

Abstract
Improving response to epidermal growth factor receptor (EGFR)-targeted therapies in patients with advanced wild-type (WT) RAS colorectal cancer (CRC) remains an unmet need. In this preclinical work, we evaluated a new therapeutic combination aimed at enhancing efficacy by targeting cancer cell metabolism in concert with EGFR. We hypothesized that combined blockade of glutamine metabolism and EGFR represents a promising treatment approach by targeting both the “fuel” and “signaling” components that these tumors need to survive. To explore this hypothesis, we combined CB-839, an inhibitor of glutaminase 1 (GLS1), the mitochondrial enzyme responsible for catalyzing conversion of glutamine to glutamate, with cetuximab, an EGFR-targeted monoclonal antibody in preclinical models of CRC. 2D and 3D in vitro assays were executed following treatment with either single agent or combination therapy. The combination of cetuximab with CB-839 resulted in reduced cell viability and demonstrated synergism in several cell lines. In vivo efficacy experiments were performed in cell-line xenograft models propagated in athymic nude mice. Tumor volumes were measured followed by immunohistochemical (IHC) analysis of proliferation (Ki67), mechanistic target of rapamycin (mTOR) signaling (pS6), and multiple mechanisms of cell death to annotate molecular determinants of response. In vivo, a significant reduction in tumor growth and reduced Ki67 and pS6 IHC staining were observed with combination therapy, which was accompanied by increased apoptosis and/or necrosis. The combination showed efficacy in cetuximab-sensitive as well as resistant models. In conclusion, this therapeutic combination represents a promising new precision medicine approach for patients with refractory metastatic WT RAS CRC.
Abbreviations: CAC, citric acid cycle; CRC, colorectal cancer; EGFR, epidermal growth factor receptor; Gln, glutamine; GLS1, glutaminase 1; Glu, glutamate; H&E, hematoxylin and eosin; IHC, immunohistochemical; mAb, monoclonal antibody; MAPK, mitogen activated protein kinase; NSCLC, non-small cell lung cancer; SD, standard deviation; WT, wild-type
Introduction
Colorectal cancer (CRC) is the third most common cancer and the third leading cause of cancer deaths in men and in women in the United States [1]. Despite advances in detection and treatment in recent years, only 39% of CRC is diagnosed at the localized stage when the survival rate is high (90%) [1]. The remaining patients are diagnosed at the regional or distant stages where the survival is significantly decreased to 71% and 14%, respectively [1]. Treatment for metastatic CRC (mCRC) frequently includes chemotherapy agents such as irinotecan or oxaliplatin combined with a fluorouracil (5-FU) and folinic acid (leucovorin) (FOLFIRI or FOLFOX regimens) [2]. Targeted therapies such as epidermal growth factor receptor (EGFR) neutralizing monoclonal antibodies (i.e. cetuximab and panitumumab) are approved for patients with advanced wild-type (WT) RAS CRC and can be used in first-line or late-line therapy [2,3]. However, in late-line therapy, only 12–17% of patients have durable responses to anti-EGFR monotherapy [4], and patients commonly, and often rapidly, acquire resistance [5]. Thus, novel therapeutic combinations are needed that enhance the efficacy of these agents.
A well-known hallmark of cancer is the emergence of altered cellular metabolism, to supply energy and building blocks for growth and proliferation [6]. The metabolic requirements of proliferating cells link signal transduction with nutrient accumulation (Fig. 1), resulting in a direct relationship between signal transduction leading to proliferation and cellular metabolism [7]. Glutamine (Gln) is a key anaplerotic substrate used by cancer cells, providing energy, carbon, and nitrogen to meet the demands of rapid and sustained growth [7,8]. In addition to glucose, cancer cells utilize Gln as a carbon source for ATP production, biosynthesis, and as a defense against reactive oxygen species (ROS) [9,10]. The first step in the metabolism of Gln is the conversion of Gln to glutamate (Glu) by a collection of mitochondrial enzymes known as glutaminases (GLS, Fig. 1) that are elevated in many solid tumors [11]. Glu is the primary nitrogen donor for the synthesis of non-essential amino acids [7], can contribute to the synthesis of glutathione [12], and can be converted to α-ketoglutarate, which enters the citric acid cycle (CAC) to generate ATP [12]. EGFR and Gln cooperate to provide the “signals” and the “fuel” required for mitogen activated protein kinase (MAPK)-dependent growth and proliferation [13,14]. Indeed, MAPK/ERK activity results in cMYC activation, which is responsible for transcribing Gln metabolism machinery, including GLS [7,8,12]. Consequently, we hypothesize that Gln-avid CRCs may respond poorly to EGFR-targeted therapy. Combining EGFR inhibitors with inhibitors targeting glutaminolysis may represent a promising approach to enhance efficacy of EGFR therapy in WT RAS CRC patients by blocking both the “signals” and “fuel” needed for survival of tumor cells.
Fig. 1.

Glutamine (Gln) and EGFR cooperate to promote growth and proliferation. Gln “fuels” the citric acid cycle (CAC) as required for signal transduction-mediated growth and proliferation. Glutamine transport is mediated through solute carrier transporters including ASCT2 (SLC1A5), a key Gln transporter in CRC, and xCT (SLC7A11), an exchanger of glutamine-derived glutamate (Glu) and cystine. Intracellular Gln is metabolized by mitochondrial enzymes, glutaminase 1 and 2 (GLS1/2), to Glu, which fuels the CAC and contributes to redox balance via glutathione biosynthesis, a process that requires exchange via xCT (SLC7A11). This work explored the combination of an EGFR neutralizing mAb, cetuximab, with GLS1 inhibition using CB-839 to enhance the anti-tumor efficacy of cetuximab.
There are two GLS isoforms: GLS1 (kidney-type) and GLS2 (liver-type). GLS1 is the predominant isoform expressed in tumor cells. To replenish CAC intermediates, the Raf/MEK/ERK signaling complex associates with and activates GLS1 [15]. Early studies silencing GLS1 activity using genetic knockdown with siRNA in vitro [[16], [17], [18], [19], [20], [21], [22]], shRNA in vitro [23] and in vivo [24], or morpholinos in vivo [25] illuminated potential anti-tumor effects of future pharmacological inhibitors in several tumor types, including lymphoma [25], glioma [[16], [17], [18],24], non-small cell lung cancer (NSCLC) [19], prostate cancer [20], pancreatic cancer [23], and breast cancer [21,22]. In CRC, glutaminase expression is significantly increased in tumors compared to normal colonic tissue [11,26]. Two recent studies report the effects of genetic knockdown of GLS1 in CRC [26,27]. In a study of oxaliplatin-resistant CRC, siRNA targeting GLS1 inhibited cell formation ability, wound healing ability, cell migration ability, and cell invasion ability and significantly increased apoptosis in vitro [27]. In another study, loss of GLS1 through shRNA silencing decreased the proliferation and viability of CRC cells in vitro and in vivo through a decrease in ATP levels [26]. Together these studies demonstrate the therapeutic potential of targeting GLS1 in CRC. Several selective small-molecule inhibitors of GLS1, have been developed [21,28,29]. These agents have been tested in a variety of cancer types including lymphoma [21,25,29], breast [21,22,28], glioma [16], pancreatic [23], lung [19], and renal [30] cancers. Pharmacological inhibition of glutaminase suppressed cell growth and induced apoptosis in human CRC cell lines, thus suggesting that glutaminase may serve as a target for CRC therapy [11,26,31,32]. One promising GLS1 inhibitor, CB-839 (Calithera Biosciences), has moved into early phase clinical studies [33]. In the first-in-human trials, CB-839 was well tolerated across multiple tumor types [[34], [35], [36], [37], [38], [39], [40], [41]]; however, CB-839 monotherapy has primarily resulted in disease control, defined as complete response (CR), partial response (PR) or stable disease (SD), with the majority of efficacy-evaluable patients demonstrating radiographic SD as defined by RECIST criteria [[39], [40], [41]]. Preclinical data combining CB-839 with traditional chemotherapies have suggested improved efficacy over single agent administration [28]; thus, Phase I/II clinical trials evaluating combination regimens of CB-839 with chemotherapies, such as paclitaxel in triple negative breast cancer (TNBC), docetaxel in KRAS-mutant NSCLC, azacitidine in Myelodysplastic Syndrome (MDS), carfilzomib in multiple myeloma, and capecitabine in CRC, have been opened and are currently ongoing (NCT03057600, NCT02071862, NCT03047993, NCT03798678, NCT02861300). Clinical trials combining CB-839 with targeted therapies, such as erlotinib in EGFR-mutant NSCLC, osimertinib in EGFR-mutant NSCLC, cabozantinib and everolimus in renal cell cancer, talazoparib in renal cell cancer, TNBC, and CRC, palbociclib in NSCLC and CRC, and nivolumab in renal cell cancer, melanoma and NSCLC, have also been started and are currently recruiting patients (NCT02071862, NCT03831932, NCT03163667, NCT03875313, NCT03965845, NCT02771626). Preliminary safety data suggests that these combinations were well-tolerated with minimal overlapping toxicities [40,41]. Capitalizing upon the synergy between EGFR signaling and Gln metabolism, recent preclinical studies have shown that inhibiting GLS1 reversed acquired resistance to erlotinib, an EGFR tyrosine kinase inhibitor, in NSCLC [42,43]. In NSCLC and other disease states, small molecule tyrosine kinase inhibitors that target the catalytic domain of EGFR are used (ie. erlotinib and gefitinib), whereas in CRC monoclonal antibodies are used to target the extracellular domain of EGFR (ie. cetuximab and panitumumab) thus preventing EGFR activation [3,44,45]. Although the mechanism of action is different, both types of EGFR-targeted therapies result in blockade of downstream signal transduction. In this work, we describe the efficacy of EGFR-targeted therapy combined with glutaminase inhibition by CB-839 in preclinical models of CRC.
Materials and ethods
General methods/reagents and supplies
Cetuximab was obtained from Eli Lilly and Company. CB-839 was obtained from Calithera Biosciences. Caco-2, LoVo, LS 174 T, and SW48 cell lines were purchased from ATCC (American Type Culture Collection) and authenticated using a commercial vendor (Genetica). The DiFi cell line was provided by Bruce Boman (University of Delaware), the HCA-7 cell line was provided by Susan Kirkland (Imperial College London), the LIM1215 cell line was provided by Robert Whitehead (Vanderbilt University), and the V9P cell line was provided by John Mariadason (Olivia Newton-John Cancer Research Institute). The HCA-7 derivatives CC, SC, SC1 and CC-CR cells (described below) and the SW48 derivative, SW48 LZRS, cells (described below) were generated and maintained in the Coffey lab. Our group has previously shown that when HCA-7 cells are cultured in 3D in type I collagen they form two types of colonies with distinct morphologies which we designated cystic colonies (HCA-7 CC) or spiky colonies (HCA-7 SC) [46]. SC1 cells, a spiky clone isolated from HCA-7 SC [46], are used in this study and are designated HCA-7 SC in this work. HCA-7 CC-CR cells, cetuximab-resistant HCA-7 CC cells, were previously generated by our group through continuous exposure of HCA-7 CC cells to cetuximab in 3D culture [47]. SW48 LZRS cells are SW48 cells transfected with an empty vector [48]. Caco-2, DiFi, HCA-7, LIM1215, LoVo, LS 174 T, and V9P cells were cultured in Dulbecco's Modified Eagle Medium (DMEM) containing 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (p/s). SW48 cells were cultured in RPMI-1640 media containing 10% FBS and 1% p/s. The cells were incubated in 5% CO2 at 37 °C. Animals were purchased from Envigo and used in accordance with Institutional and Federal guidelines.
2D cell viability assays
Cell viability was evaluated using a commercially available sequential-reagent-addition fluorescent and bioluminescent assay (MultiTox Glo, Promega Corp., #G9270) in 96-well plate format according to the manufacturers protocol. The cells were exposed to vehicle, single agent cetuximab or CB-839 or combination cetuximab and CB-839. The concentrations of the agents were either 5 or 10 μg/mL or 0.01 μg cetuximab, either 250 nM or 1 μM CB-839, and either 5 μg/mL cetuximab and 250 nM CB-839, 10 μg/mL cetuximab and 1 μM CB-839, or 0.01 μg cetuximab and 1 μM CB-839. The cells were treated for 48 h. Subsequently, the MultiTox Glo cytotoxicity assay was performed and the plates were read using a plate reader (BioTek Synergy 4) with standard settings. Each set of conditions was performed in either triplicate or quadruplicate. Shown are data for the live cell fluorescence assay with the following treatments: SW48 and HCA-7 cells were exposed to vehicle, 5 μg/mL cetuximab, 250 nM CB-839, or 5 μg/mL cetuximab and 250 nM CB-839; V9P, LoVo, and LS 174 T cells were exposed to vehicle, 10 μg/mL cetuximab, 1 μM CB-839, or 10 μg/mL cetuximab and 1 μM CB-839; and DiFi and LIM1215 cells were exposed to vehicle, 0.01 μg cetuximab, 1 μM CB-839, or 0.01 μg cetuximab and 1 μM CB-839.
The combination indices (CIs) of the drugs were determined using CompuSyn software [49,50]. Cell lines V9P, LoVo, LS 174 T, LIM1215, DiFi, SW48 and HCA-7 were exposed to vehicle, 1, 5, or 10 μg/mL cetuximab, 0.25, 1, or 5 μM CB-839, or combination (5 μg/mL cetuximab and 0.25 μM CB-839 or 10 μg/mL cetuximab and 1 μM CB-839). Viability was determined using the MultiTox Glo assay as described above. Synergism or antagonism are defined as previously described where CI < 1, =1, and >1 indicate synergism, additive effect, and antagonism, respectively [49].
3D cell assays
3D collagen cultures were prepared as previously described [46,47]. Briefly, the 3D collagen cultures were set up using 3 layers of type I collagen (PureCol, Advanced BioMatrix) in 12-well dishes in triplicate. Top and bottom layers consisted of 400 μL per well of collagen diluted to 2 mg/mL in DMEM containing 10% FBS. The middle layers consisted of 400 μL per well of 2 mg/mL collagen in medium plus FBS and 5000 cells/mL in single-cell suspension. 400 μL of medium with or without cetuximab and/or CB-839 was added on top of each collagen sandwich. HCA-7 SC and HCA-7 CC-CR cells were exposed to vehicle, 3 μg/mL cetuximab, 0.5 μM CB-839, or 3 μg/mL cetuximab and 0.5 μM CB-839. SW48 LZRS cells were exposed to vehicle, 5 μg/mL cetuximab, 0.02 μM CB-839, or 5 μg/mL cetuximab and 0.02 μM CB-839. V9P cells were exposed to vehicle, 0.01 μg/mL cetuximab, 1 μM CB-839, or 0.01 μg/mL cetuximab and 1 μM CB-839. Medium was changed every 2–3 days. Colonies were counted using an Oxford Optronix GelCount after 14–29 days. The assay was replicated at least two times biologically with three technical replicates.
In vivo experiments
All animal procedures were in compliance with the Guide for the Care and Use of Laboratory Animal Resources (1996), National Research Council and approved by the Vanderbilt University Institutional Animal Care and Use Committee. 5–6-week old female athymic nude mice (Hsd: Athymic Nude-Foxn1nu, Envigo, #6903) were injected subcutaneously with 2 × 106 SW48, Caco-2, HCA-7 or HCA-7 CC-CR cells. Treatment was started when the tumor volume reached 200–250 mm3 for SW48 tumors, 250–300 mm3 for Caco-2 tumors, 150–300 mm3 for HCA-7 tumors or 100–150 mm3 for HCA-7 CC-CR tumors. Mice were randomly assigned to treatment cohorts. Mice were treated with either vehicle control (PBS), cetuximab, CB-839, or cetuximab and CB-839. Cetuximab was administered at 1 mg/kg for SW48, Caco-2, and HCA-7 tumors or at 20 mg/kg for HCA-7 CC-CR tumors every 3 days for a total of 7 treatments. CB-839 was administered at 200 mg/kg orally every 12 h. Tumor volumes were measured using either an automated tumor scanner (Peira TM900) or manually by calipers every third day and quantified using the formula V = W ∗ L ∗ H/2.
Immunohistochemistry (IHC)
Animals were sacrificed and tumor tissue samples were fixed in 10% formalin for 24 h then stored in 70% EtOH/PBS. Tissues were sectioned (5 μm thickness) and evaluated for expression of Ki67, phospho-S6 (pS6) and cleaved caspase-3. For Ki67 and pS6, all staining was performed by hand. Antigen retrieval was performed on all samples in pH 6.0 Citrate Buffer for 20 min at 104 °C using a pressure cooker followed by cool down for 10 min at room temperature. For Ki67 analysis, samples were blocked with Mouse IgG Blocking Reagent (Vector Labs, #MKB-2213) for 60 min, followed by quenching with 0.03% H2O2 with sodium azide for 5 min. The samples were incubated with Ki67 mouse monoclonal antibody (Vector Laboratories, #VP-K452) at a dilution of 1:100 for 60 min. Detection was performed by incubating with Dako K4007 Envision + System HRP labelled polymer Anti-mouse for 20 min followed by 3, 3′-diaminobenzidine (DAB+) chromogen for 5 min. For pS6 analysis, samples were quenched with 0.03% H2O2 with sodium azide for 5 min followed by blocking with Serum-free Protein Block (Dako, #X0909) for 15 min. The samples were incubated with pS6 Ribosomal Protein rabbit monoclonal antibody (Cell Signaling, #4858) at a dilution of 1:500 overnight. Detection was performed by incubating with Dako K4011 Envision + System HRP labelled polymer Anti-rabbit for 30 min followed by 3, 3′-diaminobenzidine (DAB+) chromogen for 5 min. All incubations were performed at room temperature. Positive tissue controls were included.
For Caspase-3 analysis, staining was performed using the Leica Bond Max IHC stainer. All steps besides dehydration, clearing, and coverslipping were performed on the Bond Max. Slides were placed on the Leica Bond Max IHC stainer and deparaffinized. Heat induced antigen retrieval was performed on the Bond Max using their Epitope Retrieval 2 solution for 20 min. Samples were blocked with Serum-free Protein Block (Dako, #X0909) for 15 min. The sections were incubated with anti-Cleaved Caspase-3 rabbit monoclonal antibody (Cell Signaling, #9664) at a dilution of 1:300 for 60 min. The Bond Refine Polymer detection system was used for visualization. Slides were then dehydrated, cleared, and coverslipped. Additional tissue slices were stained using hematoxylin and eosin (H&E). H&E was performed using the Thermo/Shandon Gemini Automated Slide Stainer. All steps except coverslipping were performed on the Gemini. Slides were deparaffinized, cleared, and hydrated. Slides were placed in hematoxylin (Richard-Allan Scientific, #7211) for 4 min. Slides were then rinsed in water for 1 min, placed in Clarifier 1 (Richard-Allan Scientific, #7401) for 1 min, rinsed in water for 1 min, placed into Bluing Reagent (Richard-Allan Scientific, #7301) for 30 s, and rinsed in water and 95% alcohol for 1 min each. Slides were submerged in Eosin-Y (Richard-Allan Scientific, #7111) for 1 min. Following staining, slides were dehydrated, cleared, and coverslipped. For Caspase-3 and H&E slides, images were captured using a high throughput Leica SCN400 Slide Scanner automated digital image system from Leica Microsystems. Whole slides were imaged at 20× magnification to a resolution of 0.5 μm/pixel.
Tissue slides from at least 3 animals per treatment group were imaged at 20× and/or 40× magnification. The number of positive cells in each field of view were counted manually from the 20× images (n = 3–15).
Statistics
All data are represented as mean ± standard deviation (SD). All statistical analyses and graphs were generated with GraphPad Prism 7 or 8. Unpaired Student's t-test was used to determine the statistical significance of differences between two independent groups of variables. For IHC comparisons between >2 groups, 1-way ANOVA with Tukey's multiple comparison test was performed. For all tests, p ≤ 0.05 was considered significant.
Results
Evaluation of treatment in 2D and 3D cell culture models in vitro
To assess the effects of combination treatment in vitro, we initially conducted viability assays using 2D cell culture systems of seven CRC cell lines (V9P, LoVo, LS 174 T, LIM1215, DiFi, SW48 and HCA-7). The panel of cell lines consist of RAS WT and mutant cell lines that demonstrate a range of sensitivity to cetuximab single agent treatment [47]. The cells were treated with either vehicle, cetuximab, CB-839, or combined cetuximab and CB-839. Combination therapy resulted in significantly decreased cell viability when compared to vehicle or single agent controls (Fig. 2, p < 0.05). The combination was able to overcome resistance to cetuximab monotherapy in a number of cell lines including two cell lines that are KRAS mutant, LoVo and LS 174 T. This indicates that this treatment may not only overcome resistance to EGFR-targeted therapy in RAS WT patients for which this single agent therapy is already approved, but it may also expand the patient population to include those with RAS mutations. Determination of the combination indices (CIs) showed that combination drug treatment resulted in an additive effect in the LIM1215 cell line (Fig. 2, caret) and synergism in several of the other CRC cell lines analyzed (Fig. 2, asterisks).
Fig. 2.
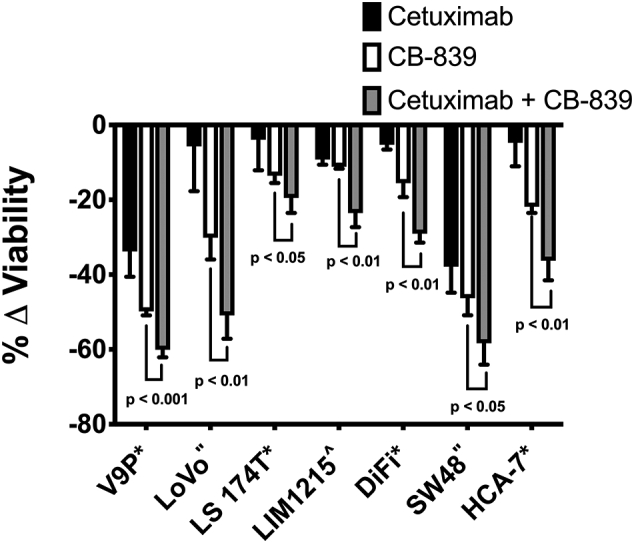
Combined CB-839/anti-EGFR mAb therapy results in cooperative efficacy in 2D culture in vitro. CRC cell lines were propagated in 2D culture. Cells were treated with either cetuximab (black), CB-839 (white), or cetuximab and CB-839 (grey). Cell viability was determined using MultiTox Glo cytotoxicity assay. Shown is the percentage change in live cells from the fluorescence assay normalized to vehicle control. CRC cell lines propagated in 2D culture exhibit reduced viability with combined therapy compared to single agents (relative to vehicle control). Error bars represent ± standard deviation (SD) (n = 3 or 4 technical replicates). Combination indices were determined and are indicated for each cell line by the following symbols: * synergism, ^ additive, and” not available.
3D cell culture systems have been demonstrated to more closely mimic in vivo behavior than 2D cultures [46]. Therefore, after establishing the efficacy of the combined treatment in 2D, we tested the combination therapy using 3D culture systems of a number of WT RAS CRC cells. We used two of the cell lines, SW48 and V9P, in which the combination was effective in 2D culture. We also tested a cell line that our group has previously shown to be intrinsically cetuximab-resistant in 3D cultures, HCA-7 SC [46], and a cell line that our group generated to be cetuximab-resistant (CR) in 3D cultures, HCA-7 CC-CR, thus exemplifying a model of acquired resistance [47]. Combined cetuximab and CB-839 treatment resulted in significantly reduced colony counts compared to vehicle or single agent controls in all of the cell lines tested (Fig. 3, p < 0.05). Interestingly, we found that combined cetuximab and CB-839 was able to overcome both intrinsic and acquired resistance to EGFR antibody treatment in HCA-7 SC and HCA-7 CC-CR cells, respectively.
Fig. 3.
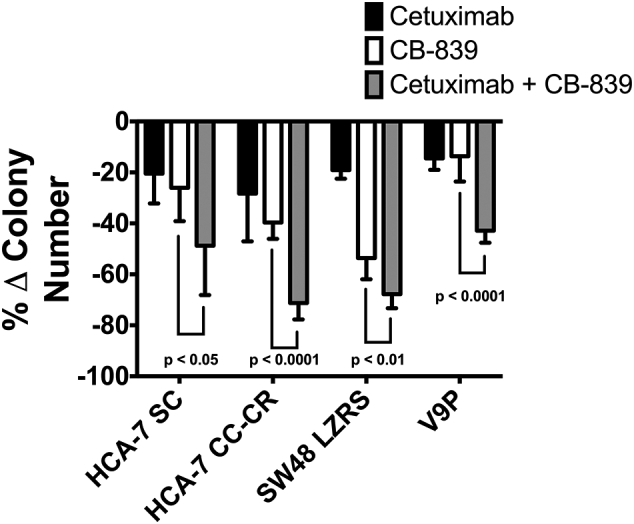
Combined CB-839/anti-EGFR mAb therapy results in cooperative efficacy in 3D culture in vitro. CRC cell lines were grown in 3D collagen culture. Cells were treated with either cetuximab (black), CB-839 (white), or cetuximab and CB-839 (grey). The number of colonies were counted. Shown is the percentage change in colony number normalized to vehicle control. Combination therapy results in decreased colonies in a 3D culture model compared to single agents (relative to vehicle control). Error bars represent ± SD (n = 2–3 biological replicates and 3 technical replicates).
In vivo efficacy studies
Having validated the therapeutic efficacy of the combination therapy in vitro, we were next interested in studying the treatment efficacy in vivo. Athymic nude mice bearing RAS WT colorectal tumor xenografts were longitudinally treated with either vehicle, CB-839, cetuximab, or combined cetuximab and CB-839 and tumor volume was monitored. Following the experiment, tumors were collected from mice from each of the groups and immunohistochemistry (IHC) was performed to evaluate molecular determinants associated with response to combined cetuximab and CB-839 treatment. In particular, mechanistic studies to determine the effects of the drugs on cell proliferation and mTOR signaling were assessed through Ki67 and pS6 IHC, respectively. In addition, to gain further insights into the mechanism of action of these drugs, analyses of apoptosis and necrosis were performed through IHC for cleaved caspase-3 and H&E staining, respectively. Initial experiments were performed using SW48 cell-line xenografts which display intrinsic cetuximab-resistance [47] (Fig. 4A and B). Tumors progressed with vehicle and single agent CB-839 treatment, while tumor growth in the cetuximab treatment group was slightly reduced. However, there was a significant reduction in tumor growth for the combination cetuximab and CB-839 treatment group in comparison to the cetuximab treatment group starting at day 18 post-treatment (p < 0.05) through day 30 (p < 0.01). Consistent with the treatment response data, quantification of IHC showed a minimal reduction (<20%) in Ki67 staining for groups treated with cetuximab and CB-839 single agents as compared to vehicle control (Fig. 4D, p < 0.05 and p < 0.01 respectively). There was a much greater reduction (≈50%) in Ki67 staining as a result of combined cetuximab and CB-839 compared to vehicle (Fig. 4C and D, p < 0.0001). No statistically significant difference was observed between cetuximab and CB-839 single agent groups; however, there was significantly reduced Ki67 staining when treated with combined cetuximab and CB-839 compared to either single agent control indicating lower proliferation in these tumors (Fig. 4D, p < 0.0001 and p < 0.0001 for cetuximab and CB-839 respectively). Further mechanistic insights were gained through evaluation of pro-survival mTOR-mediated signaling, as determined by pS6. Evaluation of pS6 IHC showed no effect of cetuximab and a modest effect (40% reduction) of CB-839 as compared to vehicle (Fig. 4E, p > 0.05 and p < 0.05 respectively). In addition, CB-839 resulted in significantly lower pS6 staining in comparison to cetuximab (p < 0.05). Interestingly, significantly reduced pS6 was observed in the group treated with combined cetuximab and CB-839 compared to vehicle (77% reduction) or single agent controls (Fig. 4C and E, p < 0.001, p < 0.001, and p < 0.05 for vehicle, cetuximab, and CB-839 respectively). No noticeable differences between treatment groups were observed in the H&E analysis of necrosis (Fig. 4F). However, an increase in cleaved caspase-3 staining was observed for all treatment groups relative to vehicle (Fig. 4G and H, p < 0.05, p < 0.0001, and p < 0.01 for cetuximab, CB-839, and combined cetuximab and CB-839 respectively) with CB-839 having the highest staining (p < 0.0001 and p < 0.001 in comparison to cetuximab and combined cetuximab and CB-839 respectively). Thus, apoptosis potentially plays a role in the reduced cell viability observed.
Fig. 4.

Combined CB-839/anti-EGFR mAb therapy overcomes intrinsic cetuximab-resistance in SW48 xenografts in vivo. (A) Mice with SW48 cell line xenografts, a model of intrinsic cetuximab resistance, were treated with either vehicle (black), CB-839 (red), cetuximab (blue) or cetuximab and CB-839 (purple) for 30 days and tumor volumes were monitored (n = 5 mice/group). (B) Representative images of mice treated with cetuximab alone (left) or cetuximab in combination with CB-839 (right). Mice progressed on single agent CB-839 or cetuximab yet exhibit reduced tumor volume with combination cetuximab and CB-839. (C) Immunohistochemistry (IHC) was performed on tumors from each of the treatment groups to evaluate Ki67 and pS6 expression. Shown are representative images (40×). The number of positive cells were counted in regions of interest on images from Ki67 IHC (D) and pS6 IHC (E) (n = 3–15 fields of view, at least 3 animals/treatment group). Reduced Ki67 and pS6 were observed when treated with CB-839 in combination with cetuximab compared to cetuximab or CB-839 alone. (F) Representative H&E images (0.46×) of tumor tissues from each of the treatment groups. (G) IHC was performed on tumors from each of the treatment groups to evaluate cleaved caspase-3 expression. Shown are representative images (20×). (H) The number of positive cells were counted in regions of interest on images from cleaved caspase-3 IHC (n = 3–15 fields of view, at least 3 animals/treatment group). Error bars represent ± SD. Statistical significance is defined as follows: *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
We also evaluated Caco-2 cell line xenografts which are partially responsive to cetuximab treatment [47] (Supporting Information Fig. S1). The mice in the cetuximab and cetuximab and CB-839 groups exhibited similar levels of tumor regression up to day 15 (Supporting Information Fig. S1A). Following 21 days of treatment, after which treatment was stopped, tumors from mice treated with cetuximab are slightly larger than tumors from mice treated with cetuximab and CB-839 (p < 0.05). Tumor sizes were monitored for an additional 27 days after the end of the treatment study. Tumors in the mice treated with single-agent cetuximab exhibited recurrence of tumor growth while the cetuximab and CB-839 cohort exhibited sustained tumor regression with no regrowth. Mice treated with the combination had significantly smaller tumors than mice treated with cetuximab alone from day 24 post-treatment (p < 0.001) through day 48 post-treatment (p < 0.0001) (Supporting Information Fig. S1A). No significant differences in necrosis (Supporting Information Fig. S1B) or apoptosis (Supporting Information Fig. S1C and S1D) were observed between treatment groups in this tumor model.
Finally, we performed in vivo experiments using HCA-7 and HCA-7 CC-CR xenografts as models of cetuximab-sensitive and cetuximab-resistant tumors respectively (Supporting Information Fig. S2 and Fig. 5). The mice were treated for a total of 21 days and tumor sizes were monitored up to 84 and 57 days after the start of treatment for HCA-7 and HCA-7 CC-CR tumors respectively (Supporting Information Fig. S2A and Fig. 5A). In both models, the mice in the cetuximab and cetuximab and CB-839 groups exhibited similar levels of tumor regression following treatment. Following the end of the treatment course, the tumors in the HCA-7 mice treated with cetuximab and cetuximab and CB-839 showed very slight tumor growth (Supporting Information Fig. S2A). Although at the end of the experiment the volumes of the tumors in mice treated with cetuximab were significantly higher than the tumors in mice treated with cetuximab and CB-839 (p < 0.01), both therapies worked well to control tumor growth. In HCA-7 CC-CR tumors, mice treated with CB-839 exhibited stable disease after treatment followed by tumor recurrence at treatment cessation (Fig. 5A). In contrast to cetuximab-treated mice bearing HCA-7 xenografts, cetuximab-treated mice bearing HCA-7 CC-CR xenografts recurred after the stop of treatment while mice treated with cetuximab and CB-839 did not (p < 0.0001). Thus, the combination is able to prevent recurrence in this model unlike single agent therapy. Quantification of IHC in HCA-7 CC-CR tumors showed no effect of cetuximab on Ki67 staining as compared to vehicle control (Fig. 5C), consistent with the tumor recurrence observed in this group. There was a significant reduction in Ki67 staining observed with CB-839 (46%) with an even more pronounced reduction (69%) as a result of combined cetuximab and CB-839 compared to vehicle (Fig. 5B and C, p < 0.0001 and p < 0.0001 respectively). Tissues from mice treated with CB-839 showed significantly less Ki67 staining than tissues from mice treated with cetuximab (Fig. 5C, p < 0.01). There was less proliferation when treated with combined cetuximab and CB-839 compared to either single agent control as indicated by reduced Ki67 staining (Fig. 5C, p < 0.0001 and p = 0.0574 for cetuximab and CB-839 respectively). Evaluation of pS6 IHC in HCA-7 CC-CR xenografts showed no effect of cetuximab or CB-839 as compared to vehicle and no differences between cetuximab and CB-839 single agent groups (Fig. 5D). However, significantly reduced pS6 was observed in the group treated with combined cetuximab and CB-839 compared to vehicle (80% reduction) or single agent controls (Fig. 5D, p < 0.01, p < 0.001, and p < 0.05 for vehicle, cetuximab, and CB-839 respectively). Analyses of necrosis and apoptosis in HCA-7 and HCA-7 CC-CR xenografts demonstrated distinctions between these tumors that help elucidate the observed differences in cetuximab sensitivity and response to the drugs. In cetuximab-sensitive HCA-7 tumors, increased necrosis was seen with cetuximab and combined cetuximab and CB-839 treatments (Supporting Information Fig. S2B). In addition, CB-839 had significantly higher cleaved caspase-3 than vehicle or cetuximab single agent groups (Supporting Information Fig. S2C and S2D, p < 0.001 and p < 0.05 respectively). Therefore, apoptosis is the mechanism behind the delayed tumor growth observed with CB-839 treatment whereas necrosis is the main mechanism of treatment response in cetuximab-treated tumors whether alone or in combination. On the other hand, in cetuximab-resistant HCA-7 CC-CR tumors, higher necrosis was only observed for the combined cetuximab and CB-839 group relative to vehicle or single agent controls (Fig. 5E). Similar to the HCA-7 tumors, there was significantly higher apoptosis in the CB-839 treatment group than vehicle, cetuximab, or combined cetuximab and CB-839 treatment groups (Fig. 5F and G, p < 0.01, p < 0.01, and p < 0.05 respectively). Thus, apoptosis and reduced proliferation appeared to be associated with response to CB-839 relative to vehicle or cetuximab single agent groups, while necrosis seemed to further account for the efficacy of the combined treatment.
Fig. 5.

Combined CB-839/anti-EGFR mAb therapy overcomes acquired cetuximab-resistance in HCA-7 CC-CR xenografts in vivo. (A) Mice with HCA-7 CC-CR cell line xenografts, a model of acquired cetuximab-resistance, were treated with either vehicle (black), CB-839 (red), cetuximab (blue) or cetuximab and CB-839 (purple) for 21 days. At this time point, treatment was stopped (dotted line). Tumor volumes were monitored to day 57 (n = 8 mice/group). Mice progressed on single agent CB-839 or cetuximab yet exhibit reduced tumor volume with combination cetuximab and CB-839. (B) IHC was performed on tumors from each of the treatment groups to evaluate Ki67 and pS6 expression. Shown are representative images (40×). The number of positive cells were counted in regions of interest on images from Ki67 IHC (C) and pS6 IHC (D) (n = 3–15 fields of view, at least 3 animals/treatment group). Reduced Ki67 and pS6 were observed when treated with CB-839 in combination with cetuximab compared to cetuximab or CB-839 alone. (E) Representative H&E images (0.46×) of tumor tissues from each of the treatment groups. (F) IHC was performed on tumors from each of the treatment groups to evaluate cleaved caspase-3 expression. Shown are representative images (20×). (G) The number of positive cells were counted in regions of interest on images from cleaved caspase-3 IHC (n = 3–15 fields of view, at least 3 animals/treatment group). Error bars represent ± SD. Statistical significance is defined as follows: #p = 0.0574, *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Discussion
Precision medicine represents a promising approach for treatment of CRC patients by taking into consideration the genetic and metabolic differences between individual tumors. By studying these differences, it may be possible to predict which patients will respond to therapy and thereby select the ideal therapeutic regimen for each patient. Patients with advanced WT RAS CRC tumors are eligible for treatment with EGFR-targeted therapy [2,3]. However, many of these patients become resistant to these treatments and their tumors recur [[2], [3], [4], [5],45]. There are multiple reported mechanisms of EGFR resistance including mainly genetic alterations [[2], [3], [4], [5],45]. We have recently identified novel modes of cetuximab-resistance, including both genetic [46] and non-genetic alterations, for instance changes in long non-coding RNA (lncRNA), that are present in both intrinsic and acquired resistance [47]. Additional ways to improve the selection of patients for treatment with anti-EGFR therapy and new treatments that increase survival are needed. To capitalize on the link between EGFR signaling and tumor glutamine metabolism in CRC, we developed a treatment strategy combining the clinically-approved EGFR-targeted monoclonal antibody, cetuximab, with a GLS1 inhibitor, CB-839. We hypothesized that this combination would enhance the efficacy of EGFR therapy by targeting the fuel and signaling needed by tumors to survive. Here we report that combination therapy with CB-839 and cetuximab results in improved efficacy compared to single agents in colorectal cancer cell lines and animal models. A synergistic effect of this combination therapy was seen in several cell lines. We observed that this combination sensitizes cells with both modes of resistance. We also note that this combination was most effective in cetuximab-resistant models as we did not observe much added benefit of the combination in cells that were already sensitive to cetuximab. This combination therapy can thus be used to treat patients whose tumors have become refractory to anti-EGFR monoclonal antibody (mAb) therapy alone. Given that we observed in vitro efficacy against CRC cell lines expressing mutant KRAS, the combination can potentially be relevant in patients with mutant RAS tumors. This should be the focus of further evaluation.
Mechanistic studies to determine the effects of the drugs on signaling pathways involved in proliferation and survival were performed in two xenograft models. As both tumors are models of cetuximab resistance, inhibiting EGFR by itself had no/minimal effect on proliferation (Ki67) and no effect on mTOR signaling (pS6), as expected. CB-839 as a single agent showed minimal changes in proliferation and a modest change in mTOR signaling in the model of intrinsic cetuximab-resistance (SW48) while in the model of acquired cetuximab-resistance (HCA-7 CC-CR), a modest decrease in proliferation and no change in mTOR signaling were observed. While outside the scope of the current work, these mechanistic observations could be due to underlying differences in the modes of resistance in these two models that affect downstream signaling pathways. In both models, the combination was more potent than either drug individually at inhibiting proliferation (Ki67) and pro-survival signaling through mTOR (pS6). These data suggest there is synergy in blocking these two pathways and future studies will further explore signaling mechanisms.
The mechanism of action for these drugs was further evaluated through assessment of necrosis and apoptosis in both cetuximab-sensitive and cetuximab-resistant xenograft models. A combination of these two modes of cell death were observed in our studies. CB-839 appears to induce cell killing through apoptosis in both cetuximab-sensitive (HCA-7) and cetuximab-resistant (SW48 and HCA-7 CC-CR) models. However, combined cetuximab and CB-839 reduced cell viability mainly through necrosis. Interestingly, when comparing HCA-7 to HCA-7 CC-CR cells, we noticed a difference in the amounts of necrosis in the cetuximab treatment group. HCA-7 CC-CR cells were derived from HCA-7 cells and were generated to be cetuximab-resistant in 3D culture [47]. While high levels of necrosis were observed for HCA-7 xenografts treated with cetuximab, there was not a difference in necrosis observed for HCA-7 CC-CR xenografts treated with cetuximab relative to vehicle. This observation can help explain the underlying mechanism for the differences in response to cetuximab among these two xenografts.
CB-839 sensitivity has been reported to correlate to a number of functional measures including glutaminase activity, the ratio of intracellular glutamate to glutamine, and the extent to which treatment promoted accumulation of cellular glutamine or depletion of cellular glutamate [19]. Analysis of additional genes and pathways may provide further insight into treatment response.
All together, these data indicate that the combination of cetuximab and CB-839 may have potential for treatment of CRC. Given the promising preclinical results reported in this paper, we have opened a clinical trial testing the combination of CB-839 with a fully humanized EGFR-mAb therapy (panitumumab) in patients with WT RAS CRC (NCT03263429). An alternative mechanism of inhibiting glutamine metabolism is through targeting of the glutamine transporter ASCT2 (SLC1A5). We have recently developed a potent and selective small molecule inhibitor of ASCT2, which we designated as V-9302 [51]. This compound inhibits glutaminolysis upstream of glutaminase and we have shown that it is more effective against CRC cancer cell lines than CB-839. Thus, V-9302 may represent a superior treatment strategy for CRC. A recent paper reports treatment of CRC by combining EGFR-inhibition by cetuximab with inhibition of SLC1A5 through either shRNA knockdown or pharmacological inhibition by l-γ-glutamyl-p-nitroanilide (GPNA) [52]. The authors observed enhanced efficacy and sensitization of resistant tumors with the combination in vitro and in vivo [52]. Building upon these promising results and in view of the 100-fold improvement in potency of V-9302 over GPNA [51], combination therapy with V-9302 and EGFR-targeted antibodies may result in even further improved efficacy. Experiments with this molecule are underway in our laboratory. In addition, we are developing PET imaging tracers as biomarkers of treatment to drugs targeting glutamine metabolism, including CB-839 and V-9302. We are exploring the combination of two PET imaging tracers that report on different aspects of glutamine metabolism, 11C-glutamine, which is taken up by the glutamine transporter ASCT2 (SLC1A5), and 18F-FSPG, which is taken up by the cystine-glutamate transporter xCT (SLC7A11), as predictive and prognostic biomarkers of tumor response. These PET tracers are being evaluated preclinically as well as in our clinical trial (NCT03263429). The ability of PET imaging prior to and following therapy to predict early response to treatment with glutamine metabolism inhibitors is being studied. The possibility of using these PET tracers to select patients who will respond to treatment with these agents is a future area of investigation.
Acknowledgments
Acknowledgments
The authors thank Frank Revetta and the Translational Pathology Shared Resource at Vanderbilt Universtiy Medical Center for assistance with the IHC staining. Whole slide imaging was performed in the Digital Histology Shared Resource at Vanderbilt University Medical Center (www.mc.vanderbilt.edu/dhsr). The Translational Pathology Shared Resource is supported by NCI/NIH Cancer Center Support Grant 2P30 CA068485-14.
Funding
This work was supported by the National Institutes of Health (NIH) National Cancer Institute (NCI) [grant numbers U24 CA220325, P50 CA236733, P30 CA068485] and the Vanderbilt Center for Molecular Probes.
Author contribution statement
Allison S. Cohen: Methodology, Formal analysis, Writing-Original Draft, Writing-Review & Editing, Visualization, Project administration Ling Geng: Investigation Ping Zhao: Investigation Allie Fu: Investigation Michael L. Schulte: Methodology, Formal analysis, Writing-Original Draft, Writing-Review & Editing, Visualization, Project administration Ramona Graves-Deal: Investigation M. Kay Washington: Methodology, Supervision Jordan Berlin: Conceptualization, Supervision Robert J. Coffey: Conceptualization, Writing-Review & Editing, Supervision, Funding acquisition H. Charles Manning: Conceptualization, Writing-Original Draft, Writing-Review & Editing, Supervision, Project administration, Funding acquisition.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.tranon.2020.100828.
Appendix A. Supplementary data
Supplementary figures
References
- 1.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2018. CA Cancer J. Clin. 2018;68(1):7–30. doi: 10.3322/caac.21442. 29313949 [DOI] [PubMed] [Google Scholar]
- 2.Moriarity A., O'Sullivan J., Kennedy J., Mehigan B., McCormick P. Current targeted therapies in the treatment of advanced colorectal cancer: a review. Ther Adv Med Oncol. 2016;8(4):276–293. doi: 10.1177/1758834016646734. (PubMed PMID: 27482287; PMCID: PMC4952023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yewale C., Baradia D., Vhora I., Patil S., Misra A. Epidermal growth factor receptor targeting in cancer: a review of trends and strategies. Biomaterials. 2013;34(34):8690–8707. doi: 10.1016/j.biomaterials.2013.07.100. (PubMed PMID: 23953842) [DOI] [PubMed] [Google Scholar]
- 4.Bertotti A., Papp E., Jones S., Adleff V., Anagnostou V., Lupo B. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature. 2015;526(7572):263–267. doi: 10.1038/nature14969. (PubMed PMID: 26416732; PMCID: PMC4878148) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Diaz L.A., Jr., Williams R.T., Wu J., Kinde I., Hecht J.R., Berlin J. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012;486(7404):537–540. doi: 10.1038/nature11219. (PubMed PMID: 22722843; PMCID: PMC3436069) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. (PubMed PMID: 21376230) [DOI] [PubMed] [Google Scholar]
- 7.Wise D.R., Thompson C.B. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci. 2010;35(8):427–433. doi: 10.1016/j.tibs.2010.05.003. (PubMed PMID: 20570523; PMCID: PMC2917518) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wise D.R., DeBerardinis R.J., Mancuso A., Sayed N., Zhang X.Y., Pfeiffer H.K. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A. 2008;105(48):18782–18787. doi: 10.1073/pnas.0810199105. (PubMed PMID: 19033189; PMCID: PMC2596212) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amores-Sanchez M.I., Medina M.A. Glutamine, as a precursor of glutathione, and oxidative stress. Mol. Genet. Metab. 1999;67(2):100–105. doi: 10.1006/mgme.1999.2857. (PubMed PMID: 10356308) [DOI] [PubMed] [Google Scholar]
- 10.Fan J., Kamphorst J.J., Mathew R., Chung M.K., White E., Shlomi T. Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol Syst Biol. 2013;9:712. doi: 10.1038/msb.2013.65. (PubMed PMID: 24301801; PMCID: PMC3882799) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang F., Zhang Q., Ma H., Lv Q., Zhang T. Expression of glutaminase is upregulated in colorectal cancer and of clinical significance. Int J Clin Exp Pathol. 2014;7(3):1093–1100. (PubMed PMID: 24696726; PMCID: PMC3971313) [PMC free article] [PubMed] [Google Scholar]
- 12.Altman B.J., Stine Z.E., Dang C.V. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. 2016;16(10):619–634. doi: 10.1038/nrc.2016.71. (PubMed PMID: 27492215; PMCID: PMC5484415) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rhoads J.M., Argenzio R.A., Chen W., Rippe R.A., Westwick J.K., Cox A.D. L-glutamine stimulates intestinal cell proliferation and activates mitogen-activated protein kinases. Am. J. Phys. 1997;272(5 Pt 1):G943–G953. doi: 10.1152/ajpgi.1997.272.5.G943. (PubMed PMID: 9176200) [DOI] [PubMed] [Google Scholar]
- 14.Ko T.C., Beauchamp R.D., Townsend C.M., Jr., Thompson J.C. Glutamine is essential for epidermal growth factor-stimulated intestinal cell proliferation. Surgery. 1993;114(2):147–153. (discussion 153-4. PubMed PMID: 7688149) [PubMed] [Google Scholar]
- 15.Thangavelu K., Pan C.Q., Karlberg T., Balaji G., Uttamchandani M., Suresh V. Structural basis for the allosteric inhibitory mechanism of human kidney-type glutaminase (KGA) and its regulation by Raf-Mek-Erk signaling in cancer cell metabolism. Proc Natl Acad Sci U S A. 2012;109(20):7705–7710. doi: 10.1073/pnas.1116573109. (PubMed PMID: 22538822; PMCID: PMC3356676) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seltzer M.J., Bennett B.D., Joshi A.D., Gao P., Thomas A.G., Ferraris D.V. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010;70(22):8981–8987. doi: 10.1158/0008-5472.CAN-10-1666. (PubMed PMID: 21045145; PMCID: PMC3058858) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Szeliga M., Bogacinska-Karas M., Rozycka A., Hilgier W., Marquez J., Albrecht J. Silencing of GLS and overexpression of GLS2 genes cooperate in decreasing the proliferation and viability of glioblastoma cells. Tumour Biol. 2014;35(3):1855–1862. doi: 10.1007/s13277-013-1247-4. (PubMed PMID: 24096582; PMCID: PMC3967065) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martin-Rufian M., Nascimento-Gomes R., Higuero A., Crisma A.R., Campos-Sandoval J.A., Gomez-Garcia M.C. Both GLS silencing and GLS2 overexpression synergize with oxidative stress against proliferation of glioma cells. J Mol Med (Berl) 2014;92(3):277–290. doi: 10.1007/s00109-013-1105-2. (Pub Med PMID: 24276018; PMCID: PMC4327995) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van den Heuvel A.P., Jing J., Wooster R.F., Bachman K.E. Analysis of glutamine dependency in non-small cell lung cancer: GLS1 splice variant GAC is essential for cancer cell growth. Cancer Biol Ther. 2012;13(12):1185–1194. doi: 10.4161/cbt.21348. (PubMed PMID: 22892846; PMCID: PMC3469476) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao P., Tchernyshyov I., Chang T.C., Lee Y.S., Kita K., Ochi T. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458(7239):762–765. doi: 10.1038/nature07823. (PubMed PMID: 19219026; PMCID: PMC2729443) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang J.B., Erickson J.W., Fuji R., Ramachandran S., Gao P., Dinavahi R. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell. 2010;18(3):207–219. doi: 10.1016/j.ccr.2010.08.009. (PubMed PMID: 20832749; PMCID: PMC3078749) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qie S., Chu C., Li W., Wang C., Sang N. ErbB2 activation upregulates glutaminase 1 expression which promotes breast cancer cell proliferation. J Cell Biochem. 2014;115(3):498–509. doi: 10.1002/jcb.24684. (PubMed PMID: 24122876; PMCID: PMC4518873) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Son J., Lyssiotis C.A., Ying H., Wang X., Hua S., Ligorio M. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496(7443):101–105. doi: 10.1038/nature12040. (PubMed PMID: 23535601; PMCID: PMC3656466) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheng T., Sudderth J., Yang C., Mullen A.R., Jin E.S., Mates J.M. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc Natl Acad Sci U S A. 2011;108(21):8674–8679. doi: 10.1073/pnas.1016627108. (PubMed PMID: 21555572; PMCID: PMC3102381) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiang Y., Stine Z.E., Xia J., Lu Y., O'Connor R.S., Altman B.J. Targeted inhibition of tumor-specific glutaminase diminishes cell-autonomous tumorigenesis. J Clin Invest. 2015;125(6):2293–2306. doi: 10.1172/JCI75836. (PubMed PMID: 25915584; PMCID: PMC4497742) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song Z., Wei B., Lu C., Li P., Chen L. Glutaminase sustains cell survival via the regulation of glycolysis and glutaminolysis in colorectal cancer. Oncol Lett. 2017;14(3):3117–3123. doi: 10.3892/ol.2017.6538. (PubMed PMID: 28928849; PMCID: PMC5588174) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu W.Q., Hu Y.Y., Lin X.P., Fan W. Knockdown of PKM2 and GLS1 expression can significantly reverse oxaliplatin-resistance in colorectal cancer cells. Oncotarget. 2017;8(27):44171–44185. doi: 10.18632/oncotarget.17396. (PubMed PMID: 28498807; PMCID: PMC5546471) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gross M.I., Demo S.D., Dennison J.B., Chen L., Chernov-Rogan T., Goyal B. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol. Cancer Ther. 2014;13(4):890–901. doi: 10.1158/1535-7163.MCT-13-0870. (PubMed PMID: 24523301) [DOI] [PubMed] [Google Scholar]
- 29.Shukla K., Ferraris D.V., Thomas A.G., Stathis M., Duvall B., Delahanty G. Design, synthesis, and pharmacological evaluation of bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide 3 (BPTES) analogs as glutaminase inhibitors. J Med Chem. 2012;55(23):10551–10563. doi: 10.1021/jm301191p. PubMed PMID: 23151085; PMCID: PMC3539823) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gameiro P.A., Yang J., Metelo A.M., Perez-Carro R., Baker R., Wang Z. In vivo HIF-mediated reductive carboxylation is regulated by citrate levels and sensitizes VHL-deficient cells to glutamine deprivation. Cell Metab. 2013;17(3):372–385. doi: 10.1016/j.cmet.2013.02.002. (PubMed PMID: 23473032; PMCID: PMC4003458) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Richard S.M., Martinez Marignac V.L. Sensitization to oxaliplatin in HCT116 and HT29 cell lines by metformin and ribavirin and differences in response to mitochondrial glutaminase inhibition. J. Cancer Res. Ther. 2015;11(2):336–340. doi: 10.4103/0973-1482.157317. (PubMed PMID: 26148596) [DOI] [PubMed] [Google Scholar]
- 32.Li J., Song P., Zhu L., Aziz N., Zhou Q., Zhang Y. Synthetic lethality of glutaminolysis inhibition, autophagy inactivation and asparagine depletion in colon cancer. Oncotarget. 2017;8(26):42664–42672. doi: 10.18632/oncotarget.16844. (PubMed PMID: 28424408; PMCID: PMC5522096) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li J., Chen C., Goyal B., Laidig G., Stanton T.F., Sjogren E.B. 2013. Heterocyclic Inhibitors of Glutaminase. [Google Scholar]
- 34.Boroughs L.K., Chen P.H., Cai L., Rodriguez M., Zhang W., Huffman K.E. Evaluation of the glutaminase inhibitor CB-839 in non-small cell lung cancer. Cancer Research. 2015;75 doi: 10.1158/1538-7445.Am2015-4710. (PubMed PMID: WOS:000371597104338) [DOI] [Google Scholar]
- 35.Harding J.J., Telli M.L., Munster P.N., Le M.H., Molineaux C., Bennett M.K. Safety and tolerability of increasing doses of CB-839, a first-in-class, orally administered small molecule inhibitor of glutaminase, in solid tumors. J Clin Oncol. 2015;33(15) doi: 10.1200/jco.2015.33.15_suppl.2512. (PubMed PMID: WOS:000358036900545) [DOI] [Google Scholar]
- 36.Konopleva M.Y., Flinn I.W., Wang E.S., DiNardo C.D., Bennett M.K., Molineaux C.J. Phase 1 study: safety and tolerability of increasing doses of Cb-839, an orally-administered small molecule inhibitor of glutaminase, in acute leukemia. Haematologica. 2015;100:378–379. (PubMed PMID: WOS:000361204902442) [Google Scholar]
- 37.Vogl D.T., Younes A., Stewart K., Orford K.W., Bennett M., Siegel D. Phase 1 study of CB-839, a first-in-class, glutaminase inhibitor in patients with multiple myeloma and lymphoma. Blood. 2015;126(23) (PubMed PMID: WOS:000368020103254) [Google Scholar]
- 38.Wang E.S., Frankfurt O., Orford K.W., Bennett M., Flinn I.W., Maris M. Phase 1 study of CB-839, a first-in-class, orally administered small molecule inhibitor of glutaminase in patients with relapsed/refractory leukemia. Blood. 2015;126(23) (PubMed PMID: WOS:000368020102078) [Google Scholar]
- 39.Meric-Bernstam F., DeMichele A., Telli M.L., Munster P., Orford K.W., Demitri G.D. 2015. In Phase 1 Study of CB-839, A First-in-Class, Orally Administered Small Molecule Inhibitor of Glutaminase in Patients With Refractory Solid Tumors. AACR-EORTC-NCI. [Google Scholar]
- 40.DeMichele A., Harding J.J., Telli M.L., Munster P.N., McKay R., Iliopoulos O. ASCO; Chicago, IL, Chicago, IL: 2016. In Phase 1 Study of CB-839, a Small Molecule Inhibitor of Glutaminase (GLS) in Combination With Paclitaxel (Pac) in Patients (pts) With Triple Negative Breast Cancer (TNBC) [Google Scholar]
- 41.Meric-Bernstam F., Tannir N.M., Mier J.W., DeMichele A., Telli M.L., Fan A.C. ASCO; 2016. In Phase 1 Study of CB-839, a Small Molecule Inhibitor of Glutaminase (GLS), Alone and in Combination With Everolimus (E) in Patients (pts) With Renal Cell Cancer (RCC) [Google Scholar]
- 42.Xie C., Jin J., Bao X., Zhan W.H., Han T.Y., Gan M. Inhibition of mitochondrial glutaminase activity reverses acquired erlotinib resistance in non-small cell lung cancer. Oncotarget. 2016;7(1):610–621. doi: 10.18632/oncotarget.6311. (PubMed PMID: 26575584; PMCID: PMC4808021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Momcilovic M., Bailey S.T., Lee J.T., Fishbein M.C., Magyar C., Braas D. Targeted inhibition of EGFR and glutaminase induces metabolic crisis in EGFR mutant lung cancer. Cell Rep. 2017;18(3):601–610. doi: 10.1016/j.celrep.2016.12.061. (PubMed PMID: 28099841; PMCID: PMC5260616) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Harari P.M. Epidermal growth factor receptor inhibition strategies in oncology. Endocr. Relat. Cancer. 2004;11(4):689–708. doi: 10.1677/erc.1.00600. (PubMed PMID: 15613446) [DOI] [PubMed] [Google Scholar]
- 45.Bertotti A., Sassi F. Molecular pathways: sensitivity and resistance to anti-EGFR antibodies. Clin. Cancer Res. 2015;21(15):3377–3383. doi: 10.1158/1078-0432.CCR-14-0848. (PubMed PMID: 26071484) [DOI] [PubMed] [Google Scholar]
- 46.Li C., Singh B., Graves-Deal R., Ma H., Starchenko A., Fry W.H. Three-dimensional culture system identifies a new mode of cetuximab resistance and disease-relevant genes in colorectal cancer. Proc Natl Acad Sci U S A. 2017;114(14):E2852–E2861. doi: 10.1073/pnas.1618297114. (PubMed PMID: 28320945; PMCID: PMC5389279) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lu Y., Zhao X., Liu Q., Li C., Graves-Deal R., Cao Z. lncRNA MIR100HG-derived miR-100 and miR-125b mediate cetuximab resistance via Wnt/beta-catenin signaling. Nat Med. 2017;23(11):1331–1341. doi: 10.1038/nm.4424. (PubMed PMID: 29035371; PMCID: PMC5961502) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kinsella T.M., Nolan G.P. Episomal vectors rapidly and stably produce high-titer recombinant retrovirus. Hum. Gene Ther. 1996;7(12):1405–1413. doi: 10.1089/hum.1996.7.12-1405. (PubMed PMID: 8844199) [DOI] [PubMed] [Google Scholar]
- 49.Chou T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006;58(3):621–681. doi: 10.1124/pr.58.3.10. (PubMed PMID: 16968952) [DOI] [PubMed] [Google Scholar]
- 50.Chou T.C., Martin N. Compusyn Software for Drug Combinations and for General Dose Effect Analysis, and User's Guide. www.combosyn.com
- 51.Schulte M.L., Fu A., Zhao P., Li J., Geng L., Smith S.T. Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat Med. 2018;24(2):194–202. doi: 10.1038/nm.4464. (PubMed PMID: 29334372; PMCID: PMC5803339) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ma H., Wu Z., Peng J., Li Y., Huang H., Liao Y. Inhibition of SLC1A5 sensitizes colorectal cancer to cetuximab. Int. J. Cancer. 2018;142(12):2578–2588. doi: 10.1002/ijc.31274. (PubMed PMID: 29363109) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figures