

Summary
Intake of fructose-containing sugars is strongly associated with metabolic syndrome. Dietary fructose is uniquely metabolized versus other sugars by fructokinase. However, the tissue-specific role of fructokinase in sugar-induced metabolic syndrome and the specific roles of glucose and fructose in driving it is not fully understood. Here, we show that in mice receiving excess fructose-glucose solutions, whole-body deletion of fructokinase, and thus full blockade of fructose metabolism, is sufficient to prevent metabolic syndrome. This protection is not only due to reduced fructose metabolism but also to decreased sugar intake. Furthermore, by using tissue-specific fructokinase-deficient mice, we determined that while sugar intake is controlled by intestinal fructokinase activity, metabolic syndrome is driven by fructose metabolism in the liver. Our findings show a two-pronged role for fructose metabolism in sugar-induced metabolic syndrome - one arm via the intestine that mediates sugar intake and a second arm in the liver that drives metabolic dysfunction.
Graphical Abstract
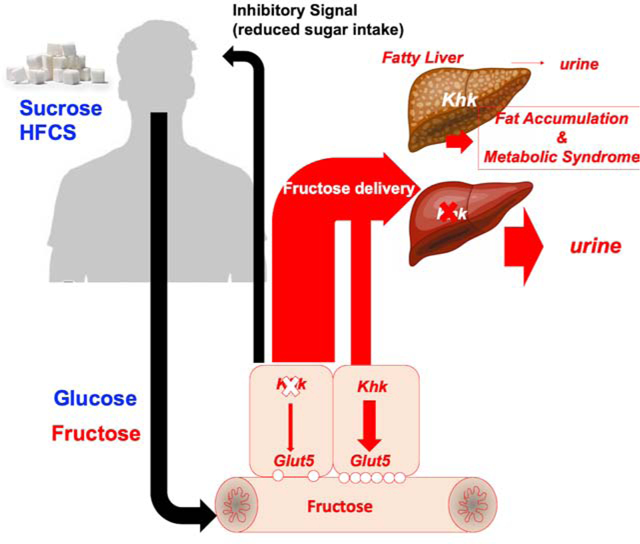
eTOC
Here, Andres-Hernando et al. demonstrate that fructose exerts differential physiological effects depending on the tissue where it is metabolized. By manipulating the expression of fructokinase, a key fructose metabolizing enzyme, they show that fructose acts in the gut to regulate sweet-taste preference and sugar intake, while its excess catabolism in the liver drives the majority of metabolic syndrome development.
Introduction
Table sugar (sucrose) and high fructose corn syrup (HFCS) are the two major added sweeteners used throughout the world. Intake of sweeteners is pervasive, being present in over 70 percent of processed foods available in grocery stores (Ng et al., 2012) and accounting for approximately one sixth of total energy intake (Yang et al., 2014). While originally viewed as simply an additional caloric source without much nutritional value (‘an empty calorie’) (White, 2008), there has been increasing evidence that the intake of these sweeteners may have a direct causal role in the epidemics of obesity and metabolic syndrome. Indeed, sugar and HFCS are epidemiologically linked with these epidemics, and experimental studies in both humans and laboratory animals have supported a key role for these sugars in inducing features of metabolic syndrome, a recognized precursor of diabetes (Goran et al., 2013; Johnson et al., 2013; Lustig et al., 2016; Schwarz et al., 2017; Schwimmer et al., 2019; Stanhope et al., 2011; Stanhope et al., 2009). Intake of these sugars is also of interest due to their relationship with other common medical illnesses, including cancer and dementia (Goncalves et al., 2019; Li et al., 2016; Stephan et al., 2010). Some studies have even ascribed increased cardiovascular mortality rates to the excessive intake of added sugars (Malik et al., 2019; Yang et al., 2014).
Both sweeteners contain glucose and fructose, bound together as a disaccharide in sucrose and mixed as free monosaccharides in HFCS. Our group, as well as others, have suggested that the primary mechanism by which added sugars cause metabolic syndrome is via the fructose component (Johnson et al., 2013; Stanhope and Havel, 2008; Vos et al., 2008; Weiss et al., 2013). We have also reported that when glucose is administered alone its ability to induce the metabolic syndrome is mediated in part by the endogenous conversion of glucose to fructose (Lanaspa et al., 2013). Fructose has a unique metabolism in which the first enzyme, fructokinase C (ketohexokinase C or KHK-C) metabolizes fructose with consumption of ATP so rapidly that a transient depletion of intracellular phosphate and ATP occurs (Maenpaa et al., 1968; van den Berghe et al., 1977). KHK-C is expressed in those organs involved in metabolizing the majority of dietary fructose, liver, small intestine and kidney cortex. However, it is important to note that there is another KHK isoform, namely KHK-A, ubiquituously expressed. Compared to KHK-C, KHK-A has lower affinity for fructose and as such it does not consume ATP so rapidly. The lowering of intracellular phosphate then activates an enzyme, AMP deaminase, that causes nucleotide degradation of AMP to IMP and uric acid (Kurtz et al., 1986; van den Berghe et al., 1977). In turn, the uric acid induces mitochondrial oxidative stress that blocks fatty acid oxidation and maintains a low ATP state (Lanaspa et al., 2012a; Lanaspa et al., 2012b). In addition, activation of the transcription factors SREBP1c and ChREBP, along with mitochondrial dysfunction, help drive de novo lipogenesis (Herman and Samuel, 2016; Kim et al., 2016; Lanaspa et al., 2012b; Lanaspa et al., 2012c), while fatty acid oxidation is also impaired due to alterations in mitochondrial function (Lanaspa et al., 2012a; Softic et al., 2019). This process activates biological processes that cause insulin resistance, fatty liver, fat accumulation and other characteristics of metabolic syndrome (Cicerchi et al., 2014; Kratzer et al., 2014; Lanaspa et al., 2012a; Lanaspa et al., 2012b; Nakagawa et al., 2006).
Several studies have shown that blocking fructose metabolism can ameliorate the development of metabolic syndrome from fructose, with or without high-fat diet feeding (Ishimoto et al., 2012; Ishimoto et al., 2013; Miller et al., 2018; Softic et al., 2017). Surprisingly, it yet to be shown that blocking fructose metabolism will block the effects of sucrose or HFCS in the absence of a high-fat diet on inducing metabolic syndrome. Furthermore, the relative roles of the liver and intestine in fructose metabolism are important because both are central metabolic organs and KHK is expressed in both tissues. Indeed, there is evidence that fructose metabolism in the gut may actually protect the body from metabolic syndrome by preventing fructose delivery to the liver (Jang et al., 2018). To address these important questions, we first derived mice in which both isoforms of fructokinase were deleted in a whole-body manner (global knockout, KHK-A/C KO) or were specifically deleted from the liver and/or intestine using the Cre-lox system. This approach allowed for specific dissection of the role of fructose vs glucose in inducing metabolic syndrome as well as distinguishing the role of the intestine from the liver in any effect of fructose.
Results
Glucose promotes fructose intake and metabolism in mice by a mechanism dependent on KHK-C.
We have previously shown that KHK blockade in mice dramatically diminishes both the intake of fructose and its metabolism (Ishimoto et al., 2012). However, fructose is rarely found alone in foods. Instead, it is normally present in combination with glucose, either as sucrose or high fructose corn syrup (HFCS). Therefore, a more clinically relevant approach would be to determine the effect of KHK blockade in response to fructose/glucose (FG) combinations. To this end, we prepared FG solutions in a ratio similar to that of HFCS (55% fructose/45% glucose) and determined the response of mice to this solution compared to mice receiving equimolar amounts of pure fructose. The addition of glucose to the fructose solution markedly increased both fluid intake (from 5.6 ± 0.3 ml fructose/day to 14.4 ± 1.4 ml FG/day, P < 0.01, Figure 1A–B) and caloric intake (from 11.46 ± 0.6 cal/day in fructose fed to 15.47 ± 1.1 ml/day in FG fed mice, P < 0.01). Furthermore, by determining fructose levels in the portal vein following an oral gavage of either fructose or FG solution (1 g/kg), we found that glucose accelerated the delivery of fructose into the liver (Figure 1C).This data is consistent with other studies that suggest that glucose can augment fructose absorption (Laughlin, 2014). As the majority of dietary fructose is metabolized in the gut and the liver, and consistent with greater exposure to fructose in FG-treated mice, we found that FG substantially up-regulated the expression of KHK in these tissues in chronically (30 weeks) exposed mice (a 92 % (P < 0.01) and a 33 % (P < 0.05) increase in the gut and liver, respectively, Figure 1D–E) thus indicating glucose enhances intake and transport of fructose.
Figure 1: Glucose potentiates fructose intake and metabolism in mice in a mechanism dependent of KHK.
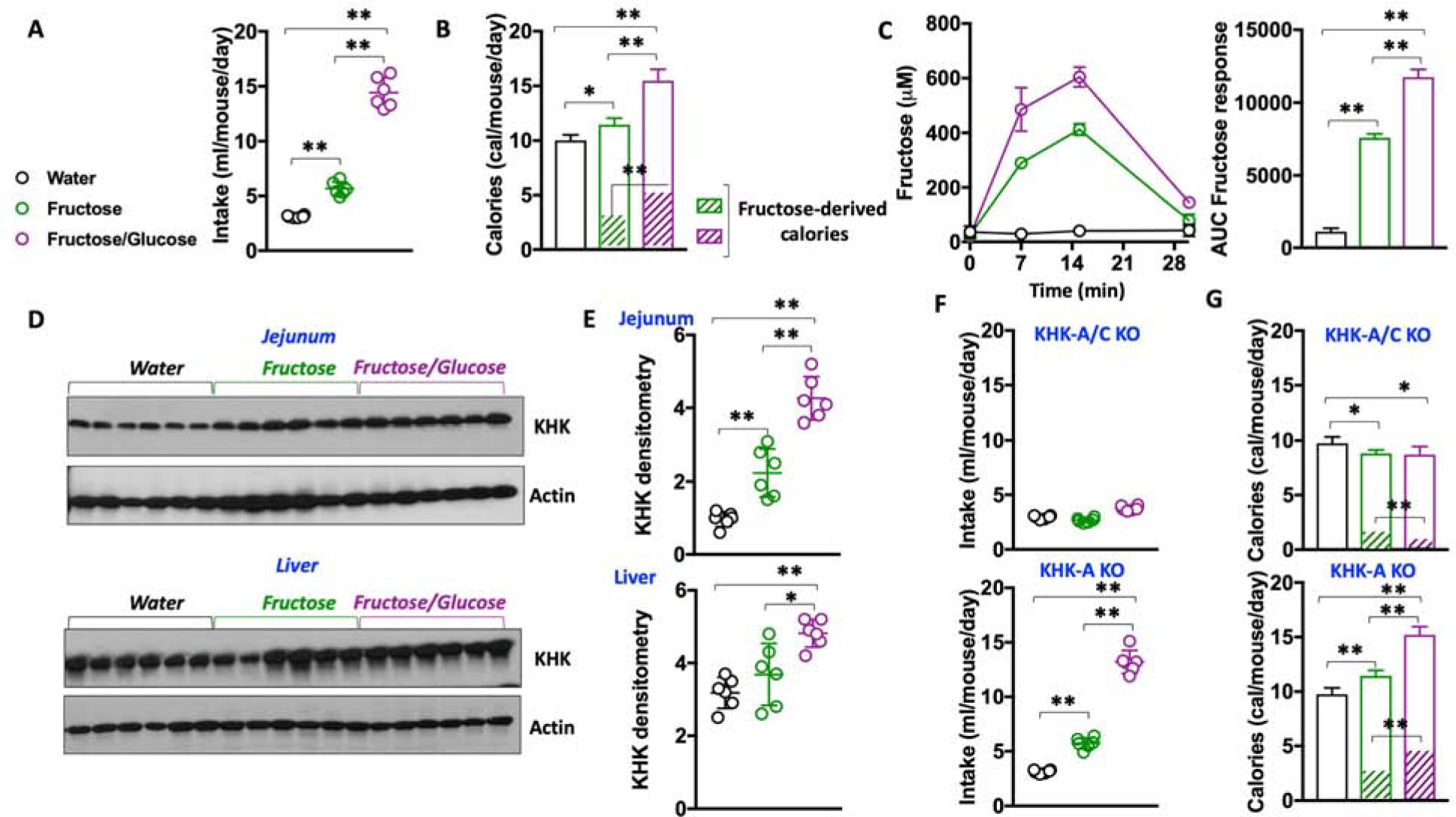
A) Intake of water (black), fructose solution (5%, green) or a 1:1 fructose/glucose mixture (FG, purple, 5% each) in naïve -never exposed to sugar- WT mice.
B) Daily caloric intake and daily fructose-derived calories from the same groups as A).
C) Fructose levels in the portal vein of WT mice after oral gavage of water (black), fructose or an equimolar concentration of 1:1 FG mixture in naïve WT mice.
D–E) A representative western blot (n = 3 total blots) and densitometry for KHK in the jejunum and liver of mice exposed to water, fructose or FG mixture.
F) Daily intake of water (black), fructose solution (5%, green) or a 1:1 FG mixture (5% each) in KHK-A/C KO -top- and KHK-A KO-bottom- mice.
G) Daily caloric intake and daily fructose-derived calories from the same groups as F).
The data in A–C) and E–G) were presented as the means ± SEM and analyzed by One Way ANOVA with Tukey post hoc analysis. *P < 0.05, **P < 0.01, n = 6 mice per group.
We then aimed to characterize the specific role of fructose metabolism in sugar and caloric intake. To this end, we analyzed the response to FG solutions in mice deficient for KHK (KHK-A/C KO). Whole body KHK-A/C KO mice demonstrated no substantial increased intake of either fructose (2.66 ± 0.2 ml/day) or FG (3.76 ± 0.2 ml/day) (Figure 1F–G), indicating that the metabolism of fructose drives the intake of fructose-containing sugars. As a result, daily caloric intake in KHK-A/C KO mice on fructose or FG did not substantially differ from mice on an unsweetened water control (9.76 ± 0.6 in water control mice versus 8.83 ± 0.3 and 8.71 ± 0.7 in fructose and FG fed mice, P>0.05) and it was lower than wild-type (WT) mice. Notably, the protective effect observed in KHK-A/C KO mice seems to be mediated by KHK-C as mice deficient only for the A isoform of KHK (KHK-A KO) demonstrated elevated intake of both sugar and overall calories (Figure 1F–G).
Blockade of fructose metabolism is sufficient to prevent sugar-induced metabolic syndrome.
To ascertain the importance of fructose metabolism in overall sugar-induced metabolic syndrome, we characterized the metabolic response of WT and global KHK-A/C KO mice chronically exposed to FG (30 weeks). In order to better characterize the response and considering that KHK-A/C KO mice consumed much lower amounts of sugar and calories than WT animals, we compared the response of WT mice fed 10% FG solutions against KHK-A/C KO mice on 30% FG. This way, we matched for both sugar and caloric intake as well as fructose-derived calories (Figure 2A–C and Supplemental Table 1). Importantly, KHK-A/C KO mice fed FG demonstrated significantly less body weight gain than the WT counterparts on FG (10.55 ± 0.9 grams in KHK-A/C KO mice fed 30% FG versus 20.31 ± 1.7 grams in WT mice on 10 % FG, P<0.01) despite receiving similar calories (Figure 2D). This marked reduction in body weight gain in KHK-A/C KO mice fed FG was associated with substantially lower liver and adipose weights at the time of killing (Supplemental Table 1). Furthermore, and unlike FG-fed WT mice, we did not observe fatty liver – a common feature of metabolic syndrome- in FG-fed KHK-A/C KO mice (Figure 2E–F) or liver injury, as demonstrated by significantly lower levels of plasma AST and ALT in the KO mice vs the WT mice (Figure 2G and Supplemental Table 1). Less epididymal adipose tissue was present in the KHK-A/C KO mice as well as fewer crown-like structures (as a measure of adipose inflammation, Figure 2H) and significantly lower plasma insulin and leptin levels (Figure 2I–J and Supplemental Table 1), all which are commonly found in subjects with metabolic syndrome. In addition, and consistent with reduced fasting insulin levels, FG-fed KHK-A/C KO mice demonstrated improved insulin sensitivity compared to WT counterparts as demonstrated by a healthier HOMA-IR index and glucose levels following an oral glucose tolerance test (Supplemental Table 1). Thus, our data indicate that the mechanism by which added sweeteners affect both intake and metabolic syndrome is due to the specific deleterious effects associated with KHK-mediated fructose metabolism.
Figure 2: Blockade of fructose metabolism is sufficient to prevent sugar-induced metabolic syndrome.
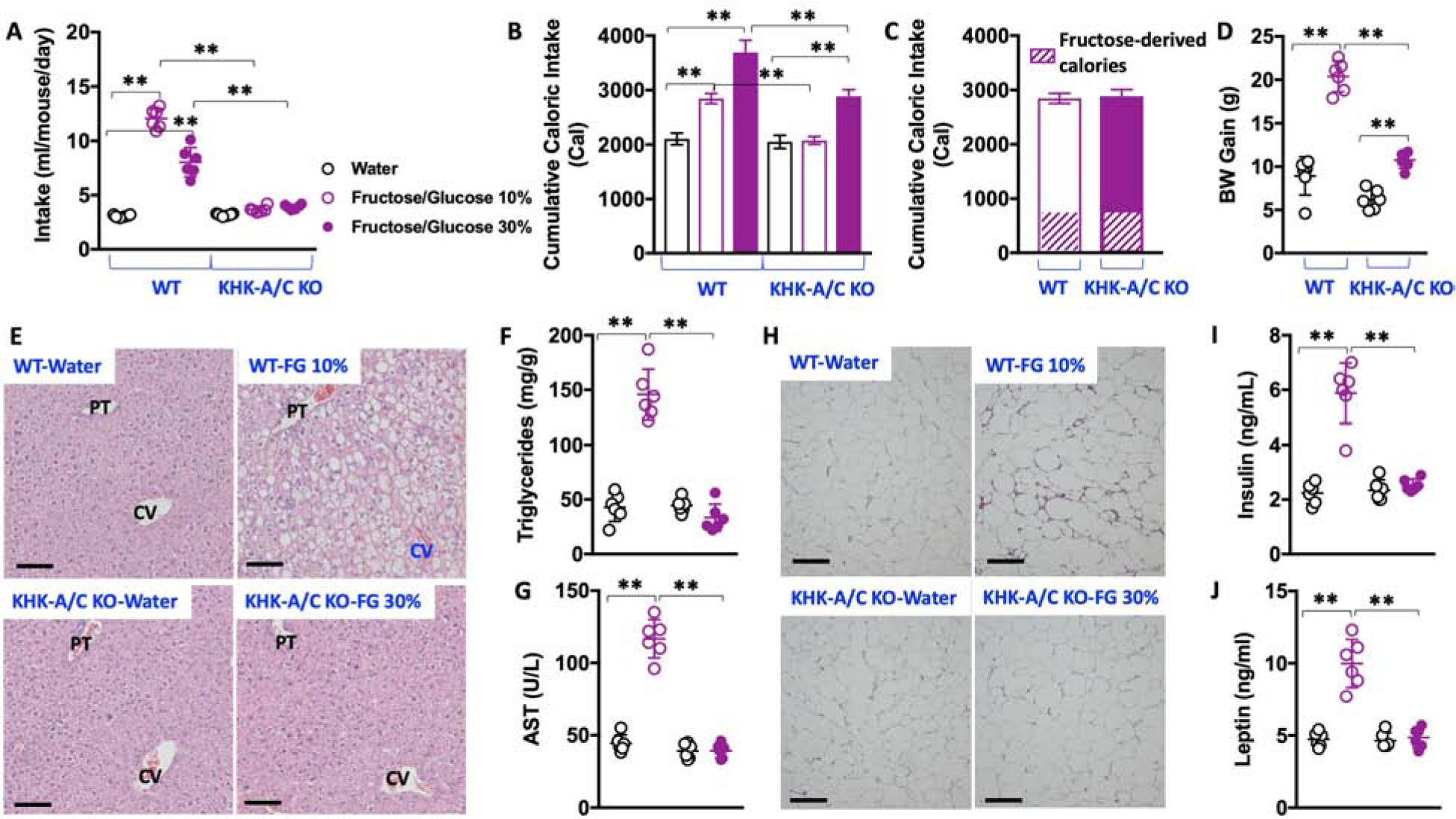
A) 30-week average intake of water (black), 10 % fructose/glucose (FG) solution (purple, clear symbols) or 30 % FG solution (purple, solid symbols) in wild type and KHK-A/C KO mice.
B) 30-week cumulative caloric intake in same groups as A).
C) 30-week cumulative caloric intake and fructose-derived calories in WT mice exposed to 10% FG and KHK-A/C KO mice exposed to 30 % FG.
D) 30-week body weight gain in WT and KHK-A/C KO mice exposed to water, 10% FG (wild type) or 30% FG (KHK-A/C KO).
E) Representative H&E images from livers of mice (n > 10 images per animal) of the same groups as in D). Size bars: 50 μM, PT: Portal triad; CV: Central Vein
F–G) Hepatic triglycerides and plasma AST levels in the same groups as in D).
H) Representative H&E images from epididymal adipose tissue of mice (n > 10 images per animal) of the same groups as in D). Size bars: 50 μM
I–J) Fasting plasma insulin and leptin levels in the same groups as in D).
The data in A–D), F–G) and I–J) were presented as the means ± SEM and analyzed by One Way ANOVA with Tukey post hoc analysis. *P < 0.05, **P < 0.01, n = 6 mice per group. See also Supplemental Table 1
Differential tissue-specific effects of KHK blockade in sugar-dependent intake and metabolic syndrome.
Similar to our previous findings with fructose (Ishimoto et al., 2012), our data here (Figure 1F–G) suggests that the KHK-C isoform is the driver of sugar intake, and not KHK-A. Therefore, to better characterize the specific role of KHK in sugar intake and metabolic syndrome, we generated conditional KHK-A/C KO mice for tissue-specific deletion of the Khk gene. To this end, and following the original approach from Diggle et al. (Diggle et al., 2009), exons 3 and 4 of the Khk gene were flanked by LoxP sequences for tissue-specific Cre-recognition and deletion of both splice variants of KHK (KHKFl/Fl, Supplemental Figure 1A). For gut and liver-specific deletion of KHK, KHKFl/Fl mice were crossed with Cre-recombinase expressing mice under the control of the villin (Cre-Vil) or the albumin (Cre-Alb) gene promoters, respectively. As shown in Supplemental Figure 1B, the insertion of a LoxP site before the exon 3a did not substantially changed the splicing of KHK-A and KHK-C in KHKFl/Fl mice. Similarly, and as shown in Supplemental Figure 1C–D, the specific deletion of KHK in either gut or liver did not result in its up-regulation or compensation in the remaining KHK-C expressing tissues.
Intestinal fructose metabolism drives sweet taste preference and thus promotes sugar intake.
KHKFl/Fl and KHKFl/FlXCre-Vil were allowed to consume water, FG 10%, or FG 30% solutions for 30 weeks and characterized as above. Similar to WT mice, KHKFl/Fl mice preferred FG solutions over drinking water (with higher preference for the lower sugar concentrations Figure 3A and Supplemental Table 2). In contrast, KHKFl/FlXCre-Vil mice showed minimal intake of either FG solution (2.65 ± 0.5 and 2.70 ± 0.1 ml/day of 10% FG and 30 % FG, respectively) indicating that fructose metabolism in the gut influences sweet taste preference in mice. As a result of lower sugar intake, 30-week cumulative caloric intake as well as fructose-derived calories was significantly lower in KHKFl/FlXCre-Vil mice than KHKFl/Fl mice (Figure 3B and Supplemental Table 2). Of interest, the observed reduced sugar intake in KHKFl/FlXCre-Vil mice is not necessarily associated with an aversion response. In two-bottle choice preference paradigms involving water versus FG KHKFl/Fl mice demonstrate a substantially higher preference for sugar than KHKFl/FlXCre-Vil mice (0.93 and 0.88 FG/water preference ratios for 10% and 30 % FG, respectively, P<0.01 versus KHKFl/FlXCre-Vil Figure 3C). The preference values in KHKFl/FlXCre-Vil mice were close to 0.5 (0.58 and 0.43 for 10% and 30% FG, respectively) indicating that these mice do not prefer nor necessarily dislike sugar, further supporting the idea that the lack of KHK in the gut significantly reduces the mouse preference for added sweeteners.
Figure 3: Intestinal KHK mediates sugar and total caloric intake but not metabolic syndrome.
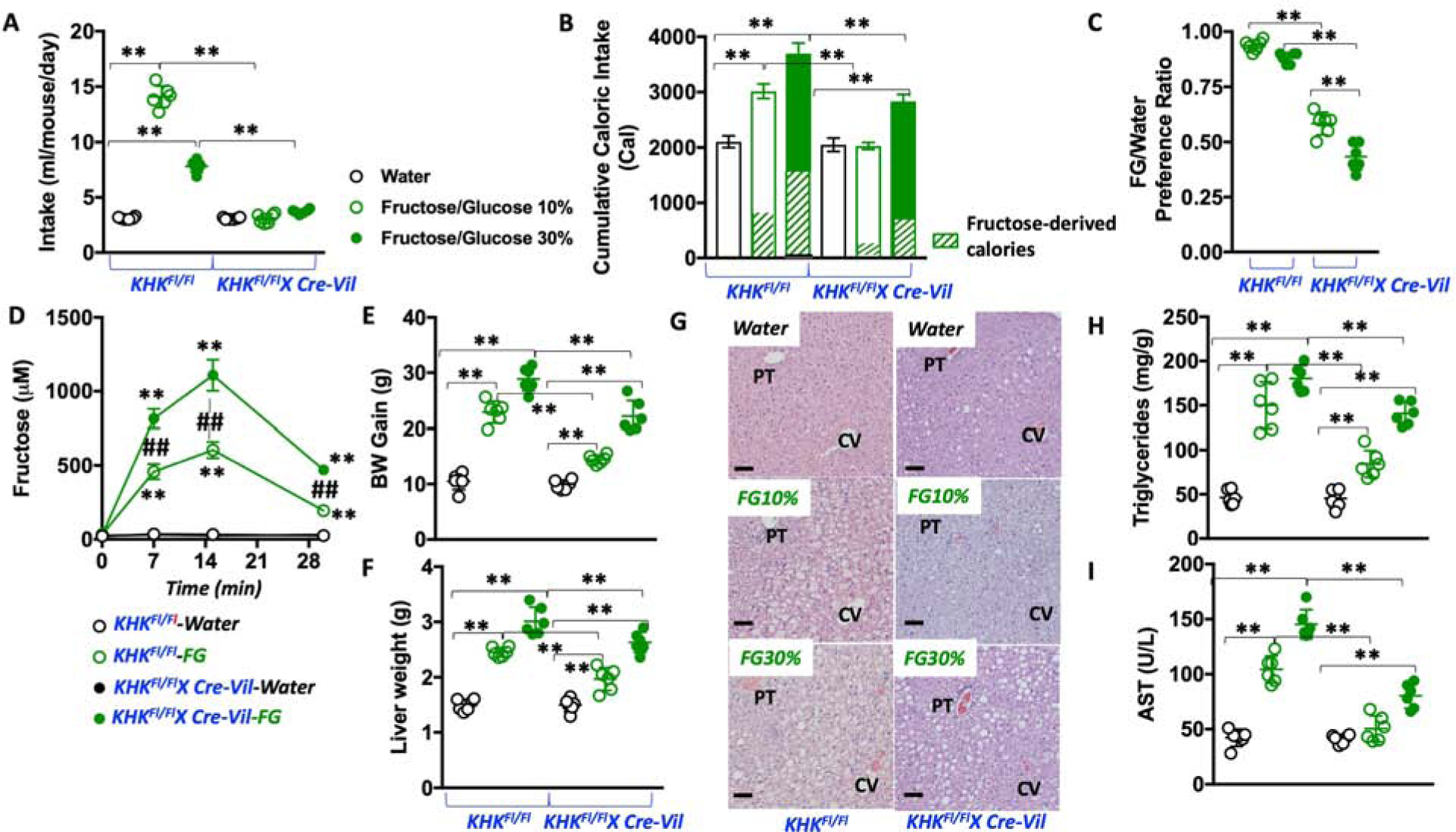
A) 30-week average daily intake of water (black), 10 % fructose/glucose (FG) solution (green, clear symbols) or 30 % FG solution (green, solid symbols) in KHKFl/Fl and KHKFl/Fl-CreVil mice.
B) 30-week cumulative caloric intake in same groups as A).
C) 10% and 30% FG over water preference ratio in KHKFl/Fl and KHKFl/Fl-CreVil mice.
D) Fructose levels in portal vein of KHKFl/Fl and KHKFl/Fl-CreVil mice after oral gavage of water (black) or an equimolar concentration of 1:1 FG mixture.
E) 30-week body weight gain in KHKFl/Fl and KHKFl/Fl-CreVil mice exposed to water, 10% FG (KHKFl/Fl) or 30% FG (KHKFl/Fl-CreVil mice).
F) 30-week liver weight in the same animal groups as in E)
G) Representative H&E images from livers of mice of the same groups as in E). (n > 10 images per animal), Size bars: 50 μM, PT: Portal triad; CV: Central Vein.
H–I) Hepatic triglycerides and plasma AST levels in the same groups as in E).
The data in A–F), and H–I) were presented as the means ± SEM and analyzed by One Way ANOVA with Tukey post hoc analysis. *P < 0.05, **P < 0.01. For panel D, ##P < 0.01 between strains at the same time point and **P < 0.01 versus time 0. n = 6 mice per group. See also Supplemental Table 2 and supplemental figure 1
It has been reported previously that intestinal fructokinase-mediated catabolism of fructose reduces the amount of the sugar that reaches the liver (Jang et al., 2018). We therefore measured portal vein levels of fructose following oral gavage with an equimolar concentration of FG in both KHKFl/Fl and KHKFl/FlXCre-Vil mice. Of interest, we observed a substantial amount of fructose in the portal vein of KHKFl/Fl mice - up to 457 and 605 μM fructose at 7 and 15 minutes post-oral gavage, respectively. However, and consistent with the finding from Jang et al. on global KHK-A/C KO mice, the specific deletion of KHK in the jejunum resulted in significantly higher fructose levels in the portal vein (816 and 1108 μM fructose at 7 and 15 minutes post-oral gavage, respectively, P<0.01 versus KHKFl/Fl) indicating that jejunum fructose metabolism determines the rate of delivery of this sugar to the liver (Figure 3D).
Based on these observations regarding the specific role of intestinal KHK in sugar intake and transport, we next compared the long-term (30 week) effect of varying FG solutions in the drinking water on the development of metabolic syndrome. Interestingly, features of metabolic syndrome occurred in both KHKFl/Fl and KHKFl/FlXCre-Vil mice. However, for each concentration of FG in the drinking water, body weight gain, fatty liver, hyperleptinemia, hyperinsulinemia and insulin resistance were significantly greater in KHKFl/Fl mice (Figure 3E–I and Supplemental Table 2). This protective effect appeared to be because KHKFl/FlXCre-Vil mice ingested less total and fructose-derived calories when compared to controls. However, and unlike global KHK-A/C KO mice, KHKFl/FlXCre-Vil that are sugar and calorie-matched with KHKFl/Fl -by comparing KHKFl/Fl on 10% FG against KHKFl/FlXCre-Vil on 30% FG-demonstrated equivalent features indicating that the blockade of intestinal metabolism of fructose does not substantially affect the development and progression of metabolic syndrome in mice.
Hepatic fructose metabolism mediates sugar-induced metabolic syndrome independently of intake.
The observation that the metabolic syndrome developed in KHKFl/FlXCre-Vil mice that imbibed the same amount of FG as the control mice raised the question of whether hepatic fructose metabolism is the main driver of sugar-induced metabolic syndrome. Importantly, and unlike intestinal KHK-deficient mice, KHKFl/FlXCre-Alb mice compared to KHKFl/Fl mice showed a similar intake and preference for sugar, as well as total caloric intake, over the 30-week study (Figure 4A–C). However, and despite similar total and fructose-derived caloric intake, liver-specific deletion of KHK resulted in a marked protection from all features of metabolic syndrome analyzed, including body weight gain, the development of fatty liver, adipose changes and inflammation, hyperinsulinemia, hyperleptinemia and insulin resistance (Figure 4D–L and Supplemental Table 3). For example, compared to WT mice, adipose accumulation in KHKFl/FlXCre-Alb mice was markedly lower (Supplemental Table 3) but more importantly, epididymal adipose inflammation was minimal in these mice (Figure 4I). Furthermore, and unlike WT animals, we did not observe whitening of subscapular brown adipose tissue (Figure 4J), suggesting the liver blockade of hepatic KHK exerted its protective effects in distant organs and that metabolic syndrome is a hepatocyte-centered condition. Consistent with decreased fructose metabolism in the liver via KHK in KHKFl/FlXCre-Alb mic, urinary fructose excretion (corrected by creatinine) is markedly higher compared to fructose-fed KHKFl/Fl mice (Figure 4M).
Figure 4: Blockade of hepatic KHK does not reduce sugar intake but completely protects against sugar-induced metabolic syndrome.
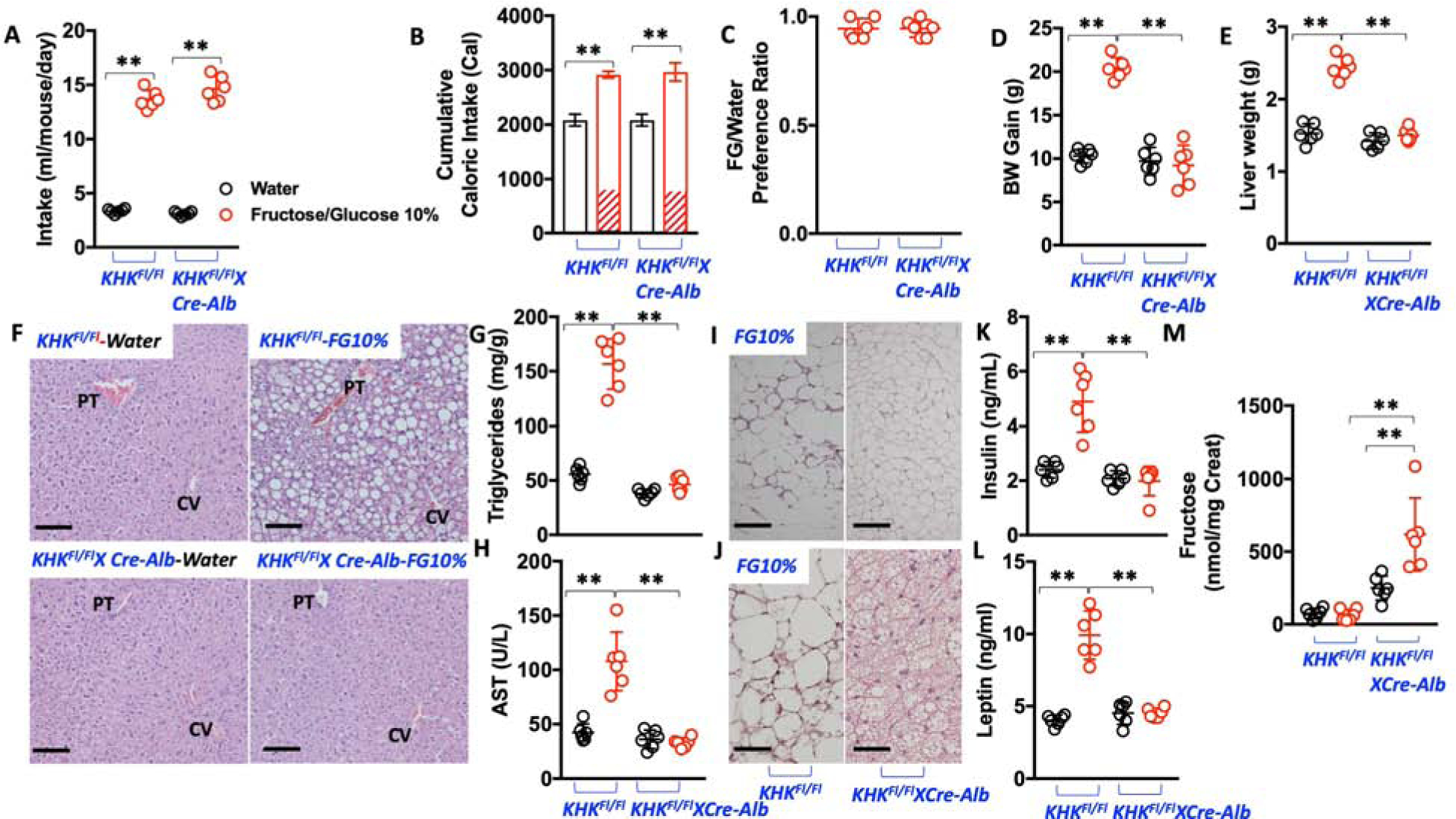
A) 30-week average daily intake of water (black) and 10 % fructose/glucose (FG) solution (red) in KHKFl/Fl and KHKFl/Fl-CreAlb mice.
B) 30-week cumulative caloric intake in same groups as A).
C) 10% FG over water preference ratio in KHKFl/Fl and KHKFl/Fl-CreAlb mice.
D) 30-week body weight gain in KHKFl/Fl and KHKFl/Fl-CreAlb mice exposed to water or 10% FG.
E) 30-week liver weight in the same animal groups as in D).
F) Representative H&E images from livers of mice of the same groups as in D). (n > 10 images per animal), Size bars: 50 μM, PT: Portal triad; CV: Central Vein.
G–H) Hepatic triglycerides and plasma AST levels in the same groups as in D).
I) Representative H&E images from epididymal adipose tissue of KHKFl/Fl and KHKFl/Fl-CreAlb mice exposed to 10% FG. (n > 10 images per animal), Size bars: 50 μM
J) Representative H&E images from subscapular -brown- adipose of KHKFl/Fl and KHKFl/Fl-CreAlb mice exposed to 10% FG. (n > 10 images per animal), Size bars: 50 μM
K–L) Fasting plasma insulin and leptin levels in the same groups as in D). .
M) Urinary fructose excretion -corrected by creatinine excretion- in spot urines from the same groups as in D).
The data in A–H), and K–M) were presented as the means ± SEM and analyzed by One Way ANOVA with Tukey post hoc analysis. *P < 0.05, **P < 0.01. n = 6 mice per group. See also Supplemental Table 3 and supplemental figure 1
Characterization of potential mechanisms driving intake of sugar that are mediated by fructose metabolism
The reduced intake of sugar observed in both global and intestinal KHK deficient mice indicate that the gut plays a key role in determining this physiological response. Even though the data here (Figure 3C) suggest the absence of a true aversion response to fructose and fructose-containing-sugars, the presence of some type of gastrointestinal (GI) discomfort associated with sugar uptake cannot be ruled out. In this regard, Glut5 has been shown to be the most important fructose transporter in the gut and its deletion in mice is associated with severe fructose malabsorption (Barone et al., 2009). Consistent with increased exposure to sugar, the expression of Glut5 in the jejunum of KHKFl/Fl and KHKFl/FlXCre-Alb mice is significantly up-regulated at 30 weeks at both the mRNA and protein levels (Figure 5A–C). In contrast, while the expression of Glut5 is substantially higher in control KHKFl/FlXCre-Vil mice, its levels are markedly lower when mice are exposed to sugar. This observation suggests that KHK and fructose metabolism in the gut is an important mechanism to maintain Glut5 expression and proper fructose uptake in sugar-exposed mice as already previously proposed (Patel et al., 2015a) and therefore, gut-specific KHK deficiency could result in GI discomfort in mice exposed to sugar.
Figure 5: Blockade of intestinal KHK decreases the expression of Glut5 in the gut of mice exposed to sugar.
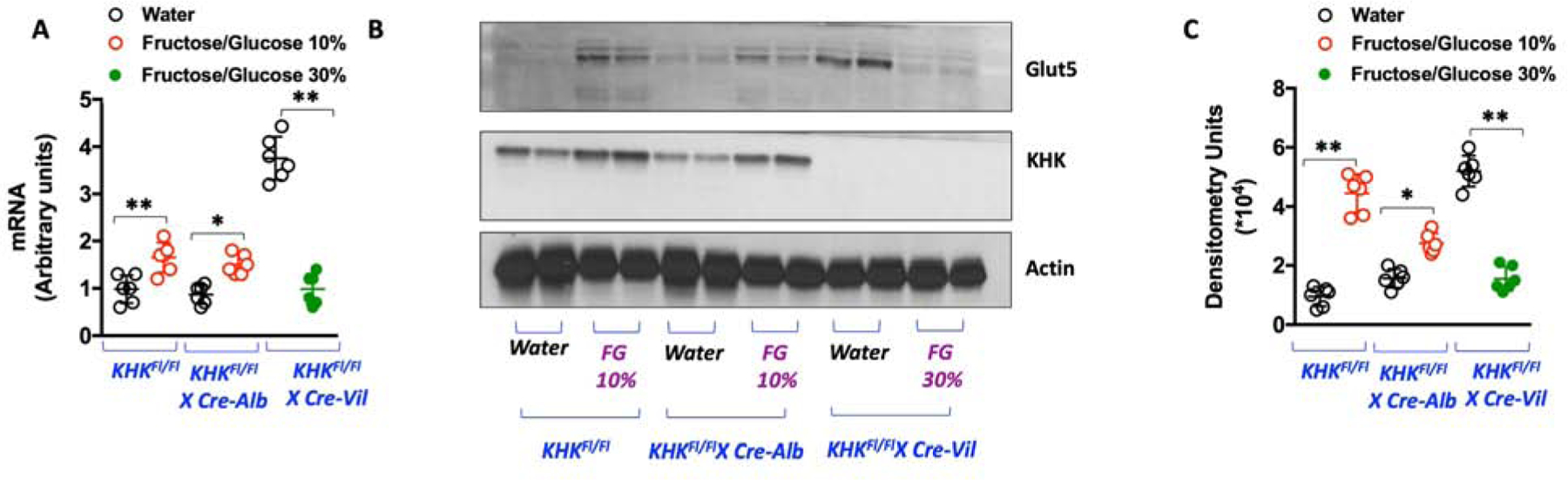
A) Glut5 (slc2a5) mRNA levels in jejunum of KHKFl/Fl, KHKFl/FlXCre-Alb and KHKFl/Fl-CreVil mice on Water or FG.
B) Representative western blot for Glut5, KHK and actin loading control in jejunum of KHKFl/Fl, KHKFl/FlXCre-Alb and KHKFl/Fl-CreVil mice on Water or FG (n= 3 total blots).
C) Densitometry for Glut5 (normalized to actin loading control) for 6 different animals in each group.
The data in A), and C) were presented as the means ± SEM and analyzed by One Way ANOVA with Tukey post hoc analysis. *P < 0.05, **P < 0.01. n = 6 mice per group. See also Supplemental Figure 2
It is important to note that the liver can also play a substantial role in regulating fructose intake. In this regard, recently, a key role has been proposed for FGF21, a liver-derived factor involved in the regulation of sweet preference intake (von Holstein-Rathlou et al., 2016). Consistent with previous reports(Soberg et al., 2017; von Holstein-Rathlou et al., 2016), exposure of WT mice to sugar results in a marked and significant up-regulation of plasma FGF21 levels (from 396 to 5326 pg/ml, P<0.01, Figure 6A). In contrast, sugar-dependent FGF21 up-regulation is substantially blunted in global KHK-A/C KO mice receiving similar amounts of sugar (from 286 to 1107 pg/ml, P<0.01). Furthermore, while plasma FGF21 levels correlate with average daily sugar intake in WT mice (R2=0.73, P=0.02), the correlation is lost when WT and global KHK-A/C KO mice are compared (Figure 6B). This would suggest that sugar-dependent production of FGF21 relies on KHK and fructose metabolism. Consistent with this notion, hepatic mRNA levels of Fgf21 are up-regulated in WT but not in global KHK-A/C KO mice (Figure 6C). Importantly and consistent with a key role for hepatic fructose metabolism in controlling FGF21 expression, FGF21 is elevated in plasma of KHKFl/Fl and KHKFl/FlXCre-Vil mice but not KHKFl/FlXCre-Alb mice (Figure 6D). The observation that both liver-specific and global KHK-A/C KO mice do not have elevated FGF21 levels suggests that FGF21 elevation is not necessarily dependent on the amount of sugar intake in mice. FGF21 levels in plasma in all mice correlate with markers of liver dysfunction like plasma AST (R2=0.86, P<0.001) and liver triglycerides (R2=0.69, P=0.03, Figure 6E–F).
Figure 6: Regulation of FGF21 by KHK and fructose metabolism.
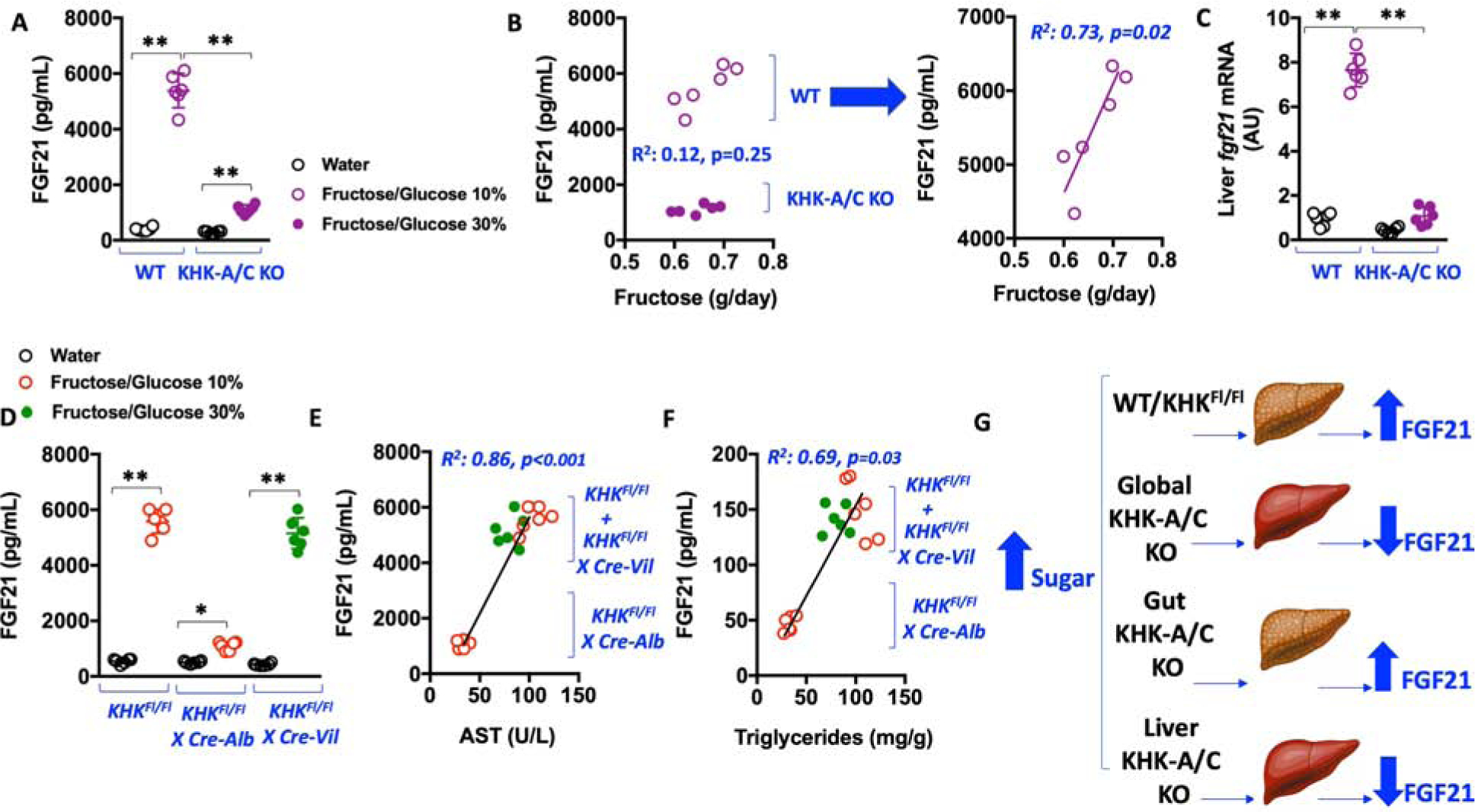
A) Plasma levels of FGF21 in wild type (WT) and global KHK-A/C KO mice on water or Fructose/Glucose solution.
B) Correlations between plasma FGF21 and daily fructose intake in WT and global KHK-A/C KO mice on Fructose/Glucose solution.
C) Hepatic mRNA levels of Fgf21 in WT and global KHK-A/C KO mice on water or Fructose/Glucose solution.
D) Plasma levels of FGF21 in KHKFl/Fl, KHKFl/FlXCre-Alb and KHKFl/Fl-CreVil mice on water or Fructose/Glucose solution. E–F) Correlations between plasma FGF21 and plasma AST or liver triglycerides in KHKFl/Fl, KHKFl/FlXCre-Alb and KHKFl/Fl-CreVil mice on FG.
G) Proposed mechanism whereby hepatic KHK induces the production and release of FGF21 independently of sugar
The data in A), C) and D) were presented as the means ± SEM and analyzed by One Way ANOVA with Tukey post hoc analysis. The data in B) and E–F) were presented as single values with Pearson r data analysis. *P < 0.05, **P < 0.01. n = 6 mice per group. See also Supplemental Tables 1–3.
Discussion
We have investigated the role of fructose metabolism in driving the metabolic syndrome, particularly in response to added sweeteners using a fructose-glucose combination that is similar in composition to soft drinks containing sucrose or HFCS. There has been controversy over the mechanisms by which soft drinks induce metabolic syndrome, with some arguing it is simply a caloric source and that there is no clear evidence that any dietary sugar has a unique impact relative to any other source of calories (Kahn and Sievenpiper, 2014; Keys, 1971), while others have focused on the high glycemic effects of glucose to stimulate insulin-mediated effects (Taubes, 2007), and yet others emphasizing the role of fructose (Lustig et al., 2012; Stanhope and Havel, 2009). Here, we document that fructose metabolism, and specifically fructokinase (KHK), is critical for how added sweeteners cause metabolic syndrome in laboratory mice. This does not rule out the importance of high glycemic carbohydrates in driving disease, as our prior work has shown that mice drinking glucose water also develop metabolic syndrome, but importantly much of these effects were due to the conversion of glucose to fructose in the intestine due to the induction of aldose reductase (Lanaspa et al., 2013). Also, because the differences in body weight between WT and KHK-A/C KO mice on FG are clear, we cannot conclude that the differences observed in other metabolic features are independent of body weight gain and thus, they are not necessarily due to KHK blockade. However, it is important to note that it has been previously reported that there is a significant difference in both liver fat content and insulin sensitivity in KHK-A/C KO mice exposed to a fructose diet for 8 weeks compared to similar weight WT animals (Miller et al., 2018). Nonetheless, our results point to a hepatocyte-centric origin of the metabolic syndrome.
We also evaluated the effect of genetically deleting fructokinase in the intestines on the development of metabolic syndrome. Firstly, we confirmed previously reported finding (Jang et al., 2018) that the absence of intestinal fructokinase results in a greater delivery of fructose to the liver compared to control mice when given equal FG loads. However, this was countered by a spontaneous reduction in FG intake in the ad libitum studies due to low expression of the fructose transporter Glut5, and hence the intestinal KHK-A/C KO was protected from developing sugar-induced metabolic syndrome compared to controls. We then performed a study in which total calories, as well as calories from FG were matched between intestinal KHK-A/C KO and control mice, and in this setting the intestinal KO mice developed similar levels of obesity and metabolic syndrome. This was surprising, as we had expected to observe an exacerbation in metabolic syndrome in gut-specific KHK-A/C KO mice compared to KHKFl/Fl on sugar.
To better understand potential reasons for this finding, we evaluated the expression of Glut5, the primary fructose transporter. Interestingly, Glut5 tended to be higher in intestinal KHK-A/C KO mice on water compared to controls, but unlike the upregulation of Glut5 that occurs with fructose exposure, the intestinal KO showed a downregulation in expression. The up-regulation of Glut5 observed in gut-specific KHK-A/C KO mice at baseline differ from that previously reported in global KHK-A/C KO mice (Patel et al., 2015b) probably due to the presence of KHK in the liver of gut-specific but not in whole-body KHK-A/C KO mice . Nevertheless, a connection between KHK and Glut5 expressions in mice has already been documented before (Patel et al., 2015a; Patel et al., 2015b). As both Glut5 and KHK expressions are regulated by the transcription factor associated to carbohydrates (ChREBP) (Lanaspa et al., 2012c; Ma et al., 2006; Uyeda and Repa, 2006), it is likely that this transcription factor is playing a key role in regulating the expression of Glut5 in sugar-exposed gut-specific KHK-A/C KO mice. Of interest, ChREBP deficiency is associated with diarrhea and fructose intolerance (Oh et al., 2018). The reduction of Glut5 expression in sugar-exposed intestinal KHK-A/C KO mice suggests that there is likely decreased absorption of fructose into the intestinal epithelial cells, but greater delivery to the liver due to no metabolism in the gut, with the net effect being a similar severity of metabolic syndrome as control mice. It also suggests that there might be more fructose that enters the distal gut. Whether this latter finding might lead to side effects leading to less intake is possible, although we did not observe diarrhea or any signs of toxicity, and the intestinal KO mice did not show signs of aversion based on two-bottle preference testing.
In contrast to the intestinal-specific KHK-A/C KO mice, the liver-specific KHK-A/C KO mice had similar preference for sugar compared to the control mice, but were completely protected from the metabolic syndrome. The observation that they were protected not simply from fatty liver but also from systemic effects (adipose and weight gain) demonstrate the importance of the liver in regulating metabolic syndrome induced by sugar. The data also confirms earlier studies showing the protective effects of fructokinase blockade on fatty liver and metabolic syndrome (Ishimoto et al., 2012; Miller et al., 2018; Softic et al., 2018), which is largely mediated by preventing fructose-induced lipogenesis, mitochondrial dysfunction and adenine nucleotide turnover (Herman and Samuel, 2016; Kim et al., 2016; Lanaspa et al., 2018a; Lanaspa et al., 2012a; Lanaspa et al., 2012b; Lanaspa et al., 2012c; Softic et al., 2019). We also examined the relationship of FGF21 in these models since FGF21 might provide protective effects. However, our data , showed that FGF21 correlated more with the presence of liver injury, and levels were lower in the liver-specific and global KHK-A/C KO mice. Thus, our data indicate that fructose metabolism via KHK is necessary for FGF21 production. As FGF21 is thought to be protective against metabolic disease, these data suggest as well that the benefits of knocking out KHK outweigh the potential detrimental effect of reduced circulating FGF21 levels. (Figure 6G). A schematic summarizing whole body and tissue specific effects of KHK blockade is shown in Figure 7.
Figure 7: Proposed mechanism for the differential effects of KHK blockade in sugar-induced metabolic syndrome.
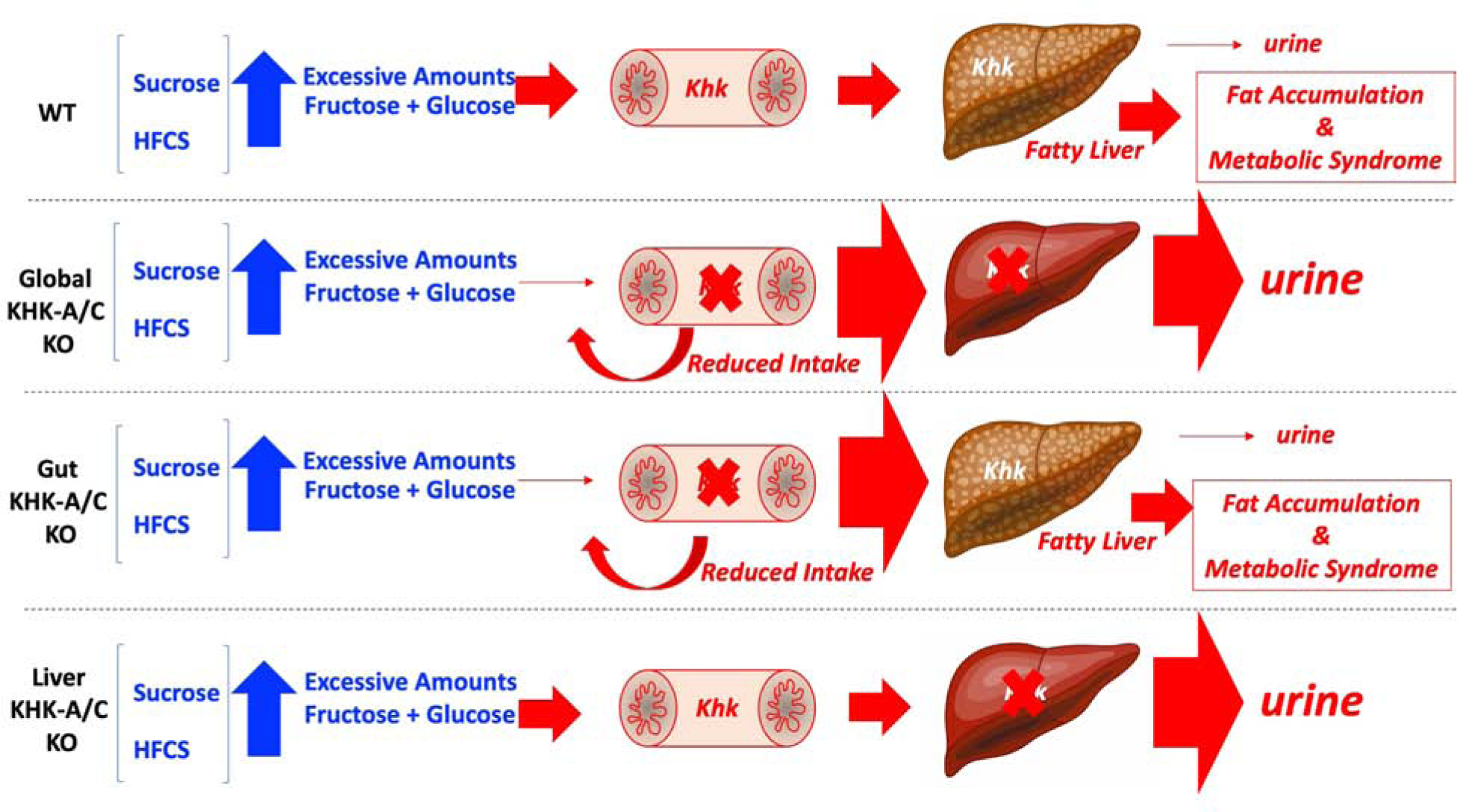
Low amounts of sugar (glucose+fructose) are rapidly cleared by the intestine and liver with little or no fructose being excreted into the urine. On the other hand, excessive amounts of sugar (fructose and glucose) induce fatty liver and metabolic syndrome in WT mice by inducing greater sugar intake thus leading to increased fructose metabolism via KHK. The absence of KHK in whole-body (global) KHK-A/C KO mice protects mice against sugar-induced metabolic syndrome by both reducing the intake of sugar and eliminating the metabolism of fructose. Intestinal KHK mediates sugar and overall caloric intake. However, in the settings of excessive or chronic exposure to fructose, the reduced intestinal clearance of fructose results in greater delivery to the liver thus promoting fatty liver, fat accumulation and metabolic syndrome. In contrast, while the blockade of hepatic KHK does not decrease sugar intake, it completely prevents fructose metabolism thus promoting its metabolism through enzymes with lower affinity for fructose (hexokinase) or its excretion into the urine.
In conclusion, added sweeteners such as sucrose and HFCS are a major staple in the western diet and are strongly associated with obesity and diabetes. Our studies suggest a key role for fructose metabolism in driving these effects. Evolutionarily and during pre-industrial time periods fructose was an important nutrient whose metabolic actions were to stimulate fat storage as a protection mechanism to periods of famine and starvation, but when taken in excess, this sugar leads to obesity and diabetes (Johnson et al., 2019). Reducing added sweeteners from the diet is recommended, as is decreasing foods with high-glycemic carbohydrates and excess salt, which can stimulate endogenous fructose production (Lanaspa et al., 2013; Lanaspa et al., 2018c). These simple dietary measures may provide important benefits in slowing the twin epidemics of obesity and diabetes.
Limitations of the Study
While the advantage of studying mice is the ability to delete genes of interest in a tissue-specific manner, a limitation of the current study is that our mouse data may not fully translate to humans. Furthermore, the study does not provide a mechanism to explain how intestinal fructokinase regulates sugar preference. However, glucose is known to be involved in preference for sugar via post-gastric signaling (Sclafani and Ackroff, 2012), so conversion of fructose to glucose by intestinal fructokinase (Jang et al., 2018) could potentially affect sugar intake. More studies will be needed to evaluate this potential mechanism, as well as understanding the role of KHK in the brain. Finally, a better understanding of how liver fructose metabolism may induce systemic effects of metabolic syndrome is needed. One potential explanation could be the production and release of lipids from the liver that are then taken up by adipose tissue, leading not only to fat accumulation but an activation of the adipokine systems.
STAR*METHODS
RESOURCE AVAILABILITY [
Lead Contact
Further information and requests for resources and reagents should be directed and will be fulfilled by the Lead Contact, Miguel A. Lanaspa (Miguel.lanaspagarcia@cuanschutz.edu).
Materials Availability
Mouse lines generated in this study are available for any researcher upon reasonable request.
Data and Code availability
This study did not generate unique datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Study Approval.
All animal experiments were conducted with adherence to the NIH Guide for the Care and Use of Laboratory Animals (National Research Council (U.S.). Committee for the Update of the Guide for the Care and Use of Laboratory Animals. et al., 2011). The animal protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Colorado (Aurora, CO).
Animals.
KHK-A/C KO (B6;129-Khktm2Dtb) and KHK-A KO (B6;129-Khktm2.1Dtb) mice were originally developed by David Bonthorn at Leeds University (UK)(Diggle et al., 2010) and were bred and maintained at the Univ. Colorado with pure C57/Bl6 for over 7 generations to ensure the mice were on the B6 genetic background. Mice with LoxP sequences flanking exons 3 and 4 of the Khk gene (KHKFl/Fl) were generated by the Genomic Core at the University of Colorado Cancer Center. Tissue-specific KHK-A/C KO mice were obtained by crossing KHKFl/Fl mice with tissue-specific Cre-recombinase expressing mice obtained from Jackson labs (Cre-Alb 003574, and Cre-Vil 004586). All experimental mice were maintained in temperature- and humidity-controlled specific pathogen-free condition on a 14-h dark/10-h light cycle and at 25°C, and allowed ad libitum access to normal laboratory chow (Harlan Teklad, #2920X). Water and food consumption was monitored daily and body weight recorded weekly for 30 weeks. Caloric intake was calculated as the sum of chow intake (3.1 cal/g) and water intake (accounting that fructose and glucose provide 4 cal/g). All experiments were conducted with adherence to the NIH Guide for the Care and Use of Laboratory Animals. In all studies, 7–10 week old male mice (n=3–7) were employed. Food consumption was monitored daily and body weight recorded. All animals in the study were phenotypically normal and generally healthy during the legthe study.
For 24 hr two-bottle preference studies, mice were housed individually and provided with two similar water bottles filled with water for acclimation for a 5-day period. For the experiment, water in one of the bottles was substituted by water containing a mixture of fructose and glucose (FG) at the indicated concentration. One day after exposure, the position of the two bottles (regular and sweet-containing water) was switched to control for side preference. The preference ratio was calculated as the ratio of volume of tastant consumed over the 2 day test period to total volume consumed, i.e. a score of 0.5 or 50% shows no preference.
For portal vein studies, same weight animals were gavaged with either 1 g/kg fructose or a combination of fructose/glucose −1 g/kg each-. Portal vein blood was collected at the indicated times on mice under isoflurane anesthesia and serum was obtained after centrifugation at 13000 rpm for 2 min at room temperature. Fructose levels were determined biochemically following manufacturer’s instructions (bioassay systems, EFRU-100).
METHOD DETAILS
Biochemical analysis.
Blood was collected in microtainer tubes (BD) from cardiac puncture of mice under isoflurane, and serum was obtained after centrifugation at 13000 rpm for 2 min at room temperature. Serum parameters was performed biochemically following manufacturer’s instruction (uric acid: Bioassay systems, DIUA-250; FGF21: R&D, MF2100, AST: Bioassay Systems, EASTR-100, ALT: Bioassay Systems, EALT-100, Insulin: Crystal Chem, 90080, Leptin: R&D, MOB00, FGF21: R&D, MF2100). Determination of parameters in tissue was performed in freeze-clamped tissues and measured biochemically following manufacturer’s protocol (Triglycerides (Liver): Bioassay Systems, ETGA-200; uric acid: Bioassay Systems DIUA-250).
Histopathology.
Formalin-fixed paraffin-embedded liver, epididymal and subscapular adipose sections were stained with hematoxylin and eosin (H&E). Histological examination was performed through an entire cross section of liver from each mouse. Images were captured on an Olympus BX51 microscope equipped with a four megapixel Macrofire digital camera (Optronics; Goleta, CA) using the PictureFrame Application 2.3 (Optronics). Composite images were assembled with the use of Adobe Photoshop. All images in each composite were handled identically.
Western blot.
Protein lysates were prepared from mouse tissue employing MAP Kinase lysis buffer as previously described(Lanaspa et al., 2007). Protein content was determined by the BCA protein assay (Pierce). Total protein (50 μg) was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (10% w/v), and transferred to PVDF membranes (BioRad). Membranes were first blocked for 1 h at 25 °C in 4% (w/v) instant milk dissolved in 0.1 % Tween-20 Tris-Buffered Saline (TTBS), incubated with primary rabbit or mouse-raised antibodies (1:1000 dilution in TTBS) KHK (Sigma HPA007040; RRID:AB_1079185), KHK-A (SAB Signalway; Cat# 21708), KHK-C (SAB Signalway; Cat# 21709), Glut5 (Millipore, 07–1406 lot 2999837, this antibody and lot has been validated using specific Glut5 KO mice, a representative western blot is shown in supplemental figure 2) and Actin (Cell Signaling 4968; RRID:2313904) and visualized using an anti-rabbit (7074; RRID:AB_2099233) or anti-mouse IgG (7076; RRID:AB_330924) horseradish-peroxidase conjugated secondary antibody (1:2000, Cell Signaling) using the HRP Immunstar® detection kit (Bio-Rad, Hercules, CA). Chemiluminescence was recorded with an Image Station 440CF and results analyzed with the 1D Image Software (Kodak Digital Science, Rochester, NY).
Insulin Tolerance Tests:
Insulin sensitivity was determined by both oral glucose and insulin tolerance tests as previously described (Lanaspa et al., 2018b).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis.
All numerical data are presented as the mean ± s.e.m. Independent replicates for each data point (n) are identified in figure legends. Data graphics and statistical analysis were performed using Prism 5 (GraphPad). Data without indications were analyzed by one-way ANOVA, Tukey post hoc test. A value of P < 0.05 was regarded as statistically significant. Animals were randomly allocated in each group using randomizer (www.randomizer.org). Power calculations for the number of animals assigned to each group were based on our previous publications and designed to observe a greater than 15% difference in body weight difference between groups. IN general, an n of 6 mice per group was used. No animals were excluded from the study and whenever possible experiments were done in a blond fashion. For example, for data analysis, except for western blot, single samples (plasma, homogenates,…) were first codified and decoded after determination. Similarly, histological records and scoring were done in a blind fashion.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse KHK | Sigma | HPA007040; RRID:AB_1079185 |
| Anti-mouse KHK-A | SAB Signalway | 21708 |
| Anti-mouse KHK-C | SAB Signalway | 21709 |
| Anti-mouse Glut5 | Millipore | Lot#2999837; 07–146: RRID:AB_10615493 |
| Anti-mouse Actin | Cell Signaling | 4968; RRID:2313904 |
| Goat Anti-Rabbit IgG HRP conjugated | Cell Signaling | 7074; RRID:AB_2099233 |
| Goat Anti-Mouse IgG HRP conjugated | Cell Signaling | 7076; RRID:AB_330924 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Fructose | Sigma | F0127 |
| Glucose | Sigma | G8270 |
| Insulin (human) | Sigma | I2643 |
| Hematoxyline | Thermo Scientific | 22–110–639 |
| Eosin-Y | Fisher Scientific | SE23–500D |
| Magnesium Chloride | Sigma | M8266 |
| Sodium Vanadate | Sigma | 590088 |
| Triton-X | Millipore | MTX15681 |
| Tween 20 | MP Biomedicals | MP1TWEEN201 |
| Critical Commercial Assays | ||
| Fructose Determination Kit | Bioassay Systems | EFRU-100 |
| Uric Acid Determination Kit | Bioassay Systems | DIUA-250 |
| Triglyceride Determination Kit | Bioassay Systems | ETGA-200 |
| AST Determination Kit | Bioassay Systems | EASTR-100 |
| ALT Determination Kit | Bioassay Systems | EALT-100 |
| Anti-Mouse FGF21 ELISA | R&D | MF2100 |
| Anti-Mouse Insulin ELISA | Crystal Chem | 90080 |
| Anti-Mouse Leptin ELISA | R&D | MOB00 |
| Experimental Models: Organisms/Strains | ||
| KHK-A KO mouse | Obtained from Dr David Bonthron at Leeds University, UK | N/A |
| KHK-A/C KO mouse | Obtained from Dr David Bonthron at Leeds University, UK | N/A |
| KHKFl/Fl | Generated in-house | N/A |
| KHKFl/FlXCreVil | Generated in-house | N/A |
| KHKFl/FlXCreAlb | Generated in-house | N/A |
| CreVil | Jackson Labs | 004586 |
| CreAlb | Jackson Labs | 003574 |
| Oligonucleotides | ||
| Primers for mouse Glut5 (slc2a5) Forward | 5’-TGTACGACACCCTACCCTACTG-3’ | N/A |
| Primers for mouse Glut5 (slc2a5) Reverse | 5′GGTTCGAAAGATAATGAACGTGT-3′ | N/A |
| Primers for mouse Actin Forward | 5’TTCTTGCAGCTCCTTCGTTGCCG3’ | N/A |
| Primers for mouse Actin reverse | 5′TCTACACGCTAGGCGTAAAGTTGG-3′ | N/A |
| Other | ||
| Glucometer | Accu-Check | Accu-Check Guide Meter |
| Glucose Strips | Accu-Check | Accu-Check Guide Test Strips |
Highlights.
Fructose metabolism is necessary for sugar-induced obesity and metabolic syndrome.
Fructose induces tissue-dependent effects on sugar intake and metabolic dysfunction
Sugar intake and preference is mediated by intestinal fructose metabolism.
Sugar-induced metabolic syndrome is mediated by hepatic fructose metabolism.
Context and Significance.
Fructose is the most typical sugar consumed by man, usually in the form of sucrose or high-fructose corn syrup. Excess ingestion, however, is believed to cause metabolic syndrome, which in lab animals can be prevented by blocking fructokinase, a key enzyme in its metabolism. Even so, to date there have been no studies showing that preventing the metabolism of fructose rather than glucose can completely block the deleterious effects of excess sugar. By using tissue-specific fructokinase knockout mice, Andres-Hernando et al. find that blocking fructose metabolism completely, or just in the liver, prevents sugar-induced metabolic syndrome, while gut fructokinase drives sugar intake. These findings illustrate that sugar-dependent metabolic syndrome is a hepatocyte-centered condition, and one that is driven by fructose and not glucose.
Acknowledgements:
We would like to thank Dr Ronaldo Ferraris (Rutgers University) and his group for providing meaningful discussions on Glut5 regulation by KHK and fructose and with samples from Glut5 knockout mice to test the efficiency of our antibody.
Funding: This work has been supported by NIH Grants 1R01DK105364-01A1 (to M.A.L. and R.J.J.), 1R01DK121496 (to M.A.L. and R.J.J.), and 1R01DK108859 (to M.A.L.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
M.A.L. and R.J.J. are inventors in two patents (13/814,568 and 62/473,005) related to the blockade of fructokinase to treat metabolic syndrome. M.A.L. and R.J.J. are founders and members of Colorado Research Partners (CRP), a LLC company dedicated to the generation of fructokinase inhibitors.
References:
- Barone S, Fussell SL, Singh AK, Lucas F, Xu J, Kim C, Wu X, Yu Y, Amlal H, Seidler U, et al. (2009). Slc2a5 (Glut5) is essential for the absorption of fructose in the intestine and generation of fructose-induced hypertension. J Biol Chem 284, 5056–5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicerchi C, Li N, Kratzer J, Garcia G, Roncal-Jimenez CA, Tanabe K, Hunter B, Rivard CJ, Sautin YY, Gaucher EA, et al. (2014). Uric acid-dependent inhibition of AMP kinase induces hepatic glucose production in diabetes and starvation: evolutionary implications of the uricase loss in hominids. FASEB J 28, 3339–3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diggle CP, Shires M, Leitch D, Brooke D, Carr IM, Markham AF, Hayward BE, Asipu A, and Bonthron DT (2009). Ketohexokinase: expression and localization of the principal fructose-metabolizing enzyme. J Histochem Cytochem 57, 763–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diggle CP, Shires M, McRae C, Crellin D, Fisher J, Carr IM, Markham AF, Hayward BE, Asipu A, and Bonthron DT (2010). Both isoforms of ketohexokinase are dispensable for normal growth and development. Physiol Genomics 42A, 235–243. [DOI] [PubMed] [Google Scholar]
- Goncalves MD, Lu C, Tutnauer J, Hartman TE, Hwang SK, Murphy CJ, Pauli C, Morris R, Taylor S, Bosch K, et al. (2019). High-fructose corn syrup enhances intestinal tumor growth in mice. Science 363, 1345–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goran MI, Ulijaszek SJ, and Ventura EE (2013). High fructose corn syrup and diabetes prevalence: a global perspective. Glob Public Health 8, 55–64. [DOI] [PubMed] [Google Scholar]
- Herman MA, and Samuel VT (2016). The Sweet Path to Metabolic Demise: Fructose and Lipid Synthesis. Trends Endocrinol Metab 27, 719–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimoto T, Lanaspa MA, Le MT, Garcia GE, Diggle CP, Maclean PS, Jackman MR, Asipu A, Roncal-Jimenez CA, Kosugi T, et al. (2012). Opposing effects of fructokinase C and A isoforms on fructose-induced metabolic syndrome in mice. Proc Natl Acad Sci U S A 109, 4320–4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimoto T, Lanaspa MA, Rivard CJ, Roncal-Jimenez CA, Orlicky DJ, Cicerchi C, McMahan RH, Abdelmalek MF, Rosen HR, Jackman MR, et al. (2013). High-fat and high-sucrose (western) diet induces steatohepatitis that is dependent on fructokinase. Hepatology 58, 1632–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang C, Hui S, Lu W, Cowan AJ, Morscher RJ, Lee G, Liu W, Tesz GJ, Birnbaum MJ, and Rabinowitz JD (2018). The Small Intestine Converts Dietary Fructose into Glucose and Organic Acids. Cell Metab 27, 351–361 e353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RJ, Nakagawa T, Sanchez-Lozada LG, Shafiu M, Sundaram S, Le M, Ishimoto T, Sautin YY, and Lanaspa MA (2013). Sugar, uric acid, and the etiology of diabetes and obesity. Diabetes 62, 3307–3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RJ, Stenvinkel P, Andrews P, Sánchez-Lozada LG, Nakagawa T, Gaucher EA, Andres-Hernando A, Rodriguez-Iturbe B, Roncal Jimenez C, Garcia G, et al. (2019). Fructose Metabolism as a Common Evolutionary Pathway of Survival associated with climate change, food shortage and droughts. J Int Med (in press). J Intern Med. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn R, and Sievenpiper JL (2014). Dietary sugar and body weight: have we reached a crisis in the epidemic of obesity and diabetes?: we have, but the pox on sugar is overwrought and overworked. Diabetes Care 37, 957–962. [DOI] [PubMed] [Google Scholar]
- Keys A (1971). Sucrose in the diet and coronary heart disease. Atherosclerosis 14, 193–202. [DOI] [PubMed] [Google Scholar]
- Kim MS, Krawczyk SA, Doridot L, Fowler AJ, Wang JX, Trauger SA, Noh HL, Kang HJ, Meissen JK, Blatnik M, et al. (2016). ChREBP regulates fructose-induced glucose production independently of insulin signaling. J Clin Invest 126, 4372–4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratzer JT, Lanaspa MA, Murphy MN, Cicerchi C, Graves CL, Tipton PA, Ortlund EA, Johnson RJ, and Gaucher EA (2014). Evolutionary history and metabolic insights of ancient mammalian uricases. Proc Natl Acad Sci U S A 111, 3763–3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz TW, Kabra PM, Booth BE, Al-Bander HA, Portale AA, Serena BG, Tsai HC, and Morris RC Jr. (1986). Liquid-chromatographic measurements of inosine, hypoxanthine, and xanthine in studies of fructose-induced degradation of adenine nucleotides in humans and rats. Clin Chem 32, 782–786. [PubMed] [Google Scholar]
- Lanaspa MA, Almeida NE, Andres-Hernando A, Rivard CJ, Capasso JM, and Berl T (2007). The tight junction protein, MUPP1, is up-regulated by hypertonicity and is important in the osmotic stress response in kidney cells. Proc Natl Acad Sci U S A 104, 13672–13677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanaspa MA, Andres-Hernando A, Orlicky DJ, Cicerchi C, Jang C, Li N, Milagres T, Kuwabara M, Wempe MF, Rabinowitz JD, et al. (2018a). Ketohexokinase C blockade ameliorates fructose-induced metabolic dysfunction in fructose-sensitive mice. J Clin Invest 128, 2226–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanaspa MA, Cicerchi C, Garcia G, Li N, Roncal-Jimenez CA, Rivard CJ, Hunter B, Andres-Hernando A, Ishimoto T, Sanchez-Lozada LG, et al. (2012a). Counteracting Roles of AMP Deaminase and AMP Kinase in the Development of Fatty Liver. PLoS ONE 7, e48801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanaspa MA, Ishimoto T, Li N, Cicerchi C, Orlicky DJ, Ruzycki P, Rivard C, Inaba S, Roncal-Jimenez CA, Bales ES, et al. (2013). Endogenous fructose production and metabolism in the liver contributes to the development of metabolic syndrome. Nat Commun 4, 2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanaspa MA, Kuwabara M, Andres-Hernando A, Li N, Cicerchi C, Jensen T, Orlicky DJ, Roncal-Jimenez CA, Ishimoto T, Nakagawa T, et al. (2018b). High salt intake causes leptin resistance and obesity in mice by stimulating endogenous fructose production and metabolism. Proc Natl Acad Sci U S A 115, 3138–3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanaspa MA, Kuwabara M, Andres-Hernando A, Li N, Cicerchi C, Jensen T, Orlicky DJ, Roncal-Jimenez CA, Ishimoto T, Nakagawa T, et al. (2018c). High salt intake causes leptin resistance and obesity in mice by stimulating endogenous fructose production and metabolism. Proc Natl Acad Sci U S A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanaspa MA, Sanchez-Lozada LG, Choi YJ, Cicerchi C, Kanbay M, Roncal-Jimenez CA, Ishimoto T, Li N, Marek G, Duranay M, et al. (2012b). Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: potential role in fructose-dependent and - independent fatty liver. J Biol Chem 287, 40732–40744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanaspa MA, Sanchez-Lozada LG, Cicerchi C, Li N, Roncal-Jimenez CA, Ishimoto T, Le M, Garcia GE, Thomas JB, Rivard CJ, et al. (2012c). Uric acid stimulates fructokinase and accelerates fructose metabolism in the development of fatty liver. PLoS One 7, e47948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laughlin MR (2014). Normal roles for dietary fructose in carbohydrate metabolism. Nutrients 6, 3117–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Qian X, Peng LX, Jiang Y, Hawke DH, Zheng Y, Xia Y, Lee JH, Cote G, Wang H, et al. (2016). A splicing switch from ketohexokinase-C to ketohexokinase-A drives hepatocellular carcinoma formation. Nat Cell Biol 18, 561–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lustig RH, Mulligan K, Noworolski SM, Tai VW, Wen MJ, Erkin-Cakmak A, Gugliucci A, and Schwarz JM (2016). Isocaloric fructose restriction and metabolic improvement in children with obesity and metabolic syndrome. Obesity (Silver Spring) 24, 453–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lustig RH, Schmidt LA, and Brindis CD (2012). Public health: The toxic truth about sugar. Nature 482, 27–29. [DOI] [PubMed] [Google Scholar]
- Ma L, Robinson LN, and Towle HC (2006). ChREBP*Mlx is the principal mediator of glucose-induced gene expression in the liver. J Biol Chem 281, 28721–28730. [DOI] [PubMed] [Google Scholar]
- Maenpaa PH, Raivio KO, and Kekomaki MP (1968). Liver adenine nucleotides: fructose-induced depletion and its effect on protein synthesis. Science 161, 1253–1254. [DOI] [PubMed] [Google Scholar]
- Malik VS, Li Y, Pan A, De Koning L, Schernhammer E, Willett WC, and Hu FB (2019). Long-Term Consumption of Sugar-Sweetened and Artificially Sweetened Beverages and Risk of Mortality in US Adults. Circulation 139, 2113–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CO, Yang X, Lu K, Cao J, Herath K, Rosahl TW, Askew R, Pavlovic G, Zhou G, Li C, et al. (2018). Ketohexokinase knockout mice, a model for essential fructosuria, exhibit altered fructose metabolism and are protected from diet-induced metabolic defects. Am J Physiol Endocrinol Metab 315, E386–E393. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Hu H, Zharikov S, Tuttle KR, Short RA, Glushakova O, Ouyang X, Feig DI, Block ER, Herrera-Acosta J, et al. (2006). A causal role for uric acid in fructose-induced metabolic syndrome. Am J Physiol Renal Physiol 290, F625–631. [DOI] [PubMed] [Google Scholar]
- National Research Council (U.S.). Committee for the Update of the Guide for the Care and Use of Laboratory Animals., Institute for Laboratory Animal Research (U.S.), and National Academies Press (U.S.) (2011). Guide for the care and use of laboratory animals. (Washington, D.C.: National Academies Press,), pp. xxv, 220 p. [Google Scholar]
- Ng SW, Slining MM, and Popkin BM (2012). Use of caloric and noncaloric sweeteners in US consumer packaged foods, 2005–2009. J Acad Nutr Diet 112, 1828–1834 e1821–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh AR, Sohn S, Lee J, Park JM, Nam KT, Hahm KB, Kim YB, Lee HJ, and Cha JY (2018). ChREBP deficiency leads to diarrhea-predominant irritable bowel syndrome. Metabolism 85, 286–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel C, Douard V, Yu S, Gao N, and Ferraris RP (2015a). Transport, metabolism, and endosomal trafficking-dependent regulation of intestinal fructose absorption. FASEB J 29, 4046–4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel C, Douard V, Yu S, Tharabenjasin P, Gao N, and Ferraris RP (2015b). Fructose-induced increases in expression of intestinal fructolytic and gluconeogenic genes are regulated by GLUT5 and KHK. Am J Physiol Regul Integr Comp Physiol 309, R499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Noworolski SM, Erkin-Cakmak A, Korn NJ, Wen MJ, Tai VW, Jones GM, Palii SP, Velasco-Alin M, Pan K, et al. (2017). Effects of Dietary Fructose Restriction on Liver Fat, De Novo Lipogenesis, and Insulin Kinetics in Children With Obesity. Gastroenterology 153, 743–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwimmer JB, Ugalde-Nicalo P, Welsh JA, Angeles JE, Cordero M, Harlow KE, Alazraki A, Durelle J, Knight-Scott J, Newton KP, et al. (2019). Effect of a Low Free Sugar Diet vs Usual Diet on Nonalcoholic Fatty Liver Disease in Adolescent Boys: A Randomized Clinical Trial. JAMA 321, 256–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sclafani A, and Ackroff K (2012). Role of gut nutrient sensing in stimulating appetite and conditioning food preferences. Am J Physiol Regul Integr Comp Physiol 302, R1119–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soberg S, Sandholt CH, Jespersen NZ, Toft U, Madsen AL, von Holstein-Rathlou S, Grevengoed TJ, Christensen KB, Bredie WLP, Potthoff MJ, et al. (2017). FGF21 Is a Sugar-Induced Hormone Associated with Sweet Intake and Preference in Humans. Cell Metab 25, 1045–1053 e1046. [DOI] [PubMed] [Google Scholar]
- Softic S, Gupta MK, Wang GX, Fujisaka S, O’Neill BT, Rao TN, Willoughby J, Harbison C, Fitzgerald K, Ilkayeva O, et al. (2017). Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling. J Clin Invest 127, 4059–4074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Softic S, Gupta MK, Wang GX, Fujisaka S, O’Neill BT, Rao TN, Willoughby J, Harbison C, Fitzgerald K, Ilkayeva O, et al. (2018). Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling. J Clin Invest 128, 1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Softic S, Meyer JG, Wang GX, Gupta MK, Batista TM, Lauritzen H, Fujisaka S, Serra D, Herrero L, Willoughby J, et al. (2019). Dietary Sugars Alter Hepatic Fatty Acid Oxidation via Transcriptional and Post-translational Modifications of Mitochondrial Proteins. Cell Metab 30, 735–753 e734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanhope KL, Bremer AA, Medici V, Nakajima K, Ito Y, Nakano T, Chen G, Fong TH, Lee V, Menorca RI, et al. (2011). Consumption of fructose and high fructose corn syrup increase postprandial triglycerides, LDL-cholesterol, and apolipoprotein-B in young men and women. J Clin Endocrinol Metab 96, E1596–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanhope KL, and Havel PJ (2008). Endocrine and metabolic effects of consuming beverages sweetened with fructose, glucose, sucrose, or high-fructose corn syrup. Am J Clin Nutr 88, 1733S–1737S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanhope KL, and Havel PJ (2009). Fructose consumption: considerations for future research on its effects on adipose distribution, lipid metabolism, and insulin sensitivity in humans. J Nutr 139, 1236S–1241S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanhope KL, Schwarz JM, Keim NL, Griffen SC, Bremer AA, Graham JL, Hatcher B, Cox CL, Dyachenko A, Zhang W, et al. (2009). Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J Clin Invest 119, 1322–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan BC, Wells JC, Brayne C, Albanese E, and Siervo M (2010). Increased fructose intake as a risk factor for dementia. J Gerontol A Biol Sci Med Sci 65, 809–814. [DOI] [PubMed] [Google Scholar]
- Taubes G (2007). Good calories, Bad Calories. (New York: Alfred A Knopf; ). [Google Scholar]
- Uyeda K, and Repa JJ (2006). Carbohydrate response element binding protein, ChREBP, a transcription factor coupling hepatic glucose utilization and lipid synthesis. Cell Metab 4, 107–110. [DOI] [PubMed] [Google Scholar]
- van den Berghe G, Bronfman M, Vanneste R, and Hers HG (1977). The mechanism of adenosine triphosphate depletion in the liver after a load of fructose. A kinetic study of liver adenylate deaminase. Biochem J 162, 601–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Holstein-Rathlou S, BonDurant LD, Peltekian L, Naber MC, Yin TC, Claflin KE, Urizar AI, Madsen AN, Ratner C, Holst B, et al. (2016). FGF21 Mediates Endocrine Control of Simple Sugar Intake and Sweet Taste Preference by the Liver. Cell Metab 23, 335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos MB, Kimmons JE, Gillespie C, Welsh J, and Blanck HM (2008). Dietary fructose consumption among US children and adults: the Third National Health and Nutrition Examination Survey. Medscape J Med 10, 160. [PMC free article] [PubMed] [Google Scholar]
- Weiss R, Bremer AA, and Lustig RH (2013). What is metabolic syndrome, and why are children getting it? Ann N Y Acad Sci 1281, 123–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JS (2008). Straight talk about high-fructose corn syrup: what it is and what it ain’t. Am J Clin Nutr 88, 1716S–1721S. [DOI] [PubMed] [Google Scholar]
- Yang Q, Zhang Z, Gregg EW, Flanders WD, Merritt R, and Hu FB (2014). Added Sugar Intake and Cardiovascular Diseases Mortality Among US Adults. JAMA Intern Med. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate unique datasets or code.