

Abstract
Background and Purpose
Febrile seizures (FS), the most common seizures in childhood and often accompanied by later epileptogenesis, are not well controlled. Inflammatory processes have been implicated in the pathophysiology of epilepsy. However, whether caspase‐1 is involved in FS generation and could be a target for the treatment of FS is still unclear.
Experimental Approach
By using pharmacological and gene intervention methods in C57BL/6J mice, we assessed the role of caspase‐1 in FS generation. We used structural virtual screening against the active site of caspase‐1, to screen compounds for druggable and safe low MW inhibitors of caspase‐1 in vitro. One compound was chosen to test in vivo for therapeutic potential, using FS models in neonatal mice and epileptogenesis in adult mice.
Key Results
In mice, levels of cleaved caspase‐1 increased prior to FS onset. Caspase‐1 −/− mice were resistant to FS and showed lower neuronal excitability than wild‐type littermates. Conversely, overexpression of caspase‐1 using in utero electroporation increased neuronal excitability and enhanced susceptibility to FS. The structural virtual screening, using molecular docking approaches for the active site of caspase‐1 of over 1 million compounds yielded CZL80, a brain‐penetrable, low MW inhibitor of caspase‐1. In neonatal mice, CZL80 markedly reduced neuronal excitability and incidence of FS generation, and, in adult mice, relieved later enhanced epileptogenic susceptibility. CZL80 was devoid of acute diazepam‐like respiratory depression and chronic liver toxicity.
Conclusion and Implications
Caspase‐1 is essential for FS generation. CZL80 is a promising low MW inhibitor of FS and later enhanced epileptogenic susceptibility.
Abbreviations
- ACSF
artificial CSF
- AHP
after‐hyperpolarization
- AP
action potential
- CZL80
3‐(3‐(thiophene‐2‐carboxamido)benzamido)benzoic acid
- FS
febrile seizures
- GS
generalized seizure
- IL‐1R
IL‐1 receptor
- IR
input resistance
- REOS
rapid elimination of swill
- MES
maximal electroshock seizures
- PBMC
peripheral blood mononuclear cell
- SE
status epilepticus
- TLE
temporal lobe epilepsy
- WT
wild‐type
1. INTRODUCTION
Febrile seizures (FS) are unpredictable convulsive events induced by fever, affecting 3%–14% of infants and children aged 6 months to 5 years (Patel et al., 2015; Verity, Butler, & Golding, 1985). Currently, there are no appropriate therapeutic options to control FS. Conventional antipyretics combined with anticonvulsant drugs, such as phenobarbital and valproic acid, are effective in alleviating fever and ceasing seizures, but potential toxicities of anti‐epileptic drugs in infants outweigh their therapeutic effects (Lux, 2010). Although intermittent treatment with diazepam is effective in reducing the risk of the first FS when given in time, this therapeutic strategy is not effective in reducing FS recurrence (Ruusuvuori et al., 2013). In addition, diazepam is not recommended because of its respiratory depression and inhibition of EEG activity (Khosroshahi, Faramarzi, Salamati, Haghighi, & Kamrani, 2011; Mula, 2014). Consequently, one third of FS patients are poorly controlled and experience recurrent or prolonged seizures, a condition of complex FS (Pust, 2004). Children with complex FS are at high risks of temporal lobe epilepsy (TLE), hippocampal or mesial temporal sclerosis or cognitive impairment in later life (Chungath & Shorvon, 2008; Feng & Chen, 2016). Thus, it is of great importance to understand the mechanism of FS generation. Ideally, such understanding will facilitate the identification of potential drug targets to prevent the occurrence of FS and later epileptogenesis.
Inflammatory processes have been implicated in the pathophysiology of FS and epilepsy (Dube et al., 2010; Saghazadeh, Gharedaghi, Meysamie, Bauer, & Rezaei, 2014; Vezzani, Maroso, Balosso, Sanchez, & Bartfai, 2011). In particular, the IL‐1 receptor (IL‐1R1) is closely involved in FS (Heida, Moshe, & Pittman, 2009; Vezzani et al., 2011). Thus, IL‐1R1 −/− mice show higher FS threshold and conversely, IL‐1β reduces FS threshold (Dube, Vezzani, Behrens, Bartfai, & Baram, 2005; Feng et al., 2016). However, low MW antagonists of IL‐1R1 are not available at present. Analyses of the crystal structures of IL‐1R1 show that the contact interfaces between IL‐1β and IL‐1R1 are much larger than those in sites binding low MW compounds (Vigers, Dripps, Edwards, & Brandhuber, 2000; Yang, 2015). Besides, the currently available IL‐1R1 antagonist (IL‐1Ra), is a 17‐kDa protein and does not easily pass through the blood brain barrier (BBB) (Skinner, Gibson, Rothwell, Pinteaux, & Penny, 2009; Sukedai et al., 2011). Caspase‐1 is an IL‐1β‐converting enzyme, which cleaves immature pro‐IL‐1β to its activated form (Schroder & Tschopp, 2010). Interestingly, cleaved caspase‐1 is up‐regulated in both epilepsy patients and animal models of TLE (Meng et al., 2014; Tan et al., 2015). However, whether cleaved caspase‐1 is involved in FS generation and is therefore a potential target in the treatment of FS is still unclear. In our present work we have tested the hypothesis that caspase‐1 may be a pharmacological target for FS and the later enhanced epileptogenic susceptibility and searched for novel caspase‐1 inhibitors, with high efficacy and low side effects, based on the structure of caspase‐1.
2. METHODS
2.1. Animals
All animal care and experimental procedures were in complete compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and carried out in accordance with the ethical guidelines of the Zhejiang University Animal Experimentation Committee (No. ZJU20160027). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010; McGrath & Lilley, 2015) and with the recommendations made by the British Journal of Pharmacology.
Mice pups of caspase‐1 knockout (Casp1 −/−, RRID:IMSR_JAX:004947) and littermate controls (wild‐type [WT], Casp1 +/+) were used in the present study (Schott et al., 2004). The Casp1 −/− mice were kindly gifted by Prof. Hu Gang (Nanjing Medical University, China) and were backcrossed onto C57BL/6J (RRID:IMSR_JAX:000664) background mice for at least three generations after introduction to our lab. Then Casp1 +/− mice were crossed with Casp1 +/− mice to generate the mice pups of WT or Casp1 −/−. Breeders were maintained in cages with a 12‐h light/dark cycle (lights on from 8:00 to 20:00) and had access to water and food ad libitum. The time of birth of pups was monitored every 12 h, and the day of birth was considered postnatal day 0 (P0). Experiments were conducted between 10:00 and 17:00hr.
2.2. Induction of experimental FS
Animals were randomized into control and different treatment groups, and the investigators were blind to the allocation of treatments. Experimental FS were induced in mice pups (male and female) at postnatal day 8 (P8), as described previously (Chen et al., 2016; Feng et al., 2016). Pups were randomly placed in an incubator chamber with different hyperthermia circumstances (38, 41 or 44°C). The standards for classifying seizure onset were as follows: 1, generalized myoclonic jerk; 2, wild running; 3, ataxic walking; 4, circling; and 5, falling, or tonic–clonic seizures of limbs (Eun, Abraham, Mlsna, Kim, & Koh, 2015). The movement of a sudden jerk was defined as the sign of the pre‐onset of an FS, which usually occurred at 35–38 min. Upon the onset of the first seizure behaviour of falling or tonic–clonic seizures of limbs (1st seizure), the latency (in minutes) to FS generation was recorded. Rectal temperature was monitored by inserting a probe (FHC, Bowdoinham, USA) into the recta at the starting time point, subsequently every 5 min, and finally at seizure onset to establish the temperature (threshold) to FS generation. If the seizure was not evoked within 55 min, then the pup was classed as ‘no seizure’ and 55 min was noted as the latency (Schuchmann et al., 2006), and the rectal temperature at this time was noted as threshold in these no‐seizure pups.
For inducing the second seizure (2nd seizure), the pups were moved to a cool surface for 2 min once the 1st seizure was evoked. Then they were moved back to the hyperthermia chamber to elicit the 2nd seizure. For inducing prolonged FS (Dai et al., 2019; Wu et al., 2017), the pups were moved to a cool surface for 2 min once seizure was evoked. Then they were moved back to the hyperthermia chamber. The procedure repeated for at least 30 min after the 1st seizure. The control pups were littermates of the experimental pups and were separated from the dams for the same duration; their core temperatures were maintained within the normal range.
2.3. Drug treatments
Pentobarbital (25 mg·kg−1; i.p.) was injected to block seizures 20 min before mice pups were placed into the hyperthermia chamber. To confirm the pro‐FS role of caspase‐1, recombinant caspase‐1 (0.5, 1 and 2 nM) was injected in the hippocampus. Ac‐YVAD‐cmk (0.01, 0.1 and 1 nM) was also intra‐hippocampally injected to inhibit caspase‐1 and compared with the effects of other caspase‐1 inhibitors. For intra‐hippocampal injection, the skull of P8 mice pups exposed under transient anaesthesia with isoflurane (4% for induction and 2% for maintenance). An injection needle was stereotaxically inserted into the dorsal hippocampus to deliver drugs. Five minutes after recovery from anaesthesia, pups were placed into the hyperthermia chambers to induce experimental FS. Vehicle (2% DMSO in saline) or solutions of CZL80 (0.0075, 0.075, 0.75 and 7.5 mg·kg−1) were intravenously injected prior to placing the animals into the hyperthermia chambers in order to test enzyme activity. We did not find any vehicle effects in our study. Vehicle (2% DMSO in saline), CZL80 (0.75 mg·kg−1, i.v.), diazepam (0.3 mg·kg−1, i.v.) and IL‐1Ra (0.35 mg·kg−1, i.v.) were administered immediately prior to placing the animals in the hyperthermia chambers, to test the experimental FS behaviour. VX‐765 (50 mg·kg−1, i.p.) was administered 1 h prior to the seizure behavioural test.
2.4. Plasmid DNA preparation
Mouse caspase‐1 cDNA was amplified from a mouse brain cDNA library by PCR with the forward primer 5′‐CCTGCTCGAGATGGCTGACAAGATCCTGAG‐3′ and the reverse primer 5′‐ATA CTG CAG TTA ATG TCC CGG GAA GAG GTA GA‐3′. The PCR product was inserted into the XhoI and PstI sites of the pCIG2 plasmid to construct the pCIG2‐Casp1 plasmid. The pCIG2‐Casp1 plasmid was sequenced to confirm its structure by BGI Group Guangdong Group Inc.
2.5. In utero electroporation
The day of mating (limited to 12 h) was defined as embryonic day 0.5 (E0.5). Pregnant mice (E13.5–E14.5) were anaesthetized with isoflurane (4% for induction and 2% for maintenance) to expose the uterus through a midline incision in the abdominal wall. Approximately 1 μl of DNA solution (1.5 μg·μl−1) mixed with Fast Green (Sigma) was injected into the lateral ventricle using pulled glass micropipettes, followed by five square pulses (35 V each, 50‐ms duration at 950‐ms intervals) delivered across the embryo's head using electrodes connected to an electroporator generator (CUY21EDIT, NEPA GENE). Throughout the procedure, embryos were kept moist by constant bathing with 37°C saline. The uterus was returned to the abdominal cavity, and the midline incision was sutured. Animals were placed in a recovery chamber until they woke up (usually 5–10 min) and were then transferred to their home cages. ‘Con’ group were exposed to all the same surgical procedures of in utero electroporation, except for the delivery of pCIG2‐Casp1 or pCIG2 plasmid.
2.6. Search for low MW caspase‐1 inhibitors
The methods of searching for low MW compounds with a particular biological activity have been described previously (Hou, Wang, Li, & Wang, 2011; Pan et al., 2015; Sun, Hou, & Zhang, 2014). The crystal structure of caspase‐1 complexed with a Cys285‐linked peptide‐like ligand (PDB code: 3NS7) (Galatsis et al., 2010) was used as the initial structure for molecular docking. The Glide module in Schrödinger was used for molecular docking. Prior to virtual screening of a large database, we at first validated the best strategy between Glide HTVS docking (high throughput virtual screening), Glide SP docking (standard precision) and Glide XP docking (extra precision), by evaluating the screening power (P value), the ranking power (Pearson correlation coefficient) and the AUC value of the receiver operating characteristic (ROC) curve of the docking scores. After a hierarchical virtual screening of the ChemBridge database (>1 million), the top 2,000 compounds derived from the XP docking score were further filtered (by Lipinski's ‘rules of five’ and rapid elimination of swill (REOS) rules) and clustered into 144 groups (by considering chemical diversity with the molecular similarity less than 0.8 based on the Tanimoto matrix calculated by the FCFP_4 fingerprints) for analysis. The compound with highest score in each group was ranked by the XP docking score (a total of 144 groups), and the highest ranked 50 compounds were purchased from Topscience for further experimental testing. A crucial aspect of docking experiments is the correct preparation of the molecular target, as many active residues were reported to be important for caspase‐1 inhibition (Ramos‐Guzman, Zinovjev, & Tunon, 2019; Wilson et al., 1994). In our study, we did not try to adjust the active residues individually in the binding site of caspase‐1, and fortunately, tight binders were screened by our protocol.
To give a general idea of caspase‐1 inhibitor design, we analysed the binding mechanisms of the four highest ranked, tight‐binding molecules (CZL80, CZL55, CZL78 and CZL06), by performing molecular dynamics (MD) simulations, molecular mechanics/generalized Born surface area (MM/GBSA) binding free energy calculations and residue decompositions for each system. The MM/GBSA methodology was applied to calculate the binding free energy, which has been widely used in characterizing drug–target interactions. Accordingly, the binding free energy of the drug–target complex can be calculated using the following equations:
| (1) |
| (2) |
| (3) |
| (4) |
where ΔG bind denotes the total free energy difference between the ligand–receptor complex (G com) and the sum of the ligand (G lig) and the receptor (G rec) individuals. The total binding free energy can be further expressed as the sum of the enthalpy part (ΔH) and the entropy part (−TΔS), where the enthalpy part is consisted of ΔE MM (gas‐phase interaction between protein and ligand, including the electrostatic energy, ΔE ele, the van der Waals energy, ΔE vdW, and the well‐cancelled internal energies, ΔE int) and ΔG sol (desolvation free energy, including the polar part, ΔG GB, and the non‐polar part, ΔG SA, of the solvent effect).
Specially, to analyse the binding mechanism of the tight binding molecules, MD simulations, MM/GBSA binding free energy calculations and decompositions were performed for each system. In the phase of system construction, the molecules were at first optimized by the Hartree–Fock method based on the 6‐31G* basis set using Gaussian 09 program. Then the electrostatic potentials were calculated by using the RESP technology embedded in antechamber module in Amber12 simulation package. Counterions of Na+ or Cl− were added where has the lowest or highest electrostatic potential to neutralize the systems. The general Amber force field (gaff) and Amber99SBnmr force field were used for the small molecules and protein, respectively. Each system was immersed in a TIP3P water box with the water molecules extended 10 Å of the solute in each direction.
Before MD simulations, each system was minimized by four steps: at first, all the heavy atoms (including the heavy atoms in protein, solvent and ions) were constrained with an elastic constant of 10 kcal·mol−1·Å2 with only hydrogen atoms set free (5,000 cycles, 2,500 cycles of steepest descent and 2,500 cycles of conjugate gradient minimization); second, the heavy atoms in solvent and ions were also set free (5,000 cycles, 2,500 cycles of steepest descent and 2,500 cycles of conjugate gradient minimization); third, the heavy atoms in the sidechain of protein were allowed to move freely as well (5,000 cycles, 2,500 cycles of steepest descent and 2,500 cycles of conjugate gradient minimization); and finally, the whole system was minimized for 10,000 steps with all the atoms free moving (5,000 cycles of steepest descent and 5,000 cycles of conjugate gradient minimization).
In the phase of MD simulation, the cut‐off was set to 10 Å for the short‐range interactions (van der Waals and electrostatic interactions), while the Particle mesh Ewald (PME) algorithm was employed to handle the long‐range electrostatics. All the covalent bonds involving hydrogen atoms were constrained with SHAKE algorithm. The time step was set to 2 fs. Each system was gradually heated from 0 to 300 K in the NVT ensemble over a period of 50 ps (with a constraint of 5 kcal·mol−1·Å2 on the backbone of the protein) and then relaxed by 50 ps in the NPT ensemble (T = 300 K and P = 1 atm, 2 kcal·mol−1·Å2 on the backbone of the protein). Finally, 30‐ns production simulations were performed for each system (without any constraint in the system). The snapshots were saved with an interval of 15 ps (a total of 2,000 frames were collected for each system).
2.7. Code availability
The Glide docking module is available in commercial version of Schrödinger (Maestro version 9.3.5, RRID:SCR_016748), and the MM/GBSA free energy calculation module is available free of charge to academic users in amber 14 simulation package (http://ambermd.org, RRID:SCR_014230).
2.8. Peripheral blood mononuclear cell isolation and culture
Informed written consent form for the use of these cell samples from humans was obtained before study, and this experiment was approved by the Medical Ethical Committee of Zhejiang University School of Medicine (No. ZJU20160027).
To assess the anti‐caspase‐1 activities of candidate compounds, we used a model of LPS‐pretreated peripheral blood mononuclear cells (PBMC) which is a commonly used model for measuring the effects of inhibitors on caspase‐1 (Wannamaker et al., 2007). Buffy coat fractions from healthy volunteer donors were the kind gift from Prof. Hu Hu (Zhejiang University, China). PBMCs were prepared from the buffy coat fractions with a Ficoll gradient using Ficoll Hypaque (Amersham Bioscience, Sweden) followed by washing twice with RPMI 1640 medium (Gibco 12633‐012). Cells were then transferred to six‐well microtitre plates at a concentration of 2 × 106 cells per well and stimulated with 5 μg·ml−1 of Escherichia coli LPS (O111:B4; Sigma‐Aldrich). Plates were incubated overnight (16–20 h) at 37°C in 5% CO2.
2.9. Caspase‐1 activity assays
Hippocampal tissues or cultured PBMCs were lysed, and caspase‐1 activity was assessed using a Caspase‐1 Colorimetric Assay Kit (Abcam, ab39470), according to the manufacturer's instructions. Fold increases in caspase‐1 activity were determined by comparing the results of the experimental group samples with those of controls or of sham groups.
2.10. Maximal electroshock seizures model
After mice pups (male and female) experienced prolonged FS at P8, as described previously (Feng et al., 2016), vehicle (2% DMSO in saline) or CZL80 (0.75 mg·kg−1) were intravenously injected immediately. When the animals were 50–55 days old, electroshocks were delivered through Rodent Shocker (Hugo Sachs Elektronik, March‐Hugstetten, Germany). Moistened with saline, the electrodes were clipped to the ears of adult mice. The maximal electroshock seizures (MES) threshold was determined by an ‘up‐and‐down’ method (Fischer, Kittner, Regenthal, Malinowska, & Schlicker, 2001): the stimulus current intensity began at 5 mA, and the current was lowered 1 mA if the preceding shock caused tonic hindlimb extension or was raised 1 mA if not. The interval between trails is about 15 min.
2.11. Intra‐hippocampal kainic acid‐induced seizures
After mice pups (male and female) experienced prolonged FS at P8, as described previously (Feng et al., 2016), vehicle (2% DMSO in saline) or CZL80 (0.75 mg·kg−1) were intravenously injected immediately. When the animals were 55–60 days old, under sodium pentobarbital anaesthesia (35 mg·kg−1, i.p.), adult mice were mounted in a stereotaxic apparatus. A cannula was implanted into hippocampal CA1 (AP: −2.0 mm, L: −1.3 mm, V: −1.6 mm) for kainic acid (KA) injection, and electrodes were implanted into hippocampal CA3 (AP: −2.9 mm, L: −3.0 mm, V: −3.2 mm) for EEG recording. The electrodes were made of stainless‐steel Teflon‐coated wires (diameter 0.12 mm; A.M. Systems, USA), and the tip separation was about 0.5 mm. The electrodes were connected to a miniature receptacle, which were attached to the skull with dental cement. After 7–10 days of recovery, KA (0.5 μg·μl−1 in saline, 0.5 μl, Abcam ab120100) was injected through the cannula over a period of 2 min in freely moving mice followed by a holding period of 1 min. Seizure score was based on Racine: (1) facial movement and daze; (2) head nodding; (3) unilateral forelimb clonus; (4) bilateral forelimb clonus and rearing; and (5) bilateral forelimb clonus with rearing and falling. The EEGs of the hippocampus were recorded 5 min before and 90 min after KA injection by a digital amplifier (NuAmps, Neuroscan System, USA). In our KA‐induced seizure model, at the initial stage after KA infusion, there are several changes in EEG, with an initial stage of discrete seizures, followed by continuous seizures. The onset of status epilepticus (SE) was defined as the appearance of continuous ictal discharge, which was defined as twofold‐baseline high amplitude discharges with frequency greater than 3 Hz (Joshi, Rajasekaran, Sun, Williamson, & Kapur, 2017; Xu et al., 2016).
2.12. Open field test
2.12.1. Acute test
Five minutes after treatment with vehicle (2% DMSO in saline, i.v.), CZL80 (7.5 mg·kg−1, i.v.) or diazepam (0.3 mg·kg−1, i.v.), the breathing rates (breaths min−1) of the pups were monitored. Additionally, locomotor behaviour was tracked automatically using an Open Field Video Tracking System: parameters including distance travelled and immobility time (sedation time) were recorded during 15‐min sessions.
2.12.2. Chronic test
During the treatment of vehicle (2% DMSO in saline, qod, i.v.) or CZL80 (7.5 mg·kg−1, qod, i.v.), the weight and breathing rates (min−1) of the mice were monitored. After 6 weeks of treatment, locomotor behaviour was tracked automatically via using an Open Field Video Tracking System: parameters including distance travelled and immobility time (sedation time) were recorded during 15‐min sessions.
2.13. Measurement of toxicity
2.13.1. Acute test
The liver function markers (ALT/AST), renal function markers (BUN, CREA) and Routine Blood Test (RBC, WBC, MONO %) were measured at 12 h after treatment of vehicle (2% DMSO in saline, i.v.), CZL80 (7.5 mg·kg−1, i.v.) or diazepam (0.3 mg·kg−1, i.v.)
2.13.2. Chronic test
These experiments were carried out by the Drug Safety Evaluation Platform in the Laboratory Animal Centre of Zhejiang University. The liver function markers (ALT/AST), renal function markers (BUN, CREA) and Routine Blood Test (RBC, WBC, MONO %) were measured after treatment with vehicle (2% DMSO in saline, qod, i.v.) or CZL80 (7.5 mg·kg−1, qod, i.v.) for 6 weeks.
2.13.3. Haematoxylin and eosin staining
These experiments were carried out by the Drug Safety Evaluation Platform in the Laboratory Animal Centre of Zhejiang University. Briefly, after deparaffinization and rehydration, 5‐μm longitudinal sections of liver and kidney were stained with haematoxylin solution for 5 min followed by five dips in 1% acid ethanol (1% HCl in 70% ethanol) and then rinsing in distilled water. The sections were then stained with eosin solution for 3 min and followed by dehydration with graded alcohol and clearing in xylene. The mounted slides were then examined and photographed using Olympus BX53 fluorescence microscope (Tokyo, Japan).
2.14. Analysis of CZL80 concentrations in serum and in brain tissue
Blood was collected from the orbit of mice pups (P8). They were killed by decapitation, and the brain tissue was rapidly removed, rinsed with cold saline solution and weighed. Serum (100 μl) or the supernatant from brain homogenates (100 μl) was shaken on a vortex mixer for 3 min. After centrifuging at 9727 x g for 10 min, 20 μl of the supernatant liquid was analysed with an Agilent 1100 HPLC system with a Cl8 column (Diamonsil, 200 × 4.6, 5 μm). The flow rate was kept constant at 1.0 ml·min−1. A mixture of acetonitrile and water (45:55; pH 2.5) was used as the mobile phase, with UV detection at 209 nm. All concentrations were calculated from a standard curve of known amounts of CZL80 added to tissue samples (R 2 > 0.99).
2.15. In vitro electrophysiology
C57BL/6J and Casp1 −/− mice pups (P8–10) were anaesthetized with diethyl ether. After decapitation, the brain was quickly placed in ice‐cold artificial CSF (ACSF) oxygenated with 95% O2 and 5% CO2. The ACSF contained the following (concentrations in mM): 119 NaCl, 2.5 KCl, 2.5 CaCl2, 1 NaH2PO4, 1.3 MgSO4, 26.2 NaHCO3 and 11 d‐glucose (pH 7.4), with osmolarity at 310–320 mOsm. Transverse hippocampal slices (300 μm thick), containing the hippocampus and parts of the adjacent cortex, were obtained with a vibratome (VT1000S, Leica instruments Ltd., Germany) and equilibrated in an incubation chamber with oxygenated ACSF for at least 1 h at 25°C. The slices were then positioned in a recording chamber and continuously perfused with oxygenated ACSF at a rate of 3–4 ml·min−1 before electrophysiological recordings were taken at 25°C.
CA1 pyramidal cells were visualized and identified by means of infrared differential interference contrast (IR‐DIC) video microscopy on an upright microscope (FN1, Nikon, Japan) and equipped with a X40/0.80 water‐immersion objective and a charge‐coupled device camera (Clara‐E; Andor Technology, UK). Patch pipettes with resistance at 5–10 MΩ were prepared from borosilicate glass (Sutter Instruments, USA) on a two‐step micropipette puller (PC‐10, Narishige Group, Japan). The pipette solution contained the following (in mM): 140 K‐gluconate, 5 NaCl, 2 Mg‐ATP, 0.2 EGTA and 10 HEPES at pH 7.28, with osmolarity at 300 mOsm.
Whole‐cell patch recordings were obtained in an interface‐type chamber at 25°C (to determine the temperature responses of the hippocampal pyramidal cells, the perfused ACSF temperature was increased from 25°C to approximately 41°C). The membrane potential was held at −60 mV using PATCHMASTER software (RRID:SCR_000034) in combination with a patch clamp amplifier (EPC10; HEKA Elektronik, Germany). The whole‐cell configuration was obtained in voltage‐clamp mode before proceeding to the current‐clamp recording mode. Resting membrane potential was recorded immediately upon switching to the current‐clamp mode. Once stable access was obtained, the threshold, amplitude and the ½ peak width of the action potentials were assessed by applying a series of 300‐ms depolarizing current pulses (increased in 5‐pA steps). Input resistance (IR) was calculated as the slope of the linear fit of the voltage–current plot. Current threshold was defined as the current that evoked the first derivative of the voltage trace. The amplitude of after‐hyperpolarization (AHP) was determined with the first spike.
2.16. Western blotting
All antibody‐based procedures used in this study comply with the recommendations made by the British Journal of Pharmacology (Alexander et al., 2018). The mouse pups were decapitated, and the hippocampus was rapidly dissected out and homogenized in RIPA buffer (20 mmol·L−1 Tris–HCl, pH 7.5, 150 mmol·L−1 NaCl, 1 mmol·L−1 EDTA, 1% Triton‐X100, 0.5% sodium deoxycholate, 1 mmol·L−1 PMSF and 10 μg·ml−1 leupeptin). Protein samples (100 μg per well) were separated using SDS‐PAGE and transferred to a nitrocellulose membrane, which was then blocked with 5% skim milk dissolved in PBS (pH 7.4) for 1 h. Then the membrane was incubated overnight at 4°C with primary antibodies against caspase‐1 (1:500; Cat# ab108362, RRID:AB_10858984) and GAPDH (1:8,000; Cat# KM9002, RRID:AB_2721026). Secondary antibodies against rabbit (1:10,000; Cat# 926‐32211, RRID:AB_621843) and mouse (1:10,000; Cat# 926‐68020, RRID:AB_10706161) were incubated for 2 h at room temperature. Blots were visualized with the Odyssey infrared imaging system (LI‐COR Biosciences) and analysed with Odyssey software. The relative OD was obtained by comparing the measured values with the mean value from the control group.
2.17. Histology
Mouse pups (P8) were anaesthetized and perfused transcardially with 4% paraformaldehyde. The brains were removed and stored in 4% paraformaldehyde at 4°C for 24 h and then in 30% sucrose for 5–7 days. The brains were coronally sectioned and examined histologically for the transfection of caspase‐1. Nuclei were counterstained with DAPI. Fluorescent images were collected on a fluorescence microscope (BX‐UCB, Olympus, Japan) using IPP software.
2.18. Data and statistical analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018). All data were collected and analysed in a blinded manner. Number of experimental replicates (n) is indicated in Figure legends and refers to the number of experimental subjects used in each experimental condition (independent observations). Data are presented as means ± SEM. No approach was used to reduce unwanted sources of variation. Statistical analyses were undertaken only when each group size has a minimum of n = 5 independent samples and performed with Prism 7 (RRID:SCR_005375) with appropriate inferential methods as indicated in each figure legend. Statistical tests were applied after testing for normal distribution. Post hoc tests were run only if F achieved P < 0.05 and there was no significant variance inhomogeneity. A small cohort of samples (n < 5) was prepared for exploratory western blot analysis and caspase‐1 activity assays in our study. No statistical methods were used to pre‐determine sample size or to randomize. The group sizes were selected according to our previous studies (Chen et al., 2020; Wang et al., 2017). Two‐tailed P < 0.05 was the threshold to determine statistical significance throughout this work.
The suppliers (with catalogue numbers) of the materials used in these experiments are as follows: Ac‐YVAD‐cmk (Bachem, N‐1330), recombinant caspase‐1 (Abcam, ab52079), diazepam (King York Amino Acid of Tianjin, China, H12020957), IL‐1Ra (Prospec‐Tany Techno Gene, CYT‐203), pentobarbital (Sigma, P3761) and VX‐765 (Selleckchem, USA, S2228). CZL 80 (ChemBridge 6626080); CZL 55 (ChemBridge 6810755); CZL 78 (ChemBridge 9198978); CZL 06 (ChemBridge 7135306) were all supplied by Topscience Co., Ltd. (Shanghai, China), with purity >95%.
2.19. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander et al., 2019).
3. RESULTS
3.1. Caspase‐1 is necessary and sufficient for FS generation
Mouse pups were placed in different hyperthermia conditions (38°C and 41°C). At 41°C, the pups displayed typical behavioural seizures with ictal activity with latency of 38.48 ± 0.36 min (Figure 1a). The sign prior to seizure is characterized as movement of a sudden jerk, which occurs 2–5 min before FS generation. The expression of cleaved caspase‐1, as well as its enzyme activity, increased to a significantly higher level just prior to seizure onset compared to the basal level (Figure 1b,c). At 38°C, no pups displayed behavioural seizure or ictal activity during 55‐min observation period (Figure S1A). In exploratory experiments, there was no difference in the expression of cleaved caspase‐1 or its enzyme activity compared to the basal level (Figure S1B,C). Of interest, cleaved caspase‐1 expression increased when the body temperature was 41°C, if seizures were blocked by pretreatment with pentobarbital (Figure S1D). This finding suggests that the increase of cleaved caspase‐1 expression was induced by hyperthermia, but not by seizure itself. Notably, we also observed increased cleaved caspase‐1 before FS recurrence (Figure S1E). Collectively, these results establish that the level of cleaved caspase‐1 increases specifically prior to FS generation in conditions of hyperthermia.
FIGURE 1.
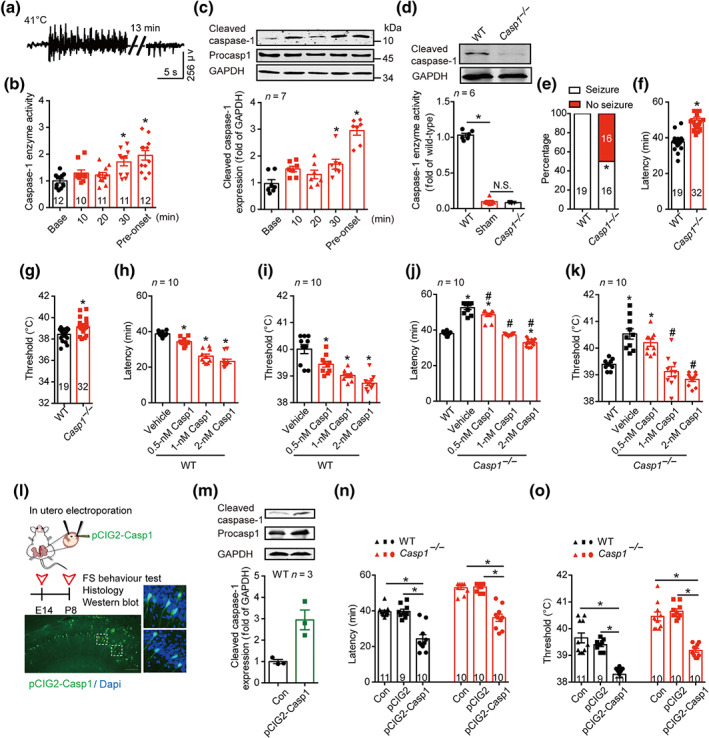
Cleaved caspase‐1 mediates FS generation. (a) Typical EEG of FS onset at 41°C. (b) The enzyme activity of caspase‐1 at different time points at 41°C. (c) The amount of caspase‐1 at different time points at 41°C, was analysed by western blotting with indicated antibodies. Data were quantified and normalized against the base. (d) Enzyme activity and protein level of caspase‐1 in WT (C57BL/6J) and Casp1 −/− mice at P8 age in basal condition. (e–g) The percentage of seizure (e), latency (f) and threshold (g) to FS generation at 41°C in WT and Casp1 −/− mice. (h–k) The latency and threshold to FS generation after intra‐hippocampal injection of caspase‐1 (0.5, 1 and 2 nM) to WT (h, i) and Casp1 −/− mice (j, k). (l) Experimental paradigm and the transfected caspase‐1 in the hippocampus after in utero electroporation of pCIG2‐Casp1. green, caspase‐1, blue, Dapi, scale bar: 100 μm. (m) The amount of caspase‐1 after in utero electroporation of pCIG2‐Casp1 was analysed by western blotting with indicated antibodies, and the data were quantified. ‘CON’ group have all same procedures of in utero electroporation, but except the delivery of pCIG2‐Casp1 plasmid. The length of procasp1 sequence is 1,533 bp, but the sequence we inserted in the pCIG2 is 1,209 bp, with the same beginning as the cleaved caspase‐1. (n, o) The latency (n) and threshold (o) to FS generation in WT and Casp1 −/− mice after in utero electroporation of pCIG2 and pCIG2‐Casp1. Data are presented as means ± SEM. *P < .05, significantly different from baseline, WT, vehicle or as indicated; # P<.05, significantly different from vehicle in i and j. One‐way ANOVA followed by Dunnett post hoc test was used for (b), (c), (h) and (i). Fisher exact test was used for (e). Unpaired t test was used for (f) and (g). Two‐way ANOVA followed by Bonferroni post hoc test was used for (d), (j), (k), (n) and (o)
To determine whether caspase‐1 is necessary to the process of FS generation, we used Casp1 −/− mice, in which the enzyme activity of caspase‐1 is absent (Figure 1d). Notably, 50% of the Casp1 −/− mice tested did not show any seizure within 55 min at 41°C (Figure 1e). Those Casp1 −/− mice who showed seizure displayed significantly longer latency and higher threshold to FS generation, compared with WT littermates (Figure 1f,g), indicating that disrupting caspase‐1 levels delayed FS generation. As Casp1 −/− mice were reported to have an abnormal expression of other genes, such as a deletion in the gene for caspase‐11 (Kayagaki et al., 2011; Man et al., 2017), we tested the effects of restoring caspase‐1 levels, on FS generation in Casp1 −/− mice. We injected exogenous recombinant caspase‐1 into the hippocampus, 5 min before FS induction, and found that this treatment dose‐dependently shortened the latency and decreased the threshold to FS generation in both WT (Figure 1h,i) and Casp1 −/− mice (Figure 1j,k). Notably, all the Casp1 −/− mice tested showed seizure behaviour within 55 min after treatment with caspase‐1 (0.5, 1 and 2 nM), and 1‐nM caspase‐1 reversed the latency and the threshold of Casp1 −/− mice to the level of WT mice (Figure 1j,k). Further, we used in utero electroporation to deliver pCIG2‐Casp1 constructs into the hippocampus of WT and Casp1 −/− mice to specifically increase or restore the expression of caspase‐1 (Figure 1l,m). On testing in our experimental FS model, we found that there was no significant difference between Con and pCIG2 groups (Figure 1n,o). Electroporation of pCIG2‐Casp1 constructs reduced the latency and decreased the threshold to FS generation in WT mice. It also reversed the latency and the threshold to FS generation of Casp1 −/− mice to the level of WT mice (Figure 1n,o). Following electroporation of pCIG2‐Casp1 constructs, all the Casp1 −/− mice experienced seizure within 55 min after FS induction. Taken together, our results demonstrated that caspase‐1 is a seizure‐promoting factor for FS generation. Therefore, caspase‐1 merits consideration as a promising target for treatment strategies aimed at FS.
3.2. Structure‐based search for low MW inhibitors for caspase‐1
At present, the most commonly used caspase‐1 inhibitors are peptides (Zhang et al., 2016). Owing to their pharmacokinetic limitations (Callus & Vaux, 2007), these peptides cannot be used in typical clinical settings. Moreover, clinical trials of both of the non‐peptide caspase‐1 inhibitors, VX‐740 and VX‐765, have been terminated because of their side effects (Vezzani et al., 2010). Thus, there is still a need to identify novel inhibitors to suppress the activity of caspase‐1. According to the binding position of the co‐crystallized ligand in 3NS7, residues of Arg179, Arg341, Arg383, Gln283 and His237 were established as selective aspartic acid recognition sites (Wilson et al., 1994) for the structure‐based virtual screening (Figure 2a, also see detail in Section 2). Based on targeting these sites, over 1 million compounds against the active pocket of caspase‐1 were screened with a molecular docking approach (Glide module in Schrödinger). Prior to virtual screening of this large database, we at first validated the best strategy among Glide HTVS docking (high throughput virtual screening), Glide SP docking (standard precision) and Glide XP docking (extra precision), by evaluating the screening power (P value), the ranking power (Pearson correlation coefficient) (Figure S2A–F) and the AUC value of the ROC curve of the docking scores (Figure S2G). After a hierarchical virtual screening of the database (Xu et al., 2014), the top 2,000 compounds derived from the docking score were further filtered (by Lipinski's ‘rules of five’ and REOS rules) (Lyne, 2002) and clustered into 144 groups (by considering chemical diversity with the molecular similarity less than 0.8 based on the Tanimoto matrix calculated by the FCFP_4 fingerprints) for analysis. The single top molecule in each group was ranked by the XP docking score (a total of 144 groups), and ultimately, 50 top‐scoring compounds (selected by docking scores, Table S1) out of the over 1 million docked were selected for further testing. Of these 50 compounds tested, 11 compounds reliably reduced the enzyme activity of caspase‐1 in vitro, compared with the control group (Table S1). We further assessed the effects of these 11 compounds on LPS‐pretreated PBMCs (Wannamaker et al., 2007). Of the 11 compounds tested, CZL80, CZL55, CZL78 and CZL06 demonstrated high inhibitory activities in this exploratory screen (Figures 2b and S3A), with IC50 values ranging from 0.01 to 11.31 μM (Figure 2c). These four compounds showed very low toxicity, and it did not affect cell viability even at a 100‐fold effective concentration (Figure S3B).
FIGURE 2.
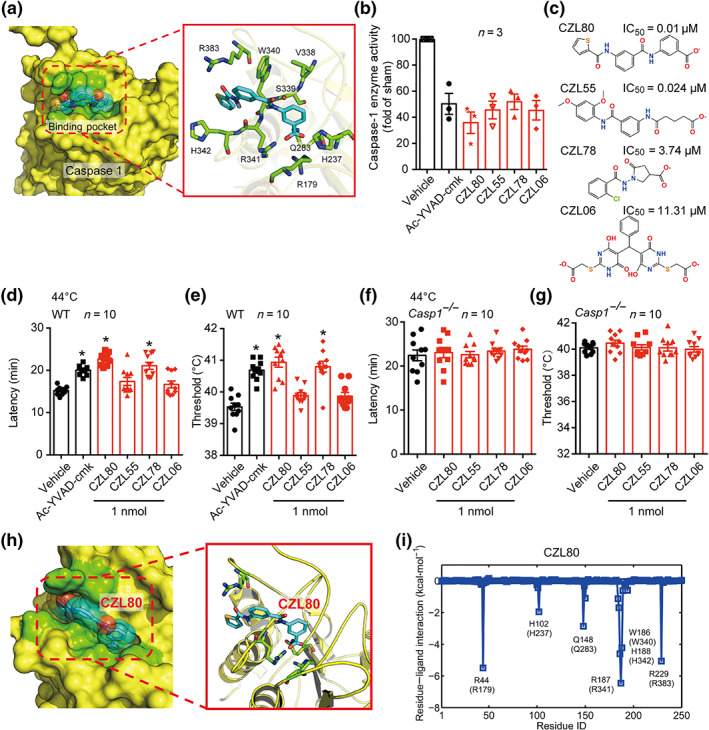
Structure‐based search for caspase‐1 inhibitors. (a) Target identification ligand binding site determination and virtual screening of active compounds using the conserved pocket residues (green stick model). (b) The enzyme activity of caspase‐1 was inhibited by the selected four compounds (CZL80, CZL55, CZL78 and CZL06) and Ac‐YVAD‐cmk, in LPS‐pretreated PBMCs, compared with the sham group. (c) The molecular structure and IC50 values of CZL80, CZL55, CZL78 and CZL06. (d–g) The latency and threshold of FS generation in WT (d, e) and Casp1 −/− (f, g) mice in hyperthermic condition, injected with vehicle, Ac‐YVAD‐cmk, CZL80, CZL55, CZL78 and CZL06 (1 nmol, i.c.v.). (h) The solvent‐accessible surfaces of CZL80 in the binding pocket of caspase‐1. The compound, the key residues and the protein backbones are shown in cyan stick model, green stick model and yellow cartoon model, respectively. (i) The corresponding hydrogen bond networks of CZL80 to the binding pocket of caspase‐1. Data are presented as means ± SEM. *P < .05, significantly different from vehicle; One‐way ANOVA followed by Dunnett post hoc test was used for (d)–(g)
Encouragingly, in the in vivo FS model, CZL80 and CZL78 (0.1 and 1 nmol, i.c.v.) significantly prolonged the latency and increased the threshold to FS generation in WT mice, while CZL55 and CZL06 showed no effect (Figures 2d,e and S3C,D). Furthermore, these four compounds (1 nmol, i.c.v.) were specific caspase‐1 inhibitors as none of them was effective in Casp1 −/− mice (Figure 2f,g). Though CZL78 significantly prolonged the latency and increased the threshold to FS generation, it is worth noting that CZL80 was more fat‐soluble (S + logP: 3.57 vs. S + logP: 0.89) and should be better able to penetrate the BBB. This supposition was confirmed by measuring the concentrations of CZL80 in serum and in brain after intravenous administration of CZL80 (Figure S4) to mice. Furthermore, CZL80 is most favourable in matching with the binding pocket of caspase‐1 with no extra fragments out of the binding position compared with other three compounds (Figures 2h and S5), and its tail inserted into the deep‐binding slot formed by Arg179, Arg341, Gln283 and Arg383 to anchor caspase‐1 (Figure 2i and Table S2). Moreover, CZL80 showed highest binding free energy (ΔG bind) among the four compounds (Table S3), fitting in more specificity regions of caspase‐1. Having confirmed this, and in consideration of the other properties reported in this subsection of the results, we selected CZL80 as a candidate molecule.
3.3. CZL80 prevents FS generation and enhanced epileptogenic susceptibility
A major goal of this study was to find a novel specific caspase‐1 inhibitor with more favourable in vivo profiles and potential clinical utility. Caspase‐1 enzyme activity was reduced following intravenous administration of CZL80 in exploratory experiments (Figure 3a). CZL80 reduced seizure incidence, prolonged seizure latency and increased threshold to FS generation in a dose‐dependent manner (Figure 3b–d). As positive controls, we used diazepam, a clinically used acute treatment for FS (Khosroshahi et al., 2011), the caspase‐1 inhibitor VX‐765 and the cytokine receptor antagonist IL‐1Ra. About 67% of the CZL80‐treated, 43% of the diazepam‐treated and 21% of the VX‐765‐treated mice pups did not experience the first FS (Figure 3e). CZL80 (0.75 mg·kg−1, i.v.), diazepam (0.3 mg·kg−1, i.v.) and VX‐765 (50 mg·kg−1, i.p.) prolonged the latency to first FS generation, while IL‐1Ra (0.35 mg·kg−1, i.v.) had no effect (Figure 3f). CZL80 also increased threshold to first FS generation (Figure 3g). The anti‐FS effect of CZL80 results from caspase‐1 inhibition as it had no effect on the latency or threshold to FS generation on Casp1 −/− mice (Figure 3h,i). Of interest, injection of CZL80 after the first FS reduced the incidence of second FS (recurrent FS) to 30% and significantly prolonged the latency and increased the threshold to the second FS generation (Figure 3j–l). In contrast, diazepam showed no effect on the second FS generation.
FIGURE 3.
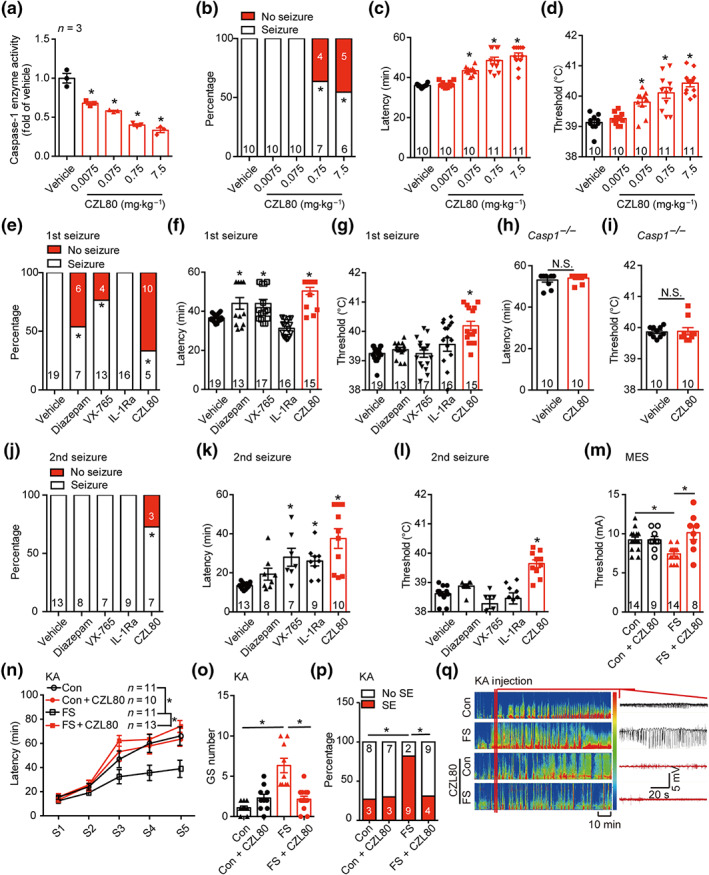
Caspase‐1 inhibitor CZL80 prevents FS generation and later enhanced epileptogenic susceptibility. (a) The enzyme activity of caspase‐1 after treatment with vehicle (2% DMSO in saline) or CZL80 (0.0075, 0.075, 0.75 and 7.5 mg·kg−1, i.v.). (b–d) The percentage of seizure (b), latency (c) and threshold (d) of FS after treatment of vehicle and CZL80 in WT mice. (e–g) The percentage of seizure (e), latency (f) and threshold (g) of FS after treatment of vehicle and CZL80 (0.75 mg·kg−1, i.v.), diazepam (0.3 mg·kg−1, i.v.), VX‐765 (50 mg·kg−1, i.p.) or IL‐1Ra (0.35 mg·kg−1, i.v.) in WT mice. (h, i) The latency (h) and threshold (i) to FS after injection of vehicle and CZL80 (0.75 mg·kg−1, i.v.) in Casp1 −/− mice. (j–l) The percentage of seizure (j), latency (k) and threshold (l) of second FS after injection of vehicle, CZL80, diazepam, VX‐765 or IL‐1Ra. (m) Threshold of MES‐induced tonic seizures in adult mice after early life prolonged FS administered with or without CZL80 (0.75 mg·kg−1, i.v.). (n–q) Seizure progression (n), number of generalized seizure (o), percentage of status epilepticus (p) and representative EEG tracing (q) in adult mice after KA‐induced seizure model in different groups. Data are presented as means ± SEM. * P < .05, significantly different from vehicle or as indicated. One‐way ANOVA followed by Dunnett post hoc test was used for (a), (c), (d), (f), (g), (k) and (l). Fisher exact test was used for (b), (e), (j) and (p). Two‐way ANOVA followed by Bonferroni post hoc test was used for (m) and (o). A general line model followed by Bonferroni post hoc test was used for (n)
As experience of FS in early life increases seizure susceptibility later in life (Dube, Brewster, Richichi, Zha, & Baram, 2007; Feng & Chen, 2016), we further investigated whether CZL80 had therapeutic effects on enhanced epileptogenic susceptibility in adult mice. We found that CZL80 administered once, immediately after prolonged FS. reduced MES‐induced adult seizure susceptibility (Figure 3m). Consistent with the data from the MES model, CZL80 also lowered the progression of KA‐induced seizure stages (Figure 3n), decreased numbers of generalized seizure (GS) (Figure 3o) and the percentage of SE (Figure 3p). Figure 3q shows the typical epileptic hippocampal discharges in different groups. Collectively, our results indicated that the caspase‐1‐specific inhibitor CZL80 delayed FS generation and blocked the enhanced epileptogenic susceptibility in adult mice, following infantile FS.
3.4. CZL80 reduces neuronal excitability
FS induce hyper‐excitability of hippocampal pyramidal cells (Koyama et al., 2012). To investigate whether caspase‐1 is involved in neuronal excitability after hyperthermia, we performed whole‐cell voltage‐clamp recording of CA1 pyramidal neurons. Electroporation of pCIG2‐Casp1 alone increased IR, lowered the action potential (AP) threshold and up‐regulated the spike number in GFP‐expressing pyramidal neurons from both WT and Casp1 −/− mice and restored the IR, AP and spike number level in Casp1 −/− neurons to normal (Figure 4a–c). At 41°C, neurons from Casp1 −/− mice and CZL80‐treated WT mice showed decreased IR, enhanced AP and reduced spike number of CA1 pyramidal neurons, compared with values from vehicle‐treated WT mice (Figure 4e–g). There was no difference in the amplitude or the ½ peak width of the action potentials. Moreover, we observed reduced amplitudes of AHP in pCIG2‐Casp1 transfected neurons and the amplitude of AHP in Casp1 −/− cells was reduced to the same level as in WT mice (Figure 4d). Neurons from Casp1 −/− mice and CZL80‐treated WT mice showed increased amplitude of AHP (Figure 4h). From these results, we concluded that CZL80 reduced neuronal excitability following a hyperthermia stimulus, through targeting caspase‐1.
FIGURE 4.
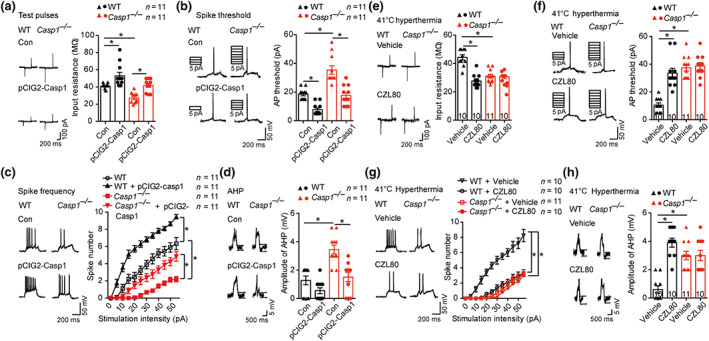
CZL80 reduces neuronal excitability. (a–d) Test pulses and input resistance (a), AP threshold (b), spike frequency (c) and amplitude of the AHP (d) in control and in utero electroporation of pCIG2‐Casp1 transfected CA1 pyramidal neurons in hippocampus slices of WT and Casp1 −/− mice. (e–h) Test pulses and input resistance (e), AP threshold (f), spike frequency (g) and amplitude of the AHP (h) in vehicle‐ and CZL80‐treated (100 μM) CA1 pyramidal neurons in hippocampus slices of WT and Casp1 −/− mice after 41°C hyperthermia stimulus. Data are presented as means ± SEM. * P < .05, significantly different as indicated.. Two‐way ANOVA followed by Bonferroni post hoc test was used for (a), (b), (d)–(f) and (h). A general line model followed by Bonferroni post hoc test was used for (c) and (g)
3.5. CZL80 has no obvious acute and chronic side effects
Based on previous pharmacological studies with diazepam and VX‐765, we further tested whether CZL80 at therapeutic doses had acute side effects. The highest therapeutic dose of CZL80 (7.5 mg·kg−1, i.v.) showed no inhibitory effects on motor activity, respiratory frequency, EEG activity, liver or renal function or biochemistry index, indicating that this low MW antagonist had few, if any, acute side effects (Figure 5a–m). This profile contrasted with that of diazepam, which at 0.3 mg·kg−1, i.v., decreased movement distance and increased the immobility time in open field test (Figure 5b–d). Diazepam also lowered the breathing rate (Figure 5e), decreased EEG activity in the low frequency range (0–4 Hz) (Figure 5g) and had an adverse effect on renal function (Figure 5l,m). This was in accordance with earlier results showing that diazepam has inhibitory effects on motor activity, the respiratory system and the CNS (Rosman et al., 1993). One possible explanation of this difference in acute side effects is that the half‐life of diazepam is much longer than that of CZL80.
FIGURE 5.
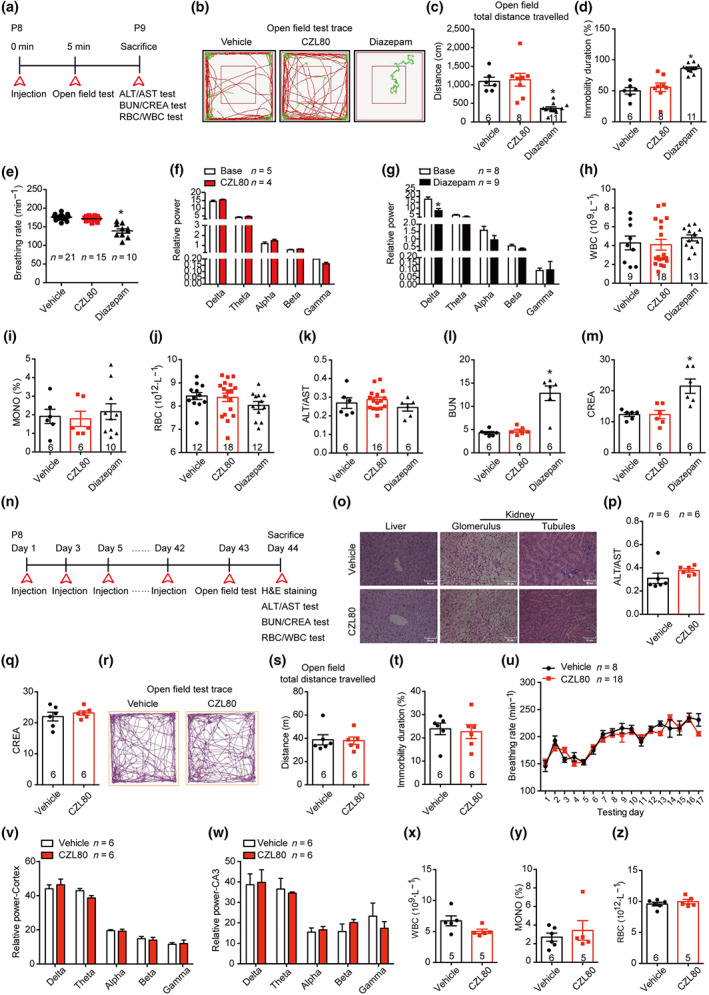
CZL80 has no obvious acute and chronic side effects. (a) Experimental paradigm of acute side effect test. (b–d) The representative tracts (b), total distance (c) and immobility duration (d) of vehicle, CZL80 (7.5 mg·kg−1, i.v.) or diazepam (0.3 mg·kg−1, i.v.)‐treated groups in open field tests. (e) The breathing rate (min−1) was tested in the vehicle, CZL80 or diazepam‐treated groups. (f, g) The EEG power in CZL80 (f) or diazepam‐treated group (g). (h–m) The WBC (h), MONO% (i), RBC (j), ALT/AST (k), BUN (l) and CREA (m) were tested in the vehicle, CZL80 and diazepam‐treated groups. (n) Experimental paradigm of chronic side effect test. (o) Representative images of morphological structure preservation of liver and kidney (Glomerulus and tubules) with HE staining in vehicle or CZL80‐treated groups, scale bar: 50 μm. (p, q) ALT/AST (p) and CREA (q) were tested in vehicle or CZL80‐treated groups. (r–t) Representative tracks (r), total distance (s) and immobility duration (t) in vehicle (2% DMSO in saline, qod for 6 weeks, i.v.) or CZL80 (7.5 mg·kg−1, qod for 6 weeks, i.v.)‐treated groups in the open field test. (u) The breathing rate was tested in vehicle or CZL80‐treated groups. (v, w) The EEG activity power of cortex (v) and hippocampal CA3 region (w) in vehicle or CZL80‐treated groups. (x–z) The WBC (x), MONO% (y) and RBC (z) were tested in vehicle or CZL80‐treated groups. Values are means ± SEM. *P < .05, significantly different from vehicle. One‐way ANOVA followed by Dunnett post hoc test was used for (c)–(e), (l) and (m). Unpaired t test was used for (g)
We also tested whether CZL80 has any chronic side effects, because clinical trials with VX‐765 were terminated due to chronic liver toxicity (Bialer et al., 2013). In our study, after treatment with vehicle (2% DMSO in saline, qod, i.v.) or CZL80 (7.5 mg·kg−1, qod, i.v.) for 6 weeks, the liver and renal function, motor activity, respiratory system, CNS and routine blood test were measured (Figure 5n–z). We observed no effects of CZL80 on the function or structure of liver and kidney. Further, CZL80 showed no chronic effect on motor activity, breathing rate, EEG activity or biochemistry index. Our results indicate that CZL80 may be a promising and effective treatment for FS and enhanced epileptogenic susceptibility in adults, with no major acute or chronic side effects.
4. DISCUSSION
Here, we showed that activated caspase‐1, which is a fever‐derived factor, increased significantly and specifically prior to FS generation. The essential pro‐convulsive role of caspase‐1 in FS generation is supported by the results of our experiments evaluating treatment with exogenous caspase‐1 and caspase‐1 overexpression; we demonstrated that caspase‐1 is a seizure‐promoting factor that makes the brain vulnerable to a hyperthermic stimulus. In contrast, Casp1 −/− mice were resistant to FS generation. Our findings provided important clues about the natural variability in the prevalence of FS, as different levels of caspase‐1, in our mouse model, modified the susceptibility to these seizures. Patients with FS have been reported to have increased expression of Casp‐1 gene relative to the general population (Sasaki, Matsuo, Maeda, Zaitsu, & Hamasaki, 2009). Further, caspase‐1 increased neuronal excitability with increased IR, decreased AP threshold and increased spike frequency, which were absent in Casp1 −/− mice. AHP is a key component in controlling the discharge of CA1 pyramidal cells and is determined by potassium currents (Kamal, Artola, Biessels, Gispen, & Ramakers, 2003; Madison & Nicoll, 1984). It is thus possible that calcium‐activated potassium currents or hyperpolarization‐activated currents (Ih) may be involved in the mechanism through which caspase‐1 causes seizures (Lancaster & Nicoll, 1987; Madison & Nicoll, 1984). Moreover, Casp1 −/− mice showed impaired processing of pro‐IL‐1β and reduced secretion of IL‐1β after LPS stimulation (Yao et al., 1999). Thus, caspase‐1 could regulate seizure generation through a IL‐1β pathway. It should be noted that the Casp1 −/− mice were also reported to have a deletion in the gene for caspase‐11 and therefore, data interpretation in our study should take this into consideration, and further study is still needed to address the role of caspase‐11 in FS. Although a caspase‐1–IL‐1β pathway has been implicated in the pathophysiology of epilepsy, our present findings demonstrated that caspase‐1 also mediates FS generation and represents a promising therapeutic target, and an inhibitor of caspase‐1 might provide a drug candidate for this therapeutic target.
At present, druggable and safe low MW antagonists targeting the caspase‐1–IL‐1β pathway are not available. Based on the structure and active sites of caspase‐1, we searched for and found a novel, selective, brain‐penetrable caspase‐1 inhibitor, CZL80. As the structure of CZL80 is more favourable in matching with the conformation of the binding pocket of caspase‐1, it is a structure‐matching compound with strong affinity to the active sites of caspase‐1. CZL80 had several other obviously appealing characteristics as a potential drug. First, it was a low MW compound and penetrated the BBB rapidly, thus it had better “druggability” compared with the previous peptide inhibitors or high MW agents such as IL‐1Ra (Sukedai et al., 2011). Second, it was a potent, selective and efficacious caspase‐1 inhibitor with high anticonvulsive effect at a low dose, compared with diazepam, VX‐765 and IL‐1Ra. Third, no major acute or chronic adverse effects were detected for CZL80 in monitoring motor activity, respiratory depression, CNS inhibition and renal or liver toxicities, suggesting that CZL80 may be a safer treatment than other commercial compounds. In addition, we have tested CZL80 at dose of 7.5 mg·kg−1 in naïve mice and found that the body temperature of CZL80‐treated mice showed no difference from that of the control groups, suggesting that CZL80 may not affect body temperature in basal condition, but rather in condition of high expression of caspase‐1. Moreover, CZL80 reduced the incidence of FS recurrence. Further, we observed increased cleaved caspase‐1 before FS recurrence, suggesting that controlling the inflammatory process in FS recurrence may be very important. In addition to the effect on the FS generation, it is still valid in preventing seizures in adults, after prolonged FS. As targeting the caspase‐1–IL‐1β pathway can affect neural excitability (Iori, Frigerio, & Vezzani, 2016; Wang & Chen, 2019), we found that CZL80 decreased neuronal excitability, suggesting the potential of CZL80 being a potential anti‐convulsant compound in other epilepsy models. What should be noted is that the pharmacokinetic data of CZL80 are not optimal and, consequently, CZL80 itself is unlikely to be used clinically but it would be a lead compound in designing a new compound with a longer half‐life.
In conclusion, our work indicates a caspase‐1‐mediated mechanism for triggering hyperthermia‐induced seizures and demonstrates new safe strategies for the acute treatment and prevention of fever‐related epileptic syndromes and the following enhanced epileptogenic susceptibility in adults.
AUTHOR CONTRIBUTIONS
Y.S.T., B.F. and Z.C. designed the study. Y.S.T. and B.F. performed animal behavioural examination, western blotting, in utero electroporation and EEG recording. Y.W. and Y.Y. performed experiments of seizure susceptibility. J.Y. and C.L.X. performed electrophysiological experiments. B.C. and Y.P.R. performed the experiment of histology. H.Y.S. and T.J.H. designed the small molecular compounds. S.L.C. contributed to the synthesis of small molecular compounds. G.H. provided mutant mice and scientific insight. Y.S.T., B.F. and Y.W. contributed to data analysis and paper writing. Y.W. and Z.C. had oversight of the project.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, Immunoblotting and Immunochemistry, and Animal Experimentation, and as recommended by funding agencies, publishers and other organizations engaged with supporting research.
Supporting information
Figure S1. Fever, but not seizure, increases the expression of cleaved caspase‐1
Figure S2. Validation of different docking approaches on the test dataset
Figure S3. Screening the top‐ranked compounds in vitro and in vivo
Figure S4. CZL80 penetrates the BBB
Figure S5. Structural and energetic representations of the 3 compounds (CZL55, CZL78, and CZL06) interacting with caspase‐1
Table S1. Detailed information of the selected 50 compounds obtained and tested in bioassays
Table S2. H‐bond occupancy details in each active compound
Table S3. Binding free energy of the four tight‐binding compounds
ACKNOWLEDGEMENTS
This work was funded by grants from the National Natural Science Foundation of China (81630098 and 81521062) and the Young Elite Scientist Sponsorship Program by CAST (2018QNRC001).
Tang Y, Feng B, Wang Y, et al. Structure‐based discovery of CZL80, a caspase‐1 inhibitor with therapeutic potential for febrile seizures and later enhanced epileptogenic susceptibility. Br J Pharmacol. 2020;177:3519–3534. 10.1111/bph.15076
Yangshun Tang, Bo Feng, Yi Wang and Huiyong Sun contributed equally to this paper.
Contributor Information
Tingjun Hou, Email: tingjunhou@zju.edu.cn.
Zhong Chen, Email: chenzhong@zju.edu.cn.
REFERENCES
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , … Sharman, J. L. (2019). The Concise Guide to PHARMACOLOGY 2019/20: Enzymes. British Journal of Pharmacology, 176(Suppl 1), S297–S396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Roberts, R. E. , Broughton, B. R. S. , Sobey, C. G. , George, C. H. , Stanford, S. C. , … Ahluwalia, A. (2018). Goals and practicalities of immunoblotting and immunohistochemistry: A guide for submission to the British Journal of Pharmacology . British Journal of Pharmacology, 175, 407–411. 10.1111/bph.14112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bialer, M. , Johannessen, S. I. , Levy, R. H. , Perucca, E. , Tomson, T. , & White, H. S. (2013). Progress report on new antiepileptic drugs: A summary of the Eleventh Eilat Conference (EILAT XI). Epilepsy Research, 103, 2–30. 10.1016/j.eplepsyres.2012.10.001 [DOI] [PubMed] [Google Scholar]
- Callus, B. A. , & Vaux, D. L. (2007). Caspase inhibitors: Viral, cellular and chemical. Cell Death and Differentiation, 14, 73–78. 10.1038/sj.cdd.4402034 [DOI] [PubMed] [Google Scholar]
- Chen, B. , Feng, B. , Tang, Y. S. , You, Y. , Wang, Y. , Hou, W. W. , … Chen, Z. (2016). Blocking GluN2B subunits reverses the enhanced seizure susceptibility after prolonged febrile seizures with a wide therapeutic time‐window. Experimental Neurology, 283, 29–38. 10.1016/j.expneurol.2016.05.034 [DOI] [PubMed] [Google Scholar]
- Chen, B. , Xu, C. , Wang, Y. , Lin, W. , Wang, Y. , Chen, L. , … Chen, Z. (2020). A disinhibitory nigra‐parafascicular pathway amplifies seizure in temporal lobe epilepsy. Nature Communications, 11, 923 10.1038/s41467-020-14648-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chungath, M. , & Shorvon, S. (2008). The mortality and morbidity of febrile seizures. Nature Clinical Practice Neurology, 4, 610–621. 10.1038/ncpneuro0922 [DOI] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175, 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai, Y. J. , Wu, D. C. , Feng, B. , Chen, B. , Tang, Y. S. , Jin, M. M. , … Chen, Z. (2019). Prolonged febrile seizures induce inheritable memory deficits in rats through DNA methylation. CNS Neuroscience & Therapeutics, 25, 601–611. 10.1111/cns.13088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dube, C. , Vezzani, A. , Behrens, M. , Bartfai, T. , & Baram, T. Z. (2005). Interleukin‐1β contributes to the generation of experimental febrile seizures. Annals of Neurology, 57, 152–155. 10.1002/ana.20358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dube, C. M. , Brewster, A. L. , Richichi, C. , Zha, Q. , & Baram, T. Z. (2007). Fever, febrile seizures and epilepsy. Trends in Neurosciences, 30, 490–496. 10.1016/j.tins.2007.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dube, C. M. , Ravizza, T. , Hamamura, M. , Zha, Q. , Keebaugh, A. , Fok, K. , … Baram, T. Z. (2010). Epileptogenesis provoked by prolonged experimental febrile seizures: Mechanisms and biomarkers. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 30, 7484–7494. 10.1523/JNEUROSCI.0551-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eun, B. L. , Abraham, J. , Mlsna, L. , Kim, M. J. , & Koh, S. (2015). Lipopolysaccharide potentiates hyperthermia‐induced seizures. Brain and Behavior: A Cognitive Neuroscience Perspective, 5, e00348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, B. , & Chen, Z. (2016). Generation of febrile seizures and subsequent epileptogenesis. Neuroscience Bulletin, 32, 481–492. 10.1007/s12264-016-0054-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, B. , Tang, Y. , Chen, B. , Xu, C. , Wang, Y. , Dai, Y. , … Chen, Z. (2016). Transient increase of interleukin‐1β after prolonged febrile seizures promotes adult epileptogenesis through long‐lasting upregulating endocannabinoid signaling. Scientific Reports, 6, 21931 10.1038/srep21931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer, W. , Kittner, H. , Regenthal, R. , Malinowska, B. , & Schlicker, E. (2001). Anticonvulsant and sodium channel blocking activity of higher doses of clenbuterol. Naunyn‐Schmiedeberg's Archives of Pharmacology, 363, 182–192. [DOI] [PubMed] [Google Scholar]
- Galatsis, P. , Caprathe, B. , Gilmore, J. , Thomas, A. , Linn, K. , Sheehan, S. , … Talanian, R. (2010). Succinic acid amides as P2‐P3 replacements for inhibitors of interleukin‐1β converting enzyme (ICE or caspase 1). Bioorganic & Medicinal Chemistry Letters, 20, 5184–5190. 10.1016/j.bmcl.2010.07.004 [DOI] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR . (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heida, J. G. , Moshe, S. L. , & Pittman, Q. J. (2009). The role of interleukin‐1β in febrile seizures. Brain & Development, 31, 388–393. 10.1016/j.braindev.2008.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou, T. , Wang, J. , Li, Y. , & Wang, W. (2011). Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. Journal of Chemical Information and Modeling, 51, 69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iori, V. , Frigerio, F. , & Vezzani, A. (2016). Modulation of neuronal excitability by immune mediators in epilepsy. Current Opinion in Pharmacology, 26, 118–123. 10.1016/j.coph.2015.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi, S. , Rajasekaran, K. , Sun, H. Y. , Williamson, J. , & Kapur, J. (2017). Enhanced AMPA receptor‐mediated neurotransmission on CA1 pyramidal neurons during status epilepticus. Neurobiology of Disease, 103, 45–53. 10.1016/j.nbd.2017.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamal, A. , Artola, A. , Biessels, G. J. , Gispen, W. H. , & Ramakers, G. M. (2003). Increased spike broadening and slow afterhyperpolarization in CA1 pyramidal cells of streptozotocin‐induced diabetic rats. Neuroscience, 118, 577–583. 10.1016/s0306-4522(02)00874-6 [DOI] [PubMed] [Google Scholar]
- Kayagaki, N. , Warming, S. , Lamkanfi, M. , Vande Walle, L. , Louie, S. , Dong, J. , … Dixit, V. M. (2011). Non‐canonical inflammasome activation targets caspase‐11. Nature, 479, 117–121. 10.1038/nature10558 [DOI] [PubMed] [Google Scholar]
- Khosroshahi, N. , Faramarzi, F. , Salamati, P. , Haghighi, S. M. , & Kamrani, K. (2011). Diazepam versus clobazam for intermittent prophylaxis of febrile seizures. Indian Journal of Pediatrics, 78, 38–40. [DOI] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. 10.1111/j.1476-5381.2010.00872.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama, R. , Tao, K. , Sasaki, T. , Ichikawa, J. , Miyamoto, D. , Muramatsu, R. , … Ikegaya, Y. (2012). GABAergic excitation after febrile seizures induces ectopic granule cells and adult epilepsy. Nature Medicine, 18, 1271–1278. 10.1038/nm.2850 [DOI] [PubMed] [Google Scholar]
- Lancaster, B. , & Nicoll, R. A. (1987). Properties of two calcium‐activated hyperpolarizations in rat hippocampal neurones. The Journal of Physiology, 389, 187–203. 10.1113/jphysiol.1987.sp016653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lux, A. L. (2010). Treatment of febrile seizures: Historical perspective, current opinions, and potential future directions. Brain & Development, 32, 42–50. [DOI] [PubMed] [Google Scholar]
- Lyne, P. D. (2002). Structure‐based virtual screening: An overview. Drug Discovery Today, 7, 1047–1055. [DOI] [PubMed] [Google Scholar]
- Madison, D. V. , & Nicoll, R. A. (1984). Control of the repetitive discharge of rat CA 1 pyramidal neurones in vitro. The Journal of Physiology, 354, 319–331. 10.1113/jphysiol.1984.sp015378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man, S. M. , Karki, R. , Briard, B. , Burton, A. , Gingras, S. , Pelletier, S. , & Kanneganti, T. D. (2017). Differential roles of caspase‐1 and caspase‐11 in infection and inflammation. Scientific Reports, 7, 45126 10.1038/srep45126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath, J. C. , & Lilley, E. (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): New requirements for publication in BJP . British Journal of Pharmacology, 172, 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng, X. F. , Tan, L. , Tan, M. S. , Jiang, T. , Tan, C. C. , Li, M. M. , … Yu, J. T. (2014). Inhibition of the NLRP3 inflammasome provides neuroprotection in rats following amygdala kindling‐induced status epilepticus. Journal of Neuroinflammation, 11, 212 10.1186/s12974-014-0212-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mula, M. (2014). The safety and tolerability of intranasal midazolam in epilepsy. Expert Review of Neurotherapeutics, 14, 735–740. 10.1586/14737175.2014.925398 [DOI] [PubMed] [Google Scholar]
- Pan, P. , Tian, S. , Sun, H. , Kong, X. , Zhou, W. , Li, D. , … Hou, T. (2015). Identification and preliminary SAR analysis of novel type‐I inhibitors of TIE‐2 via structure‐based virtual screening and biological evaluation in in vitro models. Journal of Chemical Information and Modeling, 55, 2693–2704. 10.1021/acs.jcim.5b00576 [DOI] [PubMed] [Google Scholar]
- Patel, N. , Ram, D. , Swiderska, N. , Mewasingh, L. D. , Newton, R. W. , & Offringa, M. (2015). Febrile seizures. BMJ, 351, h4240. [DOI] [PubMed] [Google Scholar]
- Pust, B. (2004). Febrile seizures—An update. Kinderkrankenschwester: Organ der Sektion Kinderkrankenpflege, 23, 328–331. [PubMed] [Google Scholar]
- Ramos‐Guzman, C. A. , Zinovjev, K. , & Tunon, I. (2019). Modeling caspase‐1 inhibition: Implications for catalytic mechanism and drug design. European Journal of Medicinal Chemistry, 169, 159–167. 10.1016/j.ejmech.2019.02.064 [DOI] [PubMed] [Google Scholar]
- Rosman, N. P. , Colton, T. , Labazzo, J. , Gilbert, P. L. , Gardella, N. B. , Kaye, E. M. , … Winter, M. R. (1993). A controlled trial of diazepam administered during febrile illnesses to prevent recurrence of febrile seizures. The New England Journal of Medicine, 329, 79–84. 10.1056/NEJM199307083290202 [DOI] [PubMed] [Google Scholar]
- Ruusuvuori, E. , Huebner, A. K. , Kirilkin, I. , Yukin, A. Y. , Blaesse, P. , Helmy, M. , … Kaila, K. (2013). Neuronal carbonic anhydrase VII provides GABAergic excitatory drive to exacerbate febrile seizures. The EMBO Journal, 32, 2275–2286. 10.1038/emboj.2013.160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saghazadeh, A. , Gharedaghi, M. , Meysamie, A. , Bauer, S. , & Rezaei, N. (2014). Proinflammatory and anti‐inflammatory cytokines in febrile seizures and epilepsy: Systematic review and meta‐analysis. Reviews in the Neurosciences, 25, 281–305. 10.1515/revneuro-2013-0045 [DOI] [PubMed] [Google Scholar]
- Sasaki, K. , Matsuo, M. , Maeda, T. , Zaitsu, M. , & Hamasaki, Y. (2009). Febrile seizures: Characterization of double‐stranded RNA‐induced gene expression. Pediatric Neurology, 41, 114–118. [DOI] [PubMed] [Google Scholar]
- Schott, W. H. , Haskell, B. D. , Tse, H. M. , Milton, M. J. , Piganelli, J. D. , Choisy‐Rossi, C. M. , … Leiter, E. H. (2004). Caspase‐1 is not required for type 1 diabetes in the NOD mouse. Diabetes, 53, 99–104. 10.2337/diabetes.53.1.99 [DOI] [PubMed] [Google Scholar]
- Schroder, K. , & Tschopp, J. (2010). The inflammasomes. Cell, 140, 821–832. [DOI] [PubMed] [Google Scholar]
- Schuchmann, S. , Schmitz, D. , Rivera, C. , Vanhatalo, S. , Salmen, B. , Mackie, K. , … Kaila, K. (2006). Experimental febrile seizures are precipitated by a hyperthermia‐induced respiratory alkalosis. Nature Medicine, 12, 817–823. 10.1038/nm1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner, R. A. , Gibson, R. M. , Rothwell, N. J. , Pinteaux, E. , & Penny, J. I. (2009). Transport of interleukin‐1 across cerebromicrovascular endothelial cells. British Journal of Pharmacology, 156, 1115–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukedai, M. , Ariyoshi, W. , Okinaga, T. , Iwanaga, K. , Habu, M. , Yoshioka, I. , … Nishihara, T. (2011). Inhibition of adjuvant arthritis in rats by electroporation with interleukin‐1 receptor antagonist. Journal of Interferon & Cytokine Research: The Official Journal of the International Society for Interferon and Cytokine Research, 31, 839–846. 10.1089/jir.2011.0024 [DOI] [PubMed] [Google Scholar]
- Sun, H. Y. , Hou, T. J. , & Zhang, H. Y. (2014). Finding chemical drugs for genetic diseases. Drug Discovery Today, 19, 1836–1840. 10.1016/j.drudis.2014.09.013 [DOI] [PubMed] [Google Scholar]
- Tan, C. C. , Zhang, J. G. , Tan, M. S. , Chen, H. , Meng, D. W. , Jiang, T. , … Tan, L. (2015). NLRP1 inflammasome is activated in patients with medial temporal lobe epilepsy and contributes to neuronal pyroptosis in amygdala kindling‐induced rat model. Journal of Neuroinflammation, 12, 18 10.1186/s12974-014-0233-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verity, C. M. , Butler, N. R. , & Golding, J. (1985). Febrile convulsions in a national cohort followed up from birth. II—Medical history and intellectual ability at 5 years of age. British Medical Journal, 290, 1311–1315. 10.1136/bmj.290.6478.1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzani, A. , Balosso, S. , Maroso, M. , Zardoni, D. , Noe, F. , & Ravizza, T. (2010). ICE/caspase 1 inhibitors and IL‐1β receptor antagonists as potential therapeutics in epilepsy. Current Opinion in Investigational Drugs, 11, 43–50. [PubMed] [Google Scholar]
- Vezzani, A. , Maroso, M. , Balosso, S. , Sanchez, M. A. , & Bartfai, T. (2011). IL‐1 receptor/Toll‐like receptor signaling in infection, inflammation, stress and neurodegeneration couples hyperexcitability and seizures. Brain, Behavior, and Immunity, 25, 1281–1289. [DOI] [PubMed] [Google Scholar]
- Vigers, G. P. , Dripps, D. J. , Edwards, C. K. 3rd , & Brandhuber, B. J. (2000). X‐ray crystal structure of a small antagonist peptide bound to interleukin‐1 receptor type 1. The Journal of Biological Chemistry, 275, 36927–36933. 10.1074/jbc.M006071200 [DOI] [PubMed] [Google Scholar]
- Wang, Y. , & Chen, Z. (2019). An update for epilepsy research and antiepileptic drug development: Toward precise circuit therapy. Pharmacology & Therapeutics, 201, 77–93. [DOI] [PubMed] [Google Scholar]
- Wang, Y. , Xu, C. , Xu, Z. , Ji, C. , Liang, J. , Wang, Y. , … Guo, Y. (2017). Depolarized GABAergic signaling in subicular microcircuits mediates generalized seizure in temporal lobe epilepsy. Neuron, 95, 92–105 e105. [DOI] [PubMed] [Google Scholar]
- Wannamaker, W. , Davies, R. , Namchuk, M. , Pollard, J. , Ford, P. , Ku, G. , … Kuida, K. (2007). (S)‐1‐((S)‐2‐{[1‐(4‐amino‐3‐chloro‐phenyl)‐methanoyl]‐amino}‐3,3‐dimethyl‐butanoy l)‐pyrrolidine‐2‐carboxylic acid ((2R,3S)‐2‐ethoxy‐5‐oxo‐tetrahydro‐furan‐3‐yl)‐amide (VX‐765), an orally available selective interleukin (IL)‐converting enzyme/caspase‐1 inhibitor, exhibits potent anti‐inflammatory activities by inhibiting the release of IL‐1β and IL‐18. The Journal of Pharmacology and Experimental Therapeutics, 321, 509–516. [DOI] [PubMed] [Google Scholar]
- Wilson, K. P. , Black, J. A. , Thomson, J. A. , Kim, E. E. , Griffith, J. P. , Navia, M. A. , … Raybuck, S. A. (1994). Structure and mechanism of interleukin‐1β converting enzyme. Nature, 370, 270–275. 10.1038/370270a0 [DOI] [PubMed] [Google Scholar]
- Wu, D. C. , Feng, B. , Dai, Y. J. , Wu, X. H. , Chen, B. , Xu, C. L. , … Chen, Z. (2017). Intergenerational transmission of enhanced seizure susceptibility after febrile seizures. eBioMedicine, 17, 206–215. 10.1016/j.ebiom.2017.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, L. , Zhang, Y. , Zheng, L. , Qiao, C. , Li, Y. , Li, D. , … Hou, T. (2014). Discovery of novel inhibitors targeting the macrophage migration inhibitory factor via structure‐based virtual screening and bioassays. Journal of Medicinal Chemistry, 57, 3737–3745. 10.1021/jm401908w [DOI] [PubMed] [Google Scholar]
- Xu, Z. H. , Wang, Y. , Tao, A. F. , Yu, J. , Wang, X. Y. , Zu, Y. Y. , … Chen, Z. (2016). Interleukin‐1 receptor is a target for adjunctive control of diazepam‐refractory status epilepticus in mice. Neuroscience, 328, 22–29. 10.1016/j.neuroscience.2016.04.036 [DOI] [PubMed] [Google Scholar]
- Yang, C. Y. (2015). Identification of potential small molecule allosteric modulator sites on IL‐1R1 ectodomain using accelerated conformational sampling method. PLoS ONE, 10, e0118671 10.1371/journal.pone.0118671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, J. H. , Ye, S. M. , Burgess, W. , Zachary, J. F. , Kelley, K. W. , & Johnson, R. W. (1999). Mice deficient in interleukin‐1β converting enzyme resist anorexia induced by central lipopolysaccharide. The American Journal of Physiology, 277, R1435–R1443. [DOI] [PubMed] [Google Scholar]
- Zhang, F. , Wang, L. , Wang, J. J. , Luo, P. F. , Wang, X. T. , & Xia, Z. F. (2016). The caspase‐1 inhibitor AC‐YVAD‐CMK attenuates acute gastric injury in mice: involvement of silencing NLRP3 inflammasome activities. Scientific Reports, 6, 24166 10.1038/srep24166 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Fever, but not seizure, increases the expression of cleaved caspase‐1
Figure S2. Validation of different docking approaches on the test dataset
Figure S3. Screening the top‐ranked compounds in vitro and in vivo
Figure S4. CZL80 penetrates the BBB
Figure S5. Structural and energetic representations of the 3 compounds (CZL55, CZL78, and CZL06) interacting with caspase‐1
Table S1. Detailed information of the selected 50 compounds obtained and tested in bioassays
Table S2. H‐bond occupancy details in each active compound
Table S3. Binding free energy of the four tight‐binding compounds