

Abstract
Hereditary hearing loss is one of the most common sensory disabilities worldwide. Mutation of POU domain class 4 transcription factor 3 (POU4F3) is considered the pathogenic cause of autosomal dominant nonsyndromic hearing loss (ADNSHL), designated as autosomal dominant nonsyndromic deafness 15. In this study, four novel variants in POU4F3, c.696G>T (p.Glu232Asp), c.325C>T (p.His109Tyr), c.635T>C (p.Leu212Pro), and c.183delG (p.Ala62Argfs∗22), were identified in four different Chinese families with ADNSHL by targeted next-generation sequencing and Sanger sequencing. Based on the American College of Medical Genetics and Genomics guidelines, c.183delG (p.Ala62Argfs∗22) is classified as a pathogenic variant, c.696G>T (p.Glu232Asp) and c.635T>C (p.Leu212Pro) are classified as likely pathogenic variants, and c.325C>T (p.His109Tyr) is classified as a variant of uncertain significance. Based on previous reports and the results of this study, we speculated that POU4F3 pathogenic variants are significant contributors to ADNSHL in the East Asian population. Therefore, screening of POU4F3 should be a routine examination for the diagnosis of hereditary hearing loss.
1. Introduction
Hearing loss is one of the most common hereditary sensory disabilities worldwide [1]. Hair cells (HCs) in the inner ear are critical for hearing ability. HCs transfer the mechanical vibration into an acoustic electrical signal, which can then be transmitted to the auditory cortex via spiral ganglion neurons (SGNs) [2]. The causes of deafness are complex, and most of the hearing loss is due to irreversible HCs loss. HCs are very sensitive and vulnerable to many stresses and damage, which can be divided mainly into genetic factors, environmental factors, ototoxic drugs, aging, inflammation, and other unknown etiologies [3–5]. Among all these factors, it is estimated that genetic factors account for more than 50% of the causes of deafness [6]. Hereditary hearing loss can be classified as syndromic hearing loss or nonsyndromic hearing loss (NSHL) according to whether the patient has other symptoms or signs, and these account for 30% and 70% of cases of hearing loss, respectively [7]. NSHL can be further divided into three categories according to the mode of inheritance: autosomal dominant nonsyndromic hearing loss (ADNSHL), autosomal recessive nonsyndromic hearing loss, and X-linked nonsyndromic hearing loss. ADNSHL accounts for 15% of cases of NSHL [8]. One of the most significant characteristics of hereditary hearing loss is a high degree of heterogeneity. To date, 49 genes related to ADNSHL, including POU domain class 4 transcription factor 3 (POU4F3) and approximately 70 other loci, have been reported (http://hereditaryhearingloss.org/).
The POU4F3 gene encodes POU4F3, a POU-domain class IV protein, has two exons, and encodes a protein of 338 amino acids that belongs to the POU-domain family of transcription factors, which are expressed specifically in inner ear hair cells and play a critical role in the maturation, differentiation, and maintenance of inner ear hair cells [9, 10]. POU4F3 contains two conserved DNA-binding domains (a POU-specific domain and a POU homeodomain), which are the main functional parts [10].
In 1998, POU4F3 was first described as a disease-causing gene within the DFNA15 locus in an Israeli Jewish family [11]. To date, 32 variants (including those in this study) and whole-gene deletion of POU4F3 have been reported to cause ADNSHL with variable ages of onset and degrees of severity in various ethnic groups, including Chinese, Japanese, Dutch, Korean, and Brazilian populations [12–24]. In 2017, Kitano et al. reported that POU4F3 variants represent the third largest cause of ADNSHL (2.5%, 15/602) in Japan and the most prevalent configuration as midfrequency hearing loss type followed by high-frequency hearing loss [14]. He et al. reported that the POU4F3 pathogenic variant is a relatively common (3/18) cause of ADNSHL among Chinese Hans [15]. Therefore, impairment of hair cells in the cochlea caused by pathogenic variants of POU4F3 has been considered as one of the major causes of sensorineural hearing loss [14].
In this study, we identified four novel variants using targeted next-generation sequencing (NGS) of a panel of 168 deafness genes from four different Chinese families suffering from ADNSHL. Among the four novel variants, three are missense variants, c.696G>T (p.Glu232Asp) detected in family A, c.325C>T (p.His109Tyr) in family B, and c.635T>C (p.Leu212Pro) in family C, and the fourth is a frameshift variant, c.183delG (p.Ala62Argfs∗22), which was identified in family D. Hearing loss in the four families analyzed in this study showed a high degree of variability, even in patients carrying the same variant within one family.
2. Materials and Methods
2.1. Subjects
Probands suffering from ADNSHL in the four families were recruited from the Chinese PLA General Hospital. The pedigrees of these four families are shown in Figures 1–4(a) In addition to the probands, three additional members of family A (II:3, II:5, and III:2), six additional members of family B (I:1, I:2, II:1, II:2, II:3, and II:4), five additional members of family C (II:1, II:2, II:5, III:6, and III:9), and four additional members of family D (II:1, II:3, II:6, and III:1) were recruited from our hospital. All of the subjects or their guardians provided written informed consent to participate in the study. This study was approved by the Ethics Research Committee of the Chinese PLA General Hospital.
Figure 1.
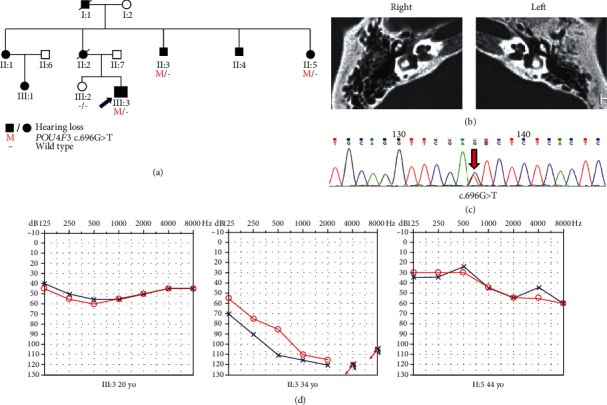
Pedigree, temporal bone CT, variant analysis, and audiogram of family A. (a) Affected subjects are denoted in black. Arrow shows the proband. (b) Temporal bone CT of the III:3 shows no structural change. (c) Chromatogram shows POU4F3 heterozygous c.696G>T detected in patients. (d) Audiograms of the affected subjects. Hearing loss appears to be highly heterogeneous (red: right ear; blue: left ear).
Figure 2.
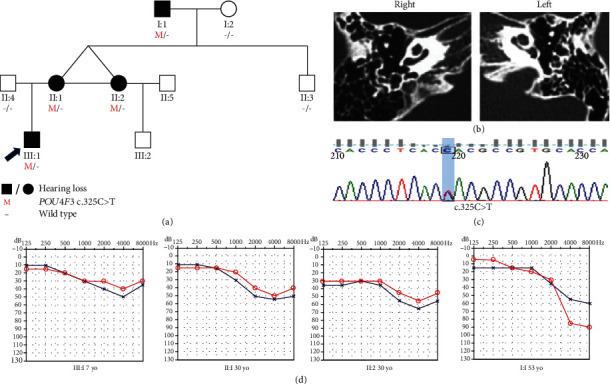
Pedigree, temporal bone CT, variant analysis, and audiogram of family B. (a) Affected subjects are denoted in black. Arrow shows the proband. (b) Temporal bone CT of the III:1 shows no structural change. (c) Chromatogram shows POU4F3 heterozygous c.325C>T detected in patients. (d) Audiograms of the affected subjects. Hearing loss appears to involve high frequency (red: right ear; blue: left ear).
Figure 3.
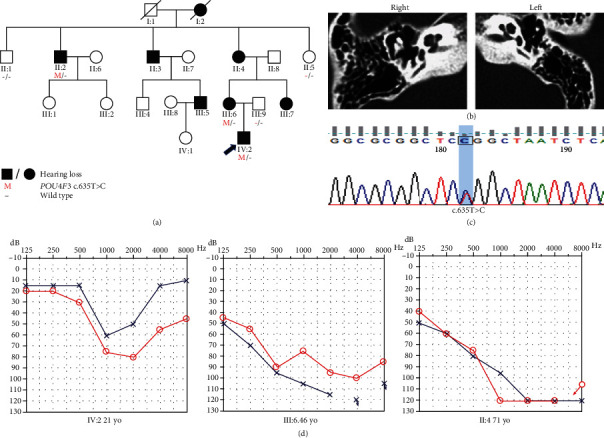
Pedigree, temporal bone CT, variant analysis, and audiogram of family C. (a) Affected subjects are denoted in black. Arrow shows the proband. (b) Temporal bone CT of the IV:2 shows no structural change. (c) Chromatogram shows POU4F3 heterozygous c.635T>C detected in patients. (d) Audiograms of the affected subjects. Audiogram configuration of IV:2 was U-shaped. Downsloping audiogram configurations were observed in III:6 and II:4 (red: right ear; blue: left ear).
Figure 4.
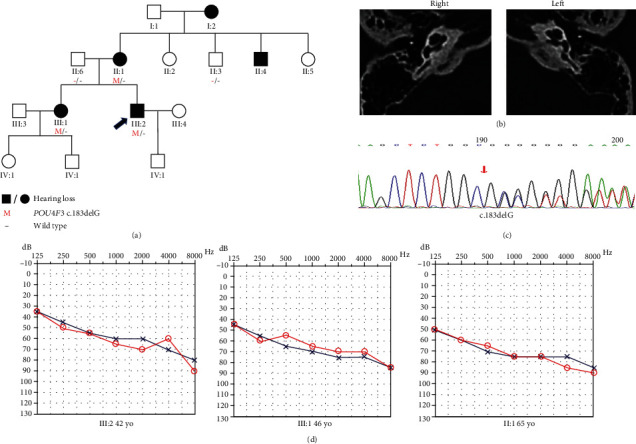
Pedigree, temporal bone CT, variant analysis, and audiogram of family D. (a) Affected subjects are denoted in black. Arrow shows the proband. (b) Temporal bone CT of the III:2 shows no structural change. (c) Chromatogram shows POU4F3 heterozygous c.183delG detected in patients. (d) Audiograms of the affected subjects (red: right ear; blue: left ear).
2.2. Clinical Information and Examination
Clinical information was obtained via multiple interviews with the subjects. Medical history was obtained using a questionnaire that elicited responses regarding the symmetry of hearing loss, subjective degree of hearing loss, use of hearing aids, age at onset, evolution, presence of tinnitus, noise exposure, medications, trauma history, and other relevant clinical manifestations. The subjects all received clinical examinations at the Department of Otorhinolaryngology, which included otoscopy, physical examination, pure tone audiometric examination (at frequencies from 125 to 8000 Hz), computed tomography scans of the temporal bone, and acoustic immittance testing. The tandem gait test was performed to evaluate the balance. The diagnosis of sensorineural hearing loss was made according to the WHO criteria based on audiometric examination performed as described previously (the methods description partly reproduces our wording) [12]. Tandem gait and Romberg tests were performed to evaluate balance.
2.3. Variant Analysis
DNA was extracted from peripheral blood samples from all subjects using a blood DNA extraction kit (TIANGEN, Beijing, China), according to the manufacturer's instructions.
The most prevalent genes related to hearing loss, including GJB2, SLC26A4, and mtDNA12SrRNA, were screened in all of the probands and Chinese controls. The probands and some of the additional family members were examined using a gene panel containing 168 genes related to deafness (Supplementary Table 1). Capture sequencing and NGS of the coding exons of the 168 deafness-related genes and their flanking 100 bp were performed on the Illumina HiSeq 2000 (Illumina, San Diego, CA, USA) using the MyGenostics gene enrichment system (MyGenostics, Boston, MA, USA).
The methods for DNA library preparation, amplification, capture, detection, sequencing, and bioinformatics analyses were described previously [12]. Nonsynonymous variants were further evaluated for candidate pathogenic variants. Variants were annotated by ANNOVAR; compared with multiple databases including gnomAD, dbSNP, and ExAC; and were predicted by the computational programs SIFT, PolyPhen-2, and MutationTaster. Potential pathogenic variants were filtered using a minimum allele frequency threshold ≤ 0.001 for dominant inheritance [25]. As POU4F3 has an autosomal dominant inheritance pattern, only heterozygous subjects were selected.
Manual classification of those variants was conducted based on American College of Medical Genetics and Genomics (ACMG)/Association for Molecular Pathology (AMP) guidelines for genetic hearing loss [26]. Sanger sequencing was performed in members of the four families, and the candidate variant of each family was cosegregated with the hearing loss phenotype.
3. Results
3.1. Families and Clinical Characteristics
The pedigrees of the four families showed autosomal dominant inheritance patterns (Figures 1–4(a)). High-resolution CTs of the temporal bone in probands of four families were normal, excluding middle- and inner-ear malformations (Figures 1–4(b)). The hearing impairments in these four families were sensorineural, postlingual, late onset, and progressive. Audiograms of some affected members of these four families are shown in Figures 1–4(d).
Family A was a three-generation Chinese family with ADNSHL and included eight affected patients (Figure 1). The ages at onset of the subjects ranged from 7 to 22 years old. The audiogram of the 20-year-old proband (III:3) with an onset age of 13 years showed all-frequency moderate hearing loss. The audiogram of II:3 showed profound hearing loss; interestingly, this subject had an onset age of 7 years old, which was the earliest in this family. The audiogram of II:5 showed a moderate level of hearing loss.
Family B was a three-generation Chinese family with ADNSHL and included four affected patients (Figure 2). The audiograms had a downsloping shape. The hearing loss in family B involved mostly high frequencies. The proband (III:1) was 7 years old with symmetric hearing loss, and the audiogram showed mild hearing impairment; thus, the proband could communicate with others normally. This family included one set of affected identical twin sisters (II:1 and II:2) who had similar audiograms but different hearing thresholds. Comparison of the audiograms of the proband and 53-year-old I:1 showed that although the hearing impairment had progressed over time, the progression was slight in the affected individual I:1 and involved mainly high frequencies.
Family C was a four-generation Chinese family with ADNSHL and included eight affected patients (Figure 3). The audiogram of proband (IV:2) was asymmetric, and hearing loss involved mainly middle frequencies. Hearing impairment in family C was postlingual, with onset in the first or second decade of life and progression to profound deafness with advancing age. The onset age of the proband was 15 years, and hearing loss was progressive. There was no history of hearing aid use or artificial cochlear implants in the proband. With regard to other auditory symptoms, the proband had complained of tinnitus. Audiograms showed that although low-frequency and high-frequency hearing were normal in the beginning, hearing ultimately deteriorated at all frequencies in the order of middle, high, and low frequencies. Downsloping audiogram configurations were observed in two subjects, who were 46 (III:6) and 69 (II:4) years old, whereas the audiogram of IV:2 was U-shaped (Figure 3(d)). Audiograms were unavailable for the other affected subjects.
Family D was a four-generation Chinese family with ADNSHL and included five affected patients (Figure 4). The audiogram of the proband (III:2) had a downsloping shape. The hearing impairment of the proband was moderate. The proband had a history of using hearing aids, but the effect was unsatisfactory. With regard to other auditory-related symptoms, individual II:1 and the proband complained of tinnitus.
3.2. Variant Identification
According to the autosomal dominant pattern of inheritance, only variants that were heterozygous in the affected siblings were selected as candidates. Four novel variants were identified using targeted NGS of 168 known deafness-related genes in the four different ADNSHL Chinese families. Among the four novel variants, three were missense variants: c.696G>T (p.Glu232Asp) detected in family A, c.325C>T (p.His109Tyr) in family B, and c.635T>C (p.Leu212Pro) in family C. The fourth variant was a frameshift variant, c.183delG (p.Ala62Argfs∗22), which was identified in family D. Sanger sequencing was performed in the other participating family members from these four families, which confirmed that these variants cosegregated with the hearing phenotypes (Figures 1–4(c)). The four variants have not been reported in previous studies and were not detected in 481 Chinese controls with normal hearing. The variants c.696G>T (p.Glu232Asp), c.635T>C (p.Leu212Pro), and c.183delG (p.Ala62Argfs∗22) are not present in the gnomAD or ExAC database, and c.325C>T (p.His109Tyr) has an allele frequency of 0.0001 in both gnomAD (Asian) and ExAC (Asian). The localizations of the four novel variants are shown in Figure 5(a). Conservation analysis was performed in the three families with missense variants (Figure 5(b)) and showed that the three variants are conserved among 11 species. Finally, the four novel variants were predicted to be deleterious by SIFT, Polyphen2, and CADD software. According to the American College of Medical Genetics and Genomics/Association for Molecular Pathology guidelines for genetic hearing loss [26, 27], c.696G>T (p.Glu232Asp) is classified as a likely pathogenic variant (PM1+PM2+PM5+PP1+PP3), c.325C>T (p.His109Tyr) is classified as a variant of uncertain significance (PP1), c.635T>C (p.Leu212Pro) is classified as a likely pathogenic variant (PM1+PM2+PP1+PP3), whereas c.183delG (p. Ala62Argfs∗22) is classified as a pathogenic variant (PVS1+PM2+ PP1) (Table 1).
Figure 5.
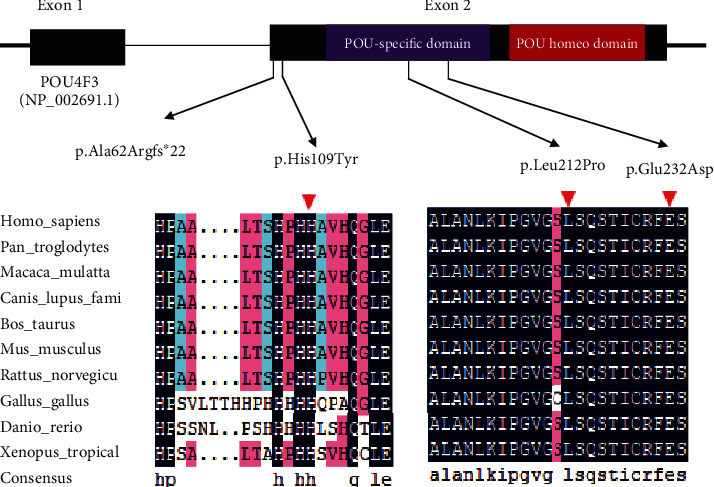
Protein structure of POU4F3 and conservation analysis. (a) Domain structure of POU4F3 showing the localization of four variants identified in this study. (b) Protein alignment showing that POU4F3 p.His109Tyr, p.Leu212Pro, and p.Glu232Asp all occur at evolutionarily conserved amino acids (shown by the red triangle) across 10 species.
Table 1.
Summary of the four POU4F3 variants identified in this study.
| Family | Nucleotide change | Amino acid change | hom/het | Allele frequency∗ | Pathogenicity | ACMG code | Computational evidence | Origin of variant | Cosegregation | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| SIFT | PolyPhen | Mutation assessor | |||||||||
| A | c.696G>T | p.(Glu232Asp) | het | 0.0001 | Likely pathogenic | PM1+PM2+PM5+PP1+PP3 | Deleterious | Probably damaging | High | De novo | Yes |
| B | c.325C>T | p.(His109Tyr) | het | — | Uncertain significance | PP1 | Tolerated | Benign | Medium | De novo | Yes |
| C | c.635T>C | p.(Leu212Pro) | het | — | Likely pathogenic | PM1+PM2+PP1+PP3 | Deleterious | Probably damaging | High | De novo | Yes |
| D | c.183delG | p.(Ala62Argfs∗22) | het | — | Pathogenic | PVS1+PM2+PP1 | De novo | Yes | |||
∗Allele frequency in East Asia reported by ExAC. hom: homozygous; het: heterozygous; —: no data. Notes: PVS1: null variant (nonsense, frameshift, canonical ±1 or 2 splice sites, initiation codon, single, or multiexon deletion) in a gene where loss of function (LOF) is a known mechanism of disease; PM1: located in a mutational hot spot and/or critical and well-established functional domain (e.g., active site of an enzyme) without benign variation; PM2: a variant is absent from a large general population or a control cohort; PM5: novel missense change at an amino acid residue where a different missense change determined to be pathogenic has been seen before; PP1: segregation of a variant in a family; PP3: multiple lines of computational evidence support a deleterious effect on the gene or gene product.
4. Discussion
In mammals' cochlea, HCs are the key cell type for hearing function, which convert the mechanical vibrations into electronic neural signals [9]. HCs are sensitive to multiple stresses and injuries and are easy to damage. While a mammal's cochlea only has very limited HC regeneration ability, most of the HC damage is permanent and irreversible [28–34]. Genetic factor accounts for 50% of sensorineural hearing loss. A genetic diagnosis is valuable for providing essential prognostic information needed for deciding optimal treatment/rehabilitation options and for genetic counseling [35]. Molecular epidemiological studies have found several common deafness genes in Chinese deafness population, such as GJB2, SLC26A4, and mtDNA12SrRNA [36]. However, genetic variants responsible for a large number of cases of hereditary hearing loss remain unknown. Next-generation sequencing has greatly increased the efficiency in screening known deafness genes for diagnostic purposes and in identifying new deafness genes [37–40].
In this study, we identified four novel variants in the POU4F3 gene, three missense variants, and one frameshift variant, which led to sensorineural hearing loss in four different Chinese families. The variabilities in onset age and severity of hearing loss in these four families demonstrated the heterogeneity of these variants both interfamilial and intrafamilial.
In 1998, POU4F3 was first discovered in an Israeli Jewish family. The results of a linkage analysis identified it as a novel independent locus for hearing loss, and the gene was designated as autosomal dominant nonsyndromic deafness 15 (DFNA15) [11]. The clinical presentation of DFNA15 is a form of progressive nonsyndromic sensorineural hearing loss with postlingual onset [13, 41]. In the present study, the earliest recorded age of hearing loss onset in affected individuals was 7 years old (III:1, family B). Among the 32 variants, 28 were reported in East Asian populations (13 in Japan, 12 in China, and 3 in Korea), and only 4 variants (2 in Netherlands, 1 in Israel, and 1 in Brazil) were reported from other areas, indicating that the POU4F3 pathogenic variant is an important contributor to ADNSHL, especially in East Asian populations (Table 2). In summary, the variant of POU4F3 is relatively common, especially in East Asian populations. Therefore, screening of POU4F3 should be a routine examination for the diagnosis of hereditary hearing loss. POU4F3 contains only two exons, making it convenient for screening.
Table 2.
Summary of all reported pathogenic variants in POU4F3.
| Number | Nucleotide change | Protein change | Exon | Domain | Onset age of hearing loss | Progression | Prevalence | Origin | Audiometric configuration | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Whole deletion of POU4F3 | 11~13 yo | Yes | N/A | Brazil | Flat and HF | Freitas et al. [13] | |||
| 2 | c.74dupA | p.His25fs∗18 | 1 | ~20 yo | Yes | 15/602 | Japan | HF | Kitano et al. [14] | |
| 3 | c.120+1G>C | 1 | 0~40 yo | Yes | 3/16 | China | Flat | He et al. [15] | ||
| 4 | c.183delG | p.A62Rfs∗22 | 2 | 25~44 yo | Yes | N/A | China | HF | This study | |
| 5 | c.191A>T | p.Asp64Val | 2 | ~30 yo | Yes | 15/602 | Japan | HF | Kitano et al. [14] | |
| 6 | c.325C>T | p.His109Tyr | 2 | 7~30 yo | Yes | N/A | China | HF | This study | |
| 7 | c.337C>T | p.Gln113Ter | 2 | 14~40 yo | Yes | N/A | China | Zhang et al. [16] | ||
| 8 | c.367delA | p.Ile123fs∗3 | 2 | ~40 yo | Yes | 15/602 | Japan | MF | Kitano et al. [14] | |
| 9 | c.427C>T | p.Gln143Ter | 2 | 3 yo | N/A | 15/602 | Japan | MF | Kitano et al. [14] | |
| 10 | c.491C>G | p.Pro164Arg | 2 | N/A | N/A | 1/6 | China | Flat and HF | Wei et al. [17] | |
| 11 | c.574G>T | p.Glu192Ter | 2 | POU | 17~30 yo | Yes | 15/602 | Japan | HF | Kitano et al. [14] |
| 12 | c.581T>A | p.Phe194Tyr | 2 | POU | 20 yo | Yes | 15/602 | Japan | HF | Kitano et al. [14] |
| 13 | c.602T>C | p.Leu201Pro | 2 | POU | >10 yo | Yes | N/A | China | MF | Gao et al. [12] |
| 14 | c.602delT | p.Leu201fs∗3 | 2 | POU | 16~30 yo | Yes | N/A | China | HF | Cai et al. [18] |
| 15 | c.603_604delGG | p.Val203Aspfs∗11 | 2 | POU | N/A | N/A | N/A | China | N/A | Yang et al. [19] |
| 16 | c.635T>C | p.Leu212Pro | 2 | POU | 10~20 yo | Yes | N/A | China | MF | This study |
| 17 | c.662_675del14 | p.Gly221Glufsf∗14 | 2 | POU | 20 yo | N/A | 1/42 | Korea | HF | Lee et al. [20] |
| 18 | c.665C>T | p.Ser222Leu | 2 | POU | 6 yo | Yes | 15/602 | Japan | HF | Kitano et al. [14] |
| 19 | c.668T>C | p.Leu223Pro | 2 | POU | 13~20 yo | Yes | N/A | Netherlands | Flat, MF, and HF | Collin et al. [21] |
| 20 | c.680delC | p.Thr227fs∗13 | 2 | POU | 0 yo | Yes | 15/602 | Japan | MF | Kitano et al. [14] |
| 21 | c.694G>A | p.Glu232Lys | 2 | POU | ~20 yo | N/A | 1/8 | Korea | HF | Baek et al. [22] |
| 22 | c.696G>T | p.Glu232Asp | 2 | POU | 7~22 yo | Yes | N/A | China | HF | This study |
| 23 | c.718A>T | p.Asn240Tyr | 2 | POU | 6 yo | Yes | 15/602 | Japan | MF | Kitano et al. [14] |
| 24 | c.841A>G | p.Ile281Val | 2 | POU homeobox | 50~54 yo | Yes | 15/602 | Japan | HF | Kitano et al. [14] |
| 25 | c.865C>T | p.Leu289Phe | 2 | POU homeobox | 13~20 yo | Yes | N/A | Netherlands | Flat, MF, and HF | Collin et al. [21] |
| 26 | c.884_891del8 | Ile295Thrfs∗5 | 2 | POU homeobox | 18~30 yo | Yes | N/A | Israel | HF | Vahava et al. [11] |
| 27 | c.896C>T | p.Pro299Leu | 2 | POU homeobox | 26~41 yo | Yes | 15/602 | Japan | MF | Kitano et al. [14] |
| 28 | c.932T>C | p.Leu311Pro | 2 | POU homeobox | 10~20 yo | Yes | 3/16 | China | HF | He et al. [2] |
| 29 | c.976A>T | p.Arg326Ter | 2 | POU homeobox | Childhood | Yes | 15/602 | Japan | HF | Kitano et al. [14] |
| 30 | c.977G>A | p.Arg326Lys | 2 | POU homeobox | 10~50 yo | N/A | N/A | Korea | HF | Kim et al. [41] |
| 31 | c.982A>G | p.Lys328Glu | 2 | POU homeobox | N/A | Yes | N/A | Taiwan | HF | Lin et al. [23] |
| 32 | c.1007delC | p.Ala336fs∗ | 2 | POU homeobox | 0 yo | Yes | 1/3 | Japan | N/A | Mutai et al. [24] |
yo: years old; HF: high frequency; MF: middle frequency; N/A: not available.
Hearing impairment involves mainly the middle frequency range (1000–2000 Hz) in a low percentage of cases of hereditary hearing loss. Kitano et al. reported that POU4F3-associated hearing loss usually presents with middle- or high-frequency hearing loss [14]. In 2018, we reported a family with middle-frequency hearing loss associated with POU4F3 c.602T>C (p.Leu201Pro) [12]. In this study, the proband in family C presented with typical middle-frequency hearing loss, and the older patients showed downsloping audiograms and mainly middle- and high-frequency hearing loss. In accordance with our previous report, we proposed that the affected frequencies of certain types of POU4F3-associated hearing loss were in the order of middle (U-shaped audiogram), high (downsloping audiogram), and low frequencies (flatter audiogram). Accordingly, the different forms of auditory configuration represented different disease phases.
POU4F3 belongs to a family of proteins characterized by a well-conserved bipartite domain [42]. The bipartite domain is comprised of a POU-specific domain (amino acids 179–256) and a POU homeodomain (amino acids 274–333) separated by a linker [43]. These two domains are responsible for the main functions of POU4F3.
However, the specific mechanisms underlying sensorineural hearing loss caused by the POU4F3 variant have remained unclear to date. Several previous studies have shown that although the wild-type POU4F3 is localized almost exclusively in the nucleus, the mutant protein is also present in both the cytoplasm and the nucleus. Cytoplasmic localization of transcription factors obviously affects their ability to activate downstream targets. Mutant proteins showed greatly reduced capability for binding to DNA as well as transcriptionally activating reporter gene expression [10, 16, 20, 21, 23]. One possible mechanism is that the variant in the POU homeodomain of POU4F3 leads to a prematurely truncated protein with loss of the second and third helices, and the third helix is crucial for high-affinity binding to DNA; thus, the target gene cannot be induced, leading to impairment of inner ear hair cells [11].
Further studies showed that POU4F3 contains two nuclear localization signals (NLSs): a monopartite NLS (amino acids 274–278) and bipartite NLS (amino acids 314–331) [10]. NLS is crucial for the trafficking of cytoplasmic proteins into the nucleus. Variant of POU4F3 results in the absence of these two NLSs, which leads to subcellular protein mislocalization. The normal wild-type protein is localized mainly in the nucleus [44]. However, transient transfection studies revealed that NLS-mutated POU4F3 proteins are localized mainly in the cytoplasm, most likely due to the absence of the NLSs. As POU4F3 proteins are transcription factors, their function requires their entry into the nucleus and binding to DNA. In addition, the mutated POU4F3 proteins have longer half-lives and much lower levels of transcriptional activity than those of the wild-type protein [11].
Although mice require only one copy of the functional POU4F3 to retain hearing [45, 46], several previous studies supported that haploinsufficiency is the most likely molecular mechanism underlying the hearing loss caused by the POU4F3 variant [13, 23, 24]. Heterozygous deletion of the entire POU4F3 has been reported in a Brazilian family with ADNSHL [13]. Another study identified an ADNSHL-associated POU4F3 heterozygous frameshift variant c.1007delC (p.Ala336fs∗), which would produce a transcript without an in-frame stop codon, and presumably, the nonstop mRNA might be degraded through nonstop decay [24]. Both variants cause the loss of one copy of POU4F3, indicating the mechanism of haploinsufficiency [47]. Also, the subcellular protein mislocalization of mutant POU4F3 shown in Lin et al. and other studies support the mechanism of haploinsufficiency [10, 16, 20]. ExAC pLI score of POU4F3 is 0.721 which is not an indication for extreme loss of function intolerance. In addition, studies showed that the pathways downstream of POU4F3 play crucial roles in the maintenance of inner ear hair cells, which also provides insight into the mechanisms underlying POU4F3 mutation-induced hearing loss. A study performed in 2004 showed that the degeneration of outer hair cells caused by the POU4F3 variant was mainly or entirely the result of inhibited expression of growth factor independence 1 (Gfi1), which is one of the target genes of POU4F3 [46]. Gfi1 not only plays a late role in the differentiation and maintenance of hair cells but also promotes the formation of hair cells in cooperation with atonal BHLH transcription factor 1 (Atoh1) [48]. In addition, another study showed that Atoh1 is upstream of POU4F3 and Gfi1 [49]. Thus, regulation of Atoh1 will affect the expression of Gfi1, and both Atoh1 and POU4F3 are required for maintenance of Gfi1 expression [50]. Another possible mechanism is that the variant of POU4F3 inhibits the expression of myosin VI, which plays a large role in the maintenance of stereocilia of hair cells that are responsible for auditory transduction [51]. Tornari et al. reported that the orphan thyroid nuclear receptor Nr2f2, which is related to the development and survival of hair cells, is a target of POU4F3 [52]. Although several downstream pathways and probable mechanisms have been reported, further studies are required to explore the mechanisms related to POU4F3.
In this study, we identified four novel variants in POU4F3 (three missense variants and one frameshift variant) involved in hearing loss. The missense variant c.696G>T (p.Glu232Asp), detected in family A, is located in the POU-specific domain, and a different missense variant at the same locus, c.694G>A (p.Glu232Lys), has been reported previously [22]. The missense variant c.696G>T (p.Glu232Asp) in POU4F3 leads to substitution of the glutamate at position 232 with an aspartic acid, which probably alters the structure of the α-helix of the POU-specific domain. The structural changes in the helix might affect the DNA-binding ability, what was probably responsible for the hearing loss in this family. The missense variant found in family C, c.635C>T (p.Leu212Pro), is also localized in the POU-specific domain, and it is possible that the mechanism of action is likely the same as described above. The missense variant observed in family B, c.325C>T (p.His109Tyr), is located in the transcriptional activation domain, which is not a functional domain, and this is likely why the hearing impairment in this family was mild. This variant is heterozygous in 0.016% (3/18,385 alleles) of East Asians, according to the gnomAD database. We speculate that its detection in the public database is due to the mild hearing loss associated with this variant. The frameshift variant, c.183delG (p.Ala62Argfs∗22), identified in family D results in a truncated protein with loss of both functional domains crucial for high-affinity binding to DNA.
5. Conclusions
In summary, four novel variants in POU4F3 were identified in four different families. These consisted of three missense variants, c.696G>T (p.Glu232Asp), c.325C>T (p.His109Tyr), and c.635C>T (p.Leu212Pro), and one frameshift variant, c.183delG (p.Ala62Argfs∗22). These variants of POU4F3 are considered to be responsible for ADNSHL, designated as DFNA15. POU4F3 variants are not rare, and therefore, screening of POU4F3 should be included in routine examinations for diagnosis of ADNSHL. Further studies are required to determine the specific mechanisms underlying hearing loss.
Acknowledgments
We sincerely thank all the family members for their participation and cooperation in this study. The English in this document has been checked by at least two professional editors, both native speakers of English. For a certificate, please seehttp://www.textcheck.com/certificate/MccWLW. This study was supported by grants from the National Key Research and Development Project of China (2016YFC1000706), National Natural Science Foundation of China (81873704), and Fostering Funds of Chinese PLA General Hospital for National Distinguished Young Scholar Science Fund (2017-JQPY-001) to Yong-Yi Yuan; a grant from the Foundation of the Second Hospital of Hebei Medical University (2h201816) to Qing-Wen Zhu; grants from the Beijing Natural Science Foundation (7192234) and National Natural Science Foundation of China (81570929) to Xue Gao; and a grant from the National Natural Science Foundation of China (81870731) to Sha-Sha Huang.
Contributor Information
Qing-Wen Zhu, Email: zqw301@163.com.
Yong-Yi Yuan, Email: yyymzh@163.com.
Data Availability
The patient's phenotype and the detected variants have been submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), and the Submission ID is SUB7170390.
Ethical Approval
This study was approved by the Ethics Committee of the Chinese People's Liberation Army General Hospital (reference number S2016-120-02).
Disclosure
The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflicts of Interest
There are no financial relationships with any organizations that might have an interest in the submitted work, and there are no other relationships or activities that could appear to have influenced the submitted work.
Authors' Contributions
Tianyi Cui and Xue Gao contributed equally to this paper.
Supplementary Materials
In this study, we identified four novel variants using NGS of a panel of 168 deafness genes from four different Chinese families suffering from ADNSHL. The 168 deafness genes of the panel are listed concretely in Supplementary Table 1.
References
- 1.Liu L., Chen Y., Qi J., et al. Wnt activation protects against neomycin-induced hair cell damage in the mouse cochlea. Cell Death & Disease. 2016;7(3):p. e2136. doi: 10.1038/cddis.2016.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.He Z., Guo L., Shu Y., et al. Autophagy protects auditory hair cells against neomycin-induced damage. Autophagy. 2017;13(11):1884–1904. doi: 10.1080/15548627.2017.1359449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu C., Cheng C., Wang Y., et al. Loss of ARHGEF6 Causes Hair Cell Stereocilia Deficits and Hearing Loss in Mice. Frontiers in Molecular Neuroscience. 2018;11:p. 362. doi: 10.3389/fnmol.2018.00362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu W., Xu X., Fan Z., et al. Wnt Signaling Activates TP53-Induced Glycolysis and Apoptosis Regulator and Protects Against Cisplatin-Induced Spiral Ganglion Neuron Damage in the Mouse Cochlea. Antioxidants & Redox Signaling. 2019;30(11):1389–1410. doi: 10.1089/ars.2017.7288. [DOI] [PubMed] [Google Scholar]
- 5.He Z. H., Zou S. Y., Li M., et al. The nuclear transcription factor FoxG1 affects the sensitivity of mimetic aging hair cells to inflammation by regulating autophagy pathways. Redox Biology. 2020;28:p. 101364. doi: 10.1016/j.redox.2019.101364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith R. J. H., Bale J. F., Jr., White K. R. Sensorineural hearing loss in children. Lancet. 2005;365(9462):879–890. doi: 10.1016/S0140-6736(05)71047-3. [DOI] [PubMed] [Google Scholar]
- 7.Kremer H. Hereditary hearing loss; about the known and the unknown. Hearing Research. 2019;376:58–68. doi: 10.1016/j.heares.2019.01.003. [DOI] [PubMed] [Google Scholar]
- 8.Shearer A. E., Hildebrand M. S., Smith R. J. H. GeneReviews®[Internet] Seattle: University of Washington; 2017. Hereditary hearing loss and deafness overview. [Google Scholar]
- 9.Cox B. C., Chai R., Lenoir A., et al. Spontaneous hair cell regeneration in the neonatal mouse cochlea in vivo. Development. 2014;141(4):816–829. doi: 10.1242/dev.103036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weiss S., Gottfried I., Mayrose I., et al. The DFNA15 deafness mutation affects POU4F3 protein stability, localization, and transcriptional activity. Molecular and Cellular Biology. 2003;23(22):7957–7964. doi: 10.1128/MCB.23.22.7957-7964.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vahava O., Morell R., Lynch E. D., et al. Mutation in transcription factor POU4F3 associated with inherited progressive hearing loss in humans. Science. 1998;279(5358):1950–1954. doi: 10.1126/science.279.5358.1950. [DOI] [PubMed] [Google Scholar]
- 12.Gao X., Xu J. C., Wang W. Q., et al. A Missense Mutation in POU4F3 Causes Midfrequency Hearing Loss in a Chinese ADNSHL Family. BioMed Research International. 2018;2018:7. doi: 10.1155/2018/5370802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Freitas É. L., Oiticica J., Silva A. G., Bittar R. S. M., Rosenberg C., Mingroni-Netto R. C. Deletion of the entire POU4F3 gene in a familial case of autosomal dominant non-syndromic hearing loss. European Journal of Medical Genetics. 2014;57(4):125–128. doi: 10.1016/j.ejmg.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 14.Kitano T., Miyagawa M., Nishio S.-y., et al. POU4F3 mutation screening in Japanese hearing loss patients: Massively parallel DNA sequencing-based analysis identified novel variants associated with autosomal dominant hearing loss. PLOS ONE. 2017;12(5):p. e0177636. doi: 10.1371/journal.pone.0177636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.He L., Pang X., Chen P., Wu H., Yang T. Mutation in the hair cell specific gene POU4F3 is a common cause for autosomal dominant nonsyndromic hearing loss in Chinese Hans. Neural Plasticity. 2016;2016:6. doi: 10.1155/2016/9890827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang C., Wang M., Xiao Y., et al. A novel nonsense mutation of POU4F3 gene causes autosomal dominant hearing loss. Neural Plasticity. 2016;2016:10. doi: 10.1155/2016/1512831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei Q., Zhu H., Qian X., et al. Targeted genomic capture and massively parallel sequencing to identify novel variants causing Chinese hereditary hearing loss. Journal of Translational Medicine. 2014;12(1):p. 311. doi: 10.1186/s12967-014-0311-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cai X. Z., Li Y., Xia L., et al. Exome sequencing identifies POU4F3 as the causative gene for a large Chinese family with non-syndromic hearing loss. Journal of Human Genetics. 2017;62(2):317–320. doi: 10.1038/jhg.2016.102. [DOI] [PubMed] [Google Scholar]
- 19.Yang T., Wei X., Chai Y., Li L., Wu H. Genetic etiology study of the non-syndromic deafness in Chinese Hans by targeted next-generation sequencing. Orphanet Journal of Rare Diseases. 2013;8(1):p. 85. doi: 10.1186/1750-1172-8-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee H. K., Park H. J., Lee K. Y., Park R., Kim U. K. A novel frameshift mutation of POU4F3 gene associated with autosomal dominant non-syndromic hearing loss. Biochemical and Biophysical Research Communications. 2010;396(3):626–630. doi: 10.1016/j.bbrc.2010.04.132. [DOI] [PubMed] [Google Scholar]
- 21.Collin R. W. J., Chellappa R., Pauw R. J., et al. Missense mutations in POU4F3 cause autosomal dominant hearing impairment DFNA15 and affect subcellular localization and DNA binding. Human Mutation. 2008;29(4):545–554. doi: 10.1002/humu.20693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baek J. I., Oh S. K., Kim D. B., et al. Targeted massive parallel sequencing: the effective detection of novel causative mutations associated with hearing loss in small families. Orphanet Journal of Rare Diseases. 2012;7(1):p. 60. doi: 10.1186/1750-1172-7-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin Y. H., Lin Y. H., Lu Y. C., et al. A novel missense variant in the nuclear localization signal of POU4F3 causes autosomal dominant non-syndromic hearing loss. Scientific Reports. 2017;7(1):p. 7551. doi: 10.1038/s41598-017-08236-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mutai H., Suzuki N., Shimizu A., et al. Diverse spectrum of rare deafness genes underlies early-childhood hearing loss in Japanese patients: a cross-sectional, multi-center next-generation sequencing study. Orphanet Journal of Rare Diseases. 2013;8(1):p. 172. doi: 10.1186/1750-1172-8-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao X., Yuan Y. Y., Lin Q. F., et al. Mutation ofIFNLR1, an interferon lambda receptor 1, is associated with autosomal-dominant non-syndromic hearing loss. Journal of Medical Genetics. 2018;55(5):298–306. doi: 10.1136/jmedgenet-2017-104954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Richards S., on behalf of the ACMG Laboratory Quality Assurance Committee, Aziz N., et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine. 2015;17(5):405–423. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oza A. M., DiStefano M. T., Hemphill S. E., et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Human Mutation. 2018;39(11):1593–1613. doi: 10.1002/humu.23630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang T., Chai R., Kim G. S., et al. Lgr5+ cells regenerate hair cells via proliferation and direct transdifferentiation in damaged neonatal mouse utricle. Nature Communications. 2015;6(1) doi: 10.1038/ncomms7613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang S., Zhang Y., Dong Y., et al. Knockdown of Foxg1 in supporting cells increases the trans-differentiation of supporting cells into hair cells in the neonatal mouse cochlea. Cellular and Molecular Life Sciences. 2020;77(7):1401–1419. doi: 10.1007/s00018-019-03291-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu X., Sun S., Qi J., et al. Bmi 1 regulates the proliferation of cochlear supporting cells via the canonical Wnt signaling pathway. Molecular Neurobiology. 2017;54(2):1326–1339. doi: 10.1007/s12035-016-9686-8. [DOI] [PubMed] [Google Scholar]
- 31.Tan F., Chu C., Qi J., et al. AAV-ie enables safe and efficient gene transfer to inner ear cells. Nature Communications. 2019;10(1):p. 3733. doi: 10.1038/s41467-019-11687-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng C., Wang Y., Guo L., et al. Age-related transcriptome changes in Sox2+ supporting cells in the mouse cochlea. Stem Cell Research & Therapy. 2019;10(1):p. 365. doi: 10.1186/s13287-019-1437-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang S., Liu D., Dong Y., et al. Frizzled-9+ supporting cells are progenitors for the generation of hair cells in the postnatal mouse cochlea. Frontiers in Molecular Neuroscience. 2019;12:p. 184. doi: 10.3389/fnmol.2019.00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yan W., Liu W., Qi J., et al. A three-dimensional culture system with Matrigel promotes purified spiral ganglion neuron survival and function in vitro. Molecular Neurobiology. 2018;55(3):2070–2084. doi: 10.1007/s12035-017-0471-0. [DOI] [PubMed] [Google Scholar]
- 35.ACMG Working Group on Update of Genetics Evaluation Guidelines for the Etiologic Diagnosis of Congenital Hearing Loss. American College of Medical Genetics and Genomics guideline for the clinical evaluation and etiologic diagnosis of hearing loss. Genetics in Medicine. 2014;16(4):347–355. doi: 10.1038/gim.2014.2. [DOI] [PubMed] [Google Scholar]
- 36.Yuan Y., You Y., Huang D., et al. Comprehensive molecular etiology analysis of nonsyndromic hearing impairment from typical areas in China. Journal of Translational Medicine. 2009;7(1):p. 79. doi: 10.1186/1479-5876-7-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin X., Tang W., Ahmad S., et al. Applications of targeted gene capture and next-generation sequencing technologies in studies of human deafness and other genetic disabilities. Hearing Research. 2012;288(1-2):67–76. doi: 10.1016/j.heares.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Idan N., Brownstein Z., Shivatzki S., Avraham K. B. Advances in genetic diagnostics for hereditary hearing loss. Journal of Basic and Clinical Physiology and Pharmacology. 2013;24(3):165–170. doi: 10.1515/jbcpp-2013-0063. [DOI] [PubMed] [Google Scholar]
- 39.Sloan-Heggen C. M., Bierer A. O., Shearer A. E., et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Human Genetics. 2016;135(4):441–450. doi: 10.1007/s00439-016-1648-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yuan Y., Li Q., Su Y., et al. Comprehensive genetic testing of Chinese SNHL patients and variants interpretation using ACMG guidelines and ethnically matched normal controls. European Journal of Human Genetics. 2020;28(2):231–243. doi: 10.1038/s41431-019-0510-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim H. J., Won H. H., Park K. J., et al. SNP linkage analysis and whole exome sequencing identify a novel POU4F3 mutation in autosomal dominant late-onset nonsyndromic hearing loss (DFNA15) PLoS One. 2013;8(11, article e79063) doi: 10.1371/journal.pone.0079063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wegner M., Drolet D. W., Rosenfeld M. G. POU-domain proteins: structure and function of developmental regulators. Current Opinion in Cell Biology. 1993;5(3):488–498. doi: 10.1016/0955-0674(93)90015-i. [DOI] [PubMed] [Google Scholar]
- 43.Herr W., Cleary M. A. The POU domain: versatility in transcriptional regulation by a flexible two-in-one DNA-binding domain. Genes & Development. 1995;9(14):1679–1693. doi: 10.1101/gad.9.14.1679. [DOI] [PubMed] [Google Scholar]
- 44.Xiang M., Gan L., Li D., et al. Essential role of POU-domain factor Brn-3c in auditory and vestibular hair cell development. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(17):9445–9450. doi: 10.1073/pnas.94.17.9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Keithley E. M., Erkman L., Bennett T., Lou L., Ryan A. F. Effects of a hair cell transcription factor, Brn-3.1, gene deletion on homozygous and heterozygous mouse cochleas in adulthood and aging. Hearing Research. 1999;134(1-2):71–76. doi: 10.1016/S0378-5955(99)00070-2. [DOI] [PubMed] [Google Scholar]
- 46.Hertzano R., Montcouquiol M., Rashi-Elkeles S., et al. Transcription profiling of inner ears from Pou4f3ddl/ddl identifies Gfi1 as a target of the Pou4f3 deafness gene. Human Molecular Genetics. 2004;13(18):2143–2153. doi: 10.1093/hmg/ddh218. [DOI] [PubMed] [Google Scholar]
- 47.Klauer A. A., van Hoof A. Degradation of mRNAs that lack a stop codon: a decade of nonstop progress. Wiley Interdisciplinary Reviews: RNA. 2012;3(5):649–660. doi: 10.1002/wrna.1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Costa A., Powell L. M., Lowell S., Jarman A. P. Atoh1 in sensory hair cell development: constraints and cofactors. Seminars in Cell & Developmental Biology. 2017;65:60–68. doi: 10.1016/j.semcdb.2016.10.003. [DOI] [PubMed] [Google Scholar]
- 49.Wallis D., Hamblen M., Zhou Y., et al. The zinc finger transcription factor Gfi1, implicated in lymphomagenesis, is required for inner ear hair cell differentiation and survival. Development. 2003;130(1):221–232. doi: 10.1242/dev.00190. [DOI] [PubMed] [Google Scholar]
- 50.Chonko K. T., Jahan I., Stone J., et al. Atoh1 directs hair cell differentiation and survival in the late embryonic mouse inner ear. Developmental Biology. 2013;381(2):401–410. doi: 10.1016/j.ydbio.2013.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ma D. B., Chen J., Xia Y., et al. Inhibition of Myo6 gene expression by co-expression of a mutant of transcription factor POU4F3 (BRN-3C) in hair cells. Molecular Medicine Reports. 2014;9(4):1185–1190. doi: 10.3892/mmr.2014.1953. [DOI] [PubMed] [Google Scholar]
- 52.Tornari C., Towers E. R., Gale J. E., Dawson S. J. Regulation of the orphan nuclear receptor Nr2f2 by the DFNA15 deafness gene Pou4f3. PLoS One. 2014;9(11, article e112247) doi: 10.1371/journal.pone.0112247. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
In this study, we identified four novel variants using NGS of a panel of 168 deafness genes from four different Chinese families suffering from ADNSHL. The 168 deafness genes of the panel are listed concretely in Supplementary Table 1.
Data Availability Statement
The patient's phenotype and the detected variants have been submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), and the Submission ID is SUB7170390.