

Abstract
Background:
Polygenic risk scores (PRS) for coronary artery disease (CAD) identify high-risk individuals more likely to benefit from primary prevention statin therapy. Whether polygenic CAD risk is captured by conventional paradigms for assessing clinical cardiovascular risk remains unclear.
Objectives:
To intersect polygenic risk with guideline-based recommendations and management patterns for CAD primary prevention.
Methods:
We applied a genome-wide CAD PRS to 47,108 individuals across three U.S. healthcare systems. We then assessed whether primary prevention patients at high polygenic risk might be distinguished on the basis of greater guideline-recommended statin eligibility and higher rates of statin therapy.
Results:
Of 47,108 study participants, mean age was 60 years, and 11,020 (23.4%) had CAD. The CAD PRS strongly associated with prevalent CAD (OR=1.4 per SD increase in PRS; p<0.0001). High polygenic risk (top 20% of PRS) conferred 1.9-fold odds of developing CAD (p<0.0001). However, among primary prevention patients (N=33,251), high polygenic risk did not correspond with increased recommendations for statin therapy - per the American College of Cardiology/American Heart Association (46.2% for those with high PRS versus 46.8% for all others, p=0.54) or U.S. Preventive Services Task Force (43.7% versus 43.7%, p=0.99) - or higher rates of statin prescriptions (25.0% versus 23.8%, p=0.04). An additional 4.1% of primary prevention patients may be recommended for statin therapy if high CAD PRS were considered a guideline-based, “risk-enhancing factor.”
Conclusions:
Current paradigms for primary cardiovascular prevention incompletely capture a polygenic susceptibility to CAD. An opportunity may exist to improve CAD prevention efforts by integrating both genetic and clinical risk.
Keywords: coronary artery disease, genetic risk, primary prevention, statin
Condensed Abstract:
Polygenic risk scores (PRS) robustly prognosticate coronary artery disease (CAD) risk in the general population, but their utility within the healthcare setting - and in the context of guideline-based primary prevention strategies - remains unexplored. We applied a CAD PRS to 47,108 patients across three distinct healthcare systems and observed strong and consistent associations with CAD. Despite a marked risk gradient across PRS strata, standard preventive recommendations and statin prescription patterns did not distinguish those at elevated polygenic risk for CAD. The findings suggest that integration of genetic risk into conventional, guideline-based approaches may inform and enhance CAD primary prevention efforts.
Introduction
Despite continued advances in medical care, cardiovascular diseases - and in particular, coronary artery disease (CAD) - remain the leading cause of death in the United States (1). Over the last three decades, professional societies have consistently recommended escalation of CAD primary prevention efforts for asymptomatic middle-aged Americans at elevated CAD risk as determined by conventional risk factors and established clinical risk scoring schema.(2–4) Most recently, the 2018 American College of Cardiology (ACC)/American Heart Association (AHA) Guidelines for Blood Cholesterol Management recommended consideration of CAD “risk-enhancing factors” to adjudicate statin suitability for patients deemed to be at intermediate clinical risk.(5) Risk-enhancers include clinical factors that independently associate with CAD and confer a near two-fold risk of disease, such as family history of premature ASCVD, history of pre-eclampsia, and lipoprotein (a) concentration > 50mg/dL.(6–8) With accruing data on the population genetic determinants of CAD, and increased availability of both healthcare-associated and consumer-driven genetic testing - the latter now pursued by over 26 million individuals - a genetic predictor of CAD may serve as another risk-enhancing factor that is both broadly available and quantifiable early in life.(9)
A CAD “polygenic risk score” (PRS) captures the net, inherited susceptibility to CAD conferred by many common genetic variants as a single, quantitative risk factor following a normal distribution. PRS that quantify a genetic predisposition to CAD have been validated in multiple population-based cohorts.(10–12) Notably, ample data suggest that CAD PRS may identify subsets of the population more likely to benefit from lifestyle modifications and from statin therapy.(13–15),(16) More recently, the use of a genome-wide set of common genetic variants improved the prognostic capabilities of CAD PRS, particularly for identifying those with the highest genetic predisposition.(17) In addition, application of a genome-wide PRS to a large, population-based cohort demonstrated the potential discriminative benefit of a genome-wide CAD PRS when added to select clinical risk factors.(18)
Genetic risk prediction is a goal of precision medicine, desired by roughly 3 in 4 Americans, and is increasingly feasible in both healthcare-related and direct-to-consumer settings.(19),(20) In addition, direct-to-consumer testing services are more commonly providing polygenic risk predictions for numerous clinical conditions, including cardiometabolic diseases.(21,22) Therefore, there is broad interest in understanding how a CAD PRS may be incorporated into current guideline-based frameworks to inform clinical care.
When considering the clinical applicability of a CAD PRS for primary prevention of CAD, several important questions remain, including: (1) Does a CAD PRS retain its predictive capacity in the healthcare setting? (2) To what extent is high polygenic risk for CAD already captured by conventional paradigms for assessing clinical risk? (3) For whom and for how many might knowledge of high polygenic CAD risk alter contemporary clinical management? We therefore applied a genome-wide PRS to approximately 50,000 patients across three different U.S. healthcare systems to assess PRS performance in the context of contemporary clinical care. We next investigated whether individuals at high polygenic risk were identified by widely used, guideline-recommended tools for clinical risk assessment, and whether they received primary prevention statin therapy within the health care system.
Methods
Study Subjects
We assessed 47,108 individuals across the Partners Healthcare Biobank, the Penn Medicine Biobank, and the Mount Sinai BioMe Biobank with genomic and clinical data, including linked electronic health record (EHR) data from hospitals affiliated with the Partners Healthcare System, the University of Pennsylvania Health System, and the Mount Sinai Health System, respectively (Supplemental Methods). All study participants provided informed consent according to local Institutional Review Board (IRB) protocols. Data analyses were supported by approved IRB protocols at Partners Healthcare, University of Pennsylvania Perelman School of Medicine, and Icahn School of Medicine at Mount Sinai.
Polygenic Risk Score Generation and Application to Biobanks
We recently developed a genome-wide PRS for CAD comprising 6,630,150 common (minor allele frequency >1%) genetic variants utilizing genetic data from a published GWAS of CAD and the LDPred computational algorithm, as previously described (Supplemental Methods) (17,23,24). We applied variant-specific weights from this score to study participants within the Partners Biobank, Penn Medicine Biobank, and BioMe Biobank using the --score function in PLINK 2.0.(25) This algorithm weights the number of risk alleles x at each variant i by its respective LDPred-adjusted per-allele CAD log odds and sums across M variants to generate a PRS ϕ for each study participant . Within each cohort, raw scores were mean centered and normalized by genetic ancestry. Pre-specified PRS strata (top 5% and top 20%) were also defined within genetic ancestries for each cohort. Ancestry groups were then combined within cohorts to minimize the effect of ancestry-specific differences in raw scores (26).
Curation of Clinical Data
Disease phenotypes were curated from the EHR within each health system. Curated disease definitions were based on presence of an International Classification of Diseases (ICD)-9/ICD-10 billing code or documented medication prescription as defined in Supplemental Table 1. Continuous traits - total cholesterol (TC), high-density lipoprotein cholesterol (HDL-C), low density lipoprotein cholesterol (LDL-C), systolic blood pressure (SBP), and body mass index (BMI) - were ascertained from the EHR in each health system; unless otherwise noted, the most recent value (prior to a known statin prescription) was reported and utilized for clinical risk estimations. Outlier values and missing data were handled as described in the Supplemental Methods. Statin prescription data were ascertained from the EHR in a manner specific to each health system.
Clinical Risk Scores and Statin Indication
For individuals without known atherosclerotic cardiovascular disease (ASCVD) - defined as CAD, peripheral artery disease (PAD), and/or cerebral atherosclerosis - the ACC/AHA PCE was applied to calculate each participant’s 10-year risk of an ASCVD event. Factors assessed in the PCE include age, gender, race, TC, HDL-C, SBP, hypertension treatment, diabetes, and smoking.(27) We first applied the PCE to those study participants with available input variables; in sensitivity analyses, we applied the PCE to all study participants using imputed trait values for any missing data (Supplemental Methods). Patients were divided into “Low”, “Borderline”, “Intermediate”, and “High” clinical risk strata using the calculated PCE score per the 2018 ACC/AHA Guidelines for Blood Cholesterol Management.(5) Statin-eligibility criteria were applied per the 2018 ACC/AHA Guidelines for Blood Cholesterol Management and the 2016 USPSTF recommendations on statin use for primary prevention (Supplemental Table 2).(28)
Statistical Analyses
To validate the association of the CAD PRS with prevalent CAD in each health care system, logistic regression was performed adjusting for age, sex, and up to ten principal components of ancestry. An additional covariate representing genotyping array was included for Partners Biobank and Penn Medicine Biobank data. Principal components were used as covariates in logistic regression analyses to increase statistical power for true relationships and to minimize confounding by ancestry.(29) Inverse-variance weighted fixed-effects meta-analysis was used to combine study-specific effect estimates. Categorical variables are reported as frequencies and proportions, and between-group differences were assessed by Chi-squared or Fisher’s exact test, as appropriate. Continuous variables are reported as means and compared with t-tests or Mann-Whitney U test, as appropriate. Cohort- and ancestry-specific risk discrimination for the CAD PRS was assessed by the area under the receiver operating characteristic curve (AUROC). All analyses were performed using R version 3.3.3 software (The R Foundation, Vienna, Austria); AUROC was calculated using the “pROC” package in R. We defined statistical significance as a two-tailed P < 0.05.
Results
Baseline Characteristics
Of the 47,108 study participants, most were women (53.5%) and of European genetic ancestry (62.0%), and mean age across all biobanks was 59.6 years. In total, there were 11,020 cases of CAD (23.4%), including 3,538 cases from the Partners Biobank (7.5%), 4,658 cases from the Penn Biobank (9.9%), and 2,824 cases from the BioMe Biobank (6.0%) (Table 1).
Table 1.
Baseline characteristics of study populations.
| Study characteristics | Partners Healthcare Biobank (N = 13,667) |
Penn Medicine Biobank (N = 9,070) |
Mt. Sinai BioMe Biobank (N = 24,371) |
|---|---|---|---|
| Age | 60 (17) | 68 (14) | 57 (18) |
| Female gender, n (%) | 7,523 (55.0%) | 3,711 (41.0%) | 13,961 (57.3%) |
| Genetic ancestry | |||
| African (AFR) | 867 (6.3%) | 1927 (21.2%) | 6,979 (28.6%) |
| Ad Mixed American (AMR) | 799 (5.9%) | ----- | 7,048 (28.9%) |
| East Asian (EAS) | 167 (1.2%) | ----- | ----- |
| European (EUR) | 11,725 (85.8%) | 7143 (78.8%) | 10,344 (42.4%) |
| South Asian (SAS) | 109 (0.8%) | ----- | ----- |
| Body mass index, kg/m2 | 31.5 (8.3) | 29.3 (6.8) | 28.3 (6.9) |
| Systolic blood pressure, mmHg | 127 (16) | 126 (19) | 127 (20) |
| Smoking status, ever, n (%) | 4,807 (35.2%) | 4,366 (48.1%) | 4,251 (17.4%) |
| Hypertension, n (%) | 7,272 (46.9%) | 6,632 (73.1%) | 9,322 (38.3%) |
| Diabetes Mellitus, n (%) | 2,966 (21.7%) | 2,868 (31.6%) | 5,204 (21.4%) |
| Coronary Artery Disease, n (%) | 3,538 (25.9%) | 4,658 (51.4%) | 2,824 (11.6%) |
| Peripheral Artery Disease, n (%) | 1,427 (10.4%) | 1,736 (19.1%) | 1,679 (6.9%) |
| Cerebrovascular disease, n (%) | 1,439 (10.5%) | 649 (7.2%) | 862 (3.5%) |
| Total Cholesterol, mg/dL | 177 (42) | 175 (40) | 179 (45) |
| LDL-C, mg/dL | 97 (34) | 99 (33) | 100 (37) |
| HDL-C, mg/dL | 56 (19) | 50 (16) | 57 (23) |
| Triglycerides, mg/dL | 120 (65) | 141 (83) | 144 (77) |
Values are presented as mean (standard deviation) unless otherwise noted. Abbreviations: HDL-C = high-density lipoprotein cholesterol; LDL-C = low-density lipoprotein cholesterol.
Validation of genome-wide CAD PRS across health care systems
The CAD PRS strongly associated with CAD across the entire study population (OR=1.42 per 1-SD increase in the PRS; 95% CI: 1.38–1.46; p<0.0001). Effect estimates were consistent across biobanks, (range: OR=1.41 to 1.45 per SD increase in PRS; Pheterogeneity=0.57)(Figure 1). AUROC ranged from 0.59 to 0.61 across sites with separation of PRS distributions between CAD cases and controls (Supplemental Table 3; Supplemental Figure 1A). Individuals in the top 20% of the PRS distribution were at 1.9-fold odds of CAD compared to the remaining 80% of the population (95% CI: 1.8–2.0 for top 20% PRS), while those in the top 5% of the PRS distribution were at 2.3-fold odds of CAD compared to the remaining 95% of the population (95% CI: 2.0–2.5 for top 5% PRS) (Supplemental Figures 2 and 3).
Figure 1. Association of coronary artery disease polygenic risk score with coronary artery disease across three healthcare systems.
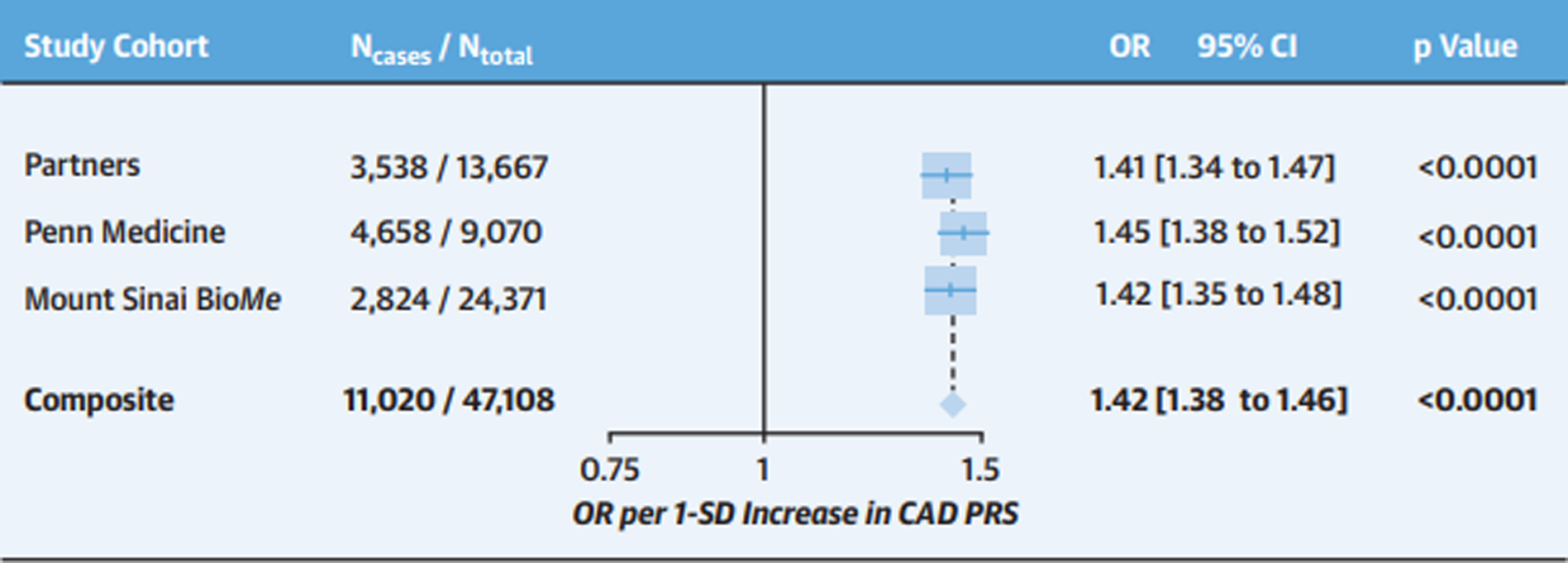
Logistic regression was used to test the association of a one standard deviation increase in a genome-wide CAD PRS with prevalent CAD in the Partners Biobank, Penn Medicine Biobank, and the Mt. Sinai BioMe Biobank. Logistic regression models were adjusted for age, sex, and the first 5 principal components of ancestry. In the Partners Biobank, an additional adjustment for genotyping array was included. Principal components of ancestry were based on observed genotypic differences across subpopulations (i.e., race or ethnicity) within each overall study. Fixed-effects meta-analysis was used to combine results across cohorts (Pheterogeneity=0.57). Cohort-specific participant numbers and CAD case counts are displayed. CAD case counts represent the sum total of disease at baseline and incident disease. Abbreviations: CAD=coronary artery disease; CI=confidence interval; OR=odds ratio; PRS=polygenic risk score; SD=standard deviation.
Clinical risk stratification and guideline-based statin eligibility by PRS strata
Subsequent analyses compared PRS strata to determine whether current, guideline-based clinical risk assessments and practice patterns adequately distinguish individuals at elevated polygenic risk for CAD. Analyses were limited to study participants without ASCVD to reflect a primary prevention cohort warranting risk stratification to guide statin suitability. We first assessed the subset of participants with complete clinical data available for PCE calculation (N=20,628) before performing a sensitivity analysis on all participants without ASCVD (N=33,251) using imputed values for missing data.
A similar proportion of patients in the top 20% and the remaining 80% of the PRS distribution were classified as “Low” (43.0% versus 40.5%), “Borderline” (8.1% versus 9.3%), “Intermediate” (27.2% versus 27.2%), and “High” risk (21.7% versus 23.0%) by the PCE (Chi-square p-value for differences between PRS strata=0.01) (Figure 2A; Supplemental Table 4A). In sensitivity analyses, we observed comparable PCE risk distributions between individuals in the top 5% and bottom 95% of the CAD PRS (Supplemental Table 4A); and between high and low PRS strata when replacing missing with imputed values to permit assessment of all patients without ASCVD (Supplemental Figure 4A; Supplemental Table 4B).
Figure 2. Guideline-based clinical risk estimation and statin-eligibility by strata of CAD PRS.
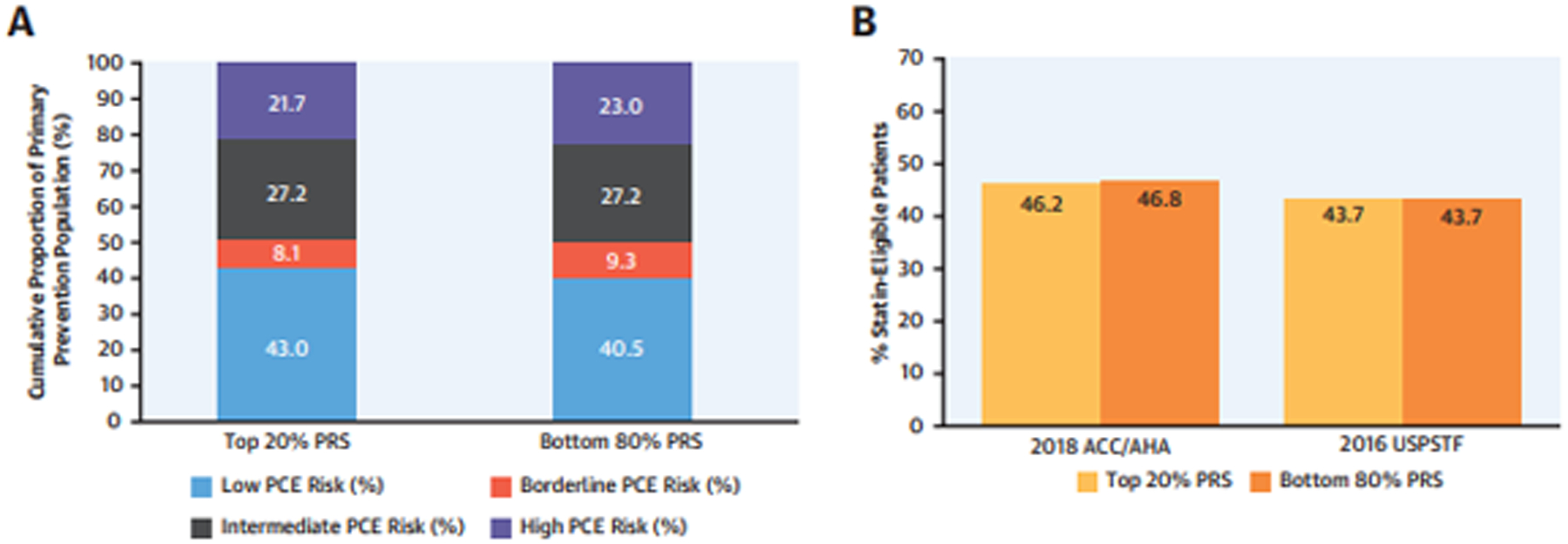
Among primary prevention patients without atherosclerotic cardiovascular disease and with available clinical data (N=20,628), individuals in the Top 20% versus the Bottom 80% of the CAD PRS were compared with respect to: (A) Clinical risk as defined by the ACC/AHA Pooled Cohort Equations (PCE); and (B) Statin-eligibility as defined by the 2018 ACC/AHA Guidelines on Blood Cholesterol Management and the 2016 USPSTF guidelines for primary prevention statin therapy. Abbreviations: ACC=American College of Cardiology; AHA=American Heart Association; CAD=coronary artery disease; PRS=polygenic risk score; USPSTF=United States Preventive Services Task Force.
Applying guideline-based criteria for statin eligibility, a comparable fraction of individuals in the top 20% versus the bottom 80% of the CAD PRS met ACC/AHA criteria (46.2% versus 46.8%; Chi-square p-value for differences between PRS strata=0.54) and USPSTF criteria (43.7% versus 43.7%; p=0.99) for primary prevention statin therapy (Figure 2B; Supplemental Table 5A and 6A). In sensitivity analyses, we observed similar rates of statin-eligibility between the top 5% and the bottom 95% of the PRS as assessed by ACC/AHA (44.3% versus 46.8%; p=0.17) and USPSTF criteria (42.4% versus 43.7%, p=0.46) (Supplemental Tables 5A and 6A); and similar rates of ACC/AHA and USPSTF statin eligibility between PRS strata after replacing missing with imputed values to permit assessment of all patients without ASCVD (Supplemental Figure 4B; Supplemental Tables 5B and 6B).
Contemporary management patterns by PRS strata
The overall statin prescription rate among study participants without ASCVD was 24.0%, with variability across healthcare systems - 25.1% in Partners Biobank, 43.1% in Penn Medicine Biobank, and 19.8% in Mt. Sinai BioMe Biobank. Among participants in the top 20% of the PRS distribution, 25.0% had at least one documented statin prescription in the EHR, comparable to the 23.8% statin prescription rate among individuals in the remaining 80% of the PRS distribution (p=0.04) (Figure 3; Supplemental Table 7). Despite site-specific differences in statin prescription rates, we observed no significant differences between those in the top 20% and bottom 80% of the CAD PRS within Partners Biobank (26.9% versus 24.7%; p=0.06), Penn Medicine Biobank (42.2% versus 43.2%, p=0.61), and the Mt. Sinai BioMe Biobank (20.9% versus 19.6%, p=0.07) (Supplemental Figure 5; Supplemental Table 7). In a secondary analysis, we observed no overall or site-specific differences in statin prescription rates between those in the top 5% and bottom 95% of the CAD PRS (Supplemental Table 7).
Figure 3. Management patterns by strata of CAD PRS.
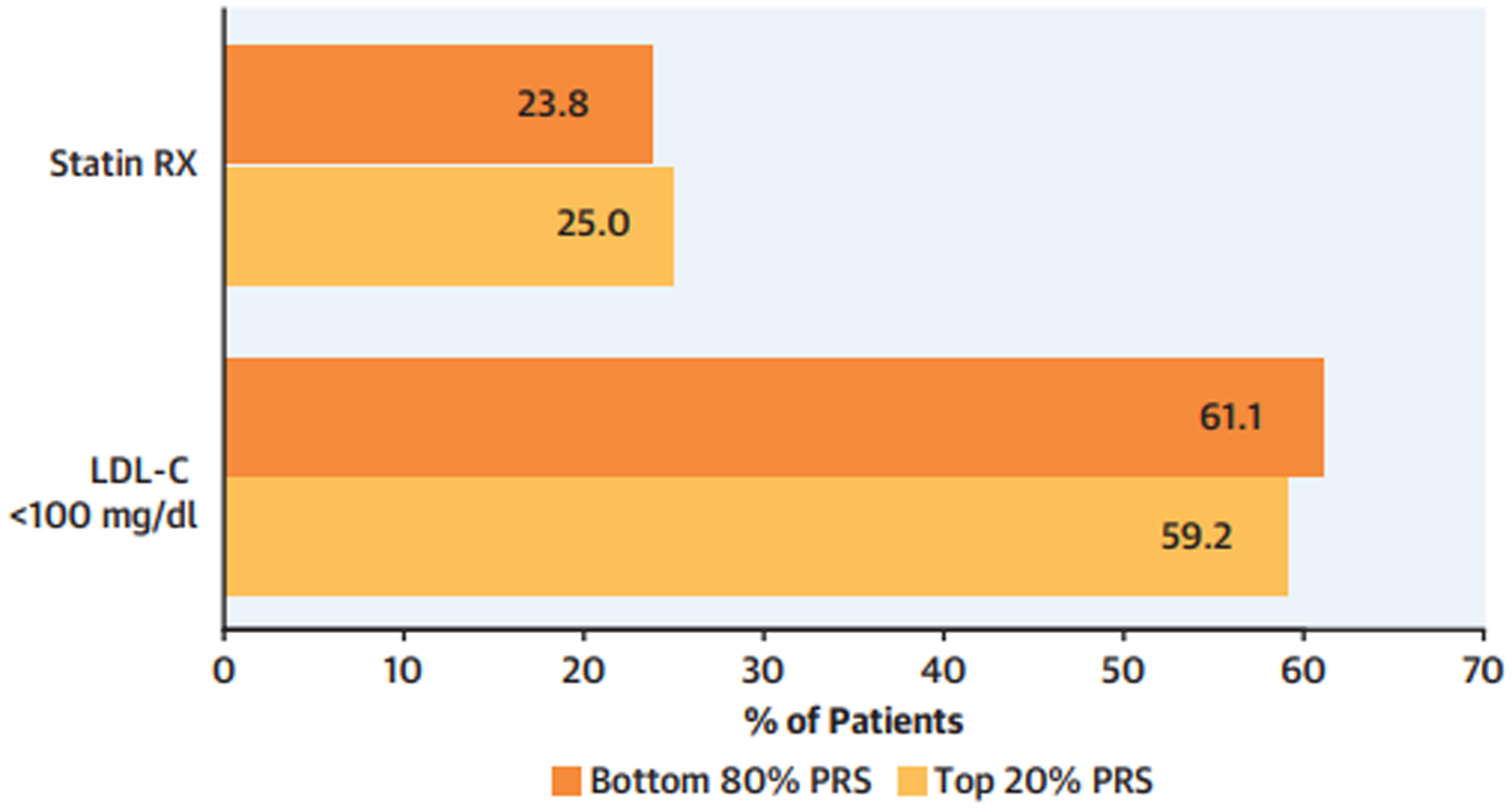
Among primary prevention patients without atherosclerotic cardiovascular disease (N=33,251), individuals in the Top 20% versus the Bottom 80% of the CAD PRS were compared with respect to rates of statin prescriptions (Statin RX), and attainment of LDL-C < 100mg/dl. Abbreviations: CAD=coronary artery disease; LDL-C=low-density lipoprotein cholesterol; PRS=polygenic risk score.
In total, 21,740 participants had at least one documented LDL-C assessment in the EHR, of which 13,205 (60.7%) had a minimum LDL-C < 100mg/dL. A similar proportion of top 20% PRS versus bottom 80% PRS individuals achieved a minimum LDL-C < 100mg/dL (59.2% versus 61.1%, p=0.03) (Figure 3; Supplemental Table 8). A secondary comparison of top 5% versus bottom 95% PRS strata showed no significant difference in the rates of attaining a minimum LDL-C < 100mg/dL (58.0% v. 60.9%, p=0.08).
PRS performance and clinical practice patterns by genetic ancestry
As recent studies have demonstrated variability in PRS performance by genetic ancestry, we assessed ancestry-specific differences in the CAD PRS, and in corresponding clinical risk stratification and management patterns. Although we observed separation of ancestry-specific PRS distributions between CAD cases and controls within each dataset (Supplemental Figure 1B), we focused our ancestry-specific analyses on Mt. Sinai BioMe, given appreciable non-European ancestry participants in this cohort. The PRS strongly associated with CAD in each of the three genetic ancestries tested within the Mt. Sinai BioMe Biobank. The strongest association was seen among individuals of European ancestry (OR=1.52 per 1-SD increase in the PRS; 95% CI: 1.46 – 1.58; p<0.0001; AUROC=0.63), followed by those of Hispanic (Admixed American) ancestry (OR=1.50 per 1-SD increase in the PRS; 95% CI: 1.44 – 1.57; p<0.0001; AUROC=0.63), and those of African ancestry (OR=1.29 per 1-SD increase in the PRS; 95% CI: 1.23 – 1.34; p<0.0001; AUROC=0.58). Across genetic ancestries, those in the top 20% of the CAD PRS distribution were at 1.6-fold (African-ancestry) to 2.1-fold (European ancestry) odds of CAD compared to the remaining 80% of the population (Supplemental Table 9). Despite ancestry-specific variability in score performance, we again observed no significant differences in PCE risk distributions or statin prescription rates between high PRS individuals and all others, irrespective of genetic ancestry (Supplemental Tables 10 and 11).
Use of the CAD PRS as a Risk-Enhancing Factor
The 2018 ACC/AHA Guidelines for Blood Cholesterol Management endorse the use of “risk-enhancing factors” to guide statin initiation in patients without ASCVD, diabetes mellitus or severe hypercholesterolemia (LDL-C > 190mg/dL) categorized as “Borderline” or “Intermediate” risk by the PCE (10-year risk of ASCVD event=5–20%). Risk-enhancing factors are those that independently associate with CAD and confer a near two-fold risk of disease. We observed a comparable level of risk among those in the top 20% of the CAD PRS - 1.9-fold odds of disease in our analysis. Therefore, in post-hoc analyses, we explored statin prescription rates among those at Borderline/Intermediate clinical risk, but high polygenic risk (top 20% of the PRS), to determine the fraction of patients for whom high polygenic risk might serve as a risk-enhancing factor for CAD and motivate primary prevention statin therapy within a guideline-supported framework.
Among participants without ASCVD, diabetes mellitus, or severe hypercholesterolemia (N=16,002), 5,890 (36.8%) were classified as Borderline or Intermediate risk by the PCE. Of these, 987 were in the top 20% of the CAD PRS (6.2% of the primary prevention population), and 652 (4.1% of the primary prevention population) did not have a statin prescription in the health care system. The percentage of patients at high polygenic risk, Borderline/Intermediate clinical risk, and without a statin prescription was comparable across the three health systems (3.5% to 4.3% of the primary prevention population) (Table 2). A sensitivity analysis replacing missing with imputed values for PCE estimates (N=27,921 patients without ASCVD, diabetes, or severe hypercholesterolemia), yielded a consistent proportion of untreated individuals at Borderline/Intermediate clinical risk, but high polygenic risk for CAD (4.1%) (Supplemental Table 12).
Table 2. CAD PRS as a guideline-based CAD risk-enhancing factor.
Primary prevention patients without atherosclerotic cardiovascular disease, diabetes mellitus, or LDL-C > 190 (N = 16,002) stratified on the basis of Borderline/Intermediate clinical risk (by Pooled-Cohort Equations), high CAD polygenic risk strata (top 20% PRS), and lack of statin therapy. Abbreviations: CAD = coronary artery disease; LDL-C = low-density lipoprotein cholesterol; PCE = Pooled-Cohort Equations; PRS = polygenic risk score.
| Partners Healthcare Biobank (N = 4,190) |
Penn Medicine Biobank (N = 1,337) |
Mt. Sinai BioMe Biobank (N = 10,475) |
Composite (N = 16,002) |
|
|---|---|---|---|---|
| Borderline / Intermediate PCE Risk, N (%) | 1,490 (36%) | 614 (46%) | 3,786 (36%) | 5,890 (37%) |
| + Top 20% CAD PRS, N (%) | 237 (5.7%) | 108 (8.1%) | 642 (6.1%) | 987 (6.2%) |
| + No Statin Prescription, N (%) | 146 (3.5%) | 53 (4.0%) | 453 (4.3%) | 652 (4.1%) |
Discussion
Application of a genome-wide CAD PRS to nearly 50,000 patients across three contemporary healthcare-associated biobanks demonstrated robust and consistent associations with CAD. For those in the top 20% of the PRS distribution, levels of risk were akin to most CAD “risk-enhancing factors” endorsed by current ACC/AHA prevention guidelines. However, guideline-based recommendations and contemporary management patterns for the primary prevention of CAD did not differ by polygenic risk strata, suggesting under-recognition of individuals at high polygenic CAD risk within U.S. healthcare systems (Central Illustration).
Central Illustration. Conventional clinical parameters do not distinguish patients at high polygenic risk for coronary artery disease.
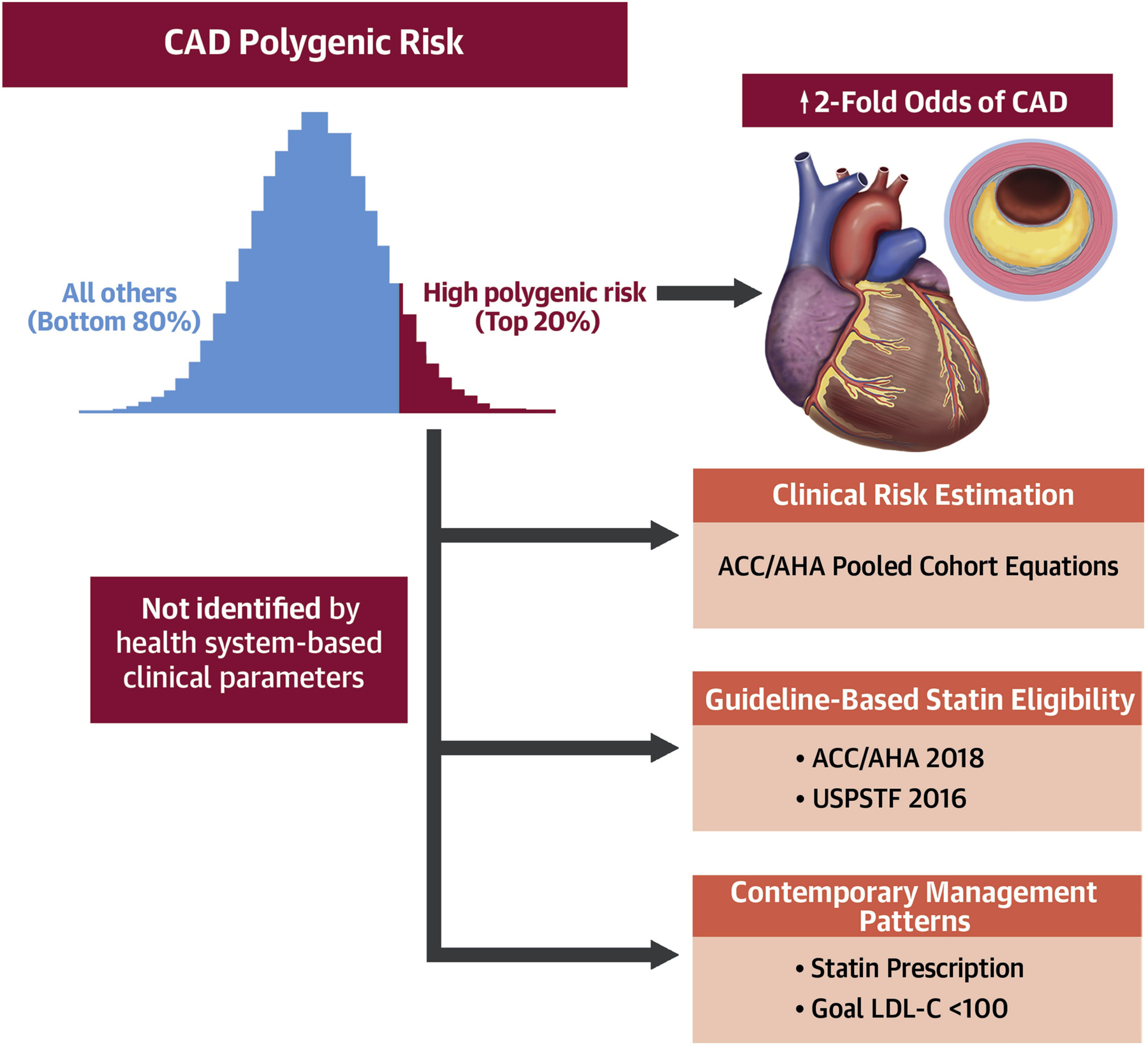
Abbreviations: ACC=American College of Cardiology; AHA=American Heart Association; CAD=coronary artery disease; LDL-C=low-density lipoprotein cholesterol; PRS=polygenic risk score; USPSTF=United States Preventive Services Task Force.
Our findings permit several conclusions. First, we confirm that a genome-wide CAD PRS associates with CAD in healthcare settings. Prior validation studies have demonstrated strong associations between PRS and CAD in prospective, population-based cohorts, particularly as PRS have been derived from progressively larger GWAS and included a greater number of genetic variants (i.e. beyond those reaching genome-wide significance). (10–12,17,18) Here, we provide consistent estimates of CAD risk in three geographically distinct healthcare systems that may inform forthcoming efforts seeking to incorporate polygenic risk into routine clinical management. With the rise of healthcare-associated biobanks and greater access to provider- and consumer-driven genetic testing, broader application of the CAD PRS within the healthcare setting may enable earlier identification and management of patients at the highest-risk for CAD.
Second, current, guideline-based strategies for estimating CAD risk do not account fully for an inherited susceptibility to CAD. Recent analyses have yielded conflicting conclusions on the clinical utility of a CAD PRS for middle-aged adults beyond the PCE. Two studies showed modest improvements in risk discrimination (C-statistic) when the CAD PRS was added to the PCE in three population-based cohorts - Atherosclerosis Risk in Communities (ARIC), Multiethnic Study of Atherosclerosis (MESA), and UK Biobank.(30,31) A third analysis found similar changes in the C-statistic in the UK Biobank and the Malmo Diet and Cancer Study (MDCS); however, this study also demonstrated a striking gradient of longitudinal risk when considering the CAD PRS within each guideline-supported PCE risk category (“Low”, “Borderline”, “Intermediate” and “High” risk). Specifically, a two- to four-fold difference in cumulative incidence was noted across PRS strata, suggesting the ability of the CAD PRS to refine long-term prognoses even within these well-established clinical risk groups.(32)
Here, we extend such assessments of risk prognostication by intersecting a CAD PRS with guideline-based recommendations and management patterns to highlight the under-recognition of these at-risk patients within U.S. healthcare systems. While a prior study demonstrated discrepancies in classification between ACC/AHA and USPSTF criteria for determining statin eligibility, in our analysis, neither criteria differentiated between strata of the CAD PRS.(33) As assessed in the context of these widely-used algorithms, we provide evidence from health systems that polygenic CAD risk is orthogonal (i.e. independent of and complementary) to risk captured by conventional clinical factors. That relevant management patterns did not differ between PRS strata suggests that contemporary clinical practice - the composite of guideline-based and non-guideline-based approaches - is not reflective of patients’ polygenic risk for CAD.
Notably, despite ancestry-specific differences in PRS performance, our study indicates that polygenic risk for CAD remains under-recognized across genetic ancestries. Recent analyses highlighted the underperformance of PRS when applied to those of non-European descent, likely due to derivation of PRS from GWAS enriched for individuals of European genetic ancestry.(34) We previously reported on the ancestry-specific differences of the CAD PRS in a multi-ethnic cohort,(35) and now recapitulate these ancestry-specific differences within the healthcare setting. Nevertheless, in the current study, the CAD PRS strongly associated with disease in individuals of African and Hispanic (Admixed American) ancestry, conferring 1.6- to 2.0-fold odds of disease among those in the top 20% of the PRS risk distribution. Furthermore, among those of non-European ancestry, the PCE did not identify individuals at high PRS nor were there more statin prescriptions among those at high PRS, similar to observations in European-ancestry participants. While additional efforts are required to develop more robust ancestry-specific PRS, our data suggest that current implementation of the CAD PRS may be of broad benefit by capturing currently unmeasured risk across diverse genetic ancestries.
Third, consideration of a CAD PRS alongside current, guideline-based practices may influence management for a subset of primary prevention patients in whom clinical uncertainty remains after implementing standard prevention algorithms. While setting an “optimal” cutpoint for a continuous marker such as the PRS is inherently subjective, in our prior, post hoc analyses of statin randomized controlled clinical trials, we demonstrated that those in the top 20% of a CAD PRS derived greater relative and absolute benefit from statin therapy.(14,15) Within the context of the 2018 ACC/AHA Blood Cholesterol Guidelines, clinical parameters that associate with a roughly two-fold risk of CAD and are orthogonal to traditional risk factors (including lipoprotein (a) concentration > 50mg/dL and high-sensitivity C-reactive protein ≥ 2.0mg/L) are incorporated as risk-enhancing factors, which are used to guide statin eligibility for patients at borderline-intermediate clinical risk.(5) Notably, in the abovementioned analysis of UK Biobank and MDCS, those in the top 20% of the CAD PRS were also at >2-fold hazards of incident disease, including among borderline-intermediate PCE risk groups.
In the current analysis, high polygenic risk again proved orthogonal to conventional clinical algorithms, and the top 20% of the CAD PRS distribution associated with a similar, near two-fold risk of CAD. Taken together across three healthcare settings, we observed that 1 in 25 primary prevention patients may newly be suitable for a statin prescription if polygenic risk were used as a CAD risk-enhancer to inform clinical decision-making for those in whom guideline-based recommendations are unclear. In a recent analysis of ~50,000 participants from the Geisinger Health System, whole exome sequencing identified ~ 1 in 625 individuals with familial hypercholesterolemia (FH) not prescribed statin therapy.(36) Coupled with this prior study, our findings support the clinical translation of human genetic data to better identify and treat both monogenic (i.e. FH) and polygenic drivers of CAD risk. As high CAD PRS is more common than FH, and less recognizable by clinical risk factors - i.e. severe hypercholesterolemia - the impact of polygenic testing for population-wide efforts at disease prevention may be particularly profound.
Accordingly, although we do not anticipate that a CAD PRS will influence management for the majority of primary prevention patients, it may indeed be helpful - and akin to other CAD risk-enhancing factors - when there is clinical uncertainty. However, a unique strength of a CAD PRS is that it may be available early in life and need only be assessed once. As 10-year absolute CAD risk increases with age and the acquisition of clinical risk factors, earlier knowledge of a CAD PRS may prompt timelier initiation of preventive strategies.
Study Limitations
First, disease phenotypes were predicated on data from the EHR, which carry the potential for phenotype misclassification. Second, clinical data were limited to that obtained within each health system, and therefore, diagnoses and medical care received elsewhere may have gone unrecognized; however, given consistent effect estimates for the association of CAD PRS with CAD across three health systems, such differences are unlikely to have influenced our findings. Third, availability of data on specific risk-enhancers - as outlined in the 2018 ACC/AHA guidelines - was limited precluding a comprehensive analysis of these factors. Fourth, in post-hoc analyses, we modeled the top 20% of the PRS as a CAD risk-enhancing factor, whereas using different PRS cutoffs would affect different proportions of the health system. However, we endeavored to inform this somewhat subjective decision using a guideline-supported framework - two-fold risk and orthogonal to traditional risk factors. While our current and prior analyses (across contemporary healthcare settings, epidemiologic cohorts, and randomized controlled trials) suggest that statins mitigate risk conferred by an elevated polygenic susceptibility to CAD, dedicated placebo-controlled, PRS-stratified statin clinical trials are required to confirm this hypothesis and to estimate treatment effect. Fifth, biobank volunteer participants may not be representative of their respective health care systems and the population at large, which may influence the generalizability of these study findings. Efforts to harmonize PRS interpretation, including healthcare system and ethnicity normalization and individual-level percentiles, are necessary to support evaluations of generalizability and clinical utility. Finally, given the varied performance of PRS across different ancestries, validation of our analysis is required in larger, non-European cohorts.
Conclusions
A genome-wide PRS demonstrated strong and consistent associations with CAD across three U.S. healthcare systems. Polygenic risk for CAD was not fully captured by guideline-based clinical risk algorithms, and was not reflected in contemporary practice patterns. Given increasing availability of a CAD PRS to healthcare-associated biobank researchers and consumers of direct-to-consumer genetic testing, prior and present observations may support focused incorporation of CAD PRS into guideline-based primary prevention algorithms. A prospective clinical trial may be warranted to determine whether targeted prevention efforts in those at high polygenic risk for CAD will improve clinical outcomes.
Supplementary Material
PERSPECTIVES.
Competency in Medical Knowledge:
Current guideline-based strategies for the primary prevention of CAD do not account for an elevated genetic predisposition to disease.
Translational Outlook:
Further investigations are required to assess whether integration of genetic risk into conventional, guideline-based approaches for CAD primary prevention will improve clinical outcomes.
Acknowledgments:
The data, analytic methods, and study materials will be made available for purposes of replicating the results, with approval from the appropriate Institutional Review Board and appropriate collaboration/data sharing agreements. Genotyping of BioMe was performed in collaboration with Regeneron Genetics Center. Regeneron Genetics Center assisted with the collection, management, and analysis of the genetic data and approval of the final manuscript; they had no input as to the design and conduct of the study, the interpretation of the data, and preparation, review, or decision to submit the manuscript for publication. Aayushee Jain B.S., Arden Moscati Ph.D.., Gillian Belbin Ph.D, Lisheng Zhou Ph.D., Michael Preuss Ph.D., Quingbin Song Ph.D., Stephane Wenric Ph.D., and Steve Ellis M.S., all of whom are affiliated with the Icahn School of Medicine at Mt. Sinai, assisted with quality control and/or file handling for the BioMe genome-wide genotyping data. Data analysis was performed by K.G.A., R.J., A.D., M.C., and K.C. P.N. (Partners), S.M.D. (Penn), and R.D. (BioMe) had full access to all of the respective data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
DISCLOSURES:
K.A. is supported by an award from the American Heart Association Institute for Precision Cardiovascular Medicine (17IFUNP33840012). P.N. is supported by grants from the National Heart, Lung, and Blood Institute (R01HL142711, R01HL148565, R01HL148050), Fondation Leducq (TNE-18CVD04), and Hassenfeld Scholar Award from the Massachusetts General Hospital. R.D is supported by R35GM124836 from the National Institute of General Medical Sciences of the National Institutes of Health, and R01HL139865 from the National Heart, Lung, Blood Institute of the National Institutes of Health. S.M.D. is supported by the U.S. Department of Veterans Affairs (IK2-CX001780). S.A.L. is supported by NIH grant 1R01HL139731 and American Heart Association 18SFRN34250007. S.T.W,, E.W.K., and J.W.S. are supported in part by an NHGRI grant supporting the eMERGE Network (U01HG008685). P.N. reports grant support from Amgen, Apple, and Boston Scientific, and is a scientific advisor to Apple. R.D. has received research support from AstraZeneca and Goldfinch Bio, not related to this work. S.M.D. has received research support to the University of Pennsylvania from CytoVas LLC and Renalytix AI, not related to this work. M.C. is supported by a grant from Bayer AG to the Broad Institute focused on the development of therapeutics for cardiovascular disease. S.A.L. receives sponsored research support from Bristol Myers Squibb / Pfizer, Bayer AG, and Boehringer Ingelheim, and has consulted for Bristol Myers Squibb / Pfizer and Bayer AG. J.W.S. is an unpaid member of the Bipolar/Depression Research Community Advisory Panel of 23andMe. This publication is solely the responsibility of the authors, and does not represent the views of the American Heart Association, National Institutes of Health/National Heart, Lung, and Blood Institute, Department of Veterans Affairs or the United States government. The remaining authors report no competing interests.
Abbreviations:
- CAD
coronary artery disease
- PRS
polygenic risk score
- ASCVD
atherosclerotic cardiovascular disease
- ACC
American College of Cardiology
- AHA
American Heart Association
- USPSTF
United States Preventive Services Task Force
- PCE
Pooled Cohort Equations
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Benjamin EJ, Muntner P, Alonso A et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation 2019;139:e56–e528. [DOI] [PubMed] [Google Scholar]
- 2.Grundy SM, Balady GJ, Criqui MH et al. Guide to primary prevention of cardiovascular diseases. A statement for healthcare professionals from the Task Force on Risk Reduction. American Heart Association Science Advisory and Coordinating Committee. Circulation 1997;95:2329–31. [DOI] [PubMed] [Google Scholar]
- 3.Grundy SM, Balady GJ, Criqui MH et al. Primary prevention of coronary heart disease: guidance from Framingham: a statement for healthcare professionals from the AHA Task Force on Risk Reduction. American Heart Association. Circulation 1998;97:1876–87. [DOI] [PubMed] [Google Scholar]
- 4.Pearson TA, Blair SN, Daniels SR et al. AHA Guidelines for Primary Prevention of Cardiovascular Disease and Stroke: 2002 Update: Consensus Panel Guide to Comprehensive Risk Reduction for Adult Patients Without Coronary or Other Atherosclerotic Vascular Diseases. American Heart Association Science Advisory and Coordinating Committee. Circulation 2002;106:388–91. [DOI] [PubMed] [Google Scholar]
- 5.Grundy SM, Stone NJ, Bailey AL et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol 2018. [Google Scholar]
- 6.Sivapalaratnam S, Boekholdt SM, Trip MD et al. Family history of premature coronary heart disease and risk prediction in the EPIC-Norfolk prospective population study. Heart 2010;96:1985–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu P, Haththotuwa R, Kwok CS et al. Preeclampsia and Future Cardiovascular Health: A Systematic Review and Meta-Analysis. Circ Cardiovasc Qual Outcomes 2017;10. [DOI] [PubMed] [Google Scholar]
- 8.Emerging Risk Factors C, Erqou S, Kaptoge S et al. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA 2009;302:412–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.More than 26 million people have taken an at-home ancestry test. https://www.technologyreview.com/s/612880/more-than-26-million-people-have-taken-an-at-home-ancestry-test/. Accessed June 11, 2019.
- 10.Ripatti S, Tikkanen E, Orho-Melander M et al. A multilocus genetic risk score for coronary heart disease: case-control and prospective cohort analyses. Lancet 2010;376:1393–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tada H, Melander O, Louie JZ et al. Risk prediction by genetic risk scores for coronary heart disease is independent of self-reported family history. Eur Heart J 2016;37:561–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abraham G, Havulinna AS, Bhalala OG et al. Genomic prediction of coronary heart disease. Eur Heart J 2016;37:3267–3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khera AV, Emdin CA, Drake I et al. Genetic Risk, Adherence to a Healthy Lifestyle, and Coronary Disease. N Engl J Med 2016;375:2349–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mega JL, Stitziel NO, Smith JG et al. Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy: an analysis of primary and secondary prevention trials. Lancet 2015;385:2264–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Natarajan P, Young R, Stitziel NO et al. Polygenic Risk Score Identifies Subgroup With Higher Burden of Atherosclerosis and Greater Relative Benefit From Statin Therapy in the Primary Prevention Setting. Circulation 2017;135:2091–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Said MA, Verweij N, van der Harst P. Associations of Combined Genetic and Lifestyle Risks With Incident Cardiovascular Disease and Diabetes in the UK Biobank Study. JAMA Cardiol 2018;3:693–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khera AV, Chaffin M, Aragam KG et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet 2018;50:1219–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inouye M, Abraham G, Nelson CP et al. Genomic Risk Prediction of Coronary Artery Disease in 480,000 Adults: Implications for Primary Prevention. J Am Coll Cardiol 2018;72:1883–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med 2015;372:793–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaufman DJ, Baker R, Milner LC, Devaney S, Hudson KL. A Survey of U.S Adults’ Opinions about Conduct of a Nationwide Precision Medicine Initiative(R) Cohort Study of Genes and Environment. PLoS One 2016;11:e0160461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Introducing the MyHeritage DNA Health+Ancestry Test. https://blog.myheritage.com/2019/05/introducing-the-myheritage-dna-health-ancestry-test/. Accessed June 11, 2019.
- 22.23andMe Offers New Genetic Report on Type 2 Diabetes. https://blog.23andme.com/health-traits/type-2-diabetes/. Accessed June 11, 2019.
- 23.Nikpay M, Goel A, Won HH et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet 2015;47:1121–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vilhjalmsson BJ, Yang J, Finucane HK et al. Modeling Linkage Disequilibrium Increases Accuracy of Polygenic Risk Scores. Am J Hum Genet 2015;97:576–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 2015;4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reisberg S, Iljasenko T, Lall K, Fischer K, Vilo J. Comparing distributions of polygenic risk scores of type 2 diabetes and coronary heart disease within different populations. PLoS One 2017;12:e0179238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goff DC Jr., Lloyd-Jones DM, Bennett G et al. 2013 ACC/AHA guideline on the assessment of cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2014;63:2935–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Force USPST, Bibbins-Domingo K, Grossman DC et al. Statin Use for the Primary Prevention of Cardiovascular Disease in Adults: US Preventive Services Task Force Recommendation Statement. JAMA 2016;316:1997–2007. [DOI] [PubMed] [Google Scholar]
- 29.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 2006;38:904–9. [DOI] [PubMed] [Google Scholar]
- 30.Mosley JD, Gupta DK, Tan J et al. Predictive Accuracy of a Polygenic Risk Score Compared With a Clinical Risk Score for Incident Coronary Heart Disease. JAMA 2020;323:627–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Elliott J, Bodinier B, Bond TA et al. Predictive Accuracy of a Polygenic Risk Score-Enhanced Prediction Model vs a Clinical Risk Score for Coronary Artery Disease. JAMA 2020;323:636–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hindy G, Aragam K, Chaffin M et al. Integration Of A Genome-wide Polygenic Score With ACC/AHA Pooled Cohorts Equation In Prediction Of Coronary Artery Disease Events In >285,000 Participants. Circulation 2019;140: A16565. [Google Scholar]
- 33.Pagidipati NJ, Navar AM, Mulder H, Sniderman AD, Peterson ED, Pencina MJ. Comparison of Recommended Eligibility for Primary Prevention Statin Therapy Based on the US Preventive Services Task Force Recommendations vs the ACC/AHA Guidelines. JAMA 2017;317:1563–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martin AR, Kanai M, Kamatani Y, Okada Y, Neale BM, Daly MJ. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat Genet 2019;51:584–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khera AV, Chaffin M, Zekavat SM et al. Whole Genome Sequencing to Characterize Monogenic and Polygenic Contributions in Patients Hospitalized with Early-Onset Myocardial Infarction. Circulation 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abul-Husn NS, Manickam K, Jones LK et al. Genetic identification of familial hypercholesterolemia within a single U.S. health care system. Science 2016;354. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.