

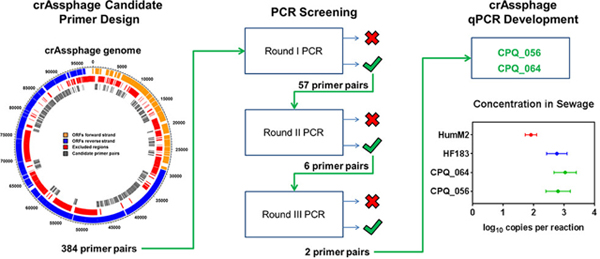
Abstract
Environmental waters are monitored for fecal pollution to protect public health and water resources. Traditionally, general fecal-indicator bacteria are used; however, they cannot distinguish human fecal waste from other animal pollution sources. Recently, a novel bacteriophage, crAssphage, was discovered by metagenomic data mining and reported to be abundant in and closely associated with human fecal waste. To confirm bioinformatic predictions, 384 primer sets were designed along the length of the crAssphage genome. Based on initial screening, two novel crAssphage qPCR assays (CPQ_056 and CPQ_064) were designed and evaluated in reference fecal samples and water matrices. The assays exhibited high specificities (98.6%) when tested against an animal fecal reference library, and crAssphage genetic markers were highly abundant in raw sewage and sewage-impacted water samples. In addition, CPQ_056 and CPQ_064 performance was compared to HF183/BacR287 and HumM2 assays in paired experiments. Findings confirm that viral crAssphage qPCR assays perform at a similar level to well-established bacterial human-associated fecal-source-identification approaches. These new viral-based assays could become important water quality management and research tools.
Graphical Abstract
Introduction
Many environmental waters are polluted with human fecal waste originating from numerous sources such as leaking sewer lines, faulty septic systems, improperly connected downspouts, and combined sewer overflows. Human fecal waste can harbor disease-causing pathogens that contribute to poor public health, reduced ecological outcomes, and economic burdens. Many public health managers rely on general fecal indicator methods (e.g., Escherichia coli and enterococci) to monitor fecal pollution levels, which do not discriminate between human and other potential animal sources of fecal pollution. General indicators provide limited information that prevents focused remediation because many areas are polluted by a combination of human, agricultural, and wildlife sources. Information on human waste (i.e., sewage) is particularly important because it may pose a greater risk to public health compared to fecal pollution from other animal sources.(1–3) To compliment general indicator measurements and better-characterize human fecal pollution, many researchers and water quality managers use fecal-source-identification technologies.
Most currently available human fecal-source-identification technologies target fecal bacteria, mainly Bacteroides species.(4) These fecal bacteria are abundant in the human gut and sewage, facilitating their detection in diluted environmental water samples. Bacterial human fecal genetic markers, such as HF183/BacR287 and HumM2, are highly human-associated and reproducible in multiple laboratory validation studies.(4–6) Although bacterial methods exist for human fecal source characterization, technologies targeting viruses are also needed.(7–9)
Enteric viruses, such as noroviruses, adenoviruses, and enteroviruses are reported to be the dominant etiological agents of waterborne and shellfish-borne disease.(10, 11) Several studies suggest enteric viruses react to environmental and waste treatment conditions in markedly different ways compared to bacterial fecal indicators.(8, 12–15) In addition, waterborne viral outbreaks have occurred when general bacterial fecal indicators are not detected or are below regulated levels.(10) Reliance solely on bacterial indicators limits the ability of water quality managers to link measures of human fecal pollution with public health risk; thus, viral human-associated technologies offer an attractive alternative to bacterial fecal-source-identification methods. Researchers have previously recognized the potential value of viral human-associated methodologies, leading to the development of technologies targeting enteroviruses,(16–20)adenoviruses,(21, 22) noroviruses,(23, 24) polyomaviruses BK and JC,(25) somatic coliphage,(26, 27) Bacteroides phages,(28–30) and pepper mild mottle viruses,(31) among others. However, a recent multiple laboratory study evaluated the performance of many of these viral methods and concluded that the technologies tested either lacked sensitivity or exhibited poor specificity, potentially limiting the suitability for widespread water quality management applications.(32) A recent comparison of human polyomavirus levels to predicted public health risk also illustrates that more-sensitive viral methodologies are needed.(33)
An ideal viral human-associated method for environmental water quality testing would target a virus that is both highly human-associated and consistently abundant in human waste sources. Recently, a novel bacteriophage, “crAssphage”, was described via metagenome cross-assembly and was predicted to be a Bacteroides phage by co-occurrence profiling.(34) The double-stranded DNA crAssphage putative genome was assembled from shotgun metagenomic libraries isolated from an individual human fecal sample.(34) Further bioinformatic testing predicted that the crAssphage genome is highly abundant and was identified in 73% of human fecal metagenomes surveyed.(34) A subsequent metagenome survey detected crAssphage in sewage from the United States and Europe, while crAssphage was absent in other environments, such as nonhuman fecal samples and water environments.(35) In addition, the crAssphage genome is estimated to be up to 10 times more abundant in sewage than other known human-associated viruses, including noroviruses and adenoviruses.(35)
Near-ubiquity across human fecal metagenomes and the high abundance compared to other sewage-derived viruses, combined with potential human specificity, motivates the development of crAssphage as a fecal-source-identification technology.(35) However, several unknown issues remain that must be addressed for the successful development of a crAssphage fecal-source-identification tool. For instance, the crAssphage genome likely represents a viral quasi-species consensus sequence compiled from a collection of DNA regions with unknown sequence variability. Furthermore, most information about the crAssphage genome has been generated from computer predictions with minimal laboratory testing to verify findings. Extensive laboratory testing of fecal samples gathered from a wide variety of animal species and sewage collected across a broad geographic range is necessary to evaluate the suitability of the crAssphage genome for human fecal-source-identification applications.
The goals of the present study are to survey the crAssphage genome for human-associated genetic regions, develop qPCR methods as potential future environmental water quality monitoring tools, and compare their performance to top performing bacterial human-associated technologies. We employed a “biased genome shotgun strategy”, in which select genetic regions of the crAssphage genome were screened using end-point PCR for highly specific and abundant human-associated fecal pollution genetic regions. These genetic regions were subsequently utilized to develop two novel qPCR fecal-source-identification assays. Findings suggest that high-throughput laboratory screening of novel virus genomes discovered through metagenomic DNA sequence mining is a successful strategy to develop host-associated qPCR methods that may be important for future research and water quality management activities.
Materials and Methods
Sample Collection
Individual fecal samples (n = 222) were collected from various locations across the continental United States, as previously described.(6) Animal fecal samples represent ten species including Anser spp. (Canada goose; n = 18), Canis familiariz (dog, n = 41), Bos taurus (cow, n = 61), Larusspp. (gull, n = 25), Equus caballus (horse, n = 20), Cervus canadensis (elk, n = 20), Gallus gallus(chicken, n = 11), Sus scrofa. (pig, n = 9), Castor canadensis (beaver, n = 8), and Odocoileus virginianus (deer, n = 9) (see Table S1 for sample details). Each fecal sample was collected from a different individual as previously described(6) and stored at −80 °C until time of DNA extraction (<18 months). Primary influent sewage samples were collected at nine geographically distributed wastewater treatment plants within the United States (Table S2) as previously described.(6)Briefly, 1 L of primary influent was collected and immediately packed in ice and shipped overnight to Cincinnati, OH for laboratory testing. DNA extraction of sewage samples was performed within 48 h of collection as described below. Finally, as a proof-of-concept pilot study, surface water samples were collected from the Heiserman Stream (East Fork Watershed, southwest OH) in close proximity to a treated sewage discharge area. These samples were collected in a sterile 1 L container, immediately stored on ice, and transported to the laboratory for DNA extraction and testing (<4 h).
DNA Extraction and Quantification
DNA was extracted from 10 mL of primary influent sewage with the QIAamp Blood Maxi Kit according to the manufacturer’s instructions, except that Buffer AVL was substituted for Buffer AL (Qiagen, Valencia, CA). DNA was extracted from individual animal fecal samples using the DNA-EZ Kit (GeneRite, North Brunswick, NJ), substituting Buffer AE (Qiagen, Valencia, CA) for the elution buffer in a modified protocol of the manufacturer’s instructions. Briefly, fecal slurries were made by adding molecular grade PBS and fecal matter to the bead mill tubes and were homogenized in a bead beater at 6 m/s for 30 s. After a prolonged centrifugation, 760 μL of binding buffer was added to recovered supernatant, and the manufacturer’s instructions were followed eluting with molecular grade water warmed to 60 °C. For environmental water samples, a 200 mL sample was concentrated to a final volume of approximately 150 μL using an automated Concentrating Pipette with a single-use ultrafiltration hollow fiber poly(ether sulfone) tip following the manufacturer’s instructions (InnovaPREP, Drexel, MO). DNA extraction of concentrate was performed with the DNA-EZ Kit (GeneRite, North Brunswick, NJ) as described above for fecal-sample processing. Water sample DNA extracts were stored at 4 °C in GeneMate Slick low-adhesion microcentrifuge tubes (ISC BioExperess, Kaysville, UT) until the time of amplification (<24 h). Fecal and primary influent sewage DNA extract concentrations were determined with a NanoDrop ND-1000 UV spectrophotometer (NanoDrop Technologies, Wilmington, DE), diluted to 0.5 ng/μL to normalize sample test quantities for performance testing, and stored in low-adhesion microcentrifuge tubes at −20 °C (<6 months). For each batch of DNA extractions, three method extraction blanks with purified water substituted for fecal, sewage, or environmental water were performed to monitor for potential contamination.
Selection of Candidate Genetic Regions for PCR-Based Assay Development
To identify candidate genetic regions for the development of human-associated fecal-source-identification methods, select portions of the putative ∼97 kbp crAssphage genome (accession code: JQ995537)(34) were identified for end-point PCR laboratory testing. Due to the reported potential for rapid DNA mutation rates in the human gut virome, such as those described for Microviridae,(36) efforts were focused on predicted coding regions to select for sequences with some evidence of genetic conservation. Metaviromic islands were also excluded due to increased genetic diversity leading to under-recruitment in metaviromes, suggesting the potential for low abundance in environmental samples.(34, 37) In addition, regions bordering modular junctions were eliminated. Finally, because orf00045 has homology with bat guano virome sequences,(35) it was not considered for human-associated crAssphage assay development.
Candidate Primer Set Design
A total of 384 end-point PCR primer pairs were designed to amplify selected crAssphage genomic regions. Primer pairs were designed and tested in silico using Primer-BLAST(38) with default parameters, except that PCR product length was constrained to 90–180 bps, and primer pair specificity was evaluated using the nr database (May, 2015). Only primer pairs that generated BLAST hits (E-value of <30 000) to crAssphage or clone DNA sequences from human gut metagenome projects were selected as candidate primer pairs. Genetic regions where no primer sets met design criteria were eliminated from further consideration.
End-Point PCR Amplifications
Each 25 μL end-point PCR amplification consisted of TaKaRa Ex Taq Hot Start PCR reagents (Clontech Laboratories), 100 nM each of the forward and reverse primers, 0.8 μg of bovine serum albumin (BSA; Sigma-Aldrich, St. Louis, MO), 2 μL of template DNA, and molecular-grade water. End-point PCR tests were performed in duplicate or in triplicate on a Tetrad 2 Thermocycler (Bio-Rad Laboratories) under the following conditions: 94 °C for 5 min followed by 40 cycles of 94 °C for 40 s, 57 °C for 1 min, and 72 °C for 30 s, followed by a final extension at 72 °C for 10 min. To monitor for potential sources of extraneous DNA during end-point PCR amplification, a minimum of two no-template controls (reactions contained additional purified water instead of template DNA) were performed with each instrument run. PCR products were verified on a 2% agarose gel with 1% lithium borate and 1X GelStar (Lonza, Rockland, ME) and visualized on a Gel Logic 100 Imaging System (Eastman Kodak Company, Rochester, NY).
Candidate End-Point PCR Primer Set Evaluation
To determine which candidate crAssphage genetic regions have human fecal-source-identification potential, all 384 primer sets were tested with end-point PCR in a three-round process. In the first round, candidate primer sets were challenged against two fecal DNA composites: sewage and nonhuman. A sewage DNA composite was created by combining primary influent sewage DNA from three geographic locales (1 ng of total DNA per reaction and 0.33 ng of DNA per reaction from each sample) and was used to identify the presence or absence of candidate genetic regions in a known human fecal pollution source. A nonhuman DNA composite was created from cow (n = 9), dog (n = 9), goose (n = 9), and pig (n = 9) fecal DNA (4 ng of total DNA per reaction and 1 ng of DNA per reaction from each animal group) and was used to determine the presence of candidate genetic regions in nontarget fecal pollution sources. All primers were tested in duplicate against DNA composites. Candidate primer sets proceeded to a second round of testing if the following criteria were met: (1) a PCR product of expected size was present when primary influent sewage DNA composite was used as template, (2) PCR product of expected size was absent when the nonhuman DNA composite was used as a template, (3) low amplification efficiency was not observed in reactions with sewage DNA composite as the template as evaluated by manual inspection, and (4) absence of any spurious PCR products noticeably different in size from the expected PCR product, including primer dimerization.
In round two, remaining candidate primer sets were challenged against diluted preparations of the primary influent sewage DNA composite (0.1, 1 × 10–2, and 1 × 10–3 ng per reaction) and a higher concentration of individual animal group composites for cow (n = 9), dog (n = 9), goose (n = 9), and pig (n = 9) using 5 ng of total DNA per reaction. Candidate primer sets proceeded to a third round of testing under the following conditions: (1) amplification of expected size in triplicate reactions when 1 × 10–2 ng of total DNA per reaction from primary influent sewage composite was used as the template, (2) absence of expected PCR product size in all reactions when a nonhuman DNA composite was used as the template, and (3) the absence of spurious PCR byproducts, including primer dimers.
Round three represented the most-rigorous performance-screening step for candidate end-point PCR primer sets. Testing began with specificity determination from an expanded fecal reference collection (n = 70 individual samples), followed by geographic distribution characterization in primary influent sewage samples collected from nine different locations and ending with limit of detection (LOD) assessment. Reference fecal samples for specificity screening included cow (n = 9), goose (n = 8), dog (n = 9), pig (n = 9), horse (n = 9), elk (n = 9), deer (n = 9), and beaver (n = 8). All candidate primer sets passing round two were challenged with individual DNA preparations at 1 ng of total DNA per reaction. Specificity was defined as the proportion of nonhuman samples testing negative for a crAssphage genetic region. Only candidate primer sets with an observed specificity of 100% proceeded to geographic distribution testing. Sewage distribution characterization entailed testing of 1 ng of total DNA per reaction isolated from nine primary influent sewage samples collected from different locations across the continental United States (Table S2). Candidate primer sets with ≥95% detection frequency were eligible for LOD assessment. LOD95 was measured based on repeated testing (40 replicates per primer set) of primary influent sewage composite serial dilutions (10, 1, 0.1, 1 × 10–2, and 1 × 10–3 ng of total DNA per reaction) consisting of equal DNA mass from samples collected from all nine geographic locales. LOD95 was defined as the lowest dilution concentration at which a minimum of 95% (38 of 40) of reactions yielded an amplification product of the expected size.
DNA Sequence Verification of Top-Performing End-Point PCR Primer Sets
To verify that candidate primer sets passing round three screening were amplifying the intended crAssphage genetic region, amplification products from the round one primary influent sewage composite and an environmental water sample with known human sewage pollution impairment (Heiserman Stream, OH) were sequenced and evaluated. PCR was performed using primer sets passing round three, using the same amplification conditions as above. PCR products were cloned into plasmid vector pCR2.1-TOPO and transformed into TOP10 chemically competent cells using the TOPO TA Cloning Kit as described by the manufacturer (Invitrogen, Thermo Fisher Scientific). Transformed E. coli colonies plated on LB plates with kanamycin and X-gal for blue and white screening were sent to GENEWIZ for sequencing (South Plainfield, NJ). Sanger sequencing was performed from transformed bacterial colonies for each primer-template combination using the M13R primer for amplification. PCR products were aligned with the previously reported crAssphage sequence (accession: JQ995537)(34) using CLC Genomics Workbench 8.5.1 (Qiagen, Valencia, CA).
CrAssphage qPCR Assay Development
Candidate primer sets passing round three end-point PCR testing were adapted to TaqMan qPCR chemistry. Primers and probes for putative human-associated crAssphage genetic regions were designed using default parameters of the Primer Express version 3.0.1 software (Thermo Fisher Scientific). Fluorogenic minor binding groove (MGB) probes were 5′ labeled with 6-carboxyfluorescein.
qPCR Amplifications
A total of five qPCR assays were used in this study: two novel crAssphage assays (this study) and two previously reported human-associated bacterial fecal-source-identification methods (HF183/BacR287 and HumM2) as well as an environmental water-sample-processing control assay (Sketa22).(5, 39, 40) Each 25 μL qPCR reaction was composed of 1× TaqMan Environmental Master Mix 2.0 (Thermo Fisher Scientific), 5 μg of BSA, 1 μM of each primer, 80 nM 6-carboxyfluorescein (FAM)-labeled probe, 80 nM VIC-labeled probe (HF183/BacR287 and HumM2 only), 2 μL of template DNA, and molecular-grade water. All qPCR tests were performed in triplicate using the QuantStudio 6 Flex Real-Time PCR System (Thermo Fisher Scientific). The thermal cycling profile for all assays was 10 min at 95 °C followed by 40 cycles of 15 s at 95 °C and 1 min at 60 °C. The threshold for qPCR assays was manually set to 0.03 (crAssphage, HF183/BacR287, and Sketa22) or 0.08 (HumM2) and quantification cycle (Cq) values were exported to Microsoft Excel. A total of six y-intercept control reactions (standard reference material at 100 copies per reaction) were performed with each instrument run to utilize a mixed calibration model approach.(41) To monitor for potential contamination, six no-template controls were performed with each instrument run. Amplification inhibition was monitored in all DNA extracts using the HF183/BacR287 and HumM2 IAC procedures as previously reported.(42)
qPCR Standard DNA Material Preparation
Standard DNA material consisted of a customized gBlock gene fragment containing target sequences for crAssphage, HF183/BacR287, and HumM2 standard curve generation and an internal amplification control (IAC) plasmid construct for HF183/BacR287 and HumM2 amplification inhibition screening (Integrated DNA Technologies, Coralville, IA).(42) Standard DNA concentrations were determined with a NanoDrop ND-1000 UV spectrophotometer (NanoDrop Technologies, Wilmington, DE). For standard curve reference material, five dilutions were prepared to contain 10 to 1 × 105 copies per 2 μL. IAC reference DNA material was prepared as previously described.(42) All reference DNA material preparations were stored in GeneMate Slick low-adhesion microcentrifuge tubes (ISC BioExpress, Kaysville, UT) at −20 °C prior to use (<3 months).
Performance Testing of crAssphage qPCR Assays
To investigate the suitability of newly developed technologies for human fecal-source-identification application, the performance of crAssphage qPCR methods was evaluated in a series of head-to-head experiments with HF183/BacR287 and HumM2 bacterial human-associated methods.(5, 39)Calibration model performance including amplification efficiency (E = 10(−1/slope) – 1), lower limit of quantification (LLOQ), and precision (percent coefficient of variation) at 10 copies per reaction were calculated from six standard curves generated from independent instrument runs. LLOQ (log10 copies per reaction) was defined as the upper bound of the 95% credible interval from repeated measures of the 10 copies per reaction standard curve dilutions. Next, the abundance of each genetic marker was measured in primary effluent sewage samples (n = 9) at a test concentration of 1 ng of total DNA per reaction (Table S2). The prevalence of putative human-associated genetic markers in nonhuman pollution sources was evaluated with a reference fecal collection consisting of 222 individual samples from 10 different animals (Table S1; test quantity of 1 ng of total DNA per reaction). Prevalence was expressed both quantitatively (log10 copies per ng of total DNA) and qualitatively (specificity = TNC/(TNC + TPI), where TNC represents the total number of negative individual samples that tested negative correctly, and TPI is the total number of individual samples that tested positive incorrectly). Finally, as a proof-of-concept pilot demonstration, genetic marker concentrations were estimated from two environmental water samples known to be impacted by human sewage pollution (Heiserman Stream, OH). Average log10 copies per ng of total DNA (sewage) and log10 copies per reaction (water) with 95% credible intervals were determined (mean Cq for each sample group and assay combination transformed using respective mixed calibration model followed by a nested analysis of variance to estimate standard deviation values) and compared to identify similarities and differences between qPCR genetic marker concentrations.
Data Analysis
Mixed-model calibration models, unknown DNA concentration estimates, and credible intervals were determined using a Monte Carlo Markov Chain (MCMC) approach.(41) MCMC calculations were performed using the publically available software WinBUGS, version 1.4.1 (http://www.mrc-bsu.cam.ac.uk/bugs).
Results
Putative Human-Associated CrAssphage Genetic Regions and Candidate Primer Set Design
A total of 46 564 bp (48%) of the crAssphage genome were selected for end-point PCR screening to identify potential human-associated genetic regions. Select genetic regions were excluded due to (1) noncoding regions (8%), (2) metaviromic island motifs (32%), (3) modular junction regions (3%), (4) evidence of similarity with nonhuman or non-crAssphage sequences (3%), and (5) regions not amenable for PCR testing based on primer design parameters (e.g., product size and melting temperature restrictions (6%)) (Figure 1). In total, 384 end-point PCR primer sets were designed with 90% coverage (41 794 bp) of select putative human-associated genetic regions (Figure 1 and Table S3).
Figure 1.
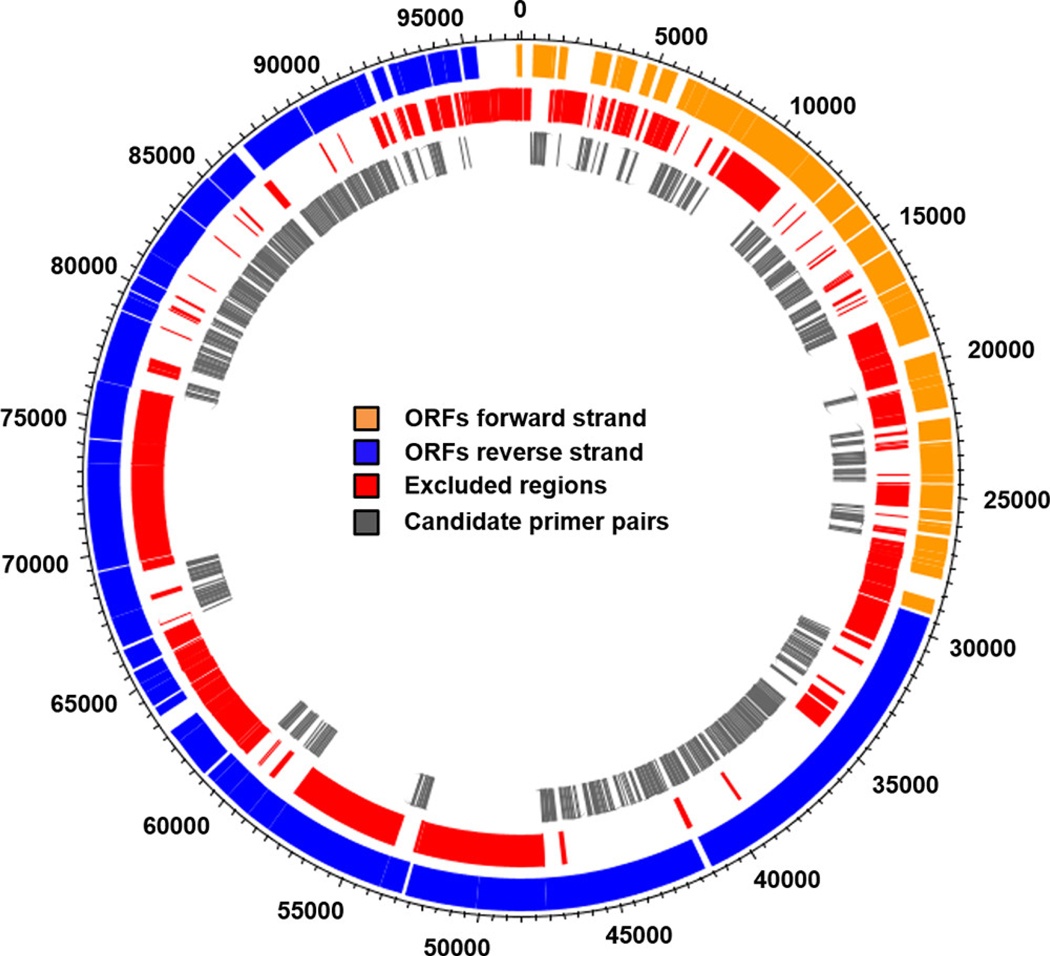
Map representation of the crAssphage genome. The outermost track represents the open reading frames (ORFs) on the forward and reverse strand of the crAssphage genome. The middle track represents the areas of the crAssphage genome that were eliminated from primer design, including noncoding regions, metaviromic islands, modular junction areas, nontarget sequence homology, and regions unsuitable for primer design. The innermost track represents the location of the 384 end-point primer pairs designed in this study and their amplification products.
Identification of Human-Associated crAssphage Genetic Regions with End-Point PCR
Candidate primer sets were subjected to three rounds of performance testing to identify human-associated crAssphage genetic regions. The first round of testing composed of testing the primers against a sewage DNA composite and nonhuman animal fecal DNA composite. Round one testing eliminated 327 (85.2%) candidate primer sets: 34.6% (n = 133) failing to yield a clearly distinct PCR product of the expected size in the sewage composite, 44.0% (n = 169) generating spurious PCR products (including primer dimerization byproducts), and 4.7% (n = 18) yielding false-positive results in nonhuman composite tests. A total of 49 (12.8%) candidate primer sets failed more than one first-round criteria. A total of 57 candidate primer sets were deemed eligible for round two testing.
In round two testing, primer sets were challenged against lower dilutions of sewage composite DNA and higher concentrations of nontarget animal fecal composite DNA. A total of 51 primer sets were eliminated due to amplification product of expected size when nonhuman samples were used as DNA template (n = 24), failure to consistently yield a PCR product of the expected size when 1 × 10–2 ng of total sewage composite DNA per reaction was used as template (n = 14), or there is evidence of spurious PCR byproducts, including primer dimerization (n = 40). A total of 27 primer sets failed more than one criteria. False positives observed with each nonhuman animal group tested were: pig (n = 15), cow (n = 15), canine (n = 6), and goose (n = 1). A total of six primer sets passed round two testing, including crAss028, crAss056, crAss064, crAss301, crAss303, and crAss375.
Round three testing included specificity determination with an expanded reference fecal collection, characterization of geographic distribution in sewage, and a limit of detection (LOD95) assessment (Table S4). Primer sets crAss056 and crAss064 exhibited the best performance with 100% specificity and 100% detection in geographic sewage samples and were subject to LOD95assessment. Both primer sets yielded an LOD95 of 1 × 10–2 ng of total DNA per reaction. Primer sets crAss064 and crAss056 were detected in 52.5% and 45% of test replicates, respectively, at a DNA template concentration of 1 × 10–3 ng per reaction.
DNA Sequencing Verification
End-point PCR products from crAss056 and crAss064 primer sets were sequenced from a primary influent sewage composite and human fecal pollution impacted environmental water sample to confirm amplification of the expected crAssphage sequences. Sequencing efforts resulted in 91 sequences (Figure S1). Alignment of crAss056 sequences indicated that 84.1% (37 of 44) of sequences exhibited 100% similarity to the corresponding reference crAssphage genome region (accession: JQ995537; 14 735 to 14 836 bp). A total of five additional variants designated B–F were observed with 1 mismatch each (99% similarity to crAssphage genetic region). Primer set crAss064 alignments yielded 74.5% (35 of 47) of sequences with 100% similarity to reported crAssphage genomic region (16 058 to 16 152 bp). Variant D was observed in 12.8% (6 of 47) of the sequences with a 1 base pair substitution. The remaining six crAss064 sequences each exhibited sequence similarities ranging from 98% (2 mismatches) to 99% (1 mismatch) designated variants B, C, and E–H.
Performance of CrAssphage qPCR Assays
Candidate primer sets crAss056 and crAss064 were adapted as CPQ_056 and CPQ_064 to TaqMan qPCR chemistry, respectively (sequences in Table 1). A series of paired experiments were performed to characterize new crAssphage qPCR assays with established HF183/BacR287 and HumM2 methods. Calibration model performance metrics are reported in Table S5 and include slope, y-intercept range, amplification efficiency (E), LLOQ range, and precision at 101copies per reaction. All assays had a range of quantification from 101–105 copies per reaction (full range of tested standard concentrations). CPQ_056 and CPQ_064 both exhibited a specificity of 98.6% cross-reacting with the same three individual samples from gull (n = 2) and dog (n = 1), while HF183/BacR287 (100%) and HumM2 (99.5%; elk = 1) yielded slightly higher performance levels. CPQ_056 and CPQ_064 target log10 copies per ng of total DNA concentrations were ≤1.33 ± 0.04 in the 2 cross-reacting gull samples and ≤2.60 ± 0.01 in 1 dog sample. HumM2 was detected in a single elk sample (1.02 ± 0.06 log10 copies per ng of total DNA). Genetic marker log10 copies per ng of total DNA concentrations in primary influent sewage samples collected from different geographic locations ranged from 1.49 ± 0.05 to 3.37 ± 0.05 (CPQ_056), 1.83 ± 0.04 to 3.47 ± 0.05 (CPQ_064), 1.55 ± 0.02 to 3.18 ± 0.02 (HF183/BacR287), and 1.13 ± 0.02 to 2.09 ± 0.02 (HumM2). Total log10 copies per reaction concentrations in polluted environmental water samples ranged from 2.12 ± 0.04 to 2.50 ± 0.04 (CPQ_056), 2.33 ± 0.03 to 2.55 ± 0.03 (CPQ_064), 2.28 ± 0.07 to 2.45 ± 0.07 (HF183/BacR287), and 1.06 ± 0.06 to 1.49 ± 0.06 (HumM2). Wastewater qPCR reactions contained 1 ng of template DNA extracted from 10 mL of wastewater, while environmental water qPCR reactions contained 2 μL of DNA extracted from a total volume of 200 mL of impacted water. A comparison of mean estimates with 95% credible intervals both indicated that primary influent sewage (Figure 2, panel A) and environmental water samples (Figure 2, panel B) show no significant difference between CPQ_056, CPQ_064, and HF183/BacR287 results, while HumM2 measurements were significantly lower (p < 0.05).
Table 1.
CrAssphage qPCR Assay Oligonucleotides and Targeted Genomic Regions
| qPCR assay | primer or probe | sequence 5′ → 3′ | genomic region |
|---|---|---|---|
| CPQ_056 | 056F1 | CAGAAGTACAAACTCCTAAAAAACGTAGAG | 14731–14856 |
| 056R1 | GATGACCAATAAACAAGCCATTAGC | ||
| 056P1 | [FAM] AATAACGATTTACGTGATGTAAC [MGB] | ||
| CPQ_064 | 064F1 | TGTATAGATGCTGCTGCAACTGTACTC | 16030–16177 |
| 064R1 | CGTTGTTTTCATCTTTATCTTGTCCAT | ||
| 064P1 | [FAM] CTGAAATTGTTCATAAGCAA [MGB] |
Figure 2.

Abundance of crAssphage and bacterial human-associated qPCR targets in primary influent sewage (panel A) and environmental water samples (panel B). Values are reported as mean log10 copies estimates per nanogram of total DNA (panel A) or per reaction (panel B) with 95% credible intervals. Sewage qPCR reactions contained 1 ng of template DNA extracted from 10 mL of wastewater, while environmental water qPCR reactions contained 2 μL of DNA extracted from a total volume of 200 mL of impacted water.
Experiment Controls
No template control amplifications indicated the absence of contamination in 99.7% of control reactions (n = 1884). All method extraction blanks were negative ensuring no contamination was introduced during sample DNA extraction procedures. All DNA preparations exhibited no evidence of amplification inhibition except three fecal sample preparations, which were discarded from the study (data not shown). All environmental water samples showed no evidence of matrix interference as determined using the Sketa22 approach (data not shown).
Discussion
Identification of Human-Associated CrAssphage Genetic Regions
Data mining of human fecal metagenomic DNA libraries recently identified a putative crAssphage genome.(34) Previously reported comparative sequence analyses suggest this genome is both highly abundant and broadly distributed in human fecal and sewage metagenomic DNA sequence libraries.(34, 35) To investigate the potential use of crAssphage for human fecal-source-identification qPCR method development, we interrogated 43% of the crAssphage genome via laboratory testing to identify candidate qPCR target genetic regions. Approximately 65% of end-point PCR candidate primer sets generated expected PCR products in primary influent sewage samples, supporting bioinformatic predictions that the crAssphage genome is widespread in United States wastewaters.(35) In contrast, 35% of candidate primer sets targeting genetic regions selected by bioinformatic analysis were not detected in sewage. No PCR product amplification from these primer sets may have been due to a lack of assay optimization (e.g., reaction mixture and thermal cycling conditions) or sequence variation at primer hybridization sites. Another plausible explanation could be low genetic conservation between individuals leading to reduced template availability in sewage DNA extracts. The crAssphage genome is reported as a consensus sequence of a quasispecies population isolated from a single fecal sample,(34)whereas sewage is typically a mixture of fecal material contributed typically from thousands of individuals. Diversity between individuals in the crAssphage genome is limited. A recent study identified that patients in China were missing one open reading frame (ORF) and had low identity to another ORF of the crAssphage genome in their samples; however, these ORFs were identified as metaviromic islands and were not interrogated for primer design within this study.(34, 43)Additional studies are warranted to characterize within and between individual sequence variation in these genetic regions.
Nearly 95% (n = 366) of end-point PCR primer sets in round one testing did not yield amplification products of the expected size with nonhuman animal sources used as DNA template. These findings support bioinformatic predictions of a close association of the crAssphage genome with human fecal waste.(34, 35) The bioinformatic analysis limited the number of false-positives observed in laboratory testing; however, some false positives were identified, suggesting that parts of the crAssphage genome share homology with gut microbiome associated microorganisms of animals tested. A total of 14 of the 18 false-positive results in round one testing were located within ORFs with reported homology to known or hypothetical proteins.(34) In addition, 169 (44%) of the primer sets yielded spurious PCR products (e.g., incorrect size and primer dimerization) during round one end-point PCR screening of sewage and fecal DNA preparations. Primer dimerization products can occur when a short region of complementary bases is shared between oligonucleotides in the same reaction, while byproducts of the incorrect size may result from the improper annealing of primers or the potential amplification of pseudogenes or conserved sequence motifs in DNA template preparations. Because the generation of spurious PCR products leads to competition for reagents between the DNA target of interest and amplification byproducts, these genetic regions were not considered for qPCR method development. However, because the current study did not employ any optimization of PCR reaction conditions, these genome regions may be human-associated and may yield adequate methods with additional optimization.
After three rounds of screening, the crAss056 and crAss064 primer sets were selected for qPCR method development. These genetic regions represent the most human-associated and abundant candidate DNA targets based on end-point PCR amplification conditions and reference fecal and sewage collections utilized in this study. Both primer sets target the forward strand of the crAssphage genome (Figure 1). Primer set crAss056 (14 712–14 860 bp) amplifies a region within orf00024, which currently has no known protein homologue.(34) Primer set crAss064 (16 038–16 177 bp) targets a region within orf00025, which was previously reported to have homology with a DNA primase–helicase protein from Veillonella sp.,(34) a bacterial genera commonly found in the intestines and oral mucosa of mammals. DNA sequencing efforts verified that crAssphage amplification products from primer sets crAss056 and crAss064 are conserved in primary influent sewage and environmental samples tested in this study (Figure S1), implying a level of genetic stability for the crAss056 and crAss064 target regions. Limited information exists on DNA mutation rates of intestinal viruses; however, several studies report considerable variation in the human gut virome in samples taken from different individuals.(36, 44, 45) In this study, we attempted to avoid genetic regions with high mutagenic or recombination potential by focusing only on predicted protein coding regions without metaviromic islands or proximity to modular junction regions. The crAssphage assays may need to be monitored in the future to ensure DNA sequence stability of the targeted gene sequences.
Performance of CrAssphage qPCR Assays
Systematic testing of 384 candidate primer sets identified two genetic regions (primer sets crAss056 and crAss064) that were selected for qPCR method development based on the study design. The performance of crAssphage CPQ_056 (based on primer set crAss056) and CPQ_064 (based on primer set crAss064) qPCR assays was evaluated through a series of paired experiments with established HF183/BacR287 and HumM2 assays. The crAssphage-based assays exhibited high calibration model performance, comparable to the performance of HF183/BacR287 and HumM2 (Table S5). In addition, the crAssphage qPCR genetic markers were present at similar concentrations to HF183/BacR287 and were significantly more abundant (p > 0.05) compared to HumM2 in primary influent sewage and impacted environmental water samples tested in this study. In contrast, a recent multiple laboratory evaluation of fecal-source-identification technologies found sensitivities ranging from 0 to 60.5% for human-associated viral and bacteriophage genetic markers in challenge samples, in contrast to much-higher levels reported for bacterial HF183SYBR (all laboratories reporting >87% sensitivity).(4, 32) The high sensitivity exhibited by the crAssphage qPCR assays in this study (100%) is only matched by the pepper mild mottle virus assay(31) and could be another useful alternative to other currently available human-associated viral methods.
In addition to exceptional sensitivity, the crAssphage qPCR assays designed in this study exhibited high specificity (98.6%) based on a fecal reference collection consisting of 222 individual samples from 10 different animal groups. All qPCR assays evaluated in this study exhibited high specificities ranging from 98.6 to 100%, well above the recommended 80% threshold for water quality management applications.(4) CrAssphage qPCR assays cross-reacted with gull (n = 2; 8%) and dog (n = 1; 2.4%) samples, both common sources in recreational and residential areas. However, the crAssphage marker concentration was often lower in nonhuman sources compared to primary influent sewage. In addition, false positives in dog and gull sources was rare, occurring in only 2.4% (1 of 41) dog samples and 8% (2 of 25) gull samples. Other human-associated methods cross-react with these same animal sources likely due to cohabitation with dogs and animal food scavenging.(6, 25, 31) HF183/BacR287 did not cross-react with any samples in the quantifiable range; however, it has been shown to cross-react with chicken and turkey at a much lower concentration than in sewage.(5) Despite the high-specificity performance of the crAssphage qPCR assays, it is recommended that specificity is confirmed with reference samples from the local area of interest before implementation.
Fecal Source Identification and the Human Fecal Viral Metagenome
This study demonstrates that viral metagenomes are a valuable source of genetic information that can be mined for host-associated sequences to develop novel fecal-source-identification technologies; however, using metagenomic sequences for method development presents several challenges. First, compiled genomes are constructed from viral quasispecies, resulting in a genome sequence with unknown variability and stability. In addition, novel genomes discovered through metagenomic sequences may lack homology with known annotated genes, resulting in poor-quality sequence annotation within compiled genomes. Hence, it is difficult to infer specificity of these sequences in silico without laboratory testing. To overcome these challenges, we performed laboratory testing of select regions of the crAssphage genome to find the most-abundant and broadly distributed human-associated genetic regions. This approach builds off of bioinformatic predictions, with extensive end-point PCR laboratory screening to narrow down regions for future genetic marker development. This strategy could also be used for other human-associated viruses; for example, bacteriophages that infect Bacteroides strain GB-124.(28, 29)Currently, no qPCR assays are available for these phages, and they may require the isolation of new hosts based on geographic distribution.(30) This approach will continue to be of use as viral metagenome mining continues to improve with additional research efforts leading to more publicly available data sets.
CrAssphage Fecal-Source-Identification Application
Findings in this study highlight the benefits of crAssphage qPCR methods for human fecal source identification. First, the abundance of the crAssphage markers in sewage and polluted environmental waters implies that it will be possible to monitor in smaller sample volumes (≤200 mL) compared to typical virus assays requiring ≥1 L. The isolation strategy used in this study allows for the simultaneous recovery of bacterial and viral genetic markers as well as the same DNA-purification technique because the crAssphage genome is dsDNA. In addition, there is some evidence of genetic stability for the crAssphage method genome regions based on Sanger sequencing in this study, further showing the potential utility of these assays. Findings also indicate that the crAssphage qPCR assays possess a strong human host association (>98%), performing on par with top bacterial human fecal-source-identification methods. Lastly, as a viral genetic marker, the crAssphage qPCR assays could be a convenient tool with which to compliment bacterial fecal pollution monitoring tools in future studies.
Despite the high performance observed with the crAssphage qPCR assays, necessary developments remain prior to application of these methods. In this study, environmental samples were processed with ultrafiltration, which is expensive and time-consuming. The concentrating pipet procedure used worked well but was only tested with a small number of samples. More research should be conducted to determine the best concentration strategies for crAssphage, including how matrix composition influences recovery efficiencies. In addition, the crAssphage qPCR assays were found to be 98.6% human-associated. This requires specificity testing to be completed in each geographic region prior to implementation, especially in areas with high densities of dogs or gulls. Due to this cross-reaction, it may be necessary to pair these methods with other established human fecal identification technologies in a toolbox approach to improve confidence in results. In addition, future studies may be needed to verify the temporal genetic stability of the crAssphage DNA target sequences, even though the results of this study suggest some level of conservation. Additional studies are necessary to understand linkages of the crAssphage methods to currently recommended fecal indicators, other fecal-source-identification targets, and pathogens with public health relevance. Lastly, the bacterial host and genome sequence variability of crAssphage remains unconfirmed. While this information is not required to exploit crAssphage for fecal source identification, this information could prove valuable to further utilize this virus for other water quality management applications. The availability of reliable viral assays that are abundant in impacted waters could have broad implications for water quality monitoring and human fecal waste treatment.
Supplementary Material
Acknowledgment
This material is based on work supported by the National Science Foundation Graduate Research Fellowship Program under grant no. 1247842 and NSF grant no. 1510925. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation. Information has been subjected to U.S. EPA peer and administrative review and has been approved for external publication. Any opinions expressed in this paper are those of the authors and do not necessarily reflect the official positions and policies of the U.S. EPA. Any mention of trade names or commercial products does not constitute endorsement or recommendation for use.
Footnotes
The authors declare the following competing financial interest(s): The primers reported in the manuscript are the subject of a patent application entitled Cross-Assembly Phage DNA Sequences, Primers and Probes for PCR-based Identification of Human Fecal Pollution Sources (Application Number: 62/386,532).
References
- 1.Soller JA; Schoen ME; Bartrand T; Ravenscroft JE; Ashbolt NJ Estimated human health risks from exposure to recreational waters impacted by human and non-human sources of faecal contamination Water Res. 2010, 44 (16) 4674–4691 DOI: 10.1016/j.watres.2010.06.049 [DOI] [PubMed] [Google Scholar]
- 2.Soller JA; Schoen ME; Varghese A; Ichida AM; Boehm AB; Eftim S; Ashbolt NJ; Ravenscroft JE Human health risk implications of multiple sources of faecal indicator bacteria in a recreational waterbody Water Res. 2014, 66, 254–264 DOI: 10.1016/j.watres.2014.08.026 [DOI] [PubMed] [Google Scholar]
- 3.Boehm A; Soller J, Risks associated with recreational waters: pathogens and fecal indicators Encyclopedia of Sustainability Science and Technology; Springer Publishing: New York, 2011. [Google Scholar]
- 4.Boehm AB; Van De Werfhorst LC; Griffith JF; Holden PA; Jay JA; Shanks OC; Wang D;Weisberg SB Performance of forty-one microbial source tracking methods: A twenty-seven lab evaluation study Water Res. 2013, 47 (18) 6812–6828 DOI: 10.1016/j.watres.2012.12.046 [DOI] [PubMed] [Google Scholar]
- 5.Green HC; Haugland RA; Varma M; Millen HT; Borchardt MA; Field KG; Walters WA;Knight R; Sivaganesan M; Kelty CA Improved HF183 quantitative real-time PCR assay for characterization of human fecal pollution in ambient surface water samples Appl. Environ. Microbiol 2014,80 (10) 3086–3094 DOI: 10.1128/AEM.04137-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shanks OC; White K; Kelty CA; Sivaganesan M; Blannon J; Meckes M; Varma M;Haugland RA Performance of PCR-based assays targeting Bacteroidales genetic markers of human fecal pollution in sewage and fecal samples Environ. Sci. Technol 2010, 44 (16) 6281–6288 DOI: 10.1021/es100311n [DOI] [PubMed] [Google Scholar]
- 7.Rodriguez-Lazaro D; Cook N; Ruggeri FM; Sellwood J; Nasser A; Nascimento MSJ;D’Agostino M; Santos R; Saiz JC; Rzeżutka A Virus hazards from food, water and other contaminated environments FEMS Microbiol. Rev 2012, 36 (4) 786–814 DOI: 10.1111/j.1574-6976.2011.00306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Okoh AI; Sibanda T; Gusha SS Inadequately treated wastewater as a source of human enteric viruses in the environment Int. J. Environ. Res. Public Health 2010, 7 (6) 2620– 2637 DOI: 10.3390/ijerph7062620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Symonds EM; Breitbart M Affordable Enteric Virus Detection Techniques Are Needed to Support Changing Paradigms in Water Quality Management Clean: Soil, Air, Water 2015, 43 (1) 8–12 DOI: 10.1002/clen.201400235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sinclair R; Jones E; Gerba CP Viruses in recreational water–borne disease outbreaks: A review J. Appl. Microbiol 2009, 107 (6) 1769–1780 DOI: 10.1111/j.1365-2672.2009.04367.x [DOI] [PubMed] [Google Scholar]
- 11.Soller JA; Bartrand T; Ashbolt NJ; Ravenscroft J; Wade TJ Estimating the primary etiologic agents in recreational freshwaters impacted by human sources of faecal contamination Water Res. 2010, 44 (16)4736–4747 DOI: 10.1016/j.watres.2010.07.064 [DOI] [PubMed] [Google Scholar]
- 12.Osuolale O; Okoh A Human enteric bacteria and viruses in five wastewater treatment plants in the Eastern Cape, South Africa J. Infect. Public Health 2017, DOI: 10.1016/j.jiph.2016.11.012 [DOI] [PubMed] [Google Scholar]
- 13.Zhou J; Wang XC; Ji Z; Xu L; Yu Z Source identification of bacterial and viral pathogens and their survival/fading in the process of wastewater treatment, reclamation, and environmental reuse World J. Microbiol. Biotechnol 2015, 31 (1) 109–120 DOI: 10.1007/s11274-014-1770-5 [DOI] [PubMed] [Google Scholar]
- 14.Qiu Y; Lee BE; Neumann N; Ashbolt N; Craik S; Maal-Bared R; Pang X Assessment of human virus removal during municipal wastewater treatment in Edmonton, Canada J. Appl. Microbiol 2015, 119 (6)1729–1739 DOI: 10.1111/jam.12971 [DOI] [PubMed] [Google Scholar]
- 15.Blatchley ER; Gong W-L; Alleman JE; Rose JB; Huffman DE; Otaki M; Lisle JT Effects of wastewater disinfection on waterborne bacteria and viruses Water Environ. Res 2007, 79 (1) 81–92 DOI: 10.2175/106143006X102024 [DOI] [PubMed] [Google Scholar]
- 16.Donaldson K; Griffin DW; Paul J Detection, quantitation and identification of enteroviruses from surface waters and sponge tissue from the Florida Keys using real-time RT–PCR Water Res. 2002, 36 (10) 2505–2514 DOI: 10.1016/S0043-1354(01)00479-1 [DOI] [PubMed] [Google Scholar]
- 17.Gregory JB; Litaker RW; Noble RT Rapid one-step quantitative reverse transcriptase PCR assay with competitive internal positive control for detection of enteroviruses in environmental samples Appl. Environ. Microbiol 2006, 72 (6) 3960–3967 DOI: 10.1128/AEM.02291-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Leon R; Shieh C; Baric R; Sobsey M Detection of enteroviruses and hepatitis A virus in environmental samples by gene probes and polymerase chain reaction In Proceedings of the 1990 Water Quality Technology Conference; American Water Works Association: Denver, CO, 1990; pp 11–15. [Google Scholar]
- 19.Fuhrman JA; Liang X; Noble RT Rapid detection of enteroviruses in small volumes of natural waters by real-time quantitative reverse transcriptase PCR Appl. Environ. Microbiol. 2005, 71 (8) 4523–4530 DOI: 10.1128/AEM.71.8.4523-4530.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Monpoeho S; Maul A; Mignotte-Cadiergues B; Schwartzbrod L; Billaudel S; Ferre V Best viral elution method available for quantification of enteroviruses in sludge by both cell culture and reverse transcription-PCR Appl. Environ. Microbiol 2001, 67 (6) 2484–2488 DOI: 10.1128/AEM.67.6.2484-2488.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jothikumar N; Cromeans TL; Hill VR; Lu X; Sobsey MD; Erdman DD Quantitative real-time PCR assays for detection of human adenoviruses and identification of serotypes 40 and 41 Appl. Environ. Microbiol 2005, 71 (6) 3131–3136 DOI: 10.1128/AEM.71.6.3131-3136.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heim A; Ebnet C; Harste G; Pring-Åkerblom P Rapid and quantitative detection of human adenovirus DNA by real–time PCR J. Med. Virol 2003, 70 (2) 228–239 DOI: 10.1002/jmv.10382 [DOI] [PubMed] [Google Scholar]
- 23.Jothikumar N; Lowther JA; Henshilwood K; Lees DN; Hill VR; Vinjé J Rapid and sensitive detection of noroviruses by using TaqMan-based one-step reverse transcription-PCR assays and application to naturally contaminated shellfish samples Appl. Environ. Microbiol 2005, 71 (4) 1870–1875 DOI: 10.1128/AEM.71.4.1870-1875.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kageyama T; Kojima S; Shinohara M; Uchida K; Fukushi S; Hoshino FB; Takeda N; Katayama K Broadly reactive and highly sensitive assay for Norwalk-like viruses based on real-time quantitative reverse transcription-PCR J. Clin. Microbiol 2003, 41 (4) 1548–1557 DOI: 10.1128/JCM.41.4.1548-1557.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McQuaig SM; Scott TM; Lukasik JO; Paul JH; Harwood VJ Quantification of human polyomaviruses JC virus and BK virus by TaqMan quantitative PCR and comparison to other water quality indicators in water and fecal samples Appl. Environ. Microbiol 2009, 75 (11) 3379–3388 DOI: 10.1128/AEM.02302-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.IOS. 10705–2: Water Quality Detection and Enumeration of Bacteriophages–Part 2: Enumeration of Somatic. Coliphages; International Organization for Standardization: Geneva, Switzerland, 2000. [Google Scholar]
- 27.EPA. Method 1602: Male-specific (F+) and Somatic Coliphage in Water by Single Agar Layer (SAL) Procedure; EPA: Washington, DC, 2001. [Google Scholar]
- 28.Ebdon J; Muniesa M; Taylor H The application of a recently isolated strain of Bacteroides (GB-124) to identify human sources of faecal pollution in a temperate river catchment Water Res. 2007, 41 (16) 3683–3690 DOI: 10.1016/j.watres.2006.12.020 [DOI] [PubMed] [Google Scholar]
- 29.Ebdon JE; Sellwood J; Shore J; Taylor HD Phages of Bacteroides (GB-124): a novel tool for viral waterborne disease control? Environ. Sci. Technol 2012, 46 (2) 1163–1169 [DOI] [PubMed] [Google Scholar]
- 30.Vijayavel K; Fujioka R; Ebdon J; Taylor H Isolation and characterization of Bacteroides host strain HB-73 used to detect sewage specific phages in Hawaii Water Res. 2010, 44 (12) 3714–3724 DOI: 10.1016/j.watres.2010.04.012 [DOI] [PubMed] [Google Scholar]
- 31.Rosario K; Symonds EM; Sinigalliano C; Stewart J; Breitbart M Pepper mild mottle virus as an indicator of fecal pollution Appl. Environ. Microbiol 2009, 75 (22) 7261–7267 DOI: 10.1128/AEM.00410-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harwood VJ; Boehm AB; Sassoubre LM; Vijayavel K; Stewart JR; Fong T-T; Caprais M-P;Converse RR; Diston D; Ebdon J Performance of viruses and bacteriophages for fecal source determination in a multi-laboratory, comparative study Water Res. 2013, 47 (18) 6929–6943 DOI: 10.1016/j.watres.2013.04.064 [DOI] [PubMed] [Google Scholar]
- 33.Staley C; Gordon KV; Schoen ME; Harwood VJ Performance of two quantitative PCR methods for microbial source tracking of human sewage and implications for microbial risk assessment in recreational waters Appl. Environ. Microbiol 2012, 78 (20) 7317–7326 DOI: 10.1128/AEM.01430-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dutilh BE; Cassman N; McNair K; Sanchez SE; Silva GG; Boling L; Barr JJ; Speth DR;Seguritan V; Aziz RK A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes Nat. Commun 2014, 5, 1–11 DOI: 10.1038/ncomms5498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stachler E; Bibby K Metagenomic Evaluation of the Highly Abundant Human Gut Bacteriophage CrAssphage for Source Tracking of Human Fecal Pollution Environ. Sci. Technol. Lett 2014, 1 (10) 405–409 DOI: 10.1021/ez500266s [DOI] [Google Scholar]
- 36.Minot S; Bryson A; Chehoud C; Wu GD; Lewis JD; Bushman FD Rapid evolution of the human gut virome Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (30) 12450–12455 DOI: 10.1073/pnas.1300833110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mizuno CM; Ghai R; Rodriguez-Valera F Evidence for metaviromic islands in marine phages Front. Microbiol 2014, 5, 1–10 DOI: 10.3389/fmicb.2014.00027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ye J; Coulouris G; Zaretskaya I; Cutcutache I; Rozen S; Madden TL Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction BMC Bioinf. 2012, 13 (1) 134 DOI: 10.1186/1471-2105-13-134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shanks OC; Kelty CA; Sivaganesan M; Varma M; Haugland RA Quantitative PCR for genetic markers of human fecal pollution Appl. Environ. Microbiol 2009, 75 (17) 5507–5513 DOI: 10.1128/AEM.00305-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haugland RA; Varma M; Sivaganesan M; Kelty C; Peed L; Shanks OC Evaluation of genetic markers from the 16S rRNA gene V2 region for use in quantitative detection of selected Bacteroidalesspecies and human fecal waste by qPCR Syst. Appl. Microbiol 2010, 33 (6) 348–357 DOI: 10.1016/j.syapm.2010.06.001 [DOI] [PubMed] [Google Scholar]
- 41.Sivaganesan M; Haugland RA; Chern EC; Shanks OC Improved strategies and optimization of calibration models for real-time PCR absolute quantification Water Res. 2010, 44 (16) 4726–4735 DOI: 10.1016/j.watres.2010.07.066 [DOI] [PubMed] [Google Scholar]
- 42.Shanks OC; Kelty CA; Oshiro R; Haugland RA; Madi T; Brooks L; Field KG; Sivaganesan M Data acceptance criteria for standardized human-associated fecal source identification quantitative real-time PCR methods Appl. Environ. Microbiol 2016, 82 (9) 2773–2782 DOI: 10.1128/AEM.03661-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liang Y; Zhang W; Tong Y; Chen S CrAssphage is not associated with diarrhoea and has high genetic diversity Epidemiol. Infect 2016, 144 (16) 3549–3553 DOI: 10.1017/S095026881600176X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reyes A; Semenkovich NP; Whiteson K; Rohwer F; Gordon JI Going viral: next-generation sequencing applied to phage populations in the human gut Nat. Rev. Microbiol 2012, 10 (9) 607–617 DOI: 10.1038/nrmicro2853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Minot S; Wu GD; Lewis JD; Bushman FD Conservation of gene cassettes among diverse viruses of the human gut PLoS One 2012, 7 (8) e42342 DOI: 10.1371/journal.pone.0042342 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.