR knockdown induced widespread mitochondrial ring/donut fragmentation. These depolarized mitochondria co-existed as a mixed population with a small number of hyperpolarized mitochondria. Hyperpolarized mitochondria have been reported to be associated with an increase in ATP, which is necessary for cells to undergo apoptosis.29-32 Since fragmentation is associated with a significant loss of respiratory capacity, we speculate that mitochondria produce ATP before depolarization and fragmentation. Interestingly, unlike corneal epithelial cells, INSR does not bind VDAC1 in bronchial epithelium. This, combined with the absence of INSR in the mitochondria, explains why bronchial epithelial cells exhibit differential responses to insulin. This also indicates that INSR localization in mitochondria and VDAC1 binding is not a universal phenomenon among cell types.
VDAC1 is known to have multiple different binding partners, including BCL-2 family members, and thus plays a major role in mediating apoptosis and survival.18 Hexokinase isoforms, which regulate the ability of BCL-2 family members to bind VDAC1, are dependent on activity from multiple kinases, including GSK3β. Our data indicate that in addition to INSR, IGF-1R, EGFR, and HGFR all translocate to the mitochondria and interact with VDAC1. Of the four receptors studied, only EGFR has been previously reported to translocate to mitochondria.33-36 The majority of this work
has been done in lung cancer models. Che et al reported that mitochondrial translocation of EGFR is mediated by EGF to regulate mitochondrial function and induce metastasis in lung cancer cells.33 Likewise, in tumor cells, mitochondrial EGFR has been correlated to cell survival.35 In agreement with this work, treatment with the EGFR inhibitor Gefitinib increased mitochondrial activity and membrane potential leading to mitochondrial-related cell death in clustered lung cancer cell cultures.37 Subcellular localization of EGFR is regulated in part by autophagy. Studies have shown that induction of autophagy not only prevents cell death in cancer but also triggers the accumulation of EGFR in mitochondria.38,39 In the corneal epithelium, EGFR is not only present in the mitochondria, but receptor accumulation is decreased by growth factor withdrawal. This contrasts with other receptors (INSR, IGF-1R) whose mitochondrial levels increase when deprived of critical growth factors. Together, these data suggest that a balance exists between the tyrosine kinase receptors that modulate mitochondrial activity and cell viability in non-tumor epithelial cells capable of growth downregulation, during normal culture and in response to stress.
Similar to INSR, IGF-1R has also been implicated in the promotion of mitochondrial homeostasis through its impact on oxidative stress. In their seminal study, Holzenberger and colleagues showed that mice heterozygous for IGF-1R have a normal metabolism but are more resistant to oxidative stress.40 More recently, in cancer cell lines, IGF-1 has been shown to promote mitochondrial turnover through an increase in mitophagy and mitochondria biogenesis. In that study, mitophagy is mediated by the mitophagy receptor protein, Bcl-2/adenovirus E1B 19-kDa-interacting protein 3 (BNIP3) and not PINK-1, as shown in this study.41
While an animal model was not included in the current study, it should be noted that our cell line findings were repeatable in primary cultures of isolated corneal epithelial cells. Isolated human corneal epithelial cells consist mostly of corneal-limbal cells and not differentiated corneal epithelial cells. Similar to primary cultures, the hTCEpi cell line was derived from a population of limbal cells and thus, in the absence of sequential high calcium/air-lifted induced differentiation, retains those characteristics in culture, making for a valid comparison. Future studies will include the use of stratified cultures in vitro and in vivo animal models to further investigate the functional significance of mitochondrial-localized INSR in the regulation of mitochondrial homeostasis.
In summary, our findings suggest that INSR plays a key role in metabolic regulation in corneal epithelial cells. This occurs through direct interactions with VDAC1 in mitochondria. Our findings further suggest that this pathway is not conserved across all human mucosal epithelial cells. Further studies are needed to determine tissue specificity for mitochondrial localization of this receptor and the impact on mitochondrial health and the pathophysiology of disease.
Supplementary Material
ACKNOWLEDGMENTS
This study was funded by NIH grants R01 EY029258 (DMR), R01 EY024546 (DMR), P30 EY030413, and S10 OD021685, an unrestricted grant from Research to Prevent Blindness, New York, NY, and a Fight for Sight postdoctoral award (RT).
Funding information
HHS ∣ NIH ∣ National Eye Institute (NEI), Grant/Award Number: EY024546, EY029258 and EY030413; Research to Prevent Blindness (RPB), Grant/Award Number: unrestricted grant; Fight for Sight (FFS)
Abbreviations:
- CCCP
Carbonyl cyanide m-chlorophenyl hydrazine
- COX IV
cytochrome C oxidase IV
- EGFR
epidermal growth factor receptor
- GLUT1
glucose uptake transporter
- GSK3β
insulin blocks glycogen synthase kinase beta
- HBECs
human telomerase immortalized bronchial epithelial cells
- HCECs
human corneal epithelial cells primary cultures
- HGFR
hepatocyte growth factor receptor
- hTCEpi cells
human telomerase immortalized corneal epithelial cells
- IGF-1R
insulin-like growth factor type 1 receptor
- INS
insulin
- INSR
insulin receptor
- KBM
keratinocyte basal media
- KGM
keratinocyte growth media
- MTG
mitotracker green
- PDI
protein disulfide-isomerase
- PINK-1
PTEN-induced kinase 1
- SOD
superoxide dismutase
- VDAC1
voltage-dependent anion channel-1
Footnotes
CONFLICT OF INTEREST
None of the authors have any conflicts of interest to disclose.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section.
REFERENCES
- 1.Laron Z Insulin-like growth factor 1 (IGF-1): a growth hormone. Mol Pathol. 2001;54:311–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cunha DA, Carneiro EM, Alves Mde C, et al. Insulin secretion by rat lacrimal glands: effects of systemic and local variables. Am J Physiol Endocrinol Metab. 2005;289:E768–E775. [DOI] [PubMed] [Google Scholar]
- 3.Friend J, Snip RC, Kiorpes TC, Thoft RA. Insulin sensitivity and sorbitol production of the normal rabbit corneal epithelium in vitro. Invest Ophthalmol Vis Sci. 1980;19:913–919. [PubMed] [Google Scholar]
- 4.Rocha EM, Cunha DA, Carneiro EM, Boschero AC, Saad MJ, Velloso LA. Identification of insulin in the tear film and insulin receptor and IGF-1 receptor on the human ocular surface. Invest Ophthalmol Vis Sci. 2002;43:963–967. [PubMed] [Google Scholar]
- 5.Rocha EM, de M. Lima MH, Carvalho CRO, Saad MJA, Velloso LA. Characterization of the insulin-signaling pathway in lacrimal and salivary glands of rats. Curr Eye Res. 2000;21:833–842. [DOI] [PubMed] [Google Scholar]
- 6.Shanley LJ, McCaig CD, Forrester JV, Zhao M. Insulin, not leptin, promotes in vitro cell migration to heal monolayer wounds in human corneal epithelium. Invest Ophthalmol Vis Sci. 2004;45:1088–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zagon IS, Klocek MS, Sassani JW, McLaughlin PJ. Use of topical insulin to normalize corneal epithelial healing in diabetes mellitus. Arch Ophthalmol. 2007;125:1082–1088. [DOI] [PubMed] [Google Scholar]
- 8.Titone R, Zhu M, Robertson DM. Insulin mediates de novo nuclear accumulation of the IGF-1/insulin Hybrid Receptor in corneal epithelial cells. Sci Rep. 2018;8:4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Titone R, Zhu M, Robertson DM. Mutual regulation between IGF-1R and IGFBP-3 in human corneal epithelial cells. J Cell Physiol. 2019;234:1426–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elrod JW, Gustafsson ÅB. Editorial overview: mitochondria—master regulators of cellular function. Curr Opin Physiol. 2018;3:iv–vi. [Google Scholar]
- 11.Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. [DOI] [PubMed] [Google Scholar]
- 12.Yaribeygi H, Atkin SL, Sahebkar A. Mitochondrial dysfunction in diabetes and the regulatory roles of antidiabetic agents on the mitochondrial function. J Cell Physiol. 2019;234(6):8402–8410. [DOI] [PubMed] [Google Scholar]
- 13.Ding W-X, Yin X-M. Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol Chem. 2012;393:547–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mariappan MM, Shetty M, Sataranatarajan K, Choudhury GG, Kasinath BS. Glycogen synthase kinase 3beta is a novel regulator of high glucose- and high insulin-induced extracellular matrix protein synthesis in renal proximal tubular epithelial cells. J Biol Chem. 2008;283:30566–30575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Y, Huang NQ, Yan F, et al. Diabetes mellitus and Alzheimer's disease: GSK-3beta as a potential link. Behav Brain Res. 2018;339:57–65. [DOI] [PubMed] [Google Scholar]
- 16.Henriksen EJ. Dysregulation of glycogen synthase kinase-3 in skeletal muscle and the etiology of insulin resistance and type 2 diabetes. Curr Diabetes Rev. 2010;6:285–293. [DOI] [PubMed] [Google Scholar]
- 17.Lin CF, Chen CL, Chiang CW, Jan MS, Huang WC, Lin YS. GSK-3beta acts downstream of PP2A and the PI 3-kinase-Akt pathway, and upstream of caspase-2 in ceramide-induced mitochondrial apoptosis. J Cell Sci. 2007;120:2935–2943. [DOI] [PubMed] [Google Scholar]
- 18.Shoshan-Barmatz V, De Pinto V, Zweckstetter M, Raviv Z, Keinan N, Arbel N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol Aspects Med. 2010;31:227–285. [DOI] [PubMed] [Google Scholar]
- 19.Yang K, Chen Z, Gao J, et al. The key roles of GSK-3β in regulating mitochondrial activity. Cell Physiol Biochem. 2017;44: 1445–1459. [DOI] [PubMed] [Google Scholar]
- 20.Das S, Wong R, Rajapakse N, Murphy E, Steenbergen C. Glycogen synthase kinase 3 inhibition slows mitochondrial adenine nucleotide transport and regulates voltage-dependent anion channel phosphorylation. Circ Res. 2008;103:983–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pastorino JG, Hoek JB. Regulation of hexokinase binding to VDAC. J Bioenerg Biomembr. 2008;40:171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu YC, Zhu M, Robertson DM. Novel nuclear localization and potential function of insulin-like growth factor-1 receptor/insulin receptor hybrid in corneal epithelial cells. PLoS ONE. 2012;7:e42483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Molina SA, Moriarty HK, Infield DT, et al. Insulin signaling via the PI3-kinase/Akt pathway regulates airway glucose uptake and barrier function in a CFTR-dependent manner. Am J Physiol Lung Cell Mol Physiol. 2017;312:L688–L702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robertson DM, Li L, Fisher S, et al. Characterization of growth and differentiation in a telomerase-immortalized human corneal epithelial cell line. Invest Ophthalmol Vis Sci. 2005;46:470–478. [DOI] [PubMed] [Google Scholar]
- 25.Ramirez RD, Sheridan S, Girard L, et al. Immortalization of human bronchial epithelial cells in the absence of viral oncoproteins. Can Res. 2004;64:9027–9034. [DOI] [PubMed] [Google Scholar]
- 26.Kim I, Lemasters JJ. Mitochondrial degradation by autophagy (mitophagy) in GFP-LC3 transgenic hepatocytes during nutrient deprivation. Am J Physiol Cell Physiol. 2011;300:C308–C317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vyssokikh M, Zorova L, Zorov D, et al. The intra-mitochondrial cytochrome c distribution varies correlated to the formation of a complex between VDAC and the adenine nucleotide translocase: this affects Bax-dependent cytochrome c release. Biochem Biophys Acta. 2004;1644:27–36. [DOI] [PubMed] [Google Scholar]
- 28.Beutner G, Ruck A, Riede B, Brdiczka D. Complexes between porin, hexokinase, mitochondrial creatine kinase and adenylate translocator display properties of the permeability transition pore. Implication for regulation of permeability transition by the kinases. Biochem Biophys Acta. 1998;1368:7–18. [DOI] [PubMed] [Google Scholar]
- 29.Giovannini C, Matarrese P, Scazzocchio B, Sanchez M, Masella R, Malorni W. Mitochondria hyperpolarization is an early event in oxidized low-density lipoprotein-induced apoptosis in Caco-2 intestinal cells. FEBS Lett. 2002;523:200–206. [DOI] [PubMed] [Google Scholar]
- 30.Apoptosis Tsujimoto Y. and necrosis: intracellular ATP level as a determinant for cell death modes. Cell Death Differ. 1997;4:429–434. [DOI] [PubMed] [Google Scholar]
- 31.Zamzami N, Marchetti P, Castedo M, et al. Reduction in mitochondrial potential constitutes an early irreversible step of programmed lymphocyte death in vivo. J Exp Med. 1995;181:1661–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zamzami N, Marchetti P, Castedo M, et al. Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J Exp Med. 1995;182:367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Che T-F, Lin C-W, Wu Y-Y, et al. Mitochondrial translocation of EGFR regulates mitochondria dynamics and promotes metastasis in NSCLC. Oncotarget. 2015;6:37349–37366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang TH, Lin YH, Yang SC, Chang PC, Wang TC, Chen CY. Tid1-S regulates the mitochondrial localization of EGFR in non-small cell lung carcinoma. Oncogenesis. 2017;6:e361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yao Y, Yan B, Wang G, et al. Mitochondrially localized EGFR is independent of its endocytosis and associates with cell viability. Acta Biochim Biophys Sin. 2010;42:763–770. [DOI] [PubMed] [Google Scholar]
- 36.Demory ML, Boerner JL, Davidson R, et al. Epidermal growth factor receptor translocation to the mitochondria: regulation and effect. J Biol Chem. 2009;284:36592–36604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takenaka T, Katayama M, Sugiyama A, et al. Gefitinib enhances mitochondrial biological functions in NSCLCs with EGFR mutations at a high cell density. Anticancer Res. 2017;37:4779–4788. [DOI] [PubMed] [Google Scholar]
- 38.Li H, You L, Xie J, Pan H, Han W. The roles of subcellularly located EGFR in autophagy. Cell Signal. 2017;35:223–230. [DOI] [PubMed] [Google Scholar]
- 39.Yue X, Song W, Zhang W, et al. Mitochondrially localized EGFR is subjected to autophagic regulation and implicated in cell survival. Autophagy. 2008;4:641–649. [DOI] [PubMed] [Google Scholar]
- 40.Holzenberger M, Dupont J, Ducos B, et al. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature. 2003;421:182–187. [DOI] [PubMed] [Google Scholar]
- 41.Lyons A, Coleman M, Riis S, et al. Insulin-like growth factor 1 signaling is essential for mitochondrial biogenesis and mitophagy in cancer cells. J Biol Chem. 2017;292:16983–16998. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.