

Abstract
目的
报告一个遗传性血小板功能障碍家系并探索其分子发病机制。
方法
收集该家系临床资料,采用二代高通量测序法进行遗传性血液病和血小板功能相关近600种基因突变筛查。采用RT-PCR及一代测序法检测先证者及其父母RASGRP2基因部分转录本序列。
结果
该遗传性血小板功能障碍家系临床表现符合变异型血小板无力症,表现为反复皮肤黏膜严重出血,先证者血小板对多种不同浓度的生理性诱聚剂反应低下甚至缺如。先证者未检出整合素αⅡbβ3基因突变,而存在RASGRP2 IVS3-1C>G纯合突变,其父母均为该位点杂合突变。先证者在扩增区检测出2种转录本,功能型RASGRP2蛋白对应的正常转录本未检出,其父母同时存在正常转录本及异常转录本,但以正常转录本为主。
结论
RASGRP2基因IVS3-1C>G纯合突变是导致该家系先证者发生血小板功能障碍性出血的原因。RASGRP2基因IVS3-1C>G突变导致RASGRP2异常剪接,发挥激活Rap1的RASGRP2蛋白缺失,进而导致血小板功能障碍。本研究为国内首次报道RASGRP2基因异常导致血小板功能障碍性出血,也是国际上首次报道该突变类型。
Keywords: 遗传性血小板功能障碍, RASGRP2基因, 剪接突变
Abstract
Objective
To review the clinical characteristics of a pedigree with inherited hemorrhagic disease to explore its molecular pathogenesis.
Methods
The clinical data of the pedigree with inherited hemorrhagic disease were collected. After extracting DNA, next generation sequencing was utilized to detect the potential gene mutation. The changes of RASGRP2 transcript of this proband and his parents were detected using RT-PCR to compare with normal control.
Results
The phenotype of the proband in this pedigree with inherited platelet dysfunction and bleeding disorder was similar to variant Glanzmann's thrombasthenia, the maximum aggregations of platelet in response to the physiological agonists including ADP, epinephrine and arachidonic acid were significantly lower, leading to severe spontaneous mucosal bleeding. Integrin αIIbβ3 gene mutation was not detected, but another gene mutation RASGRP2 IVS3-1 stood out. The mutation was homozygous in the proband and heterozygosis in both of his parents. Two transcript types were detected in the proband, without transcripts coding functional RASGRP2 protein, however, his parents had functional transcripts and abnormal transcripts, with the normal transcripts in the majority.
Conclusion
The RASGRP2 IVS3-1 gene mutation was responsible for the inherited hemorrhagic disease. The RASGRP2 IVS3-1 gene mutation led to abnormal alternative splicing, without formation of functional RASGRP2 protein. The RASGRP2 protein is at the nexus of calcium-dependent platelet activation and hemostasis after damage of blood vessels. Spontaneous mucosal bleeding was a result of the lack of the functional RASGRP2 protein. This was the first report of RASGRP2 gene mutation resulting in bleeding disorder in China, and also the first report of the mutation type of RASGRP2 IVS3-1.
Keywords: Inherited platelet function disorders, RASGRP2 gene, Splicing mutation
遗传性血小板功能障碍性疾病(IPFD)是一组异质性疾病,根据发病机制不同,IPFD主要包括血小板表面糖蛋白异常、血小板内颗粒异常、血小板信号转导及分泌功能异常及血小板促凝活性异常等[1]–[3],其中信号转导异常最常见,但由于认识不足及技术受限,大多数信号转导异常导致的出血性疾病难以作出临床诊断。本研究报道1个临床表现符合变异型遗传性血小板无力症的遗传性血小板功能障碍家系,并对其分子发病机制进行探索。发现其分子发病机制为RASGRP2基因发生剪接位点的突变,而RASGRP2通过激活Rap1在血小板激活过程中发挥关键作用[4]–[5]。目前国际上关于RASGRP2基因突变导致的血小板功能障碍性出血报道较少,国内尚无相关报道,本研究在国内首次报道RASGRP2基因突变导致血小板功能障碍性出血,也是在国际上首次报道该突变类型。
病例资料
一、临床表现及家系资料
先证者,男,9岁,来自云南省,苗族,自3岁开始反复发生皮肤瘀点瘀斑及双侧鼻出血,每次鼻出血时间长达数小时,余无其他部位明显出血史。先证者父母非近亲婚配,且均无出血症状。先证者父亲的2个姐姐及1个哥哥有与先证者相似出血症状,分别于1、2、4岁死亡。家系其他成员无异常出血史(图1)。先证者及其父母血小板计数、血块收缩试验正常,凝血因子Ⅱ、Ⅴ、Ⅶ、Ⅷ、Ⅸ、Ⅹ、Ⅺ、Ⅻ活性及血管性血友病因子(VWF)、纤维蛋白原含量均正常。由于长期反复出血,先证者就诊时存在中度贫血(HGB 64 g/L),铁蛋白6.83 ng/ml(参考范围23.00~336.00 ng/ml)。先证者血小板大小、形态正常,散在分布,无成簇现象。先证者血小板对不同生理性诱聚剂聚集反应均低下甚至缺如(图2),其父母血小板聚集功能基本正常。
图1. 遗传性血小板功能障碍家系系谱图.
图2. 先证者(A)及正常对照(B)血小板聚集曲线.
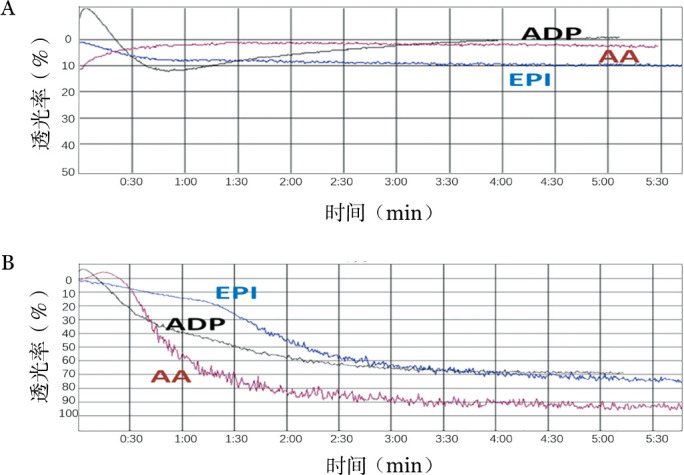
ADP:10 µmol/L二磷酸腺苷;EPI:5 µmol/L肾上腺素;AA:150 mg/L花生四烯酸
二、血小板整合素αⅡbβ3表达及基因突变情况
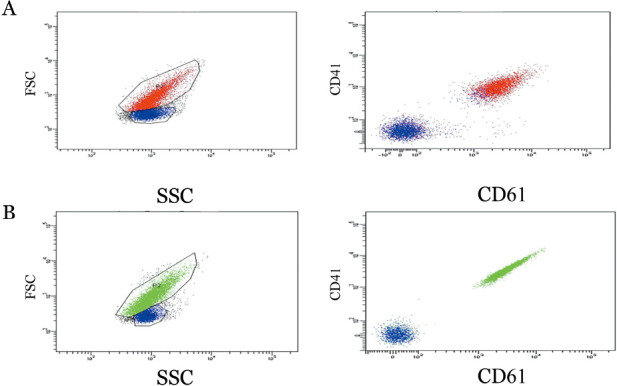
流式细胞术检测先证者及其父母血小板表面CD41(αⅡb)及CD61(β3)表达量基本正常(图3)。二代高通量测序亦未发现αⅡbβ3基因突变。
图3. 流式细胞术检测先证者(A)及正常对照(B)血小板表面CD41(αⅡb)及CD61(β3)表达.
三、基因分析
1.高通量二代测序结果:应用二代高通量测序筛查遗传性疾病和与血小板功能疾病相关的近600种基因(海斯特医学检验公司),其中包括αⅡbβ3基因、PTGS1基因、TBXA2R基因、GPⅠb基因、Kindlin-3基因、P2RY12基因等,共发现以下8种基因变异(表1)。其中PPOX基因编码原卟啉原氧化酶,该基因突变可引起变异性血卟啉病(VP)[6];RFXANK基因突变可导致MHC Ⅱ类分子缺乏,进而导致免疫缺陷[7],但这2种基因变异在国际千人基因组中检出比例均超过1%,且该先证者均无相关症状,考虑为基因多态性。C7基因编码补体C7,其缺陷可导致萘瑟菌感染[8];CTNS基因缺陷可导致溶酶体膜转运缺陷引起胱氨酸贮积症[9];PRF1基因编码穿孔素蛋白前体,其缺陷可导致家族性噬血细胞综合征(FHL2)[10],以上这几种疾病均为常染色体隐性遗传性疾病,编码基因杂合突变不致病,且本研究中先证者亦无以上疾病的临床表现。RPL32为核糖体蛋白编码基因,该基因突变可导致先天性纯红细胞再生障碍性贫血[11];VHL基因为抑癌基因,该基因异常可导致VHL综合征[12],是一种家族性肿瘤综合征,以上2种疾病为常染色体显性遗传,先证者无相关疾病表现,且以上疾病临床表现中亦无出血症状。
表1. 二代测序结果.
| 染色体 | 突变前碱基 | 突变后碱基 | 基因 | 突变类型 | 基因组中频率 | 类型 |
| 11 | C | G | RASGRP2 | 剪接突变 | N* | 纯合 |
| 5 | G | T | C7 | 非同义点突变 | N* | 杂合 |
| 17 | A | T | CTNS | 非同义点突变 | N* | 杂合 |
| 1 | G | A | PPOX | 非同义点突变 | 0.05 | 纯合 |
| 10 | G | A | PRF1 | 非同义点突变 | N* | 杂合 |
| 19 | C | G | RFXANK | 非同义点突变 | 0.05 | 纯合 |
| 3 | C | T | RPL32 | 非同义点突变 | 0.0009 | 杂合 |
| 3 | G | A | VHL | 非同义点突变 | N* | 杂合 |
注:*国际千人基因组中未检出
由此,上述基因突变并非该血小板功能障碍的致病原因,而RASGRP2蛋白在血小板激活中起关键作用,且我们发现的RASGRP2 IVS3-1C>G位于剪接受体位点,通过剪接预测软件human splicing finder进行剪接预测分析,结果显示野生型剪接受体位点被破坏,该突变很有可能导致mRNA的剪接异常。因此考虑该家系致病基因可能为RASGRP2基因突变。
2.RASGRP2基因Sanger测序结果:PCR扩增突变位点所在区域DNA片段后行一代Sanger测序再次验证先证者RASGRP2基因突变,并验证先证者父母该位点基因突变情况。NCBI中查询RASGRP2基因2号内含子及3号外显子之间的碱基序列为GGAGTCATGTGACTCC,而测序结果显示先证者存在RASGRP2基因IVS3-1C>G纯合突变,其父母分别为该位点的杂合突变(图4)。
图4. 先证者及其父母RASGRP2基因Sanger测序图.

A:野生型;B:先证者;C:先证者父母
3.外周血RASGRP2转录本测序结果:扩增先证者、先证者父母、正常人外显子2-外显子5 cDNA片段,PCR产物琼脂糖凝胶电泳结果如图5所示,目的条带处先证者、先证者父母、正常对照均为两条带。NCBI中记录RASGRP2基因存在4种转录本,即2、3、4、5号转录本,在我们扩增区域2、3、4号转录本序列相同,而5号转录本比2、3、4号转录本在3号外显子序列中少80 bp。测序结果经过与RASGRP2不同转录本对照分析,发现先证者扩增区未检测出正常的2、3、4号转录本对应序列,扩增区存在2种类型转录本片段:3号外显子缺失80 bp(5号转录本)、3号外显子前面插入7 bp的内含子序列。而先证者父亲、母亲及对照扩增区均以正常2、3、4号转录本片段序列为主。
图5. 先证者、先证者父母、正常对照转录本片段PCR电泳图.
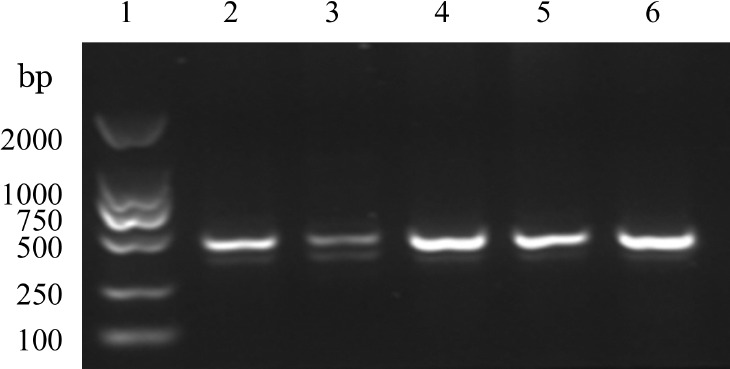
1:Marker;2:父亲;3:先证者;4:母亲;5、6:对照
讨论
遗传性血小板无力症(Glanzmann's thrombasthenia, GT)是发病率最高的IPFD,其中Ⅰ、Ⅱ型为血小板表面整合素αⅡbβ3数量减少,Ⅲ型为αⅡbβ3功能异常,又称变异型[3]。根据先证者临床特点分析,考虑该先证者符合血小板无力症,血小板无力症致病原因为整合素αⅡbβ3基因纯合突变或双重杂合突变,但二代测序结果显示先证者αⅡbβ3基因无突变。
血小板表面整合素αⅡbβ3通过一系列信号通路激活后参与血小板聚集,该先证者可能原因为αⅡbβ3上游信号分子的异常而导致αⅡbβ3激活异常,进而血小板聚集功能障碍。RASGRP2蛋白在血小板激活中起关键作用,因此我们将致病基因锁定于RASGRP2基因IVS3-1C>G上,该突变为2号内含子剪接受体位点的纯合突变,目前尚无该突变类型的相关报道。
在剪接位置的核苷酸信号指引着剪接的精确位置,当内含子发生突变会造成错误的剪接,剪接突变常导致编码蛋白的异常[13]–[14]。我们发现的RASGRP2 IVS3-1C>G位于剪接受体位点,通过剪接预测软件human splicing finder进行剪接预测分析,结果显示野生型剪接受体位点被破坏,因此该突变很有可能导致mRNA的剪接异常。
RASGRP2基因在NCBI中记录有4种转录本,2、3、4号转录本在突变位点附近序列相同,而5号转录本在此位置与2、3、4号转录本相比,在3号外显子序列中少80 bp。2、3、4号转录本对应RASGRP2蛋白亚型a,5号转录本对应蛋白亚型b。我们设计这几种转录本的公共引物扩增2号外显子和5号外显子之间片段并进行测序,测序结果显示先证者该片段存在2种转录本序列,分别为3号外显子缺失80 bp外显子及3号外显子前保留7 bp内含子,而未检测出正常的2、3、4号转录本片段。正常对照及其父母中均以正常2、3、4号转录本的序列为主。
在2、3、4号转录本中,插入7 bp的内含子序列后,从起始密码子ATG开始翻译到27个氨基酸时,TGA终止密码子使翻译提前终止进而形成截短蛋白;缺失80 bp序列后至第81个氨基酸时,翻译也提前终止形成截短蛋白。显然这两种截短蛋白都不能发挥RASGRP2蛋白正常功能。
5号转录本可编码RASGRP2蛋白亚型b,该亚型与亚型a的区别在于亚型b缺少REM功能域。由于该蛋白亚型近期才被模拟出来,尚无针对其功能的相关文献报道。但针对RASGRP2蛋白a亚型的研究发现,该蛋白包括REM、CDC25、EF臂、C1端4个功能域,RASGRP2蛋白需定位于胞膜后才能发挥激活Rap1功能,而招募到胞膜这一过程是通过REM结构域与细胞骨架蛋白F-actin相互作用实现的[15]–[16]。因此,我们认为患儿体内仅有的RASGRP2蛋白b亚型不能单独行使激活Rap1的功能。
血小板黏附、聚集、释放是经过一系列信号转导来实现的,RASGRP2主要通过激活Rap1参与血小板由内向外信号转导进而激活αⅡbβ3发挥作用[17]。体外及体内试验均证实,敲除RASGRP2的小鼠血小板不能促使血流状态下胶原表面血栓的形成[5],[18]。因此RASGRP2蛋白缺失可导致先证者在血管受损时血小板功能障碍而无法正常止血。
目前,RASGRP2基因突变导致的遗传性血小板功能障碍性出血的报道较少,Canault等[19]于2014年首次报道RASGRP2基因G742T突变单独导致一个家系3个同胞的遗传性出血性疾病的发生。到目前为止,共报道了16种致病RASGRP2基因突变[20]–[23],以纯合突变为主,也包括双重杂合突变。这些突变均位于外显子,分别影响RASGRP2蛋白的表达及相关功能域的功能,但突变类型与先证者出血严重程度关系似乎不大。目前尚无内含子位置基因突变的相关报道。
综上所述,通过表型检测、基因诊断、mRNA测序,我们对一个遗传性血小板功能障碍家系进行了研究,初步阐明了RASGRP2 IVS3-1C>G突变导致mRNA异常转录,形成异常转录本。此异常的mRNA由于带有提前终止密码子形成截短蛋白,无法发挥正常RASGRP2功能进而导致血小板功能障碍。经过检索,该家系为国内首次报道RASGRP2突变导致遗传性血小板功能障碍,且RASGRP2基因IVS3-1C>G突变类型为国际首次报道。
References
- 1.Nurden AT, Nurden P. Congenital platelet disorders and understanding of platelet function[J] Br J Haematol. 2014;165(2):165–178. doi: 10.1111/bjh.12662. [DOI] [PubMed] [Google Scholar]
- 2.Rao AK. Inherited platelet function disorders: overview and disorders of granules, secretion, and signal transduction[J] Hematol Oncol Clin North Am. 2013;27(3):585–611. doi: 10.1016/j.hoc.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 3.Dizkucukkaya R. Inherited platelet disorders including Glanzmann thrombasthenia and Bernard-Soulier syndrome[J] Hematology. 2013;2013(1):268–275. doi: 10.1182/asheducation-2013.1.268. [DOI] [PubMed] [Google Scholar]
- 4.Chrzanowska-Wodnicka M, Smyth SS, Schoenwaelder SM, et al. Rap1b is required for normal platelet function and hemostasis in mice[J] J Clin Invest. 2005;115(3):680–687. doi: 10.1172/JCI22973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crittenden JR, Bergmeier W, Zhang Y, et al. CalDAG-GEFI integrates signaling for platelet aggregation and thrombus formation[J] Nat Med. 2004;10(9):982–986. doi: 10.1038/nm1098. [DOI] [PubMed] [Google Scholar]
- 6.Novel human pathological mutations. Gene symbol: PPOX. Disease: porphyria, variegate[J] Hum Genet. 2009;125(3):333–352. [PubMed] [Google Scholar]
- 7.Hanna S, Etzioni A. MHC class I and II deficiencies[J] Allergol Immunopathol (Madr) 2014;134(2):269–275. doi: 10.1016/j.aller. [DOI] [PubMed] [Google Scholar]
- 8.Rameixwelti M, Regnier CH, Bienaime F, et al. Hereditary complement C7 deficiency in nine families: subtotal C7 deficiency revisited.[J] Eur J Immunol. 2007;37(5):1377–1385. doi: 10.1002/eji.200636812. [DOI] [PubMed] [Google Scholar]
- 9.Town MM, Jean G, Cherqui S, et al. A novel gene encoding an integral membrane protein is mutated in nephropathic cystinosis[J] Nat Genet. 1998;18(4):319–324. doi: 10.1038/ng0498-319. [DOI] [PubMed] [Google Scholar]
- 10.Trizzino A, Stadt UZ, Ueda I, et al. Genotype-phenotype study of familial haemophagocytic lymphohistiocytosis due to perforin mutations[J] J Med Genet. 2007;45(1):15–21. doi: 10.1136/jmedgenet-2018-105362. [DOI] [PubMed] [Google Scholar]
- 11.Farrar JE, Dahl N. Untangling the Phenotypic Heterogeneity of Diamond Blackfan Anemia[J] Semin Hematol. 2011;48(2):124–135. doi: 10.1053/j.seminhematol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim JJ, Rini BI, Hansel DE, et al. Von Hippel Lindau syndrome.[J] Adv Exp Med Biol. 2010:228–249. doi: 10.1007/978-1-4419-6448-9_22. [DOI] [PubMed] [Google Scholar]
- 13.Krawczak M, Thomas NS, Hundrieser B, et al. Single base-pair substitutions in exon-intron junctions of human genes: nature, distribution, and consequences for mRNA splicing[J] Hum Mutat. 2007;28(2):150–158. doi: 10.1002/humu.20400. [DOI] [PubMed] [Google Scholar]
- 14.Kurmangaliyev YZ, Gelfand MS. Computational analysis of splicing errors and mutations in human transcripts[J] BMC Genomics. 2008;9:13. doi: 10.1186/1471-2164-9-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clyde-Smith J, Silins G, Gartside M, et al. Characterization of RasGRP2, a plasma membrane-targeted, dual specificity Ras/Rap exchange factor[J] J Biol Chem. 2000;275(41):32260–32267. doi: 10.1074/jbc.M006087200. [DOI] [PubMed] [Google Scholar]
- 16.Caloca MJ, Zugaza JL, Vicente-Manzanares M, et al. F-actin-dependent translocation of the Rap1 GDP/GTP exchange factor RasGRP2[J] J Biol Chem. 2004;279(19):20435–20446. doi: 10.1074/jbc.M313013200. [DOI] [PubMed] [Google Scholar]
- 17.陆 四. 血小板膜蛋白受体信号转导通路的研究进展[J] 医学综述. 2014;20(21):3851–3854. [Google Scholar]
- 18.Bergmeier W, Goerge T, Wang HW, et al. Mice lacking the signaling molecule CalDAG-GEFI represent a model for leukocyte adhesion deficiency type III[J] J Clin Invest. 2007;117(6):1699–1707. doi: 10.1172/JCI30575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Canault M, Ghalloussi D, Grosdidier C, et al. Human CalDAG-GEFI gene (RASGRP2) mutation affects platelet function and causes severe bleeding[J] J Exp Med. 2014;211(7):1349–1362. doi: 10.1084/jem.20130477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Westbury SK, Canault M, Greene D, et al. Expanded repertoire of RASGRP2 variants responsible for platelet dysfunction and severe bleeding[J] Blood. 2017;130(8):1026–1030. doi: 10.1182/blood-2017-03-776773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lozano ML, Cook A, Bastida JM, et al. Novel mutations in RASGRP2, which encodes CalDAG-GEFI, abrogate Rap1 activation, causing platelet dysfunction[J] Blood. 2016;128(9):1282–1289. doi: 10.1182/blood-2015-11-683102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sevivas T, Bastida JM, Paul DS, et al. Identification of two novel mutations in RASGRP2 affecting platelet CalDAG-GEFI expression and function in patients with bleeding diathesis[J] Platelets. 2018;29(2):192–195. doi: 10.1080/09537104.2017.1336214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kato H, Nakazawa Y, Kurokawa Y, et al. Human CalDAG-GEFI deficiency increases bleeding and delays αIIbβ3 activation[J] Blood. 2016;128(23):2729–2733. doi: 10.1182/blood-2016-03-704825. [DOI] [PubMed] [Google Scholar]