

Graphical abstract

Abbreviations: NSAID, Non-steroidal anti-inflammatory drug; PGHS, Prostaglandin-endoperoxide synthase; PG, Prostaglandin; FDA, US Food and Drug Administration; IC, Inhibitory concentration; P450, Cytochromes P450; PLA, Phospholipase; Tx, Thromboxane; LTE, Leukotriene; MOS, Mitochondrial oxidative stress; ETC, Electron transport chain; O2.-, Superoxide; CVD, Cardiovascular disease; TNF-α, Tumor necrosis factor-alpha; NF-κB, Nuclear factor kappa-light-chain-enhancer of activated B cells; ICAM-1, Intercellular adhesion molecule 1; MAPK, Mitogen activated protein kinase; PKC, Protein kinase C; DRP1, Dynamin-related protein 1; LPS, Lipopolysaccharide; HMGB1, High mobility group box 1; DAMP, Damage associated molecular pattern; MEDAL, Multinational Etoricoxib and Diclofenac Arthritis Long-term trial; GI, Gastrointestinal; MI, Myocardial infarction; HF, Heart failure; VIGOR, Vioxx Gastrointestinal Outcomes Research; APPROVe, Adenomatous Polyp PRevention On Vioxx trial; PRECISION, Prospective Evaluation of Celecoxib Integrated Safety versus Ibuprofen or Naproxen; NANSAIDs, Non aspirin NSAIDs; ADMA, Asymmetric dimethyl arginine; eNOS, Endothelial nitric oxide synthase; CKD, Chronic kidney disease; AKI, Acute kidney injury; GFR, Glomerular filtration rate; PPAR, Peroxisome proliferator-activated receptor; CRESCENT, Celecoxib Rofecoxib Efficiency and Safety in Cormorbidities Evaluation Trial; ICH, Intracerebral haemorrhage; AERD, Aspirin-exacerbated respiratory disease; CAP, Community Acquired Pneumonia; Cys-LTEs, Cysteinyl leukotrienes
Keywords: NSAID, Cyclooxygenase, Prostaglandin, Inflammation, Mitochondria, Organ damage, Apoptosis, Gastropathy
Abstract
Owing to the efficacy in reducing pain and inflammation, non-steroidal anti-inflammatory drugs (NSAIDs) are amongst the most popularly used medicines confirming their position in the WHO’s Model List of Essential Medicines. With escalating musculoskeletal complications, as evident from 2016 Global Burden of Disease data, NSAID usage is evidently unavoidable. Apart from analgesic, anti-inflammatory and antipyretic efficacies, NSAIDs are further documented to offer protection against diverse critical disorders including cancer and heart attacks. However, data from multiple placebo-controlled trials and meta-analyses studies alarmingly signify the adverse effects of NSAIDs in gastrointestinal, cardiovascular, hepatic, renal, cerebral and pulmonary complications. Although extensive research has elucidated the mechanisms underlying the clinical hazards of NSAIDs, no review has extensively collated the outcomes on various multiorgan toxicities of these drugs together. In this regard, the present review provides a comprehensive insight of the existing knowledge and recent developments on NSAID-induced organ damage. It precisely encompasses the current understanding of structure, classification and mode of action of NSAIDs while reiterating on the emerging instances of NSAID drug repurposing along with pharmacophore modification aimed at safer usage of NSAIDs where toxic effects are tamed without compromising the clinical benefits. The review does not intend to vilify these ‘wonder drugs’; rather provides a careful understanding of their side-effects which would be beneficial in evaluating the risk–benefit threshold while rationally using NSAIDs at safer dose and duration.
1. Introduction
Since the isolation of salicyclate from willow bark in around 1830s, followed by the discovery of aspirin (acetyl salicyclate) by Felix Hoffman of Bayer industry, Germany, in 1897, non steroidal anti-inflammatory drugs (NSAIDs) have been enjoying a blockbuster status in the pharmaceutical industry [1]. In around 500 BCE, even before conceptualization of clinical trials and scientific knowledge, Hippocrates, wrote about the potential of willow bark and leaves in curing pain and fever. Thereafter, generations of scientists, chemists and science enthusiasts have extensively worked and ushered the development of these ‘wonder drugs’. At present, NSAIDs are among the most popular over-the-counter drugs across the world, constituting 5% of all the prescribed medicines [2]. Traditionally NSAIDs were classified on the basis of their chemical characteristics wherein most of the popular NSAIDs are categorized as major derivatives of salicylic acid, acetic acid, enolic acid, anthranilic acid or propionic acid. However, with the advancement of scientific knowledge the classification has also been shifted based on their selectivity for inhibiting cyclooxygenase/prostaglandin-endoperoxide synthase (PGHS) enzymes which are the major targets of these drugs. In addition, a classification system has also been formulated to categorize NSAIDs on the basis of their half-life. Nevertheless, despite the interclass diversity, their functions are relatively similar. NSAIDs are mostly used for the treatment of patients suffering from pain and inflammatory conditions such as chronic pain, osteoarthritis, rheumatoid arthritis, postoperative surgical conditions, menstrual cramps and even used extensively as analgesics and anti-pyretics [3], [4]. Moreover, the superiority of typical NSAIDs (with potent anti-inflammatory action) over acetaminophen (atypical NSAID devoid of any anti-inflammatory action, but with weak, non-specific cyclooxygenase/PGHS-inhibitory and antipyretic action) [5], [6] and other antipyretic analgesics like opioids has already been established. Blinded, randomized control trials have demonstrated the efficacy of NSAIDs over acetaminophen [7]. Several opioid-addiction problems actually initiate with the therapeutic use of opioid-based painkillers. Therefore, opiods are far more dangerous than NSAIDs owing to the non-addictive nature of the latter drugs [8]. Regarding other effects, a recent study on amyloid beta-induced experimental Alzheimer’s disease (AD) in mice has shown that mefenamic acid, a fenamate NSAID used to relieve period pain, could offer protection by suppressing neuroinflammation and memory loss [9]. However, incidents of cognitive impairment in aspirin users and risk of dementia in elderly people also warn about the safety concerns and plausible neurotoxic effects of NSAIDs in AD patients [10], [11]. Notably, the protective role of NSAIDs has been significantly implicated in various cancers [12]. In addition, there are some speculations regarding the use of NSAIDs against coronavirus [13]. In spite of this extensive therapeutic utility, the NSAIDs are infamous for multiple severe side effects including gastrointestinal toxicities, cardiovascular risks, renal injuries, and hepatotoxicity as well as hypertension and other minor disorders [14], [15], [16], [17]. The general mode of action of NSAIDs involves the inhibition of cyclooxygenase/prostaglandin-endoperoxide synthase (PGHS-1 and PGHS-2), regulatory enzymes, involved in the biosynthesis of prostaglandin (PG) which is strongly implicated in inflammation. PGHS-1 is believed to be a housekeeping gene engaged in multiple biological functions including protection of gastric mucosa while PGHS-2 is responsible for inflammation. Some NSAIDs are non-specific inhibitors of both the enzymes while others are specific, notably “coxibs” that specifically inhibit PGHS-2. Previously it was believed that PGHS-1 inhibitors, by inhibiting PG synthesis in gastric mucosa, can indulge in gastrointestinal complications. However, contrary to this, a substantial amount of evidence has emerged in recent years [18], [19], [20], [21]. At one point of time, a shift to coxibs was thought to be effective. Since PGHS-2 is inducible and is not directly related to gastric mucosal defence, rather engaged in inflammation, inhibition of PGHS-2 by coxibs were supposed to be rather safe and devoid of gastrointestinal and other complications [22]. But emerging evidences have challenged this theory too. In line with that, in 2000–03 several reports began to emerge regarding the cardiovascular adverse effects of PGHS-2 inhibitors and subsequent placebo controlled trials also showed that those inhibitors were linked to increased risk of atherothrombotic vascular events [15], [16], [23]. Moreover, meta-analyses and randomized trials have further confirmed these findings which led to the withdrawal of several PGHS-2 inhibitors by US Food and Drug Administration (FDA) [15]. Concurrently with the evolution and establishment of PGHS/PG-dependent mode of NSAID toxicity, a second school of opinions advocating the direct effect of NSAIDs on mitochondria followed by generation of cellular oxidative stress and apoptosis has also strengthened its foothold as an independent pathway of NSAID-induced cytopathology [24]. Other than gastrointestinal and cardiovascular complications, habitual use of NSAIDs are also associated with nephrotoxicity and eventual renal failure [25] together with other transient effects on water and electrolyte balance. In addition, idiosyncratic drug toxicity by NSAIDs has also been significantly attributed to multiple incidences of hepatotoxicity [26]. However, the exact mechanism involved in inflicting these adverse effects by NSAIDs is still elusive. All these data together eventually challenge the long-standing reputation of these ‘wonder drugs’ in relation to their rampant medicinal use. Since, in the current context, NSAID use has become almost unavoidable, a relatively safe usage of these drugs is the need of time which can avert the risk of organ failure. To this end, precise understanding of the molecular mechanism and signaling pathways involved in NSAID therapy is quintessential. In addition, chemical modification of FDA approved NSAIDs, by pharmacophore modification, aimed at reducing their toxic effects and improving their bioavailability without compromising the therapeutic effects is strongly advocated. Extensive studies have been already undertaken to analyse the effects of NSAIDs at the molecular level besides designing new combinatorial formulations in the form of NSAID-prodrugs with enhanced efficacy. Although there are discrete individual reports of organ-specific impact of these drugs where both PGHS/PG-dependent as well as independent effects of NSAIDs have been implicated in their cytotoxic actions, till date no comprehensive document is available which encompasses and accounts the outcome of different randomized trials, meta-analyses, systemic reviews along with the mode of action of NSAIDs and its effects on multiple organs in a coherent manner. In this article, we have tried to provide an overall idea about the structure and function of NSAIDs with special emphasis on organ damage as well as indicated the possible strategies of using these drugs in a safer approach by discussing the previous contributions concurrent with recent understanding of the field.
2. Chemistry and classification of NSAIDs
NSAIDs encompass a large class of drugs with extreme structural and functional diversity. These are mostly weak organic acids (comprising of an acidic moiety along with an aromatic functional group). In terms of functional diversity, isoform-specific selectivity for PGHS inhibition forms the basis of differentiation; while a third classification takes into account the bioavailability of the NSAIDs in the serum, accounting for the pharmacokinetic aspects of NSAID action in the systemic context. In the following section, a brief description about the classification of NSAIDs is presented.
2.1. Classification of NSAIDs based on structure
Based on their chemical structure, NSAIDs can be broadly classified into salicylates, aryl and heteroarylacetic acid derivatives, indole/indene acetic acid derivatives, anthranilates and oxicams (enol acids) (Fig. 1 ) [27]. The general structure of a typical NSAID consists of an acidic moiety (carboxylic acid, enols) attached to a planar aromatic functional group. Salicylates were the first identified NSAIDs following extraction of salicylic acid from willow bark [1]. They are actually the derivatives of 2-hydroxybenzoic acid (salicylic acid) [28]. Initially, salicylic acid was used medicinally in the form of sodium salt; later, this compound got replaced therapeutically by the acetylated derivative, acetylsalicylic acid (ASA) or aspirin. Therapeutic utility was enhanced by the esterification of the phenolic hydroxyl group, as in aspirin, or substitution of a hydrophobic/lipophilic group at C-5 as in diflunisal [28]. After salicylates, aryl or heteroaryl acetic acid derivatives constitute an important class of NSAIDs. Ibuprofen, fenoprofen, naproxen and oxaprozin are some structural derivatives of aryl or heteroaryl acetic acid which comprise some of the most popular NSAIDs. The next category of NSAIDs is indole or indene acetic acid which includes popular pain killers including indomethacin and sulindac. Moving further, anthranilates are another class of NSAIDs which are N-aryl substituted derivatives of anthranilic acid. Diclofenac, the derivative of 2-arylacetic acid, is the most widely used anthranilate NSAID found in diverse formulations including pain killer tablets, injections, topical ointments and fast acting sprays. Mefenamic acid and meclofenamic acid are also derived from anthranilic acid. Finally, oxicams, mainly consisting of piroxicam and meloxicam comprise the last structural category of NSAIDs, characterized by the presence of 4-hydroxybenzothiazine heterocycle [28]. In addition to the structural classification, NSAIDs have been also categorized based on their PGHS-specific inhibitory activity as well as half life in serum.
Fig. 1.

Classification of non steroidal anti-inflammatory drugs (NSAIDs) based on structure.
2.2. Classification of NSAIDs based on the type of PGHS interaction and selectivity
Bioconversion of arachidonic acid (AA) into inflammatory prostanoids (prostaglandins and prostacyclin), is mediated by PGHS enzymes; mainly PGHS-1 and PGHS-2 which are inhibited by NSAIDs. The potency of NSAIDs in inhibiting PGHS-1 and PGHS-2 are often compared on the basis of their IC50 values (the concentration of NSAIDs that inhibits 50% activity of the enzyme) in the in vitro assay system. Of all the assay systems that have been documented, human whole blood assay is the most widely accepted [29], [30]. Here, it is worthwhile to mention that almost all the NSAIDs variably inhibit both the PGHS isoforms at their therapeutic doses. Thus, on the basis of PGHS selectivity, an inhibitory ratio is determined which allows a classification of NSAIDs. The inhibitory ratio is based on the PGHS-1 IC50/ PGHS-2 IC50 [29], [31], [32]. If the ratio is 1, then both the PGHS enzymes are equally inhibited by the concerned NSAID, if the ratio is less than 1, it means that the concerned NSAID is less selective for PGHS-2 compared to PGHS-1 and in case of ratio greater than 1, the NSAID is preferentially selective towards PGHS-2 [29], [30], [31], [32]. It is presumed that side effects of NSAIDs (such as GI toxicity) are associated with PGHS-1 inhibition while therapeutic effect (anti-inflammatory) is correlated with that of PGHS-2 and often a high level of PG suppression is needed for therapeutic relevance; however this simplistic view has been questioned recently [29]. In general, NSAIDs are therapeutically employed at doses that generate more than 50% reduction of PG production. In this context, it would be important to check the extent to which PGHS-1 gets inhibited at the same concentration of NSAID that is required for inhibiting 80% of PGHS-2 activity. However, in case of diclofenac, the concentration which inhibits 80% of PGHS-2 activity can also inhibit almost 70% of PGHS-1 activity at the same time. So, therapeutic dose (80% inhibition of PGHS-2) can even lead to toxicity (70% inhibition of PGHS-1). Hence, in this scenario, when relative selectivity varies within a narrow range, other variables including consumed dose and plasma half-life should be considered. For example, piroxicam which has long plasma half life and correlated with GI toxicity in vivo, did not show notable PGHS-1 selectivity in the in vitro assay [29]. So, it is clear that the relative potency of NSAIDs vary with their dose, concentration, plasma half life. Therefore, IC80 value seems to be clinically more relevant in comparing NSAIDs’ inhibitory potencies against PGHS-1 and PGHS-2. Now, on the basis of the potencies to inhibit PGHS isoforms, NSAIDs can be divided into four main categories (Table 1 ): (i) non-selective, complete inhibitors of both PGHS-1 and PGHS-2 (ii) complete inhibitors of PGHS-1 and PGHS-2, although with specific preference for PGHS-2 (iii) strong inhibitors of PGHS-2, although with weak inhibiting action against PGHS-1 (iv) weak inhibitors of both PGHS-1 and PGHS-2 [29]. However, in terms of kinetics, NSAID interactions with both the PGHS isoforms can be also used for their classification which is as follows: freely reversible interaction (ibuprofen), slowly reversible interaction (indomethacin, diclofenac, celecoxib) and irreversible interaction (aspirin) [32].
Table 1.
Categorization of NSAIDs based on PGHS-selective inhibitory action.
| Categories | PGHS isoform selectivity | Representative NSAIDs |
|---|---|---|
| Category 1 | PGHS-1 and PGHS-2 | Indomethacin, Aspirin, Diclofenac, Naproxen, Ibuprofen |
| Category 2 | 5-50 fold selectivity for PGHS-2 | Meloxicam, Celecoxib, Nimesulide, Etodolac |
| Category 3 | Greater than 50 fold selectivity for PGHS-2 | NS-398 |
| Category 4 | Poor selectivity for PGHS-1 and PGHS-2 | Sulfasalazine, Sodium salicyclate, Nabumetone, |
2.3. Classification of NSAIDs on the basis of plasma half-life (t½)
NSAIDs can also be divided into short-acting (plasma half-life less than 6 h) such as aspirin, diclofenac and ibuprofen and long-acting (half-life approximately greater than 10 h) such as naproxen, celecoxib. Those with short half-life, like ibuprofen, exhibit quick onset of action and thus suitable for acute pain. While NSAID like naproxen has a longer half-life and effective for treating chronic conditions [33].
3. Pharmacology of NSAIDs
More than 90% of the NSAIDS are highly bound to plasma proteins. These drugs normally exhibit considerably well bioavailability in monogastric species upon oral, subcutaneous and intramuscular administration due to their moderate to high lipid solubility (an aspect that also allows their penetration of blood–brain barrier) [34]. Direct urinary excretion of the parent drug is low due to high binding with plasma proteins; hepatic metabolism clears out the NSAIDs from the body as inactive metabolites that are excreted in urine and bile. Drug elimination by clearance and terminal half life is highly species specific. The microsomal enzymes cytochrome P450 (CYP)-containing-mixed-function-oxidase system is responsible for most of the NSAIDs metabolism wherein CYP2C9 is the most important oxidase primarily responsible for metabolism of a wide range of NSAIDs including celecoxib, indomethacin, diclofenac, flurbiprofen, ibuprofen, naproxen and enolic acids like piroxicam and tenoxicam. Notably, allelic variations in this protein (including CYP2C9*2 and CYP2C9*3) affect the pharmacotherapeutic efficacy of the medicine in patient-specific manner [35]. Hence, pharmacogenomic variability of P450 in a population also correlates well with varying degree of metabolism of the drugs among individuals or ethnic groups [36]. It has been observed that CYP2C9 predominantly contributes to ibuprofen clearance through formation of 3-hydroxyibuprofen and 2-hydroxyibuprofen [37]. Apart from ibuprofen, the action of CYP2C9 on other target NSAIDs has been reported wherein diclofenac is metabolized to 4′-hydroxydiclofenac, flurbiprofen is metabolized to 4′-hydroxiflurbiprofen and naproxen is metabolized to 6-desmethylnaproxen [35]. Other than CYP2C9, NSAIDs are also metabolized by other cytosolic hepatic enzymes of phase I metabolism including CYP3A4, CYP2C19, CYP2C8 through oxidative transformations. This is followed by phase II conjugation reactions involving glucuronidation by uridine 5′-diphosphoglucuronosyltransferases (including UGT1A1, UGT2B7, UGT1A9 and UGT2B4) and ultimately biliary excretion of the resultant glucuronides and reactive metabolites by ABCC2 efflux transporters expressed on the apical canalicular membranes of hepatocytes and renal tubular epithelial cells [38], [39], [40]. In the aforesaid context, diclofenac metabolism by UGT2B7, ABCC2 and CYP2C8 has been also linked with hepatotoxicity due to allelic variations in these enzymes notably, UGT2B7 allele [38], [41]. In regards to ibuprofen metabolism, at high concentrations, CYP3A4 catalyzes 2-hydroxylation for systemic clearance while 3-OH-ibuprofen (formed by CYP2C9) gets further transformed to carboxy-ibuprofen by cytosolic dehydrogenases [37]. Thus, NSAIDs may undergo either CYP-mediated phase I followed by UGT-mediated phase II metabolism or direct UGT-mediated glucuronidation (phase II metabolism) [5] before ultimate renal excretion mediated by efflux transporters. Around 10–15% of absorbed ibuprofen is glucuronidated directly to ibuprofen-acyl glucuronide by multiple UGTs including UGT1A9, UGT2B17, UGT1A3 and UGT2B7 [42]. Although binding to plasma proteins hinder the ready passage of NSAIDs to the interstitial and transcellular fluids from the plasma, the same aspect facilitates their penetrance and persistence in the inflammatory exudates, thereby allowing for anti-inflammatory actions. Inherent differences in the pharmacokinetic properties of NSAIDs with chiral centers in their structure (including arylpropionates and etodolac) further demand specific attention before prescribing a suitable dosage for administration. This is due to the structural and hence pharmacokinetic differences between the enantiomers of same NSAIDs along with their potential conversion from R(-) to S(+) form or vice versa, in vivo, that ultimately dictate their specificities in terms of systemic clearance, elimination half lives and distribution volumes of a particular enantiomer in the circulation [34]. PGHS inhibition predominantly accounts for the pharmacodynamic and toxicodynamic actions of NSAIDs [34]; although mitochondrial toxicity and adverse drug-drug interactions also significantly contribute to NSAID actions on the system. While PGHS-1 inhibition primarily contributes to the toxic side effects of these drugs, PGHS-2 inhibition underlies most of the therapeutic effects [34]; although instances of PGHS-2 inhibition associated cardiotoxicity strongly warrants the risk of coxib usage.
4. Therapeutic uses and repurposing of NSAIDs
With the discovery of aspirin’s mechanism of action by John Vane (Noble Prize 1982), understanding of NSAIDs increased over the years, thereby escalating the abilities to develop novel anti-inflammatory therapeutics. Owing to the analgesic, anti-pyretic, anti-inflammatory activities these drugs are extensively used in treating pain, fever and inflammation in rheumatic disorders, osteoarthritis and dysmenorrhoea [36], [43], [44], [45]. They are also implicated in sports medicine and popular among sports persons and soldiers [46], [47], [48], [49]. Owing to the increasing demand for repurposing of already FDA-approved cheaper drugs in treating new diseases (in addition to the canonical purposes), and considering the multiple effects of NSAIDs, these drugs are now also aimed for repurposing against other serious health implications. Use of aspirin as a cardioprotective agent for treating atherosclerosis can be considered as the oldest and most classical example of using an anti-inflammatory drug for cardiovascular disease. Exploiting the same rationale of NSAID-induced PGHS inhibition and PG depletion, these drugs are gaining immense importance as new generation anti-cancer agents [50]. Other than PGHS-2 inhibition, induction of tumor cell apoptosis, downregulation of epidermal growth factor receptor, attenuation of neoangiogenesis and protection from DNA damage are some of the strategies by which coxib and non-coxib NSAIDs like aspirin offer chemo preventive as well as therapeutic effect against various cancers [51]. Evidences of reduced risk for developing cancers (by long term NSAID-users) as well as regression of tumor mass upon treatment with NSAIDs (used in combination chemotherapy regimen) clearly indicate the preventive as well as therapeutic potencies [52], [53]. Because, inflammatory microenvironment is highly conducive for survival and proliferation of malignant cells along with evasion of anti-tumor immune response and resistance to chemotherapeutic drugs, NSAIDs strike at the base of these evasive strategies by depleting the inflammatory milieu thereby culling the cancer cells [54]. Several studies have convincingly proved the efficacy of NSAIDs in direct chemotherapy, combination chemotherapy and even neoadjuvant chemotherapy owing to their multiple anticancer actions which include, but not essentially limited to, blocking cell proliferation of diverse cancer cell types, preventing tumor angiogenesis and metastasis, reducing chemo and radio resistance and of course inducing apoptosis [55]. Platelet aggregation and adherence to tumor cells aid in immune evasion of malignant cells thereby facilitating metastasis. NSAIDs significantly interfere with these processes. As a novel strategy of potential anti-cancer therapy, sulindac, aspirin, naproxen and other NSAIDs have been used to target cancer-associated epigenetic processes and microRNA (miRNA) expression [56], [57]. A system-biological approach was also used to construct an NSAID-effect model based on the PGHS-pathway to identify miRNA biomarkers relevant to tumors. The outcome of this study could be important in development of personalized medicine [58]. In regards to the PGHS-2 and its impact on cancer, coxibs have been found to prevent chemo resistance in breast cancer cells through blocking PGHS-2-dependent-PGE2 production and inflammation-associated carcinogenesis [59], [60]. Notably, celecoxib has been found to target breast cancer stem cells, prevent carcinogenic epithelial to mesenchymal transition and block metastasis via PGE2 depletion and Wnt signaling downregulation [61]. Although PGHS-2 inhibition seems a potential strategy for cancer prevention by coxibs, serious concerns regarding the long term cardiovascular safety of coxibs used in Adenomatous Polyp Prevention on Vioxx (APPROVe, rofecoxib) and Adenoma Prevention with Celecoxib (APC, celecoxib) trials (which aimed at testing the preventive effect of coxibs on recurrence of colorectal polyps) got highlighted. A 2–3 fold elevation in cardiovascular risk among patients taking 25 mg of rofecoxib in the APPROVe trial led to premature termination of the study along with subsequent withdrawal of Vioxx from the market [62]. The cardiovascular risk of selective PGHS-2 inhibitors therefore overshadowed the anti-cancer potency of some of these drugs; although PGHS-2 still remained as a potential anti-neoplastic target. In this regard, compared to coxibs, aspirin has proved to be a rather safe NSAID with significant efficacy against diverse tumors. Aspirin-induced PGHS-1 inhibition and associated TxA2 depletion blocks platelet aggregation and adherence to tumor cells, thereby increasing chemo sensitivity of tumors. Aspirin, which has shown promising preclinical results in preventing cancer is now being investigated in an Add-Aspirin phase III, double-blind, randomized trial with four non-metastatic solid-tumour cohorts (breast, colorectal, gastro-oesophageal and prostrate) to check the recurrence and survival of the patients with those tumours after standard aspirin use [63]. The interpretation of the preplanned analysis of feasibility of Add-Aspirin Trial indicates well-tolerability of aspirin after radical radiotherapy [64]. It is noteworthy to mention that despite its anti-cancer effect the risk of serious bleeding due to aspirin persists. This has been well addressed in the Add-Aspirin study where several special strategies were taken to reduce bleeding with aspirin, such as exclusion of subjects susceptible to bleeding, randomization of volunteers with age above 75 years to 100 mg aspirin or placebo due to their dose and age dependent vulnerability to bleeding and also use of proton pump inhibitors (PPI) for participants post partial gastrectomy or oesophagectomy [64]. Another randomized phase III placebo-controlled trial is also undergoing to check the potential of aspirin (300 mg daily) in preventing the recurrence of cancer in human epidermal growth factor receptor 2 (HER2) negative breast cancer patients post chemotherapy, surgery and/or radiation therapy (ClinicalTrials.gov Identifier: NCT02927249). In addition, a 10-year population cohort study (aspirin user is age-sex matched to non-user with a ratio of 1:2) in Honk Kong depicted significant reduction of different cancers (liver, stomach, colorectum, lung, oesophagus, pancreas, leukaemia), except breast cancer, in long-term aspirin users compared with those who have not been prescribed aspirin [65]. Other systemic reviews and cohort studies are also in agreement with this observation [66], [67]. Apart from cancer, NSAIDs have been also repurposed for other clinical complications which include diflunisal-induced osteoprotection against staphylococcal osteomyelitis [68], mefenamic acid-mediated protection against schistosomiasis [69], aspirin and ibuprofen-mediated cryptococcal cell death [70], oxyphenbutazone-induced sensitization of Mycobacterium tuberculosis to host-derived factors and exogenous antimycobacterial compounds [71] and piroxicam-induced dipeptidyl peptidase-4 inhibition as an alternative strategy for regulating glucose metabolism in diabetes mellitus [72]. While observations from drug repurposing studies in pre-clinical and research settings are highly encouraging, further exploration and extensive validations are mandatory before repurposing of NSAIDs in clinical settings. Recently, induction of PGHS-2 has been also linked with seizures and PGHS-2 inhibitors have been proposed as potential therapeutic option, targeting PGHS-2 mediated neuroinflammation during epilepsy [73]. In this regard, mefenamic acid has been linked to neuroprotection and prevention of cognitive impairment in mice by preventing amyloid beta-induced NLRP3/IL-1β-dependent inflammosome activation, neuroinflammation and memory loss suggesting its putative effect against AD [9], [74], [75]. In contrast to aforesaid, instances of NSAID-associated cognitive problems and risk of dementia in elderly people raise multiple concerns about the safety profiles of NSAIDs for using against AD [10], [11], [76], [77]. The complex associations (both positive and negative) of AD with NSAID-use therefore demands precise randomized clinical trials taking into account the specific NSAIDs used by patients, duration, dose, past history of cognitive defects and other relevant confounders in order to define safety profiles of NSAIDs in AD. Despite these complex and contradictory effects on cognitivefunctions, NSAIDs have been positively implicated in post-surgical complications and in treating burn patients [78], [79]. Furthermore, in the COVID-19 background, owing to a previous report of indomethacin in preventing RNA synthesis of coronavirus, a lot of speculations are flying around the therapeutic use of NSAIDs against COVID-19 [80]. A schematic representation of the diverse canonical and emerging applications of NSAIDs has been presented (Fig. 2 )
Fig. 2.
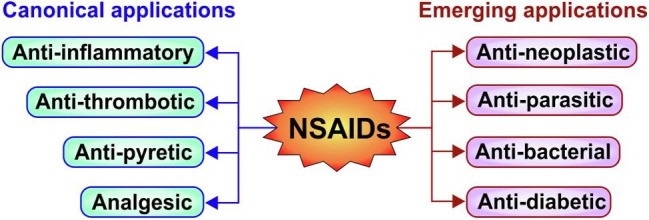
Classical applications and emerging uses of NSAIDs.
Since, NSAIDs are unfortunately associated with number of serious complications making different organs vulnerable to damage, a thorough understanding about their diverse subcellular effects and mode of action are extremely essential.
5. Mode of action of NSAIDs
There are several schools of opinions which tend to categorize the NSAID actions based on major subcellular targets. PGHS dependent and independent pathways of action are the two most widely accepted mechanisms by which NSAIDs are reported to act. While the first mode relies on the action of NSAIDs on the production and abundance of prostanoids (the major inflammatory mediators of the system) to regulate tissue inflammation, the second mode of action is dependent on the toxic action of NSAIDs against the cells specially the subcellular bioenergetic work horses, mitochondria. Before delving deeper into the organ-specific action of these drugs, it is essential to provide a comprehensive idea about the two basic mechanisms behind NSAIDs-related organ damage.
5.1. NSAID-PGHS-prostanoid axis
Although human beings have been consuming NSAIDs in different forms for more than 3,500 years, it was not until John Vane in 1971, when cyclooxygenase 1 enzyme (PGHS-1) was identified as the molecular target of NSAIDs which proved instrumental in elucidating the molecular mechanism of NSAID action [81], [82]. Vane’s pioneering work also revealed that these chemically varied drugs, by inhibiting PGHS-1, actually reduced PG formation [81], [83], [84], [85]. His elucidation was followed twenty years later by the identification of a second cyclooxygenase enzyme named PGHS-2 which is also associated with prostaglandin synthesis like PGHS-1 [86], [87]. Other than PGHS-1 and 2, Chandrasekharan et.al in 2002 described a third distinct cyclooxygenase isozyme, cyclooxygenase-3, in brain-tissue of dogs, which is a splice variant of PGHS-1 and showed some sensitivity to paracetamol [88]. Although, this sensitivity has not been properly justified in humans [89]. However, in this review the main focus would remain on PGHS-1 and PGHS-2, the two main pharmacological targets of NSAIDs. Both these isoforms reside predominantly in the endoplasmic reticulum and are identical in length with just over 600 amino acids (having 63% homology). The molecular weights of the enzymes are approximately 71 kDa and they are bisfunctional in catalytic activity having both cyclooxygenase and peroxidase functions [90], [91]. The active site of both the isoforms are at the end of a hydrophobic channel. NSAIDs prevent the access of the arachidonic acid (AA), the substrate of PGHS, to this channel. PGHS-1 and PGHS-2 channels differ with non-selective NSAIDs having access to both the channels; which is not the case for PGHS-2 [19]. The principal difference between the isoenzyme is the substitution of isoleucine 523 in PGHS-1 with valine in PGHS-2 [92]. As mentioned earlier, these two isoforms are responsible for catalyzing the rate-limiting step of prostanoid biosynthesis and are negatively regulated by NSAIDs (Fig. 3 ). Prostanoids are actually the eicosanoids. The major eicosanoids consist of PGs, thromboxanes (Txs), leukotrienes (LTEs) and lipoxins (LXs). Of these PGs and Txs are collectively called prostanoids [93]. PGHS-1 is constitutively expressed in most cells, and produces prostanoids for housekeeping purposes such as gastric mucosal protection, regulation of acid secretion, and homeostasis as well as maintaining renal functions. On the other hand, PGHS-2, induced by inflammatory cytokines, mitogens, endotoxins and tumor-promoters, produces prostanoids during inflammation and cancer [94]. PGHS-2 has been mostly linked with immune cells including leukocytes and macrophages as well as fibroblasts, chondrocytes, endothelial and mesangial cells and notably the ensuing inflammatory sites in the body [95], [96]. However accumulated evidence now clearly suggests that PGHS-2 is also constitutively expressed in a number of tissues including brain, gut, lung, thymus and kidney wherein it produces prostanoids [97]. Chemically, prostanoids are bioactive lipid molecules which mediate autocrine and paracrine action via high-affinity G protein coupled receptors [81]. They are derived from 20 carbon ω-6 fatty acid, AA, by the action of PGHS on AA [98]. Since AA is highly reactive and sensitive to oxidation, it is not found freely in cells; rather stored in membrane phospholipids mainly as phosphatidylcholine, phosphatidylethanolamine and phosphatidylinositol. The phospholipases (PLA2) liberate them from the membrane phospholipids. Subsequently free AA is first oxidized to PGG2 (endoperoxide) by the cyclooxygenase activity followed by peroxidation to form PGH2 (precursor of prostanoids) by PGHS enzymes (Fig. 3). PGH2 is the precursor of series-2 prostaglandins or inflammatory prostanoids. Notably, PGHS enzymes can also convert other polyunsaturated fatty acid (PUFA) such as dihomo-γ-linolenic acid (DGLA) to series-1 prostaglandin precursor PGH1 (generally considered as anti-inflammatory PGs), eicosapentaenoic acid (EPA) to series-3 prostaglandin precursor PGH3 and linolenic acids to 9- and 13-hydroxyoctadecaienoic acids (HODE) [99]. Other than by PGHS, PUFA such as AA can be further metabolized through lipoxygenase (LOX) or epoxygenase to form other eicosanoids. The structural and functional distinctions of the PGHS isoforms is attributed to expressional dichotomy of the genes in the physiological and pathological contexts as well as differences in the requirement of substrate concentrations and range of substrate specificity. Notably, PGHS-2 has wider substrate specificity, with ability to metabolize AA ester and amide derivatives and oxygenate diverse PUFAs unlike PGHS-1 [62]. Further, the retention of AA-oxygenation ability of aspirin-treated PGHS-2 also contributes to the difference in PGHS isoform-specific function and holds physiological relevance owing to the anti-inflammatory property of polyhydroxylated lipids produced in the process [96]. As already stated, the series-2 prostaglandin precursor, PGH2 gives rise to a number of prostanoids, such as PGD2, PGE2 PGF2α, PGI2 and thromboxane A2 (TxA2) [94], through tissue-specific isomerisation to produce the diverse prostanoids which exert wide range of biological effects upon interaction with specific G-protein coupled receptors [96]. The role of these prostanoids is therefore also tissue specific and hence determines the pathophysiological outcomes in a context-dependent manner. While PGD2, PGE2 and PGI2 are potent vasodilators in cardiovascular system, TxA2 exhibit vasoconstriction in the same system. TxA2 plays a significant role in platelet aggregation, whereas PGI2 displays anticoagulant properties. In airways, TxA2 and PGF2α act as bronchoconstrictors while PGI2 and PGE2 are bronchodilators. PGE2, PGF2α and PGI2 also give protection to gastric mucosa. Moreover, PGE2 and PGI2 are responsible for renal blood flow and diuresis in compromised kidneys. In relation to inflammatory response of body, PGE2 is the most important PG [81]. Regarding the adverse effects of NSAIDs, it is very important to understand the role of the two PGHS isoforms in producing these prostanoids as signaling mediators since the effect of PGHS isoforms could be counter intuitive. For example, in kidney while PGI2 produced by PGHS-2 affects renin secretion, PGE2 produced by PGHS-1 regulates glomerular filtration [85]. Essentially, all the biological activities of these prostanoids depend on their binding to G protein-coupled cell-surface receptors for example FP, IP, and TP receptors which are meant for PGF2, PGI2 and TxA2 respectively [100], [101]. The receptors for PGD2 are DP1 and DP2 or CRTH2 (chemoattractant receptor-homologous molecule) receptors [100], [101]. On the other hand PGE2 has four subtypes of receptors named EP1-EP4 [100], [101]. Here, it is important to note that although, there is structural difference between PGHS-1 and PGHS-2 the enzymatic steps involved in the conversion of AA to prostanoids by both these isoforms are identical [98].
Fig. 3.

NSAID-PGHS-prostanoid axis. PGHS-1/2 isoenzyme mediated prostanoid biosynthesis from arachidonic acid. Arachidonic acid is produced from phospholipids of the plasma membrane under the action of phospholipase A2. In addition to prostaglandin (PG) and thromboxane (Tx) formation by PGHS isoforms in a cell and tissue-specific manner, leukotrienes (LTEs) are other immune mediators which are produced by the enzyme 5- lipoxygenase (5-LO). Each prostanoid interacts with its specific receptor as indicated in the figure.
5.2. NSAIDs, mitochondrial oxidative stress and apoptosis
As already mentioned above, despite their clinical relevance, NSAIDs are cytotoxic compounds. NSAIDs can exert activation of mitochondrial oxidative stress (MOS), a cytopathological condition characterized by severe mitochondrial damage due to activation of detrimental redox-active chain reactions followed by severe bioenergetic crisis and eventual cell death. Mitochondrial electron transport chain (ETC) complex-I is the major target of NSAIDs (Fig. 4 ). Notably, diclofenac has been found to be most potent among the NSAIDs in inhibiting ETC complex I activity, thereby leading to electron leakage from the respiratory chain [102]. The leaked e- causes partial reduction of molecular oxygen to form superoxide, O2 .-, the progenitor reactive oxygen species (ROS) [103]. Intra-mitochondrial O2 .-, which is membrane impermeable, is immediately converted to H2O2 by the mitochondrial super oxide dismutase, SOD2, as a protective response against oxidative stress. However, being membrane permeable, H2O2 escapes the mitochondria (before subsequent neutralization) causing oxidative damage to cellular macromolecules including DNA, protein, lipids and carbohydrates, rendering them functionally inactive. While inside mitochondria, O2 .- damages various Fe-S cluster proteins including aconitase and cytochrome c, leading to Fe2+ release which in turn reacts with H2O2 via Fenton reaction to produce hydroxyl radical (.OH), the most damaging ROS [104]. All these events perturb the cellular redox homeostasis leading to the activation of intrinsic pathway of apoptosis. Elevated ROS also triggers mitochondrial depolarization and detrimentally shifts the mitochondrial structural dynamic balance towards hyperfission [105]. NSAIDs further enhance ubiquitination-dependent mitochondrial clearance. In course, the damaged mitochondrial membrane proteins release cytochrome c which moves on to form the apoptosome complex required for activating the executioner caspases leading to nuclear DNA damage and eventual cell death. During these processes, damaged mitochondrial DNA is often released from the necrotic cells which act as damage associated molecular patterns to attract immune cells for phagocytic clearance of the dying cells. In this way MOS leads to tissue inflammation which operates in a positive feedback loop during chronic insults leading to organ damage. The cytotoxic actions of NSAID also include arrested cell proliferation, activation of multiple death pathways including extrinsic apoptosis [106], [107] and most notably increasing the sensitivity of cancer cells to apoptosis while overcoming chemo and radioresistance along with inhibiting tumor-associated angiogenesis [108], [109]. Within the purview of this NSAID-PGHS-prostanoid axis and NSAID-MOS-apoptosis axis, we can now proceed to discuss the basic mechanism involved in NSAIDs-mediated damage to different organs.
Fig. 4.

Overview of NSAID-induced mitochondrial reactive oxidants production leading to cellular pathology. NSAID triggers electron leakage from complex I of the electron transport chain (ETC) leading to incomplete reduction of molecular oxygen followed by superoxide (O2.-) production. O2.- is the precursor of most reactive oxygen species (ROS); itself rapidly being converted to H2O2 by superoxide dismutase (SOD2). In the presence of transitional metals (such as Fe2+), H2O2 can be converted to hydroxyl radical (HO·). H2O2 may be scavenged by glutathione peroxidise (GPx) in presence of GSH. The oxidised glutathione (GSSG) is reduced back to GSH by glutathione reductase (GR) in presence of NADPH. Amplification of ROS causes fall of mitochondrial membrane potential (ΔΨm). Reactive radicals react with biomolecules like membrane lipids, proteins and mtDNA producing lipid peroxides, protein carbonyls and DNA damage. Cytochrome C gets released from damaged mitochondrial to cytosol through outer mitochondrial membrane (OMM) permeabilization and activate apoptotic pathway. All these events together contribute to a wide range of cellular pathologies. Dotted arrows indicate detrimental reactions involving free radicals.
6. NSAIDs and organ damage
With their antinociceptive, anti-inflammatory and anticancer properties, NSAIDs which are friends in need often become formidable foes (owing to their non specific cytotoxic effects), leading to multiple organ pathologies. Here, we will focus on the detrimental impact of NSAIDs on 6 major organs whose coordinated functioning is pivotal for maintaining healthy life. A schematic representation summarizing the underlying cytotoxic effects of NSAIDs on human body is presented (Fig. 5 ).
Fig. 5.

Effect of NSAIDs on different target organs. The action of NSAIDs on major organs including stomach, small intestine, heart, liver, kidney, respiratory tract and brain is mainly mediated through PGHS-dependent prostanoid modulation and alteration of mitochondrial functional integrity leading to mitochondrial oxidative stress (MOS) generation, depolarization of mitochondrial transmembrane potential (ΔΨm) and consequent cell death. However, in heart, low dose aspirin actually offers cardioprotection through anti-thrombotic effect. Upward arrows indicate upregulation/elevation; downward arrows indicate downregulation/depletion.
6.1. Risk to the gastric mucosal and small bowel injuries
In the treatment of chronic inflammatory conditions by NSAIDs, development of gastric mucosal injury creates the major limitation. Prevalence of gastric mucosal lesions has been observed in patients, with rheumatoid arthritis or pain, who have taken NSAIDs [110], [111]. A prospective, cross-sectional multi-centred study in India, which included 8 medical colleges across the country, reported NSAID-related GI complications in 30.08% cases [112]. In a study performed in Pakistan, analysing 820 patients who have undergone upper gastrointestinal (GI) endoscopy (1998–2000), it was found that 14.7% patients reflected NSAIDs-related peptic ulcer. Notably, duodenal ulcer (65.3%) was found more frequently than gastric ulcer (42.3%) [111]. In all the cases the duration and dose of NSAID usage were the major determinants of the severity of complications [112]. The risk of GI complications by NSAIDs increases by 2.5–5.0% in patients with a previous history of GI incidents [113]. In United States, in 2000, more than 111 million prescriptions were written for NSAIDs with an approximate cost of $4.8 billion [114]. Since these drugs have a wide range of applications, the US Food and Drug Administration (FDA) issued a health advisory which stated that NSAIDs should be used at the lowest effective dose and for the minimum duration consistent with individual patient treatment goals [114]. However, several studies have linked relatively short-time use of NSAIDs to risk of GI, CVD and renal complications [114]. Initially, it was believed that traditional NSAIDs inhibit gastroprotective PGHS-1-derived PGE2 and PGI2 thereby inflicting injury in the gastric mucosa [115]. This instigated the development of PGHS-2 selective NSAIDS [115]. PGHS-2 is generally induced during inflammation [116]. So, the idea was, PGHS-2 specific NSAIDs would take care of the pain and inflammation while not interfering with PGHS-1 pathway. However, this simplistic view has been challenged by overwhelming evidence, one of which shows that PGHS-2 also plays a significant role in resolving gastric mucosal inflammation and healing the ulcers [20]. Subsequently, in a meta-analysis study by Coxib and traditional NSAID Trialists’ (CNT) Collaboration, which comprised of 124,514 participants, the CNT researchers compared the NSAID-induced GI complications among various NSAIDs including naproxen, diclofenac, ibuprofen and PGHS-2 inhibitors such as rofecoxib, lumiracoxib, etoricoxib, valdecoxib, GW40368. They found an increased risk of upper GI complications across all the medicines analyzed [117]. On top of that, since the beginning of this millennium, a number of randomized control trials (RCT), aimed at evaluating GI complications of NSAIDs, have linked PGHS-2 selective NSAIDs with cardiovascular diseases (CVD) (detailed in the next section) which led to the withdrawal of a number of NSAIDs [118], [119]. Thus ‘shift-to-PGHS-2’ strategy did not give expected results. Now, it is really important to verify the notion whether PGHS-1 inhibition is really the actual perpetrator. Surprisingly, one study has shown that in PGHS-1 knock-out mice, aspirin predominantly induced gastric mucosal injury possibly by a PG-independent mechanism, questioning the previous understanding of PGHS-1-mediated gastroprotection [21]. In addition, aspirin increased gastric mucosal PGE2 expression in PGHS-1 knock-out mice, probably through induction of PGHS-2. Now, it is clear that PGHS-1 pathway is not the only pathway underlying NSAID-toxicity in the gastric mucosa. Hence, the question is what is that PG-independent mechanism? However, in that study the authors also proposed reduced surface hydrophobicity as a possible explanation for the damage [21]. NSAIDs, which are weak acids, have been reported to interact with the mucosal cell surface phospholipid bilayer and reduce hydrophobicity in the gastroduodenal mucosa. This compromises the hydrophobic lining, thereby exposing the mucosa to luminal aggressive factors (including gastric acid). A study has shown that when aspirin was co-administered with a phospholipid, there was reduced gastric injury even after treatment with otherwise toxic combination of aspirin and a PGHS-2 selective agent [19]. However, disruption of phospholipid bilayer alone is still not sufficient to explain the severity of NSAID-induced gastric toxicity. In this regard, recently NSAID-mediated uncoupling of oxidative phosphorylation, MOS and apoptosis has been implicated. Owing to ‘ion trapping’ during absorption, micromolar to millimolar concentration of NSAIDs can uncouple oxidative phosphorylation, thereby destabilizing ATP production in the mitochondria leading to bioenergetic crisis [19]. Electron microscopic studies and sub-cellular organelle marker enzyme analysis revealed the uncoupling effect of NSAIDs in both in vitro and in vivo [120] pre-clinical settings of experimental gastropathy. In a dose dependent manner, NSAIDs were found to inhibit electron transfer through complex I [102], [120]. Furthermore, electron paramagnetic studies using sub-mitochondrial particles, revealed that indomethacin, a very well known non-selective NSAID, could bind to a site near Complex I and ubiquinone thereby generating radical species [121]. Accumulated evidence has supported this finding by demonstrating amplification of ROS in the gastric mucosa with severe MOS and apoptosis [104], [122], [123], [124], [125], [126], [127]. Further studies with antioxidant-therapy have found to ameliorate NSAID-inflicted gastric mucosal complications indicating a crucial role of oxidative stress in the pathophysiology [128], [129], [130]. In addition to apoptosis and MOS, NSAIDs can also unleash myriad of pro-inflammatory factors (TNF-α, NF-κB, ICAM-1) in the gastric mucosa leading to profound inflammation [115], [131], [132]. Very recently, an anti-inflammatory molecule, polydeoxyribonucleotide (PDRN) has been found to reduce NSAID-induced gastropathy by reducing pro-inflammatory cytokines and apoptosis via MAPK/NF-κB signaling pathway [131]. Moreover, due to the inhibition of PGHS isoforms, AA metabolism probably shifts towards 5-lipoxygenase pathway producing leukotrienes which can also induce inflammation in the gastric mucosa [18]. Lately, it has been also shown that via PKCζ-P38-DRP1 pathway, indomethacin triggered the subcellular activation of mitochondrial hyper-fission which is critical in contributing to gastric mucosal injury [105]. Having discussed all these mechanisms, it is important to mention that gastric acid secretion along with inhibition of PGHS-derived PGs may be attributed to the aggravation of the pathology inflicted by MOS. Conforming to the aforementioned; the most critical consequence of PGHS-inhibition could be perturbation of microcirculation [19]. Other than PGs, nitric oxide (NO) also plays a significant role in maintaining intestinal microcirculation. However, a long-term clinical trial did not significantly show any difference between AZD3582 (PGHS-inhibiting nitric oxide donator) versus NSAID in osteoarthritis patients [133]. In addition, neutrophil infiltration has been also implicated in the mechanically compromised micro vascular blood flow [19]. However, vascular effects could not be considered as the primary event in NSAID-mediated gastric mucosal damage [19]. Since gastric mucosal lining is constantly exposed to gastric acid, a role of acid should also be discussed in regard to NSAID-toxicity. The “ion trapping theory” demonstrates that the acidic pH of the gastric lumen facilitates passive cellular diffusion of the non-ionized, lipid soluble NSAIDs followed by re-conversion into ionized and lipophobic form within the neutral cytosolic pH of the gastric mucosal cell where they are assumed to induce subsequent toxicity [24]. In addition, the gastric acid synergistically acts as an aggravating necrotic factor to further erode the mucosa thereby worsening the gastric consequences of long term NSAIDs-treatment [19]. In relationtomucosalerosion, it is noteworthy that NSAIDS also prevent re-epithelialization of the injured mucosal layer [134]. As a remedial measure, clinicians mostly depend on histamine H2 receptor antagonists, proton pump inhibitors (PPIs) and antacids which predominantly act by suppressing gastric acid secretion and neutralizing luminal acidity respectively [115]. There is also evidence that PPIs like omeprazole exert anti-oxidant effect at a dose much lower than their acid suppressing dose [126], thereby indicating a dual effect of PPI-based treatment. However, complications of excessive gastric acid suppression, like dyspepsia, luminal dysbiosis, gastric microfloral imbalance, vitamin B12 deficiency, pernicious anemia, osteoarthritis, malabsorption of certain drugs active in the acidic microenvironment and even gastric parietal cell hyperplasia and gastric cancer staunchly warrant against rampant use of PPIs and histamine receptor blockers [104]. In fact, a recent study also indicated that PPIs can actually exacerbate indomethacin-induced small intestinal damage by detrimentally altering intestinal microbiome composition [135]. Hence, gastric acid is in no way a physiological evil and opting for anti-oxidant-based therapeutic strategy to prevent the gastric mucosa from NSAID-induced cytopathies is a rather safer approach than indiscriminate usage of acid suppressors.
In addition to upper gut complications, detrimental impact of NSAIDs on lower gastrointestinal tract is equally crucial in the risk evaluation of these drugs. In fact, over the past decade hospitalizations due to NSAID-associated lower gut complications have increased compared to the upper gut pathologies [136], thereby underscoring the relevance of NSAID-enteropathy manifested as iron deficiency anemia, protein loss and hypoalbuminemia, indigestion, and abdominal pain. Severe complications include luminal perforation, occult bleeding, intestinal stricture, obstructions and ulcers [137]. Advent of modern imaging techniques including capsule endoscopy and double balloon endoscopy have eased the detection of myriad enteric lesions including erosions, petechiae, varix, reddened folds, loss of villi and ulcers [137], [138]. Incidentally, enteric injuries are evident in approximately 71% of chronic NSAID users [139], while prevalence rate of direct mucosal breach has been found in around 50% of chronic NSAID users [140]. Mode of NSAID absorption within the enterocytes is more direct and different from “trapping theory” applicable in the gastric cells. However, both NSAID-mediated PG depletion and mitochondrial pathology have been found to be instrumental in the early stage of injury leading to impairment of intestinal mucosal barrier function. As a consequence, the invasion of gram negative enterobacteria initiate the inflammatory cascade wherein Toll like receptor-4 (TLR-4)-induced proinflammatory cytokine upsurge along with NLRP3 inflammosome activation attract the neutrophils eventually leading to oxidative burst, chronic inflammation, apoptosis and ultimately intestinal ulceration [140]. Although, the pathogenesis of enteropathy differs with that of gastropathy in the later stages, uncoupling of mitochondrial oxidative phosphorylation, ATP deficiency, elevation of cytosolic Ca2+ and Na+/K+ imbalance and consequent induction of apoptosis are some of the hall mark and common events triggered in both these gut compartments by NSAIDs. As a result the barrier function is severely compromised due to osmotic imbalance of enteric epithelial cells leading to increased permeability and invasion by luminal aggressive and noxious factors including bile acids, pancreatic secretions, hydrolytic/proteolytic enzymes and gram negative bacteria. Intercellular tight junctions are severely compromised by the aforesaid events along with aggravation due to apoptotic loss of enterocytes [24] and proteolytic disruption of intestinal capillaries. Bacterial LPS along with TNF-α and High mobility group box 1 (HMGB1), a DAMP released from necrotic enterocytes [141] are chemotactic signals for macrophage and neutrophil infiltration to the injured mucosa. Intestinal pathogens and bile acids further strengthen inflammation and the process proceeds in a feedback loop leading to severe tissue injury in chronic NSAID users. Bile acids are especially instrumental factors that highly aggravate NSAID-induced damage as there are reports showing that NSAID alone can trigger less macroscopic injuries in spite of increasing membrane permeability and inflammation in bile duct-ligated rats [142]. An interesting study revealed that insoluble dietary fibers and gliadin (contained in wheat gluten) lead to intestinal epithelial injury and increase in intestinal permeability mainly when mucus levels are low due to NSAID usage [143], [144]. It was evident from the post hoc analysis of the data, obtained from Multinational Etoricoxib and Diclofenac Arthritis Long-term (MEDAL) trial that small intestinal complications accounted for 40% of severe gastrointestinal complications in patients using NSAIDs [145]. Regarding the relevance of NSAIDs in inducing enteropathy, it is a noteworthy fact that the difficulty in identifying the primary site of injury in the distal small intestine is one of the major drawbacks in diagnosis of enteropathy. However recently, exfoliated cell transcriptome (exfoliome), which closely resembles small intestinal mucosal-transcriptome, has been recognized as a promising tool in non-invasive scrutinizing of NSAID enteropathy [146]. Having stated the direct action of NSAIDs on gastrointestinal cellular integrity, the role of gut microbiota, in indirectly regulating NSAID-induced enteropathy, is worth highlighting as a promising field to explore. There are already preliminary indications that probiotic treatment can modulate intestinal gram negative bacteria-induced pathogenesis wherein protective action of some bacteria like Lactobacillus casei, L. gasseri and Bifidobacterium breve Bif195 against NSAIDs is already evident [147], [148], [149]; although establishing specific and precise patterns of microbial profile among regular NSAID users is yet elusive. All these, demonstrate the possible underlying mechanisms by which NSAIDS affect the upper and lower GI tracts thereby warranting effective use of NSAIDs without GI complications.
6.2. Risk of cardiovascular disease
Cardiovascular diseases (CVD), which include events like myocardial infarction (MI), stroke, hypertension and heart failure, to name a few, are one of the major causes of morbidity and mortality across the world (2020 Heart Disease and Stroke Statistical Update Fact Sheet At-a-Glance). During 2014–15, in the United States, the estimated average annual direct and indirect expenses due to CVD and stroke was $351.2 billion which reconfirms the severity of the disease and the economic burden on society [150]. So, it would be highly relevant to enquire the link between one of the leading causes of death globally i.e. CVD and one of the most-prescribed-drugs on the planet i.e. NSAIDs. Over the years, a large number of scientific reports have associated NSAIDs with CVD. Although, aspirin has been known to be effective in secondary prevention of MI and stroke, it does not depict the actual connection between CVD and NSAIDs. Since traditional NSAIDs including aspirin (targeting PGHS-1, thereby suppressing PGE2 & PGI2 in the GIS tract and TxA2 in the platelets) inflict severe gastropathy, it was the ‘gastrointestinal safety concern’ which motivated the development of PGHS-2 specific NSAIDs. Moreover, the analgesic efficacy of NSAIDs was largely attributed to the suppression of PGHS-2 derived PGE2 & PGI2 [151]. As a result, despite the fact that PGHS-2 also implies some ‘mucosal defence’, the competition for getting approval of PGHS-2 inhibitors gained momentum [20], [152]. With the development of coxibs, it was thought that now gastric complications could be safely taken care off without compromising the analgesic efficacy of NSAIDs. With this view, after the introduction of the first coxib, a number of RCT were arranged to evaluate their gastrointestinal tolerability. However, the first Trial in 2000, the Vioxx Gastrointestinal Outcomes Research (VIGOR) Trial, showed a link between PGHS-2-specific NSAIDs and CVD risk. This TRIAL, including 8076 patients with a history of osteoarthritis or rheumatoid arthritis and being treated with rofecoxib or naproxen, showed a five fold increase in MI events in patients treated with rofecoxib compared to naproxen. In September 2004, Merck recalled rofecoxib (Vioxx) from the market and later US-FDA had to issue a black box warning against valdecoxib (Bextra) [118], [119]. Further, the VIGOR Trail findings got support from Adenomatous Polyp PRevention On Vioxx (APPROVe) trial, which was primarily aimed at studying the prevention of intestinal polyps by rofecoxib. This study, where 2586 patients were treated with either rofecoxib or placebo, inferred that long term use of rofecoxib is associated with twice the risk of heart attack or stroke versus placebo [118], [119]. Recently, the results of PRECISION (Prospective Evaluation of Celecoxib Integrated Safety versus Ibuprofen or Naproxen) Trial, funded by Pfizer, concluded that in regard to cardiovascular hazards, there exists no significant difference between these three NSAIDs [153]. A number of other trials have also linked the risk of CVD with NSAIDs [118]. Albeit, these RCTs seemed to provide ‘gold-standard’ evidence; still their interpretations are not beyond the scope of interrogation in the scientific community. G.A FitzGerald has pointed out a number of issues regarding the interpretation of PRECISION trial. He suggested a more intense phenotypic study at individual level to identify factors that differentially predispose to benefits versus risks, thereby being more precise about NSAID usage [154]. Hence, it is clear that whatever be the issues regarding the nuances of the interpretation of various outcomes of different RCTs, there is no doubt about the link between NSAID-use and compromised cardiovascular health. In line with this, Garcia-Rodriguez et.al, in 2008, reported a positive correlation between increasing PGHS-2 inhibitory potency with increasing CVD incidents [155]. On the contrary to those trials which used patients with long term use of NSAIDs, studies have raised doubts on the risk of coronary events due to NSAID usage at lower doses for short durations which are practically the case for over the counter use or prescription-based intake of NSAIDs for managing acute pain or fever [156].
At this point it is however noteworthy that, following all the trials, meta-analyses and cohort studies traditional NSAIDs (nonspecific PGHS inhibitors) have also been negatively linked to CVDs other than the coxibs [157]. Nevertheless, lower doses of aspirin were found to offer protective effects through secondary prevention of CVD. Low dose aspirin (≥30 mg/day) can significantly prevent platelet aggregation without affecting important endothelial cell functions. It was Dr. Lawrence Craven who, in the early 20th century, conducted some studies to prove his hypothesis that aspirin prevents coronary thrombosis [158]. Further studies revealed that antithrombotic effects of aspirin were due to acetylation of PGHS-1 at a serine 530 in platelets (the similar acetylation site on PGHS-2 being Ser 516; although platelets only express PGHS-1) which prevented the interaction of AA with PGHS-1, thus limiting the synthesis of TxA2 [158]. On one hand, PGHS-1 is the major source of TxA2 production in platelets leading to proaggregation and vasoconstriction; on the other hand, both PGHS-1 constitutively synthesized in the arterial wall and endothelium, are involved in the production of the vasodilator and anti-aggregant PGI2. A healthy equilibrium exists through balanced inhibition of TxA2 and PGI2. Inhibition of PGHS-1 in platelets creates a disruption of the equilibrium in favour of PGI2, with a cardioprotective anti-aggregant effect, manifested due to low dose-use of aspirin [85], [156], [159]. Since the plasma half-life of aspirin is 20 min, the enucleate platelets cannot produce any new PGHS-1; so the effect of aspirin remains as long as the platelets survive (10 days) [160]. Unlike platelets, in endothelial cells the PGHS-1 is restored within a short period of time after being inhibited by aspirin thus without much affecting the PGI2 production [161]. Since enhanced calcium is required for platelet aggregation or vessels to contract, PGI2 exerts it anti-thrombotic effect by activating PKA through IP receptors which in turn cause profound reduction of intracellular calcium in platelets or vascular smooth muscle cells by a mechanism still unknown [98]. However, non-aspirin NSAIDs (NANSAIDs) can cause incomplete and reversible inhibition of PGHS-1 in platelets [162]. Hence, effect of aspirin in secondary prevention of CVD is mediated through irreversible inhibition of PGHS-1-derived TxA2 in platelets and not through PGHS-2 [156]. Regarding primary prevention of CVD by aspirin, the current guidelines are very close to what Dr. Craven recommended for the primary prevention of CVD by aspirin in males aged between 45 and 65 years [157], [158]. A recent meta-analysis based report has pointed out an insufficient benefit-risk ratio for aspirin-mediated primary prevention of CVD [163] which was supported by further studies [164]. The risk of ‘bleeding’ such as intracerebral haemorrhage or gastrointestinal bleeding is, however, one of the key limitations of aspirin use in CVD [163]. Since the efficacy of aspirin against MI and ischemic stroke (secondary prevention) is indisputable over the years, doctors have to evaluate the risk–benefit balance while prescribing it. However, very recently G. Ravach et.al mentioned that according to European Cardiovascular prevention recommendations, aspirin should be prohibited from prescriptions for the primary prevention of CVD [164]. All these issues have made cardiovascular safety of NSAIDs a highly controversial subject [157]. Although aspirin stands out alone among other NSAIDs in giving cardio-protection, a number of pathophysiological mechanisms have been proposed to explain non-aspirin NSAID-associated CVD. One key explanation postulated that inhibition of the endothelial PGI2 which normally prevents thrombosis, by NSAIDs, could be responsible for the onset of CVDs. Recently it is believed that, in endothelial cells, primarily, PGHS-1 plays the pivotal role in PGI2 biosynthesis replacing the previous concept of PGHS-2 mediated PGI2 production in those cells. Therefore, non-specific NSAIDs can block PGHS-1 mediated production of cardio-protective PGI2 at therapeutic doses [98]. However, it is noteworthy that, while deletion of TP (TxA2) receptors minimizes atherogenesis, deletion of IP (PGI2) receptors enhances atherogenesis which indicates that selective inhibition of PGHS-2 can also disturb the physiological balance between PGI2 and TxA2, thereby increasing the risk of CVD [152], [165]. Despite this observation, the role of PGHS-2 in PGI2 production in endothelial cells is controversial. Then how do PGHS-2 specific NSAIDs induce CVD? It is important to note that, the previously predominant hypothesis of PGHS-2 mediated PGI2 production in endothelial cells, which fitted well with ‘PGHS-2-CVD’ link, was actually based on two major observations [98]. First being the decrease in urinary metabolite of PGI2 (2,3-dinor-6-keto-PGF1α) in subjects treated with PGHS-2 selective NSAIDs and the second being PGHS-2 gene deletion in endothelial cells of mice causing thrombosis. However, later these observations were challenged by evidence of PGHS-1 being responsible solely for the physiological synthesis of PGI2 in endothelium [98]. Hence, the link between PGHS-2 and CVD is still elusive. Growing evidence supports the presence of a constitutively expressed PGHS-2 in the kidney or systemic vascular endothelium which is responsible for cardio-protective effect of PGHS-2 [98]. In the context of inter-organ communication, it is known that response of one organ to the surrounding changes may affect or modulate the performance of other organs. In line with that, a kidney-heart link has been proposed. Hence, inhibiting renal PGHS-2 by NSAIDs could increase asymmetric dimethyl arginine (ADMA), an endogenous eNOS inhibitor, which eventually could block eNOS, minimizing the protective effect of NO and thereby increasing hypertension, atherosclerosis and thrombosis [98], [166]. However, contrary to this, in relation to ADMA, another group has proposed that this increased level of ADMA is actually a result of PGHS-2 deletion or inhibition-associated renal dysfunction. No alteration of ADMA was found in the context of normal renal function. Now, in relation to NO, initially it was assumed that vascular NO might compensate for the reduction in PGI2; but subsequent studies have disapproved this assumption [167]. Surprisingly, it was reported in a PGHS-2 depletion study that endothelial NO synthase expression is actually regulated by PGHS-2 mediated PGI2 production [168], which in turn likely magnifies the effect of PGHS-2 inhibition [167], [168]. Moreover, PGHS-2 inhibition has also been associated with sodium and water retention, augmenting heart failure and hypertension as well as adverse ventricular remodelling [157]. So, even after so many years of research, the role of PGHS-2 and its cell specific expression is one of the most enigmatic topics of this field. Other than PGHS-prostanoid pathway, MOS has been also implicated in the pathogenesis of CVD [92]. Since cardiac cells are highly active, myocardial energy demand is predominantly met by the ATP produced in huge amount by the cardiomyocyte mitochondria. Inhibition of PGHS can lead to accumulation of AA which in turn enhances cardiac mitochondrial dysfunction. In an in vitro study with bovine heart mitochondria, AA was found to inhibit complex I and III, thereby generating increased ROS and subsequently releasing cytochrome C, which is the rate limiting event in apoptosis [165]. In addition, the mitochondrial antioxidant superoxide dismutase (SOD2) has been largely associated with the prevention of atherosclerotic lesion through protecting mtDNA [165]. In another study, diclofenac was found to induce ROS (detected with increased H2DCFDA fluorescence), decrease proteasomal activity (indicated by reduced β5 proteasome activity, increased polyubiquitination) and enhance apoptosis (detected by increased caspase 3 activity) unlike aspirin which did not show any effect on ROS and cell viability. In the same study, the generation of ROS has been mechanistically linked to diclofenac-mediated disruption of mitochondrial complex III, and fall of mitochondrial membrane potential [169]. Recently, diclofenac and celecoxib-treated cardiomyocyte microarray analyses revealed differential gene expression (around 2-fold increase of 32 genes in diclofenac and 560 genes in celecoxib while around 2-fold decrease in 424 genes for diclofenac and 1718 genes in celecoxib). Interestingly, genes associated with calcium and potassium signaling were found to be significantly affected by NSAIDs along with involvement of major signaling pathways including apoptosis [170]. In NSAID-treated cardiomyocytes, pathway analyses suggested changes in gene expression pattern in major cellular pathways such as apoptotic, signal transduction and transcription [170]. Hence, it indicates as well as essentially warrants that role of PG-independent pathway, specially MOS in CVD, needs to be deeply explored in the context of NSAID treatment. Although, hitherto, NSAID-mediated inhibition of PGHS-derived PGI2 is still the dominant mechanism by which NSAIDs affect cardiac health while contributing to CVD [167].
6.3. Risk of renal injury
In addition to CVD and GI complications, NSAIDs also carry a significant risk of kidney damage [171] which includes multiple nephrological complications like acute kidney injury (AKI), and chronic kidney disease (CKD) encompassing electrolyte imbalance, glomerulonephritis, renal papillary necrosis, fluid retention-induced hypertension, renal tubular acidosis, hyponatremia and hyperkalemia to name a few [172], [173]. While high dose of NSAIDs has been associated with AKI, long term NSAID-use also severely increase the risk of CKD. NSAIDs come next to aminoglycosides as the most common cause of nephrotoxicity and acute renal failure [174]. Annually 2.5 million Americans suffer from NSAID-related renal complications [175]. Consumption of NSAIDs by individuals suffering from chronic pain, rheumatoid arthritis, osteoarthritis and other musculoskeletal disorders render them more susceptible to nephrotoxicity [171]. Significant risk of renal failure was evaluated in an older study with inpatient mean age 15.2 ± 2.3 years (ten females and five males) [176]. 15% cases of AKI have been found associated with NSAID usage, with 25% greater propensity of contraction in patients older than 65 years [175]. In addition, recent reports have documented the risk of over-the-counter NSAID usage in pediatric population leading to acute renal failure [173], [177]. Thus, it reveals the risk of using these drugs even at therapeutic dose. In a recent histopathological study on rat, meloxicam has been linked to nephrotoxicity along with hepatotoxicity, and gastric metaplasia [178]. Given that kidney is the organ for drug excretion, receiving almost 25% of the cardiac output, the renal arterioles and glomerular capillaries are significantly vulnerable to NSAIDs [171]. Regarding the mode of action, invariably, PGHS-derived prostanoid inhibition by NSAIDs plays a pivotal role in the progression of nephrotoxicity. PGHS-1 is synthesized constitutively in the collecting ducts and in diverse cells of the Bowman’s capsule. On the other hand, medullary interstitial cells of renal papillae, epithelial cell lining of the ascending loop of Henle and cells of macula densa exhibit PGHS-2 expression [175]. PGHS-1 primarily controls the glomerular filtration rate (GFR), while PGHS-2 is important for sodium and water retention. Blocking PGHS enzymes in turn blocks PG production, of which PGE2 (tubular) and PGI2 (vascular) are important [179]. Notably, PGs and their suppression by selective and non-selective PGHS-inhibitors have been also associated with renal development [175]. PGs, with their vasodilatation effect, control pre-glomerular resistance, maintain GFR and preserve renal blood flow especially in fluid-depleted state [171], [174]. Localized PG synthesis (PGI2, PGE2, PGD2) causes vasodilatation of afferent arteriole, increases renal perfusion together with redistribution of the cortical flow to the nephron in juxta-medullary region [171], [174]. PGE2 and PGF2α prevent sodium and chloride transport in thick ascending limb of the loop of Henle and colleting ducts. While PGE1 antagonizes vasopressin, PGI2 maintains glomerular filtration. During volume depletion, the renin-angiotensin-aldosterone axis is activated, leading to vasoconstriction and increased reabsorption of sodium and chloride along with increased sympathetic blood flow, further enhancing the vascular tone [174]. In this setting of decreased perfusion pressure, PGs are responsible for vasodilatation of afferent arterioles, thereby maintaining normal GFR. This action of PGs is further reinforced by angiotensin II, norepinephrine, endothelin and vasopressin [175]. During NSAID-mediated inhibition of PGHS-derived PG production, normal GFR is compromised, eventually leading to ischemic injury and acute tubular necrosis [175]. PGs in the macula densa cells impose a check on the renin secretion which in turn would enhance under PGHS inhibition, eventually intensifying the renin-angiotensin-aldosterone pathway and ultimately leading to sodium reabsorption, hypertension and oedema [175]. AKI is aggravated when NSAIDs are used in the background of heart failure, or usage of angiotensin-converting enzyme inhibitors, angiotensin receptor blockers as well as intravascular volume depletion to name a few [175], [180]. Having said about the mechanistic basis of NSAID-induced nephrotoxicity, precise mention about some of the typical and prominent nephrological complications is essential to project the cytotoxic spectrum of these drugs in the renal context. Interstitial Nephritis: Approximately 15% of the cases of acute kidney injury are acute interstitial nephritis which is characterised by poor renal function as a result of acute inflammation and edema of renal interstitium. Both PGHS-specific and non-specific NSAIDs are one of the major causes of this situation [175]. Minimal Change Disease: In this case, PGHS-inhibition by NSAIDs favours the conversion of AA to leukotriene which eventually activates helper T lymphocytes to infiltrate glomeruli causing podocyte injury leading to minimal change disease (MCD) [175]. Papillary necrosis: PGs maintain adequate perfusion and oxygenation in the renal medullary and papillary regions. Inhibition of PGHS by NSAIDs inflicts ischemia and necrosis in the medullary and papillary regions thereby causing renal failure [175]. In addition to the aforesaid, some of the prominent renal complications associated with ionic imbalance, experienced as a side effect of rampant NSAID usage essentially deserves special mention. Hyponatremia: The antidiuretic hormone, ADH, or vasopressin is regulated by PGs. In presence of NSAIDs, PG-mediated ADH’s role of free-water clearance is prohibited, thereby forcing the nephron to absorb free water leading to hyponatremia and associated renal pathology [175]. Hyperkalemia: There are two predominant mechanisms regulating NSAID-induced hyperkalemia; either causing reduced secretion of potassium in the principal cells of collecting duct or by compromising GFR, thereby resulting in attenuated potassium secretion through sodium–potassium exchanger which in turn aggravates the renal complications [175]. Renal Tubular Acidosis: Prolonged hyperkalemia is associated with decreased net renal acid excretion in the distal nephron leading to renal tubular acidosis type 4 (hyperchloremic metabolic acidosis) and consequent nephritis [175]. Hypertension: In a patient with CKD, within 1–2 weeks of taking NSAIDs, hypertension may develop leading to heart failure (HF). NSAID-mediated inhibition of PGs, causing sodium retention and vasoconstriction, leads to edema and high blood pressure. Under normal condition a healthy kidney can respond to such a situation by minimizing the clinical side effects thereby maintaining a steady-state homeostasis. However, in a patient suffering from CKD, the fluid load will exacerbate the edema and hypertension leading to HF. Another mechanism involves NSAID-mediated blocking of glucuronidation of aldosterone by human kidney which eventually increases the level of circulating aldosterone, ultimately leading to hypertension, edema and HF. In the previous section, the role of constitutive PGHS-2 in renal medulla has been mentioned in relation to CVD. This constitutive PGHS-2 has been shown to be under transcriptional control of NFAT (nuclear factor of activated T cells). It has been proposed that in kidney, the constitutive PGHS-2 regulates renal blood flow by PGI2-controlled PPAR-β/δ (peroxisome proliferator-activated receptor β/δ)-mediated vasodilation pathway. NSAIDs prevent this blood flow by blocking PGHS-2, thereby inducing hypertension [181]. In a recent study, ambulatory blood pressure and GFR were compared for two drugs, diclofenac and rofecoxib, wherein it was found that 24-hour systolic blood pressure (BP) was increased and GFR was decreased by diclofenac compared to celecoxib. In the Celecoxib Rofecoxib Efficiency and Safety in Cormorbidities Evaluation Trial (CRESCENT), the systolic BP was evaluated for monitoring the effect of NSAIDs in subjects treated with antihypertensive therapy. The study documented that a 25 mg daily dose of rofecoxib resulted in 24-hour systolic pressure greater than 135 mm Hg in 30% patients compared to 16% and 19% of patients randomly assigned to celecoxib and naproxen respectively [175], [182]. Therefore, it is incumbent upon the clinicians to prescribe the selective and non-selective NSAIDs with caution. Since a lower dose of NSAID can also induce renal failure, there is actually no safer dose of these drugs for patients with CKD. Because CKD patients are often on medications like ACEs, ARBs and diuretics, which in fact can interact with NSAIDS worsening the situation, clinicians should be utterly careful in treating these patients with NSAIDs. Among the patients with CKD, in primary care, use of NSAIDs is variable and relatively high which essentially demand for thorough research specifically aimed at strategies to reduce NSAID-use in this patient population [183]. Regarding the PGHS-independent mechanism of NSAID-induced renal damage (including both AKI and CKD), oxidative stress and inflammation has been associated [161], [184]. Cells found in kidney vasculature such as vascular smooth muscle cells, endothelial cells, adventitial fibroblast and inflammatory cells are able to produce ROS. NADPH oxidase, along with MOS, majorly contributes to ROS production in kidney [184]. In an old study, indomethacin was found to inflict mitochondrial abnormalities in the proximal tubules with evidence of oxidative stress (lipid peroxidation), mitochondrial dysfunction (fall of respiratory control ratio) and neutrophil infiltration (characterized by increased myeloperoxidase activity) [185]. Diclofenac, on the other hand, induced oxidative stress, renal cell apoptosis and upregulation of pro-inflammatory cytokines (NF-κB) in AKI as evident from a recent study [186].
Thus, both PGHS-dependent and independent pathways play significant role in kidney injury. However, it is important to note that, young patients without renal diseases or comorbidities are not under great risk of NSAIDs; although, there is no place for complacency.
6.4. Risk of hepatotoxicity
Hepatotoxicity is another serious complication that has been linked to NSAIDs although the incidences are less frequent compared to that of gastrointestinal damage, CVD and renal insufficiency. In line with that, a recent study in Brazil, conducted in the primary health care units, inferred drug-induced-liver-injury as a rare event [187]. However, the seriousness of the issue lies elsewhere. Considering the case of USA, where approximately 30 million Americans take NSAIDs each year, it is clear that a huge number of individuals are actually exposed to the potential threat of NSAID-induced liver injury [188]. Hence, studies on the role of NSAIDs in hepatotoxicity are scientifically pertinent. Since premarketing studies are insufficient, hepatotoxicity is mostly encountered in the post-marketing studies [189], [190]. A number of drugs have been withdrawn from the market due to hepatotoxicity which includes NSAIDs like bromfenac (1999) and lumiracoxib (2008) [189], [191]. Liver injury caused by most NSAIDs is likely to be idiosyncratic and clinically apparent liver injury is rare (1–10 cases approximately per 100,000 prescriptions). Although two NSAIDs, sulindac and diclofenac, are most commonly linked to hepatotoxicity [188], [191], [192], at least in rare cases, virtually all the extensively used NSAIDs are clinically linked to apparent drug-induced liver injury [188], [189]. However, in a very recent systemic review, ibuprofen has been regarded safe in relation to hepatotoxicity despite a number of published reports previously linking it otherwise [193]. Several other systemic reviews are also available which have evaluated the risk of NSAIDs in hepatotoxicity [189], [191], [194], [195]. Notably, PGHS-2 specific NSAIDs are less frequent in causing hepatotoxicity compared to non-selective NSAIDs [194]. It is important to realize that NSAIDs can actually lead to two major clinical patterns of hepatotoxicity; first being acute hepatitis (characterized by jaundice, nausea and fever together with elevated serum levels of transaminases) while the other being chronic active hepatitis (characterized by serological and histopathological abnormalities) [192]. Analyzing the published studies, aspartate transaminase (AST) and alanine transaminase (ALT) could be considered as the two most common biomarkers linked with hepatotoxicity. Nevertheless, serological abnormalities in alkaline phosphatase and total bilirubin levels are also reported [191]. Regarding the hepatotoxic mode of action of NSAIDs, a number of mechanisms have been implicated depending on the concerned drug [189], [192]. However, two predominant mechanisms have been identified; hypersensitivity and metabolic aberration [192]. Patients who have suffered hepatotoxicity from one NSAID, often experience the same reaction in case the drug is re-initiated or a sister drug with identical structure is given. Hypersensitivity reactions are characterized by elevated anti-nuclear factor or anti-smooth muscle antibody titres, eosinophilia and lymphadenopathy [192]. On the other hand, metabolic aberrations could be due to genetic polymorphisms and varied susceptibility to a spectrum of drugs as evident from in vitro studies which showed phenotypic variability of aceclofenac-metabolism amongst donor liver cells [192]. Studies with isolated rat liver mitochondria or rat hepatocytes have demonstrated that diphenylamine, which is structurally identical with NSAIDs, actually acts as an uncoupler of oxidative phosphorylation thereby disrupting ATP formation leading to hepatocyte bioenergetic crisis and death. Mitochondrial swelling, fall of mitochondrial membrane potential and diminished cellular ATP were observed after incubation of mitochondria with diphenylamine, mefenamic acid and diclofenac [196]. In case of naproxen, ferrous iron (Fe2+) release is responsible for naproxen-induced lipid peroxidation in rat liver microsomes [197]. Diclofenac was found to be more toxic in drug metabolizing cells (isolated primary hepatocytes) compared to non-metabolizing cells (HepG2, FaO). The toxicity may be due to diclofenac-induced mitochondrial impairment along with a pointless and superfluous consumption of NADPH during the reduction of N,5-hydroxydiclofenac to 5-hydroxydiclofenac; which is otherwise only oxidized to N,5-hydroxydiclofenac [197]. Mitochondrial permeability transition followed by production of reactive oxygen species (ROS), mitochondrial swelling, oxidation of NADP, proteins and thiols are also linked to diclofenac-induced liver injury [198]. Similar to diclofenac, oxidative stress is also connected with salicylic acid-induced hepatotoxicity [199]. Notably, genetic predisposition has been an important factor in diclofenac-induced idiosyncratic hepatotoxicity [38]. Polymorphism in genes for UGT2B7 and CYP2C8 (which forms reactive diclofenac metabolites) and in ABCC2 (encoding the transporter MRP2 that helps in biliary excretion) may predispose to the formation and accumulation of diclofenac metabolites which are implicated in hepatotoxicity [38]. On the other hand, sulindac can induce cholestasis by competitively inhibiting canalicular bile salt transport [200]. In one study with Wistar rats, naproxen has been linked to increased ROS (indicated by enhanced lipid peroxidation and reduced GSH) and hepatotoxicity. In the same study, naproxen also induced an adaptive response to ROS by increasing SOD and catalase. Interestingly, this increased ROS was supposed to be the underlying cause for significant genotoxic effect and fall of liver function exerted by naproxen [201]. Although, less frequent, coxibs may also cause liver damage via PG-dependent pathway. This hypothesis is supported by the proposed mechanism that implies the inhibition of PGHS-2-derived PGE2, by coxibs, which can then reduce the anti-apoptotic mitochondrial protein Bcl-2 (which offers protection against bile acid-induced apoptosis), leading to hepatocyte apoptosis [202]. Hence, it can be summarized that increased reporting, surveillance and awareness of NSAID-related hepatotoxicity will help the physicians and hepatologists to precisely assess the hepatotoxic potential of NSAIDs during prescription of these unavoidable drugs.
6.5. Risk of intracerebral haemorrhage
Bleeding within brain tissue leads to intracerebral haemorrhage (ICH), a severe type of stroke (haemorrhagic stroke, HS) [203]. Stroke events are characterized as either ischemic (blood supply to brain is blocked) or hemorrhagic (bleeding), of which ischemic strokes (IS) are more predominant [204]. A multinational European database study has shown that IS risk differs among individual NSAIDs and specifically it was found to be higher in patients with a previous history of IS or transient ischemic attack [205]. Since NSAIDs can predominantly cause gastrointestinal bleeding, risk of ICH cannot be excluded. Understanding the effect of NSAIDs on platelet-function suggests that NSAIDs can lead to enhanced risk of ICH via regulation of thrombosis. The antithrombotic effect of aspirin is linked to irreversible and almost complete inhibition of platelet PGHS activity which results in a reduction of thrombotic events, thereby increasing the chances of HS [206]. A relationship between NANSAIDs and HS (including subarachnoid haemorrhagic stroke and ICH stroke) was explored in a nationwide multicenter case control study in Korea [206]. In this study the potential confounders were adjusted. Within 14 days, proportion of NANSAIDs exposure was 2.9% for HS patients while 2.0% for controls. Comparing NANSAID-users with nonusers, the adjusted odds ratios of stroke was 1.12 for all HS, 1.03 for subarachnoid haemorrhage and 1.19 for ICH. Thus the study concludes that there is no significant rise of HS among NANSAIDs users [206]. The explanation for the probable no-antithrombotic effect of non-selective NSAIDs, other than aspirin, could be attributed either to incomplete/reversible inhibition of PGHS-1 in platelets or concomitant prevention of PGHS-2-derived PGI2 synthesis in endothelial cells; although the latter notion is still yet elusive [162]. However, Islam et.al in 2018, published a meta-analysis of observational studies including articles from 1999 to 2017. The study significantly linked NSAIDs-use with higher risk of developing HS [207]. In line with that, in a recent study in March 2019, L. Ge et.al have demonstrated that long term use of aspirin and clopidogrel (antithrombotic drug) increases the risk of cerebral microbleeds which is a marker of ICH [208]. Later in the same year, Paciaroni et. al. have shown a lower risk of bleeding event for clopidogrel monotherapy compared to aspirin [208]. Having said this, it should be mentioned that the advantage of aspirin in secondary prevention of IS is greater over its risk for ICH [160]. Since a large population is exposed to NSAIDs, these drugs should be prescribed judiciously by the clinicians keeping in mind the risk–benefit thresholds which determine the safety profiles.
6.6. Risk of respiratory tract inflammation and infection
It was until 1922, the correlation between asthma, nasal polyposis and aspirin sensitivity (Samter’s Triad) was not known [209]. Later, it was found that about 7% of asthma patients experience bronchospasms after taking aspirin or other PGHS-1 inhibiting NSAIDs which could be potentially fatal [210]. This condition, characterised by asthma, chronic rhinitis, nasal polyps and acute respiratory tract reaction, in response to aspirin and NSAIDs is referred to as aspirin-exacerbated respiratory disease (AERD). Recollections of patients have made it clear that 50% of AERD occurred after viral respiratory tract infections [210]. In cells, such as mast cells, a precarious balance is maintained between cysteinyl leukotrienes (Cys-LTEs), product of 5-lipoxygenase pathway, and prostanoids, products of PGHS pathways. NSAID-induced inhibition of PGHS-1, coupled with excessive production of leukotrienes disrupts the physiologic brake and triggers the release of plethora of proinflammatory cytokines (such as IFN-γ, IL-4) responsible for the pathophysiology of the AERD [209]. It is important to note that of all the leukotrienes (LTEs), elevated cysteinyl LTE4 has been suggested to play a significant role in the pathophysiology of AERD. Presence of LTE4 increases airway responsiveness to histamine, a phenomenon not observed with LTD4 and LTC4 indicating the presence of specific LTE4 receptor [210]. Regarding the prostanoids, PGE2 generally exerts an inhibitory effect on mast cells and eosinophils to prevent LTE release. When PGE2 is prevented by NSAID treatment, the PGE2-inhibitory control on mast cell gets lost, unleashing histamine and LTEs by mast cells, a hallmark event in AERD [209], [210]. Furthermore, during AERD, downregulation of anti-inflammatory lipoxins, which normally compete for Cys-LTEs-receptors and prevent expression of proinflammatory cytokines (IFN-γ, IL-5, IL-6), also contributes to the pathophysiology [209]. In addition, the role of PGD2, type II inflammation and group 2 innate lymphoid cells are also implicated in AERD [211]. On the other hand, PGHS-2 specific inhibitors, which are larger molecules and cannot fit to PGHS-1 channels, are not responsible for respiratory reactions in patients with AERD [210]. PGHS-2 inhibitors are therefore unable to affect the constitutive activity of PGHS-1 and production of PGE2 by mast cells, platelets, basophils and eosinophils [210]. Nevertheless, meloxicam, which is one of the two PGHS-2 inhibitors available in US market (other than celecoxib), can cause mild symptoms of respiratory reaction in AERD patients at 15 mg dose, behaving as a partial PGHS-1 inhibitor [210]. So shifting to PGHS-2 from PGHS-1 could be a better strategy; however PGHS-2 is not available without prescriptions [210]. Other than its role in AERD, NSAIDs are known to be frequently used in Community Acquired Pneumonia (CAP) or acute respiratory tract infections [212], [213]; although NSAIDs have never been found to significantly reduce total symptoms or duration of acute respiratory infections. A nationwide case cross-over study showed that NSAID use during acute respiratory infection was associated with increased risk of MI [213]. In relation to CAP, different surveys and observational studies have also shown that half of the patients of CAP were treated with NSAIDs [212]. However, this extensive use of NSAIDs was neither approved by medical guidelines or any clinical data in pneumonia patients. From the mechanistic point of view NSAIDs rather worsen the situation by preventing the recruitment and functioning of polymorphonuclear neutrophils [212]. Ibuprofen was found to inhibit TNF-α and NF-κB transcriptional activity, thereby limiting pro-inflammatory cytokine production [214]. Therefore, it may be presumed that NSAIDs could enhance pulmonary susceptibility to secondary infection to CAP. However, conflicting results were also obtained when role of NSAIDs were evaluated in animal models of pulmonary bacterial infection [212]. So, pre-existing medication and conditions should be considered before prescribing a drug. In one study, in human airway epithelial cells Parainfluenza virus (PIV3) reduced PGE2 generation. However, celecoxib treatment on PIV3-infected airway epithelial cells significantly aggravated the PGE2 reduction along with reduced PGHS-2 expression [215]. In the current perspective of COVID-19 outbreak, several speculations are popping up regarding the use of NSAIDs. Although indomethacin has been shown previously to block coronavirus RNA synthesis in dogs, in a PGHS-independent pathway, still there are no direct data advocating clinical use of NSAIDs against the recent COVID-19 situation [13]; even though, the nucleocapsid protein of the SARS coronavirus, which was responsible for 2003 outbreak, was found to interact with PGHS-2 promoter along with up regulating its expression, thereby suggesting a putative role of NSAIDs in virus prevention [216]. However, use of NSAIDs against COVID-19 is still elusive [217]. It is important to state that in the midst of this topsy-turvy situation, where the world is desperately seeking for effective COVID-19 antidote, recently a warning regarding a probable adverse effect of ibuprofen in aggravating the current coronavirus infection has also come up [80]; although clinicians have also assured patients from not withdrawing this drug who are already using it for other reasons just out of the fear of increased COVID-19 risk. Nevertheless, it has been also mentioned that patients suffering from COVID-19 should not be administered NSAIDs, as treatment, unless a strong evidence of NSAIDs against COVID-19 is obtained, because that might predispose those patients with other NSAID-associated comorbidities [80].
7. Pharmacophore modification
Since NSAIDs are associated with severe toxicities, newer approaches or alternative strategies were taken for “pharmacophore modification” to tame down the toxic side effects without compromising their beneficial health effects. In this regard, recently, prodrugs are gaining immense popularity. These are bioreversible derivatives of pharmacologically active agents which undergo chemical transformation and/or enzymatic cleavage, in vivo, to release the parent drug which then exhibits the desired pharmacological impact. The non-parent moiety may/may not be pharmaceutically active in a context specific manner [218]. Emerging documents on NSAID prodrugs have pointed towards a promising field where pharmacological research can be logically channelized. Interestingly, the palatable form of salicyclic acid i.e. acetylsalicyclic acid or aspirin itself acts as a prodrug. In vivo, aspirin liberates salicyclic acid to elicit the anti-inflammatory action while the acetyl group particularly induces PGHS inactivation. A similar mode of action is actually found in several ‘aspirin like drugs’ including indomethacin, ibuprofen and naproxen. Thus, irreversible acetylation of serine residue in PGHS-1, mediated by the acetyl group, is one of the ways aspirin exert its therapeutic effect; although few complications like GI toxicity, disruption of platelet functions, renal failure still accompany, leaving room for further chemical modification of this unavoidable NSAID into a more potent prodrug to use it effectively. Several aspirin prodrugs have been reported such as aspirin triglycerides, cyclic aspirin triglycerides, methysulfonylmethyl and nitro aspirin [218]. On the other hand, non acetylated salicyclates are also another class of prodrugs (salsalate and choline magnesium trisalicyclate), considered as viable options in perioperative conditions owing to minimum hemorrhagic effects, negligible PGHS-1 inhibition, no effect on renal blood flow and platelet functions [219]. A furoxan-containing ester of aspirin was found to show significant anti-inflammatory and anti-platelet activity in vivo, although with reduced gastric toxicity [220]. In general, for synthesis of NSAID prodrugs, focus has been laid on masking its acidic functional group or the carboxyl moiety since reports have already linked it to the toxic actions of NSAIDs. For instance, mitochondrial-uncoupling potential of NSAIDs is inversely correlated with drug pKa. In this regard, modification of the carboxyl moiety of flurbiprofen (dimero-flurbiprofen and nitrobutyl flurbiprofen) led to reduced mitochondrial uncoupling [221]. In addition, coupling of sulfonamides with free carboxylic group of NSAIDs (flurbiprofen, ibuprofen, diclofenac) was also found to increase their PGHS-2 selectivity while providing relief in gastric ulceration [222], thereby highlighting avoidance of NSAID-gastrotoxicity. However, over time, designing of NSAID prodrug has evolved from ‘just masking the carboxylic group’ to meticulously designed conjugates bearing moieties aimed at specific goals, including antioxidant activity, increasing water solubility and dissolution, NO release, hydrogen sulphide (H2S) release, acetylcholine esterase inhibitory (AChEI) activity and site specific delivery [220]. For example, many NSAID prodrugs, synthesized by coupling an antioxidant moiety through amide linkage, as well as several mutual prodrugs (both moieties are pharmacologically active) made with naturally found phenolic antioxidants including guaiacol, thymol, eugenol and other alcoholic compounds have yielded gastro-sparing results; although without compromising the typical functions of NSAIDs [223]. On the other hand, PGHS-2 inhibiting prodrugs, such as apricoxib (marketed by Tragara Pharmaceuticals) were made to improve oral bioavailability or increase water solubility for parenteral use [220]. The characteristic features of PGHS-2 inhibitors include a para-sulfonamide (SO2NH2) or a para methanesulfonyl (SO2Me) pharmacophore on one of the aryl rings which bestow PGHS-2-active-site-specificity to the drug [224]. Apricoxib, mentioned above is an anticancer NSAID, having 1,2-diphenyl moiety attached to a central 5-membered pyrrole ring together with a para-SO2NH2 PGHS-2-specific pharmacophore [224]. Moving further, it is worth mentioning that owing to the importance of NO and H2S as two vital endogenous gaseous signaling molecules involved in different physiological functions, a group of NSAID prodrugs have been made which release either NO or H2S or both the gastro-transmitters. These compounds have supposed potential in treating different complications. A new class of drug called ‘cyclooxygenase inhibitor nitric oxide donor’ (CINOD) was developed by adding NO generating group to the parent NSAID with an ester linkage. Naproxcinod, (the first CINOD), has undergone phase-II and III clinical trials; but also got disapproved by FDA owing to the lack of long-term controlled studies [225], [226]. Moreover, NCX-4016 (NO-aspirin), although forbidden from production (since 2007) due to generating mutagenic metabolites, has showed reduced gastro-toxicity in phase-I trials compared to aspirin [227]. As mentioned above, in a phase-II trial, the primary end point of 6 week endoscopic gastroduodenal ulcer did not reveal significant difference between naproxen and AZD3582 (naproxcinod) at doses effective in treating osteoarthritis; however, the secondary endpoints favoured AZD3582 [133]. It is worthwhile to note that often the NO-NSAIDs have intrinsic pharmacological activity which may not have been predicted during the prodrug designing [220]. P2026, a NO-diclofenac in which the diclofenac moiety is linked to the –ONO2 (nitrate) group via alkyl spacer, showed no gastric toxicity. In addition, another NO-releasing diclofenac derivative with a benzofuroxan moiety has been also synthesised, whereupon it was found to offer anti-inflammatory action devoid of gastrointestinal toxicity [222]. While the organic nitrate derivatives release NO only after metabolic reduction, in a NONOate derivative of NO-NSAID, NO is released spontaneously in physiological media [220]. Moreover, many of the NO-NSAIDs have been progressed through clinical trials for evaluating their effect in cardiovascular safety and pain-killing attribute [133], [227]. Other than NO, H2S also plays a significant role in mucosal defence by inhibiting leucocyte adherence on vascular endothelium, neutralizing gastric acid and enhancing gastric mucosal blood flow. Based on these ideas, researchers have developed a number of H2S-releasing NSAIDs (HS-NSAID) of which a number of molecules have been already progressed to clinical trials such as H2S-releasing naproxen, ATB-346 [228]. This is supported by a phase-II-B study, where subjects with ATB-346 (250 mg once daily) showed lesser ulcers per subject compared to naproxen (550 mg twice daily) and also lesser incidence of large ulcers (≥5 mm diameter) and marked reduction of GI complication [229]. Interestingly, ATB-337, another HS-NSAID, has been made out of linking DTT to diclofenac where DTT acts as H2S donor. HS-NSAIDs including HS-sulindac, HS-ibuprofen and HS-aspirin have also been reported to act as anticancer agents. Moving further, NBS-1120, which releases H2S, NO and aspirin, simultaneously, has been attributed with the most potent anti-cancer potency [230]. Since H2S has been linked to cardiovascular homeostasis and normal physiology of kidney, H2S prodrugs can be also effective against CVD and renal failure [231], [232]. Another promising group of NSAID-prodrugs are phospho-NSAIDs, such as phospho-aspirin (MDC-22), phospho-sulindac (OXT-328) and phospho-ibuprofen (MDC-917) which probably exert their action via COX-independent pathway [220]. In addition, NSAID prodrugs with anticholinergic agents, acetylcholinesterase inhibitory activity (AChEI-NSAIDs) and chemically stable nitroxides, 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) and 4-hydroxy-TEMPO (TEMPOL), have also been designed and tested for their efficacy, albeit with reduced toxicity [220]. Most of these NSAID prodrugs are endowed with anti-inflammatory potential as tested in in vitro andinvivo studies and clinical trials; however, the gastro-toxic effect of their respective parent NSAIDs are rationally bypassed thereby improving their utility. A schematic representation of some common NSAID prodrugs has been presented (Fig. 6 ). Other than these few examples, a myriad of other NSAID-associated prodrugs, mutual prodrugs and hybrid molecules have been prepared and tested and many more are in the pipeline [218], [223]. Hence, it is evident that prodrugs can be useful to manage the gruesome challenges of therapeutic handling of NSAIDs. However, extensive research is strongly advocated to fine tune the actual therapeutic potential of these molecules.
Fig. 6.

Structural representation of some NSAID prodrugs.
8. Conclusion
Thus, it is evident that the success of NSAIDs comes with a huge cost. NSAIDs are associated with 30% hospital admissions for preventable adverse drug reactions [233]. However, it is not our motto to project NSAIDs in a negative perspective; rather, presenting a comprehensive view of the susceptibility of some major organs to an almost unavoidable drug rampantly used in the day-to-day life. This would increase the awareness in prudent usage of these drugs. Reports of randomized clinical trials, meta-analysis, cohort studies, systemic reviews and observational studies would lead to understanding the actual drug-related risk while mechanistic details will provide indications about the potential therapeutic candidates/targets. However, various aspects of NSAID-induced organ damage still need to be investigated which should also include other organs and systems of the body. A number of areas need to be explored from mechanistic point of view, such as the link between ROS and PGs, role of PGHS-2 in CVD, role of mitochondria and other organelles in NSAID-pathophysiology along with genetic polymorphisms plausibly linked with NSAID-susceptibility. Population based studies may prove helpful in answering complex gene-driven effects on xenobiotics metabolism in the context of NSAIDs. With respect to age, older individuals are at a higher baseline risk of cardiovascular, GI, renal and hepatic complications. So a special strategy should be developed regarding the usage of NSAIDs by the older people [233]. Answers to these issues will definitely advance the current strategies of NSAID-related risk management, albeit in a more rationally organised manner while maintaining high safety profiles of these unavoidable ‘wonder drugs’ in days to come.
CRediT authorship contribution statement
Samik Bindu: Conceptualization, Writing - original draft, Writing - review & editing. Somnath Mazumder: Writing - original draft, Writing - review & editing. Uday Bandyopadhyay: Conceptualization, Writing - review & editing, Supervision, Project administration, Funding acquisition.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
SB acknowledges research grants from Early Career Research Award, SERB (Department of Science and Technology, Govt. of India) and Higher Education, Science & Technology and Biotechnology (Dept. of Science and Technology, Govt. of West Bengal). UB acknowledges J.C. Bose National Fellowship (Department of Science and Technology, Govt. of India) to perform this work.
References
- 1.Montinari M.R., Minelli S., De Caterina R. The first 3500years of aspirin history from its roots - A concise summary. Vasc.Pharmacol. 2019;113:1–8. doi: 10.1016/j.vph.2018.10.008. [DOI] [PubMed] [Google Scholar]
- 2.Tsutsumi S., Gotoh T., Tomisato W., Mima S., Hoshino T., Hwang H.J., Takenaka H., Tsuchiya T., Mori M., Mizushima T. Endoplasmic reticulum stress response is involved in nonsteroidal anti-inflammatory drug-induced apoptosis. Cell Death Differ. 2004;11(9):1009–1016. doi: 10.1038/sj.cdd.4401436. [DOI] [PubMed] [Google Scholar]
- 3.Gupta A., Bah M. NSAIDs in the Treatment of Postoperative Pain. Curr. Pain Headache Rep. 2016;20(11):62. doi: 10.1007/s11916-016-0591-7. [DOI] [PubMed] [Google Scholar]
- 4.Budoff P.W. Use of mefenamic acid in the treatment of primary dysmenorrhea. JAMA. 1979;241(25):2713–2716. [PubMed] [Google Scholar]
- 5.R.W.U. Daniel E. Furst, Shraddha Prakash Nonsteroidal Anti-Inflammatory Drugs, Disease-Modifying Antirheumatic Drugs, Nonopioid Analgesics, & Drugs Used in Gout, in: B.G. Katzung (Ed.), Basic & Clinical Pharmacology, The McGraw-Hill Companies, Inc.2012, pp. 635-657.
- 6.E.M.S. Tilo Grosser, and Garret A. FitzGerald, Pharmacotherapy of Inflammation, Fever, Pain, and Gout, in: L.L. Brunton (Ed.), GOODMAN & GILMAN'S THE PHARMACOLOGICAL BASIS OF THERAPEUTICS, McGraw-Hill Education2018, pp. 685-709.
- 7.Ong C.K., Lirk P., Tan C.H., Seymour R.A. An evidence-based update on nonsteroidal anti-inflammatory drugs. Clin. Med. Res. 2007;5(1):19–34. doi: 10.3121/cmr.2007.698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dhalla I.A., Gomes T., Mamdani M.M., Juurlink D.N. Opioids versus nonsteroidal anti-inflammatory drugs in noncancer pain. Can. Fam. Physician. 2012;58(1):30. [PMC free article] [PubMed] [Google Scholar]
- 9.Daniels M.J., Rivers-Auty J., Schilling T., Spencer N.G., Watremez W., Fasolino V., Booth S.J., White C.S., Baldwin A.G., Freeman S., Wong R., Latta C., Yu S., Jackson J., Fischer N., Koziel V., Pillot T., Bagnall J., Allan S.M., Paszek P., Galea J., Harte M.K., Eder C., Lawrence C.B., Brough D. Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer's disease in rodent models. Nat. Commun. 2016;7:12504. doi: 10.1038/ncomms12504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wichmann M.A., Cruickshanks K.J., Carlsson C.M., Chappell R., Fischer M.E., Klein B.E., Klein R., Schubert C.R. NSAID Use and Incident Cognitive Impairment in a Population-based Cohort. Alzheimer Dis. Assoc. Disord. 2016;30(2):105–112. doi: 10.1097/WAD.0000000000000098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Breitner J.C., Haneuse S.J., Walker R., Dublin S., Crane P.K., Gray S.L., Larson E.B. Risk of dementia and AD with prior exposure to NSAIDs in an elderly community-based cohort. Neurology. 2009;72(22):1899–1905. doi: 10.1212/WNL.0b013e3181a18691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rao C.V., Rivenson A., Simi B., Zang E., Kelloff G., Steele V., Reddy B.S. Chemoprevention of colon carcinogenesis by sulindac, a nonsteroidal anti-inflammatory agent. Cancer Res. 1995;55(7):1464–1472. [PubMed] [Google Scholar]
- 13.Amici C., Di Caro A., Ciucci A., Chiappa L., Castilletti C., Martella V., Decaro N., Buonavoglia C., Capobianchi M.R., Santoro M.G. Indomethacin has a potent antiviral activity against SARS coronavirus. Antivir Ther. 2006;11(8):1021–1030. [PubMed] [Google Scholar]
- 14.James D.S. The multisystem adverse effects of NSAID therapy. J. Am. Osteopath. Assoc. 1999;99(11 Suppl):S1–S7. doi: 10.7556/jaoa.1999.99.11.S1. [DOI] [PubMed] [Google Scholar]
- 15.Hooper W.P., Gerber H. Monte Carlo simulations of laser-generated sea surface aureole. Appl. Opt. 1988;27(24):5111–5118. doi: 10.1364/AO.27.005111. [DOI] [PubMed] [Google Scholar]
- 16.Arfe A., Scotti L., Varas-Lorenzo C., Nicotra F., Zambon A., Kollhorst B., Schink T., Garbe E., Herings R., Straatman H., Schade R., Villa M., Lucchi S., Valkhoff V., Romio S., Thiessard F., Schuemie M., Pariente A., Sturkenboom M., Corrao G. Non-steroidal anti-inflammatory drugs and risk of heart failure in four European countries: nested case-control study. BMJ. 2016;354 doi: 10.1136/bmj.i4857. [DOI] [PubMed] [Google Scholar]
- 17.Dreischulte T., Morales D.R., Bell S., Guthrie B. Combined use of nonsteroidal anti-inflammatory drugs with diuretics and/or renin-angiotensin system inhibitors in the community increases the risk of acute kidney injury. Kidney Int. 2015;88(2):396–403. doi: 10.1038/ki.2015.101. [DOI] [PubMed] [Google Scholar]
- 18.Becker J.C., Domschke W., Pohle T. Current approaches to prevent NSAID-induced gastropathy–COX selectivity and beyond. Br. J. Clin. Pharmacol. 2004;58(6):587–600. doi: 10.1111/j.1365-2125.2004.02198.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bjarnason I., Scarpignato C., Holmgren E., Olszewski M., Rainsford K.D., Lanas A. Mechanisms of Damage to the Gastrointestinal Tract From Nonsteroidal Anti-Inflammatory Drugs. Gastroenterology. 2018;154(3):500–514. doi: 10.1053/j.gastro.2017.10.049. [DOI] [PubMed] [Google Scholar]
- 20.Wallace J.L., Devchand P.R. Emerging roles for cyclooxygenase-2 in gastrointestinal mucosal defense. Br. J. Pharmacol. 2005;145(3):275–282. doi: 10.1038/sj.bjp.0706201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Darling R.L., Romero J.J., Dial E.J., Akunda J.K., Langenbach R., Lichtenberger L.M. The effects of aspirin on gastric mucosal integrity, surface hydrophobicity, and prostaglandin metabolism in cyclooxygenase knockout mice. Gastroenterology. 2004;127(1):94–104. doi: 10.1053/j.gastro.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 22.Jana N.R. NSAIDs and apoptosis. Cell. Mol. Life Sci. 2008;65(9):1295–1301. doi: 10.1007/s00018-008-7511-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bombardier C., Laine L., Reicin A., Shapiro D., Burgos-Vargas R., Davis B., Day R., Ferraz M.B., Hawkey C.J., Hochberg M.C., Kvien T.K., Schnitzer T.J. Comparison of Upper Gastrointestinal Toxicity of Rofecoxib and Naproxen in Patients with Rheumatoid Arthritis. N. Engl. J. Med. 2000;343(21):1520–1528. doi: 10.1056/NEJM200011233432103. [DOI] [PubMed] [Google Scholar]
- 24.Matsui H., Shimokawa O., Kaneko T., Nagano Y., Rai K., Hyodo I. The pathophysiology of non-steroidal anti-inflammatory drug (NSAID)-induced mucosal injuries in stomach and small intestine. J Clin Biochem Nutr. 2011;48(2):107–111. doi: 10.3164/jcbn.10-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murray M.D., Brater D.C. Renal toxicity of the nonsteroidal anti-inflammatory drugs. Annu. Rev. Pharmacol. Toxicol. 1993;33:435–465. doi: 10.1146/annurev.pa.33.040193.002251. [DOI] [PubMed] [Google Scholar]
- 26.Boelsterli U.A. Diclofenac-induced liver injury: a paradigm of idiosyncratic drug toxicity. Toxicol. Appl. Pharmacol. 2003;192(3):307–322. doi: 10.1016/s0041-008x(03)00368-5. [DOI] [PubMed] [Google Scholar]
- 27.Kowalski M.L., Makowska J.S. Seven steps to the diagnosis of NSAIDs hypersensitivity: how to apply a new classification in real practice? Allergy Asthma Immunol Res. 2015;7(4):312–320. doi: 10.4168/aair.2015.7.4.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.J.O.B.F.J.R.A. Chicoine, Non‐steroidal Anti‐inflammatory Drugs in: J.F.K.J.D.M.J. Wang (Ed.), Chemical Analysis of Non‐antimicrobial Veterinary Drug Residues in Food, Wiley Online Library2016, pp. 427-496.
- 29.Warner T.D., Giuliano F., Vojnovic I., Bukasa A., Mitchell J.A., Vane J.R. Nonsteroid drug selectivities for cyclo-oxygenase-1 rather than cyclo-oxygenase-2 are associated with human gastrointestinal toxicity: a full in vitro analysis. Proc Natl Acad Sci U S A. 1999;96(13):7563–7568. doi: 10.1073/pnas.96.13.7563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tacconelli S., Bruno A., Grande R., Ballerini P., Patrignani P. Nonsteroidal anti-inflammatory drugs and cardiovascular safety - translating pharmacological data into clinical readouts. Expert Opin Drug Saf. 2017;16(7):791–807. doi: 10.1080/14740338.2017.1338272. [DOI] [PubMed] [Google Scholar]
- 31.Rao P., Knaus E.E. Evolution of nonsteroidal anti-inflammatory drugs (NSAIDs): cyclooxygenase (COX) inhibition and beyond. J. Pharm. Pharm. Sci. 2008;11(2):81s–110s. doi: 10.18433/j3t886. [DOI] [PubMed] [Google Scholar]
- 32.Wong R.S.Y. Role of Nonsteroidal Anti-Inflammatory Drugs (NSAIDs) in Cancer Prevention and Cancer Promotion. Adv. Pharmacol. Sci. 2019;2019:3418975. doi: 10.1155/2019/3418975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Conaghan P.G. A turbulent decade for NSAIDs: update on current concepts of classification, epidemiology, comparative efficacy, and toxicity. Rheumatol. Int. 2012;32(6):1491–1502. doi: 10.1007/s00296-011-2263-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lees P., Landoni M.F., Giraudel J., Toutain P.L. Pharmacodynamics and pharmacokinetics of nonsteroidal anti-inflammatory drugs in species of veterinary interest. J. Vet. Pharmacol. Ther. 2004;27(6):479–490. doi: 10.1111/j.1365-2885.2004.00617.x. [DOI] [PubMed] [Google Scholar]
- 35.Wyatt J.E., Pettit W.L., Harirforoosh S. Pharmacogenetics of nonsteroidal anti-inflammatory drugs. Pharmacogenomics J. 2012;12(6):462–467. doi: 10.1038/tpj.2012.40. [DOI] [PubMed] [Google Scholar]
- 36.Crofford L.J. Use of NSAIDs in treating patients with arthritis. Arthritis Res. Ther. 2013;15(Suppl 3):S2. doi: 10.1186/ar4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mazaleuskaya L.L., Theken K.N., Gong L., Thorn C.F., FitzGerald G.A., Altman R.B., Klein T.E. PharmGKB summary: ibuprofen pathways. Pharmacogenet. Genom. 2015;25(2):96–106. doi: 10.1097/FPC.0000000000000113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Daly A.K., Aithal G.P., Leathart J.B., Swainsbury R.A., Dang T.S., Day C.P. Genetic susceptibility to diclofenac-induced hepatotoxicity: contribution of UGT2B7, CYP2C8, and ABCC2 genotypes. Gastroenterology. 2007;132(1):272–281. doi: 10.1053/j.gastro.2006.11.023. [DOI] [PubMed] [Google Scholar]
- 39.Jemnitz K., Heredi-Szabo K., Janossy J., Ioja E., Vereczkey L., Krajcsi P. ABCC2/Abcc2: a multispecific transporter with dominant excretory functions. Drug Metab. Rev. 2010;42(3):402–436. doi: 10.3109/03602530903491741. [DOI] [PubMed] [Google Scholar]
- 40.Jedlitschky G., Hoffmann U., Kroemer H.K. Structure and function of the MRP2 (ABCC2) protein and its role in drug disposition. Expert Opin. Drug Metab. Toxicol. 2006;2(3):351–366. doi: 10.1517/17425255.2.3.351. [DOI] [PubMed] [Google Scholar]
- 41.Meunier L., Larrey D. Recent Advances in Hepatotoxicity of Non Steroidal Anti-Inflammatory Drugs. Ann. Hepatol. 2018;17(2):187–191. doi: 10.5604/01.3001.0010.8633. [DOI] [PubMed] [Google Scholar]
- 42.Kuehl G.E., Lampe J.W., Potter J.D., Bigler J. Glucuronidation of nonsteroidal anti-inflammatory drugs: identifying the enzymes responsible in human liver microsomes. Drug Metab. Dispos. 2005;33(7):1027–1035. doi: 10.1124/dmd.104.002527. [DOI] [PubMed] [Google Scholar]
- 43.Wong R.S.Y. Disease-Modifying Effects of Long-Term and Continuous Use of Nonsteroidal Anti-Inflammatory Drugs (NSAIDs) in Spondyloarthritis. Adv Pharmacol Sci. 2019;2019:5324170. doi: 10.1155/2019/5324170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cooper C., Chapurlat R., Al-Daghri N., Herrero-Beaumont G., Bruyere O., Rannou F., Roth R., Uebelhart D., Reginster J.Y. Safety of Oral Non-Selective Non-Steroidal Anti-Inflammatory Drugs in Osteoarthritis: What Does the Literature Say? Drugs Aging. 2019;36(Suppl 1):15–24. doi: 10.1007/s40266-019-00660-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.J. Marjoribanks M. Proctor C. Farquhar R.S. Derks Nonsteroidal anti-inflammatory drugs for dysmenorrhoea Cochrane Database Syst Rev (1) 2010 CD001751. [DOI] [PubMed]
- 46.Lundberg T.R., Howatson G. Analgesic and anti-inflammatory drugs in sports: Implications for exercise performance and training adaptations. Scand. J. Med. Sci. Sports. 2018;28(11):2252–2262. doi: 10.1111/sms.13275. [DOI] [PubMed] [Google Scholar]
- 47.Steckling F.M., Lima F.D., Farinha J.B., Rosa P.C., Royes L.F.F., Cuevas M.J., Bresciani G., Soares F.A., Gonzalez-Gallego J., Barcelos R.P. Diclofenac attenuates inflammation through TLR4 pathway and improves exercise performance after exhaustive swimming. Scand. J. Med. Sci. Sports. 2020;30(2):264–271. doi: 10.1111/sms.13579. [DOI] [PubMed] [Google Scholar]
- 48.Gorski T., Cadore E.L., Pinto S.S., da Silva E.M., Correa C.S., Beltrami F.G., Kruel L.F. Use of NSAIDs in triathletes: prevalence, level of awareness and reasons for use. Br. J. Sports Med. 2011;45(2):85–90. doi: 10.1136/bjsm.2009.062166. [DOI] [PubMed] [Google Scholar]
- 49.Walker L.A., Zambraski E.J., Williams R.F. Widespread Use of Prescription Nonsteroidal Anti-Inflammatory Drugs Among U.S. Army Active Duty Soldiers. Mil. Med. 2017;182(3):e1709–e1712. doi: 10.7205/MILMED-D-16-00183. [DOI] [PubMed] [Google Scholar]
- 50.Pantziarka P., Sukhatme V., Bouche G., Meheus L., Sukhatme V.P. Repurposing Drugs in Oncology (ReDO)-diclofenac as an anti-cancer agent. Ecancermedicalscience. 2016;10:610. doi: 10.3332/ecancer.2016.610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang Z., Chen F., Shang L. Advances in antitumor effects of NSAIDs. Cancer Manag. Res. 2018;10:4631–4640. doi: 10.2147/CMAR.S175212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thun M.J., Henley S.J., Patrono C. Nonsteroidal anti-inflammatory drugs as anticancer agents: mechanistic, pharmacologic, and clinical issues. J. Natl. Cancer Inst. 2002;94(4):252–266. doi: 10.1093/jnci/94.4.252. [DOI] [PubMed] [Google Scholar]
- 53.Ricchi P., Zarrilli R., Di Palma A., Acquaviva A.M. Nonsteroidal anti-inflammatory drugs in colorectal cancer: from prevention to therapy. Br. J. Cancer. 2003;88(6):803–807. doi: 10.1038/sj.bjc.6600829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pallu R. Anti-inflammatory Drugs for Cancer Prevention and Treatment: Emerging Options. J. Cancer Sci. Ther. 2012;4(12) [Google Scholar]
- 55.Hilovska L., Jendzelovsky R., Fedorocko P. Potency of non-steroidal anti-inflammatory drugs in chemotherapy. Mol. Clin. Oncol. 2015;3(1):3–12. doi: 10.3892/mco.2014.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ma R., Yi B., Piazza G.A., Xi Y. Mechanistic Role of MicroRNA in Cancer Chemoprevention by Nonsteroidal Anti-inflammatory Drugs. Curr. Pharmacol Rep. 2015;1(3):154–160. doi: 10.1007/s40495-014-0011-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Izzotti A., Balansky R., Ganchev G., Iltcheva M., Longobardi M., Pulliero A., Camoirano A., D'Agostini F., Geretto M., Micale R.T., La Maestra S., Miller M.S., Steele V.E., De Flora S. Early and late effects of aspirin and naproxen on microRNAs in the lung and blood of mice, either unexposed or exposed to cigarette smoke. Oncotarget. 2017;8(49):85716–85748. doi: 10.18632/oncotarget.20464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li J., Mansmann U.R. Modeling of non-steroidal anti-inflammatory drug effect within signaling pathways and miRNA-regulation pathways. PLoS ONE. 2013;8(8) doi: 10.1371/journal.pone.0072477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ishida J., Konishi M., Ebner N., Springer J. Repurposing of approved cardiovascular drugs. J. Transl. Med. 2016;14:269. doi: 10.1186/s12967-016-1031-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.M.C. Zatelli, A. Luchin, F. Tagliati, S. Leoni, D. Piccin, M. Bondanelli, R. Rossi, E.C. degli Uberti, Cyclooxygenase-2 inhibitors prevent the development of chemoresistance phenotype in a breast cancer cell line by inhibiting glycoprotein p-170 expression, Endocr Relat Cancer 14(4) (2007) 1029-38. [DOI] [PubMed]
- 61.Huang C., Chen Y., Liu H., Yang J., Song X., Zhao J., He N., Zhou C.J., Wang Y., Dong Q. Celecoxib targets breast cancer stem cells by inhibiting the synthesis of prostaglandin E2 and down-regulating the Wnt pathway activity. Oncotarget. 2017;8(70):115254–115269. doi: 10.18632/oncotarget.23250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Blobaum A.L., Marnett L.J. Structural and functional basis of cyclooxygenase inhibition. J. Med. Chem. 2007;50(7):1425–1441. doi: 10.1021/jm0613166. [DOI] [PubMed] [Google Scholar]
- 63.Coyle C., Cafferty F.H., Rowley S., MacKenzie M., Berkman L., Gupta S., Pramesh C.S., Gilbert D., Kynaston H., Cameron D., Wilson R.H., Ring A., Langley R.E. ADD-ASPIRIN: A phase III, double-blind, placebo controlled, randomised trial assessing the effects of aspirin on disease recurrence and survival after primary therapy in common non-metastatic solid tumours. Contemp. Clin Trials. 2016;51:56–64. doi: 10.1016/j.cct.2016.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Joharatnam-Hogan N., Cafferty F., Hubner R., Swinson D., Sothi S., Gupta K., Falk S., Patel K., Warner N., Kunene V., Rowley S., Khabra K., Underwood T., Jankowski J., Bridgewater J., Crossley A., Henson V., Berkman L., Gilbert D., Kynaston H., Ring A., Cameron D., Din F., Graham J., Iveson T., Adams R., Thomas A., Wilson R., Pramesh C.S., Langley R. Aspirin as an adjuvant treatment for cancer: feasibility results from the Add-Aspirin randomised trial. Lancet Gastroenterol. Hepatol. 2019;4(11):854–862. doi: 10.1016/S2468-1253(19)30289-4. [DOI] [PubMed] [Google Scholar]
- 65.Tsoi K.K.F., Ho J.M.W., Chan F.C.H., Sung J.J.Y. Long-term use of low-dose aspirin for cancer prevention: A 10-year population cohort study in Hong Kong. Int. J. Cancer. 2019;145(1):267–273. doi: 10.1002/ijc.32083. [DOI] [PubMed] [Google Scholar]
- 66.Magee D.J., Jhanji S., Poulogiannis G., Farquhar-Smith P., Brown M.R.D. Nonsteroidal anti-inflammatory drugs and pain in cancer patients: a systematic review and reappraisal of the evidence. Br. J. Anaesth. 2019;123(2):e412–e423. doi: 10.1016/j.bja.2019.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shebl F.M., Hsing A.W., Park Y., Hollenbeck A.R., Chu L.W., Meyer T.E., Koshiol J. Non-steroidal anti-inflammatory drugs use is associated with reduced risk of inflammation-associated cancers: NIH-AARP study. PLoS ONE. 2014;9(12) doi: 10.1371/journal.pone.0114633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hendrix A.S., Spoonmore T.J., Wilde A.D., Putnam N.E., Hammer N.D., Snyder D.J., Guelcher S.A., Skaar E.P., Cassat J.E. Repurposing the Nonsteroidal Anti-inflammatory Drug Diflunisal as an Osteoprotective, Antivirulence Therapy for Staphylococcus aureus Osteomyelitis. Antimicrob. Agents Chemother. 2016;60(9):5322–5330. doi: 10.1128/AAC.00834-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lago E.M., Silva M.P., Queiroz T.G., Mazloum S.F., Rodrigues V.C., Carnauba P.U., Pinto P.L., Rocha J.A., Ferreira L.L.G., Andricopulo A.D., de Moraes J. Phenotypic screening of nonsteroidal anti-inflammatory drugs identified mefenamic acid as a drug for the treatment of schistosomiasis. EBioMedicine. 2019;43:370–379. doi: 10.1016/j.ebiom.2019.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ogundeji A.O., Pohl C.H., Sebolai O.M. Repurposing of Aspirin and Ibuprofen as Candidate Anti-Cryptococcus Drugs. Antimicrob. Agents Chemother. 2016;60(8):4799–4808. doi: 10.1128/AAC.02810-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gold B., Pingle M., Brickner S.J., Shah N., Roberts J., Rundell M., Bracken W.C., Warrier T., Somersan S., Venugopal A., Darby C., Jiang X., Warren J.D., Fernandez J., Ouerfelli O., Nuermberger E.L., Cunningham-Bussel A., Rath P., Chidawanyika T., Deng H., Realubit R., Glickman J.F., Nathan C.F. Nonsteroidal anti-inflammatory drug sensitizes Mycobacterium tuberculosis to endogenous and exogenous antimicrobials. Proc Natl Acad Sci U S A. 2012;109(40):16004–16011. doi: 10.1073/pnas.1214188109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chittepu V., Kalhotra P., Osorio-Gallardo T., Gallardo-Velazquez T., Osorio-Revilla G. Repurposing of FDA-Approved NSAIDs for DPP-4 Inhibition as an Alternative for Diabetes Mellitus Treatment: Computational and in Vitro Study. Pharmaceutics. 2019;11(5) doi: 10.3390/pharmaceutics11050238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rawat C., Kukal S., Dahiya U.R., Kukreti R. Cyclooxygenase-2 (COX-2) inhibitors: future therapeutic strategies for epilepsy management. J. Neuroinflammation. 2019;16(1):197. doi: 10.1186/s12974-019-1592-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Joo Y., Kim H.S., Woo R.S., Park C.H., Shin K.Y., Lee J.P., Chang K.A., Kim S., Suh Y.H. Mefenamic acid shows neuroprotective effects and improves cognitive impairment in in vitro and in vivo Alzheimer's disease models. Mol. Pharmacol. 2006;69(1):76–84. doi: 10.1124/mol.105.015206. [DOI] [PubMed] [Google Scholar]
- 75.Wongrakpanich S., Wongrakpanich A., Melhado K., Rangaswami J. A Comprehensive Review of Non-Steroidal Anti-Inflammatory Drug Use in The Elderly. Aging Dis. 2018;9(1):143–150. doi: 10.14336/AD.2017.0306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Thal L.J., Ferris S.H., Kirby L., Block G.A., Lines C.R., Yuen E., Assaid C., Nessly M.L., Norman B.A., Baranak C.C., Reines S.A. A randomized, double-blind, study of rofecoxib in patients with mild cognitive impairment. Neuropsychopharmacology. 2005;30(6):1204–1215. doi: 10.1038/sj.npp.1300690. [DOI] [PubMed] [Google Scholar]
- 77.Cunningham C., Skelly D.T. Non-steroidal anti-inflammatory drugs and cognitive function: are prostaglandins at the heart of cognitive impairment in dementia and delirium? J. Neuroimmune Pharmacol. 2012;7(1):60–73. doi: 10.1007/s11481-011-9312-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhao-Fleming H., Hand A., Zhang K., Polak R., Northcut A., Jacob D., Dissanaike S., Rumbaugh K.P. Effect of non-steroidal anti-inflammatory drugs on post-surgical complications against the backdrop of the opioid crisis. Burns Trauma. 2018;6:25. doi: 10.1186/s41038-018-0128-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Griggs C., Goverman J., Bittner E.A., Levi B. Sedation and Pain Management in Burn Patients. Clin. Plast. Surg. 2017;44(3):535–540. doi: 10.1016/j.cps.2017.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.FitzGerald G.A. Misguided drug advice for COVID-19. Science. 2020;367(6485):1434. doi: 10.1126/science.abb8034. [DOI] [PubMed] [Google Scholar]
- 81.Zarghi A., Arfaei S. Selective COX-2 Inhibitors: A Review of Their Structure-Activity Relationships. Iran J. Pharm. Res. 2011;10(4):655–683. [PMC free article] [PubMed] [Google Scholar]
- 82.Vane J.R. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat. New. Biol. 1971;231(25):232–235. doi: 10.1038/newbio231232a0. [DOI] [PubMed] [Google Scholar]
- 83.Warner T.D., Mitchell J.A. Cyclooxygenase-3 (COX-3): filling in the gaps toward a COX continuum? Proc. Natl. Acad. Sci. U S A. 2002;99(21):13371–13373. doi: 10.1073/pnas.222543099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vane J.R. The fight against rheumatism: from willow bark to COX-1 sparing drugs. J. Physiol. Pharmacol. 2000;51(4 Pt 1):573–586. [PubMed] [Google Scholar]
- 85.Varga Z., Sabzwari S.R.A., Vargova V. Cardiovascular Risk of Nonsteroidal Anti-Inflammatory Drugs: An Under-Recognized Public Health Issue. Cureus. 2017;9(4) doi: 10.7759/cureus.1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Simmons D.L., Levy D.B., Yannoni Y., Erikson R.L. Identification of a phorbol ester-repressible v-src-inducible gene. Proc. Natl. Acad. Sci. U S A. 1989;86(4):1178–1182. doi: 10.1073/pnas.86.4.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Xie W.L., Chipman J.G., Robertson D.L., Erikson R.L., Simmons D.L. Expression of a mitogen-responsive gene encoding prostaglandin synthase is regulated by mRNA splicing. Proc. Natl. Acad. Sci. U S A. 1991;88(7):2692–2696. doi: 10.1073/pnas.88.7.2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chandrasekharan N.V., Dai H., Roos K.L., Evanson N.K., Tomsik J., Elton T.S., Simmons D.L. COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: cloning, structure, and expression. Proc. Natl. Acad. Sci. U S A. 2002;99(21):13926–13931. doi: 10.1073/pnas.162468699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jozwiak-Bebenista M., Nowak J.Z. Paracetamol: mechanism of action, applications and safety concern. Acta Pol. Pharm. 2014;71(1):11–23. [PubMed] [Google Scholar]
- 90.Vane J.R., Bakhle Y.S., Botting R.M. Cyclooxygenases 1 and 2. Annu. Rev. Pharmacol. Toxicol. 1998;38:97–120. doi: 10.1146/annurev.pharmtox.38.1.97. [DOI] [PubMed] [Google Scholar]
- 91.Vane J.R. Introduction: mechanism of action of NSAIDs. Br. J. Rheumatol. 1996;35(Suppl 1):1–3. doi: 10.1093/rheumatology/35.suppl_1.1. [DOI] [PubMed] [Google Scholar]
- 92.Ghosh R., Alajbegovic A., Gomes A.V. NSAIDs and Cardiovascular Diseases: Role of Reactive Oxygen Species. Oxid. Med. Cell. Longev. 2015;2015 doi: 10.1155/2015/536962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.M.W. King, Lipids: The Eicosanoids: Prostaglandins, Leukotrienes, and Thromboxanes, Integrative medical biochemistry : examination and board review, New York : McGraw-Hill Education/Medical2014.
- 94.Smyth E.M., Grosser T., Wang M., Yu Y., FitzGerald G.A. Prostanoids in health and disease. J. Lipid Res. 2009;50(Suppl):S423–S428. doi: 10.1194/jlr.R800094-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gierse J.K., McDonald J.J., Hauser S.D., Rangwala S.H., Koboldt C.M., Seibert K. A single amino acid difference between cyclooxygenase-1 (COX-1) and -2 (COX-2) reverses the selectivity of COX-2 specific inhibitors. J. Biol. Chem. 1996;271(26):15810–15814. doi: 10.1074/jbc.271.26.15810. [DOI] [PubMed] [Google Scholar]
- 96.Rouzer C.A., Marnett L.J. Cyclooxygenases: structural and functional insights. J. Lipid Res. 2009;50(Suppl):S29–S34. doi: 10.1194/jlr.R800042-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kirkby N.S., Zaiss A.K., Urquhart P., Jiao J., Austin P.J., Al-Yamani M., Lundberg M.H., MacKenzie L.S., Warner T.D., Nicolaou A., Herschman H.R., Mitchell J.A. LC-MS/MS confirms that COX-1 drives vascular prostacyclin whilst gene expression pattern reveals non-vascular sites of COX-2 expression. PLoS ONE. 2013;8(7) doi: 10.1371/journal.pone.0069524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mitchell J.A., Kirkby N.S. Eicosanoids, prostacyclin and cyclooxygenase in the cardiovascular system. Br. J. Pharmacol. 2019;176(8):1038–1050. doi: 10.1111/bph.14167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Allaj V., Guo C., Nie D. Non-steroid anti-inflammatory drugs, prostaglandins, and cancer. Cell Biosci. 2013;3(1):8. doi: 10.1186/2045-3701-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Breyer R.M., Bagdassarian C.K., Myers S.A., Breyer M.D. Prostanoid receptors: subtypes and signaling. Annu. Rev. Pharmacol. Toxicol. 2001;41:661–690. doi: 10.1146/annurev.pharmtox.41.1.661. [DOI] [PubMed] [Google Scholar]
- 101.Harizi H. The immunobiology of prostanoid receptor signaling in connecting innate and adaptive immunity. Biomed Res. Int. 2013;2013 doi: 10.1155/2013/683405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sandoval-Acuna C., Lopez-Alarcon C., Aliaga M.E., Speisky H. Inhibition of mitochondrial complex I by various non-steroidal anti-inflammatory drugs and its protection by quercetin via a coenzyme Q-like action. Chem. Biol. Interact. 2012;199(1):18–28. doi: 10.1016/j.cbi.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 103.Murphy M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009;417(1):1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mazumder S., De R., Sarkar S., Siddiqui A.A., Saha S.J., Banerjee C., Iqbal M.S., Nag S., Debsharma S., Bandyopadhyay U. Selective scavenging of intra-mitochondrial superoxide corrects diclofenac-induced mitochondrial dysfunction and gastric injury: A novel gastroprotective mechanism independent of gastric acid suppression. Biochem. Pharmacol. 2016;121:33–51. doi: 10.1016/j.bcp.2016.09.027. [DOI] [PubMed] [Google Scholar]
- 105.Mazumder S., De R., Debsharma S., Bindu S., Maity P., Sarkar S., Saha S.J., Siddiqui A.A., Banerjee C., Nag S., Saha D., Pramanik S., Mitra K., Bandyopadhyay U. Indomethacin impairs mitochondrial dynamics by activating the PKCzeta-p38-DRP1 pathway and inducing apoptosis in gastric cancer and normal mucosal cells. J. Biol. Chem. 2019;294(20):8238–8258. doi: 10.1074/jbc.RA118.004415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Braun F.K., Al-Yacoub N., Plotz M., Mobs M., Sterry W., Eberle J. Nonsteroidal anti-inflammatory drugs induce apoptosis in cutaneous T-cell lymphoma cells and enhance their sensitivity for TNF-related apoptosis-inducing ligand. J, Invest. Dermatol. 2012;132(2):429–439. doi: 10.1038/jid.2011.316. [DOI] [PubMed] [Google Scholar]
- 107.Farrugia G., Balzan R. The proapoptotic effect of traditional and novel nonsteroidal anti-inflammatory drugs in mammalian and yeast cells. Oxid Med Cell Longev. 2013;2013 doi: 10.1155/2013/504230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rayburn E.R., Ezell S.J., Zhang R. Anti-Inflammatory Agents for Cancer Therapy. Mol Cell Pharmacol. 2009;1(1):29–43. doi: 10.4255/mcpharmacol.09.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rosas C., Sinning M., Ferreira A., Fuenzalida M., Lemus D. Celecoxib decreases growth and angiogenesis and promotes apoptosis in a tumor cell line resistant to chemotherapy. Biol. Res. 2014;47:27. doi: 10.1186/0717-6287-47-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tsujimoto S., Mokuda S., Matoba K., Yamada A., Jouyama K., Murata Y., Ozaki Y., Ito T., Nomura S., Okuda Y. The prevalence of endoscopic gastric mucosal damage in patients with rheumatoid arthritis. PLoS ONE. 2018;13(7) doi: 10.1371/journal.pone.0200023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hamid S., Yakoob J., Jafri W., Islam S., Abid S., Islam M. Frequency of NSAID induced peptic ulcer disease. J. Pak. Med. Assoc. 2006;56(5):218–222. [PubMed] [Google Scholar]
- 112.Chatterjee S., Dureja G.P., Kadhe G., Mane A., Phansalkar A.A., Sawant S., Kapatkar V. Cross-Sectional Study for Prevalence of Non-Steroidal Anti-Inflammatory Drug-Induced Gastrointestinal Cardiac and Renal Complications in India: Interim Report. Gastroenterology Res. 2015;8(3–4):216–221. doi: 10.14740/gr658w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Dubois R.W., Melmed G.Y., Henning J.M., Laine L. Guidelines for the appropriate use of non-steroidal anti-inflammatory drugs, cyclo-oxygenase-2-specific inhibitors and proton pump inhibitors in patients requiring chronic anti-inflammatory therapy. Aliment. Pharmacol. Ther. 2004;19(2):197–208. doi: 10.1111/j.0269-2813.2004.01834.x. [DOI] [PubMed] [Google Scholar]
- 114.Fine M. Quantifying the impact of NSAID-associated adverse events. Am. J. Manag. Care. 2013;19(14 Suppl):s267–s272. [PubMed] [Google Scholar]
- 115.Sinha M., Gautam L., Shukla P.K., Kaur P., Sharma S., Singh T.P. Current perspectives in NSAID-induced gastropathy. Mediators Inflamm. 2013;2013 doi: 10.1155/2013/258209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Claria J. Cyclooxygenase-2 biology. Curr. Pharm. Des. 2003;9(27):2177–2190. doi: 10.2174/1381612033454054. [DOI] [PubMed] [Google Scholar]
- 117.Bhala N., Emberson J., Merhi A., Abramson S., Arber N., Baron J.A., Bombardier C., Cannon C., Farkouh M.E., FitzGerald G.A., Goss P., Halls H., Hawk E., Hawkey C., Hennekens C., Hochberg M., Holland L.E., Kearney P.M., Laine L., Lanas A., Lance P., Laupacis A., Oates J., Patrono C., Schnitzer T.J., Solomon S., Tugwell P., Wilson K., Wittes J., Baigent C. Vascular and upper gastrointestinal effects of non-steroidal anti-inflammatory drugs: meta-analyses of individual participant data from randomised trials. Lancet. 2013;382(9894):769–779. doi: 10.1016/S0140-6736(13)60900-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Salvo F., Antoniazzi S., Duong M., Molimard M., Bazin F., Fourrier-Reglat A., Pariente A., Moore N. Cardiovascular events associated with the long-term use of NSAIDs: a review of randomized controlled trials and observational studies. Expert. Opin. Drug. Saf. 2014;13(5):573–585. doi: 10.1517/14740338.2014.907792. [DOI] [PubMed] [Google Scholar]
- 119.Pirlamarla P., Bond R.M. FDA labeling of NSAIDs: Review of nonsteroidal anti-inflammatory drugs in cardiovascular disease. Trends Cardiovasc. Med. 2016;26(8):675–680. doi: 10.1016/j.tcm.2016.04.011. [DOI] [PubMed] [Google Scholar]
- 120.Somasundaram S., Rafi S., Hayllar J., Sigthorsson G., Jacob M., Price A.B., Macpherson A., Mahmod T., Scott D., Wrigglesworth J.M., Bjarnason I. Mitochondrial damage: a possible mechanism of the “topical” phase of NSAID induced injury to the rat intestine. Gut. 1997;41(3):344–353. doi: 10.1136/gut.41.3.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Somasundaram S., Simpson R., Rafi S., Shergill J.K., Bjarnason I., Wrigglesworth J. 2, 4-diamino-6- hydroxy pyrimidine inhibits NSAIDs induced nitrosyl-complex EPR signals and ulcer in rat jejunum. BMC Gastroenterol. 2002;2:8. doi: 10.1186/1471-230X-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Maity P., Bindu S., Dey S., Goyal M., Alam A., Pal C., Mitra K., Bandyopadhyay U. Indomethacin, a non-steroidal anti-inflammatory drug, develops gastropathy by inducing reactive oxygen species-mediated mitochondrial pathology and associated apoptosis in gastric mucosa: a novel role of mitochondrial aconitase oxidation. J. Biol. Chem. 2009;284(5):3058–3068. doi: 10.1074/jbc.M805329200. [DOI] [PubMed] [Google Scholar]
- 123.Maity P., Bindu S., Dey S., Goyal M., Alam A., Pal C., Reiter R., Bandyopadhyay U. Melatonin reduces indomethacin-induced gastric mucosal cell apoptosis by preventing mitochondrial oxidative stress and the activation of mitochondrial pathway of apoptosis. J. Pineal Res. 2009;46(3):314–323. doi: 10.1111/j.1600-079X.2009.00663.x. [DOI] [PubMed] [Google Scholar]
- 124.Pal C., Bindu S., Dey S., Alam A., Goyal M., Iqbal M.S., Maity P., Adhikari S.S., Bandyopadhyay U. Gallic acid prevents nonsteroidal anti-inflammatory drug-induced gastropathy in rat by blocking oxidative stress and apoptosis. Free Radic. Biol. Med. 2010;49(2):258–267. doi: 10.1016/j.freeradbiomed.2010.04.013. [DOI] [PubMed] [Google Scholar]
- 125.Chattopadhyay I., Bandyopadhyay U., Biswas K., Maity P., Banerjee R.K. Indomethacin inactivates gastric peroxidase to induce reactive-oxygen-mediated gastric mucosal injury and curcumin protects it by preventing peroxidase inactivation and scavenging reactive oxygen. Free Radic. Biol. Med. 2006;40(8):1397–1408. doi: 10.1016/j.freeradbiomed.2005.12.016. [DOI] [PubMed] [Google Scholar]
- 126.Biswas K., Bandyopadhyay U., Chattopadhyay I., Varadaraj A., Ali E., Banerjee R.K. A novel antioxidant and antiapoptotic role of omeprazole to block gastric ulcer through scavenging of hydroxyl radical. J. Biol. Chem. 2003;278(13):10993–11001. doi: 10.1074/jbc.M210328200. [DOI] [PubMed] [Google Scholar]
- 127.Bandyopadhyay U., Biswas K., Chatterjee R., Bandyopadhyay D., Chattopadhyay I., Ganguly C.K., Chakraborty T., Bhattacharya K., Banerjee R.K. Gastroprotective effect of Neem (Azadirachta indica) bark extract: possible involvement of H(+)-K(+)-ATPase inhibition and scavenging of hydroxyl radical. Life Sci. 2002;71(24):2845–2865. doi: 10.1016/s0024-3205(02)02143-4. [DOI] [PubMed] [Google Scholar]
- 128.Chattopadhyay I., Nandi B., Chatterjee R., Biswas K., Bandyopadhyay U., Banerjee R.K. Mechanism of antiulcer effect of Neem (Azadirachta indica) leaf extract: effect on H+-K+-ATPase, oxidative damage and apoptosis. Inflammopharmacology. 2004;12(2):153–176. doi: 10.1163/1568560041352257. [DOI] [PubMed] [Google Scholar]
- 129.Fu Y., Wu H.Q., Cui H.L., Li Y.Y., Li C.Z. Gastroprotective and anti-ulcer effects of oxymatrine against several gastric ulcer models in rats: Possible roles of antioxidant, antiinflammatory, and prosurvival mechanisms. Phytother. Res. 2018;32(10):2047–2058. doi: 10.1002/ptr.6148. [DOI] [PubMed] [Google Scholar]
- 130.Hegab R.N., II, Abd-Ellatif M.T. Sadek, The gastroprotective effect of N-acetylcysteine and genistein in indomethacin-induced gastric injury in rats. Can. J. Physiol. Pharmacol. 2018;96(11):1161–1170. doi: 10.1139/cjpp-2017-0730. [DOI] [PubMed] [Google Scholar]
- 131.Ko I.G., Jin J.J., Hwang L., Kim S.H., Kim C.J., Han J.H., Kwak M.S., Yoon J.Y., Jeon J.W. Evaluating the mucoprotective effect of polydeoxyribonucleotide against indomethacin-induced gastropathy via the MAPK/NF-kappaB signaling pathway in rats. Eur. J. Pharmacol. 2020;874 doi: 10.1016/j.ejphar.2020.172952. [DOI] [PubMed] [Google Scholar]
- 132.Bindu S., Mazumder S., Dey S., Pal C., Goyal M., Alam A., Iqbal M.S., Sarkar S., Azhar Siddiqui A., Banerjee C., Bandyopadhyay U. Nonsteroidal anti-inflammatory drug induces proinflammatory damage in gastric mucosa through NF-kappaB activation and neutrophil infiltration: anti-inflammatory role of heme oxygenase-1 against nonsteroidal anti-inflammatory drug. Free Radic. Biol. Med. 2013;65:456–467. doi: 10.1016/j.freeradbiomed.2013.07.027. [DOI] [PubMed] [Google Scholar]
- 133.Lohmander L.S., McKeith D., Svensson O., Malmenas M., Bolin L., Kalla A., Genti G., Szechinski J., Ramos-Remus C. A randomised, placebo controlled, comparative trial of the gastrointestinal safety and efficacy of AZD3582 versus naproxen in osteoarthritis. Ann. Rheum. Dis. 2005;64(3):449–456. doi: 10.1136/ard.2004.023572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Pai R., Szabo I.L., Giap A.Q., Kawanaka H., Tarnawski A.S. Nonsteroidal anti-inflammatory drugs inhibit re-epithelialization of wounded gastric monolayers by interfering with actin Src, FAK, and tensin signaling. Life Sci. 2001;69(25–26):3055–3071. doi: 10.1016/s0024-3205(01)01412-6. [DOI] [PubMed] [Google Scholar]
- 135.Nadatani Y., Watanabe T., Suda W., Nakata A., Matsumoto Y., Kosaka S., Higashimori A., Otani K., Hosomi S., Tanaka F., Nagami Y., Kamata N., Taira K., Yamagami H., Tanigawa T., Hattori M., Fujiwara Y. Gastric acid inhibitor aggravates indomethacin-induced small intestinal injury via reducing Lactobacillus johnsonii. Sci. Rep. 2019;9(1):17490. doi: 10.1038/s41598-019-53559-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sostres C., Gargallo C.J., Lanas A. Nonsteroidal anti-inflammatory drugs and upper and lower gastrointestinal mucosal damage. Arthritis Res. Ther. 2013;15(Suppl 3):S3. doi: 10.1186/ar4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Shin S.J., Noh C.K., Lim S.G., Lee K.M., Lee K.J. Non-steroidal anti-inflammatory drug-induced enteropathy. Intest. Res. 2017;15(4):446–455. doi: 10.5217/ir.2017.15.4.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Maiden L., Thjodleifsson B., Theodors A., Gonzalez J., Bjarnason I. A quantitative analysis of NSAID-induced small bowel pathology by capsule enteroscopy. Gastroenterology. 2005;128(5):1172–1178. doi: 10.1053/j.gastro.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 139.Graham D.Y., Opekun A.R., Willingham F.F., Qureshi W.A. Visible small-intestinal mucosal injury in chronic NSAID users. Clin. Gastroenterol Hepatol. 2005;3(1):55–59. doi: 10.1016/s1542-3565(04)00603-2. [DOI] [PubMed] [Google Scholar]
- 140.Watanabe T., Fujiwara Y., Chan F.K.L. Current knowledge on non-steroidal anti-inflammatory drug-induced small-bowel damage: a comprehensive review. J. Gastroenterol. 2019 doi: 10.1007/s00535-019-01657-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Nadatani Y., Watanabe T., Tanigawa T., Machida H., Okazaki H., Yamagami H., Watanabe K., Tominaga K., Fujiwara Y., Arakawa T. High mobility group box 1 promotes small intestinal damage induced by nonsteroidal anti-inflammatory drugs through Toll-like receptor 4. Am. J. Pathol. 2012;181(1):98–110. doi: 10.1016/j.ajpath.2012.03.039. [DOI] [PubMed] [Google Scholar]
- 142.Jacob M., Foster R., Sigthorsson G., Simpson R., Bjarnason I. Role of bile in pathogenesis of indomethacin-induced enteropathy. Arch. Toxicol. 2007;81(4):291–298. doi: 10.1007/s00204-006-0149-2. [DOI] [PubMed] [Google Scholar]
- 143.Satoh H., Shiotani S., Otsuka N., Hatao K., Nishimura S. Role of dietary fibres, intestinal hypermotility and leukotrienes in the pathogenesis of NSAID-induced small intestinal ulcers in cats. Gut. 2009;58(12):1590–1596. doi: 10.1136/gut.2008.156596. [DOI] [PubMed] [Google Scholar]
- 144.Shimada S., Tanigawa T., Watanabe T., Nakata A., Sugimura N., Itani S., Higashimori A., Nadatani Y., Otani K., Taira K., Hosomi S., Nagami Y., Tanaka F., Kamata N., Yamagami H., Shiba M., Fujiwara Y. Involvement of gliadin, a component of wheat gluten, in increased intestinal permeability leading to non-steroidal anti-inflammatory drug-induced small-intestinal damage. PLoS ONE. 2019;14(2) doi: 10.1371/journal.pone.0211436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Laine L., Curtis S.P., Langman M., Jensen D.M., Cryer B., Kaur A., Cannon C.P. Lower gastrointestinal events in a double-blind trial of the cyclo-oxygenase-2 selective inhibitor etoricoxib and the traditional nonsteroidal anti-inflammatory drug diclofenac. Gastroenterology. 2008;135(5):1517–1525. doi: 10.1053/j.gastro.2008.07.067. [DOI] [PubMed] [Google Scholar]
- 146.Whitfield-Cargile C.M., Cohen N.D., He K., Ivanov I., Goldsby J.S., Chamoun-Emanuelli A., Weeks B.R., Davidson L.A., Chapkin R.S. The non-invasive exfoliated transcriptome (exfoliome) reflects the tissue-level transcriptome in a mouse model of NSAID enteropathy. Sci. Rep. 2017;7(1):14687. doi: 10.1038/s41598-017-13999-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Endo H., Higurashi T., Hosono K., Sakai E., Sekino Y., Iida H., Sakamoto Y., Koide T., Takahashi H., Yoneda M., Tokoro C., Inamori M., Abe Y., Nakajima A. Efficacy of Lactobacillus casei treatment on small bowel injury in chronic low-dose aspirin users: a pilot randomized controlled study. J. Gastroenterol. 2011;46(7):894–905. doi: 10.1007/s00535-011-0410-1. [DOI] [PubMed] [Google Scholar]
- 148.Suzuki T., Masui A., Nakamura J., Shiozawa H., Aoki J., Nakae H., Tsuda S., Imai J., Hideki O., Matsushima M., Mine T., Tamura A., Ohtsu T., Asami Y., Takagi A. Yogurt Containing Lactobacillus gasseri Mitigates Aspirin-Induced Small Bowel Injuries: A Prospective Randomized, Double-Blind, Placebo-Controlled Trial. Digestion. 2017;95(1):49–54. doi: 10.1159/000452361. [DOI] [PubMed] [Google Scholar]
- 149.B. Mortensen C. Murphy J. O'Grady M. Lucey G. Elsafi L. Barry V. Westphal A. Wellejus O. Lukjancenko A.C. Eklund H.B. Nielsen A. Baker A. Damholt J.E.T. van Hylckama Vlieg F. Shanahan M. Buckley Bifidobacteriumbreve Bif195 Protects Against Small-Intestinal Damage Caused by Acetylsalicylic Acid in Healthy Volunteers Gastroenterology 157 3 2019 pp. 637–646 e4. [DOI] [PubMed]
- 150.Benjamin E.J., Muntner P., Alonso A., Bittencourt M.S., Callaway C.W., Carson A.P., Chamberlain A.M., Chang A.R., Cheng S., Das S.R., Delling F.N., Djousse L., Elkind M.S.V., Ferguson J.F., Fornage M., Jordan L.C., Khan S.S., Kissela B.M., Knutson K.L., Kwan T.W., Lackland D.T., Lewis T.T., Lichtman J.H., Longenecker C.T., Loop M.S., Lutsey P.L., Martin S.S., Matsushita K., Moran A.E., Mussolino M.E., O'Flaherty M., Pandey A., Perak A.M., Rosamond W.D., Roth G.A., Sampson U.K.A., Satou G.M., Schroeder E.B., Shah S.H., Spartano N.L., Stokes A., Tirschwell D.L., Tsao C.W., Turakhia M.P., VanWagner L.B., Wilkins J.T., Wong S.S., Virani S.S. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation. 2019;139(10):e56–e528. doi: 10.1161/CIR.0000000000000659. [DOI] [PubMed] [Google Scholar]
- 151.Grosser T., Yu Y., Fitzgerald G.A. Emotion recollected in tranquility: lessons learned from the COX-2 saga. Annu. Rev. Med. 2010;61:17–33. doi: 10.1146/annurev-med-011209-153129. [DOI] [PubMed] [Google Scholar]
- 152.Grosser T., Fries S., FitzGerald G.A. Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J. Clin. Invest. 2006;116(1):4–15. doi: 10.1172/JCI27291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Nissen S.E., Yeomans N.D., Solomon D.H., Luscher T.F., Libby P., Husni M.E., Graham D.Y., Borer J.S., Wisniewski L.M., Wolski K.E., Wang Q., Menon V., Ruschitzka F., Gaffney M., Beckerman B., Berger M.F., Bao W., Lincoff A.M. Cardiovascular Safety of Celecoxib, Naproxen, or Ibuprofen for Arthritis. N. Engl. J. Med. 2016;375(26):2519–2529. doi: 10.1056/NEJMoa1611593. [DOI] [PubMed] [Google Scholar]
- 154.FitzGerald G.A. Imprecision: Limitations to Interpretation of a Large Randomized Clinical Trial. Circulation. 2017;135(2):113–115. doi: 10.1161/CIRCULATIONAHA.116.026324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Garcia Rodriguez L.A., Tacconelli S., Patrignani P. Role of dose potency in the prediction of risk of myocardial infarction associated with nonsteroidal anti-inflammatory drugs in the general population. J. Am. Coll. Cardiol. 2008;52(20):1628–1636. doi: 10.1016/j.jacc.2008.08.041. [DOI] [PubMed] [Google Scholar]
- 156.Moore N. Coronary Risks Associated with Diclofenac and Other NSAIDs: An Update. Drug Saf. 2020;43(4):301–318. doi: 10.1007/s40264-019-00900-8. [DOI] [PubMed] [Google Scholar]
- 157.F. Marsico, S. Paolillo, P.P. Filardi, NSAIDs and cardiovascular risk, J Cardiovasc Med (Hagerstown) 18 Suppl 1: Special Issue on The State of the Art for the Practicing Cardiologist: The 2016 Conoscere E Curare Il Cuore (CCC) Proceedings from the CLI Foundation (2017) e40-e43. [DOI] [PubMed]
- 158.Miner J., Hoffhines A. The discovery of aspirin's antithrombotic effects. Tex. Heart Inst. J. 2007;34(2):179–186. [PMC free article] [PubMed] [Google Scholar]
- 159.Belton O., Byrne D., Kearney D., Leahy A., Fitzgerald D.J. Cyclooxygenase-1 and -2-dependent prostacyclin formation in patients with atherosclerosis. Circulation. 2000;102(8):840–845. doi: 10.1161/01.cir.102.8.840. [DOI] [PubMed] [Google Scholar]
- 160.Behrouz R., Miller C.M. Aspirin and intracerebral hemorrhage: Where are we now? Neurol. Clin. Pract. 2015;5(1):11–16. doi: 10.1212/CPJ.0000000000000089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Rapa S.F., Di Iorio B.R., Campiglia P., Heidland A., Marzocco S. Inflammation and Oxidative Stress in Chronic Kidney Disease-Potential Therapeutic Role of Minerals, Vitamins and Plant-Derived Metabolites. Int. J. Mol. Sci. 2019;21(1) doi: 10.3390/ijms21010263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Bak S., Andersen M., Tsiropoulos I., Garcia Rodriguez L.A., Hallas J., Christensen K., Gaist D. Risk of stroke associated with nonsteroidal anti-inflammatory drugs: a nested case-control study. Stroke. 2003;34(2):379–386. [PubMed] [Google Scholar]
- 163.Gelbenegger G., Postula M., Pecen L., Halvorsen S., Lesiak M., Schoergenhofer C., Jilma B., Hengstenberg C., Siller-Matula J.M. Aspirin for primary prevention of cardiovascular disease: a meta-analysis with a particular focus on subgroups. BMC Med. 2019;17(1):198. doi: 10.1186/s12916-019-1428-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ravach G., Fournier S., Mazzolai L., Nanchen D. Aspirin for primary cardiovascular prevention : the end of an era ? Rev. Med. Suisse. 2020;16(684):459–462. [PubMed] [Google Scholar]
- 165.Fosslien E. Cardiovascular complications of non-steroidal anti-inflammatory drugs. Ann. Clin. Lab. Sci. 2005;35(4):347–385. [PubMed] [Google Scholar]
- 166.Ahmetaj-Shala B., Kirkby N.S., Knowles R., Al'Yamani M., Mazi S., Wang Z., Tucker A.T., Mackenzie L.S., Armstrong P.C., Nusing R.M., Tomlinson J.A., Warner T.D., Leiper J., Mitchell J.A. Reply to letter regarding article, “evidence that links loss of cyclooxygenase-2 with increased asymmetric dimethylarginine: novel explanation of cardiovascular side effects associated with anti-inflammatory drugs”. Circulation. 2015;132(17):e213–e214. doi: 10.1161/CIRCULATIONAHA.115.017260. [DOI] [PubMed] [Google Scholar]
- 167.Ricciotti E., Castro C., Tang S.Y., Briggs W.T.E., West J.A., Malik D., Rhoades S.D., Meng H., Li X., Lahens N.F., Sparks J.A., Karlson E.W., Weljie A.M., Griffin J.L., FitzGerald G.A. Cyclooxygenase-2, Asymmetric Dimethylarginine, and the Cardiovascular Hazard From Nonsteroidal Anti-Inflammatory Drugs. Circulation. 2018;138(21):2367–2378. doi: 10.1161/CIRCULATIONAHA.118.033540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Yu Y., Ricciotti E., Scalia R., Tang S.Y., Grant G., Yu Z., Landesberg G., Crichton I., Wu W., Pure E., Funk C.D., FitzGerald G.A. Vascular COX-2 modulates blood pressure and thrombosis in mice. Sci Transl Med 4(132) 2012:132ra54. doi: 10.1126/scitranslmed.3003787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Ghosh R., Goswami S.K., Feitoza L., Hammock B., Gomes A.V. Diclofenac induces proteasome and mitochondrial dysfunction in murine cardiomyocytes and hearts. Int. J. Cardiol. 2016;223:923–935. doi: 10.1016/j.ijcard.2016.08.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Oh C.K., Ahmed M.S.F., Xie C., Ryu J.H., Choi J., Chung J.S., Roh G.S., Kim H.J., Cho G.J., Choi W.S., Kang S.S., Lee D.H. Microarray analysis of NSAIDs-treated cardiomyocytes to search for genes involved in COX-2 inhibitor cardiotoxicity. Genet. Mol. Res. 2018;17(4):GMR18068. [Google Scholar]
- 171.Lucas G.N.C., Leitao A.C.C., Alencar R.L., Xavier R.M.F., Daher E.F., Silva Junior G.B.D. Pathophysiological aspects of nephropathy caused by non-steroidal anti-inflammatory drugs. J. Bras. Nefrol. 2019;41(1):124–130. doi: 10.1590/2175-8239-JBN-2018-0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Y. Ziya Sener, M. Oksul, Effects of NSAIDs on kidney functions and cardiovascular system, J Clin Hypertens (Greenwich) 22(2) (2020) 302. [DOI] [PMC free article] [PubMed]
- 173.Bensman A. Non-steroidal Anti-inflammatory Drugs (NSAIDs) Systemic Use: The Risk of Renal Failure. Front. Pediatr. 2019;7:517. doi: 10.3389/fped.2019.00517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Ejaz P., Bhojani K., Joshi V.R. NSAIDs and kidney. J. Assoc. Physicians India. 2004;52:632–640. [PubMed] [Google Scholar]
- 175.Rahman S., Malcoun A. Nonsteroidal antiinflammatory drugs, cyclooxygenase-2, and the kidneys. Prim. Care. 2014;41(4):803–821. doi: 10.1016/j.pop.2014.09.001. [DOI] [PubMed] [Google Scholar]
- 176.Dixit M., Doan T., Kirschner R., Dixit N. Significant Acute Kidney Injury Due to Non-steroidal Anti-inflammatory Drugs: Inpatient Setting. Pharmaceuticals (Basel) 2010;3(4):1279–1285. doi: 10.3390/ph3041279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Clave S., Rousset-Rouviere C., Daniel L., Tsimaratos M. The Invisible Threat of Non-steroidal Anti-inflammatory Drugs for Kidneys. Front. Pediatr. 2019;7:520. doi: 10.3389/fped.2019.00520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Burukoglu D., Baycu C., Taplamacioglu F., Sahin E., Bektur E. Effects of nonsteroidal anti-inflammatory meloxicam on stomach, kidney, and liver of rats. Toxicol. Ind. Health. 2016;32(6):980–986. doi: 10.1177/0748233714538484. [DOI] [PubMed] [Google Scholar]
- 179.MacIntyre D.E., Bushfield M., MacMillan L.J., Moffat K.J., Murdoch F.A., Thomson A., Rossi A.G., McNicol A. Receptors and receptor mechanisms of endogenous platelet stimulatory and inhibitory mediators. Agents Actions Suppl. 1986;20:45–62. [PubMed] [Google Scholar]
- 180.Lapi F., Azoulay L., Yin H., Nessim S.J., Suissa S. Concurrent use of diuretics, angiotensin converting enzyme inhibitors, and angiotensin receptor blockers with non-steroidal anti-inflammatory drugs and risk of acute kidney injury: nested case-control study. BMJ. 2013;346 doi: 10.1136/bmj.e8525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Correction to: Cyclooxygenase-2 Selectively Controls Renal Blood Flow Through a Novel PPARbeta/delta-Dependent Vasodilator Pathway, Hypertension 71(3) (2018) e10. [DOI] [PMC free article] [PubMed]
- 182.Sowers J.R., White W.B., Pitt B., Whelton A., Simon L.S., Winer N., Kivitz A., van Ingen H., Brabant T., Fort J.G. The Effects of cyclooxygenase-2 inhibitors and nonsteroidal anti-inflammatory therapy on 24-hour blood pressure in patients with hypertension, osteoarthritis, and type 2 diabetes mellitus. Arch. Intern. Med. 2005;165(2):161–168. doi: 10.1001/archinte.165.2.161. [DOI] [PubMed] [Google Scholar]
- 183.Lefebvre C., Hindie J., Zappitelli M., Platt R.W., Filion K.B. Non-steroidal anti-inflammatory drugs in chronic kidney disease: a systematic review of prescription practices and use in primary care. Clin Kidney J. 2020;13(1):63–71. doi: 10.1093/ckj/sfz054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Ratliff B.B., Abdulmahdi W., Pawar R., Wolin M.S. Oxidant Mechanisms in Renal Injury and Disease. Antioxid. Redox Signal. 2016;25(3):119–146. doi: 10.1089/ars.2016.6665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Basivireddy J., Jacob M., Pulimood A.B., Balasubramanian K.A. Indomethacin-induced renal damage: role of oxygen free radicals. Biochem. Pharmacol. 2004;67(3):587–599. doi: 10.1016/j.bcp.2003.09.023. [DOI] [PubMed] [Google Scholar]
- 186.Fattori V., Borghi S.M., Guazelli C.F.S., Giroldo A.C., Crespigio J., Bussmann A.J.C., Coelho-Silva L., Ludwig N.G., Mazzuco T.L., Casagrande R., Verri W.A., Jr. Vinpocetine reduces diclofenac-induced acute kidney injury through inhibition of oxidative stress, apoptosis, cytokine production, and NF-kappaB activation in mice. Pharmacol. Res. 2017;120:10–22. doi: 10.1016/j.phrs.2016.12.039. [DOI] [PubMed] [Google Scholar]
- 187.Prado N., Messias G.C., Santos Junior G.O., Nunes V.S., Schinonni M.I., Parana R. Prospective Monitoring of Drug Use: Drug-Induced Liver Injury in a Primary Healthcare Center. Arq. Gastroenterol. 2019;56(4):390–393. doi: 10.1590/S0004-2803.201900000-73. [DOI] [PubMed] [Google Scholar]
- 188.Nonsteroidal Antiinflammatory Drugs (NSAIDs), (2012). [PubMed]
- 189.Bessone F. Non-steroidal anti-inflammatory drugs: What is the actual risk of liver damage? World J. Gastroenterol. 2010;16(45):5651–5661. doi: 10.3748/wjg.v16.i45.5651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Lee W.M. Drug-induced hepatotoxicity. N. Engl. J. Med. 2003;349(5):474–485. doi: 10.1056/NEJMra021844. [DOI] [PubMed] [Google Scholar]
- 191.Sriuttha P., Sirichanchuen B., Permsuwan U. Hepatotoxicity of Nonsteroidal Anti-Inflammatory Drugs: A Systematic Review of Randomized Controlled Trials. Int J Hepatol. 2018;2018:5253623. doi: 10.1155/2018/5253623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.O'Connor N., Dargan P.I., Jones A.L. Hepatocellular damage from non-steroidal anti-inflammatory drugs. QJM. 2003;96(11):787–791. doi: 10.1093/qjmed/hcg138. [DOI] [PubMed] [Google Scholar]
- 193.Zoubek M.E., Lucena M.I., Andrade R.J., Stephens C. Systematic review: ibuprofen-induced liver injury. Aliment. Pharmacol. Ther. 2020;51(6):603–611. doi: 10.1111/apt.15645. [DOI] [PubMed] [Google Scholar]
- 194.Bessone F., Hernandez N., Roma M.G., Ridruejo E., Mendizabal M., Medina-Caliz I., Robles-Diaz M., Lucena M.I., Andrade R.J. Hepatotoxicity induced by coxibs: how concerned should we be? Expert Opin Drug Saf. 2016;15(11):1463–1475. doi: 10.1080/14740338.2016.1225719. [DOI] [PubMed] [Google Scholar]
- 195.Kwon J., Kim S., Yoo H., Lee E. Nimesulide-induced hepatotoxicity: A systematic review and meta-analysis. PLoS ONE. 2019;14(1) doi: 10.1371/journal.pone.0209264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Masubuchi Y., Yamada S., Horie T. Possible mechanism of hepatocyte injury induced by diphenylamine and its structurally related nonsteroidal anti-inflammatory drugs. J. Pharmacol. Exp. Ther. 2000;292(3):982–987. [PubMed] [Google Scholar]
- 197.Bort R., Ponsoda X., Jover R., Gomez-Lechon M.J., Castell J.V. Diclofenac toxicity to hepatocytes: a role for drug metabolism in cell toxicity. J. Pharmacol. Exp. Ther. 1999;288(1):65–72. [PubMed] [Google Scholar]
- 198.Masubuchi Y., Nakayama S., Horie T. Role of mitochondrial permeability transition in diclofenac-induced hepatocyte injury in rats. Hepatology. 2002;35(3):544–551. doi: 10.1053/jhep.2002.31871. [DOI] [PubMed] [Google Scholar]
- 199.Doi H., Horie T. Salicylic acid-induced hepatotoxicity triggered by oxidative stress. Chem. Biol. Interact. 2010;183(3):363–368. doi: 10.1016/j.cbi.2009.11.024. [DOI] [PubMed] [Google Scholar]
- 200.Bolder U., Trang N.V., Hagey L.R., Schteingart C.D., Ton-Nu H.T., Cerre C., Elferink R.P., Hofmann A.F. Sulindac is excreted into bile by a canalicular bile salt pump and undergoes a cholehepatic circulation in rats. Gastroenterology. 1999;117(4):962–971. doi: 10.1016/s0016-5085(99)70356-2. [DOI] [PubMed] [Google Scholar]
- 201.Ahmad M.H., Fatima M., Hossain M., Mondal A.C. Evaluation of naproxen-induced oxidative stress, hepatotoxicity and in-vivo genotoxicity in male Wistar rats. J. Pharm. Anal. 2018;8(6):400–406. doi: 10.1016/j.jpha.2018.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Souto E.O., Miyoshi H., Dubois R.N., Gores G.J. Kupffer cell-derived cyclooxygenase-2 regulates hepatocyte Bcl-2 expression in choledocho-venous fistula rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2001;280(5):G805–G811. doi: 10.1152/ajpgi.2001.280.5.G805. [DOI] [PubMed] [Google Scholar]
- 203.Rymer M.M. Hemorrhagic stroke: intracerebral hemorrhage. Mo. Med. 2011;108(1):50–54. [PMC free article] [PubMed] [Google Scholar]
- 204.Park K., Bavry A.A. Risk of stroke associated with nonsteroidal anti-inflammatory drugs. Vasc Health Risk Manag. 2014;10:25–32. doi: 10.2147/VHRM.S54159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.T. Schink, B. Kollhorst, C. Varas Lorenzo, A. Arfe, R. Herings, S. Lucchi, S. Romio, R. Schade, M.J. Schuemie, H. Straatman, V. Valkhoff, M. Villa, M. Sturkenboom, E. Garbe, Risk of ischemic stroke and the use of individual non-steroidal anti-inflammatory drugs: A multi-country European database study within the SOS Project, PLoS One 13(9) (2018) e0203362. [DOI] [PMC free article] [PubMed]
- 206.Choi N.K., Park B.J., Jeong S.W., Yu K.H., Yoon B.W. Nonaspirin nonsteroidal anti-inflammatory drugs and hemorrhagic stroke risk: the Acute Brain Bleeding Analysis study. Stroke. 2008;39(3):845–849. doi: 10.1161/STROKEAHA.107.497040. [DOI] [PubMed] [Google Scholar]
- 207.Islam M.M., Poly T.N., Walther B.A., Yang H.C., Lin M.C., Li Y.C. Risk of Hemorrhagic Stroke in Patients Exposed to Nonsteroidal Anti-Inflammatory Drugs: A Meta-Analysis of Observational Studies. Neuroepidemiology. 2018;51(3–4):166–176. doi: 10.1159/000490741. [DOI] [PubMed] [Google Scholar]
- 208.L. Ge, X. Ouyang, C. Ban, H. Yu, Q. Wu, H. Wu, J. Liang, Cerebral microbleeds in patients with ischemic cerebrovascular disease taking aspirin or clopidogrel, Medicine (Baltimore) 98(9) (2019) e14685. [DOI] [PMC free article] [PubMed]
- 209.Li K.L., Lee A.Y., Abuzeid W.M. Aspirin Exacerbated Respiratory Disease: Epidemiology, Pathophysiology, and. Management, Med Sci (Basel) 2019;7(3) doi: 10.3390/medsci7030045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.White A.A., Stevenson D.D. Aspirin-Exacerbated Respiratory Disease. N. Engl. J. Med. 2018;379(11):1060–1070. doi: 10.1056/NEJMra1712125. [DOI] [PubMed] [Google Scholar]
- 211.White A.A., Doherty T.A. Role of group 2 innate lymphocytes in aspirin-exacerbated respiratory disease pathogenesis. Am J Rhinol Allergy. 2018;32(1):7–11. doi: 10.2500/ajra.2018.32.4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Voiriot G., Philippot Q., Elabbadi A., Elbim C., Chalumeau M., Fartoukh M. Risks Related to the Use of Non-Steroidal Anti-Inflammatory Drugs in Community-Acquired Pneumonia in Adult and Pediatric Patients. J Clin Med. 2019;8(6) doi: 10.3390/jcm8060786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Wen Y.C., Hsiao F.Y., Chan K.A., Lin Z.F., Shen L.J., Fang C.C. Acute Respiratory Infection and Use of Nonsteroidal Anti-Inflammatory Drugs on Risk of Acute Myocardial Infarction: A Nationwide Case-Crossover Study. J. Infect. Dis. 2017;215(4):503–509. doi: 10.1093/infdis/jiw603. [DOI] [PubMed] [Google Scholar]
- 214.Dauletbaev N., Lam J., Eklove D., Iskandar M., Lands L.C. Ibuprofen modulates NF-kB activity but not IL-8 production in cystic fibrosis respiratory epithelial cells. Respiration. 2010;79(3):234–242. doi: 10.1159/000255342. [DOI] [PubMed] [Google Scholar]
- 215.Lewandowska-Polak A., Brauncajs M., Jarzebska M., Pawelczyk M., Kurowski M., Makowska J., Kowalski M.L. Parainfluenza virus infection enhances NSAIDs-induced inhibition of PGE2 generation and COX-2 expression in human airway epithelial cells. Adv. Med. Sci. 2019;64(2):338–343. doi: 10.1016/j.advms.2019.04.004. [DOI] [PubMed] [Google Scholar]
- 216.Yan X., Hao Q., Mu Y., Timani K.A., Ye L., Zhu Y., Wu J. Nucleocapsid protein of SARS-CoV activates the expression of cyclooxygenase-2 by binding directly to regulatory elements for nuclear factor-kappa B and CCAAT/enhancer binding protein. Int. J. Biochem. Cell Biol. 2006;38(8):1417–1428. doi: 10.1016/j.biocel.2006.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 217.Russell B., Moss C., Rigg A., Van Hemelrijck M. COVID-19 and treatment with NSAIDs and corticosteroids: should we be limiting their use in the clinical setting? Ecancermedicalscience. 2020;14:1023. doi: 10.3332/ecancer.2020.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Jaya PreethiPeesa P.R.Y. Arun Rasheed, Venkata Basaveswara Rao Mandava, A perspective review on role of novel NSAID prodrugs in the management of acute inflammation. J. Acute Dis. 2016;5(5):364–381. [Google Scholar]
- 219.T.J.A. Maureen Younan Jeffrey, Fudin, A Practical Approach to Discontinuing NSAID Therapy Prior to a Procedure (Accessed 10 2013 13).
- 220.Qandil A.M. Prodrugs of nonsteroidal anti-inflammatory drugs (NSAIDs), more than meets the eye: a critical review. Int. J. Mol. Sci. 2012;13(12):17244–17274. doi: 10.3390/ijms131217244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Mahmud T., Rafi S.S., Scott D.L., Wrigglesworth J.M., Bjarnason I. Nonsteroidal antiinflammatory drugs and uncoupling of mitochondrial oxidative phosphorylation. Arthritis Rheum. 1996;39(12):1998–2003. doi: 10.1002/art.1780391208. [DOI] [PubMed] [Google Scholar]
- 222.Makhija D.T., Somani R.R., Chavan A.V. Synthesis and pharmacological evaluation of antiinflammatory mutual amide prodrugs. Indian J Pharm Sci. 2013;75(3):353–357. doi: 10.4103/0250-474X.117399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Shah K., Gupta J.K., Chauhan N.S., Upmanyu N., Shrivastava S.K., Mishra P. Prodrugs of NSAIDs: A Review. Open Med. Chem. J. 2017;11:146–195. doi: 10.2174/1874104501711010146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Rao P.P., Kabir S.N., Mohamed T. Nonsteroidal Anti-Inflammatory Drugs (NSAIDs): Progress in Small Molecule Drug Development. Pharmaceuticals (Basel) 2010;3(5):1530–1549. doi: 10.3390/ph3051530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Fiorucci S., Santucci L. Hydrogen sulfide-based therapies: focus on H2S releasing NSAIDs. Inflamm. Allergy Drug Targets. 2011;10(2):133–140. doi: 10.2174/187152811794776213. [DOI] [PubMed] [Google Scholar]
- 226.Wallace J.L. Nitric oxide in the gastrointestinal tract: opportunities for drug development. Br. J. Pharmacol. 2019;176(2):147–154. doi: 10.1111/bph.14527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Fiorucci S., Santucci L., Distrutti E. NSAIDs, coxibs, CINOD and H2S-releasing NSAIDs: what lies beyond the horizon. Dig Liver Dis. 2007;39(12):1043–1051. doi: 10.1016/j.dld.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 228.Wallace J.L., Vaughan D., Dicay M., MacNaughton W.K., de Nucci G. Hydrogen Sulfide-Releasing Therapeutics: Translation to the Clinic. Antioxid. Redox Signal. 2018;28(16):1533–1540. doi: 10.1089/ars.2017.7068. [DOI] [PubMed] [Google Scholar]
- 229.Wallace J.L., Nagy P., Feener T.D., Allain T., Ditroi T., Vaughan D.J., Muscara M.N., de Nucci G., Buret A.G. A proof-of-concept, Phase 2 clinical trial of the gastrointestinal safety of a hydrogen sulfide-releasing anti-inflammatory drug. Br. J. Pharmacol. 2020;177(4):769–777. doi: 10.1111/bph.14641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Zheng Y., Ji X., Ji K., Wang B. Hydrogen sulfide prodrugs-a review. Acta Pharm Sin B. 2015;5(5):367–377. doi: 10.1016/j.apsb.2015.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Pan L.L., Qin M., Liu X.H., Zhu Y.Z. The Role of Hydrogen Sulfide on Cardiovascular Homeostasis: An Overview with Update on Immunomodulation. Front. Pharmacol. 2017;8:686. doi: 10.3389/fphar.2017.00686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Lobb I., Sonke E., Aboalsamh G., Sener A. Hydrogen sulphide and the kidney: important roles in renal physiology and pathogenesis and treatment of kidney injury and disease. Nitric Oxide. 2015;46:55–65. doi: 10.1016/j.niox.2014.10.004. [DOI] [PubMed] [Google Scholar]
- 233.Davis A., Robson J. The dangers of NSAIDs: look both ways. Br. J. Gen. Pract. 2016;66(645):172–173. doi: 10.3399/bjgp16X684433. [DOI] [PMC free article] [PubMed] [Google Scholar]