

Abstract
The onsite next generation sequencing (NGS) of Ebola virus (EBOV) genomes during the 2013–2016 Ebola epidemic in Western Africa provides an opportunity to trace the origin, transmission, and evolution of this virus. Herein, we have diagnosed a cohort of EBOV patients in Sierra Leone in 2015, during the late phase of the outbreak. The surviving EBOV patients had a recovery process characterized by decreasing viremia, fever, and biochemical parameters. EBOV genomes sequenced through the longitudinal blood samples of these patients showed dynamic intra-host substitutions of the virus during acute infection, including the previously described short stretches of 13 serial T>C mutations. Remarkably, within individual patients, samples collected during the early phase of infection possessed Ts at these nucleotide sites, whereas they were replaced by Cs in samples collected in the later phase, suggesting that these short stretches of T>C mutations could emerge independently. In addition, up to a total of 35 nucleotide sites spanning the EBOV genome were mutated coincidently. Our study showed the dynamic intra-host adaptation of EBOV during patient recovery and gave more insight into the complex EBOV-host interactions.
Keywords: Ebola virus, Intra-host Nucleotide Variation, Adaptation, Genome sequencing, Clinical manifestations, Blood biochemistry
Highlights
Scientific question
During 2013–2016 epidemic of Ebola virus disease (EVD) in Western Africa, the Ebola virus (EBOV) genomes generated from the patient specimens based on next-generation sequencing platforms in the field helped to quickly trace the origin, evolution, and transmission of EBOV. In particular, intra-host single nucleotide variations (iSNVs) appeared during the course of the epidemic across the EBOV genome. Whether and how these iSNVs could emerge from a single patient during the disease progression, are still largely unknown.
Evidence before this study
Some of the iSNVs, such as short stretches of intra-hostT to C (T>C) mutations were shared by two or more patients, which represented a combination of human-to-human transmission and recurrent mutations. Furthermore, iSNVs could appear in the key sites, such as the B cell epitopes of GP and non-coding regions across the EBOV genome, which may influence the transcription level of an adjacent gene.
New findings
In a cohort of EBOV-infected patients in the Ebola Treatment Units (ETUs) in Freetown, Sierra Leone, the recovery processes of the patients were represented by the dynamic viremia and biochemical features. By utilizing longitudinally collected samples during the recovery process, we successfully generated series of virus genomes from the patients. We observed coincident emergence of serial nucleotide variations, including the previously defined short stretch of T>C mutations during the recovery process. Phylogenetic analyses showed that after the T>C mutations occurred, all strains possessing such mutations were grouped together, but were not clustered together with their earlier sequences without such mutations from the same patient. Our results suggested such T>C mutations could arise independently within single patients, reflecting the host-adaptation of EBOV during infection.
Significance of the study
Our data indicate that short stretches of T>C substitutions are part of the convergent evolution during the infection process of EVD patients, shedding light on the dynamic intra-host genomic variation of EBOV during the 2013–2016 epidemic.
Alt-text: Unlabelled Box
1. Introduction
The 2014–2016 epidemic of Ebola virus disease (EVD) in Western Africa is the largest EVD outbreak to date, with 28,616 confirmed, probable, and suspected cases and 11,310 deaths as of June 10, 2016 (http://who.int/csr/disease/ebola/en/). During this epidemic, the ability to perform next-generation sequencing of Ebola virus (EBOV) from patient specimens in the field [1,2] has generated EBOV genomes, which helped to trace the origin, evolution, and transmission of EBOV in West Africa [[3], [4], [5], [6], [7], [8], [9]]. Based on the phylogenetic analysis, all of the EBOV in this outbreak can be traced back to a few cases in Guéckédou, Guinea. Subsequently, the virus spread to neighboring countries, such as Sierra Leone, Liberia, Nigeria, Senegal, and Mali [4,10,11].
Along with their geographical spread in West Africa, EBOV formed different lineages [12]. These lineages are characterized with different single nucleotide polymorphisms (SNPs) emerging from different time points during the outbreak [1,[4], [5], [6],8,13]. These SNPs occur in both the non-coding and encoding regions, containing indispensable phylogenetic and evolutionary information [3,7,14]. Previous research demonstrated that the hotspots for non-synonymous substitutions are likely located in regions with a lower level of functional constraint of the encoded viral proteins [3]. Moreover, the intra-host selection for EBOV to escape from a developing humoral immune response may drive the diversifying selection of glycoprotein (GP) mucin-like domain, as shown by the enrichment of mutations within the B-cell epitopes of GP [4], although this was not observed in Ni's study [15]. On the other hand, short stretches of intra-hostT to C (T>C) mutations were also observed [1,4]. This was speculated as a result of adenosine deaminases acting on RNA (ADARs), which is yet unclear [16]. It has been reported that viruses with the 13 T>C mutations (genome positions 5512–5631) continued to circulate in the Magazine Wharf area, Freetown, Sierra Leone, causing several infections [17].
In particular, intra-host single nucleotide variations (iSNVs) appeared during the course of the epidemic, within the B cell epitopes of GP and non-coding regions across the EBOV genome [4,5]. Interestingly, several iSNVs were shared by two or more patients, which represented a combination of human-to-human transmission and recurrent mutations [4]. These iSNVs were used to estimate the effective viral population size within a single patient, during a transmission bottleneck [18], and to identify human-to-human transmission chains [4,5,15]. Furthermore, two iSNVs were able to influence the transcription level of an adjacent gene of nucleocapsid protein (NP) by two-fold [15]. Recent studies also showed that, during the epidemic, the EBOV isolates from early in the outbreak with amino acid substitutions in the GP protein possessed increased tropism for human cells, indicating human adaptation of Ebola virus during human-to-human transmission [[19], [20], [21]]. However, only a few of the previous studies described longitudinal sequence data from a single patient [5,22], and the intra-host dynamic evolution of EBOV during disease progression is still largely unknown.
At the Sierra Leone-China Friendship Biological Safety Laboratory (SLE-CHN Bio-safety Lab) [23], we cared for a cohort of EBOV-infected patients (n = 29) in the Ebola Treatment Units (ETUs) in Freetown, Sierra Leone, from mid-March to late June 2015. The dynamic viremia [24] and biochemical features during disease progression were characterized. By utilizing a deep sequencing platform in the field[25], we successfully generated virus genomes from longitudinally collected blood samples from some of the patients. Surprisingly, we observed coincident emergence of serial nucleotide variations, including the previously defined short stretch of 13 T>C mutations during the recovery process. In a single patient, genome sequences obtained from samples during earlier stages of the acute infection phase possessed Ts at the 13 T>C positions, whereas Cs were found from samples collected during the recovery process. Phylogenetic analyses showed that after the T>C mutations occurred, all strains possessing such mutations were grouped together, but were not clustered together with their earlier sequences without such T>C mutations from the same patient. Our results suggested that such T>C mutations could arise independently within single patients, reflecting the host-adaptation of EBOV during infection.
2. Materials and methods
2.1. Patients
We undertook a cohort study of patients admitted to Jui Ebola treatment centre (Sierra Leone-China Friendship Hospital) between March 2015, and June 2015 [24]. We used a standard case definition consistent with WHO guidelines. All patients were included in the study except for those who died upon arrival, or those who had no blood results within 24 h of admission. Primary survival outcome measure was collected from the EBOV treatment centre.
2.2. Laboratory diagnosis and blood analysis
Samples were tested at the on-site laboratory SLE-CHNBio-safety Lab for the presence of EBOV RNA by real-timeRT-PCR against the glycoprotein (GP) and nucleoprotein (NP) gene targets [24], after inactivation and manual RNA extraction. Positive results were reported as cycle threshold values. All patients received a RT-PCR test upon admission. The Piccolo Express system (Abaxis, CA, USA) was used to generate metabolic and liver function profiles. Amylyte8 was used to assay for blood biochemistry and liver function profile in the same day as the sample collection.
2.3. Data collection and ethics statements
All data were collected as part of routine patient care, and recorded on standardized forms, which were kept securely. The clinical data extracted for research purposes were anonymized and stored on a password-protected database. The Sierra Leone ethics and scientific review committee provided approval for the study.
This work was conducted as part of the surveillance and public health response to contain the EVD outbreak in Sierra Leone. Blood samples from suspected individuals and oropharyngeal swab samples from corpses were collected for EVD testing and outbreak surveillance, with a waiver to provide written informed consent during the EVD outbreak under the agreement between the Sierra Leone and Chinese governments. The activities were coordinated by the Emergency Operations Centre in the charge of Sierra Leone Ministry of Health and Sanitation and WHO. All the information regarding individual persons has been anonymized in the report.
2.4. Genome sequencing and assembly
The genome sequencing was performed as described previously [1]. Briefly, RNA samples extracted from whole blood (two positive oropharyngeal swab samples with high Ct values were not involved) from EVD patients were reverse transcribed to cDNA. PCR amplifications were performed with 19 EBOV-specific primer pairs with overlaps. Amplicons from one patient were pooled for library preparation. NGS was performed using the BGISEQ-100 (Ion Proton) platform. All sequenced reads were filtered to remove the low quality and short reads. The genome sequences of the viruses were assembled by mapping the filtered reads to the 2014 EBOV consensus sequence (GenBank: KT013255) using Roche 454 Newbler version 2.9 (Roche), and the mutation site was manually checked with original sequencing data.
Clean reads were mapped to the EBOV genome (KJ660346) by TMAP3.4.1. We then scanned the EBOV genome site-by-site and determined the nucleotides for each genomic site according to mapping results. Finally, the ratios of four nucleotides at each site were obtained. Among the samples sequenced, 18 full-length or nearly full-length EBOV genomes were successfully generated in this study (Table S1). The other samples with only short partial of the EBOV genomes were not analyzed in this study.
2.5. Sequence analyses
All of the mutation sites were counted for each released EBOV full-length genome, using the Zaire ebolavirus isolate H.sapiens-wt/GIN/2014/Makona-Kissidougou-C15 (GenBank accession No. KJ660346) as the reference sequence. The existence of substitutions within a genome was screened within the 1078 EBOV genomes publicly available from GenBank. A “C strain” was defined as the existence of 3 or more T-C substitutions within a genome window width of 400 nt.
Our dataset included 18 full-length or nearly full-length EBOV genomes sequenced in this study and 1078 EBOV genomespublicly available from GenBank. Amaximum-likelihood phylogenetic tree was inferred using the software RAxML, with the GTRGAMMA model and 1000 bootstrap replicates.
2.6. Accession numbers
The genomes and NGS data of the 18 EBOV viruses were deposited in GenBank with the access numbers MF599504–MF599522 (Table S1).
3. Results
3.1. Clinical characteristics of the EBOV patients
From March 11th to June 28th, 2015, 473 patients who had symptoms meeting the definition of suspected EVD were admitted to the Sierra Leone-China Friendship Hospital in Freetown. The blood specimens were collected and delivered by the Sample Center of Ministry of Health and Sanitation (MOHS), Sierra Leone. The SLE-CHN Bio-safety Lab tested the blood samples for EBOV through a double-channelreal-time RT-PCR detection kit targeting both GP and NP genes of EBOV. A total of 29 (6.1%) patients were confirmed to have EVD (Table 1 ). The first blood samples collected from these patients after hospitalization were tested for laboratory confirmation, and the Ct values from the GP and NP channels matched well with a high linear correlation (Figure 1A). Of these 29 patients, the most common clinical features at presentation included fever in 23 patients (79.3%; mean temperature, 38.4 °C) and gastrointestinal symptoms, e.g. abdominal pain, in 20 patients (69.0%) and anorexia/loss of appetite in 18 patients (62.1%) (Table 1). Ocular signs were also common in the patients (13 with conjunctivitis [44.8%] and 9 with pain behind the eyes [31.0%]).
Table 1.
Baseline information of the patients at the date of hospitalization.
| Characteristics | Values (N = 29) |
|---|---|
| Male (%) | 12 (41.4) |
| Age in years, (mean ± SD) | 32.1 ± 17.6 |
| Temperature | 38.4 ± 1.3 |
| CT value | 28.4 ± 6.7 |
| Comorbidities (%) | |
| Fever | 23 (79.3) |
| Vomiting/nausea | 16 (55.2) |
| Diarrhea | 15 (51.7) |
| Intense fatigue/general weakness | 16 (55.2) |
| Anorexia/loss of appetite | 18 (62.1) |
| Abdominal pain | 20 (69.0) |
| Chest pain | 5 (17.2) |
| Muscle pain | 12 (41.4) |
| Joint pain | 16 (55.2) |
| Headache | 13 (44.8) |
| Cough | 9 (31.0) |
| Difficulty breathing | 9 (31.0) |
| Difficulty swallowing | 17 (58.6) |
| Sore throat | 9 (31.0) |
| Jaundice | 4 (13.8) |
| Conjunctivitis | 13 (44.8) |
| Skin rash | 9 (31.0) |
| Hiccups | 8 (27.6) |
| Pain behind eyes | 9 (31.0) |
| Coma | 4 (13.8) |
| Confusion | 2 (6.9) |
Figure 1.
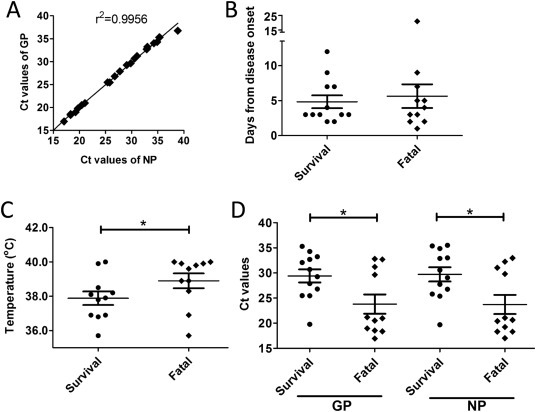
The diagnosis and initial clinical features of EBOV patients at hospitalization.
A. Correlation of the Ct values from RT-PCR testing in patient blood samples for EBOV GP and NP. B. Average days from symptom onset to initial sampling, as categorized by survival and fatal cases. C. Comparison of temperatures during initial sampling between survival and fatal cases. D. Ct values between survival and fatal cases.
The mean age of the patients was 32 years (range, 10 to 73) and 12 patients (41.4%) were male (Table 1). The case fatality rate of the 29 EVD patients was 41.4% (12/29), similar to the overall ratio of 39.5% (11,310/28,616) during this EBOV epidemic. There was no significant difference (P = 0.67) in the average interval from symptom onset to presentation between survivors and non-survivors(Figure 1B). The mean body temperature on the day of hospitalization was 37.9 °C among survivors, significantly lower (P = 0.048) than that of non-survivors, 38.9 °C (Figure 1C). The initial viremia of survivors and non-survivors was also significantly different, with non-survivors possessing lower mean Ct values for both GP (23.8 for non-survivors vs 29.4 for survivors, P = 0.023) and NP (23.8 for non-survivors vs 29.7 for survivors, P = 0.017) (Figure 1D).
3.2. The kinetics of viremia and biochemical features during EVD process
Blood samples from 12 patients (11 survivors and 1 fatality 10180) were available for longitudinal collection during the hospitalization. As in the previous study [26], the viremia of the survivors ameliorated after the presentation, as revealed by increased Ct values (Figure 2 ). Interestingly, in our study, two different trends in the variation of body temperatures of the patients after hospitalization were observed. Consistent with viremia, body temperatures of patients 4781, 8941, 9180, and 18797 decreased after hospitalization. On the other hand, in patients 6733, 8099, 18791, 18824, 18851, 18889, and 18898, a transient increase in body temperature after hospitalization was detected. Especially in patient 8099, the Ct value had a dramatic decrease from 29.6 on day 7 (hospitalization) to 19.0 on day 9 (2 days after hospitalization), and the body temperature of the patient elevated from 36.9 °C on day 7 to 38.9 °C on day 9. On day 11, the viremia and fever of this patient decreased. In contrast to the survivors, the moribund patient 10180 had a continuous viremia and the body temperature increased from 37.9 °C on day 21 to 39.8 °C on day 27. The patient died on day 30.
Figure 2.
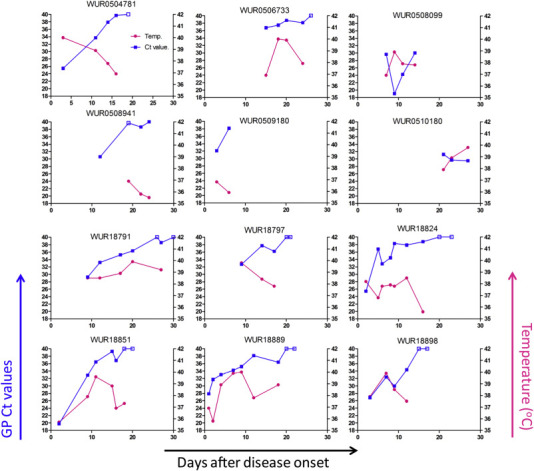
The longitudinal clinic variation of the EBOV patients during the disease process.
The EBOV shedding and body temperature. The EBOV shedding was represented by the Ct values (blue) tested by RT-PCR targeting EBOV GP in blood. The closed boxes indicate the Ct ≤ 38 (Ebola RNA Positive); while the open boxes denote the Ct>38 or no Ct available (Ebola RNA Negative).When there are two longitudinally-collected blood samples with Ebola RNA Negative, the patient will be discharged. The corresponding temperatures (purple) during the blood sampling.
Blood biochemical parameters were also tested in the SLE-CHN Bio-safety Lab in the field (Figure 3 ). Hematological and biochemical abnormalities were observed in the patients upon hospitalization, which was also reported in the previous studies (Biochemical testing in a laboratory tent and semi-intensive care of Ebola patients on-site in a remote part of Guinea: a paradigm shift based on a bleach-sensitive point-of-care device. Clinical, virological, and biological parameters associated with outcomes of Ebola virus infection in Macenta, Guinea.). Hyperglycemia occurred in patients 18791, 18824, 18851 upon hospitalization and ameliorated afterwards. Abnormal concentrations of blood urea nitrogen, potassium, sodium, and chlorine were observed in the patients, especially in patients 18791 and 18824. Hematological abnormalities detected during the disease progression included reduced concentrations of hemoglobin and hematocrit. These biochemical abnormalities alleviated gradually during the recovery process.
Figure 3.
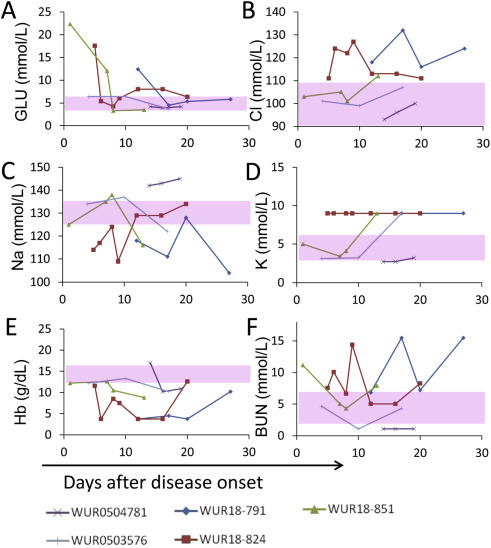
Blood biochemistry of Ebola patients.
Blood parameters were tested at the SLE-CHN Bio-safety Lab in Sierra Leone. The range of values in healthy individuals is denoted as a pink area.
3.3. Coincident substitutions during the EVD recovery process
Finally, 14 full-length and four nearly full-length EBOV genomes from 12 patients were obtained based on the NGS platform in the field. Through analysis of the NGS data of longitudinally collected blood samples from the same patient, we found that the previously reported short stretch of T>C mutations within positions 5512–5631 appeared in the late blood samples of the patients 18791 (day 20) and 18797 (days 14 and 17), while these patients still possessed Ts in early blood samples (day 12 for patient 18791 and day 9 for 18797) (Figure 4A). In particular, in the day 9 sample of patient 18797, almost no Cs were observed at the 13 positions, whereas Cs accounted for approximately 25% on day 14 and ~100% on day 17 at all of the 13 T>C nucleotide sites (Figure 4A), indicating that day 14 may be an intermediate status between days 9 and 17. The similar ratios of T:C at the different T>C nucleotide sites on the same day may indicate that these T>C mutations occurred concurrently, though not all these mutations can be found on the same nucleic acid strands (reads). However, in patient 18889, dominant percentages of Cs (>86%) were already observed on day 5, and continued increasing in percentage on day 7 (~99%) (Figure 4A).
Figure 4.
The coincident transition of the 35 coincident nucleotide variations of EBOV during the recovery process.
A. Only nucleotide positions with >100 reads were estimated. The red and black lines below the genome structure were used to highlight the specific genomic regions of EBOV with iSNVs. B–D. The similar substitution trends of the EBOV iSNV in longitudinally collected blood samples of patients 18791, 18797 and 18889. The substitution ratio of each point = the reads of the dominant iSNV at the later time/the reads of the dominant iSNV at the early time of the disease. The 50% substitution (ratio = 1) was shown in purple dashed line. The list of the iSNV sites is present in Table2. The exceptional sites which do not have the substitution trends are shown in Supplementary Figure S1.
A. Only nucleotide positions with >100 reads were estimated. The red and black lines below the genome structure were used to highlight the specific genomic regions of EBOV with iSNVs. B–D. The similar substitution trends of the EBOV iSNV in longitudinally collected blood samples of patients 18791, 18797 and 18889. The substitution ratio of each point = the reads of the dominant iSNV at the later time/the reads of the dominant iSNV at the early time of the disease. The 50% substitution (ratio = 1) was shown in purple dashed line. The list of the iSNV sites is present in Table 2. The exceptional sites which do not have the substitution trends are shown in Supplementary Figure S1.
Meanwhile, we found 22 additional coincident substitutions distributed across the entire EBOV genome (Table2 and Figure 4), which has the similar ratios for the substitutions as in the short stretch of the 13 T>C mutations between positions 5512 and 5631. This may also indicate that all these substitutions were correlated to each other. These nucleotide variations not only included T>C substitutions, but also other types of substitutions, e.g. C>T, G>A, A>G, A>T,T>A, and even one T>Deletion mutation at site 10133 (TableS2). These nucleotide variations occurred coincidently among different patients during the acute infection phase.
Table 2.
The linked and convergent substitutions of Ebola viruses emerged during human infection.
| Number | Site | Type | Former | Latter | 18791 |
18797 |
18889 |
C stains (5512–5631)h | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Day 12 | Day 20 | Figure | Day 9 | Day 14 | Day 17 | Figure | Day 5 | Day 7 | Figure | ||||||
| 1 | 1958 | NP(S497P)a | T | C | 0.01b | 289.00b | 1c | 0.01 | 207.00 | 1000.00e | 1 | 1103.31 | 660.36 | 0d | 8/8 |
| 2 | 2370 | NP(A634V) | C | T | 0.00 | 48.50 | 1 | 0.00 | 2.82 | N/Af | 1 | 4.89 | 76.14 | 1 | 8/8 |
| 3 | 4384 | VP35–VP40g | A | T | 0.00 | 26.08 | 1 | 0.00 | 2.11 | 17.27 | 1 | 2.04 | 28.00 | 1 | 7/8 |
| 4 | 5512 | VP40~GP | T | C | 0.00 | 865.00 | 1 | 0.00 | 3.06 | 76.20 | 1 | 4.21 | 346.40 | 1 | 7/8 |
| 5 | 5519 | VP40~GP | T | C | 0.00 | 43.50 | 1 | 0.00 | 2.22 | 26.00 | 1 | 3.90 | 48.13 | 1 | 8/8 |
| 6 | 5522 | VP40~GP | T | C | 0.00 | 23.24 | 1 | 0.00 | 2.47 | 35.00 | 1 | 4.19 | 38.47 | 1 | 8/8 |
| 7 | 5523 | VP40~GP | T | C | 0.00 | 135.33 | 1 | 0.00 | 2.50 | 69.80 | 1 | 4.25 | 516.36 | 1 | 8/8 |
| 8 | 5548 | VP40~GP | G | A | 0.02 | 873.00 | 1 | 0.01 | 92.50 | 347.00 | 1 | 431.71 | 631.98 | 0 | 8/8 |
| 9 | 5553 | VP40~GP | T | C | 0.00 | 137.83 | 1 | 0.00 | 3.38 | 108.33 | 1 | 5.47 | 388.47 | 1 | 8/8 |
| 10 | 5566 | VP40~GP | T | C | 0.00 | 199.50 | 1 | 0.00 | 2.94 | 150.00 | 1 | 5.43 | 195.73 | 1 | 8/8 |
| 11 | 5587 | VP40~GP | T | C | 0.00 | 43.67 | 1 | 0.00 | 3.07 | 1000.00 | 1 | 5.68 | 75.12 | 1 | 8/8 |
| 12 | 5594 | VP40~GP | T | C | 0.00 | 231.00 | 1 | 0.01 | 2.13 | 121.00 | 1 | 5.36 | 344.04 | 1 | 8/8 |
| 13 | 5595 | VP40~GP | T | C | 0.00 | 59.17 | 1 | 0.00 | 1.82 | 49.40 | 1 | 5.16 | 172.81 | 1 | 8/8 |
| 14 | 5596 | VP40~GP | T | C | 0.00 | 92.00 | 1 | 0.00 | 1.76 | 27.78 | 1 | 5.06 | 79.54 | 1 | 8/8 |
| 15 | 5604 | VP40~GP | T | C | 0.00 | 140.50 | 1 | 0.00 | 2.35 | 37.00 | 1 | 6.12 | 468.33 | 1 | 8/8 |
| 16 | 5616 | VP40~GP | T | C | 0.00 | 40.62 | 1 | 0.00 | 2.87 | 59.40 | 1 | 5.70 | 67.39 | 1 | 8/8 |
| 17 | 5631 | VP40~GP | T | C | 0.00 | 119.00 | 1 | 0.01 | 2.56 | 103.33 | 1 | 5.77 | 315.38 | 1 | 8/8 |
| 18 | 5849 | VP40~GP | T | C | 190.00 | 85.00 | 0d | 163.22 | 3.20 | 44.91 | 0 | 3.74 | 137.60 | 1 | 8/8 |
| 19 | 5880 | VP40~GP | C | T | 0.00 | 161.70 | 1 | 0.00 | 2.14 | 113.29 | 1 | 2.08 | 441.73 | 1 | 7/8 |
| 20 | 7267 | GP(R410S) | T | A | 0.00 | N/A | N/A | 0.00 | 57.00 | 1000.00 | 1 | 1281.65 | 2044.63 | 0 | 0/8 |
| 21 | 7352 | GP(E439K) | G | A | 0.03 | N/A | N/A | 0.03 | 17.33 | 1000.00 | 1 | 1102.30 | 495.67 | 0 | 8/8 |
| 22 | 8044 | GP(F669F) | C | T | 0.00 | 3.55 | 1 | 0.00 | 2.87 | 24.68 | 1 | 3.60 | 225.08 | 1 | 7/8 |
| 23 | 9426 | VP30–VP24 | C | T | 0.01 | 1000.00 | 1 | 0.01 | 1000.00 | 1000.00 | 1 | 792.13 | 473.41 | 0 | 8/8 |
| 24 | 10133 | VP30–VP24 | T | Del | 0.00 | 1000.00 | 1 | 0.01 | 0.97 | 3.95 | 1 | 1.65 | 7.45 | 1 | 6/8 |
| 25 | 10273 | VP30–VP24 | G | A | 0.11 | 116.00 | 1 | 0.12 | 173.00 | 202.50 | 1 | 330.20 | 317.62 | 0 | 8/8 |
| 26 | 10431 | VP24(F29F) | C | T | 0.00 | 30.33 | 1 | 0.00 | 2.91 | 125.67 | 1 | 2.70 | 321.75 | 1 | 7/8 |
| 27 | 10466 | VP24(Y41C) | A | G | 0.00 | 28.33 | 1 | 0.00 | 2.11 | 50.57 | 1 | 2.53 | 382.21 | 1 | 7/8 |
| 28 | 11407 | VP24~L | C | T | 113.88 | N/A | N/A | 133.23 | 1.86 | 17.14 | 0 | 2.44 | 91.21 | 1 | 8/8 |
| 29 | 13350 | L(END591Q) | T | C | 0.00 | N/A | N/A | 0.00 | 16.24 | 72.00 | 1 | 17.68 | 321.12 | 1 | 7/8 |
| 30 | 13557 | L(E657K) | G | A | 0.00 | N/A | N/A | 0.00 | 13.98 | 71.33 | 1 | 16.96 | 466.17 | 1 | 7/8 |
| 31 | 15048 | L(L1157P) | T | C | 0.01 | N/A | N/A | 0.01 | 1000.00 | 219.33 | 1 | 1216.72 | 937.44 | 0 | 8/8 |
| 32 | 15226 | L(P1216P) | T | C | 0.00 | N/A | N/A | 0.00 | 3.49 | 34.14 | 1 | 3.91 | 90.47 | 1 | 7/8 |
| 33 | 16894 | L(A1773T) | G | A | 0.00 | 34.50 | 1 | 0.00 | 77.00 | 74.25 | 1 | 93.06 | 61.84 | 0 | 8/8 |
| 34 | 18078 | L(H2187H) | T | C | 0.00 | 16.50 | 1 | 0.00 | 1.75 | N/A | 1 | 2.68 | 23.47 | 1 | 8/8 |
| 35 | 18319 | 5′-UTR | T | C | 0.00 | 9.00 | 1 | 0.00 | 2.35 | N/A | 1 | 2.64 | 27.41 | 1 | 8/8 |
The nonsynonymous mutations are emphasized by bold fonts.
The ratio of the reads of Latter/Former.
“1” means that the corresponding data of the survivor was shown in Figure 4B–D.
“0” means that the corresponding data of the survivor was shown in Supplementary Figure S1.
The ratio is termed as “1000” when the read of the former nucleotide is 0.
“N/A” means that the reading depths of the NGS was lower than 50.
“VP35–VP40” means that the substitutions locate in the non-coding region between VP35 and VP40.
“C stains (5512–5631)” indicate the 8 stains from publicly released full-length EBOV genome sequences from GenBank, including KT961624, KP759706, KU296370, KU296319, LN877955, KT357858, KT357859 and KT357860. These 8 strains possess the short stretch of T>C mutations within positions 5512–5631. Furthermore, these strains also have the linked mutations in other sites. The proportions of the strains which possess the mutations were shown in fractions.
The intra-host adaptation of EBOV was also illustrated as the continuous change of iSNV substitution ratios (the ratios of latter dominant iSNV/former dominant iSNV, for instance, ratios of C/T for the 13 T>C nucleotide sites) across longitudinal sampling points in patients 18791, 18797, and 18889 (Figures 4B–D). The iSNVs at most sites have a synchronized change in the three patients during infection. The substitution ratios for patients 18791 increased from 0.0021 (median, with lower quartile [Q1] = 0.0012 and upper quartile [Q3] = 0.0042) on day 12 to 59.17 (median, with Q1 = 30.33 and Q3 = 140.5) on day 20 (Figure 4B). For patient 18797, the substitution ratios increased from 0.0023 (median, with Q1 = 0.0013 and Q3 = 0.0062) on day 9 to 75.22 (median, with Q1 = 40.1 and Q3 = 189.375) on day 17, with a transition ratio of 2.87 (median, with Q1 = 2.22 and Q3 = 16.24) on day 14 (Figure 4C). For patient 18889, substitution ratios remained >1, indicating a dominant role of the latter iSNV on both day 5 and day 7 (Figure 4D), however, the increasing trend of the iSNV substitution ratios could still be observed from day 5 to day 7.
However, there were still some exceptional sites which had consistent iSNVs during the disease process (Supplementary Figure S1). All exceptional sites possessed the latter form at the target sites (e.g. Site 5849 of viral genome had already mutated to C in patient 18791 on day 12), indicating early substitution events at these sites.
3.4. Phylogenetic association of the T>C strains
Phylogenetic analysis of the 18 novel and 1078 publicly released full-length EBOV genome sequences from GenBank showed that the novel EBOV sequences did not cluster together (Figure 5A). Alternatively, they formed four small independent clusters scattered across lineage SL3, which was circulating during the late stage of the outbreak in 2015.
Figure 5.
Phylogenetic analysis of the newly characterized and public full-length EBOV genome sequences.
A. Sequences characterized in the present study are marked in red. B. All of the strains with serial T>C mutations are marked in blue. The numbers of serial T>C mutations are given. C. Detailed phylogenetic relationships of the strains with 13 serial T>C mutations. In this panel, sequences in the same color are obtained from the same patient at different time points. Sequences marked with black stars are reported in this paper.
We further analyzed all EBOV genomes available online and identified the strains with multiple T>C substitutions within a short genomic region in different positions of the genomes (termed as “C strains”) (Figure 5B and Table S3). A total of 49 strains, including six sequenced in this study, possessed multiple T>C nucleotide substitutions (Table S3). These “C strains” could be classified into at least 24 different types according to the number of T>C substitutions and their positions (Figure 5B). Interestingly, apart from the well-known stretch with 13 serial T>C mutations at genome position 5512–5631, stretches with 11 and 12 serial T>C mutations were also found in the intergenic region between VP40 and GP in strains G4415.1 (GenBank No. KR105247) and LIBR10051 (GenBank No. KT725368) (TableS3), respectively. To study the phylogenetic association of these “C strains”, we mapped them onto the tree (Figure 5B and Supplementary Figure S2). However, these 49 “C strains” scattered across the entire tree, with little phylogenetic association (Figure 5B), indicating multiple independent origins of these T>C substitutions. However, some strains possessing the same T>C substitution type were clustered together (Supplementary Figure S2). Based on current evidence, human-to-human transmission is the most plausible reason for these cases.
In particular, ten strains possessed the 13 T>C mutations within positions 5512–5631 (Figure 5C). The prototype strain with the 13 T>C mutations was J0169 (KP759706), sequenced from the Magazine Wharf area, Freetown, Sierra Leone in November 2014 [17]. It has been reported that J0169-like EBOV continued to circulate in this region and had infected at least three additional patients by July 2015. However, the strain sequenced from patient 18797 on days 14 and 17 possessed the 13 T>C mutations and clustered together with other such “C strains”. Surprisingly, on day 9 of patient 18797, the virus possessed Ts at all of the 13 positions, and did not fall within this cluster. This was also observed with patient 18791 (Figure 5C). Virus samples collected on day 12 from patient 18791 was located in a different cluster as the virus sequenced from the day 20 sample.
3.5. Co-current substitutions within the publicly released EBOV genomes
We further identified eight EBOV strains sharing the previously described 13 T>C mutations within positions 5512–5631 (the last column in Table 2). We called all the other viruses (1070 strains) as “T strains (5512–5631)”. Interestingly, we found that almost all 35 coincident substitutions that happened in our longitudinally sequenced EBOV genomes (Table 2) had already mutated in these eight “C strains (5512–5631)”. In contrast, none of the other EBOV “T strains (5512–5631)” without the 13 T>C mutations at position 5512–5631have these substitutions. These data suggest that all substitutions distributed within the whole genome of EBOV, including the short stretch of T>C mutations within position 5512–5631, had a coincident evolution trend during the recovery process of the patients.
4. Discussion
In this study, we sequenced longitudinally-collected blood samples from EBOV patients based on an in-fieldNGS platform at the SLE-CHNBio-safety Lab during the 2014–2016 EBOV epidemic. Although more a thousand of EBOV genomes have been sequenced during the 2014–2016 EBOV epidemic in West Africa, most of the viruses were from different patients during the early days of the acute phase during the infection [[1], [2], [3], [4], [5],7,8,13,17]. Phylogenetic analysis showed that the 18 genomes sequenced in this study were not grouped together. This suggested that in the late stages of the 2014–2016 EBOV outbreak, the virus became diversified and various minor EBOV lineages had been co-circulating in Sierra Leone.
Previous phylogenetic studies indicated that SNPs carried by different EBOV lineages were important molecular markers to study the virus transmission among humans [1,4,13]. Short stretches of the T>C substitutions appeared sporadically without phylogenetic association as shown in our analysis (Figure 5B). Although the exact reason is yet unclear, this was speculated to be the result of adenosine deaminases acting on RNA (ADARs) [16]. Thus, it is understandable that the T>C substitutions can occur in both the coding regions and the non-coding regions. In the present study, we found several related iSNVs across the EBOV genome occurring in the late acute phase of EBOV infection, including a short stretch of 13 T>C mutations within positions 5512–5631. However, these mutations were not found in samples collected during the early infection phase. In one patient, we even observed the intermediate stage for these T>C mutations. These data suggest that the 13 serial T>C mutations were emerged coincidently, and that the mutations could arise independently in different EBOV patients. Since only a few T>C mutations are able to change the transcription level of adjacent genes [15], such serial T>C mutations in our study, though some in non-coding regions, might be functional during EBOV infection in humans, which should be a subject of further investigation. Meanwhile,further analysis should be performed to investigate whether all these mutations can be found on the same nucleic acid strands (reads).
Phylogenetic analysis showed that earlier samples without the 13 T>C mutations were not grouped together, whereas later samples with the 13 T>C mutations were clustered together and formed a separate cluster. We also identified a total of 49 strains with serial T>C substitutions that belonged to 24 different T>C mutation types, with different number of T>C substitutions and/or within different genomic regions. Generally, strains possessing different T>C mutation types were not clustered together, but scattered across the tree. Based on current evidence, there was no direct phylogenetic association between these different mutation types. This once again revealed that these various T>C mutation types could arise independently. However, some of the T>C substitutions were indeed shared by a few strains, which were clustered together in the tree. Thus, the transmission hypothesis cannot be fully rejected yet [17].
This study is the first showing that linked and coincident mutations observed in EBOV could arise independently during human infections. It can be rationally proposed that the “T strains” are the dominant circulating EBOVs during the epidemic (Figure 6 ). Most of the cases are infected via contact with the patients during the early stages of acute infection. During the late stages of the infection, the “C strains” emerge, although the virus titer has become lower during this period. Transmission may also occur rarely between close contacts at this stage of disease. This may explain why the “C strains” could be found in such few patients in the acute infection phase. Meanwhile, the T>C mutation happened at different time points of the patients after EBOV infection, which may be related with disease progress or prognosis. For patient 18791, the corresponding sites for these T>C were still T, while the T>C substitution was completed on day 5 of the infection of patient 18889. The fever duration and the viremia duration of patient 18791 were longer than 18889. However, the relationship of the occurrence time of these mutations with disease progress or prognosis should be further investigated.
Figure 6.
The schematic diagram for the dynamic intra-host substitution and inter-host transmission of EBOV.
Base on the dynamic adaptation of Ebola virus in the patients we described herein, we further proposed the schematic model for the relationship of the human to human transmission and the dynamic adaptation of the Ebola viruses. Panel A shows the disease process of one survived patient A from the preclinical period to symptom presentation, and then to recovery. During this process, the dominant viruses in the patient are the T strain which possess the former nucleic acids in the 35 sites (Table 2), e.g. T in the 13 T>C stretch (5512–5631). There also will be emergence of the C strains during the recovery process, which possess the latter nucleic acids in the 35 sites (Table 2), e.g. C in the 13 T>C stretch (5512–5631) due to unknown reasons as we indicated in our patients. T strain virus dominates in the acute detoxification period of patients and certain patients died during this period. Thus in the general human to human transmission of Ebola virus (patient A to patient C), the transmitted viruses are T strain virus, which takes the dominates in the 1078 EBOV genomes of the West Africa outbreak publicly available from GenBank. However, we cannot exclude the possibility of the sporadic transmission of C strains in humans with close contact (A to B). Patient B, who was infected by the C strain of Ebola virus, may have a mixed quasi-species of the virus during the diseases process. This possibility requires further exploration.
Overall, our data indicate that short stretches of T>C substitutions are part of the linked and convergent evolution during the infection process of EVD patients, shedding light on the dynamic intra-host adaptation of EBOV during the 2014–2016 EBOV epidemic.
The following are the supplementary data related to this article.
The information of the virus genomes.
Number of different intra-host substitutions.
Summary of T-to-C mutation strains.
Exceptional mutations which do not have the substitution trends in the 35 linked nucleotide variations of EBOV in different patients.
Detailed phylogenetic tree of the newly characterized and existing full-lengthEBOV genome sequences.
Acknowledgments
Acknowledgements
Hundreds of Chinese and Sierra Leonean staff on-site took part in the sample collection, transportation, detection, and data analysis. We especially acknowledge the excellent work of four previous detection teams of ChinaCDC (mobile and fixed labs) and the essential support (materials and reagents) of the National Institute for Viral Disease Control and Prevention, China CDC. We also express our deep condolences to the family and associates of Dr. Abdul Kamara of the Ministry of Health and Sanitation, Sierra Leone, who contributed a lot to this project but succumbed after the successful control of Ebola epidemic in Sierra Leone.
This work was supported by the Megaproject for Infectious Disease Research of China(2016ZX10004222-003), the research of Ebola pathogen from the National Natural Science Foundation of China (NSFC, 81590763), National Key Research and Development Program of China(2016YFC1200200 to Y. Shu), the Distinguished Young Scientist Program of the NSFC (81525017 to Y. Shu), the Excellent Young Scientist Program of the NSFC(81822040 to W.J. Liu), the Taishan Scholar Project of Shandong Province(ts201511056 to W. Shi). G.F. Gao is a primary principal investigator of the NSFC Innovative Research Group (81621091).
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Author contributions
W.J. Liu, W. Shi, W. Zhu, C. Jin, S. Zou, J. Wang, Y. Ke, X. Li, and M. Liu performed the experiments. C. Chen, C. Geng, W. Xie, H. Jiang, I.L. Kamara, and A. Kamara provided the necessary materials. W.J. Liu, W. Shi, T. Hu, H. Fan, Y. Tong, X. Zhao, W. Chen, Y. Zhao, and D. Liu performed the data analysis. G. Wong, M. Lebby, B. Kargbo, X. Qiu, Y. Wang, X. Liang, M. Liang, X. Dong, and G. Wu contributed to fruitful discussions and key ideas. W.J. Liu, W. Shi, G.F. Gao, and Y. Shu contributed to the overall concept, experimental design and hypothesis, and wrote the manuscript.
References
- 1.Tong Y.G., Shi W.F., Liu D., Qian J., Liang L., Bo X.C., Liu J., Ren H.G., Fan H., Ni M., Sun Y., Jin Y., Teng Y., Li Z., Kargbo D., Dafae F., Kanu A., Chen C.C., Lan Z.H., Jiang H., Luo Y., Lu H.J., Zhang X.G., Yang F., Hu Y., Cao Y.X., Deng Y.Q., Su H.X., Sun Y., Liu W.S., Wang Z., Wang C.Y., Bu Z.Y., Guo Z.D., Zhang L.B., Nie W.M., Bai C.Q., Sun C.H., An X.P., Xu P.S., Zhang X.L., Huang Y., Mi Z.Q., Yu D., Yao H.W., Feng Y., Xia Z.P., Zheng X.X., Yang S.T., Lu B., et al. Genetic diversity and evolutionary dynamics of Ebola virus in Sierra Leone. Nature. 2015;524:93–96. doi: 10.1038/nature14490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Quick J., Loman N.J., Duraffour S., Simpson J.T., Severi E., Cowley L., Bore J.A., Koundouno R., Dudas G., Mikhail A., Ouedraogo N., Afrough B., Bah A., Baum J.H., Becker-Ziaja B., Boettcher J.P., Cabeza-Cabrerizo M., Camino-Sanchez A., Carter L.L., Doerrbecker J., Enkirch T., Garcia-Dorival I., Hetzelt N., Hinzmann J., Holm T., Kafetzopoulou L.E., Koropogui M., Kosgey A., Kuisma E., Logue C.H., Mazzarelli A., Meisel S., Mertens M., Michel J., Ngabo D., Nitzsche K., Pallasch E., Patrono L.V., Portmann J., Repits J.G., Rickett N.Y., Sachse A., Singethan K., Vitoriano I., Yemanaberhan R.L., Zekeng E.G., Racine T., Bello A., Sall A.A., Faye O., et al. Real-time, portable genome sequencing for Ebola surveillance. Nature. 2016;530:228–232. doi: 10.1038/nature16996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ladner J.T., Wiley M.R., Mate S., Dudas G., Prieto K., Lovett S., Nagle E.R., Beitzel B., Gilbert M.L., Fakoli L., Diclaro J.W., II, Schoepp R.J., Fair J., Kuhn J.H., Hensley L.E., Park D.J., Sabeti P.C., Rambaut A., Sanchez-Lockhart M., Bolay F.K., Kugelman J.R., Palacios G. Evolution and spread of Ebola virus in Liberia, 2014–2015. Cell Host Microbe. 2015;18:659–669. doi: 10.1016/j.chom.2015.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park D.J., Dudas G., Wohl S., Goba A., Whitmer S.L., Andersen K.G., Sealfon R.S., Ladner J.T., Kugelman J.R., Matranga C.B., Winnicki S.M., Qu J., Gire S.K., Gladden-Young A., Jalloh S., Nosamiefan D., Yozwiak N.L., Moses L.M., Jiang P.P., Lin A.E., Schaffner S.F., Bird B., Towner J., Mamoh M., Gbakie M., Kanneh L., Kargbo D., Massally J.L., Kamara F.K., Konuwa E., Sellu J., Jalloh A.A., Mustapha I., Foday M., Yillah M., Erickson B.R., Sealy T., Blau D., Paddock C., Brault A., Amman B., Basile J., Bearden S., Belser J., Bergeron E., Campbell S., Chakrabarti A., Dodd K., Flint M., Gibbons A., et al. Ebola virus epidemiology, transmission, and evolution during seven months in Sierra Leone. Cell. 2015;161:1516–1526. doi: 10.1016/j.cell.2015.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gire S.K., Goba A., Andersen K.G., Sealfon R.S., Park D.J., Kanneh L., Jalloh S., Momoh M., Fullah M., Dudas G., Wohl S., Moses L.M., Yozwiak N.L., Winnicki S., Matranga C.B., Malboeuf C.M., Qu J., Gladden A.D., Schaffner S.F., Yang X., Jiang P.P., Nekoui M., Colubri A., Coomber M.R., Fonnie M., Moigboi A., Gbakie M., Kamara F.K., Tucker V., Konuwa E., Saffa S., Sellu J., Jalloh A.A., Kovoma A., Koninga J., Mustapha I., Kargbo K., Foday M., Yillah M., Kanneh F., Robert W., Massally J.L., Chapman S.B., Bochicchio J., Murphy C., Nusbaum C., Young S., Birren B.W., Grant D.S., Scheiffelin J.S., et al. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science. 2014;345:1369–1372. doi: 10.1126/science.1259657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoenen T., Safronetz D., Groseth A., Wollenberg K.R., Koita O.A., Diarra B., Fall I.S., Haidara F.C., Diallo F., Sanogo M., Sarro Y.S., Kone A., Togo A.C., Traore A., Kodio M., Dosseh A., Rosenke K., de Wit E., Feldmann F., Ebihara H., Munster V.J., Zoon K.C., Feldmann H., Sow S. Mutation rate and genotype variation of Ebola virus from Mali case sequences. Science. 2015;348:117–119. doi: 10.1126/science.aaa5646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kugelman J.R., Wiley M.R., Mate S., Ladner J.T., Beitzel B., Fakoli L., Taweh F., Prieto K., Diclaro J.W., Minogue T., Schoepp R.J., Schaecher K.E., Pettitt J., Bateman S., Fair J., Kuhn J.H., Hensley L., Park D.J., Sabeti P.C., Sanchez-Lockhart M., Bolay F.K., Palacios G., Diseases USAMRIoI, National Institutes of H, Integrated Research Facility-Frederick Ebola Response T Monitoring of Ebola virus Makona evolution through establishment of advanced genomic capability in Liberia. Emerg. Infect. Dis. 2015;21:1135–1143. doi: 10.3201/eid2107.150522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Simon-Loriere E., Faye O., Faye O., Koivogui L., Magassouba N., Keita S., Thiberge J.M., Diancourt L., Bouchier C., Vandenbogaert M., Caro V., Fall G., Buchmann J.P., Matranga C.B., Sabeti P.C., Manuguerra J.C., Holmes E.C., Sall A.A. Distinct lineages of Ebola virus in Guinea during the 2014 West African epidemic. Nature. 2015;524:102–104. doi: 10.1038/nature14612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dudas G., Carvalho L.M., Bedford T., Tatem A.J., Baele G., Faria N.R., Park D.J., Ladner J.T., Arias A., Asogun D., Bielejec F., Caddy S.L., Cotten M., D'Ambrozio J., Dellicour S., Di Caro A., Diclaro J.W., Duraffour S., Elmore M.J., Fakoli L.S., Faye O., Gilbert M.L., Gevao S.M., Gire S., Gladden-Young A., Gnirke A., Goba A., Grant D.S., Haagmans B.L., Hiscox J.A., Jah U., Kugelman J.R., Liu D., Lu J., Malboeuf C.M., Mate S., Matthews D.A., Matranga C.B., Meredith L.W., Qu J., Quick J., Pas S.D., Phan M.V.T., Pollakis G., Reusken C.B., Sanchez-Lockhart M., Schaffner S.F., Schieffelin J.S., Sealfon R.S., Simon-Loriere E., et al. Virus genomes reveal factors that spread and sustained the Ebola epidemic. Nature. 2017;544:309–315. doi: 10.1038/nature22040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ka D., Fall G., Diallo V.C., Faye O., Fortes L.D., Faye O., Bah E.I., Diallo K.M., Balique F., Ndour C.T., Seydi M., Sall A.A. Ebola virus imported from Guinea to Senegal, 2014. Emerg. Infect. Dis. 2017;23:1026–1028. doi: 10.3201/eid2306.161092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Team WHOER Ebola virus disease in West Africa—the first 9 months of the epidemic and forward projections. N. Engl. J. Med. 2014;371:1481–1495. doi: 10.1056/NEJMoa1411100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baize S., Pannetier D., Oestereich L., Rieger T., Koivogui L., Magassouba N., Soropogui B., Sow M.S., Keita S., De Clerck H., Tiffany A., Dominguez G., Loua M., Traore A., Kolie M., Malano E.R., Heleze E., Bocquin A., Mely S., Raoul H., Caro V., Cadar D., Gabriel M., Pahlmann M., Tappe D., Schmidt-Chanasit J., Impouma B., Diallo A.K., Formenty P., Van Herp M., Gunther S. Emergence of Zaire Ebola virus disease in Guinea. N. Engl. J. Med. 2014;371:1418–1425. doi: 10.1056/NEJMoa1404505. [DOI] [PubMed] [Google Scholar]
- 13.Holmes E.C., Dudas G., Rambaut A., Andersen K.G. The evolution of Ebola virus: Insights from the 2013–2016 epidemic. Nature. 2016;538:193–200. doi: 10.1038/nature19790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang X., Zhang Z., Zhuang D., Carr M.J., Zhang R., Lv Q., Shi W. Non-coding regions of the Ebola virus genome contain indispensable phylogenetic and evolutionary information. Sci. China Life Sci. 2015;58:682–686. doi: 10.1007/s11427-015-4857-9. [DOI] [PubMed] [Google Scholar]
- 15.Ni M., Chen C., Qian J., Xiao H.X., Shi W.F., Luo Y., Wang H.Y., Li Z., Wu J., Xu P.S., Chen S.H., Wong G., Bi Y., Xia Z.P., Li W., Lu H.J., Ma J., Tong Y.G., Zeng H., Wang S.Q., Gao G.F., Bo X.C., Liu D. Intra-host dynamics of Ebola virus during 2014. Nat. Microbiol. 2016;1:16151. doi: 10.1038/nmicrobiol.2016.151. [DOI] [PubMed] [Google Scholar]
- 16.Gelinas J.F., Clerzius G., Shaw E., Gatignol A. Enhancement of replication of RNA viruses by ADAR1 via RNA editing and inhibition of RNA-activated protein kinase. J. Virol. 2011;85:8460–8466. doi: 10.1128/JVI.00240-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smits S.L., Pas S.D., Reusken C.B., Haagmans B.L., Pertile P., Cancedda C., Dierberg K., Wurie I., Kamara A., Kargbo D., Caddy S.L., Arias A., Thorne L., Lu J., Jah U., Goodfellow I., Koopmans M.P. Genotypic anomaly in Ebola virus strains circulating in Magazine Wharf area, Freetown, Sierra Leone, 2015. Euro Surveill. 2015;20 doi: 10.2807/1560-7917.ES.2015.20.40.30035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Emmett K.J., Lee A., Khiabanian H., Rabadan R. High-resolution genomic surveillance of 2014 Ebolavirus using shared subclonal variants. PLoS Curr. 2015;7 doi: 10.1371/currents.outbreaks.c7fd7946ba606c982668a96bcba43c90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Urbanowicz R.A., McClure C.P., Sakuntabhai A., Sall A.A., Kobinger G., Muller M.A., Holmes E.C., Rey F.A., Simon-Loriere E., Ball J.K. Human adaptation of Ebola virus during the West African outbreak. Cell. 2016;167:1079–1087. doi: 10.1016/j.cell.2016.10.013. (e1075) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diehl W.E., Lin A.E., Grubaugh N.D., Carvalho L.M., Kim K., Kyawe P.P., McCauley S.M., Donnard E., Kucukural A., McDonel P., Schaffner S.F., Garber M., Rambaut A., Andersen K.G., Sabeti P.C., Luban J. Ebola virus glycoprotein with increased infectivity dominated the 2013–2016 epidemic. Cell. 2016;167:1088–1098. doi: 10.1016/j.cell.2016.10.014. (e1086) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dietzel E., Schudt G., Krahling V., Matrosovich M., Becker S. Functional characterization of adaptive mutations during the West African Ebola virus outbreak. J. Virol. 2017;91:e01913–e01916. doi: 10.1128/JVI.01913-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Whitmer S.L., Albarino C., Shepard S.S., Dudas G., Sheth M., Brown S.C., Cannon D., Erickson B.R., Gibbons A., Schuh A., Sealy T., Ervin E., Frace M., Uyeki T.M., Nichol S.T., Stroher U. Preliminary evaluation of the effect of investigational Ebola virus disease treatments on viral genome sequences. J. Infect. Dis. 2016;214:S333–S341. doi: 10.1093/infdis/jiw177. [DOI] [PubMed] [Google Scholar]
- 23.Gao G.F., Feng Y. On the ground in Sierra Leone. Science. 2014;346:666. doi: 10.1126/science.346.6209.666. [DOI] [PubMed] [Google Scholar]
- 24.Wang Q., Zhang Y., Wang H.Y., Du H.J., Nie K., Song J.D., Xiao K., Lei W.W., Guo J.Q., Wei H.J., Cai K., Wang Y.H., Wu J., Bangura G., Kamara I.L., Dong X.P. Detection and analysis of Ebola virus in Sierra Leone-China Friendship Biosafety Laboratory from March 11 to April 20, 2015. Biomed. Environ. Sci. 2016;29:443–447. doi: 10.3967/bes2016.057. [DOI] [PubMed] [Google Scholar]
- 25.Liu W.J. On the ground in Western Africa: from the outbreak to the elapse of Ebola. Protein Cell. 2016 doi: 10.1007/s13238-016-0305-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de La Vega M.A., Caleo G., Audet J., Qiu X., Kozak R.A., Brooks J.I., Kern S., Wolz A., Sprecher A., Greig J., Lokuge K., Kargbo D.K., Kargbo B., Di Caro A., Grolla A., Kobasa D., Strong J.E., Ippolito G., Van Herp M., Kobinger G.P. Ebola viral load at diagnosis associates with patient outcome and outbreak evolution. J. Clin. Invest. 2015;125:4421–4428. doi: 10.1172/JCI83162. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The information of the virus genomes.
Number of different intra-host substitutions.
Summary of T-to-C mutation strains.
Exceptional mutations which do not have the substitution trends in the 35 linked nucleotide variations of EBOV in different patients.
Detailed phylogenetic tree of the newly characterized and existing full-lengthEBOV genome sequences.