

Abstract
Introduction
Poly(ADP-ribose) polymerase-1 (PARP-1) is an abundant enzyme in the cell nucleus that regulates genome repair by binding to DNA damage sites and creating the poly(ADP-ribose) posttranslational modification. PARP-1 hyperactivity leads to cell stress/death associated with many prominent diseases (e.g. cardiovascular disease and several common neurodegenerative disorders). PARP-1 has notably emerged as an effective clinical target for a growing list of cancers. Clinical PARP-1 inhibitor (PARPi) compounds all bind at the same location at the catalytic center of the enzyme to block the binding of substrate NAD+ and prevent poly(ADP-ribose) production, yet they exhibit vastly different outcomes in tumor cell killing and efficacy in the clinic – a paradox that has confounded the development of PARPi. The resolution of this paradox likely lies in the realization that the most effective PARPi compounds trap PARP-1 at the site of a DNA break, generating a lesion that becomes cytotoxic especially in tumor cells with deficiencies in the repair of DNA strand breaks.
Rationale
The molecular roots of PARP-1 trapping on DNA remain poorly understood. We focused on the retention of PARP-1 on damaged DNA using purified components, examining a panel of PARPi that included those currently approved for clinical use. Solution biophysical approaches, especially hydrogen/deuterium exchange-mass spectrometry (HXMS), combined with X-ray structures and a battery of biochemical assays were used to interrogate the molecular impact of PARPi binding to PARP-1 engaged on DNA damage. Structure-guided modification of PARPi through medicinal chemistry was combined with chromatin fractionation to monitor trapped PARP-1 and with cell survival assays to assess PARPi efficacy, in order to probe the molecular underpinnings of the variable outcomes between clinical PARPi.
Results
HXMS experiments revealed that a critical allosteric regulatory domain of PARP-1, the helical domain (HD), is impacted in distinct ways depending on the particular PARPi engaged in the NAD+-binding site adjacent to the HD. Certain PARPi destabilized specific HD regions, others have no effect on the HD, while others actually stabilized regions of the HD. PARPi that destabilized the HD increased PARP-1 affinity for DNA and retained PARP-1 on DNA breaks. Conversely, PARPi that stabilized the HD decreased PARP-1 affinity for DNA breaks. PARPi molecules were thus classified into three types: Type I, allosteric pro-retention on DNA; Type II, non-allosteric; and Type III, allosteric pro-release from DNA. X-ray structure analysis identified PARPi contacts with the HD structural element helix αF, which was established to be the discriminating factor between the types of PARPi. Type I PARPi contact helix αF to initiate an allosteric chain reaction that travels ~40 Å through the multi-domain PARP-1 molecule and culminates in increased DNA binding affinity. Structure-guided mutagenesis of helix αF disrupted PARPi contacts and abrogated the allosteric effects of a Type I inhibitor, transforming it into a non-allosteric, Type II inhibitor. Other mutations that disrupted PARP-1 allostery, including one identified in a de novo PARPi resistant patient with ovarian cancer, also prevented Type I PARPi from retaining PARP-1 on a DNA break. Type III PARPi influenced PARP-1 allostery in a manner that reduced DNA binding and favored DNA release. Structure-inspired modification of a pro-release (Type III) inhibitor converted it to a pro-retention (Type I) inhibitor that conferred potent PARP-1 trapping within the cellular context and increased its ability to kill cancer cells.
Conclusion
Our findings establish the impact of clinical PARPi on PARP-1 allostery and demonstrate that allostery plays a critical role in cellular PARP-1 trapping and can increase potency toward cancer cell killing. The results illuminate the molecular basis for the fine-tuning of PARP-1 to achieve allosteric effects. In contrast to cancer, other diseases would seem to benefit from PARP-1 inhibition but not cell death (e.g. cardiovascular disease). Our studies provide the molecular understanding to create tunable PARPi for clinical applications where PARP-1 trapping is either desirable or undesirable in specific patients.
Graphical Abstract
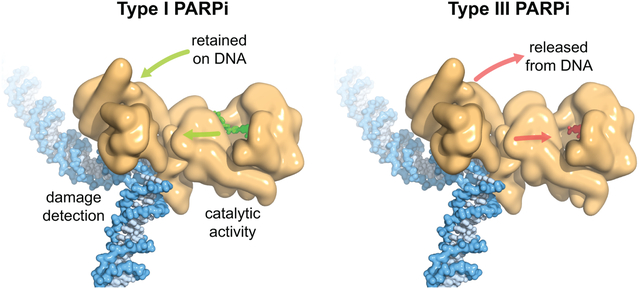
PARPi impact on PARP-1 allostery
PARP-1 (tan) uses multiple domains to detect DNA breaks, and DNA damage detection is allosterically coupled to poly(ADP-ribose) production. PARPi bind to the catalytic domain to inhibit PARP-1 activity. Type I PARPi influence PARP-1 allostery and retain PARP-1 on DNA (left, UKTT15 in green), whereas Type III PARPi perturb PARP-1 allostery and release PARP-1 from DNA (right, veliparib in red). Type II PARPi do not influence PARP-1 allostery.
The success of poly(ADP-ribose) polymerase-1 (PARP-1) inhibitors (PARPi) to treat cancer relates to their ability to trap PARP-1 at the site of a DNA break. Although different forms of PARPi all target the catalytic center of the enzyme, they have variable abilities to trap PARP-1. We find that several structurally distinct PARPi drive PARP-1 allostery to promote release from a DNA break. Other inhibitors drive allostery to retain PARP-1 on a DNA break. Further, we generate a new PARPi compound, converting an allosteric pro-release to a pro-retention compound and increasing its ability to kill cancer cells. These developments are pertinent to clinical applications where PARP-1 trapping is either desirable or undesirable.
Poly(ADP-ribose) polymerase-1 (PARP-1) is an abundant nuclear protein that monitors DNA damage arising from natural chromosomal processes and environmental insults, binding to single- and double-stranded DNA breaks (SSB and DSB) within seconds of their creation (1–3). Upon binding to a break, PARP-1 autoinhibition is relieved through an allosteric activation mechanism (4–6), leading to ~1000-fold increase in utilization of substrate NAD+ to build chains of poly(ADP-ribose) (PAR) on protein substrates including PARP-1 itself (automodification), and histones (heteromodification) in nucleosomes proximal to the break (1, 7, 8). The high density of PAR chains signals the presence and location of the DNA break, leading to rapid recruitment of components of the DNA damage repair machinery. PARP-1 must then release the DNA break to allow subsequent DNA repair reactions (9). PARP-1 also binds to non-damaged chromatin near promoter regions and uses its enzymatic activity to regulate gene expression (10–12). The function of PARP-1 is only dispensable for cell viability because of redundant activity of a less abundant closely related protein, PARP-2 (13).
A number of different PARP inhibitors (PARPi) have been approved for the treatment of breast or ovarian cancers that have defects in DNA repair by homologous recombination, including those with either BRCA1 or BRCA2 gene mutations (14–18). A major paradox has emerged: while all the different PARPi compounds are high-affinity NAD+ competitive inhibitors, they exhibit vastly different outcomes in tumor cell killing (19). The ability to “trap” PARP-1 on DNA might be the resolution to this paradox (20, 21). When PARP-1 is trapped at a DNA break, the lesion becomes cytotoxic (22, 23). The molecular basis of PARP-1 trapping remains poorly understood. The inhibitory potency, the affinity for PARP-1, and the half-life of the compound vary between PARPi, all of which might play a role in its trapping ability (21, 24–26). One model proposes that some PARPi are substantially better than others at trapping PARP-1 because they induce a reverse allosteric communication through the PARP-1 molecule from the NAD+/PARPi binding site to the DNA binding domains, leading to increased affinity for DNA and retention on the break (21, 27). An NAD+ analog indeed drives allosteric communication within the PARP-1 molecule, promoting retention on single strand break (SSB) DNA (6), suggesting that such allosteric communication could be transmitted by PARPi when engaging the NAD+-binding site. Here, we interrogate this model and define the molecular impact of PARPi binding to PARP-1 engaging a SSB.
Different PARPi drive diverse allosteric changes in PARP-1
We employed hydrogen/deuterium exchange-mass spectrometry (HXMS) to measure the protein backbone dynamics of PARP-1 (Fig. 1A)(6). We included in our study the non-clinical PARPi, EB-47, because it mimics NAD+ binding (28, 29), and binds with a high affinity (nM range) comparable to clinical PARPi (27, 28). The HXMS experiments were performed over a time course (101-105 s; Figs. 1A,B and S1). We found that EB-47 impacts PARP-1 allostery similar to the NAD+ analog benzamide adenine dinucleotide (BAD)(6), including a highly localized destabilization of the helical domain (HD), and stabilization of other contact points (Figs. 1B,C [region iv], S1A, S2B, S3A, and S4). Specifically, EB-47 binding leads to increased stability (i.e. protection from hydrogen/deuterium exchange) at the interfaces of PARP-1 domains, indicating a stronger interaction between these domains (Figs. 1B,C [regions i, ii, and iii], S1A, S2B, and S5). In general, peptides in the stabilized contact points require 10 times longer (Fig. S3B) to achieve the same level of deuteration as in the absence of EB-47, clearly indicating stabilization of domain interactions and their contacts with DNA. EB-47 thus provides a second example, after the NAD+ analog BAD, of a molecule that engages the PARP-1 active site and allosterically influences PARP-1 DNA binding domains.
Figure 1. PARPi drive distinct allosteric effects through the multi-domain architecture of PARP-1, culminating in increases or decreases in affinity for an SSB.
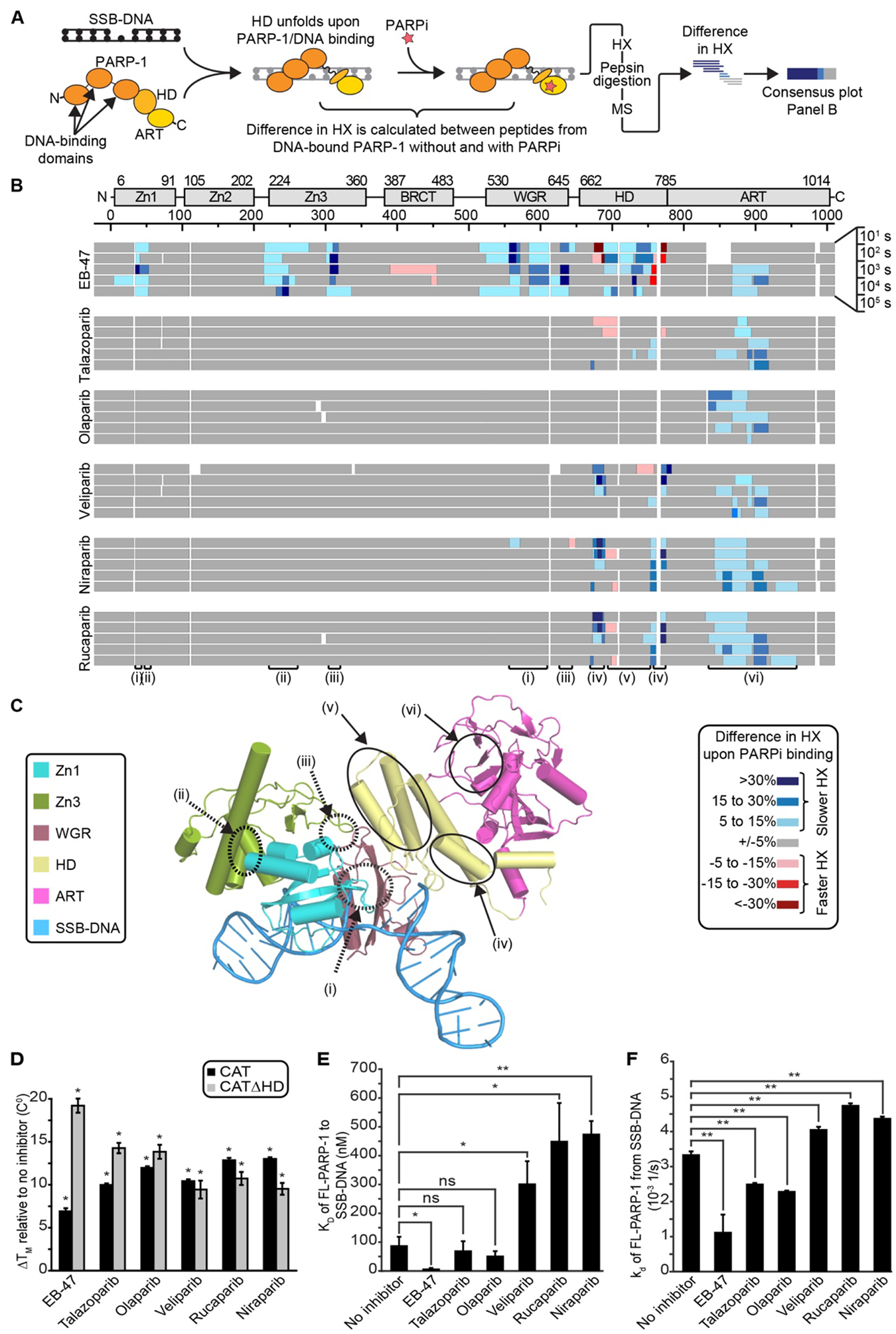
(A) Schematic of HXMS experiments with different forms of PARPi. (B) The percentage difference of HX with and without the indicated form of PARPi is calculated for each of >150 unique partially overlapping peptides for each indicated timepoint (see also Fig. S1), and then the consensus at each amino acid positions is plotted. The color key for binning of HX differences is shown to the lower right of the panel. Small white regions indicate gaps in peptide coverage. Roman numerals indicate regions highlighted in the structural model in panel C. (C) Combined model of the crystal structure of the PARP-1 (Zn1, Zn3, WGR-CAT)/DNA complex, and the NMR structure of the Zn1-Zn2/SSB-DNA complex (39), highlighting numbered regions of interest and colored by domain. Zn2 was omitted for clarity. HD, helical domain; ART, ADP-ribosyl transferase; Zn1/Zn2/Zn3, zinc finger domains; WGR, tryptophan-glycine-arginine domain. (D) DSF thermal stability of CAT and CATΔHD in the presence of the indicated forms of PARPi. (E) Apparent equilibrium binding affinity KD of PARP-1 to SSB-DNA measured by FP in the absence or presence of the indicated form of PARPi. (F) Dissociation rate constant kd of PARP-1 from SSB-DNA measured by SPR in the absence or presence of the indicated PARPi. For panels D-F, error bars represent s.d. from at least three measurements (see also Table S1). The results of student’s T-tests are shown (* represents p values <0.01, ** represents p values <0.001; ns, represents not significant).
We next looked for changes in PARP-1 allostery in the presence of five clinical PARPi (talazoparib, olaparib, veliparib, niraparib, and rucaparib)(Fig. 1B). Olaparib showed no change in allosteric communication or in the DNA binding domains (Figs. 1B, S1, and S2). Talazoparib conferred minor increases in exchange in the helical domain (HD) in the αC and αF helices. In contrast, niraparib, rucaparib, and veliparib showed stronger allosteric effects to the HD. However, the precise HD location and nature of the HX change is opposite from what occurs upon EB-47 binding (Figs. 1B, S1, and S2). Further, there is no stabilization of interdomain contacts or domain-DNA contacts by niraparib, rucaparib, or veliparib (Figs. 1B,C [regions i, ii, and iii], S1, S2, and S3B). Under our conditions >99% of PARP-1 was expected to be inhibitor-bound based on established PARPi dissociation constants (19, 28, 30, 31), and inhibitor saturation was experimentally confirmed since each inhibitor drove stabilization of the already well-folded CAT domain at later timepoints (i.e. 104 s; Figs. 1B,C [region vi] and S1). Moreover, niraparib, rucaparib, or veliparib binding stabilized the same HD region that is destabilized when NAD+ or BAD binds. Therefore, binding affinity differences between PARPi cannot explain their widely different abilities to influence PARP-1 allostery.
DNA-binding domain stabilization corresponds to increased affinity for DNA breaks
We directly test whether the different types of allosteric communication invoked by each PARPi influenced PARP-1 affinity for a DNA break. We first compared PARPi binding to the PARP-1 CAT domain and a CAT domain bearing a deletion of the HD (CATΔHD), since the HD can regulate access to the NAD+ binding site (6). All PARPi in our study, including EB-47, bound to PARP-1 CAT and to CATΔHD (Fig. 1D), in contrast to NAD+ (or BAD) that only bind to CATΔHD (6)(see also Fig. S6). We assessed PARPi influence on PARP-1 DNA binding, and found that EB-47 caused a large increase in SSB DNA binding affinity, while talazoparib and olaparib caused only a small increase in affinity (Fig. 1E). In contrast, veliparib, rucaparib, and niraparib all caused a 3–5-fold decrease in affinity (Fig. 1E). We used SPR to assess PARPi influence on the dissociation kinetics of PARP-1 from a DNA break (Figs. 1F and S7). The dissociation rate constants (kd) for PARP-1 in the presence of PARPi (Figs. 1F and S7G and Table S1) followed the same trend as our DNA binding assay (Fig. 1E): EB-47 markedly slowed release from DNA, talazoparib and olaparib slowed release to a smaller extent, and veliparib, rucaparib, and niraparib slightly accelerated release.
Classification of PARPi by their allosteric effects on PARP-1
The different PARPi tested divide into three types (Fig. 2A) based on their allosteric effects that we measured through HXMS and DNA binding studies: Type I (EB-47 and BAD), Type II (talazoparib and olaparib), and Type III (rucaparib, niraparib, and veliparib). Each of the tested clinical PARPi have the potential to trap PARP-1 on DNA based on their potency for inhibiting PARP-1 catalytic automodification that is required for rapid release of PARP-1 from an SSB (Fig. 2A, gray shading)(21, 22, 27). On top of catalytic inhibition, Type I inhibitors have a strong allosteric effect, driving instability in the HD (Figs. 1B and 2B). HD instability leads to allosteric changes that culminate in stronger contacts with the DNA break and slower release (Fig. 1B,E,F). Type II inhibitors are relatively silent/neutral toward PARP-1 allostery, producing very small increases in affinity for a DNA break. Type III inhibitors stabilize αB and αF helices while leading to increased dynamics in αC and αD helices (Fig. 2B,C). Thus, Type III inhibitors also induce HD changes, but in an opposite nature to Type I inhibitors (Fig. 2B,C), instead driving release from the DNA break.
Figure 2. Three distinct types of PARPi based on the presence and outcome of allosteric changes in PARP-1.
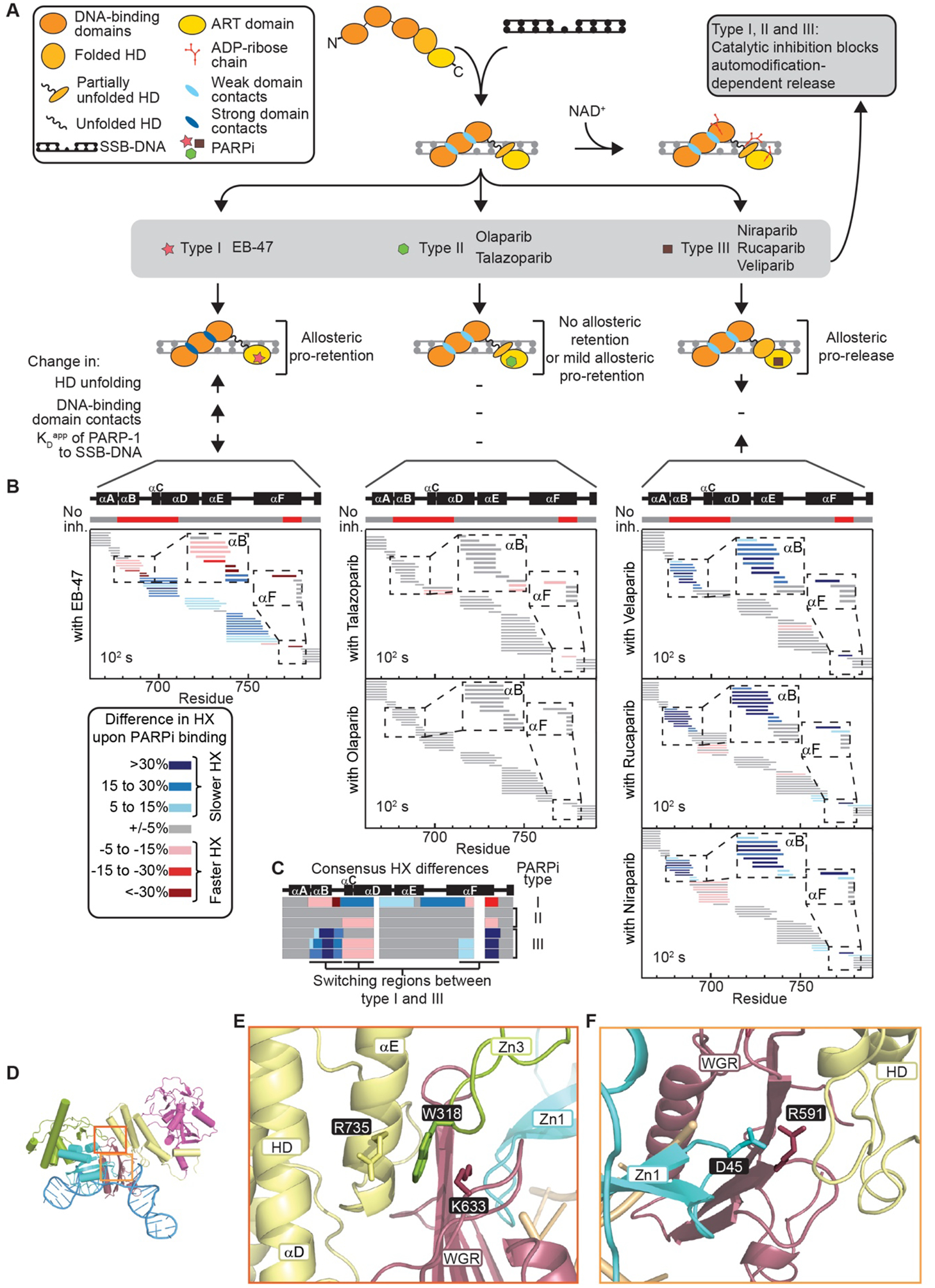
(A) Classification of PARPi into three types based on their mechanistic effects on the PARP-1/SSB-DNA complex. (B) Percent difference in HX upon binding to the indicated form of PARPi at 102 s in the HD region of PARP-1. The horizontal bar on top of the plots indicates in red the regions with faster HX when PARP-1 binds to SSB-DNA (“No inh.” – no inhibitor). Each small horizontal bar represents a peptide in our data set in the indicated region of PARP-1 with coloring indicating the HX difference upon binding each inhibitor. (C) A plot of the consensus HX behavior over the region shown in panel B is shown for each of the inhibitors, highlighting the regions where there is reciprocal behavior in Type I versus Type III PARPi. (D-F) PARP-1 structure (D) highlighting the allosteric pathway and key amino acids involved in interdomain interactions between WGR-Zn3-HD (E) and Zn1-WGR-HD domains (F).
Type I PARPi require the PARP-1 allosteric communication pathway
We disrupted key PARP-1 domain contacts to verify that Type I PARPi act by influencing the PARP-1 allosteric network. We chose two mutants at the connection points between the HD and the DNA binding domains: W318R (disrupts Zn3/HD interaction; Fig. 2D,E)(4, 5, 32), and R591C (disrupts Zn1/WGR/HD interaction; Fig. 2D,F)(5). Further, R591C was recently identified in a patient with de novo PARPi resistance and shown to reduce the extent to which talazoparib “traps” PARP-1 at the site of damaged DNA (33). EB-47 binding led to destabilization of the αB and αF helices of the HD for both mutants, similar to WT PARP-1 (Figs. 3 and S8). However, unlike WT PARP-1, the allosteric connection to the DNA binding domains was broken in both mutants (Figs. 3A,B, S8A,B, and S9). Moreover, we found that while EB-47 increased the retention of WT PARP-1 on DNA, both W318R and R591C were unaffected by EB-47 treatment (Figs. 3C, S8C, and S10). Together, these experiments support the hypothesis that Type I PARPi directly engage the PARP-1 allosteric pathway to drive pro-retention on SSB DNA.
Figure 3. Reverse allostery conferred by a Type I PARPi initiates via contact with the HD.

(A) Percent difference in HX upon binding EB-47 is calculated for each peptide in R591C and D766/770A mutant versions of PARP-1 and displayed in a similar manner as in Fig. 2B, except that the entirety of the protein is shown. A plot of the consensus HX differences for WT PARP-1 is shown on the top, and the consensus plot is also shown for R591C and D766/770A. (B) HX of a specific peptide from the αB helix of the HD for the WT (left), R591C (middle) and D766/770A (right) versions of PARP-1 in the indicated conditions. Each experiment was performed in triplicate and * indicates timepoints with a p-value <0.01 for the t-test between PARP-1/SSB-DNA experiments with or without EB-47. Error bars represent s.d. from three measurements. (C) FP DNA competition experiments with WT, R591C, and D766/770A versions of PARP-1 bound to SSB-DNA with or without EB-47. Each experiment was performed three times. Student’s t-test was performed at 60 s and 300 s to compare the percentage of DNA-bound PARP-1 between samples with or without EB-47 for each version of protein. The p-value was <0.001 at both times for the wtPARP-1, but it was not significant (>0.01) for all the mutant samples. (D) Crystal structure of PARP-1 CAT domain in complex with EB-47 (see also Table S2). D766 and D770 amino acids are shown to highlight the clash between EB-47 and αF helix of the HD (see also Fig. S13).
PARPi contact with the HD modulates PARP-1 SSB retention
We determined a crystal structure of Type I PARPi, EB-47, bound to the CAT domain of PARP-1, revealing that it clashes with the HD near two aspartic acid residues, D766/770, on helix αF (Figs. 3D). When these EB-47 contacting residues were mutated to alanines (D766/770A), EB-47 no longer influenced PARP-1 allostery in HX experiments (Fig. 3A,B). Neither EB-47 binding nor the DNA binding ability of the D766/770A mutant was perturbed (Figs. S11 and S12). The D766/770A mutant of PARP-1 was released from a DNA break in a manner that was insensitive to EB-47, in contrast to WT PARP-1 (Fig. 3C). Thus, disruption of specific HD contacts abolishes EB-47 ability to influence PARP-1 allostery and retention on a DNA break, essentially converting EB-47 from a Type I to a Type II PARPi.
Converting a Type III PARPi into a Type I PARPi
The low cellular PARP-1 trapping capacity of Type III PARPi veliparib is generally thought to be at the heart of its weaker single agent activity (relative to talazoparib and olaparib) in ovarian cancer (22, 34, 35). Veliparib is predicted to lack HD contacts when bound to PARP-1 (Fig. 4A). We developed a series of veliparib variants (design and synthesis to be published elsewhere) to introduce HD contacts that might influence PARP-1 allostery and retention on a DNA break. One such compound, UKTT15 (Fig. 4B), was found to cause strong PARP-1 retention on a DNA break (Figs. 4C and S7G and Table S1), and a 3.5-fold increase in PARP-1 affinity for DNA relative to no inhibitor (Fig. 4D). We found that UKTT15 yielded profound changes to the HX behavior of PARP-1, relative to those measured in the presence of veliparib (Fig. 4E). In particular, HX was increased in the same locations as the increases observed with Type I PARPi (Fig. 1B,C [region iv]). Moreover, there were decreases in HX in the interdomain contacts of PARP-1, extending to the DNA binding domains (Figs. 1C [regions i, ii, and iii] and 4E), also consistent with Type I PARPi behavior.
Figure 4. Converting a Type III PARPi to Type I.
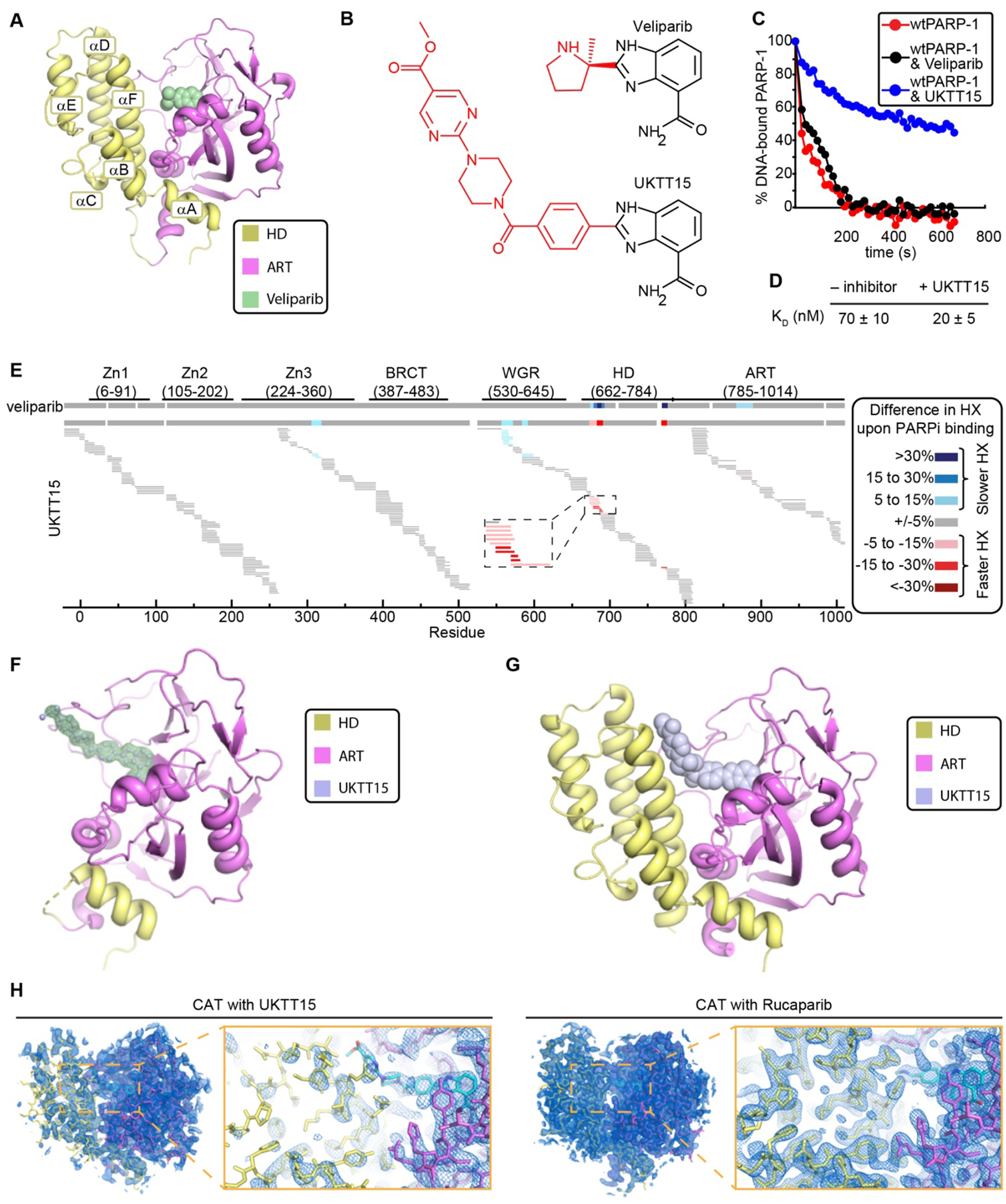
(A) Crystal structure of PARP-1 CAT domain in complex with veliparib (PDB code 2RD6). (B) Chemical structures of veliparib and UKTT15. (C) FP DNA competition experiments with WT PARP-1 in complex with SSB-DNA in the presence or absence of the indicated compounds. Each experiment was performed three times. A student’s t-test of the release difference caused by the presence of PARPi was calculated at 60 s and 300 s and had a p-value <0.01 at both times for UKTT15, but was not significant (>0.01) for veliparib. (D) KD measurements derived from FP DNA binding assay for WT PARP-1 and SSB-DNA in the presence or absence of UKTT15. (E) Percentage HX difference for each peptide of the WT PARP-1/SSB-DNA complex upon binding of UKTT15 inhibitor (data presented as in Fig. 3A). The consensus HX difference plot for the binding of the parent compound, veliparib, is shown on top for comparison. (F) Crystal structure of PARP-1 CATΔHD in complex with UKTT15. A weighted FO-FC difference electron density map (green) is shown contoured at 3s around UKTT15 (light blue sticks), illustrating the density present prior to the modeling of UKTT15. (G) Crystal structure of PARP-1 CAT domain in complex with UKTT15 (light blue spheres), in which the compound adopts a similar overall conformation as that observed in panel F (see Fig. S13D for comparison). (H) Weighted 2FO-FC electron density maps contoured at 1σ with the designated CAT/PARPi complexes overlaid, and zoomed views around the PARPi/αF helix region. The ART and HD domains are well represented in the electron density of the CAT/rucaparib complex, whereas in the CAT/UKTT15 complex the HD density is weak relative to the ART, most likely reflecting a high level of mobility associated with binding UKTT15.
We determined a crystal structure of CATΔHD bound to UKTT15 (Fig. 4F). UKTT15 engages the PARP-1 NAD+-binding pocket in a manner similar to veliparib; however, the additional chemical groups significantly extend the UKTT15 structure (Fig. 4F and Table S2). A crystal structure of PARP-1 CAT bound to UKTT15 was also determined (Fig. 4G and Table S2), illustrating how the HD is perturbed in accommodating the extended structure of UKTT15 (Figs. 4H, S13C, and S14). The CAT/EB-47 and CAT/UKTT15 complexes both exhibit HD structural perturbations that were not present in a CAT/rucaparib complex crystallized under the same conditions and with the same overall packing arrangement (Figs. 4H and S6A). The CAT/EB-47 and CAT/UKTT15 structures thus provide a high-resolution view of the initial allosteric effects imposed by these specific PARPi structures, with the common theme of steric clashes with the HD (Fig. S14). Despite similar allosteric influences on PARP-1, UKTT15 and EB-47 form distinct contacts with helix αF of the HD. Indeed, the PARP-1 mutant D766/770A that was resistant to EB-47 influence on DNA break retention was still retained on DNA in the presence of UKTT15 (Fig. S15). HX analysis was also consistent with the HD contact differences between UKTT15 and EB-47. UKTT15 contacts the N-terminal portion of helix αF, and this region exhibited faster hydrogen/deuterium exchange in the presence of EB-47 than in the presence of UKTT15 (Figs. S2B and S16A).
We tested whether the Type I PARPi behavior of UKTT15, which is opposite to the Type III behavior of veliparib, would translate to better PARP-1 trapping in cells. Chromatin fractionation assays monitor PARP-1 trapping in cells and have established that veliparib is a poor PARP-1 trapper (21, 27, 36). Compared to veliparib, UKTT15 exhibited an increased ability to trap PARP-1 on DNA in MMS-treated CAPAN-1 cells (Fig. 5A), a pancreatic cancer cell line carrying a deleterious mutation in the BRCA2 gene. The level of UKTT15-mediated trapping did not approach the level reached by talazoparib, which was reported to be the most efficient trapper of the clinical inhibitors (27). The efficient trapping of talazoparib is likely due to its highly potent catalytic inhibition (24), and we have shown that talazoparib lacks strong allosteric retention (Fig. 2A). The increased trapping ability of UKTT15 relative to veliparib is not due to higher catalytic inhibitory potency. We found that veliparib and UKTT15 have similar IC50 values (1.5 and 2.6 nM, respectively, not shown), and UKTT15 was not better than veliparib at inhibiting ADP-ribosylation activity in cells (Fig. 5B). Consistent with increased cellular trapping potential, UKTT15 treatment led to an increase in the PARP-1-GFP residence time at sites of UV laser-induced damage, comparable to that caused by talazoparib treatment and in a manner that was dose dependent (Fig. 5C,D). In contrast, veliparib led only to a marginal increase of the residence time (Figs. 5D and S17).
Figure 5. Toxic effect of PARPi in cancer cells can be tuned through modulation of allosteric SSB-DNA retention without altering enzymatic inhibition.

(A) Chromatin fractionation assay for cells in the presence of increasing concentration of veliparib, talazoparib or UKTT15 (1, 10 and 25 μM) and treated with 0.01% MMS. (B) PAR levels in cells treated with the inhibitor veliparib, talazoparib or UKTT15 at 0.01, 0.1, 1 and 10 μM in the presence of 0.01% MMS. (C) Kinetics of PARP-1 trapping at sites of DNA damage in cells. CAL51 PARP-1−/− cells were transfected with PARP-1-GFP and exposed to localized microirradiation. After microirradiation, PARP-1 localization was monitored over time. The experiment was performed in the presence of talazoparib, veliparib, or UKTT15, as well as a DMSO control representing no inhibitor. Student’s t-test was performed at the 150 s time point, comparing the control (DMSO) to the indicated inhibitors, and p-value was <0.0001 in all cases. (D) Dose dependence of kinetics of PARP-1 trapping at sites of DNA damage in cells in presence of the indicated concentrations of UKTT15. Student’s t-test was performed at the 150 s time point, comparing the control (DMSO) to the indicated concentrations of UKTT15, and p-value was <0.0001 in all cases. (E) Survival assay for SUM149PT-BRCA1mut and SUM149PT-BRCA1rev cells. Students t-test was used to calculate p-score between mutant and reverted cell lines for both PARPi. Each concentration point with statistically significant difference (p-value<0.01) is denoted with * of corresponding color. P-values were also calculated between assays using SUM149PT-BRCA1mut cells with different inhibitors and statistically significant differences were indicated with cyan asterisk. (F) Survival assays for the parental or PARP-1−/− cells in the presence of indicated concentrations of UKTT15. Students t-test was used to calculate p-score between parental and PARP-1−/− cell lines.
We next tested whether the increased trapping ability of UKTT15 compared to veliparib improved its ability to kill cancer cells. Indeed, UKTT15 was more efficient than veliparib at killing CAPAN-1 cells in a cell survival experiment (Fig. S18A,B). We also assessed the BRCA selectivity of UKTT15 in a SUM149PT-BRCA1rev cell line model, where the deleterious BRCA1 mutation is reverted to form a functional BRCA1 allele (37, 38). UKTT15 showed killing of the SUM149-BRCA1mut cells at a lower concentration compared to the SUM149PT-BRCA1rev cells (Fig. 5E). A similar result was obtained for veliparib, albeit at higher concentrations of drug (Fig. 5E). These results suggest that veliparib and UKTT15 have similar specificities for killing BRCA deficient cells, but that UKTT15 can achieve this synthetic lethality at a lower drug concentration. The effective UKTT15 concentration is thus more in the range of niraparib, rucaparib, and olaparib but still higher than talazoparib (Fig. S19). In addition, cell killing at low UKTT15 concentrations required PARP-1 expression (Figs. 5F and S20), indicating that it maintains on-target PARP-1 specificity at those low concentrations. Taking our biophysical (Fig. 4) and biochemical (Fig. 5) data into consideration, we conclude that the increased potency of UKTT15 for cell killing relative to veliparib is due to its increased pro-SSB DNA retention behavior on SSB DNA. Thus, our efforts with chemical derivatives/variants of veliparib provide a clear example of structure-based modification of PARPi to influence its allosteric behavior.
Discussion
Our work directly addresses the impact that clinical inhibitors have on PARP-1 allostery and trapping potential. The concept of “reverse allostery” was first proposed to explain why certain PARPi exhibit greater efficiency in moving PARP-1 to an insoluble chromatin-bound nuclear fraction of cells – a redistribution termed PARP-1 trapping (21). However, none of the clinical PARPi exhibited reverse allostery, and Type III inhibitors actually influenced PARP-1 allostery in a manner that was not envisioned – weakening of PARP-1 interaction with DNA damage. Our results with UKTT15 already demonstrate that reverse allostery can indeed play a role in cellular trapping of PARP-1 and can increase PARPi potency toward killing cancer cells. In addition to specificity, catalytic inhibition potency, and pharmacodynamics/pharmacokinetics, reverse allostery thus represents a fourth arm in assessing and developing new compounds that could be better forms of PARPi. Fine tuning of reverse allostery and trapping could also benefit the development of PARPi for treatment of other diseases, such as cardiovascular disease that benefit from PARP-1 inhibition but not cell death, where a Type III inhibitor with less potent trapping might be best to consider.
While we show that reverse allostery plays an important role in determining the overall efficiency of some inhibitors, other factors come into play to explain the trapping potency of some Type II and Type III inhibitors (Fig. 6). For instance, talazoparib and olaparib are more efficient at trapping PARP-1 on DNA, not because of a reverse allosteric effect, but rather due to the absence of the allosteric pro-release effect that is observed for some other PARPi. The lack of an influence on PARP-1 allostery by talazoparib and olaparib is thus combined with a different level of inhibitory potency and binding attributes that collectively enhance PARP-1 trapping efficiency. Our study gives a clear indication that the mode of PARPi binding can also influence persistence at DNA breaks through modulation of PARP-1 allostery, and this contributes to the ultimate ranking of trapping potential for PARPi.
Figure 6. Model for allosteric and non-allosteric contributions to cellular toxicity by Type I, II, and III PARPi.

PARP-1 trapping in a cancer cell generates a lesion that becomes toxic over time. The effectiveness of enzymatic inhibition is a key contributor for all three PARPi Types (vertical dimension of plot). The three PARPi Types vary by how they influence the allosteric communication that culminates in a propensity to either be released or retained at the site of broken DNA (horizontal dimension). Tuning PARPi towards pro-retention drives trapping and cancer cell killing. Tuning towards pro-release may limit trapping and preserve cell viability, which is relevant for PARPi use in other disease contexts (e.g. cardiovascular disease and several common neurodegenerative diseases).
In closing, our study provides fundamental insights into PARP-1 allostery and the influence of PARPi on the multi-domain structure of PARP-1 in complex with a DNA break. These insights have allowed a classification of PARPi based on the extent to which and the manner in which they perturb PARP-1 allostery and in turn interaction with DNA damage. We have connected this classification to differences in cellular trapping of PARP-1 and cellular toxicity. The potential to now create tunable PARPi will lead to improved clinical outcomes depending upon whether PARP-1 trapping is either desirable or undesirable in specific patients.
Materials and methods
HXMS measurements
HXMS experiments were performed by mixing PARP-1 (2.6 μM), SSB-DNA (5 μM) and an inhibitor (5.2 μM) where applicable. Deuterium on-exchange was carried out at RT by mixing 5 μL of each sample with 15 μL of deuterium on-exchange buffer (10 mM HEPES, pD 7.0, 150 mM NaCl, in D2O) yielding a final D2O concentration of 75%. Exchange reaction was quenched by mixing sample (20 μL) with 30 μL of ice-cold quench buffer and freezing in liquid nitrogen until further use. For the MS analysis, each sample (50 μL) was melted on ice, digested using pepsin column and separated on C18 analytical column. The effluent was electrosprayed into the mass spectrometer. Resulting MS data was analyzed to calculate the deuteration level in each peptide.
Differential scanning fluorimetry (DSF)
Protein melting temperature (TM) was measured using DSF. ΔTM values were calculated by subtracting the TM determined for the protein in the absence of compound from theT M determined in the presence of compound.
Fluorescence polarization
DNA competition assays and DNA binding affinity measurements were performed using FAM-labeled SSB-DNA, PARP-1, unlabeled SSB-DNA and an inhibitor where applicable.
Surface plasmon resonance (SPR)
SPR experiments were performed to assess binding specificity, kinetics, and affinity of PARP-1 towards SSB-DNA in presence of different inhibitors.
Chromatin fractionation
Chromatin fractionation assays measured the levels of chromatin-bound PARP-1 in the presence or absence of different PARPi.
Measurement of PAR in cells
PAR levels in the presence of different PARPi were measured by western blot using the anti-pan-ADP-ribose binding reagent.
Cell survival assay
Cell survival assays were performed using different PARPi in CAPAN-1 and SUM149PT cells.
Microirradiation assays
Microirradiation assays were used to analyze PARP-1 localization and dissipation at DNA damage site in cells.
Protein crystallization and structure determination
PARP-1 CATΔHD or PARP-1 CAT WT were crystallized in complex with the indicated PARPi compounds and the resulting X-ray diffraction data were used to determine structures by molecular replacement using existing models in the PDB.
Supplementary Material
Acknowledgments
We thank J. Dawicki-McKenna (Penn) and L. Mayne (Penn) for discussion and assistance, J. Brody (Thomas Jefferson University) for providing CAPAN-1 cells, and R. Greenberg (Penn) for providing SUM149PT cells and for manuscript feedback.
Funding: We acknowledge support from the Canadian Institute of Health Research (BMA342854 to J.M.P), the US National Institutes of Health (R35GM130302 to B.E.B. and F32GM128265 to L.Z.), a Basser Center for BRCA Award (to B.E.B.), the Department of Pharmaceutical Sciences and seed grant program of St. John’s University (579-1110-6709 to T.T.T.), and Breast Cancer Now (CTR-Q4-Y2 to C.J.L.) and Cancer Research UK (CRUK/A14276 to C.J.L.). The McGill SPR-MS Facility thanks the Canada Foundation for Innovation (CFI) for infrastructure support. Beamlines 8.3.1 and 12.3.1 of the Advanced Light Source, a DOE Office of Science User Facility under Contract No. DE-AC02-05CH11231, is supported in part by the ALS-ENABLE program funded by the National Institutes of Health (P30 GM124169-01). Efforts to apply crystallography to characterize eukaryotic pathways relevant to human cancers are supported in part by National Cancer Institute grant Structural Biology of DNA Repair (SBDR) CA92584.
Footnotes
Competing interests: C.J.L. received research funding from: AstraZeneca, Merck KGaA, Artios. C.J.L. received consultancy, SAB membership or honoraria payments from: Sun Pharma, GLG, Merck KGaA, Vertex, AstraZeneca, Tango, 3rd Rock, Ono Pharma, Artios. C.J.L. has stock in Tango. C.J.L. is also a named inventor on patents describing the use of DNA repair inhibitors and stands to gain from the development as part of the ICR “Rewards to Inventors” scheme. T.T.T., J.M.P., and B.E.B. are co-founders of Hysplex, LLC, with interests in PARPi development.
Data and materials availability: The atomic coordinates and structure factor amplitudes have been deposited in the Protein Data Bank under the accession codes 6NTU, 6VKO, 6VKQ, and 6VKK, and image files have been deposited in SBGrid Data Bank. All other data needed to evaluate our conclusions are included in the main paper or in the supplementary materials.
References and Notes
- 1.D’Amours D, Desnoyers S, D’Silva I, Poirier GG, Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J 342 (Pt 2), 249–268 (1999). [PMC free article] [PubMed] [Google Scholar]
- 2.Gupte R, Liu Z, Kraus WL, PARPs and ADP-ribosylation: recent advances linking molecular functions to biological outcomes. Genes Dev 31, 101–126 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pascal JM, Ellenberger T, The rise and fall of poly(ADP-ribose): An enzymatic perspective. DNA Repair. 32, 10–16 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dawicki-McKenna JM, Langelier M-F, DeNizio JE, Riccio AA, Cao CD, Karch KR, McCauley M, Steffen JD, Black BE, Pascal JM, PARP-1 Activation Requires Local Unfolding of an Autoinhibitory Domain. Mol. Cell. 60, 755–768 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Langelier M-F, Planck JL, Roy S, Pascal JM, Structural basis for DNA damage-dependent poly(ADP-ribosyl)ation by human PARP-1. Science. 336, 728–732 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Langelier M-F, Zandarashvili L, Aguiar PM, Black BE, Pascal JM, NAD+ analog reveals PARP-1 substrate-blocking mechanism and allosteric communication from catalytic center to DNA-binding domains. Nat. Commun 9, 844 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jagtap P, Szabó C, Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat. Rev. Drug Discov 4, 421–440 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Martinez-Zamudio R, Ha HC, Histone ADP-ribosylation facilitates gene transcription by directly remodeling nucleosomes. Mol. Cell. Biol 32, 2490–2502 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Satoh MS, Lindahl T, Role of poly(ADP-ribose) formation in DNA repair. Nature 356, 356–358 (1992). [DOI] [PubMed] [Google Scholar]
- 10.Gibson BA, Zhang Y, Jiang H, Hussey KM, Shrimp JH, Lin H, Schwede F, Yu Y, Kraus WL, Chemical genetic discovery of PARP targets reveals a role for PARP-1 in transcription elongation. Science. 353, 45–50 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim MY, Mauro S, Gévry N, Lis JT, Kraus WL, NAD+-dependent modulation of chromatin structure and transcription by nucleosome binding properties of PARP-1. Cell. 119, 803–814 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Tulin A, Spradling A, Chromatin loosening by poly(ADP)-ribose polymerase (PARP) at Drosophila puff loci. Science. 299, 560–562 (2003). [DOI] [PubMed] [Google Scholar]
- 13.Ménissier de Murcia J, Ricoul M, Tartier L, Niedergang C, Huber A, Dantzer F, Schreiber V, Amé J-C, Dierich A, LeMeur M, Sabatier L, Chambon P, de Murcia G, Functional interaction between PARP-1 and PARP-2 in chromosome stability and embryonic development in mouse. EMBO J. 22, 2255–2263 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dedes KJ, Wilkerson PM, Wetterskog D, Weigelt B, Ashworth A, Reis-Filho JS, Synthetic lethality of PARP inhibition in cancers lacking BRCA1 and BRCA2 mutations. Cell Cycle Georget. Tex 10, 1192–1199 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lord CJ, Ashworth A, PARP inhibitors: Synthetic lethality in the clinic. Science. 355, 1152–1158 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lord CJ, McDonald S, Swift S, Turner NC, Ashworth A, A high-throughput RNA interference screen for DNA repair determinants of PARP inhibitor sensitivity. DNA Repair. 7, 2010–2019 (2008). [DOI] [PubMed] [Google Scholar]
- 17.Mizuarai S, Kotani H, Synthetic lethal interactions for the development of cancer therapeutics: biological and methodological advancements. Hum. Genet 128, 567–575 (2010). [DOI] [PubMed] [Google Scholar]
- 18.Shaheen M, Allen C, Nickoloff JA, Hromas R, Synthetic lethality: exploiting the addiction of cancer to DNA repair. Blood. 117, 6074–6082 (2011). [DOI] [PubMed] [Google Scholar]
- 19.Shen Y, Rehman FL, Feng Y, Boshuizen J, Bajrami I, Elliott R, Wang B, Lord CJ, Post LE, Ashworth A, BMN 673, a novel and highly potent PARP1/2 inhibitor for the treatment of human cancers with DNA repair deficiency. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res 19, 5003–5015 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ashworth A, Lord CJ, Synthetic lethal therapies for cancer: what’s next after PARP inhibitors? Nat. Rev. Clin. Oncol (2018). [DOI] [PubMed] [Google Scholar]
- 21.Murai J, Huang SN, Das BB, Renaud A, Zhang Y, Doroshow JH, Ji J, Takeda S, Pommier Y, Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res 72, 5588–5599 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pommier Y, O’Connor MJ, de Bono J, Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med 8, 362ps17 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Schoonen PM, Talens F, Stok C, Gogola E, Heijink AM, Bouwman P, Foijer F, Tarsounas M, Blatter S, Jonkers J, Rottenberg S, van Vugt MATM, Progression through mitosis promotes PARP inhibitor-induced cytotoxicity in homologous recombination-deficient cancer cells. Nat. Commun 8, 15981 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hopkins TA, Shi Y, Rodriguez LE, Solomon LR, Donawho CK, DiGiammarino EL, Panchal SC, Wilsbacher JL, Gao W, Olson AM, Stolarik DF, Osterling DJ, Johnson EF, Maag D, Mechanistic Dissection of PARP1 Trapping and the Impact on In Vivo Tolerability and Efficacy of PARP Inhibitors. Mol. Cancer Res. MCR 13, 1465–1477 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Owonikoko TK, Zhang G, Deng X, Rossi MR, Switchenko JM, Doho GH, Chen Z, Kim S, Strychor S, Christner SM, Beumer J, Li C, Yue P, Chen A, Sica GL, Ramalingam SS, Kowalski J, Khuri FR, Sun S-Y, Poly (ADP) ribose polymerase enzyme inhibitor, veliparib, potentiates chemotherapy and radiation in vitro and in vivo in small cell lung cancer. Cancer Med 3, 1579–1594 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.ZEJULA (niraparib) capsules, for oral use Initial U.S https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/208447lbl.pdf (2017).
- 27.Murai J, Huang S-YN, Renaud A, Zhang Y, Ji J, Takeda S, Morris J, Teicher B, Doroshow JH, Pommier Y, Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol. Cancer Ther 13, 433–443 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jagtap PG, Southan GJ, Baloglu E, Ram S, Mabley JG, Marton A, Salzman A, Szabó C, The discovery and synthesis of novel adenosine substituted 2,3-dihydro-1H-isoindol-1-ones: potent inhibitors of poly(ADP-ribose) polymerase-1 (PARP-1). Bioorg. Med. Chem. Lett 14, 81–85 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Qiu W, Lam R, Voytyuk O, Romanov V, Gordon R, Gebremeskel S, Vodsedalek J, Thompson C, Beletskaya I, Battaile KP, Pai EF, Rottapel R, Chirgadze NY, Insights into the binding of PARP inhibitors to the catalytic domain of human tankyrase-2. Acta Crystallogr. D Biol. Crystallogr 70, 2740–2753 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG, PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer. 10, 293–301 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen Y, Aoyagi-Scharber M, Wang B, Trapping Poly(ADP-Ribose) Polymerase. J. Pharmacol. Exp. Ther 353, 446–457 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Langelier M-F, Ruhl DD, Planck JL, Kraus WL, Pascal JM, The Zn3 domain of human poly(ADP-ribose) polymerase-1 (PARP-1) functions in both DNA-dependent poly(ADP-ribose) synthesis activity and chromatin compaction. J. Biol. Chem 285, 18877–18887 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pettitt SJ, Krastev DB, Brandsma I, Dréan A, Song F, Aleksandrov R, Harrell MI, Menon M, Brough R, Campbell J, Frankum J, Ranes M, Pemberton HN, Rafiq R, Fenwick K, Swain A, Guettler S, Lee J-M, Swisher EM, Stoynov S, Yusa K, Ashworth A, Lord CJ, Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat. Commun 9, 1849 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Coleman RL, Sill MW, Bell-McGuinn K, Aghajanian C, Gray HJ, Tewari KS, Rubin SC, Rutherford TJ, Chan JK, Chen A, Swisher EM, A phase II evaluation of the potent, highly selective PARP inhibitor veliparib in the treatment of persistent or recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer in patients who carry a germline BRCA1 or BRCA2 mutation - An NRG Oncology/Gynecologic Oncology Group study. Gynecol. Oncol 137, 386–391 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Loibl S, O’Shaughnessy J, Untch M, Sikov WM, Rugo HS, McKee MD, Huober J, Golshan M, von Minckwitz G, Maag D, Sullivan D, Wolmark N, McIntyre K, Ponce Lorenzo JJ, Metzger Filho O, Rastogi P, Symmans WF, Liu X, Geyer CE, Addition of the PARP inhibitor veliparib plus carboplatin or carboplatin alone to standard neoadjuvant chemotherapy in triple-negative breast cancer (BrighTNess): a randomised, phase 3 trial. Lancet Oncol 19, 497–509 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Murai J, Zhang Y, Morris J, Ji J, Takeda S, Doroshow JH, Pommier Y, Rationale for poly(ADP-ribose) polymerase (PARP) inhibitors in combination therapy with camptothecins or temozolomide based on PARP trapping versus catalytic inhibition. J. Pharmacol. Exp. Ther 349, 408–416 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chabanon RM, Muirhead G, Krastev DB, Adam J, Morel D, Garrido M, Lamb A, Hénon C, Dorvault N, Rouanne M, Marlow R, Bajrami I, Cardeñosa ML, Konde A, Besse B, Ashworth A, Pettitt SJ, Haider S, Marabelle A, Tutt AN, Soria J-C, Lord CJ, Postel-Vinay S, PARP inhibition enhances tumor cell-intrinsic immunity in ERCC1-deficient non-small cell lung cancer. J. Clin. Invest 129, 1211–1228 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dréan A, Williamson CT, Brough R, Brandsma I, Menon M, Konde A, Garcia-Murillas I, Pemberton HN, Frankum J, Rafiq R, Badham N, Campbell J, Gulati A, Turner NC, Pettitt SJ, Ashworth A, Lord CJ, Modeling Therapy Resistance in BRCA1/2-Mutant Cancers. Mol. Cancer Ther 16, 2022–2034 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eustermann S, Wu W-F, Langelier M-F, Yang J-C, Easton LE, Riccio AA, Pascal JM, Neuhaus D, Structural Basis of Detection and Signaling of DNA Single-Strand Breaks by Human PARP-1. Mol. Cell. 60, 742–754 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Penning TD, Zhu G-D, Gandhi VB, Gong J, Liu X, Shi Y, Klinghofer V, Johnson EF, Donawho CK, Frost DJ, Bontcheva-Diaz V, Bouska JJ, Osterling DJ, Olson AM, Marsh KC, Luo Y, Giranda VL, Discovery of the Poly(ADP-ribose) polymerase (PARP) inhibitor 2-[(R)-2-methylpyrrolidin-2-yl]-1H-benzimidazole-4-carboxamide (ABT-888) for the treatment of cancer. J. Med. Chem 52, 514–523 (2009). [DOI] [PubMed] [Google Scholar]
- 41.Hopkins TA, Ainsworth WB, Ellis PA, Donawho CK, DiGiammarino EL, Panchal SC, Abraham VC, Algire MA, Shi Y, Olson AM, Johnson EF, Wilsbacher JL, Maag D, PARP1 Trapping by PARP Inhibitors Drives Cytotoxicity in Both Cancer Cells and Healthy Bone Marrow. Mol. Cancer Res. MCR (2018). [DOI] [PubMed] [Google Scholar]
- 42.Liu L, Kong M, Gassman NR, Freudenthal BD, Prasad R, Zhen S, Watkins SC, Wilson SH, Van Houten B, PARP1 changes from three-dimensional DNA damage searching to one-dimensional diffusion after auto-PARylation or in the presence of APE1. Nucleic Acids Res 45, 12834–12847 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Langelier M-F, Riccio AA, Pascal JM, PARP-2 and PARP-3 are selectively activated by 5’ phosphorylated DNA breaks through an allosteric regulatory mechanism shared with PARP-1. Nucleic Acids Res 42, 7762–7775 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Myszka DG, Improving biosensor analysis. J. Mol. Recognit. JMR. 12, 279–284 (1999). [DOI] [PubMed] [Google Scholar]
- 45.Karlsson R, Katsamba PS, Nordin H, Pol E, Myszka DG, Analyzing a kinetic titration series using affinity biosensors. Anal. Biochem 349, 136–147 (2006). [DOI] [PubMed] [Google Scholar]
- 46.Mortusewicz O, Schermelleh L, Walter J, Cardoso MC, Leonhardt H, Recruitment of DNA methyltransferase I to DNA repair sites. Proc. Natl. Acad. Sci. U. S. A 102, 8905–8909 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kabsch W, XDS. Acta Crystallogr. D Biol. Crystallogr 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ, Phaser crystallographic software. J. Appl. Crystallogr 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH, PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS, Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr 67, 235–242 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Emsley P, Lohkamp B, Scott WG, Cowtan K, Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Murshudov GN, Skubák P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA, REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr 67, 355–367 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Langelier M-F, Pascal JM, PARP-1 mechanism for coupling DNA damage detection to poly(ADP-ribose) synthesis. Curr. Opin. Struct. Biol 23, 134–143 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Langelier M-F, Planck JL, Roy S, Pascal JM, Crystal structures of poly(ADP-ribose) polymerase-1 (PARP-1) zinc fingers bound to DNA: structural and functional insights into DNA-dependent PARP-1 activity. J. Biol. Chem 286, 10690–10701 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Krastev DB, Pettitt SJ, Campbell J, Song F, Tanos BE, Stoynov SS, Ashworth A, Lord CJ, Coupling bimolecular PARylation biosensors with genetic screens to identify PARylation targets. Nat. Commun 9, 2016 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.