

近年来,无论是获得性凝血因子Ⅷ(FⅧ)/凝血因子Ⅸ(FⅨ)抑制物(也称获得性血友病)还是血友病患者合并抑制物均有增多的趋势。前者是非血友病患者产生的抗FⅧ/FⅨ自身抗体,后者是血友病患者接受外源性凝血因子产品输注后产生的抗FⅧ/FⅨ同种抗体。重型血友病A患者抑制物发生率约为30%,非重型为3%~13%,而血友病B患者为1%~6%[1]。为了规范国内同行的诊疗行为并为有关部门制定政策提供依据,我们曾先后制订了相关中国专家共识[2]–[3]。为进一步提高我们对FⅧ/FⅨ抑制物的认识,做到发现及时、处理正确,特制订此指南供国内同行参考。
FⅧ/FⅨ抑制物滴度:不同稀释度的患者血浆与正常血浆等量混合,孵育2 h,测定残余FⅧ/FⅨ活性。能使正常血浆FⅧ/FⅨ活性减少50%时,FⅧ/FⅨ抑制物的含量为1个Bethesda单位(BU),此时患者血浆稀释度的倒数即为抑制物滴度,以“BU/ml血浆”表示。2001年国际血栓与止血学会(ISTH)规定:抑制物滴度>5 BU/ml为高滴度抑制物,抑制物滴度≤5 BU/ml为低滴度抑制物
高反应者:输注凝血因子后抑制物滴度升高至5 BU/ml以上的患者。
低反应者:输注凝血因子后抑制物滴度仍小于5 BU/ml的患者。
暴露天数:患者实际接受凝血因子替代治疗的天数之和,不包括其间的间隔天数。
二、FⅧ/FⅨ抑制物实验室诊断
1.抑制物筛选(以FⅧ为例)[6]–[7]:采用部分激活的凝血活酶时间(APTT)纠正试验,用正常混合血浆(至少20人份健康人血浆)和患者血浆按1∶1混合,即刻测定混合后的APTT,并与正常混合血浆和患者血浆的APTT进行比较,若不能纠正应考虑可能存在抑制物[纠正:超过正常混合血浆5 s以内(或延长<15%)或在实验室正常参考范围内;不纠正:超过正常混合血浆5 s以上(或延长>15%)或高于实验室正常参考范围。由于试验的复杂性,在判定纠正与否方面没有统一的共识,因此每个实验室需根据自身的经验建立相应的指南和纠正的标准与相应的解释][1]。由于FⅧ抑制物有时间依赖性,因此若患者延长的APTT即刻被正常混合血浆纠正,则需要进一步进行孵育后的纠正试验。具体步骤见图1。而若将正常混合血浆与患者血浆以1∶4混合进行上述试验,有助于发现微量抑制物。
图1. 部分激活的凝血活酶时间(APTT)纠正试验.

结果解释:①APTT3、APTT6与APTT7均纠正提示凝血因子缺乏,不存在抑制物;②APTT3与APTT7均纠正、APTT6延长,提示抑制物存在且为时间依赖性;③APTT3、APTT6与APTT7均不纠正、APTT6比APTT7延长不超过10%~15%,提示存在抑制物,但抑制作用不存在时间依赖性;若APTT6比APTT7延长超过10%~15%,则抑制作用有时间依赖性;④APTT4、APTT5、APTT6分别为APTT1、APTT2、APTT3的平行对照
2.抑制物的滴度(以FⅧ为例)[5]–[6]:确诊抑制物必须测定抑制物滴度。以Bethesda试验为例,将用pH7.4的咪唑缓冲液稀释的不同稀释度(1∶1、1∶2、1∶4等)的患者血浆与正常混合血浆(Nijmegen试验中用咪唑缓冲液处理过的正常混合血浆)等量混合,37 °C孵育2 h后,测定每份混合血浆的FⅧ活性,用该值除以用咪唑缓冲液与正常混合血浆等量混合(Nijmegen试验中用无抑制物的乏FⅧ血浆与正常混合血浆等量混合)于37 °C孵育2 h后的对照血浆的FⅧ活性,便得到残留的FⅧ活性的百分比,用该值通过抑制物滴度标准表可查得对应的抗体滴度。具体步骤见图2。如果在1~4周内连续两次用Bethesda法或者Nijmegen法检测发现患者抑制物滴度≥0.6 BU/ml则为阳性[2],[4]。若患者输注凝血因子,在检测抑制物滴度前应有48 h(FⅧ)或72 h(FⅨ)的洗脱期;若患者需要持续输注凝血因子,不能经历足够的洗脱期或者患者凝血因子活性>5%,可利用免疫球蛋白的耐热性在进行滴度检测之前对患者血浆进行加热(58 °C加热90 min)以灭活血浆中残留的凝血因子,降低因FⅧ/FⅨ活性过高对检测结果造成的干扰[4]–[6]。
图2. 经典Bethesda法检测抑制物滴度.
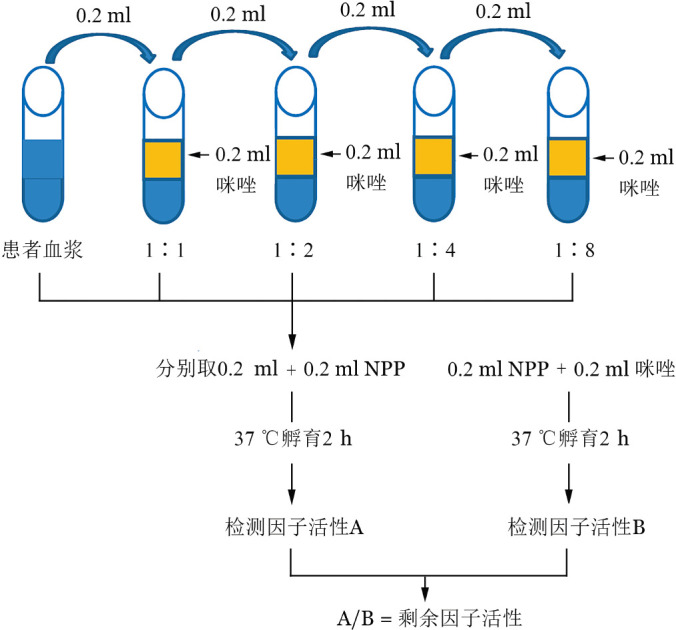
NPP:正常混合血浆;滴度计算:选取最接近50%的剩余因子活性值,由抑制物滴度标准表查得相对应的滴度数,该滴度数值乘以该样本的稀释倍数即为最终抑制物滴度
3.抗磷脂抗体:抗磷脂抗体也可引起APTT延长,且纠正试验不能纠正。虽然大部分凝血因子抑制物(尤其FⅧ)表现为时间依赖性,但也有少部分(10%~15%)狼疮抗凝物(LA)可出现时间依赖性,因此,时间依赖性的抑制特性并不能区分凝血因子抑制物和LA。LA还会干扰凝血因子抑制物的定量检测[6],[8]。因此,凝血因子抑制物需要与抗磷脂抗体鉴别。有以下两种方法用于检测抗磷脂抗体:①凝固法检测LA[8]:稀释蝰蛇毒试验(DRVVT)是诊断LA最具特异性的检测方法,LA通过干扰凝血酶原复合物(PCC)的生成而使DRVVT延长,通过加入的过量磷脂使延长的DRVVT得到纠正。由于LA的异质性,一种LA检测试剂很难检测出所有的LA,所以ISTH推荐应用两种不同凝固途径的试剂对LA进行检测,其中DRVVT为首选,第二种试剂为对LA敏感的APTT试剂(含有少量磷脂,并以硅土为激活剂)。只要其中一种试剂结果为阳性则提示存在LA。②免疫法检测抗心磷脂抗体和抗β-2糖蛋白Ⅰ抗体[9]:应用ELISA或自动检测系统(如化学发光法)定量检测抗心磷脂抗体(IgA、IgG、IgM)和抗β-2糖蛋白Ⅰ抗体(IgA、IgG、IgM),其滴度>99的百分位数(或验证过的适用于本实验室的厂家提供的cut-off值)
以上结果若为阳性,均需要12周后复查,明确是否为持续性。
三、FⅧ/FⅨ抑制物(同种抗体)的危险因素
抑制物发生的危险因素包括遗传因素和非遗传因素。遗传因素主要有基因突变、种族和家族史等;非遗传因素包括外伤史、暴露日、输注剂量、凝血因子产品种类及治疗策略等。遗传因素是抑制物产生的基础和前提,非遗传因素是抑制物产生的触发因素,二者共同参与了抑制物的发生,也决定了抑制物的严重程度和持续时间。
(一)抑制物产生的遗传因素
1.种族与家族史[1]:非洲裔、拉丁裔美国人FⅧ抑制物发生率高于白种美国人。现认为重组FⅧ(rFⅧ)产品基因型(FⅧ单体型)可能与黑种人患者FⅧ基因型不匹配有关。同时有抑制物家族史的血友病A患者,其抑制物的发生率是无家族史的3倍多;双胞胎间抑制物的发生率较一般的同胞兄弟间更高。
2.基因突变类型[1],[5],[10]:FⅧ基因位于X染色体长臂末端:(Xq28),其编码合成一条肽链,以A1-A2-B-A3-C1-C2的方式排列。与重型血友病患者产生抑制物密切相关的主要突变类型包括大片段缺失、无义突变、22号内含子倒位,其次为小片段缺失和插入、错义突变等。大片段缺失、无义突变、22号内含子倒位导致FⅧ蛋白产生缺失,不容易诱导免疫耐受,输注FⅧ后易产生抑制物。而小片段缺失和插入、错义突变等能够产生部分FⅧ蛋白,所以能够诱导免疫耐受,使得抑制物发生率较低。对于非重型血友病患者,发生在轻链部(A3、C1、C2结构域)的错义突变容易形成抑制物,错义突变导致蛋白构象变化而影响FⅧ免疫源性。小片段插入和缺失通过导致移码突变和提前产生终止密码子影响蛋白构象。但发生在ploy-A尾结构的小片段插入和缺失却与低抑制物发生风险相关。血友病B有大片段基因缺失或者无效突变的患者抑制物发生率高。因此,新诊断的血友病患者都必须进行基因突变检测,以便指导临床治疗。
(二)抑制物产生的非遗传因素
1.暴露日:现有资料表明,对于重型血友病抑制物阳性患者而言,50%的患者FⅧ/FⅨ抑制物发生在开始接受治疗后的前20个暴露日,95%发生在前50个暴露日。因此,应该在这段时间加强抑制物的筛查,以便及早发现。对于儿童患者,建议在首次接受凝血因子产品后的前20个暴露日每5个暴露日检测1次,在21~50个暴露日内每10个暴露日检测1次,此后每年至少检测2次,直至150个暴露日[2]。对于成年人而言,60岁以后发生率随年龄增加而增高。对于因为病情需要的初治患儿(PUP),使用凝血因子产品前必须与患儿父母或者监护人进行充分沟通,以便其知晓抑制物发生的风险[5]。对于轻/中型血友病患者而言,抑制物发生率随着暴露日增加而逐渐增高,50个暴露日时的发生率为6.7%,100个暴露日时为13.3%[11]。
2.治疗方式[1],[11]:对于PUP而言,强化治疗更容易形成抑制物,RODIN研究发现,严重出血和外伤时使用强化或大剂量持续FⅧ输注容易形成抑制物。连续强化治疗超过5 d抑制物发生率是强化治疗1 d或2 d的3.3倍。RODIN研究表明在20个暴露日内,按需治疗与预防治疗的抑制物发生率相同,超过20个暴露日后,预防治疗组抑制物发生率明显低于按需治疗组。
3.FⅧ制剂类型:现有数据表明,含有VWF的血源性FⅧ具有较轻的免疫源性,特别是PUP,有利于免疫耐受的建立。
迄今为止,共有5项大样本临床研究对比了PUP使用血源性FⅧ制剂和重组FⅧ制剂抑制物的发生率,发现使用血源性FⅧ制剂抑制物发生率较低。目前唯一的一项全球多中心随机对照临床研究结果表明,使用rFⅧ制剂的PUP抑制物发生率是使用血源性含VWF的FⅧ制剂患者的1.87倍[12]。进一步对该研究的235例入组患者根据基因突变进行发生抑制物的危险度分层,197例为无效突变(包括大片段缺失、无义突变、22号内含子倒位、移码突变、1号内含子倒位),为高危组,38例为非无效突变(包括错义突变、剪切点突变等),为低危组。高危组65例(38%)发生抑制物,低危组7例(24%)发生抑制物。高危组接受血源性FⅧ制剂患者的抑制物发生率为31%,接受重组FⅧ制剂患者的抑制物发生率为47%;低危组接受血源性FⅧ制剂患者没有抑制物发生,接受重组FⅧ制剂患者的抑制物发生率为43%。组内分析表明接受血源性FⅧ制剂患者低危组和高危组的抑制物发生率有显著差异,而接受重组FⅧ制剂患者的抑制物发生率低危组和高危组则没有差别[10]。
四、血友病A合并FⅧ抑制物的处理
对于FⅧ抑制物患者的治疗原则主要有两个方面:控制出血和清除抑制物。具体治疗的选择取决于患者入院时的临床表现和抑制物的滴度水平以及患者的经济条件[1]。
1.出血的治疗:出血是血友病抑制物形成最主要的并发症,临床观察到患者出血较以往增多、常规剂量FⅧ制剂输注止血疗效下降或无效;轻/中型血友病患者出血表现加重(如出现严重的自发关节和肌肉出血),要考虑到抑制物产生的可能。对于低滴度患者(≤5 BU/ml,约占25%),无明显出血时可以继续观察,部分患者的抑制物可于6个月内自行消失。对于无重要脏器出血、出血量不大的患者,加大剂量的FⅧ替代治疗仍有效。大剂量FⅧ虽可产生免疫记忆以至抗体滴度升高,导致高反应型抑制物产生,但大剂量FⅧ的输注仍是治疗急性出血(特别是出血量较大时)的最有效方法。
对于抑制物滴度>5 BU/ml(约占75%)的患者或诱导免疫耐受治疗(ITI)失败或ITI治疗中的出血患者,需立即采用“旁路途径”的方式止血。可供选择的“旁路途径”药物包括基因重组活化凝血因子Ⅶ(rFⅦa)及活化凝血酶原复合物(aPCC)(国内无此药物)。建议采用rFⅦa 90 µg/kg每2~4 h静脉注射1次,也可以270 µg/kg单次给药[5]。国外学者临床上也采用aPCC 50~70 IU/kg每12~24 h给药1次,止血效果大致相同。在没有上述药物时,可以考虑使用PCC 50~100 IU·kg−1·d−1。对单一旁路途径无效者,可采用aPCC与rFⅦa序贯疗法,即在12 h内1次aPCC与1~2次rFⅦa交替给药,每3~6 h 1次,给药剂量在单一旁路途径给药基础上根据患者情况调整。两种药物连用时应严密监测,以防止发生血栓。
2.抑制物清除治疗:ITI是目前主要的清除重型血友病伴抑制物的治疗方案。对于确诊为抑制物阳性且预防治疗(20~50 IU/kg,隔日1次)疗效不佳的患者应该立即开始ITI[13]–[14]。对于低滴度血友病A患者,ITI总体有效率近100%,高滴度血友病A患者ITI总体有效率约70%。德国学者认为含有VWF的血浆源性FⅧ比rFⅧ的ITI成功率高,但文献荟萃分析的结果显示血浆源性FⅧ和rFⅧ的ITI成功率并无差别[11]。
ITI治疗方案主要有以下三种[13]–[14]:①Bonn方案:开始时FⅧ用量为100 IU/kg每12 h 1次,同时使用aPCC 50 IU/kg或rFⅦa每日2次。等到抑制物滴度下降,FⅧ活性开始恢复后停用aPCC或rFⅦa,FⅧ用量改为150 IU/kg每12 h 1次,直到抑制物消失。②Van Creveld方案:FⅧ25~50 IU/kg隔日1次输注,根据抑制物滴度下降和FⅧ活性恢复情况逐渐减少FⅧ用量,直至与原来的预防治疗剂量一样。从经济的角度来说,该方案在我国更具可行性。③Malmö方案:在Bonn方案的基础上联合免疫抑制治疗(口服泼尼松50~150 mg/d,环磷酰胺12~15 mg·kg−1·d−1×2 d→2~3 mg·kg−1·d−1,共8~10 d,同时加用静脉丙种球蛋白0.4 g·kg−1·d−1×5 d)。抑制物滴度>10 BU/ml的患者,在开始治疗前使用免疫吸附方法(蛋白A层析柱)使抑制物滴度低于10 BU/ml。2009年我们对Malmö方案进行了改良(延长泼尼松和环磷酰胺的使用时间,不使用静脉丙种球蛋白和免疫吸附),并在国内首次获得成功[15]。目前主张在一线ITI方案中不联合使用免疫抑制剂和静脉丙种球蛋白[11],[14]。
ITI疗效评估:①完全耐受:抑制物持续阴性(<0.6 BU/ml)且FⅧ回收率>66%、FⅧ半衰期>6 h。②部分耐受:抑制物滴度<5 BU/ml,虽然FⅧ回收率<66%和(或)半衰期<6 h,但是使用FⅧ治疗可以阻止出血。③无效:不能达到完全或部分耐受。一般来说,在3~6个月内抑制物滴度下降不足20%,或经过3~5年的ITI后抑制物滴度仍>5 BU/ml提示ITI无效[2],[13]–[14]。
一旦开始ITI,不宜随便中止,以免影响后续ITI的疗效。开始ITI后,应该每周检测1次抑制物滴度,如果抑制物滴度升高或半年内抑制物滴度下降小于20%,应逐步增加ITI剂量直至200 IU·kg−1·d−1;如果剂量已经达到200 IU·kg−1·d−1,建议改为二线方案[14]。
ITI的二线方案,目前国际上无共识。目前使用人源CD20单抗以选择性清除B细胞以达到免疫抑制作用,但是人源CD20单抗近期疗效和远期并发症还需要更多的临床治疗数据[2],[12]–[13]。
ITI疗效的预测[11]:目前认为有如下指标的患者ITI疗效可能较好:①开始ITI之前抑制物滴度<10 BU/ml;②抑制物滴度历史峰值<200 BU/ml;③ITI期间抑制物滴度峰值<100 BU/ml;④从诊断到开始ITI的时间<5年;⑤ITI开始后没有间断。而有如下指标的患者ITI疗效可能较差:①开始ITI之前抑制物滴度>10 BU/ml;②抑制物滴度历史峰值>200 BU/ml;③ITI期间抑制物滴度峰值>100 BU/ml;④从诊断到开始ITI的时间>5年;⑤ITI开始后间断>2周。
五、血友病B合并FⅨ抑制物的处理
FⅨ抑制物的产生往往伴随着过敏反应的出现。因此,在患者接受FⅨ替代治疗过程中,即使出现轻微的过敏反应,也应该进行抑制物筛查。此外,长期接受大剂量FⅨ替代治疗可能引起不可逆的肾病综合征[1]。
关于血友病B伴抑制物的治疗选择,可采用以下方法:①针对低滴度抑制物患者,若FⅨ替代治疗有效、无过敏反应,可选择大剂量FⅨ继续替代治疗;②针对高滴度抑制物、无FⅨ过敏反应的患者可选择ITI治疗。由于血友病B伴抑制物发生率低,目前尚无统一治疗方案,多采用类似血友病A的治疗方案,文献报道的治疗剂量25~200 IU/d,且无特定因子产品的推荐。血友病B伴抑制物患者治疗效果不及血友病A,总体有效率13%~31%,并且可能会出现肾病综合征(19%)、过敏反应(60%~63%)等不良事件,导致成功率进一步降低,甚至无法继续进行ITI治疗。血友病B伴抑制物ITI治疗效果不佳,可选择联合应用免疫抑制剂(糖皮质激素、静脉丙种球蛋白、环孢素A、吗替麦考酚酯、利妥昔单抗等)[1],[5],[16]。
六、获得性血友病合并FⅧ抑制物的处理
由于获得性血友病病情凶险,一旦确诊后应立即给予及时恰当的治疗措施,所有患者应立即采取免疫抑制治疗以清除FⅧ抑制物,达到彻底治愈目的。本病的治疗分为止血治疗和清除抑制物治疗两部分。因为抑制物水平并不能预测出血的危险,所以不能用抑制物水平来判定是否应该采取免疫抑制治疗。抑制物水平唯一的参考价值是对免疫抑制治疗反应的预测。因大多数获得性血友病患者是老年人,可能伴发其他疾病,在积极治疗原发病的基础上,治疗方案应在快速清除抑制物以降低出血风险和免疫抑制治疗所带来的不良反应之间进行权衡[3],[17]–[18]。
(一)止血治疗
1.一般止血治疗措施:一旦确诊,应该立即采取措施防止发生大出血。考虑止血治疗潜在不良反应,尤其对伴发合并症的老年患者,应该仔细权衡治疗的风险,采取个体化治疗措施。
对于皮肤瘀斑,只需要采取密切观察而不需要特殊的治疗。对于腹膜后和咽后间隙出血、伴或不伴筋膜室综合征的肌肉出血、颅内出血、胃十二指肠出血、肺出血、手术后出血、严重的血尿和多部位出血,应予积极止血治疗[2],[17]–[18]。
2.FⅧ抑制物的旁路治疗[3],[17]–[18]:获得性血友病一线止血治疗药物包括rFⅦa和aPCC。rFⅦa推荐剂量为90 µg/kg每2~3 h静脉注射1次,直至出血控制。aPCC 50~100 IU/kg每8~12 h静脉注射1次,直至出血控制,最大剂量不超过200 IU·kg−1·d−1。由于我国尚无aPCC制剂供应,如果无法获得rFⅦa,可以考虑使用国产PCC止血。由于PCC中含有痕量的FⅧ,部分患者使用PCC后可能会引起FⅧ抑制物水平升高。尚无任何实验室检查能够判定临床治疗效果,因此疗效判定必须基于对临床出血症状的评估。
目前关于rFⅦa和aPCC诱发血栓形成的证据尚有限。在老年和有冠心病史或有血栓并发症危险因素的患者中使用要谨慎。
3.FⅧ浓缩剂:当抑制物滴度≤5 BU/ml,出血表现或者潜在出血较小并且无旁路治疗制剂时,建议选用FⅧ制剂。尚无前瞻性、随机、对照临床研究证实该治疗方案在AHA中的有效性。
4.1-去氨基-8-D-精氨酸加压素(DDAVP):DDAVP适用于轻微出血和抑制物滴度≤5 BU/ml,推荐剂量为0.3 µg/kg。应注意此药的不良反应,尤其是获得性血友病老年患者。重复注射DDAVP有可能发生水肿、持续的低钠血症和抽搐等不良反应。妊娠患者及2岁以下儿童禁用。
5.其他:在特殊情况下(包括难治性出血事件或者需要外科干预时),可使用血浆置换或者免疫吸附法快速除去血浆中的抑制物以达到有效止血。某些部位出血(如鼻出血、口腔溃疡、皮肤缺损和外科手术部位出血)可以用凝血酶或者纤维胶人工辅助止血。
(二)抑制物清除
一线方案包括单用糖皮质激素、糖皮质激素和环磷酰胺联合使用。泼尼松1 mg·kg−1·d−1口服4~6周或联合环磷酰胺1.5~2 mg·kg−1·d−1最长6周。若治疗4~6周后无反应,应该考虑单用利妥昔单抗或联合糖皮质激素作为替代治疗方案。上述治疗无效可考虑使用其他免疫抑制剂(硫唑嘌呤、长春新碱、吗替麦考酚酯和环孢素A等)[3],[17]–[18]。
在治疗成功后或者换二线治疗以后,应该尽快减停糖皮质激素。环磷酰胺的使用应该根据血常规进行调整并且不超过6周,因为继续治疗会增加不良反应的发生。对于出现不明原因的发热、严重感染以及产后哺乳的女性患者禁用环磷酰胺或其他烷化剂治疗[3],[17]–[18]。
(三)疗效判断[3]治疗有效性的评价应该基于出血是否控制,如血肿的大小、血红蛋白/红细胞压积的稳定和血肿引起的疼痛程度来判定。监测应该包括:物理检查、血常规、APTT、FⅧ活性和FⅧ抑制物滴度。在治疗的前6周,门诊患者应该每周检查1次,住院患者每周检查2次。
疗效判断[19]:完全缓解:抑制物滴度<0.6 BU/ml且FⅧ水平>50 IU/dl。部分缓解:抑制物滴度≥0.6 BU/ml但比诊断时降低,和(或)FⅧ水平<50 IU/dl。无效:治疗后抑制物滴度与治疗前相同或增高。
完全缓解后随访,最初6个月内每月复查1次APTT和FⅧ活性;6~12个期间为每2~3个月复查1次;第2年每半年检查1次,条件允许可适度延长。
Funding Statement
基金项目:“十三五”国家重点研发计划精准医学研究重点专项(2016YFC0901503);中国医学科学院医学与健康科技创新工程重大协同创新项目(2016-I2M-1-002)
References
- 1.Giangrande PLF, Hermans C, O'Mahony B, et al. European principles of inhibitor management in patients with haemophilia[J] Orphanet J Rare Dis. 2018;13(1):66. doi: 10.1186/s13023-018-0800-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.中华医学会血液学分会血栓与止血学组, 中国血友病协作组. 血友病诊断与治疗中国专家共识(2017年版)[J] 中华血液学杂志. 2017;38(5):364–370. doi: 10.3760/cma.j.issn.0253-2727.2017.05.002. [DOI] [Google Scholar]
- 3.中华医学会血液学分会血栓与止血学组, 中国血友病协作组. 获得性血友病A诊断与治疗中国专家共识[J] 中华血液学杂志. 2014;35(6):575–576. doi: 10.3760/cma.j.issn.0253-2727.2014.06.026. [DOI] [Google Scholar]
- 4.Blanchette VS, Srivastava A. Definitions in hemophilia: resolved and unresolved issues[J] Semin Thromb Hemost. 2015;41(8):819–825. doi: 10.1055/s-0035-1564800. [DOI] [PubMed] [Google Scholar]
- 5.Collins PW, Chalmers E, Hart DP, et al. Diagnosis and treatment of factor VIII and IX inhibitors in congenital haemophilia: (4th edition)[J] Br J Haematol. 2013;160(2):153–170. doi: 10.1111/bjh.12091. [DOI] [PubMed] [Google Scholar]
- 6.Verbruggen B, van Heerde WL, Laros-van Gorkom BA. Improvements in factor VIII inhibitor detection: from Bethesda to Nijmegen[J] Semin Thromb Hemost. 2009;35(8):752–759. doi: 10.1055/s-0029-1245107. [DOI] [PubMed] [Google Scholar]
- 7.Kershaw G, Favaloro EJ. Laboratory identification of factor inhibitors: an update[J] Pathology. 2012;44(4):293–302. doi: 10.1097/PAT.0b013e328353254d. [DOI] [PubMed] [Google Scholar]
- 8.Pengo V, Tripodi A, Reber G, et al. Update of the guidelines for lupus anticoagulant detection[J] J Thromb Haemost. 2009;7(10):1737–1740. doi: 10.1111/j.1538-7836.2009.03555.x. [DOI] [PubMed] [Google Scholar]
- 9.Devreese KM, Pierangeli SS, de Laat B, et al. Testing for antiphospholipid antibodies with solid phase assays: guidance from the SSC of the ISTH[J] J Thromb Haemost. 2014;12(5):792–795. doi: 10.1111/jth.12537. [DOI] [PubMed] [Google Scholar]
- 10.Rosendaal FR, Palla R, Garagiola I, et al. Genetic risk stratifica-tion to reduce inhibitor development in the early treatment of hemophilia A: a SIPPET analysis[J] Blood. 2017;130(15):1757–1759. doi: 10.1182/blood-2017-06-791756. [DOI] [PubMed] [Google Scholar]
- 11.Ljung RCR. How I manage patients with inherited haemophilia A and B and factor inhibitors[J] Br J Haematol. 2018;180(4):501–510. doi: 10.1111/bjh.15053. [DOI] [PubMed] [Google Scholar]
- 12.Peyvandi F, Mannucci PM, Garagiola I, et al. A randomized trial of factor VIII and neutralizing antibodies in hemophilia A[J] N Engl J Med. 2016;374(21):2054–2064. doi: 10.1056/NEJMoa1516437. [DOI] [PubMed] [Google Scholar]
- 13.Brackmann HH, White GC, 2nd, Berntorp E, et al. Immune tolerance induction: what have we learned over time?[J] Haemophilia. 2018;Suppl 3:3–14. doi: 10.1111/hae.13445. [DOI] [PubMed] [Google Scholar]
- 14.Collins P, Chalmers E, Alamelu J, et al. First-line immune tolerance induction for children with severe haemophilia A: a protocol from the UK Haemophilia Centre Doctors' Organisation Inhibitor and Paediatric Working Parties[J] Haemophilia. 2017;23(5):654–659. doi: 10.1111/hae.13264. [DOI] [PubMed] [Google Scholar]
- 15.张 磊, 薛 峰, 刘 晓帆, et al. 血友病A伴抑制物一例及其免疫耐受诱导治疗探讨[J] 中华血液学杂志. 2010;31(9):577–580. doi: 10.3760/cma.j.issn.0253-2727.2010.09.001. [DOI] [Google Scholar]
- 16.薛 峰, 刘 葳, 陈 云飞, et al. 凝血酶原复合物联合小剂量利妥昔单抗治疗血友病B伴抑制物[J] 中华血液学杂志. 2017;38(9):749–753. doi: 10.3760/cma.j.issn.0253-2727.2017.09.004. [DOI] [Google Scholar]
- 17.Kruse-Jarres R, Kempton CL, Baudo F, et al. Acquired hemophilia A: updated review of evidence and treatment guidance[J] Am J Hematol. 2017;92(7):695–705. doi: 10.1002/ajh.24777. [DOI] [PubMed] [Google Scholar]
- 18.Yang Y, Xue F, Shi H, et al. Acquired hemophilia a: retrospec-tive analysis of 49 cases from a single Chinese hemophilia center[J] Clin Appl Thromb Hemost. 2015;21(1):35–40. doi: 10.1177/1076029613488937. [DOI] [PubMed] [Google Scholar]
- 19.Borg JY, Négrier C, Durieu I, et al. FEIBA in the treatment of acquired haemophilia A: Results from the prospective multicentre French ‘FEIBA dans l'hémophilie A acquise’(FEIBHAC) registry[J] Haemophilia. 2015;21(3):330–337. doi: 10.1111/hae.12574. [DOI] [PubMed] [Google Scholar]