

Abstract
The close phylogenetic relationship between Ciona robusta and vertebrates makes it a powerful model for studying innate immunity and the evolution of immune genes. To elucidate the nature and dynamics of the immune response, the molecular mechanisms by which bacterial infection is detected and translated into inflammation and how potential pattern recognition receptors (PRRs) are involved in pathogen recognition in tunicate C. robusta (formerly known as Ciona intestinalis), we applied an approach combining bacterial infections, next-generation sequencing, qRT-PCR, bioinformatics and in silico analyses (criteria of a p-value < 0.05 and FDR < 0.05). A STRING analysis indicated a functional link between components of the Tlr/MyD88-dependent signalling pathway (Tlr2, MyD88, and Irak4) and components of the Nf-κB signalling pathway (Nf-κB, IκBα, and Ikkα) (p-value < 0.05, FDR < 0.05). A qRT-PCR analysis of immune genes selected from transcriptome data revealed Mif as more frequently expressed in the inflammatory response than inflammation mediator or effector molecules (e.g., Il-17s, Tnf-α, Tgf-β, Mmp9, Tlrs, MyD88, Irak4, Nf-κB, and galectins), suggesting close interplay between Mif cytokines and Nf-κB signalling pathway components in the biphasic activation of the inflammatory response. An in silico analyses of the 3′-UTR of Tlr2, MyD88, IκBα, Ikk, and Nf-κB transcripts showed the presence of GAIT elements, which are known to play key roles in the regulation of immune gene-specific translation in humans. These findings provide a new level of understanding of the mechanisms involved in the regulation of the C. robusta inflammatory response induced by LPS and suggest that in C. robusta, as in humans, a complex transcriptional and post-transcriptional control mechanism is involved in the regulation of several inflammatory genes.
Subject terms: Inflammation, Computational biology and bioinformatics, Cellular signalling networks, Computational models, Gene ontology, High-throughput screening, Phylogeny
Introduction
The innate immune system is an integrated system of biological processes and structures in vertebrates, invertebrates and plants that recognise and fight pathogens1,2. This defence mechanism relies on several classes of immune receptors, collectively named pattern recognition receptors (PRRs), which sense pathogen-associated molecular patterns (PAMPs) and induce sophisticated signalling and behaviours, based on dynamic feedback-regulated interactions among a number of components (genes, transcripts, metabolites and proteins). PAMPs include lipopolysaccharides (LPSs), bacterial lipoproteins and lipopeptides, peptidoglycans, mannose-rich glycans, flagellin, bacterial and viral nucleic acids, double stranded (ds) and single stranded (ss) RNA3. Among the PRRs, Toll-like receptors (TLRs) the most ancient family, recognize the most extensive spectrum of pathogens4.
In recent decades, Ciona robusta, the closest living relative of vertebrates, has become a model in various fields of biology, serving as a particularly powerful model for studying innate immunity5–8. These chordate invertebrates are equipped with an innate immune system that trigger humoral and cellular responses9. It has been widely proven that in C. robusta10–13, LPS induces an inflammatory response in the pharynx (a haematopoietic organ)14–18. C. robusta has three TLR genes, and only two, called Tlr1 and Tlr2, have been cloned19. They present “hybrid” functionality, reflecting mammalian TLRs, and are canonically divided into two subfamilies that primarily recognise related PAMPs: (i) TLR1, TLR2, TLR4 and TLR6 recognise lipids, whereas (ii) TLR3, TLR7, TLR8 and TLR9 recognise nucleic acids20,21. TLR signalling leads to cell signalling cascades through the myeloid differentiation primary response 88 protein (MyD88), the first molecule activated in the TLR signalling pathway22,23 and a central node in inflammatory pathways24. MyD88 binds IL-1R-associated kinase family (IRAK) members through one of its well-conserved Toll-interleukin-1-receptor domains, thus leading to the activation of nuclear factor-kappa B (NF-κB)25, which, in turn, leads to the expression of proinflammatory cytokines, such as tumour necrosis factor-α (TNF-α) and interferon (IFN)21. The NF-κB transcription factor family, which is highly conserved from Drosophila to humans, includes five members, RelA/p65, c-Rel, RelB, NF-κB1 (p50), and NF-κB2 (p52), which form homo- and heterodimers. A highly conserved DNA-binding/dimerization domain called the Rel homology domain (RHD) is involved in the formation of Rel/NF-κB dimer proteins25,26. Normally, NF-κB proteins are stored and sequestered in the cytoplasm by κB family members, characterised by Ankyrin (ANK) repeats and a solenoid fold, which are usually implicated in specific protein–protein interactions25,26. Two major signalling pathways are involved in the activation of NF-κB: the canonical and non-canonical (or alternative) pathways are both involved in regulating immune and inflammatory responses27–29. In the canonical pathway in the absence of stimuli, NF-κB transcription factors are bound to IκBs, sequestered in the cytoplasm, with consistently inactive transcription. An appropriate stimulus activates a protein kinase complex (IKK), consisting of IKKα, IKKβ and IKKγ in humans, and in turn phosphorylates IκB, leading to complex degradation and allowing the translocation of NF-κB complexes (predominantly the p50/RelA dimer) from the cytoplasm to the nucleus30,31. Non-canonical NF-κB activation does not require IκBα degradation but depends on the processing of the NF-κB2 precursor protein p100, which has yet to be identified in invertebrate organisms27. In C. robusta, one gene encodes the respective Nf-κB, IκB, and Ikk proteins, and two different isoforms of Rel have been identified32,33.
In recent years, the number of functions attributed to the human macrophage migration inhibitory factor (MIF) cytokine has increased significantly, positioning MIF as among the most frequently expressed proteins in the inflammatory process34,35. MIF is produced and stored intracellularly in different cells of the immune system, including monocytes, macrophages and T cells, in the pituitary gland36. MIF has received substantial attention as a mediator of innate and adaptive immune responses37, and it has been implicated in many infectious, inflammatory and immune diseases, including rheumatoid arthritis, atherosclerosis and tumorigenesis38–41. MIF effects are mediated via an autocrine/paracrine signalling pathways, leading to the activation of ERK1/ERK2 MAP kinases, triggering pro-inflammatory gene (e.g., TNFα, IL-1, IL-6, IL-8, and IL-12) and matrix metalloprotease gene expression and upregulating TLR4 expression35,41–44. Recently, two macrophage migration inhibitory factor (Mif) genes in C. robusta have been cloned: Mif1 and Mif245.
Although genes known to be involved in the immune response, such as TLR, NF-κB and MIF, are expressed in C. robusta, the wide-ranging nature and temporal dynamics of immune signalling in C. robusta during LPS exposure in vivo remain unclear.
In the present study, using an in vivo LPS exposure strategy, next-generation sequencing (NGS) and qRT-PCR combined with bioinformatics and in silico analyses, we compared whole pharynx transcripts from naïve and LPS-exposed C. robusta. This combined approach provided the first indication of the involvement of the canonical Nf-κB pathway in inflammation, with the qRT-PCR analysis specifically suggesting interplay between Mif1 and Nf-κB signalling pathway components through the Tlr/MyD88-dependent pathway (including Tlr2, MyD88, and Irak4) during the biphasic activation of the inflammatory response upon LPS exposure. Furthermore, the presence of cis-acting GAIT elements in the 3′ untranslated region (UTR) of Tlr2, MyD88, IκB, Ikkα, and Nf-κB suggests the presence of both transcriptional and posttranscriptional control mechanisms involved in gene expression during the LPS-mediated inflammatory response of C. robusta.
Results
Transcriptomic analysis of Ciona robusta highlights the effects of LPS on transcripts involved in inflammation
To profile the C. robusta inflammatory response under LPS exposure in vivo, we investigated the differential gene expression in its immunocompetent organ (pharynx) under physiological and challenged conditions (4-h LPS exposure) using NGS transcriptomic analysis. Transcriptomic analysis under physiological conditions revealed 16,504 detected sequences, including protein-coding transcripts, non-protein-coding transcripts, isoforms, novel transcripts and protein-RNA interaction sites (data not shown). All 16,504 genes produced by sequencing were annotated using the Ensembl database (ensemble.org), and for each of them, log-fold changes, p-values and adjusted p-values were calculated (data not shown). With the log-fold change set at 1.5, 1,227 genes were found to be differentially expressed (p-value threshold < 0.05, and adjusted p-value < 0.05). A comparison between the basal gene expression of three specimens showed physiological inter-individual variability (Fig. 1a, compared with controls, columns 4, 5 and 6), but a large set of responsive genes (1,227) were found to be reproducibly regulated after 4 h of LPS exposure in vivo (p-value ≥ 1.5-fold higher or lower than controls) (Fig. 1a, columns 1, 2 and 3). Notably, the transcript levels of 447 target genes were found to be downregulated in response to LPS, whereas 780 genes were upregulated (Fig. 1b). For functional annotations, unigenes (1,227) were aligned to Gene Ontology (GO) terms based on the Protein ANalysis THrough Evolutionary Relationships (PANTHER) (pantherdb.org) classification system for gene ontology annotations, classified into three subcategories: (i) GO molecular functions (Fig. 2a); (ii) GO cellular components (Fig. 2b); (iii) GO biological processes (Fig. 2c).
Figure 1.

(a) Heatmaps of 1,227 differentially expressed unigenes (DEGs) in C. robusta after 4 h of LPS treatment. The figure shows LPS-treated (columns 1, 2 and 3) and untreated samples (columns 4, 5 and 6) in triplicate. (b) Volcano plot of differentially expressed DEGs in the C. robusta pharynx based on the comparison of the 4-h-LPS-treated to the untreated libraries. The expression level for each unigene is included in the plot. Blue points represent DEGs (up- and downregulated); red points represent non-DEGs. The y-axis indicates − log10 (p-value), and the x-axis indicates log2 (fold change). Genes significantly (p-value < 0.05) up- or downregulated (log2-fold change < 1.5 or log2-fold change > 1.5) are displayed in blue.
Figure 2.

Gene enrichment analysis of differentially expressed genes, according to the three representative classes of GO: (a) molecular functions; (b) cellular components; (c) biological processes, for which the overrepresentation of immune system progress is shown in red. GO classes were filtered for p-value < 0.05 and FDR correction < 0.05 tests, according to the PANTHER GO-slim analysis tool.
Genes responsive to LPS were found to be predominantly involved in molecular functions leading to gene expression and regulation (e.g., transcription regulatory region DNA binding, DNA binding, RNA binding), transporter activities (e.g., amino acid transmembrane transporter activity) and monosaccharide-binding activities (Fig. 2a). Gene expression programmes, which establish and maintain specific cell states, are controlled by transcription factors, cofactors, chromatin regulators and noncoding RNAs that interact with regulatory regions. Notably, genes involved in DNA-binding transcription activity were found to be significantly downregulated (3.5-fold decrease) upon LPS exposure, whereas those involved in amino acid transmembrane transporter and monosaccharide-binding activities were found to be upregulated (1.5- and 2.5-fold increase) (Fig. 2a). Concordantly, genes specific to cellular components were expressed at lower levels than the respective control group (e.g., processes involving the Golgi apparatus, nucleoplasm, chromosome, chromatin) (Fig. 2b). Within the biological processes category, the RNA-processing group was found to be strongly reduced (3.8-fold decrease) (Fig. 2c). On the other hand, the response to xenobiotic stimuli and drug processes was found to be moderately increased, whereas organic acid metabolic processes and the immune system processes were found to be slightly increased. The pathway analysis of differentially expressed unigenes with PANTHER revealed different pathways grouped into different categories (e.g., inflammation and immune response, growth factor/growth factor receptor-mediated pathways and intracellular signalling activation, apoptosis and p53 regulation, and metabolism). The first categories listed herein, according to GO analysis results, suggest a functional link among LPS inflammatory processes, cytokines, Tgf-β and Toll receptor signalling pathways (Table 1). STRING-protein–protein interaction networks functional analysis (string-db.org) was used to predict interactions among Tlr/MyD88-dependent signalling pathways and components of Nf-κB signalling by generating network clustered that are visualised according to the k-means algorithm (clustering analysis). The STRING analysis showed a greater functional link between Tlr2 and MyD88 (interaction score of 0.946) than was shown for Tlr1 and MyD88 (interaction score of 0.795) and suggested a strong functional link between Tlrs/Myd88 and Nf-κB, which was confirmed by the identification of two distinct functional clusters: the first included Tlr1, Tlr2, MyD88 and Irak4, and the second included Nf-κB, IκB, IKKα and Rel 1 (Fig. 3).
Table 1.
Pathway analysis obtained with PANTHER tool: pathways were grouped in eight different categories, based on their potential relation.
| Pathway id | Pathway name | Pathway category |
|---|---|---|
| P00031 | Inflammation mediated by chemokine and cytokine signaling pathway | Inflammation and immune response |
| P00047 | PDGF signaling pathway | |
| P00052 | TGF-β signaling pathway | |
| P00054 | Toll receptor signaling pathway | |
| P00057 | Wnt signaling pathway | |
| P06211 | BMP signaling pathway | |
| P06215 | MYO signaling pathway | |
| P06217 | Drosophila melanogaster Toll pathway | |
| P00053 | T cell activation | |
| P00010 | B cell activation | |
| P06959 | CCKR signaling | Growth factors/growth factors receptors mediated pathways and intracellular signaling activation |
| P00012 | Cadherin signaling pathway | |
| P00018 | EGF receptor signaling pathway | |
| P00019 | Endothelin signaling pathway | |
| P00034 | Integrin signaling pathway | |
| P00021 | FGF signaling pathway | |
| P00056 | VEGF signaling pathway | |
| P04393 | Ras Pathway | |
| P00032 | Insulin/IGF pathway-mitogen activated protein kinase kinase/MAP kinase cascade | |
| P00032 | Insulin/IGF pathway-protein kinase B signaling cascade | |
| P00016 | Cytoskeletal regulation by Rho GTPase | Molecular switches |
| P00026 | Heterotrimeric G-protein signaling pathway-Gi α and Gs alpha mediated pathway | |
| P00027 | Heterotrimeric G-protein signaling pathway-Gq alpha and Go alpha mediated pathway | |
| P00028 | Heterotrimeric G-protein signaling pathway-rod outer segment photo transduction | |
| P00005 | Angiogenesis | Angiogenesis |
| P00011 | Blood coagulation | |
| P00006 | Apoptosis signaling pathway | Apoptosis and p53 regulation |
| P00059 | p53 pathway | |
| P04397 | p53 pathway feedback loops 2 | |
| P05731 | p53 pathway by glucose deprivation | |
| P00030 | Hypoxia response via HIF activation | |
| P00014 | Cholesterol biosynthesis | Metabolism |
| P00024 | Glycolysis | |
| P00032 | Insulin/IGFpathway-mitogen activated protein kinase kinase/MAP kinase cascade | |
| P00032 | Insulin/IGFpathway-protein kinase B signaling cascade | |
| P00037 | Ionotropic glutamate receptor pathway | |
| P00039 | Metabotropic glutamate receptor group III pathway | |
| P02744 | Fructose galactose metabolism | |
| P02730 | Asparagine and aspartate biosynthesis | |
| P02762 | Pentose phosphate pathway | |
| P00042 | Muscarinic acetylcholine receptor 1 and 3 signaling pathway | Nervous system |
| P00043 | Muscarinic acetylcholine receptor 2 and 4 signaling pathway | |
| P00044 | Nicotinic acetylcholine receptor signaling pathway | |
| P00003 | Alzheimer disease-amyloid secretase pathway | |
| P00004 | Alzheimer disease-presenilin pathway | |
| P00007 | Axon guidance mediated by semaphorins | |
| P00008 | Axon guidance mediated by Slit/Robo | |
| P00009 | Axon guidance mediated by netrin | |
| P00049 | Parkinson disease | |
| P00029 | Huntington | |
| P05731 | GABA-B receptor II signaling | |
| P05912 | Dopamine receptor mediated signaling pathway | |
| P00001 | Adrenaline and noradrenaline biosynthesis | |
| P06212 | DPP-SCW signaling pathway | Drosophila melanogaster development related |
| P06213 | DPP signaling pathway | |
| P06214 | GBB signaling pathway | |
| P06216 | SCW signaling pathway | |
| P00045 | Notch signaling pathway |
Figure 3.
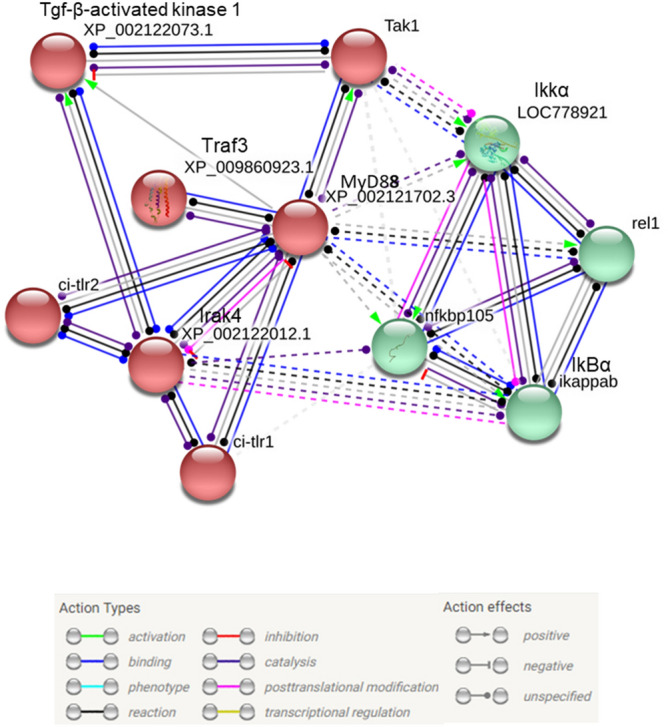
The Tlr-MyD88-dependent and Nf-κB pathways in C. robusta: protein–protein interaction network (PPIN) analysis of linked proteins by STRING. Nodes represent proteins, and edges represent direct and indirect interactions between proteins. Interesting edges are indicated by dashed lines. Clustering analysis was performed using the k-means algorithm, and two different clusters are identified in red and green. Edges represent predicted molecular action between network molecules (action types and effects are shown in the figure).
Phylogenetic and in silico analyses of the Tlr/MyD88-dependent and Nf-κB canonical signalling pathways
As previously noted, C. robusta possesses three TLR genes, and only two, called Tlr1 and Tlr2, have been cloned19. Tlr proteins present the “hybrid” functionality of mammalian TLRs: Tlr1 (NP_001159599.2) shows 23% identity and 42% sequence similarity with the structurally and functionally well-characterised human TLR5, whereas Tlr2 (NP_001159600.1) shows 27% identity and 43% sequence similarity with human TLR8. In silico analysis was performed using the RegRNA2.0 database (https://regrna.mbc.nctu.edu.tw/html/prediction.html) to identify functional RNA motifs and elements in the 3′ (UTR) involved in Tlr mRNA post-transcriptional regulation. We found that the Tlr1 3′-UTR contains a γ-interferon activated inhibitor of translation (GAIT) element, a Mos polyadenylation response element (MOS-PRE) (Fig. 4a). GAIT elements are implicated in several immune-related mRNAs, showing important roles in gene-specific translation control in innate immunity; MOS-PRE is involved in both the translational repression of mRNA stored in immature oocytes and translational induction in maturing oocytes in X. laevis; in contrast, the Tlr2 3′-UTR contains a GAIT element, a Musashi-binding element (MBE), previously identified as being involved in mRNA translational control during cell cycle progression, and an AU rich element (ARE) (Fig. 4a).
Figure 4.

(a) Analysis of 3′-UTR mRNA sequences of Tlr1, Tlr2 and MyD88 using the RegRNA web tool: MOS-PRE (polyadenylation response element), GAIT (interferon-γ-activated inhibitor of translation), ARE (AU-rich element), MBE (Musashi-binding element); (b) phylogenetic tree of the MyD88 family of vertebrates and invertebrates. The tree was constructed using the neighbour-joining method and bootstrap analysis. The bootstrap value indicates the number of particular node occurrences per 1,000 trees, as generated by bootstrapping the sequences. The bar indicates the number of amino acid residue substitutions per site.
The C. robusta MyD88 gene has not yet been cloned, and to isolate full-length mRNA, we used a RACE 5′ and 3′ strategy. MyD88 mRNA presents a 5′-UTR of 57 bp, an ORF of 1,210 bp and a 3′-UTR of 593 bp, which contains an MBE and GAIT element (Fig. 4a). It encodes a 403 amino acid polypeptide with a predicted molecular size of 45.169 kDa and a pI of 5.15 (Supplementary Fig. 1).
In silico analysis of the MyD88 amino acid sequence performed using both the Delta-Blast algorithm and the Simple Modular Architecture Research Tool (SMART) (smart.embl-heidelberg.de) jointly predicted the presence of specific structural domains: a DEATH domain (from 105 to 196 aa) and a Toll/interleukin-1 receptor (TIR) homology domain (from 265 to 399 aa) (Supplementary Fig. 1).
The C. robusta myD88-deduced amino acid sequences were examined in GenBank through Basic Local Alignment Search Tool (BLAST) (blast.ncbi.nlm.nih.gov) analysis and showed 28% identity and 48% sequence similarity with human MyD88. Molecular Evolutionary Genetics Analysis (MEGA X) software was used to build a phylogenetic tree by the neighbour-joining method (Fig. 4b). Phylogenetic analyses of vertebrate and invertebrate MyD88 proteins supported the idea of a conserved evolution from a common MyD88 ancestral gene among invertebrates, protochordates and vertebrates. The functionally and structurally conserved MyD88 motifs highlighted by CLC workbench 6.4 are shown in the supplementary material (Supplementary Fig. 2).
STRING-protein–protein interaction networks functional analysis suggested that in C. robusta, the major signalling pathways involved in the activation of Nf-κB were canonical pathways including Ikkα and IκBα proteins. In C. robusta, Nf-κB (NP_001071772.1) shows 53.36% identity with the structurally and functionally characterised NF-κB1 in humans and 41.49% identity with the Dorsal protein of the Rel subfamily present in D. melanogaster. In silico analysis using the SMART tool was performed to identify protein domains in C. robusta Nf-κB, H. sapiens NF-κB and D. melanogaster Dorsal proteins (Fig. 5a). For C. robusta Nf-κB, the search revealed a Rel homology domain (RHD) (26–215 aa), an IPT domain (222–324 aa), eight Ankyrin domains (797–1,046 aa) and a DEATH domain (1,083–1,170 aa), the same as H. sapiens NF-κB but with a different number of ANK domains (the tunicate Nf-κB has eight, and the human NF-κB has six); in D. melanogaster Dorsal, with a RHD and an IPT domain are present. Figure 5a shows the homodimer molecular model of C. robusta Nf-κB constructed according to the homology-modelling process performed on the basis of the known crystal structure of H. sapiens NF-κB1 (1svc.1.B) (Fig. 5a), generated from the super-imposition of the 2–326 residue sequence, shows 59.81% identity with the template. The phylogenetic tree representing the vertebrate and invertebrate NF-κB family members suggests that C. robusta Nf-κB is an orthologue of vertebrate NF-κB and that, in vertebrates, a single gene was duplicated into two (NF-κB1 and NF-κB2) after the divergence of the tunicate and vertebrate lineages (Fig. 5b).
Figure 5.

(a) Comparison of domain organisation in the D. melanogaster Dorsal, H. sapiens NF-κB1 and C. robusta Nf-κB proteins. The RHD domain is in blue, the ANK domain is in green, and the IPT and DEATH domains are in orange. Three-dimensional structures of C. robusta Nf-κB. (b) Phylogenetic tree of NF-κB and Rel family members in vertebrates and invertebrates. (c) Comparison of domain organisation in D. melanogaster Ikk, C. robusta Ikkα and H. sapiens IKKα. The Pkinase-Tyr domain is in orange, the PHB domain is in grey, and the IKKβ/NEMO-binding domain is in blue. Three-dimensional structures of C. robusta Ikkα. (d) Phylogenetic tree of IKK family members in vertebrates and invertebrates. (e) Comparison of domain organisation in the Cactus protein of D. melanogaster, H. sapiens and C. robusta IκBα. The ANK domain is in green. Three-dimensional structures of C. robusta IκBα. (f) Phylogenetic tree of IκB family members in vertebrates and invertebrates. The trees were constructed by the neighbour-joining method and bootstrap analysis. The bootstrap value indicates the number of particular node occurrences per 1,000 trees, as generated by bootstrapping the sequences. The bar indicates the number of amino acid residue substitutions per site. Nf-κB, Ikkα, and IkBα of C. robusta are highlighted in light blue boxes.
In humans, the activation of the NF-κB “canonical pathway” activates the protein kinase complex IKK30,31; in C. robusta, only one member of the Ikkα (XP_002125567.1) and IκBα (NP_001071739.1) families was identified. C. robusta Ikkα shows 31.53% identity with human IKKα and 32.62% identity with D. melanogaster Ikk. SMART analysis identified one PKinase-Tyr domain in C. robusta Ikkα (17–261 aa), H. sapiens IKKα and D. melanogaster Ikk (Fig. 5c). Moreover, H. sapiens IKKα presents a PHB and an IKKα/NEMO-binding domain, both of which are involved in the formation of the IKK complex and are not present in C. robusta Ikkα or D. melanogaster Ikk. Figure 5c shows the homodimer molecular model of C. robusta Ikkα based on the known crystal structure of the Mus musculus serine/threonine-protein kinase (4jlc.1.A) generated from the super-imposition of the 12–79 residue sequence that shows 21.90% identity with the template. The phylogenetic analysis results support that supposition that C. robusta Ikkα is an orthologue of vertebrate IKKα genes (Fig. 5d). Finally, C. robusta IκBα (NP_001071739.1) shows 35.77% identity with H. sapiens IκBα and 31.60% identity with the Cactus protein of D. melanogaster family members and contains six ANK domains and tandem repeated modules of a 29-residue motif with a canonical helix-loop-helix-β-hairpin/loop folded structure (ANK1: 128–157; ANK2: 164–193; ANK3: 197–226; ANK4: 244–274; ANK5: 279–308; and ANK6: 312–341) (Fig. 5e), differing from H. sapiens IκBα and D. melanogaster Cactus, which each has five ANK domains. Figure 5e shows the homodimer molecular model of C. robusta IκBα based on the known crystal structure of H. sapiens Tankyrase-1 (3utm.1.A) generated from the super-imposition of the 123–367 residue sequence that shows 23.65% identity with the IkBα protein template. The phylogenetic analysis results support the supposition that C. robusta IκBα (Fig. 5f) is an orthologue of vertebrate IκBα genes and has amino-terminal signal-responsive regions containing conserved serine residues (e.g., Ser84 and Ser88) that are phosphorylated in an 83-DSPGXXSP-89 amino acid sequence. An in silico analysis was performed based on RegRNA2.0 databases to identify functional RNA motifs and elements in the 3′-UTR of the Nf-κB, Ikkα and IκBα genes with identified GAIT elements (data not shown).
Analyses of the expression of Tlr/MyD88-dependent and Nf-κB canonical signalling pathways and immune-related genes under LPS exposure
Analyses of the time-course expression of Tlr/MyD88-dependent and Nf-κB canonical signalling pathway immune-related genes in the pharynx inflammatory response induced by LPS in C. robusta were performed at time points from 0 to 72 h post-LPS challenge by qRT-PCR (Fig. 6). The heatmap shows that a large portion of the transcripts was significantly modulated in response to LPS during the 72-h period of LPS exposure (p-value < 0.05). Based on the expression patterns of the transcripts, two major clusters were highlighted: the first includes proinflammatory cytokines and components of MyD88/Nf-κB signalling, and the second comprises the Mif2 cytokine IκBα and a few inflammation effectors (e.g., lectins, lysozymes, and Po). Specifically, the heatmap highlighted that the inflammatory cytokines Mif1, Il-17–1, Il-17–3, and Tnf-α and the metalloprotease Mmp9 were upregulated between 1 and 4 h of LPS exposure (p-value < 0.05). Tnf-α reached its maximum expression level after 2 h of LPS exposure. Notably, Tlr2, MyD88, Irak4, Nf-κB and transforming growth factor β (Tgf-β) transcripts displayed a significant increase after 4 h of LPS exposure (p-value < 0.05). On the other hand, after 8 h of exposure, Il-17–1, Il-17–2, and Mmp9 levels began to increase s they had at 1 h. Tlr2, MyD88, Irak4, Nf-κB and Tgf-β experienced a second significant increase after 48 h of exposure (p-value < 0.05). Our findings suggest biphasic activation of the inflammatory response upon LPS exposure, with a first wave of activation from 0 to 12 h and a second wave of activation from 12 to 72 h.
Figure 6.
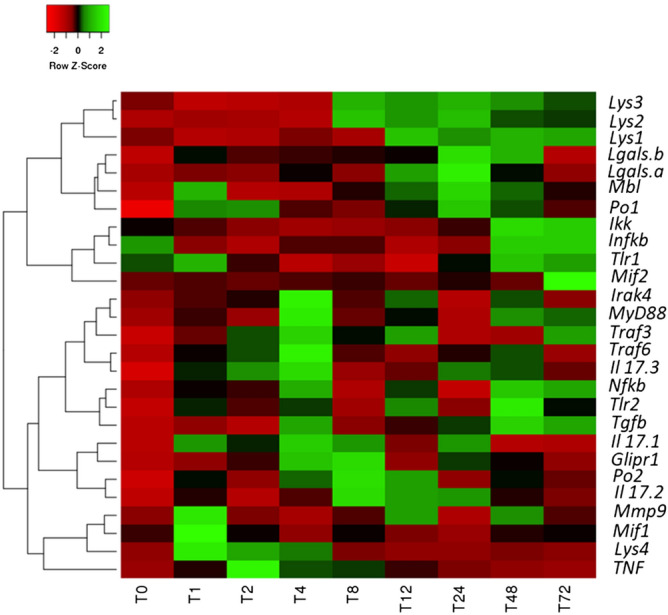
Heatmap based on the qRT-PCR analysis of the differentially expressed Tlr/MyD88-dependent and Nf-κB pathway and immuno-related genes at different times of exposure to LPS (1–72 h). Time course of gene expression in the pharynx of C. robusta exposed to LPS compared with the gene expression in untreated ascidians. Values are presented as the means ± SD. Statistical differences were estimated by one-way ANOVA and Tukey's t-test. The level of significance was set at a p-value ≤ 0.05 (N = 4).
Discussion
In this study, we found that the innate immune signalling pathway activated by LPS in C. robusta is evolutionarily conserved and involves both the Tlr and Mif signalling pathways, findings in agreement with reports highlighting the key immune signalling pathways activated against invading microbial pathogens and other potential threats to a mammalian host46. In general, these findings are not surprising, since this invertebrate occupies a key phylogenetic position in chordate evolution and is considered to be a member of the sister group of vertebrates5–7. In our model, the Mif signalling pathway seems to be activated as the first line of defence against the bacterial endotoxin LPS and appears to be a major regulator of Il-17s, Tgf-β and Mmp9 transcription, as these levels were found to be upregulated after 1 h of exposure. In contrast, the Nf-κB signalling pathway seems to be recruited a few hours later to activate Tgf-β through the involvement of Tlr2, MyD88 and Irak4.
In humans, LPS induces TLR4 activation through binding with three proteins (the lipopolysaccharide-binding protein LBP, the cluster differentiation antigen CD14, and the myeloid differentiation protein receptor MD-2)47. In humans, MD-2 is a component of the MD-2-related lipid-recognition (ML) superfamily, which contains a large set of genes encoding proteins such as MD-1, MD-2, and Niemann-Pick type C2 (NPC2) protein. The ascidian C. robusta possesses two Tlr-related genes presenting “hybrid” biological and immunological functionality of mammalian TLRs19, but LPS significantly activates only one of them, Tlr2; on the other hand, no MD-2 orthologue was found in the genome of C. robusta. The subtractive hybridization strategy allowed the identification of Npc2 mRNA in C. robusta, an homologue of ML superfamily components, preferentially expressed in haemocytes inside the vessel lumen of the pharynx48. This finding supports the hypothesis that the recognition of LPS by TLR4 through MD-2 binding may have been gained early during the evolution of vertebrates and that C. robusta may respond to LPS through a complex with alternative LPS sensors (i.e., the Ncp2 protein) developed before the differentiation of the MD-2 protein in vertebrates. LPS activation of Tlr2 likely regulates the Rel/Nf-κB signal transduction pathway, and we hypothesise that Tlr1 in is involved in the signalling that activates Mif1 in C. robusta. Notably, the molecular mechanisms underlying Tlr ligand recognition and signal transduction are distinct from those of vertebrate TLRs19. The qRT-PCR results suggest an intriguing possibility of biphasic action of the Nf-κB and Mif signalling pathways during the 72 h of exposure to LPS, suggesting their different roles in regulating immune cell behaviour, respectively, with a pro-inflammatory action period (from 0 to 12 h) and an anti-inflammatory action period at which time homeostasis is restored (from 12 to 72 h).
Recently, a few studies have indicated that, in humans, intracellular MIF is involved in the induction of NF-κB activity49, reinforcing the idea of a close relationship between Mifs, Tlrs and Nf-κB in C. robusta. In agreement, Roger et al.50 found that MIF upregulates Tlr4 expression in mouse macrophages and activates the MAPK and NF-κB signalling pathways in immortalised cell lines and mouse macrophages46. In humans, the upregulation of MIF during inflammation and under other stress conditions is primarily regulated at the level of MIF release rather than transcriptional induction51, which explains its rapid activation. In C. robusta, Mif2 is not regulated by LPS, and its 3′-UTR mRNA has a CPE element, confirming that it can be stored in the cytoplasm for rapid activation by a cytoplasmic polyadenylation mechanism; instead, Mif1 is upregulated after LPS stimulation and does not have a 3′-UTR CPE45. Moreover, the in silico analysis of the 3′-UTR of Tlr1 revealed a MOS-PRE that includes the 5′ sequence of the CPE52. Both the CPE and MOS-PRE enable translational repression or activation in response to specific stimuli; therefore, we can hypothesise that, following LPS stimulation, Mif1 and Tlr1 transcripts gain poly(A) tails, which activate their translation and allow the rapid and abundant expression of proteins for their prompt response.
In humans, MIF can regulate TNF and other cytokines by affecting the expression of TLR4, p53, ERK, c-Jun-N-terminal kinase, p38, and MAPK phosphatase-153. On the other hand, the stimulation of dendritic cells with TLR4 ligands elicits high levels of MIF production, and secreted MIF acts as an autocrine/paracrine enhancer of TNF production. Our findings support the notion that there is interplay between Mifs and Nf-κB pathway components in C. robusta, meaning that they have an effect on each other. Specifically, we speculate that to actively maintain the immune response and increase its efficiency, Mif2 and Tlr1 are involved in a rapid response (within a few minutes) and stimulate the transcription of Mmp9 and the pro-inflammatory cytokines Mif1, Tgf-β, Il-17 and Tnf-α, which, in turn, activate Nf-κB through Tlr2, eliciting renewed Mif2 activation until immune response resolution. In silico analyses of the cis-regulatory elements of the 3′-UTR in C. robusta based on RegRNA 2.0 revealed a GAIT element in Tlr2, MyD88, IκB, Ikkα, and Nf-κB. In humans, GAIT elements are implicated in several immune-related mRNAs, showing an important role in gene-specific translation control in innate immunity54. In C. robusta, GAIT elements have already been identified in Cap55 and Tnf-α56 and in Mif mRNAs45. Notably, in C. robusta, Mifs also seem to regulate Mmp9, which has several important immune functions, such as extracellular matrix degradation and Tnf-α and Tgf-β activation57. In humans, TGF-β is a crucial enforcer of immune homeostasis and tolerance, inhibiting the expansion and function of many immune system components. It can be viewed as immunosuppressive and plays important roles both during the initial phase of immune injury and during the later remodelling phase58. In agreement with these findings, Tgf-β was overexpressed in C. robusta after 4 h of LPS exposure and later, from 12 to 72 h, thus regulating the resolution of the inflammatory response.
Finally, we propose a novel schematic model representing the putative interplay between the Mif and Nf-κB pathway components (Fig. 7). To the best of our knowledge, this is the first study to show evidence of interplay between the Mif and Nf-κB pathways using a marine invertebrate model through a wide-ranging approach. Further studies in this direction are needed to cover knowledge gaps regarding the hierarchically organised set of molecular, cellular and organismal networks involved in universal immune interactions with pathogens and subsequent intracellular signal transduction.
Figure 7.

Schematic model representing the putative interplay between the Mif and Nf-κB pathways in C. robusta. After an external stimulus such as LPS treatment, Tlrs activated intracellular signalling such as that by MyD88 and Irak4 proteins. Mif1 is involved in the activation of the Nf-κB transcription factor through the downregulation of the inhibitory IκB protein and the upregulation of Tlr2. In the cytoplasm, the signalling cascade activated by MyD88 causes Nf-κB activation through Ikk molecules. IκBα is then ubiquitinated, and Nf-κB is translocated to the nucleus, where it activates mRNA transcription of inflammatory genes. Cytokines, metalloproteases, adhesion molecules and other inflammatory proteins are then produced, activating the inflammatory response.
Methods
Tunicates and LPS injection
Molecular studies have led to the hypothesis that Ciona intestinalis constitutes a compilation of species rather than a single species10–13. Accordingly, our model organism, originally from the Mediterranean Sea and formerly classified as C. intestinalis, corresponds to C. robusta10–13.
C. robusta specimens were collected from Sciacca Harbour (Sicily, Italy) and were acclimatised and maintained under controlled temperature conditions (15 °C) in tanks supplied with flow-through oxygenated seawater. The animals were fed every 2 days with Coraliquid marine invertebrate food (Sera Heinsberg, Germany). An LPS solution (Escherichia coli 055:B5, LPS, SIGMA-ALDRICH, Germany) was prepared in sterile salt medium (12 mM CaCl2, 11 mM KCl, 26 mM MgCl2, 43 mM Tris HCl, 0.4 M NaCl, pH 8.0). One hundred microlitres of the LPS-containing suspension was injected into the tunic matrix surrounding the pharynx wall (median body region) at a final LPS concentration of 100 μg. C. robusta not exposed to LPS (naïve) were used as controls. Fragments of pharynx tissue (200 mg) explanted at various times (from 1 to 72 h) were immediately soaked in RNAlater tissue collection solution (AMBION, Austin, TX) and stored at − 80 °C. Total RNA extraction was performed using an RNAqueous-Midi kit purification system (AMBION, Austin, TX).
Transcriptomics
The RNA purity and quality of total RNA extracted from the pharynx of C. robusta that were naïve (N = 3) and that were exposed to LPS for 4 h (N = 3) were examined by NanoDrop and Agilent RNA 6,000 Nano kits on an Agilent 2,100 Bioanalyzer (AGILENT, USA), respectively. High-quality RNA samples (A260/A280 = 1.9–2.1, RIN ≥ 7) were used for cDNA library construction. RNA sequencing (RNA-Seq) was performed by BMR Genomics (Padua, Italy) on an Illumina platform in a single-end format 75 bp (1 × 75 bp) containing ~ 40 million ± 10% of reads/sample. An analysis of differentially expressed genes was performed by BMR Genomics (Padua, Italy) using edgeR software, which estimates the negative binomial variance parameter globally across all genes. All data from the edgeR analysis were normalised by setting the false discovery rate (FDR) to ≤ 0.05 and the absolute value of the log fold change (logFC) to 1.5. A heatmap and a volcano plot were generated to visualise the results indicating the genes differentially expressed between the exposed samples and controls. The heatmap was produced using a heatmapper tool (https://www.heatmapper.ca). Complete linkage clustering was applied, and Spearman’s rank correlation was used as the method of distance measurement. In the volcano plot, the log2CPM, which represents the log base 2 of the count per million (CPM) for each transcript, was calculated by the CPM function. Negative log values represent transcripts presenting a CPM lower than 1, and positive log values represent those presenting a CPM ≥ 1.
Gene functional enrichment analysis and immune network construction
The Clusters of Orthologous Genes (COG) database (geneontology.org) was used to perform a Gene Ontology (GO) enrichment analysis of a set of C. robusta transcripts produced by NGS. The ontology analysis was divided into three subcategories: (i) molecular functions (MF); (ii) cellular components (CC); (iii) and biological pathways (BP). The Protein Analysis Through Evolutionary Relationships (PANTHER GO-slim analysis tool) System connected to the COG database was utilised to expand the annotation data of the gene and protein families obtained from GO. A GO analysis was performed by selecting the PANTHER “GO-slim” analysis mode, as it is more reliable and accurate than the GO annotation mode “GO-complete”. Different parameters used to measure the functional role of the list of genes were taken into account and reported as fold enrichment. In addition to p-value evaluation (p-value < 0.05), false discovery rate (FDR < 0.05) control procedures were also considered. The COG database was also used to perform pathway analysis of the NGS data. The Search Tool for the Retrieval of Interacting Genes/Protein (STRING) database, which allows the visualisation of complex networks (through clustering analysis), was used to retrieve the predicted interactions for the identified immune-related proteins. Levels of significance were set at a p-value < 0.05; the log2-fold change was </> 1.5. Significant data (proteins significantly modulated) were clustered according to the k-means algorithm, an unsupervised clustering algorithm based on an adjacency matrix.
Cloning and bioinformatics analyses of MyD88
The Ensembl automatic annotation of the genome sequence (https://www.ensembl.org/) was used to identify the following sequence: MyD88 (ENSCING00000017616). MyD88 cDNA was obtained using the GeneRacer kit (INVITROGEN, USA), as reported in Vizzini et al.45. In short, 5′- and 3′-RACE was directed by the listed primers reported in supplementary Table 1; the overlapping RACE was cloned into a pCRII vector (TA cloning kit, INVITROGEN, USA) and sequenced. cDNA was amplified by polymerase chain reaction (PCR), and the amplicon was cloned into a pCRII vector (TA cloning kit, INVITROGEN, USA) and sequenced. The full-length MyD88 cDNA was analysed with the ExPASy translation tool (https://web.expasy.org/translate/) to obtain the open reading frame (ORF) regions, the leader and trailer sequences (UTRs) and the nucleotide sequence, which was translated into a protein sequence. The protein sequence was located using the Basic Local Alignment Search Tool (BLAST, NCBI) to identify known protein sequences that are homologous to MyD88, and the physical/chemical parameters (e.g., molecular mass and isoelectric point) were estimated by the Prot-Param tool in ExPASy (https://www.expasy.org/tool/protparam/). The NCBI Conserved Domain database (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) was used to predict the domain architecture, and other conserved domains and functional motifs were determined by the PROSITE database (https://prosite.expasy.org/scanprosite/). The SignalP 4.0 server (https://www.cbs.dtu.dk/services/SignalP/) was used to predict the putative cleavage site of the signal peptide, and CLC Genomics Workbench software (Version 7.0.0) was used for the multiple sequence alignment.
Structural and phylogenetic analyses
As reported in Vizzini et al.45, secondary structures were evaluated using Polyview software (https://polyview.cchmc.org), and 3D structures were predicted using Phyre2 (Protein Homology/analogY Recognition Engine V 2.0; https://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index). Structures were validated by Ramachandran plot analysis (https://mordred.bioc.cam.ac.uk/∼rapper/rampage.php). Homology modelling was performed on the basis of the known crystal structures. Phylogenetic trees were designed in MEGA X maintaining the bootstrap value of 1,000 bootstrap iterations (neighbour-joining method). The accession numbers are listed in Table 2. The Regulatory RNA Motifs and Elements Finder tool (https://regrna.mbc.nctu.edu.tw/html/prediction.html) was used to characterise the 3′-UTRs.
Table 2.
Accession numbers.
| Name | GenBank no. |
|---|---|
| Salmo salar MyD88 | ABV59003.1 |
| Oncorhynchus mykiss MyD88 | NP_001117893.1 |
| Xenopus laevis MyD88 | NP_001081001.1 |
| Paralichthys olivaceus MyD88 | XP_019943061.1 |
| Branchiostoma belcheri MyD88 | XP_019623569.1 |
| Gallus gallus MyD88 | NP_001292020.1 |
| Latimeria chalumnae MyD88 | XP_005994282.1 |
| Sus scrofa MyD88 | NP_001093393.1 |
| Danio rerio MyD88 | NP_997979.2 |
| Bos taurus MyD88 | NP_001014404.1 |
| Homo sapiens MyD88 | AAC50954.1 |
| Mus musculus MyD88 | AAC53013.1 |
| Drosophila melanogaster MyD88 | NP_610479.1 |
| Haliotis diversicolor MyD88 | AHK60398.1 |
| Mytilus galloprovincialis MyD88c | AGG10812.1 |
| Mytilus galloprovincialis MyD88a | AFR54116.1 |
| Mytilus galloprovincialis MyD88b | AGG10811.1 |
| Amphimedon queenslandica MyD88 | ADR78337.1 |
| Culex quinquefasciatus MyD88 | EDS31829.1 |
| Hydra vulgaris MyD88 | CDG67778.1 |
| Spodoptera frugiperda MyD88 | AFK24444.1 |
| Ciona IKKα | NP_001071740.1 |
| Homo sapiens IKKα | NP_001269.3 |
| Mus musculus IKKα | NP_031726.2 |
| Danio rerio Ikkα | NP_956611.1 |
| Xenopus laevis Ikkα | NP_001086127.1 |
| Rattus norvegicus IKKα | NP_001101058.1 |
| Gallus gallus IKKα | NP_001012922.1 |
| Rattus norvegicus IKKβ | NP_445807.2 |
| Mus musculus IKKβ | NP_001153246.1 |
| Danio rerio Ikkβ | NP_001116737.1 |
| Homo sapiens IKKβ | NP_001547.1 |
| Homo sapiens TBK1 | NP_037386.1 |
| Mus musculus TBK1 | NP_062760.3 |
| Danio rerio Tbk1 | NP_001038213.2 |
| Xenopus laevis Tbk1 | NP_001086516.1 |
| Ciona Tbk1 | XP_002125567.1 |
| Homo sapiens IkBα | NP_065390.1 |
| Sus scrofa IkBα | CAA84619.1 |
| Rattus norvegicus IkBα | Q63746.1 |
| Gallus gallus IkBα | Q91974 |
| Drosophila melanogaster Cactus | AAA85908.1 |
| Mus musculus IkBβ | NP_001293151.1 |
| Homo sapiens IkBβ | AAD08997.1 |
| Rattus norvegicus IkBγ | Q6TMG5.1 |
| Bos taurus IkBγ | CAC93688.1 |
| Homo sapiens IkBγ | AAC36330.1 |
| Mus musculus IkBγ | O88522.2 |
| Sus scrofa IkBγ | A9QT41.1 |
| Mus musculus IkBε | NP_032716.2 |
| Homo sapiens IkBε | NP_004547.3 |
| Salmo salar IkBε | ACN11190.1 |
| Haliotis diversicolor IkB | AHM27300 |
| Ciona robusta IkB | NP_001071739.1 |
| Homo sapiens Ankyrin | AAA51732.1 |
| Mus musculus Ankyrin | AAA37236.1 |
| Drosophila melanogaster Ankyrin | AAC37208.1 |
| Homo sapiens c-Rel | CAA52954.1 |
| Mus musculus c-Rel | CAA42817.1 |
| Gallus gallus c-Rel | NP_001161198.1 |
| Xenopus laevis Rel2 | NP_001079306.1 |
| Homo sapiens RelA | AAH33522.1 |
| Mus musculus RelA | AAF82405.1 |
| Xenopus laevis Rel1 | Q04865.1 |
| Gallus gallus RelA | XP_025001046.1 |
| Homo sapiens I-Rel | AAA36127.1 |
| Mus musculus RelB | NP_033072.2 |
| Xenopus laevis RelB | NP_001079335.1 |
| Drosophila melanogaster Dorsal | NP_724052.1 |
| Drosophila melanogaster Dif | AAA28465.1 |
| Homo sapiens NF-kB1 | CAB94757.1 |
| Gallus gallus NF-kB1 | XP_015140904.1 |
| Homo sapiens NF-kB2 | NP_001309863.1 |
| Gallus gallus NF-kB2 | NP_989744.1 |
| Drosophila melanogaster Relish | NP_477094.1 |
| Ciona robusta Nf-kB1 | Q4H348 |
| Ciona robusta Rel1 | NP_001029013.1 |
| Ciona robusta Rel1-A | XP_026692312.1 |
qRT-PCR
The differential expression of 27 LPS immune responsive genes was studied by qRT-PCR using the SYBR-Green method and the specific sets of primers listed in Table 3. qRT-PCR analysis was performed using an APPLIED BIOSYSTEMS 7500 Real-time PCR system45. Differential expression was determine in a 25 µl PCR mixture containing 2 μl of cDNA converted from 250 ng of total RNA, 300 nM primer (forward and reverse), and 12.5 μl of Power SYBR-Green PCR MasterMix (APPLIED BIOSYSTEMS). Amplification specificity was tested by real-time PCR melting analysis. To obtain sample quantification, the 2−ΔΔCt method was used, and the relative changes in gene expression were analysed as described in the APPLIED BIOSYSTEMS Use Bulletin N.2 (P/N 4303859). The transcript levels from different tissues were normalised to that of actin to compensate for variations in the amount of RNA input. Relative expression was determined by dividing the normalised value of the target gene in each tissue by the normalised value obtained from the untreated tissue. To examine the time course of the response, LPS-treated ascidians (N = 4) were examined at incremental post-inoculation time points (1, 2, 4, 8, 12, 24, 48, and 72 h). Untreated ascidians (naïve) (N = 4) were used as controls.
Table 3.
Primers used for qRT-PCR.
| Gene | Primer sequence (5′–3′) |
|---|---|
| Mif1 |
5′-GCTTGCAGCGCTTTTGATG-3′ 5′-AAACGGGTTCCAGAAACTCCTAA-3′ |
| Mif2 |
5′-CCATGAAGCAACGAGGGAAA-3′ 5′-TTCTTGGCTGCGAGTTGGT-3′ |
| Lys-g1 |
5′-AACTTTGTATGGACGCTGCTG-3′ 5′-GCCCCTGCACGACTTTCA-3′ |
| Lys-g2 |
5′-CACGGTGCCGACACAAAGT-3′ 5′-GCGCCTTGTAAAATGTGATCTC-3′ |
| Lys-g3 |
5′-GCAAGCCGCGAAAGCA-3′ 5′-TCACCAAGCCCGTCTTTGTC-3′ |
| Lys-g4 |
5′-CGGCGTAGCCATCGCTTA-3′ 5′-CGGTGGTGTCAGTGTTGTAGAT-3′ |
| Gal-a |
5′-TGTTGAATGGCTTCCACTTGTT-3′ 5′-TCGGATGTTACGCAGAGGTTT-3′ |
| Gal-b |
5′-CTCGGTGTATGTGAACGATGTTC-3′ 5′-CGGTAGAGCGGCACCTTGT-3′ |
| Mbl |
5′-AGCCTTGACGTTCGCAGAGT-3′ 5′-AGCCTTGACGTTCGCAGAGT-3′ |
| Po1 |
5′-ATACCCGGACAAGATCACCATG-3′ 5′-TGGAGAGGTTCTCAGCTGCTTC-3′ |
| Po2 |
5′-CCCCTATTAGAGTGAATGGCCA-3′ 5′-CAAAGAGATCCACTGGTGCAGA-3′ |
| Glipr1 |
5′- GTATCTCCGCGAAGAGTTGG-3′ 5′-TCGGTATAACGTCGCCTCT-3′ |
| Mmp9 |
5′- GACGAGTTCCGCGTAACGTT-3′ 5′- ATGGAATACCGTGCTCTTGTAGGA-3′ |
| Tgf-β |
5′-TTTCAGGGACCCAAAAACGA-3′ 5′-GCCAGCTATAATGACATCCAAGGT-3′ |
| Tnf-α |
5′-GCCTCCCATAGACCGTTGTTAA-3′ 5′-CGGGACACCTTCAGCACAT-3′ |
| Il-17–1 |
5′- GCCGGGAACGTGACAGAA-3′ 5′-GGCATGTTGATTGCGACCTT-3′ |
| Il-17–2 |
5′-GTGTAGCGGGTGCATTGCT-3′ 5′-GGCACCGACTTCCCAACA-3′ |
| Il-17–3 |
5′-CAAAGCGGAGCCTTCAATGT-3′ 5′-GCTTCTTTGCTCGACACTTGTG-3′ |
| Tlr1 |
5′-GCTATCGAGAACCCGCCATA-3′ 5′-AATCACGGGAAGGAAAGCAA-3′ |
| Tlr2 |
5′-CCTGCTTCAAAACCTCCAATCT-3′ 5′-TTTAAGGAAAACGGGTTATTGTACTATG-3′ |
| MyD88 |
5′-TCGGAATGCCTAGAAGTGATTTCT-3′ 5′-TGCGTGAGTCGAAACGTAGATG-3′ |
| Irak4 |
5′-TCCCCCATACACCCTGTACCT-3′ 5′-GCGACTTAAATCCAAGCGAAA-3′ |
| Traf3 |
5′-ACGGGTGAGGTCCGTTAGG-3′ 5′-TGCGGTAATGGGCCAAGT-3′ |
| Traf6 |
5′-TAAACCGCATCCCGGTACTT-3′ 5′-GCGGGTTTTATCCAAATTTACG-3′ |
| IkBα |
5′-TTATGTGGCACTTGGTATGGAGTATT-3′ 5′-CCGGCTGAGTCAGGAACTTC-3′ |
| Ikkα |
5′-AGGCGGTGGCTGAACTGA-3′ 5′-TTCCCCATCGAACAGAAGAAGT-3′ |
| Nf-kB |
5′-GCCGACGTACTGCTTTGCA-3′ 5′-GCCAGCCACCACGATGTT-3′ |
| Actin |
5′-TGATGTTGCCGCACTCGTA-3′ 5′-TCGACAATGGATCCGGT-3′ |
Statistical methods
Statistical assessments of GO term enrichment and pathway analyses were performed by Fisher's exact test in combination with a robust false discovery rate (FDR) correction for multiple testing.
The row p-value and FDR threshold were set as < 0.05.
Minitab 17 statistical software was used for the qRT-PCR data analysis. Statistical differences were estimated by one-way ANOVA, and the significance of differences among groups was determined by Tukey's t-test. The level of significance was set at a p-value ≤ 0.05. The data are presented as the means ± SD (N = 4).
Supplementary information
Author contributions
V.A., A.B., A.V. contributed conception and design of the study and performed the experiments. L.L.P., A.U. performed bioinformatics analysis. A.P. and A.V. analyzed the data and wrote the manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-020-68339-x.
References
- 1.Buchmann K. Evolution of innate immunity: clues from invertebrates via fish to mammals. Front. Immunol. 2014;23:5–459. doi: 10.3389/fimmu.2014.00459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jones JDG, Dangl JL. The plant immune system. Nature. 2006;444:323–329. doi: 10.1038/nature05286. [DOI] [PubMed] [Google Scholar]
- 3.Janeway CA. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb. Symp. Quan. Biol. 1989;54:1–13. doi: 10.1101/SQB.1989.054.01.003. [DOI] [PubMed] [Google Scholar]
- 4.Nie L, Cai SY, Shao JZ, Chen J. Toll-like receptors, associated biological roles, and signaling networks in non-mammals. Front. Immunol. 2018;2:9–1523. doi: 10.3389/fimmu.2018.01523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Delsuc F, Brinkmann H, Chourrout D, Philippe H. Tunicates and not cephalochordates are the closest living relatives of vertebrates. Nature. 2006;439:965–968. doi: 10.1038/nature04336. [DOI] [PubMed] [Google Scholar]
- 6.Tsagkogeorga G, Turon X, Hopcroft RR, Tilak MK, Feldstein T, Shenkar N, Loya Y, Huchon D, Douzery EJ, Delsuc F. An updated 18S rRNA phylogeny of tunicates based on mixture and secondary structure models. BMC Evol. Biol. 2009;9:187. doi: 10.1186/1471-2148-9-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zeng L, Swalla BJ. Molecular phylogeny of the protochordates: chordate evolution. Can. J. Zool. 2005;83:24–33. doi: 10.1139/z05-010. [DOI] [Google Scholar]
- 8.Satoh N. The ascidian tadpole larva: comparative molecular development and genomics. Nat. Rev. Genet. 2003;4:285–295. doi: 10.1038/nrg1042. [DOI] [PubMed] [Google Scholar]
- 9.Khalturin K, Panzer Z, Cooper MD, Bosch TC. Recognition strategies in the innate immune system of ancestral chordates. Mol. Immunol. 2004;41:1077–1087. doi: 10.1016/j.molimm.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 10.Brunetti R, Gissi C, Pennati R, Manni L. Morphological evidence that the molecularly determined Ciona intestinalis type A and type B are different species: Ciona robusta and Ciona intestinalis. J. Zool. Syst. Evol. Res. 2015;53:186–193. doi: 10.1111/jzs.12101. [DOI] [Google Scholar]
- 11.Pennati R, Ficetola GF, Brunetti R, Caicci F, Gasparini F, Griggio F, Sato A, Stach T, Kaul-Strehlow S, Gissi C, Manni L. Morphological differences between larvae of the Ciona intestinalis species complex: hints for a valid taxonomic definition of distinct species. PLoS ONE. 2015;10:e0122879. doi: 10.1371/journal.pone.0122879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Caputi L, Andreakis N, Mastrototaro F, Cirino P, Vassillo M, Sordino P. Cryptic speciation in a model invertebrate chordate. PNAS. 2007;104:9364–9369. doi: 10.1073/pnas.0610158104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iannelli F, Pesole G, Sordino P, Gissi C. Mitogenomics reveals two cryptic species in Ciona intestinalis. Trends Genet. 2007;23:419–422. doi: 10.1016/j.tig.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 14.Vizzini A. Gene expression and regulation of molecules involved in pharynx inflammatory response induced by LPS in Ciona intestinalis. ISJ. 2017;14:119–128. [Google Scholar]
- 15.Parrinello N, Vizzini A, Arizza V, Salerno G, Parrinello D, Cammarata M, Giaramita FT, Vazzana M. Enhanced expression of a cloned and sequenced Ciona intestinalis TNFα-like (CiTNFα) gene during the LPS-induced inflammatory response. Cell. Tissue Res. 2008;334:305–317. doi: 10.1007/s00441-008-0695-4. [DOI] [PubMed] [Google Scholar]
- 16.Vizzini A, Di Falco F, Parrinello D, Sanfratello MA, Mazzarella C, Parrinello N, Cammarata M. Ciona intestinalis interleukin 17-like genes expression is upregulated by LPS challenge. Dev. Comp. Immunol. 2015;48:129–137. doi: 10.1016/j.dci.2014.09.014. [DOI] [PubMed] [Google Scholar]
- 17.Vizzini A, Di Falco F, Parrinello D, Sanfratello MA, Cammarata M. Transforming growth factor β (CiTGF-β) gene expression is induced in the inflammatory reaction of Ciona intestinalis. Dev. Comp. Immunol. 2016;55:102–110. doi: 10.1016/j.dci.2015.10.013. [DOI] [PubMed] [Google Scholar]
- 18.Vizzini A, Parrinello D, Sanfratello MA, Salerno G, Cammarata M, Parrinello N. Inducible galectins are expressed in the inflamed pharynx of the ascidian Ciona intestinalis. Fish Shellfish Immunol. 2012;32:101–109. doi: 10.1016/j.fsi.2011.10.028. [DOI] [PubMed] [Google Scholar]
- 19.Sasaki N, Ogasawara M, Sekiguchi T, Kusumoto S, Satake H. Toll-like receptors of the Ascidian Ciona intestinalis prototypes with hybrid functionalities of vertebrate toll-like receptors. J. Biol. Chem. 2009;284:27336–27343. doi: 10.1074/jbc.M109.032433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.2183/pjab.85.143. [DOI] [PubMed] [Google Scholar]
- 21.Ozinsky A, Underhill DM, Fontenot JD, Hajjar AM, Smith KD, Wilson CB, Schroeder L, Aderem A. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between toll-like receptors. Proc. Natl. Acad. Sci. USA. 2000;97:13766–13771. doi: 10.1073/pnas.250476497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muzio M, Ni J, Feng P, Dixit VM. IRAK (Pelle) family member IRAK-2 and MyD88 as proximal mediators of IL-1 signalling. Science. 1997;278:1612–1615. doi: 10.1126/science.278.5343.1612. [DOI] [PubMed] [Google Scholar]
- 23.Wesche H, Henzel WJ, Shillinglaw W, Li S, Cao Z. MyD88: an adapter that recruits IRAK to the IL-1 receptor complex. Immunity. 1997;7:837–847. doi: 10.1016/s1074-7613(00)80402-1. [DOI] [PubMed] [Google Scholar]
- 24.Häcker H, Redecke V, Blagoev B, Kratchmarova I, Hsu L-C, Wang GG, Kamps MP, Raz E, Wagner H, Häcker G. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature. 2006;439:204. doi: 10.1038/nature04369. [DOI] [PubMed] [Google Scholar]
- 25.Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009;1:a000034. doi: 10.1101/cshperspect.a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun SC, Chang JH, Jin J. Regulation of nuclear factor-kappaB in autoimmunity. Trends Immunol. 2013;34:282–289. doi: 10.1016/j.it.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun SC. Non-canonical NF-kappaB signaling pathway. Cell Res. 2011;21:71–85. doi: 10.1038/cr.2010.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beinke S, Ley SC. Functions of NF-kappaB1 and NF-kappaB2 in immune cell biology. Biochem. J. 2004;382:393–409. doi: 10.1042/BJ20040544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu. Rev. Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 30.Zhang H, Sun SC. NF-kappaB in inflammation and renal diseases. Cell. Biosci. 2015;5:63. doi: 10.1186/s13578-015-0056-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun SC, Ley SC. New insights into NF-kappaB regulation and function. Trends Immunol. 2008;29:469–478. doi: 10.1016/j.it.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shimada M, Satoh N, Yokosawa H. Involvement of Rel/NF-κB in regulation of ascidian notochord formation. Dev. Growth Differ. 2001;43:145–154. doi: 10.1046/j.1440-169X.2001.00560.x. [DOI] [PubMed] [Google Scholar]
- 33.Kawai N, Takahashi H, Nishida H, Yokosawa H. Regulation of NF-kappaB/Rel by IkappaB is essential for ascidian notochord formation. Dev. Biol. 2005;277:80–91. doi: 10.1016/j.ydbio.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 34.Bucala R. Signal transduction. A most interesting factor. Nature. 2000;408:146–147. doi: 10.1038/35041654. [DOI] [PubMed] [Google Scholar]
- 35.Calandra T, Roger T. Macrophage migration inhibitory factor: a regulator of innate immunity. Nat. Rev. Immunol. 2003;3:791–800. doi: 10.1038/nri1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mun SH, Oh D, Lee SK. Macrophage migration inhibitory factor downregulated the RANKL-RANK signaling pathway by activating Lyn tyrosine kinase in mouse models. Arthr. Rheum. 2014;66:2482–2493. doi: 10.1002/art.38723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Flaster H, Bernhagen J, Calandra T, Bucala R. The macrophage migration inhibitory factor-glucocorticoid dyad: regulation of inflammation and immunity. Mol. Endocrinol. 2007;21:1267–1280. doi: 10.1210/me.2007-0065. [DOI] [PubMed] [Google Scholar]
- 38.Bucala R, Donnelly SC. Macrophage migration inhibitory factor: a probable link between inflammation and cancer. Immunity. 2007;26:281–285. doi: 10.1016/j.immuni.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 39.Bernhagen J, et al. MIF is a non cognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat. Med. 2007;13:587–596. doi: 10.1038/nm1567. [DOI] [PubMed] [Google Scholar]
- 40.Mikulowska A, Metz CN, Bucala R, Holmdahl R. Macrophage migration inhibitory factor is involved in the pathogenesis of collagen type II-induced arthritis in mice. J. Immunol. 1997;158:5514–5517. [PubMed] [Google Scholar]
- 41.Bozza MT, Martins YC, Carneiro LA, Paiva CN. Macrophage migration inhibitory factor in protozoan infections. J. Parasitol. Res. 2012 doi: 10.1155/2012/413052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lue H, Kleemann R, Calandra T, Roger T, Bernhagen J. Macrophage migration inhibitory factor (MIF): mechanisms of action and role in disease. Microb. Infect. 2002;4:449–460. doi: 10.1016/s1286-4579(02)01560-5. [DOI] [PubMed] [Google Scholar]
- 43.Leng L, Bucala R. Insight into the biology of macrophage migration inhibitory factor (MIF) revealed by the cloning of its cell surface receptor. Cell. Res. 2006;16:162–168. doi: 10.1038/sj.cr.7310022. [DOI] [PubMed] [Google Scholar]
- 44.Sparkesa A, De Baetseliera P, Roelantsc K, De Treza C, Mageza S, Van Ginderachtera JA, Raesa G, Bucala R, Stijlemansa B. The non-mammalian MIF superfamily. Immunobiology. 2017;222:473–482. doi: 10.1016/j.imbio.2016.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vizzini A, Parisi MG, Di Falco F, Cardinale L, Cammarata M, Arizza V. Identification of CPE and GAIT elements in 3'UTR of macrophage migration inhibitory factor (MIF) involved in inflammatory response induced by LPS in Ciona robusta. Mol. Immunol. 2018;9:66–74. doi: 10.1016/j.molimm.2018.04.009. [DOI] [PubMed] [Google Scholar]
- 46.Alibashe-Ahmed M, Roger T, Serre-Beinier V, Berishvili E, Reith W, Bosco D, Berney T. Macrophage migration inhibitory factor regulates TLR4 expression and modulates TCR/CD3-mediated activation in CD4+ T lymphocytes. Sci. Rep. 2019;9:9380. doi: 10.1038/s41598-019-45260-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Park S, Lee J-O. Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp. Mol. Med. 2013;45:e66. doi: 10.1038/emm.2013.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vizzini A, Bonura A, Longo V, Sanfratello MA, Parrinello D, Cammarata M, Colombo P. Isolation of a novel LPS-induced component of the ML superfamily in Ciona intestinalis. Dev. Comp. Immunol. 2015;53:70–78. doi: 10.1016/j.dci.2015.06.018. [DOI] [PubMed] [Google Scholar]
- 49.Kim MJ, et al. Macrophage migration inhibitory factor interacts with thioredoxin–interacting protein and induces NF-κB activity. Cell. Signal. 2017;34:110–120. doi: 10.1016/j.cellsig.2017.03.007. [DOI] [PubMed] [Google Scholar]
- 50.Roger T, David J, Glauser MP, Calandra T. MIF regulates innate immune responses through modulation of Toll-like receptor 4. Nature. 2001;414:920–924. doi: 10.1038/414920a. [DOI] [PubMed] [Google Scholar]
- 51.Kapurniotu A, Gokce O, Bernhagen J. The multitasking potential of alarmins and atypical chemokines. Front. Med. 2019;6:3. doi: 10.3389/fmed.2019.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weill L, Belloc E, Bava FA, Méndez R. Translational control by changes in poly(A) tail length: recycling mRNAs. Nat. Struct. Mol. Biol. 2012;19:577–585. doi: 10.1038/nsmb.2311. [DOI] [PubMed] [Google Scholar]
- 53.Lang T, et al. Macrophage migration inhibitory factor is required for NLRP3 inflammasome activation. Nat. Commun. 2018;9:2223. doi: 10.1038/s41467-018-04581-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vyas K, Chaudhuri S, Leaman DW, Komar AA, Musiyenko A, Barik S, Mazumder B. Genome-wide polysome profiling reveals an inflammation-responsive posttranscriptional operon in gamma interferon-activated monocytes. Mol. Cell. Biol. 2009;29:458–470. doi: 10.1128/MCB.00824-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vizzini A, Bonura A, Longo V, Sanfratello MA, Parrinello D, Cammarata M, et al. LPS injection reprograms the expression and the 3’ UTR of a CAP gene by alternative polyadenylation and the formation of a GAIT element in Ciona intestinalis. Mol. Immunol. 2016;77:174–183. doi: 10.1016/j.molimm.2016.08.004. [DOI] [PubMed] [Google Scholar]
- 56.Vizzini A, Parisi MG, Cardinale L, Testasecca L, Cammarata M. Evolution of Ciona intestinalis tumor necrosis factor alpha (CiTNFα): polymorphism, tissues expression, and 3D modeling. Dev. Comp. Immunol. 2017;67:107–116. doi: 10.1016/j.dci.2016.11.005. [DOI] [PubMed] [Google Scholar]
- 57.Cancemi P, Di Falco F, Feo S, Arizza V, Vizzini A. The gelatinase MMP-9like is involved in regulation of LPS inflammatory response in Ciona robusta. Fish Shellfish Immunol. 2019;86:213–2222019. doi: 10.1016/j.fsi.2018.11.028. [DOI] [PubMed] [Google Scholar]
- 58.Pintavorn P, Ballermann BJ. TGF-β and the endothelium during immune injury. Kidney Int. 1997;51:1401–1412. doi: 10.1038/ki.1997.192. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.