

Abstract
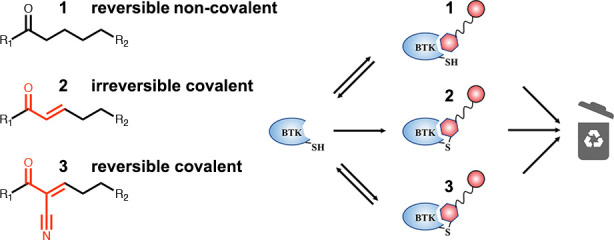
Proteolysis targeting chimeras (PROTACs) represent an exciting inhibitory modality with many advantages, including substoichiometric degradation of targets. Their scope, though, is still limited to date by the requirement for a sufficiently potent target binder. A solution that proved useful in tackling challenging targets is the use of electrophiles to allow irreversible binding to the target. However, such binding will negate the catalytic nature of PROTACs. Reversible covalent PROTACs potentially offer the best of both worlds. They possess the potency and selectivity associated with the formation of the covalent bond, while being able to dissociate and regenerate once the protein target is degraded. Using Bruton’s tyrosine kinase (BTK) as a clinically relevant model system, we show efficient degradation by noncovalent, irreversible covalent, and reversible covalent PROTACs, with <10 nM DC50’s and >85% degradation. Our data suggest that part of the degradation by our irreversible covalent PROTACs is driven by reversible binding prior to covalent bond formation, while the reversible covalent PROTACs drive degradation primarily by covalent engagement. The PROTACs showed enhanced inhibition of B cell activation compared to ibrutinib and exhibit potent degradation of BTK in patient-derived primary chronic lymphocytic leukemia cells. The most potent reversible covalent PROTAC, RC-3, exhibited enhanced selectivity toward BTK compared to noncovalent and irreversible covalent PROTACs. These compounds may pave the way for the design of covalent PROTACs for a wide variety of challenging targets.
Introduction
Proteolysis targeting chimeras (PROTACs) are receiving increasing attention as a new therapeutic modality, as was recently underscored by the first PROTAC, ARV-110, to enter clinical trials.1 PROTACs are composed of a protein target binding moiety, a linker, and an E3 ubiquitin ligase binder.2,3 Upon binding, the PROTAC induces the formation of a ternary complex between the target and E3 ligase,4−7 resulting in the ubiquitination and degradation of the target. Compared to traditional inhibition of the target protein, targeted degradation has several important advantages, including the elimination of all levels of protein function, enhanced selectivity,8−12 longer duration of action due to the need to resynthesize the target,13 and degradation by substoichiometric amounts of PROTAC.14
Efficient degradation typically requires high affinity binding to the target as well as optimized linker geometry, to optimize the ternary complex formation. However, many targets such as transcription factors,15,16 protein–protein interfaces,17,18 or challenging enzyme classes such as GTPases19 are recalcitrant to ligand discovery. This limits the applicability of PROTACs against such targets. A possible solution to this problem is to introduce an electrophile that will allow covalent binding to the target. However, irreversible binding may reduce potency by negating the catalytic nature of the PROTAC activity. While several covalent PROTACs have been developed and degrade their target successfully,20−22 there are examples in which the introduction of irreversible binding reduces the potency of PROTACs.23,24
Theoretically, reversible covalent PROTACs can benefit from the enhanced potency, selectivity, and long duration of action that accompany covalent bond formation,25−27 without compromising the substoichiometric activity of PROTACs. In this work we set out to test this hypothesis by the design of cyanoacrylamide-based reversible covalent PROTACs. To this end, we selected Bruton’s tyrosine kinase (BTK), which is an established target for noncovalent PROTACs,23,28−32 and systematically tested a series of reversible covalent PROTACs along with their irreversible covalent and noncovalent PROTAC analogues. Our work resulted in a highly potent and selective reversible covalent PROTAC (RC-3), as well as insights into the effect of covalent bond formation kinetics on the degradation by covalent PROTACs.
Results
We devised a modular scheme for the synthesis of cyanoacrylamide-based PROTACs (see Methods and SI). Using this route, we synthesized a series of 12 reversible covalent PROTACs targeting cysteine 481 in BTK (Supporting Table 1). These are based on the scaffold of the covalent BTK binder—ibrutinib—as the protein targeting moiety33 and PEG-based linkers with varying length (Figure 1). We used two approaches: in the first, we synthesized an alkyne-functionalized BTK-binding cyanoacrylamide and an amine-functionalized E3 binder and linked them in one-pot reactions using azide-PEG-NHS esters of varying lengths. In the second approach, we directly functionalized thalidomide with various PEGs and formed the cyanoacrylamide in a final condensation step.
Figure 1.
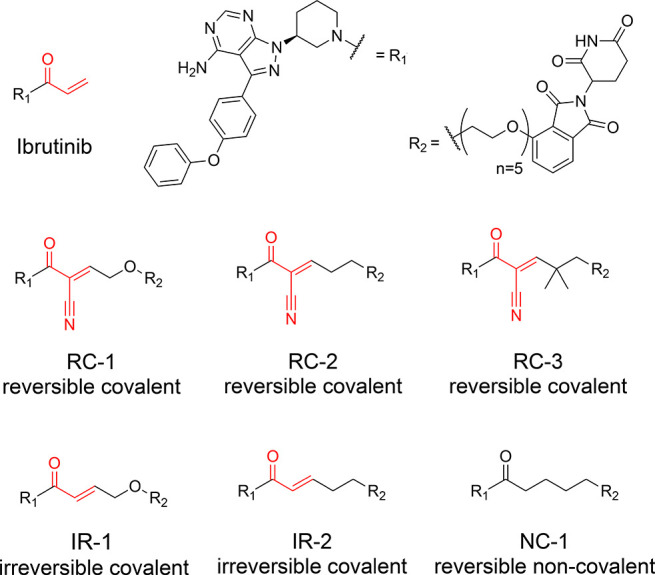
Structures of reversible covalent, irreversible covalent, and noncovalent BTK PROTACs described in this study. The electrophilic moieties are highlighted in red.
We incubated K562 or Mino cells with 1 μM of the compounds for 24 h and measured the abundance of BTK by Western blot (Supporting Figure 1). Several of the tested PROTACs displayed clear BTK degradation and could serve as potential starting points for optimization. We selected compound RC-1, which is based on a PEG6 linker and displayed consistent prominent levels of degradation in both cell lines, as a starting point for this study. On the basis of compound RC-1 we synthesized additional compounds (Figure 1), RC-2 with a CH2 group replacing the oxygen nearest the β-carbon, their analogous acrylamides IR-1 and IR-2, and the noncovalent analogue NC-1. We also synthesized the cyanoacrylamide RC-3 (Figure 1), replacing the Cα hydrogens with methyl groups. Similar dimethylated cyanoacrylamides were reported to have improved cellular permeability.34
We evaluated the ability of the compounds to induce degradation of BTK in human cell lines. We incubated Mino cells with the compounds and followed BTK levels after 24 h by Western blot (Figure 2, Supporting Figure 2). The noncovalent PROTAC NC-1 showed the highest degradation potency with DC50 = 2.2 nM (maximal degradation Dmax = 97%). The irreversible acrylamides IR-1 and IR-2 and the cyanoacrylamide RC-3 followed closely with DC50’s under 10 nM and Dmax near 90%. The cyanoacrylamides RC-1 and RC-2 were less potent. Similar trends were observed in Ramos cells (Supporting Figure 2A). We conducted metabolomics studies to estimate if cellular penetration and stability may contribute to the relative potencies (Supporting Table 2). NC-1 and RC-3 reached an effective concentration that was ∼2 times higher than IR-2 and ∼10 times higher than RC-2. Therefore, the lower potency of RC-2 can at least in part be explained by lower permeability or stability.
Figure 2.

Efficient BTK degradation in cells. (A) Western blot evaluation of BTK levels in Mino cells in response to various concentrations of RC-2, IR-2, NC-1, and RC-3, after 24 h of incubation. (B) Quantification of BTK levels in (A) by normalization to the β-actin house-keeping gene in Mino cells. DC50 and Dmax were calculated by fitting the data to a second-order polynomial using Prism software. (C) Summary of the DC50 and Dmax values for the PROTACs in Mino cells described in (A) and Supporting Figure 2.
We followed the rate of BTK degradation facilitated by this compound series via a time course experiment in Ramos and Mino cells (Supporting Figure 3). The rates of degradation correlated well with the DC50 values observed after 24 h, with NC-1, IR-1, IR-2, and RC-3 degrading BTK within 2–4 h, while RC-2 and RC-1 required 6–24 h to reach maximum degradation.
To validate the mechanism of PROTAC-mediated degradation of BTK, Mino cells were pretreated for 2 h with either ibrutinib or thalidomide-OH and subsequently treated with the PROTACs for an additional 24 h. Both ibrutinib pretreatment and thalidomide-OH hindered BTK degradation (Figure 3A). In contrast to the covalent PROTACs, degradation by the noncovalent NC-1 was only slightly hindered by thalidomide. In addition, RC-1m, a methylated thalidomide analogue of RC-1, no longer able to bind CRBN, lost all activity (Supporting Figure 4), further suggesting CRBN-mediated degradation. We treated Mino cells with bortezomib, a proteasome inhibitor,35 for 1 h before treatment with the PROTACs and assessed BTK levels after an additional 4 h. Bortezomib significantly inhibited degradation, suggesting proteasome-dependent degradation (Figure 3B).
Figure 3.

PROTAC-mediated BTK degradation is hindered by ibrutinib, thalidomide, and by proteasome inhibition. (A) Mino cells were either pretreated for 2 h with ibrutinib/thalidomide-OH or untreated, before treatment with a BTK PROTAC for 24 h. Subsequently BTK levels were measured via Western blot. (B) Mino cells were treated for 1 h with bortezomib to inhibit proteasome-dependent degradation; then PROTACs were added for 4 h, followed by measuring BTK levels via Western blot.
To assess the PROTACs’ efficiency in a clinically relevant model, we tested their ability to induce BTK degradation in primary cells from chronic lymphocytic leukemia (CLL) patients. The PROTACs displayed potent degradation with DC50’s < 100 nM, with NC-1 and IR-2 reaching higher degradation levels than RC-2 and RC-3 (Figure 4).
Figure 4.

Degradation of BTK in patient-derived CLL cells. Patient-derived primary CLL cells were treated with BTK PROTACs for 18 h, followed by measuring BTK levels via Western blot. M-IGHV/UM-IGHV: mutated/unmutated immunoglobulin heavy chain variable region (IGHV) gene.
While we observed potent degradation by covalent PROTACs, it was still not clear if and how the covalent bond contributes to the degradation process. Due to the high noncovalent binding affinity of ibrutinib to BTK, it is possible that the degradation is induced primarily by reversible binding that occurs prior to covalent bond formation. A second related question was, what is the dissociation rate of the covalent complexes formed by the cyanoacrylamides, and whether this rate can support catalytic degradation to the same degree as noncovalent binding. To answer these questions, we performed several experiments to evaluate the formation of covalent complexes with BTK at the time scale and concentration range observed for degradation, as well as their dissociation kinetics.
First, we tested the degradation activity of the PROTACs against overexpressed wild-type BTK and the C481S mutant, which cannot form covalent complexes (Figure 5). As expected, the degradation by noncovalent NC-1 was only mildly affected by the mutation. However, the covalent PROTACs IR-1, IR-2, and RC-2 also showed low sensitivity to the mutation. In contrast, degradation by RC-3 was severely impaired by the mutation, indicating an important role for covalent engagement in the degradation process by RC-3.
Figure 5.

Degradation of overexpressed BTK and BTK C481S in U2OS cells. (A) Transfected U2OS cells were treated with 100 nM PROTAC for 24 h, followed by measuring BTK levels via Western blot. (B) Quantification of normalized BTK levels in (A).
Second, we tested the ability of the compounds to covalently bind and inhibit BTK. We performed an in vitro kinase activity assay with both wild-type BTK and the C481S mutant (Figure 6A,B). The assay was performed with a preincubation period of 2 h, equivalent to the time scale of degradation induced by the PROTACs in cells (Supporting Figure 3). As expected, ibrutinib was both highly potent against the WT and sensitive to the mutation, with a 74-fold reduction in potency, indicating efficient covalent engagement. The noncovalent NC-1 showed very mild sensitivity to the mutation, with a <2-fold reduction in affinity. The acrylamide IR-2 did not inhibit BTK more potently than NC-1 and also showed only slight sensitivity to the mutation, indicating inefficient covalent bond formation, possibly due to the lowered reactivity of the β-substituted acrylamide. On the other hand, the cyanoacrylamides RC-2 and RC-3 were an order of magnitude more potent than NC-1 and also highly sensitive to the mutation with 68-fold and >1000-fold reduction in potency, respectively, indicating rapid covalent binding to BTK on the time scale of degradation. All the PROTACs tested except RC-3 exhibited sub-100 nM binding to BTK even after mutation of C481. This may explain why only degradation by RC-3 was significantly impaired by the mutation.
Figure 6.

All PROTACs are potent BTK inhibitors invitro, and the cyanoacrylamides show slow dissociation kinetics and sensitivity to the C481S mutation. (A) In vitro kinase activity assay using wild-type BTK (0.6 nM BTK, 5 μM ATP). (B) Summary of IC50 values for the PROTACs against wild-type BTK and C481S BTK. (C) Time course LC-MS binding assay (3 μM compound + 2 μM BTK at room temperature). (D) Ibrutinib competition assay validates reversible binding by cyanoacrylamides; 40 μM ibrutinib was added to the preformed complex and incubated at 37 °C, and the different species were quantified by LC-MS.
We also used LC-MS to directly observe the formation of covalent complexes with recombinant BTK and measure their dissociation kinetics (Figure 6C, D, Supporting Figure 5A). LC-MS measurements with 2 μM BTK and 3 μM compound indicated covalent labeling by all compounds except NC-1, with RC-3 forming the complex extremely fast, followed by RC-2 and IR-2, in agreement with the data from the kinase activity assay. We should note that the preincubation of the compounds with 5 mM GSH did not significantly affect protein binding of reversible or irreversible covalent binders (Supporting Figure 5B).
To test whether the formation of the covalent adducts is reversible and estimate the time scale of the exchange, we added 40 μM ibrutinib to the samples after formation of the adducts and incubated at 37 °C. For the acrylamides IR-1 and IR-2, no ibrutinib adduct was observed even after 28 h of incubation. However, only 80–85% of protein appeared to be labeled by the PROTACs (Supporting Figure 5A). This may indicate that in fact IR-1/2 have stably labeled 100% of the protein, thereby preventing ibrutinib binding, but dissociation during the separation or ionization process on the LC-MS may have generated the observed free protein peak.
In contrast, for the cyanoacrylamides RC-2 and RC-3, the addition of ibrutinib led to the gradual displacement of the PROTAC by ibrutinib, confirming the reversibility of the cyanoacrylamide covalent binding. The exchange of the cyanoacrylamide was slow, on the order of 10–20 h. The noncovalent PROTAC NC-1 forms no covalent adduct and is rapidly exchanged by ibrutinib (100% ibrutinib labeling by 4 μM in 1 h at room temperature; Supporting Figure 5A), indicating very rapid binding and dissociation kinetics.
To assess their proteomic selectivity, we incubated the PROTACs at 50 or 100 nM for 24 h with Ramos cells and followed the change in protein abundance via quantitative label-free proteomics (Figure 7, Supporting Figure 6). In agreement with the Western blot analysis, BTK was efficiently degraded by all the PROTACs we tested. NC-1, IR-1, IR-2, and RC-2 also degraded the known ibrutinib off-targets CSK, LYN, and BLK,33 while several other off-targets, such as LCK and PLK1, were not significantly affected. NC-1 showed the highest degradation potency against BTK, in agreement with Western blot analysis. On the other hand, the only significant off-target of RC-3 was BLK (a covalent off-target of ibrutinib) with no activity against the noncovalent off-targets CSK and LYN, representing enhanced selectivity and in agreement with the reduced noncovalent affinity of RC-3 to BTK. No other significant off-targets were detected consistently (Supporting Data Set 1).
Figure 7.
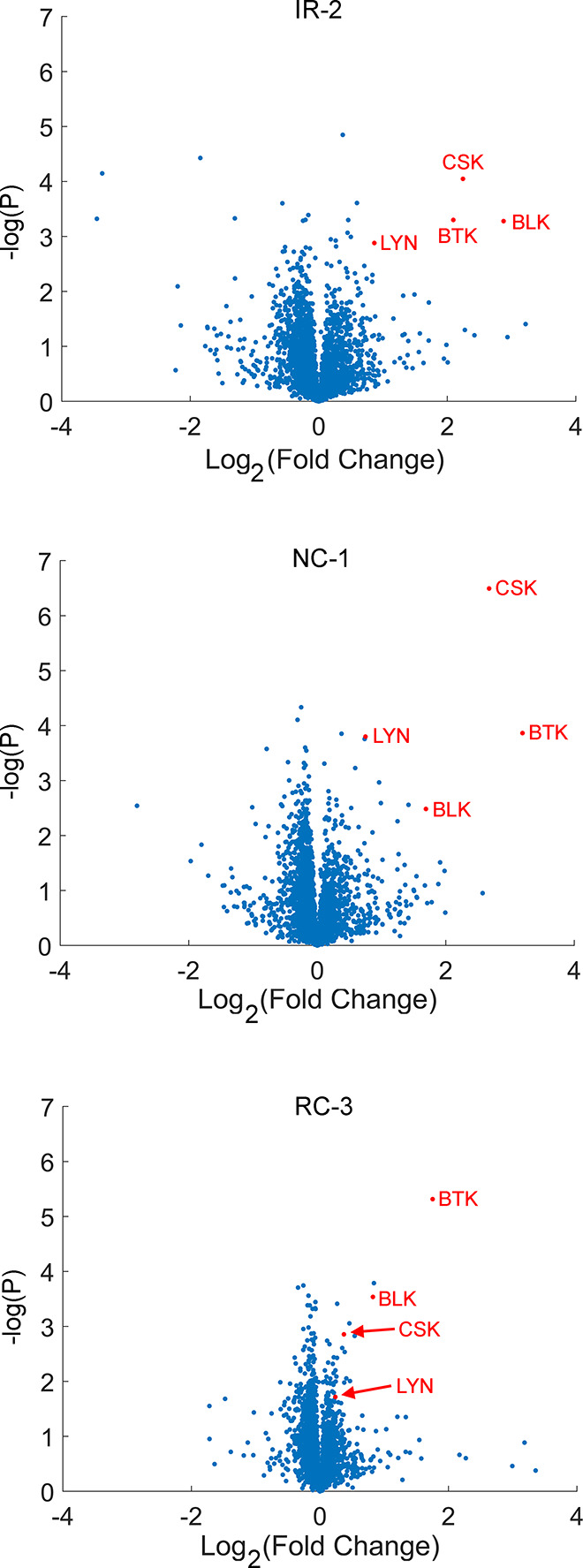
Proteomic analysis reveals high selectivity for both covalent and noncovalent BTK PROTACs. Ramos cells were incubated with each PROTAC (50 nM) or DMSO in quadruplicates for 24 h and were then subjected to label-free quantitative proteomics analysis. Each graph plots the Log2 fold-change of proteins in the treated samples compared to the DMSO controls (x-axis) vs the −log(p-value) of that comparison in a Student’s t test (y-axis).
Lastly, we assessed the ability of the PROTACs to abrogate the activation of primary mouse B cells in response to B cell receptor stimulation. For this purpose, primary B cells were treated with anti-IgM for 18 h,36,37 followed by staining for CD86, a B cell activation surface marker (Figure 8; Supporting Figure 7). The inhibition of B cell activation correlated well with the BTK degradation activity, with NC-1 and IR-2 showing the strongest effect, followed by RC-3 and RC-2. NC-1 and IR-2 displayed superior inhibition compared to ibrutinib, underscoring the benefit of targeted degradation compared to inhibition alone. The cyanoacrylamides RC-3 and RC-2 required higher concentrations to reach maximal activity but also displayed superior activity to ibrutinib at 1 μM.
Figure 8.
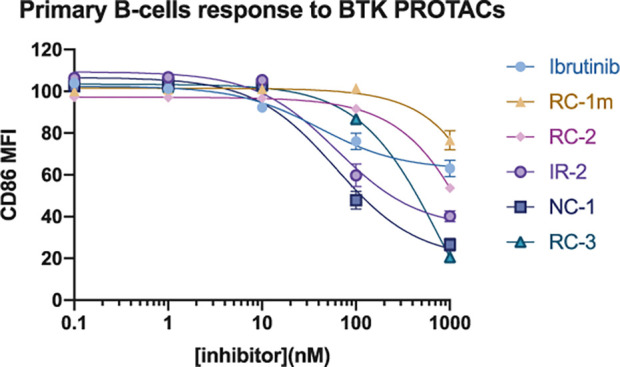
PROTACs inhibit B cell receptor signaling more potently than ibrutinib. Dose−response curves for B cell response after anti-IgM-induced activation and treatment with BTK PROTACs or ibrutinib for 24 h. The y-axis shows normalized CD86 mean fluorescence intensity, where 100% activation is cells stimulated with anti-IgM and 0% activation is unstimulated cells.
Discussion
The motivation for developing reversible covalent PROTACs lies in the combination of the advantages encompassed by covalent binding, such as increased potency and selectivity, while maintaining the reversibility that is considered important for the catalytic nature of PROTAC efficacy. Several previous studies reported noncovalent PROTACs against BTK,28−32 and some indicated that irreversible binding might be detrimental to the activity of covalent PROTACs.23 In this work we tested whether cyanoacrylamide reversible covalent binders could serve as potent PROTACs. Our results show that both acrylamides and cyanoacrylamides can function as potent and selective PROTACs, including in patient-derived cell lines (Figure 4), with the irreversible IR-2 being among the most potent BTK PROTACs reported to date. Still, the noncovalent PROTAC NC-1 outperformed IR-2.
Since ibrutinib displays nM binding even without covalent bond formation38 (Figure 6B), noncovalent BTK PROTACs can be very potent,23,39,28 and adding irreversible covalent binding would primarily be expected to reduce potency due to the loss of catalysis. Indeed, the noncovalent NC-1 was the most potent PROTAC we tested, similarly to Tinworth et al.23 However, very potent degradation was also observed with acrylamide PROTACs such as IR-2. We observed that IR-2 forms covalent bonds slowly relative to the rate of degradation, most likely due to the lower reactivity of substituted acrylamides, and therefore much of its activity may have been derived from reversible binding. Tinworth et al.23 tested irreversible covalent PROTACs based on CRBN and IAP binders, which were inactive and were also substituted acrylamides. These PROTACs harbored a piperazine moiety in the linker, attached one carbon away from the acrylamide group, which may affect reactivity and PROTAC binding. However, in vitro kinase assays using wild-type and mutant BTK had similar results to those reported here, with their acrylamide PROTAC exhibiting essentially the same IC50 toward BTK as the noncovalent counterpart. Therefore, the covalent PROTACs tested by Tinworth et al. most likely also have formed covalent bonds inefficiently, and their inactivity may have resulted from issues such as permeability, stability, or an unfavorable geometry. Conversely, Xue et al.22 recently developed unsubstituted acrylamide BTK PROTACs that covalently engaged BTK and degraded it in the cell, albeit not to 100%. Along with our study this indicates that measurement of the relative rates of covalent bond formation and degradation is needed to estimate how covalent binding affects PROTAC activity.
In parallel to this publication, Guo et al.34 have also reported cyanoacrylamide-based BTK degraders, using a different linker design. For that series of PROTACs the cyanoacrylamides were much more potent than the equivalent noncovalent and acrylamide PROTACs, which they attribute to significantly higher cell penetration of the cyanoacrylamides. Their study thus supports the use of reversible covalent PROTACs but makes it difficult to draw conclusions regarding the role of the covalent bond in the degradation. Here, RC-3 and NC-1 penetrated the cells to a similar degree, and both bind BTK reversibly, with RC-3 showing much better IC50 (Figure 6A). However, NC-1 is still a more efficient BTK degrader. We suggest two hypotheses for this discrepancy: First, the noncovalent NC-1 has a much less rigid linker than IR-2 and RC-3, with free rotation around the bond proximal to the amide linkage. This flexibility may aid the PROTAC in adopting the optimal configuration for the ternary complex formation and for ubiquitination or increase the stability of the interaction of the BTK-PROTAC complex with the E3 ligase,5,7 which is likely more relevant to degradation efficiency and may also explain the ability of NC-1 to compete with thalidomide (Figure 3A) compared to the other PROTACs. Second, the noncovalent NC-1 has a rapid binding and dissociation equilibrium: in the presence of preincubated NC-1, ibrutinib labels BTK fully within 1 h (Supporting Figure 4). Therefore, NC-1 can bind BTK in the cell, promote the formation of the ternary complex to induce ubiquitination, and quickly dissociate to bind more BTK molecules, even before the ubiquitinated BTK undergoes proteasomal degradation. The cyanoacrylamides tested here dissociate in time scales of 10–20 h, similar to the residence times observed for other cyanoacrylamide inhibitors.32 Therefore, they can only be recycled after the bound BTK molecule has been degraded, resulting in less efficient catalysis.
While RC-3 was not as potent as NC-1 in BTK degradation, it did have a significant advantage in selectivity. The addition of the cyanoacrylamide with the geminal dimethyl group greatly diminished the reversible binding affinity (which was observed for other cyanoacrylamide inhibitors of BTK27), while maintaining potent covalent binding. This significantly reduced the activity against the noncovalent off-targets LYN and CSK.
We conclude that reversible covalent PROTACs hold promise for selective degradation of challenging targets for which no high-affinity reversible ligand is available, and these are the targets where the benefits of covalent PROTACs are likely to be most evident.
Methods
General Outline of Reversible Covalent PROTAC Synthesis
To synthesize reversible covalent PROTACs, we prepared PEG-monotosylates of different lengths and coupled them to 4-OH-thalidomide to generate thalidomide-PEG-OH constructs (Supporting Information). These were oxidized to aldehydes, followed by an aldol condensation with the BTK inhibitor cyanoacetate to generate the cyanoacrylates. During the condensation, the ether linkage nearest the cyanoacrylate in RC-1 and RC0a-j was frequently cleaved and higher molecular adducts were formed, as observed by LC/MS measurements. In the synthesis of RC-2 and RC-3 (where the last ether linkage was replaced with a CH2 group or C(CH3)2), the condensation was considerably slower with reduced unwanted side reactions and higher yield (see Supporting Information for synthesis procedures). 1H and 13C NMR spectra were recorded on a 11.7 T Bruker AVANCE III HD spectrometer. Chemical shifts are reported in ppm on the δ scale downfield from TMS and are calibrated according to the deuterated solvents (see Supporting Information).
Western Blotting
Ramos (ATCC, CRL-1596), Mino (ATCC, CRL-3000), or K562 (NCI-60) cell lines were counted and diluted to 106 cell/mL, using 1 mL per well in a 24-well plate, and U2OS (ATCC HTB-96) cells overexpressing human BTK WT or C481S mutant were grown using 2 mL in a six-well plate (see Supporting Information). Cells were incubated with 1% DMSO or compound in indicated concentrations for 24 h unless indicated differently, or cells were left untreated. Lysates were prepared as previously described,29 and samples were measured for total protein quantification by bicinchoninic acid (BCA) assay (#23225 ThermoFisher Scientific) supplemented with 4× loading buffer including 20 mM DTT, heated to 70 °C for 10 min, loaded into 4% SDS-PAGE gel, run for 45 min at 140 mV, then transferred into a nitrocellulose membrane (Biorad) using a Trans-Blot Turbo transfer system (Biorad). The membrane was stained with Ponceau (Sigma) to validate transfer for 10 min in gentle agitation then destained for 1 h with MQ water. The membrane was blocked with Licor blocking buffer (LIC927-70001) for 1 h, washed three times for 5 min with TBS-T, incubated with primary antibody BTK (D3H5) rabbit mAb (CST; 8547 S) overnight (16 h) at 4 °C, washed three times for 5 min with TBS-T, and incubated with primary antibody against β-actin (CST; 3700) for 1 h at 25 °C. Membrane was washed three times for 5 min with TBS-T, incubated with fluorescent secondary antibodies anti-mouse IgG Alexa Fluor 647 (CST: 4410S) and anti-rabbit-IgG IRDye 800(LICOR: 926-32211) for 1 h, then washed three times for 5 min with TBS-T, dried, and immediately imaged and analyzed using Licor Odyssey CLx. Prism (GraphPad) software was used to calculate degradation levels. We used second-order polynomial fits to estimate DC50 and Dmax values.
In Vitro Activity Assays for BTK (Carried Out by Nanosyn, Santa Clara, CA, USA)
Test compounds were diluted in DMSO to a final concentration that ranged from 2 μM to 11.3 pM, while the final concentration of DMSO in all assays was kept at 1%. The compounds were incubated with BTK for 2 h in a 2× buffer containing the following: 1.2 nM BTK, 100 mM HEPES pH = 7.5, 10 mM MgCl2, 2 mM DTT, 0.1% BSA, 0.01% Triton X-100, 20 μM sodium orthovanadate, and 20 μM beta-glycerophosphate. Reaction was initiated by 2-fold dilution into a solution containing 5 μM ATP (50 μM for C481S) and substrate. The reference compound staurosporine was tested in a similar manner.
In Vitro BTK Binding Assays
Binding experiments were performed in Tris 20 mM pH = 8, 50 mM NaCl, and 1 mM DTT. BTK kinase domain was diluted to 2 μM in buffer, and 3 μM PROTAC was added by adding 1/100th volume from a 300 μM solution. The PROTACs were incubated with BTK at room temperature for various times. For testing by LCMS, 24 μL of the solution was mixed with 6 μL of 2.4% formic acid, and 10 μL was injected to LCMS.
For the binding reversibility experiments, the PROTACs were incubated with BTK for 2 h at room temperature, followed by addition of 40 μM ibrutinib (by addition of 1/100th volume of a 4 mM solution in DMSO). The samples were incubated with ibrutinib at 37 °C for various times and tested by LCMS as described before. For the noncovalent PROTAC NC-1, ibrutinib was added to the complex at a concentration of 4 μM and incubated for 1 h at room temperature.
For binding experiments in the presence of glutathione, freshly dissolved reduced glutathione was incubated at 6.14 mM with 4 μM compounds for 30 min at room temperature in Tris 20 mM pH = 8 and 50 mM NaCl. At this point BTK was added to a concentration of 2 μM (diluting the GSH to 5 mM and the compounds to 3.25 μM), and LC-MS was used as described previously to follow the covalent labeling of BTK.
For data analysis, the raw spectra were deconvoluted using a 27 000:37 000 Da window and 1 Da resolution. The signal from masses 27 000:30 000 and 34 000:37 000 (which contained no peaks) was averaged and subtracted from the whole signal. The peaks of each species were integrated using a 100 Da window in every direction (reducing the window down to 10 Da did not change the results significantly).
Acknowledgments
We thank Dr. Haim Barr and Dr. Christian Dubiella for critical reading of the manuscript. S.A. is the incumbent of the Prof. David Casson Research Fellowship; N.L. is the incumbent of the Alan and Laraine Fischer Career Development Chair. N.L. would like to acknowledge funding from the Israel Science Foundation (grant no. 2462/19), The Rising Tide Foundation, The Israel Cancer Research Fund, the Israeli Ministry of Science Technology (grant no. 3-14763), and the Moross Integrated Cancer Center. N.L. is also supported by the Helen and Martin Kimmel Center for Molecular Design, Joel and Mady Dukler Fund for Cancer Research, the Estate of Emile Mimran and Virgin JustGiving, and the George Schwartzman Fund. R.G. was supported by the state of Israel, ministry of Aliyah, Center for Integration in Science. Y.H. is supported by grants from the Israeli Science Foundation (1707/19), Israeli Cancer Association, and Sackler Faculty of Medicine, Tel-Aviv University. We thank Dr. Ville Paavilainen for the BTK plasmid. The proteomics work was supported by the De Botton Protein Profiling Institute of the Nancy and Stephen Grand Israel National Center for Personalized Medicine, Weizmann Institute of Science. We thank Dr. Sergey Malitsky from the Life Sciences Core Facilities at Weizmann Institute of Science for use of the LC-MS/MS instrument for metabolomics measurements.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.9b13907.
Additional information including detailed experimental methods, description of the reversible covalent BTK PROTAC library and its degradation results in two cell lines, BTK degradation by PROTACs in Ramos cells, data on cellular penetration of PROTACs, time dependency of BTK degradation, validation of CRBN-mediated degradation, kinetic studies of BTK labeling using LC/MS, GSH effect on labeling, proteomics selectivity analysis for the PROTACs at additional concentrations, B cell receptor signaling inhibition data, detailed synthetic protocols for preparation of compounds with high-resolution mass spectrometry and NMR analysis PDF)
Proteomics data set file: full lists of proteins identified in the proteomics, with quantification of the changes in abundance and p-values derived from t tests (XLSX)
Author Contributions
¶ R. Gabizon and A. Shraga contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Mullard A.First Targeted Protein Degrader Hits the Clinic. Nat. Rev. Drug Discovery 2019, 10.1038/d41573-019-00043-6. [DOI] [PubMed] [Google Scholar]
- Toure M.; Crews C. M. Small-Molecule PROTACS: New Approaches to Protein Degradation. Angew. Chem., Int. Ed. 2016, 55 (6), 1966–1973. 10.1002/anie.201507978. [DOI] [PubMed] [Google Scholar]
- Pettersson M.; Crews C. M. Proteolysis TArgeting Chimeras (PROTACs) — Past, Present and Future. Drug Discovery Today: Technol. 2019, 31, 15–27. 10.1016/j.ddtec.2019.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith B. E.; Wang S. L.; Jaime-Figueroa S.; Harbin A.; Wang J.; Hamman B. D.; Crews C. M. Differential PROTAC Substrate Specificity Dictated by Orientation of Recruited E3 Ligase. Nat. Commun. 2019, 10 (1), 1–13. 10.1038/s41467-018-08027-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy M. J.; Winkler S.; Hughes S. J.; Whitworth C.; Galant M.; Farnaby W.; Rumpel K.; Ciulli A. SPR-Measured Dissociation Kinetics of PROTAC Ternary Complexes Influence Target Degradation Rate. ACS Chem. Biol. 2019, 14 (3), 361–368. 10.1021/acschembio.9b00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadd M. S.; Testa A.; Lucas X.; Chan K.-H.; Chen W.; Lamont D. J.; Zengerle M.; Ciulli A. Structural Basis of PROTAC Cooperative Recognition for Selective Protein Degradation. Nat. Chem. Biol. 2017, 13 (5), 514–521. 10.1038/nchembio.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riching K. M.; Mahan S.; Corona C. R.; McDougall M.; Vasta J. D.; Robers M. B.; Urh M.; Daniels D. L. Quantitative Live-Cell Kinetic Degradation and Mechanistic Profiling of PROTAC Mode of Action. ACS Chem. Biol. 2018, 13 (9), 2758–2770. 10.1021/acschembio.8b00692. [DOI] [PubMed] [Google Scholar]
- Bondeson D. P.; Smith B. E.; Burslem G. M.; Buhimschi A. D.; Hines J.; Jaime-Figueroa S.; Wang J.; Hamman B. D.; Ishchenko A.; Crews C. M. Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell Chem. Biol. 2018, 25 (1), 78–87. 10.1016/j.chembiol.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand M.; Jiang B.; Bauer S.; Donovan K. A.; Liang Y.; Wang E. S.; Nowak R. P.; Yuan J. C.; Zhang T.; Kwiatkowski N.; Müller A. C.; Fischer E. S.; Gray N. S.; Winter G. E. Homolog-Selective Degradation as a Strategy to Probe the Function of CDK6 in AML. Cell Chem. Biol. 2019, 26 (2), 300–306. 10.1016/j.chembiol.2018.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang B.; Wang E. S.; Donovan K. A.; Liang Y.; Fischer E. S.; Zhang T.; Gray N. S. Development of Dual and Selective Degraders of Cyclin-Dependent Kinases 4 and 6. Angew. Chem., Int. Ed. 2019, 58 (19), 6321–6326. 10.1002/anie.201901336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H.-T.; Dobrovolsky D.; Paulk J.; Yang G.; Weisberg E. L.; Doctor Z. M.; Buckley D. L.; Cho J.-H.; Ko E.; Jang J.; Shi K.; Choi H. G.; Griffin J. D.; Li Y.; Treon S. P.; Fischer E. S.; Bradner J. E.; Tan L.; Gray N. S. A Chemoproteomic Approach to Query the Degradable Kinome Using a Multi-Kinase Degrader. Cell Chem. Biol. 2018, 25 (1), 88–99. 10.1016/j.chembiol.2017.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zengerle M.; Chan K.-H.; Ciulli A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 2015, 10 (8), 1770–1777. 10.1021/acschembio.5b00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You I.; Erickson E. C.; Donovan K. A.; Eleuteri N. A.; Fischer E. S.; Gray N. S.; Toker A. Discovery of an AKT Degrader with Prolonged Inhibition of Downstream Signaling. Cell Chem. Biol. 2020, 27, 1–8. 10.1016/j.chembiol.2019.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson D. P.; Mares A.; Smith I. E. D.; Ko E.; Campos S.; Miah A. H.; Mulholland K. E.; Routly N.; Buckley D. L.; Gustafson J. L.; Zinn N.; Grandi P.; Shimamura S.; Bergamini G.; Faelth-Savitski M.; Bantscheff M.; Cox C.; Gordon D. A.; Willard R. R.; Flanagan J. J.; Casillas L. N.; Votta B. J.; den Besten W.; Famm K.; Kruidenier L.; Carter P. S.; Harling J. D.; Churcher I.; Crews C. M. Catalytic in Vivo Protein Knockdown by Small-Molecule PROTACs. Nat. Chem. Biol. 2015, 11 (8), 611–617. 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehler A. N. A Complex Task? Direct Modulation of Transcription Factors with Small Molecules. Curr. Opin. Chem. Biol. 2010, 14 (3), 331–340. 10.1016/j.cbpa.2010.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan P.; Donev R.; Hewamana S. Targeting Transcription Factors for Therapeutic Benefit. Mol. BioSyst. 2008, 4 (9), 909–919. 10.1039/b801920g. [DOI] [PubMed] [Google Scholar]
- Scott D. E.; Bayly A. R.; Abell C.; Skidmore J. Small Molecules, Big Targets: Drug Discovery Faces the Protein-Protein Interaction Challenge. Nat. Rev. Drug Discovery 2016, 15 (8), 533–550. 10.1038/nrd.2016.29. [DOI] [PubMed] [Google Scholar]
- Arkin M. R.; Tang Y.; Wells J. A. Small-Molecule Inhibitors of Protein-Protein Interactions: Progressing toward the Reality. Chem. Biol. 2014, 21 (9), 1102–1114. 10.1016/j.chembiol.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray J. L.; von Delft F.; Brennan P.. Targeting the Small GTPase Superfamily through Their Regulatory Proteins. Angew. Chem., Int. Ed. 2020. 59, 6342. 10.1002/anie.201900585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebraud H.; Wright D. J.; Johnson C. N.; Heightman T. D. Protein Degradation by In-Cell Self-Assembly of Proteolysis Targeting Chimeras. ACS Cent. Sci. 2016, 2 (12), 927–934. 10.1021/acscentsci.6b00280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovell H.; Testa A.; Maniaci C.; Zhou H.; Prescott A. R.; Macartney T.; Ciulli A.; Alessi D. R. Rapid and Reversible Knockdown of Endogenously Tagged Endosomal Proteins via an Optimized HaloPROTAC Degrader. ACS Chem. Biol. 2019, 14 (5), 882–892. 10.1021/acschembio.8b01016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue G.; Chen J.; Liu L.; Zhou D.; Zuo Y.; Fu T.; Pan Z. Protein Degradation through Covalent Inhibitor-Based PROTACs. Chem. Commun. 2020, 56 (10), 1521–1524. 10.1039/C9CC08238G. [DOI] [PubMed] [Google Scholar]
- Tinworth C. P.; Lithgow H.; Dittus L.; Bassi Z. I.; Hughes S. E.; Muelbaier M.; Dai H.; Smith I. E. D.; Kerr W. J.; Burley G. A.; Bantscheff M.; Harling J. D. PROTAC-Mediated Degradation of Bruton’s Tyrosine Kinase Is Inhibited by Covalent Binding. ACS Chem. Biol. 2019, 14 (3), 342–347. 10.1021/acschembio.8b01094. [DOI] [PubMed] [Google Scholar]
- Burslem G. M.; Smith B. E.; Lai A. C.; Jaime-Figueroa S.; McQuaid D. C.; Bondeson D. P.; Toure M.; Dong H.; Qian Y.; Wang J.; Crew A. P.; Hines J.; Crews C. M. The Advantages of Targeted Protein Degradation Over Inhibition: An RTK Case Study. Cell Chem. Biol. 2018, 25 (1), 67–77. 10.1016/j.chembiol.2017.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafimova I. M.; Pufall M. A.; Krishnan S.; Duda K.; Cohen M. S.; Maglathlin R. L.; McFarland J. M.; Miller R. M.; Frödin M.; Taunton J. Reversible Targeting of Noncatalytic Cysteines with Chemically Tuned Electrophiles. Nat. Chem. Biol. 2012, 8 (5), 471–476. 10.1038/nchembio.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindo N.; Fuchida H.; Sato M.; Watari K.; Shibata T.; Kuwata K.; Miura C.; Okamoto K.; Hatsuyama Y.; Tokunaga K.; Sakamoto S.; Morimoto S.; Abe Y.; Shiroishi M.; Caaveiro J. M. M.; Ueda T.; Tamura T.; Matsunaga N.; Nakao T.; Koyanagi S.; Ohdo S.; Yamaguchi Y.; Hamachi I.; Ono M.; Ojida A. Selective and Reversible Modification of Kinase Cysteines with Chlorofluoroacetamides. Nat. Chem. Biol. 2019, 15 (3), 250–258. 10.1038/s41589-018-0204-3. [DOI] [PubMed] [Google Scholar]
- Bradshaw J. M.; McFarland J. M.; Paavilainen V. O.; Bisconte A.; Tam D.; Phan V. T.; Romanov S.; Finkle D.; Shu J.; Patel V.; Ton T.; Li X.; Loughhead D. G.; Nunn P. A.; Karr D. E.; Gerritsen M. E.; Funk J. O.; Owens T. D.; Verner E.; Brameld K. A.; Hill R. J.; Goldstein D. M.; Taunton J. Prolonged and Tunable Residence Time Using Reversible Covalent Kinase Inhibitors. Nat. Chem. Biol. 2015, 11 (7), 525–531. 10.1038/nchembio.1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y.; Zhao X.; Ding N.; Gao H.; Wu Y.; Yang Y.; Zhao M.; Hwang J.; Song Y.; Liu W.; Rao Y. PROTAC-Induced BTK Degradation as a Novel Therapy for Mutated BTK C481S Induced Ibrutinib-Resistant B-Cell Malignancies. Cell Res. 2018, 28 (7), 779–781. 10.1038/s41422-018-0055-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorba A.; Nguyen C.; Xu Y.; Starr J.; Borzilleri K.; Smith J.; Zhu H.; Farley K. A.; Ding W.; Schiemer J.; Feng X.; Chang J. S.; Uccello D. P.; Young J. A.; Garcia-Irrizary C. N.; Czabaniuk L.; Schuff B.; Oliver R.; Montgomery J.; Hayward M. M.; Coe J.; Chen J.; Niosi M.; Luthra S.; Shah J. C.; El-Kattan A.; Qiu X.; West G. M.; Noe M. C.; Shanmugasundaram V.; Gilbert A. M.; Brown M. F.; Calabrese M. F. Delineating the Role of Cooperativity in the Design of Potent PROTACs for BTK. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (31), E7285–E7292. 10.1073/pnas.1803662115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhimschi A. D.; Armstrong H. A.; Toure M.; Jaime-Figueroa S.; Chen T. L.; Lehman A. M.; Woyach J. A.; Johnson A. J.; Byrd J. C.; Crews C. M. Targeting the C481S Ibrutinib-Resistance Mutation in Bruton’s Tyrosine Kinase Using PROTAC-Mediated Degradation. Biochemistry 2018, 57 (26), 3564–3575. 10.1021/acs.biochem.8b00391. [DOI] [PubMed] [Google Scholar]
- Krajcovicova S.; Jorda R.; Hendrychova D.; Krystof V.; Soural M. Solid-Phase Synthesis for Thalidomide-Based Proteolysis-Targeting Chimeras (PROTAC). Chem. Commun. (Cambridge, U. K.) 2019, 55 (7), 929–932. 10.1039/C8CC08716D. [DOI] [PubMed] [Google Scholar]
- Dobrovolsky D.; Wang E. S.; Morrow S.; Leahy C.; Faust T.; Nowak R. P.; Donovan K. A.; Yang G.; Li Z.; Fischer E. S.; Treon S. P.; Weinstock D. M.; Gray N. S. Bruton Tyrosine Kinase Degradation as a Therapeutic Strategy for Cancer. Blood 2019, 133 (9), 952–961. 10.1182/blood-2018-07-862953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honigberg L. A.; Smith A. M.; Sirisawad M.; Verner E.; Loury D.; Chang B.; Li S.; Pan Z.; Thamm D. H.; Miller R. A.; Buggy J. J. The Bruton Tyrosine Kinase Inhibitor PCI-32765 Blocks B-Cell Activation and Is Efficacious in Models of Autoimmune Disease and B-Cell Malignancy. Proc. Natl. Acad. Sci. U. S. A. 2010, 107 (29), 13075–13080. 10.1073/pnas.1004594107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W.-H.; Qi X.; Liu Y.; Chung C.-I.; Bai F.; Lin X.; Wang L.; Chen J.; Nomie K. J.; Li F.; Wang M. L. M. C.; Shu X.; Onuchic J. N.; Woyach J. A.; Wang M. L. M. C.; Wang J.. Enhancing Intracellular Concentration and Target Engagement of PROTACs with Reversible Covalent Chemistry. bioRxiv 2019, 10.1101/2019.12.30.873588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teicher B. A.; Ara G.; Herbst R.; Palombella V. J.; Adams J. The Proteasome Inhibitor PS-341 in Cancer Therapy. Clin. Cancer Res. 1999, 5 (9), 2638–2645. [PubMed] [Google Scholar]
- Pal Singh S.; Dammeijer F.; Hendriks R. W. Role of Bruton’s Tyrosine Kinase in B Cells and Malignancies. Mol. Cancer 2018, 17 (1), 1–23. 10.1186/s12943-018-0779-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendriks R. W.; Yuvaraj S.; Kil L. P. Targeting Bruton’s Tyrosine Kinase in B Cell Malignancies. Nat. Rev. Cancer 2014, 14 (4), 219–232. 10.1038/nrc3702. [DOI] [PubMed] [Google Scholar]
- Pan Z.; Scheerens H.; Li S.-J.; Schultz B. E.; Sprengeler P. A.; Burrill L. C.; Mendonca R. V.; Sweeney M. D.; Scott K. C. K.; Grothaus P. G.; Jeffery D. A.; Spoerke J. M.; Honigberg L. A.; Young P. R.; Dalrymple S. A.; Palmer J. T. Discovery of Selective Irreversible Inhibitors for Bruton’s Tyrosine Kinase. ChemMedChem 2007, 2 (1), 58–61. 10.1002/cmdc.200600221. [DOI] [PubMed] [Google Scholar]
- Jaime-Figueroa S.; Buhimschi A. D.; Toure M.; Hines J.; Crews C. M. Design, Synthesis and Biological Evaluation of Proteolysis Targeting Chimeras (PROTACs) as a BTK Degraders with Improved Pharmacokinetic Properties. Bioorg. Med. Chem. Lett. 2020, 30 (3), 126877. 10.1016/j.bmcl.2019.126877. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.