

Abstract
The detoxification of ammonia to urea requires a functional hepatic urea cycle, which consists of six enzymes and two mitochondrial membrane transporters. The initial step of the urea cycle is catalyzed by carbamyl phosphate synthetase 1 (CPS1). CPS1 deficiency (CPS1D) is a rare autosomal recessive disorder. N-Carbamylglutamate (NCG), a deacylase-resistant analogue of N-acetylglutamate, can activate CPS1. We describe the therapeutic course of a patient suffering from neonatal onset CPS1D with compound heterozygosity for the c.2359C > T (p.Arg787*) and c.3559G > T (p.Val1187Phe) variants in CPS1, treated with NCG. She presented with hyperammonemia, which reached 944 μmol/L at the age of 2 days. The ammonia concentration decreased after treatment with continuous hemodiafiltration, NCG, sodium benzoate, sodium phenylbutyrate, L-arginine, vitamin cocktail (vitamin B1, vitamin B12, vitamin C, vitamin E, biotin), l-carnitine, coenzyme Q10, and parenteral nutrition. Her ammonia and glutamine levels remained low; thus, protein intake was increased to 1.2 g/kg/day. Furthermore, the amount of sodium benzoate and sodium phenylbutyrate were reduced. She remained metabolically stable and experienced no metabolic crisis following treatment with oral NCG, sodium benzoate, sodium phenylbutyrate, citrulline, vitamin cocktail, l-carnitine, and coenzyme Q10 until she underwent liver transplantation at 207 days of age. She had no neurological complications at the age of 15 months. Ammonia and glutamine levels of the patient were successfully maintained at a low level via NCG treatment with increased protein intake, which led to normal neurological development. Thus, undiagnosed urea cycle disorders should be treated rapidly with acute therapy including NCG, which should be maintained until a genetic diagnosis is reached. It is essential to prevent metabolic crises in patients with CPS1D until liver transplantation to improve their prognoses.
Keywords: N-carbamylglutamate, Carbamyl phosphate synthetase 1 deficiency, Urea cycle disorder, Hyperammonemia, Liver transplantation, Acute therapy
1. Introduction1
The urea cycle consists of six enzymes and two mitochondrial membrane transporters that are required for the conversion of ammonia to urea [1]. The initial step of the urea cycle is catalyzed by carbamyl phosphate synthetase 1 (CPS1) and is the rate-limiting step of the cycle [2]. CPS1 deficiency (CPS1D) is a rare autosomal recessive genetic disorder characterized by hyperammonemia [2]. The incidence rate of CPS1D is approximately 1 in 800,000 children in Japan [3]. Hyperammonemia caused by urea cycle disorder (UCD) requires prompt treatment by protein withdrawal, prevention of catabolism, administration of ammonia scavengers, hemofiltration and administration of L-arginine or L-citrulline. In cases of N-acetyl-L-glutamate synthase (NAGS) deficiency, N-carbamylglutamate (NCG) may be required [4]. Maintenance treatment after amelioration of the condition is aimed at preventing decompensation via protein restriction, and treatment with oral ammonia scavengers, NCG, and arginine or citrulline until liver transplantation can be performed.
Hyperammonemia associated with UCDs, including CPS1D, compromises cognitive status and is life-threatening. The extent of developmental delay is related to the peak concentration and duration of hyperammonemia [5,6]. A previous case study reported the natural course of neonatal onset UCD, including 12 patients with CPS1D [7]. Medication was administered to all 12 patients; however, a third of the patients died (4 of 12) during or immediately after the neonatal period [7].
NCG is a deacylase-resistant synthetic analogue of NAG, an essential activator of CPS1. NCG is used to treat primary NAGS deficiency and hyperammonemia associated with organic acidemias such as methylmalonic aciduria, propionic aciduria, and isovaleric aciduria [8,9]. There are few reports of administering NCG for treating CPS1D. Limited evidence suggests that application of NCG in CPS1D may benefit some patients [2,10]. Here, we report a patient with CPS1D who received early oral NCG therapy in conjunction with standard treatment. The treatment prevented hyperammonemia in the patient until liver transplantation surgery.
2. Patient report
A female patient was born as the first child to healthy, non-consanguineous parents, after 37 weeks 6 days of gestation, weighing 2864 g with an Apgar score of 8/9. Vomiting, poor suckling, and an episode of apnea were observed on day 1. On day 3, severe apnea was observed. She was admitted to a neonatal intensive care unit, and ventilation support with intubation was initiated. Laboratory investigations on admission revealed hyperammonemia, low blood urea nitrogen levels, high lactic acid, normal anion gap, and metabolic acidosis (NH3 944 μmol/L [reference range (RR): 50–150 μmol/], blood urea nitrogen 5.2 mg/dL [RR: 6.0–20.0 mg/dL], lactic acid 5.5 mmol/L [RR: 0.8–2.8 mmol/L], pH 7.23 [RR: 7.35–7.45], pCO2 42.2 mmHg [RR: 35.0–45.0 mmHg], HCO3–17.7 mEq/L [RR: 22.0–26.0 mEq/L], Base Excess −9.6 mEq/L [RR: −5 − +5 mEq/L], Anion Gap 9.3 mEq/L [RR: 8–16 mEq/L]). Acylcarnitine concentration was within normal range and urine organic acid analysis revealed no orotic acid or citrulline. Amino acid analysis revealed an elevated level of glutamine (7592 μmol/L; RR: 422–704 μmol/L). We suspected CPS1 or NAGS deficiency and conducted a genetic analysis. The patient was moved to another neonatal intensive care unit equipped for dialysis to treat hyperammonemia and was treated with protein withdrawal, vitamin cocktail (vitamin B1, vitamin B12, vitamin C, vitamin E, biotin), l-carnitine, coenzyme Q10, and parenteral nutrition with glucose and lipid therapy. On day 4, treatment for undiagnosed urea cycle disorder was initiated with continuous hemodiafiltration, and administration of sodium benzoate (250 mg/kg/day), L-arginine hydrochloride (250 mg/kg/day), sodium phenylbutyrate (250 mg/kg/day), and NCG (100 mg/kg/day). The clinical course is shown in Fig. 1. Her ammonia levels decreased immediately after initiation of acute therapy and remained within the normal range. Plasma glutamine was significantly reduced and was within normal range after the start of treatment. On day 7, we initiated total parenteral nutrition with a protein intake of 0.6 g/kg/day, as her ammonia levels slightly increased due to hypercatabolism. We started enteral feeding on day 8, and gradually increased protein intake to 0.8 g/kg/day after 2 weeks. We further increased protein intake to 1.0–1.2 g/kg/day by her discharge on day 113. Brain magnetic resonance imaging at day 38 revealed no abnormal findings (Fig. 2).
Fig. 1.

Treatment and clinical course over time.
The first onset of hyperammonemia was identified on day 2. Ammonia and plasma glutamine levels decreased immediately after initiation of acute therapy and remained within the normal range. As her ammonia levels slightly increased due to hypercatabolism, we initiated total parenteral nutrition. We started enteral alimentation on day 8, and gradually increased protein intake until she was discharged from the hospital. Due to excessive excretion of glutamine, we reduced the dose of sodium phenylbutyrate and sodium benzoate. The dose of NCG was maintained at a constant level relative to the patient's body weight. We restarted sodium benzoate treatment when her glutamine levels increased again. She remained metabolically stable and did not experience a metabolic crisis throughout her treatment.
NCG, N-carbamylglutamate; CHDF, continuous hemodiafiltration.
Fig. 2.

Brain magnetic resonance imaging (MRI) on day 38 after birth.
Brain MRI on day 38 revealed no abnormalities.
Axi, axial (horizontal) plane; Sag, sagittal plane; T1WI, T1 weighted image; T2WI, T2 weighted image.
At day 42, genetic analysis revealed no NAGS gene variant and two variants in CPS1 gene, c.2359C > T (p.Arg787*) and c.3559G > T (p.Val1187Phe). The former variant has been reported as a pathogenic variant which was inherited from the patient's father, whereas the latter, inherited from her mother, has not been previously reported [11]. In silico functional prediction algorithms revealed the following results: SIFT predicted this variant to be deleterious with a score of 0, PolyPhen-2 predicted it to be probably damaging with a score of 0.985, PredictSNP predicted the variant to have a deleterious effect with a 76% expected accuracy. Based on her condition, which was suggestive of CPS1D, we concluded that this variant was pathogenic.
Western blotting of CPS1 was carried out after SDS-PAGE, as reported using the liver tissues of the patient after liver transplantation and revealed decreased expression of CPS1 (Fig. 3) [12].
Fig. 3.
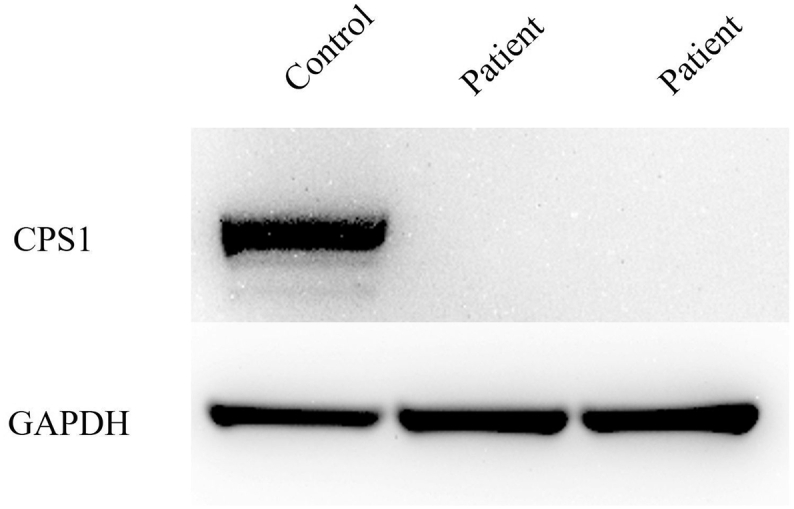
Western blot analysis of CPS1 and GAPDH(loading control) after SDS-PAGE using liver tissues collected from the patient and control.
We used anti-CPS1 anti-body, (rabbit polyclonal, Protein Tech) for the Western blot analysis.
CPS1, carbamyl phosphate synthetase 1; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
In CPS1 protein, the protein substitution Val1187Phe locates within the L3, carbamate phosphorylation domain [16]. The results of subsequent molecular structural analysis predicted that the protein substitution caused by the variant c.3559G > T (p.Val1187Phe) might have an impact on the binding of ADP to CPS1 (Fig. 4).
Fig. 4.


Representation of 3-D structures correspond to the Protein Data Bank (PDB; http://www.rcsb.org/pdb/) file 5DOU for NAG-activated, adenosine-5′-diphosphate (ADP)-bound human CPS1.
(A) Protein structure of ADP ligand-bound monomeric CPS1 (ribbon representation with domains color-coded as per de Cima et al. [16]) with V1187 highlighted in cyan. ADP is shown in space-filling mode (green - carbon; blue - nitrogen; red – oxygen; orange - phosphorous).
(B) Zoom-in view of the L3 domain (orange) highlighting position of V1187 (cyan). V1187 is surface-located, proximal to the N-terminus of a β-strand that forms part of a large β-sheet. Although the β-sheet is adjacent to the ADP binding site, V1187 is distal from ADP. V1187 is not located in the vicinity of active sites, substrate binding sites or the tunnel that connects two CPS1 active sites.
(C) The hydrophobic side chain of V1187 (cyan) is buried in a largely hydrophobic pocket which also includes, clockwise from V1187, F1240 (part of β-sheet), T1227 (on α-helix), M1178 (part of β-sheet), and L1296 (on proximal loop) all in green. The V to F substitution is likely to be tolerated, stabilizing interactions within this pocket. However, the bulkier side chain of the phenylalanine could perturb the positioning of the β-sheet region, which may have an impact on the binding of ADP.
(D) Structure of two molecules of CPS1 juxtaposed to form the dimer interface seen in the asymmetric unit of the crystal. One monomer is color coded as per Fig. 4A, the other colored blue. The position of the V1187 in both monomers (labelled) is distal from the dimer interface and hence unlikely to impact on dimerization of CPS1.
Her ammonia levels remained within normal range, and her glutamine levels dropped to near the lower limit of the normal range during hospitalization. We assumed that phenylbutyrate treatment caused the decrease in plasma glutamine, thus we reduced the dose of sodium phenylbutyrate to 100 mg/kg/day starting on day 72. For the same reason, we also weaned her off sodium benzoate and stopped this treatment on day 94. She was discharged from the hospital on day 113. We increased the dose of NCG to be consistent with her body weight gain at days 113, 134, and 155. We restarted sodium benzoate treatment since her glutamine level had increased to 828 μmol/L on day 113. She remained metabolically stable and experienced no metabolic crisis until the liver transplantation on day 207. At 15 months of age, no neurological complications have been observed.
3. Discussion
Patients with neonatal onset CPS1D have a lower survival rate than those with other UCDs in Japan [5]. Given this poor prognosis, a novel therapeutic approach is urgently required. Earlier and more aggressive interventions are required to improve patient outcomes. NCG must be considered as a prospective treatment based on previous reports of CPS1D patients who have responded to it.
The first paper discussing the potential efficacy of NCG in the kinetic variant or partial CPS1D was published in 1985 [13]. NCG was authorized in 2003 for treating hyperammonemia caused by NAGS deficiency and other organic acidurias, but not for treating hyperammonemia caused by CPS1D [8]. However, there have been some reports on the potential of NCG in treating CPS1D. Guffon et al. performed a study involving NCG therapeutic testing in patients with hyperammonemia, who were suspected to be suffering from UCD [14]. In that study, two CPS1D patients responded positively to NCG [14]. Furthermore, Ah Mew et al. indicated that NCG treatment decreased plasma ammonia [10].
Guidelines for the diagnosis and management of UCDs in Europe recommend administration of NCG for hyperammonemia associated with undiagnosed UCD [4]. Consistent with the European guidelines, Japanese guidelines for UCD also propose administration of NCG to patients with undiagnosed UCD [15]. In this case, we promptly initiated NCG administration after diagnosis of hyperammonemia according to the Japanese guidelines for treating undiagnosed UCD.
A previous study evaluated the effectiveness of NCG in 5 patients with late-onset CPS1D; NCG decreased ammonia in all patients [10]. One patient was treated with NCG for an extended period and was able to discontinue sodium phenylbutyrate. There were some non-responsive patients, suggesting that the efficacy of NCG is mutation-specific [10].
Ammonia and glutamine levels of our patient remained near the lower limit, which has not been previously observed with treatments other than NCG. Additionally, we were able to increase protein intake and decrease sodium phenylbutyrate and sodium benzoate. We speculated that our patient had responded to NCG treatment, and thus we continued the treatment.
The protein structure and function of CPS1 has been determined by De Cima et al. [16]. Yap et al. reported that structural changes in CPS1 protein caused by mutations in CPS1, decrease the stability of the protein or lower the affinity of the mutant enzyme for NAG [17]. Furthermore, they found that NCG may improve these conditions [17]. The quantity of CPS1 protein encoded by CPS1 missense variants may be highly diverse [18]. NCG can have different effects, including adverse effects on ureagenesis, depending on the type of CPS1 mutation [17]. Genetic analysis and evaluation of ammonia levels after initiation of NCG treatment are important. We maintained our patient's metabolic status without hyperammonemia until liver transplantation, leading to a good prognosis. We speculate that CPS1 activity in our patient might remain since the amino acid change is likely to only affect ADP binding. Using NCG to treat patients with CPS1D may be effective if CPS1 activity is not entirely compromised.
Ethics approval
This is an observational retrospective patient report that did not involve any research-based patient intervention. All interventions were intended to diagnose and treat the patient. No aspect of the case report is in contradiction with the Helsinki Declaration of 1975, as revised in 2000.
Animal rights
This report does not contain any studies with animal subjects.
Submission declaration and verification
This report has not been published previously and is not under consideration for publication elsewhere. Publication of this report is approved by all authors.
Use of inclusive language
We understand the use of inclusive language and this report is free from bias.
Funding
Medical writing support was funded by Recordati Rare Diseases.
Patient consent
Written informed consent for the publication of these data was obtained from the patient's mother.
Author contributions
All authors participated in the interpretation of the patient's course, and in the critical revision, and approval of the final version of the manuscript. YS drafted the original article. YS, MO-T and MS performed the western blot analysis. KM supervised the study.
Declaration of Competing Interest
Authors declare no conflict of interest.
Acknowledgements
We would like to thank Editage (www.editage.com) for English language editing, Lisa O'Rourke, PhD for medical writing support, and Chi-Chuan Lin and John Ladbury (University of Leeds, UK) for assistance with the structural analysis.
Footnotes
Abbreviations: CPS1, carbamyl phosphate synthetase 1; CPS1D, CPS1 deficiency; NCG, N-Carbamylglutamate; UCD, urea cycle disorder; NAG, N-acetyl-L-glutamate; NAGS, NAG synthase
References
- 1.Nettesheim S., Kolker S., Karall D. Incidence, disease onset and short-term outcome in urea cycle disorders-cross-border surveillance in Germany, Austria, and Switzerland. Orphanet. J. Rare Dis. 2017;12 doi: 10.1186/s13023-017-0661-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Diez-Fernandez C., Häberle J. Targeting CPS1 in the treatment of carbamoyl phosphate synthetase 1 (CPS1) deficiency, a urea cycle disorder. Expert Opin. Ther. Tar. 2017;21:391–399. doi: 10.1080/14728222.2017.1294685. [DOI] [PubMed] [Google Scholar]
- 3.Nagata N., Matsuda I., Oyanagi K. Estimated frequency of urea cycle enzymopathies in Japan. Am. J. Med. Genet. 1991;39:228–229. doi: 10.1002/ajmg.1320390226. [DOI] [PubMed] [Google Scholar]
- 4.Häberle J., Boddaert N., Burlina A. Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet. J. Rare Dis. 2012;7:8–12. doi: 10.1186/1750-1172-7-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kido J., Nakamura K., Mitsubuchi H. Long-term outcome and intervention of urea cycle disorders in Japan. J. Inherit. Metab. Dis. 2012;35:777–785. doi: 10.1007/s10545-011-9427-0. [DOI] [PubMed] [Google Scholar]
- 6.Bachmann C. Outcome and survival of 88 patients with urea cycle disorders: a retrospective evaluation. Eur. J. Pediatr. 2003;162:410–416. doi: 10.1007/s00431-003-1188-9. [DOI] [PubMed] [Google Scholar]
- 7.Unsinn C., Das A., Valayannopoulos V. Clinical course of 63 patients with neonatal onset urea cycle disorders in the years 2001–2013. Orphanet. J. Rare Dis. 2016;11:3–6. doi: 10.1186/s13023-016-0493-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Recordati Rare Diseases Carbaglu 200 Mg Dispersible Tablets Summary of Product Characteristics. 2019. https://www.medicines.org.uk/emc/medicine/20794
- 9.Baumgartner M.R., Hörster F., Dionisi-Vici C. Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J. Rare Dis. 2014;9:9–15. doi: 10.1186/s13023-014-0130-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ah Mew N, McCarter R, Daikhin Y, et al., Augmenting ureagenesis in patients with partial carbamyl phosphate synthetase 1 deficiency with N-carbamyl-L-glutamate. J. Pediatr. 2014;165:401–403.e403. doi: 10.1016/j.jpeds.2014.04.012. [DOI] [PMC free article] [PubMed]
- 11.Kurokawa K., Yorifuji T., Kawai M. Molecular and clinical analyses of Japanese patients with carbamoylphosphate synthetase 1 (CPS1) deficiency. J. Hum. Genet. 2007;52:349–354. doi: 10.1007/s10038-007-0122-9. [DOI] [PubMed] [Google Scholar]
- 12.Diez-Fernandez C., Martínez A.I., Pekkala S. Molecular characterization of carbamoyl-phosphate synthetase (CPS1) deficiency using human recombinant CPS1 as a key tool. Human Mutat. 2013;34:1149–1159. doi: 10.1002/humu.22349. [DOI] [PubMed] [Google Scholar]
- 13.O’Connor J.E., Jordá A., Grisolía S. Acute and chronic effects of carbamyl glutamate on blood urea and ammonia. Eur. J. Pediatr. 1985;143:196–197. doi: 10.1007/bf00442137. [DOI] [PubMed] [Google Scholar]
- 14.Guffon N., Schiff M., Cheillan D. Neonatal hyperammonemia: the N-carbamoyl-L-glutamic acid test. J. Pediatr. 2005;147:260–262. doi: 10.1016/j.jpeds.2005.04.059. [DOI] [PubMed] [Google Scholar]
- 15.Japanese Society for Inherited Metabolic Diseases . Newborn mass screening disease practice guideline. First edition. Diagnosis and Treatment Inc; Japan: 2019. Urea cycle disorder; pp. 67–92. [Google Scholar]
- 16.De Cima S., Polo L.M., Díez-Fernández C. Structure of human carbamoyl phosphate synthetase: deciphering the on/off switch of human ureagenesis. Sci. Rep. 2015;23 doi: 10.1038/srep16950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yap S., Gougeard N., Hart A.R. N-carbamoylglutamate-responsive carbamoyl phosphate synthetase 1 (CPS1) deficiency: a patient with a novel CPS1 mutation and an experimental study on the mutation’s effects. JIMD Rep. 2019;48:36–44. doi: 10.1002/jmd2.12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Díez-Fernández C., Hu L., Cervera J. Understanding carbamoyl phosphate synthetase (CPS1) deficiency by using the recombinantly purified human enzyme: effects of CPS1 mutations that concentrate in a central domain of unknown function. Molec. Genet. Metab. 2014;112:123–132. doi: 10.1016/j.ymgme.2014.04.003. [DOI] [PubMed] [Google Scholar]