

Abstract
The ATP-dependent BAF chromatin remodeling complex plays a critical role in gene regulation by modulating chromatin architecture, and is frequently mutated in cancer. Indeed, subunits of the BAF complex are found to be mutated in >20% of human tumors. The mechanism by which BAF properly navigates chromatin is not fully understood, but is thought to involve a multivalent network of histone and DNA contacts. We previously identified a composite domain in the BRG1 ATPase subunit that is capable of associating with both histones and DNA in a multivalent manner. Mapping the DNA binding pocket revealed that it contains several cancer mutations. Here, we utilize SELEX-seq to investigate the DNA specificity of this composite domain and NMR spectroscopy and molecular modelling to determine the structural basis of DNA binding. Finally, we demonstrate that cancer mutations in this domain alter the mode of DNA association.
Introduction
The eukaryotic genome exists in the nucleus in the form of chromatin. At its most basic level chromatin is composed of iterative subunits known as nucleosomes[1], which consist of an octamer of histones (two each of H2A, H2B, H3, and H4), wrapped by ~147 base pairs of DNA. Chromatin structure needs to be spatially and temporally remodeled for all DNA templated processes. This is in large part aided by ATP-dependent remodeling complexes. These can be categorized into four families and include the SWI/SNF, ISWI, IN080, and CHD families of remodelers. The BRG1/BRM Associated Factors complex (BAF complex) is the human homologue of yeast SWI/SNF. BAF is highly polymorphic, forming several different canonical and non-canonical complexes in embryonic stem cells as well as upon differentiation [2-4]. Additional heterogeneity is seen depending on developmental stage and cell type. BAF is important for sliding and ejecting histones in a variety of cellular processes, including transcription regulation and DNA repair [5-8].
BAF complexes are tailored for regulation at specific chromatin regions. The exact mechanism by which BAF is recruited to these regions is not fully understood. It is in part mediated by association with transcription factors. In addition to this, its many subunits harbor a number of histone and DNA binding domains [9]. These domains are thought to form a large multivalent network of chromatin contacts, that determine its association with nucleosome substrates, and thus regulate remodeling activity. These include readout of histone post-translational modifications as well as specific and non-specific recognition of DNA elements. However, the details of these interactions are still being uncovered, as is the mechanism by which they coordinate to promote proper recruitment and function.
BAF subunits are mutated in greater than 20% of all human cancers sequenced to date, making it one of the most frequently mutated chromatin regulatory complexes[8]. Mutations are found throughout most of the subunits, but are especially enriched in BAF250A and BRG1. While most mutations lead to loss of function of that subunit, the mutated complexes are potentially good therapeutic targets[10]. Thus, an understanding of the mechanism by which BAF associates with chromatin is critical both for a fundamental understanding of the complex as well as for determining etiology of disease and in the development of pharmaceutic interventions.
The BAF complex contains one of two possible catalytic subunits, either BRG1 or BRM. These are mutually exclusive ATPases in the BAF complex. Although highly homologous, they cannot substitute for one another in vivo [11,12]. This is in part due to association with distinct transcription factors through their unique N-terminal domains. BRG1-containing BAF is critical for development, with mutation of the ATP binding site in BRG1 being embryonic lethal in mice [13]. Both BRG1 and BRM contain a highly conserved bromodomain (BD) at the C-terminus (Figure 1A), the presence of which is unique among remodeler ATPases. Both bromodomains (BDs] have previously been characterized to bind acetylated-lysine on histone tails, with a moderate preference for histone H3 acetylated at lysine 14 (H3K14ac) [14-17]. In addition to its histone binding activity, we have recently discovered that both BDs can associate with DNA [17]. Nucleic acid binding activity has only been demonstrated for a few BDs[17-19]. However, we have predicted that ~30% of known BDs will bind to nucleic acids [17], highlighting the importance of better understanding this function.
Figure 1. DNA specificity of the BRG1 ATBD.

(A) Domain architecture of the human BRG1 subunit of the BAF complex. The AT-hook and bromodomain (BD) are highlighted in blue. (B) Schematic representation of SELEX-seq followed by a construction of a position specific affinity matrix and converted into the affinity logo containing AATTAAAT. (C) Specificity was confirmed through competitive binding assays analyzed by EMSA. A 15bp double-stranded DNA containing either the SELEX determined DNA sequence (SELEX-DNA: CCTCAATTAAATCTC) or a non-consensus sequence (nc-DNA: CCTCAGTCGGTCGTA) was used. The complex of ATBD and Cy5-labeled SELEX-DNA (bound) was competed against unlabeled SELEX-DNA (top) or nc-DNA (bottom). (D) Dissociation (Kd) values were determined by BLI. Shown is a set of representative traces in a BLI binding assay (top) for 15μM, 8μM, 4μM, 2μM, 1μM, 0.5μM, and 0.25μM (from top to bottom) of protein. Calculated Kd values are in the table below, values reported are the average of 3 runs and standard deviations.
Previous studies indicate that the importance of the BRG1 BD is context dependent. Histone binding activity is only essential for global chromatin association in response to histone hyper-acetylation [20,21] , and DNA binding activity does not alter the affinity of BRG1 for bulk chromatin[17]. Small molecule inhibition of the BD/histone interaction showed no effect on BAF dependent cancer proliferation, and its deletion did not affect co-activation of transcription mediated by the glucocorticoid receptor [21-23] . However, ESCs treated with the BRG1 BD inhibitor, PFI-3, showed a reduced stemness potential [18]. In addition, both the acetyl-lysine and DNA binding pockets are conserved and mutated in cancer. These context dependent functions could include roles in nucleosome affinity, and/or regulation of ATPase or remodeling activity.
Ten residues N-terminal to the BD in both BRG1 and BRM is an AT-hook (Figure 1A). AT-hooks, described for the first time in the context of the high mobility group (HMG) proteins[24], are short motifs of between nine and ten amino acids that are arginine/lysine rich. A central consensus sequence of glycine-arginine-proline (GRP) allows the motif to insert into the minor groove of DNA. In addition, the extended AT-hook has been shown to bind to RNA [25]. AT-hooks prefer AT-rich DNA sequences as this leads to narrowing of the minor groove and a more focused electrostatic potential [26,27]. In a genome wide study by Aravind et al. [28], many examples were found of an AT-hook existing just adjacent to another DNA-binding module, and it was suggested that this is a common auxiliary motif cooperating with adjacent structured DNA binding domains. Similar to the BD, the role of the AT-hook in BAF function has not been elucidated. However, it is also highly conserved, and known to be mutated in cancer. We have recently shown that the tandem AT-hook and BD (AT-BD) function as a composite DNA binding module within the BAF complex, associating with DNA in a multivalent manner, with low micromolar affinity[17]. We demonstrated that the AT-BD can bind both naked and nucleosomal DNA. However, many questions remain including whether this module has sequence specificity, and the molecular basis of binding to DNA. Here, we utilize SELEX-seq to investigate the sequence specificity of the BRG1 AT-BD and NMR spectroscopy and molecular modelling to uncover the structural basis of DNA binding.
Materials and Methods
Protein Expression and Purification
Human BRG1 AT-BD (residues 1434-1569) or BD (residues 1454-1569) were cloned into a pGEX6p-1 vector (GE Healthcare) from full length BRG1 cDNA obtained from GE Open Biosytems. Proteins were expressed in BL21 (DE3) E. coli cells (New England Biolabs, Ipswich, MA) in LB or M9 minimal media supplemented with vitamins (Centrum), 1 g/L 15NH4Cl and 1g/L 12C or 13C D-glucose. When the culture reached OD600=1, protein expression was induced with 0.16 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) for 16 hours at 28°C. Cells were lysed with a homogenizer (Avestin Emulsiflex C3) in resuspension lysis buffer containing 20 mM Tris pH 7.5, 500 mM KCl, 2.5 mM MgSO4, 0.5% triton, 3mM DTT, 1mg/ml lysozyme, DNaseI, and a protease inhibitor cocktail tablet (Roche). The lysate was cleared through centrifugation for 45 min at 12,000·g. The resulting supernatant was incubated with glutathione agarose resin (ThermoFisher Scientific) for one hour. The resin was washed thoroughly with buffer containing 50 mM Potassium phosphate pH 7.0, 50 mM KCl, and 2 mM DTT (final buffer). The GST-tag was cleaved off with PreScission Protease, and the cleaved protein was further purified using FPLC first with cation exchange (Source-S, GE Healthcare), followed by gel filtration (Superdex 75, GE Healthcare).
Selective Evolution of Ligand by Exponential Enrichment (SELEX)
| Oligos used: | ||
|---|---|---|
| SELEX library | Library | 5’ GTTCAGAGTTCTACAGTCCGACGATC (N23)TGGAATTCTCGGGTGCCAAGG 3’ |
| TSSR0 | amplification | 5’ GTTCAGAGTTCTACAGTCCGACG 3’ |
| TSSR1 | / sequencing | 5’ CCTTGGCACCCGAGAATTCCA 3’ |
| Cy5-TSSR1 | 5’ Cy5-CCTTGGCACCCGAGAATTCCA 3’ | |
| TSSR2 | 5’ AATGATACGGCGACCACCGAGATCTACACGTTCAGAGTTCTACAGTCCGA 3’ | |
| TSSR-RPIX* | 5’ CAAGCAGAAGACGGCATACGAGATXXXXXXGTGACTGGAGTTCCTTGGCACCCGAGAATTCCA 3’ | |
Available TSSR-RPIX with different barcodes (RPI1 - RPI48) can be found in Illumina Custom Sequence letter, 2012, Sep 7th. XXXXXX: 6bp barcode for Illumina sequencing.
SELEX libraries consist of a 15-base pair (bp) randomized region flanked by TruSeq Small RNA adapter sequences. The 62 bp libraries were ordered as a single strand in the 1 μM desalted format from Integrated DNA Technologies using the hand mixing option for the randomized region to ensure maximal randomization. The second strand was synthesized by Klenow extension using the Cy5-TSSR1. A 100μl reaction containing 2μM library, 5 μM Cy5-TSSR1, 150 μM dNTPs in New England Biolabs (NEB) buffer 2 was incubated at 94°C for 3 minutes, then cooled to 37°C over 45 minutes. After addition of 24 units of Klenow fragment, the reactions were incubated at 37°C for 60 minutes, 72°C for 20 minutes, and then cooled to 10°C over 45 minutes. The dsDNA library was purified using a MinElute column (Qiagen) using the standard protocol, and then quantified by absorbance at 260 nm.
SELEX-seq was then performed as previously described[29], but with a few modifications. For SELEX, the bound fraction of library was separated from free by electrophoretic mobility shift assay (EMSA). In a 125μl reaction, 0.5 μM AT-BD was incubated with 2μM library in binding buffer (20 mM TrisHCl, pH 8.0, 150 mM KCl, 5% Glycerol, 1 mM EDTA, 5 mM MgCl2, 40 ng/μl Poly(dIdC) (Sigma: P4925), 200 ng/μl BSA, 1 mM DTT, 200 mg/ml Ficoll PM400 (Sigma: F4375)) at 4°C for 1 hour. Reactions were then run on a non-denaturing PAGE (10% 75:1 acrylamide:bis-aciylamide, 0.2X TBE) for 50 minutes in 0.2X TBE at 125 V. Free and bound bands were visualized by Cy5 fluorescence (GE LAS4000 imager). The shifted bands were excised, and the DNA recovered by electroelution (Novagen D-tube dialyzer, 3.5 kDa) in 0.2X TBE. The recovered library was purified (Qiagen MinElute PCR cleanup) and eluted (10 mM TrisHCl, pH 8.0) in a final volume of 180μl. In order to maximally amplify the library while minimizing artifacts, recovered library was quantified by qPCR, divided into 85 reaction of 100 μl, and amplified using Phusion (NEB). The number cycles depended on how much DNA was present and was calculated as the number of cycles for 90% signal in qPCR plus one additional cycle. Three rounds of selection were performed in total.
Adapters with unique barcodes were added to the libraries barcodes by amplifying input (Round 0 or R0) and Round 3 (R3) using TSSR2 and TSSR-RPIX. For each library, 400ng of library was amplified for 2 rounds using standard Phusion (NEB) PCR conditions. The 133bp final library was then separated from the 63 bp input on a 16% native PAGE (0.5X TBE, 19:1 acrylamide/bis-acrylamide), excised, and recovered by electroelution. Libraries were then checked for quality and quantified by Bioanalyzed (Agilent Technologies), then sequenced to a depth of ~10 million reads/library using single-end 50bp sequencing.
The sequenced libraries were initially processed using the R package SELEX (R Core Team)(http://bioconductor.org/packages/SELEX). To correct for biased sampling from the input library, a markov model was first determined (6th order), then constructed for R0 using the selex.mm(). Next, the optimal motif length was defined by determining the Kullback-Leibler divergence for motifs of increasing size using selex.infogain(). The opt6imal motif length proved to be 10bp.
Using the R package SelexGLM (https://www.bioconductor.org/), an affinity table was first constructed for k=10 using selex.affinities (). An initial position specific affinity matrix (PSAM) was constructed (BAWTTWAWTS) and used as a seed. The subsequent iterative procedure alternated between two steps. First, the current PSAM was used to find the position/direction of highest affinity on either strand, the optimal “view” on the probe. If that optimal affinity was larger than 95% of the sum over all positions (including the top position), the probe was used in the analysis; otherwise, it was ignored. The set of optimal positions in each of the accepted probes was used to define a design matrix containing the base identity at each position relative to the start of the optimal view. Using the probe counts as independent variables, the logarithm of the expected probe frequency in R0 according to the Markov model as offset, and a logarithmic link function, a fit was performed using the glm () function. The regression coefficients were interpreted as free-energy differences ΔΔG. All computational figure panels in this paper were produced fully automatically from the raw sequencing data using R scripts that use the SELEX and SelexGLM packages.
SELEX-DNA and nc-DNA Preparation and Purification
Oligonucleotides for BLI, NMR, and gel shift experiments were purchased from Integrated DNA Technologies (IDT) (see below). Forward and reverse strands were resuspended in 50 mM potassium phosphate pH 7.0, 50 mM KCl, and 2 mM EDTA, in nuclease free water, mixed at equimolar ratios and annealed by heating to 94 °C for 10 min before being slowly cooled to room temperature. The annealed strands were then further purified by gel filtration chromatography (Superdex 75, GE Healthcare).
| Experiment, type sequence | DNA substrate used |
|---|---|
| NMR, EMSA, SELEX-DNA | 5’-CCTCAATTAAATCTC -3’ 3’-GGAGTTAATTTAGAG-5’ |
| NMR, EMSA, nc-DNA | 5’-CCTCAGTCGGTCGTA-3’ 3’-GGAGTCAGCCAGCAT-5’ |
| Competition, SELEX-DNA | 5’-Cy5-CCT CAATTAAATCTC -3’ 3’-GGAGTTAATTTAGAG -5’ |
| BLI, biotinylated SELEX-DNA | 5’-5Biot-CCTCAATTAAATCTC -3’ 3’-GGAGTTAATTTAGAG -5’ |
| BLI, biotinylated ncDNA | 5’-5Biot-CCTCAGTCGGTCGTA-3’ 3’-GGAGTCAGCCAGCAT-5’ |
List of DNA substrates and complementary strands.
NMR Data Collection and Analysis
Spectra were collected on a 500 MHz Bruker Avance II, Bruker Avance NEO 600 MHz, or an 800 MHz Bruker Avance II equipped with a cryoprobe. Data processing was performed in NMRPipe[30] and assignments completed in CcpNnmr analysis[31]. Buffer A contains 50 mM potassium phosphate, pH 7.0, 50 mM KCl, 2 mM DTT, 2 mM EDTA in 8% D2O.
NMR Assignments
In order to assign the backbone resonances of BRG1 AT-BD, HNCACB and CBCA(CO)NH, spectra were collected at 500 MHz at 25°C on a sample containing 570 μM of 15N-13C ATBD in buffer A. Data were processed in NMRPipe and analyzed in CcpNmr analysis. The HNCACB was collected using 40 scans and 80 and 86 complex points in the 15N and 13C dimensions respectively. For the CBCA(CO)NH, data were collected using 32 scans and 80 and 80 complex points in the 15N and 13C dimensions.
For side chain assignments of the AT-BD, H(CCO)NH and CC(CO)NH spectra were collected at 500 MHz at 25 °C using 570 μM 15N-13C ATBD in buffer A, and an HCCH-TOCSY was collected at 600 MHz at 25°C. The H(CCO)NH was collected using 32 scans and 70 and 110 complex points in the 15N and 1H dimensions respectively. The CC(CO)NH was collected using 48 scans and 70 and 68 complex points in the 15N and 13C dimensions respectively. The HCCH-TOCSY was collected using 32 scans and 80 and 84 complex points in the 1H and 13C dimensions respectively. The HBHACONH was collected using a sample containing 900 μM 15N-13C ATBD in the same buffer at 600 MHz at 25°C, using 48 scans and 60 and 100 complex points in the 15N and 1H dimensions respectively. To determine the side-chain assignments in the DNA-bound state, chemical shifts were tracked upon titration of SELEX-DNA in a 13C-HMQC. Sequential 13C-HMQC spectra were collected at 800 MHz at 25°C on a 200 μM sample of 15N-13C ATBD in buffer prepared in 99.8% D2O, 50 mM potassium phosphate pH 7.0, 50 mM KCl, and increasing amounts of SELEX-DNA at protein-DNA ratios of 1:0, 1:0.1, 1:0.3, 1:0.5, 1:0.9, 1:1.65. Data were processed in NMRPipe, analyzed in CcpNmr analysis.
To assign the non-exchangeable protons of the SELEX-DNA, homonuclear TOCSY and homonuclear NOESY spectra were collected at 800 MHz at 25°C using 3.2 mM double stranded SELEX-DNA in buffer containing 99.8% D2O, 50 mM potassium phosphate, 50 mM KCl, 2 mM EDTA. The propagation of non-exchangeable proton assignments from the apo to the fully saturated DNA was carried out by collecting four spectra using w1 12C-filtered, w2 12C-filtered experiments at 800 MHz at 25 °C using 4 different samples in the same buffer as described above. Each sample contained 1.2 mM of unlabeled SELEX-DNA as apo or in complex with 15N-13C ATBD. The ratios of DNA:Protein were 1:0, 1:0.15, 1:0.5, 1:1.65.
To assign the exchangeable protons of DNA, along with the H2 of adenines, two homonuclear NOESY spectra were collected encompassing the imino region of DNA at 800 MHz at 25 °C using 5 mM of SELEX-DNA on aqueous buffer (H2O) with 50 mM potassium buffer, 50 mM KCl, 2 mM EDTA.
NMR titrations
Titrations of AT-BD or BD with DNA were carried out by collecting sequential 1H,15N-HSQC spectra on 15N-AT-BD or 15N-BD upon addition of increasing concentrations of SELEX-DNA or nc-DNA. Protein and DNA samples were prepared in buffer A. Spectra were collected at 800 MHz at 25°C on 100μM AT-BD at protein:DNA ratios of 1:0, 1:0.1, 1:0.3, 1:0.5, 1:0.9, 1:1.5, 1:2.1, 1:2.7 for SELEX-DNA and 1:0, 1:0.1, 1:0.3, 1:0.5, 1:1.2, 1:1.9, 1:2.8, 1:3.7 for nc-DNA. For the titration into BD alone, spectra were collected at 800 MHz at 25°C on a 100 μM sample of 15N-BD at proteimDNA ratios of 1:0, 1:0.3, 1:0.9, 1:1.5, 1:3.3, 1:6.6, 1:13.2 for SELEX-DNA and 1:0, 1:0.3, 1:0.9, 1:1.5, 1:3.3 1:6.6, 1:13.2 for nc-DNA. Data were processed in NMRPipe and analyzed in CcpNmr analysis. The normalized chemical shift perturbations (CSPs or Δδ) were calculated as:
where ΔδH and ΔδN are the change in the 1H and 15N chemical shift respectively between the apo state and the titration point.
To monitor the binding of DNA to ATBD, a titration was performed by collecting 1D proton spectra of the imino region of unlabeled SELEX-DNA were collected at 800 MHz at 25°C in the presence of increasing amounts of unlabeled AT-BD. Protein and DNA were prepared in same buffer containing 50 mM potassium phosphate pH 7.0, 50 mM KCl, 2 mM EDTA, 2mM DTT. The ratios of DNA:protein were 1:0, 1:0.5, 1:0.1, 1:0.2, 1:0.3, 1:0.5, 1:0.9, 1:1.3, 1:1.7, 1:2.1, 1:2.5, 1:3.3.
Intermolecular NOESY
To identify the binding interface between the AT-BD and SELEX-DNA an intermolecular NOESY (Nuclear Overhauser Enhancement Spectroscopy) was collected at 800 MHz at 25°C on a 580 μM sample of 15N-13C ATBD in the presence of 2.2 mM SELEX-DNA. The NOESY was collected in 99.8% D2O and 50 mM potassium phosphate, 50 mM KCl, 2 mM EDTA and 2 mM DTT. NOEs between the 13C/15N-labeled AT-BD and unlabeled SELEX-DNA were detected using 13C-edited and 12C-filtered 3D NOESY experiments with a mixing time of 150 milliseconds[32].
Biolayer Interferometry (BLI)
BLI experiments were carried out by tethering double stranded SELEX-DNA or nc-DNA biotinylated at the 5’ end of the forward strand (15 bp each) on a streptavidin sensor. Interference was assayed against increasing concentrations of either AT-BD as analyte in solution. Experiments were carried out in triplicate using an Octet RED96 Biolayer Interferometry (BLI) System (Pall ForteBio, Menlo Park, CA) at 26°C shaking at 1000 RPM. Black 96 well microplates (Greiner bio-one Germany) and Streptavidin sensors (SA, Dip and Read biosensors, ForteBio, California) were used for all experiments. Protein and DNA samples were prepared in 50 mM potassium phosphate, pH 7.0, 50 mM KCl, 0.04% Tween20 and 2 mM EDTA. Each well contained a 200 μL sample, and all probes were pre-soaked in buffer for 45 minutes before each experiment.
The binding assay consisted of a temperature pre-equilibration for 10 min at 26°C, followed by buffer equilibration step of all probes in buffer for 300 s, loading of the biotinylated DNA (50 nM) for 300 s, followed by a baseline step in buffer for 300 s. Following baseline, association was assessed against either 0.25μM, 0.5μM, 1μM, 2μM, 4μM, 8μM, or 15μM ATBD or BD for 300s followed by a dissociation step in buffer for 600 s. Each experiment had a reference well (to correct for buffer drift). Initial trials were performed using a reference sensor to subtract non-specific binding, but no significant non-specific binding was detected. Data was collected using a data acquisition rate of 10 Hz. Steady state analysis was used to determine Kd values. Data fitting was performed using the ForteBio Data Analysis 9.0 software. During processing the data, reference wells were subtracted, and data were aligned to the last 10 s of the baseline step, inter-step correction was aligned to dissociation, and Savitzky-Golay filtering was also applied. The fitting was performed by choosing global fitting to R equilibrium option in a 1:1 binding mode which plots the response at equilibrium as a function of the analyte concentration using a single binding site model.
EMSA and Competition Assay with 15 base pairs DNA
Electrophoretic Mobility Assays (EMSAs) were carried out on SELEX-DNA or nc-DNA in the presence of increasing amounts of wild-type or mutant AT-BD or BD. Samples were run on a 10% 75:1 acrylamide:bisacrylamide gel. Gels were casted and run in 0.2X TBE buffer, for 50 minutes at constant 125 V at 4°C. The sample load in each well was 5 μL from a binding reaction containing a volume of 10 μL with 20 mM potassium phosphate, pH 7.0, 80 mM KCl, 2mM EDTA, 2mM DTT. The final concentration of DNA in each well was 53 nM and increasing protein concentrations of 0.04875μM, 0.0975μM, 0.195μM, 0.39μM, 0.781μM, 1.56μM, 3.125μM, 6.25μM, 12.5μM, 25μM, 50μM, and 100μM. Staining of the gel was performed with SYBR Green (ThermoFisher Scientific, Waltham, MA). Visualization of gels was performed in an ImageQuant LAS 4000 imager.
Competition Assays were performed on a 10% 19:1 acrylamide:bisaciylamide gel. Gels were cast and run in 0.2X TBE buffer, for 50 minutes at constant 125 V at 4 °C. Each lane was loaded with 5 μL of a 10 μL binding reaction containing a preformed complex with 0.781μM of BRG1 ATBD in 25 mM potassium phosphate, pH 7.0, 25 mM KCl, 2mM EDTA, 2mM DTT, and 10nM of cy5-labeled SELEX-DNA and using unlabeled SELEX-DNA or nc-DNA as competitor in increasing concentrations of 0.05μM, 0.2μM, 0.4μM, 0.6μM, 1.0μM, 2.0μM, 4.0μM, 8.0μM, 12.0μM, 15.0 μM, 40.0μM. Visualization of gels was performed in an ImageQuant LAS 4000 imager with the Cy5 fluorescence option.
EMSA of WT and mutants of BRG1 ATBD with Widom 601and quantitation
Electrophoretic Mobility Shift Assays (EMSA) of WT BRG1 ATBD or a mutant with DNA Widom 601 sequence or NCPs were carried out using a 5% native gel with a ratio 75:1 acrylamide:bisacrylamide. Gels were cast and pre-run in 0.2X TBE buffer for 20 minutes at constant 125 V with the gel box in an ice water bath and inside a cold room (4 °C). With samples loaded, the gels were run for 50 minutes, at 125 V and at 4 °C. The sample loaded in each well was the entire binding reaction containing a volume of 10 μL. Each well contained a final concentration of 10 mM potassium phosphate, pH 7.0, 10 mM KCl, 2mM EDTA, 2mM DTT. Each binding reaction was prepared using 5 μL (50% of final volume) of a loading dye. The loading dye was prepared by mixing 100 mg sucrose in 1 ml of 0.5 X TE buffer and adding 20 μL of bromophenol blue solution (0.1% w/v in water). Final concentration of Widom 601 or NCP in each well was 150 nM and increasing protein concentrations of 0, 0.150μM, 0.6μM, 0.9μM, 1.5μM, 2.25μM, 3.0μM, 4.5 μM in each well, along with a lane containing 100 bp ladder. Staining of the gel was performed using 10 μL of (10mg/mL) stock solution of ethidium bromide (Bio-rad) dissolved in 50 μL of 0.2X TBE buffer. The gel was stained in this solution for 5 minutes immediately after the voltage stopped. Visualization of gels was performed in an ImageQuant LAS 4000 imager.
The percent of DNA bound was calculated as
The DNA band intensity on the gel was quantified using ImageJ. Binding curves for all titrations were fitted using a nonlinear least-squares analysis to a single site binding model in GraphPad Prism:
Where [L] is the concentration of protein, [D] is the concentration of the DNA and Rmax is the percent of DNA bound at saturation.
HADDOCK
The crystallographic structure of the BRG1 BD was obtained from the Protein Data Bank, entry 2GRC[15]. Missing residues and the AT-hook motif KKQKKRGRPPAEKLS were reconstructed in silico by using the Modeller program[33] in the UCSF Chimera suite, with the most extended AT-hook structure taken for further analysis,. The DNA sequence 5’- CCTCAATTAAATCTC - 3’ was built using 3D-DART [34].
Molecular docking of the BRG1 AT-BD to SELEX-DNA was carried out with the HADDOCK webserver[35]. The active and fully flexible residues chosen to bias the docking were located in the ZA loop, AB loop, N-terminal, and the complete AT-hook, which were chosen based chemical shift perturbations. All DNA base pairs were considered as active residues to allow the AT-BD system to sample all possible likely conformations along the linear DNA sequence. HADDOCK calculations in which only the residues and DNA base pairs identified by NOE experiments were defined as active residues were carried out and resulted in similar binding poses. The docking process consisted of 100 cycles of rigid body energy minimization (EM), semi-flexible simulated annealing, and explicit solvent refinement. In particular, during the rigid-body EM 1000 structures were generated and subsequently reduced to 200 during the semi-flexible and water refinement steps. The top ten docking conformations as determined by their HADDOCK score were used for MD simulations.
Molecular Dynamics Simulations
For each of the 10 AT-BD/SELEX-DNA complex conformations selected from the HADDOCK docking, three MD simulations were performed. The simulation package GROMACS 2016.4 [36] was used in combination with the AMBER14SB force field [37]. Based on its good agreement with experimental results [38,39], the BSC1 force field was used to simulate the DNA. In each simulation, the complex was solvated in a cubic box with a minimum distance of 10 Å to the nearest box edge and surrounded by approximately 15000 water molecules described by the TIP3P water model [40]. System charges were neutralized and Na+ and Cl− ions were added to reach 150 mM salt concentration. Systems were minimized for 1000 steps with the steepest descendent method. Complexes were then equilibrated for 100 ps at constant volume and temperature and for 1 ns at constant pressure and temperature. Production simulations were carried out for 100 ns in the NPT ensemble, using a Parrinello-Rahman barostat [41] with a time constant of 2.0 ps to control the pressure and a V-rescale thermostat with a time constant 0.1 ps. Electrostatic interactions were treated with the Particle-Mesh Ewald (PME) method [42] and 9 Å cut-off. An integration time step of 2 fs was used.
Among the conformations obtained through MD simulations, five were selected for further analysis based on how closely they reproduced experimental NOE distance restraints. The systems were refined based on these distances by the addition of distance restraints between the DNA and the AT-BD. This was done in three stages, with the first being a 10 ns simulation in the NVT ensemble and a distance restraint of 1000 kJ mol−1 nm−2 between Arg1443 NH2 and 1A5 N1, the second a 10 ns NPT simulation with distance restraints of 1000 kJ mol−1 nm−2 between Arg1443 CD and 1A9 C2, Arg1443 CD and 2A8 C2, Arg1445 CD and 1A6, Arg1445 CD and 2A9 C2, and the third a 10 ns NPT simulation with additional restraints between Leu1488 CD1 and 2T7 C7, Tyr1498 CE1 and 2T7 C7. Following these three restrained simulation stages, 500 ns of unrestrained simulations were performed.
Structure equilibrations were evaluated by analyzing the root mean square deviations (RMSD). In the calculation of atomic RMSD protein rotation and translation were eliminated by a mass-weighted least squares fit with respect to the reference starting structure. VMD [43] was used for analysis of the hydrogen bonds and for trajectory visualization. In particular, hydrogen bonds were calculated between residue pairs with occupancies computed as the percent of MD frames with bond donor-acceptor distance and angle cutoffs of less than 3.5 Å and 30 degrees. Contact frequency maps were calculated for every frame of the equilibrated portion of the trajectories with GROMACS mdmat (trunc distance 4.2 Å). Do_x3DNA and dnaMD tools [44,45] were used to analyze global and local DNA properties during MD simulation.
Results
BRG1 AT-BD has preference for a 10-basepair AT-rich sequence
We previously found that the BRG1 BD can associate with DNA, and that the BD and adjacent AT-hook act as a composite domain to bind DNA in a multivalent manner [17]. Though we found a preference for AT-containing DNA, only two DNA sequences were tested in that seminal work. We therefore performed Systematic Evolution of Ligands by Exponential enrichment followed by deep sequencing (SELEX-seq, Figure 1B) [46] to measure the sequence preference of AT-BD in a comprehensive and unbiased fashion.
Having previously observed that the AT-BD occludes a single helical turn of double stranded (ds) DNA (~10bp), we sought to determine whether this composite domain has a preference for sequences of this length or longer[17]. Accordingly, we incubated AT-BD with a randomized 15 base pair library and separated bound from free by electrophoretic mobility shift assay (EMSA). The bound sequences were isolated by excising the shifted band from the gel, amplifying the sequences, and repeating twice, for a total of three rounds of enrichment (Figure 1B). Using the SELEX package[46], we found that information gain reached a maximum with a binding site of 10bp (Supplementary Figure S1A), providing strong evidence that the 10bp footprint is accurate. We then used the SelexGLM package [29] to calculate a position-specific affinity matrix over a lObp footprint, which was converted to logo format using MATRIXReduce[47]. The most favorable motif is composed of an asymmetric AT-rich core 8bp sequence (AATTAAAT) (Figure 1B, right, Supplementary Figure S1C), with slight preference for G at the first and C at the last position. Interestingly, the AT-BD appears able to accommodate either As or Ts, with little penalty for substitution of one over the other within the core motif, except at position 7.
To validate the preference for the SELEX-determined sequence we carried out electromobility shift assays (EMSAs) with the AT-BD and 15 bp double-stranded DNA containing either a non-consensus sequence (nc-DNA: 5’-CCTCAGTCGGTCGTA- 3’) or the SELEX determined consensus sequence (SELEX-DNA: 5’- CCTCAATTAAATCTC -3’). Binding of AT-BD to SELEX-DNA led to discretely shifted bands for the complex at lower AT-BD concentrations (Supplementary Figure S1B, see lanes 5 and 6) with less discrete higher-order complexes evident at higher AT-BD concentrations. In contrast, EMSAs for nc-DNA:AT-BD complexes ran as smears for all concentrations tested, reflecting a higher off-rate and likely less stable complex (Supplementary Figure S1B). EMSAs of the BD alone with SELEX-DNA ran as smears starting only at higher concentrations of BD, confirming that both the AT-hook and BD are important for robust interaction (Supplementary Figure S1B). In addition, we performed competition assays using Cy5-labeled SELEX-DNA (Figure 1C), and either unlabeled SELEX-DNA or unlabeled nc-DNA as a competitor. Titration of the unlabeled SELEX-DNA competitor led to complete disappearance of the Cy5-SELEX-DNA-AT-BD complex (Figure 1C, top) whereas titration of nc-DNA was not able to efficiently compete off the Cy5-labeled SELEX-DNA (Figure 1C, bottom). Together this indicates a higher affinity, more stable complex with SELEX-DNA relative to nc-DNA.
To quantify the increase in affinity for the preferred sequence we utilized biolayer interferometry (BLI) (Figure 1D). Fast kinetics of binding precluded reliable fitting of kinetic parameters, however consistent Kds were obtained through steady state analysis of the level of response as a function of AT-BD concentration. This yielded Kd=4.9μM for nc-DNA and Kd=1.8μM for SELEX-DNA, or a ~3-fold greater affinity for the SELEX-DNA than for the non-consensus DNA sequence (Figure 1D).
Chemical shift mapping reveals a unique binding mode to SELEX-DNA
To gain insight in the molecular basis of complex formation with SELEX-DNA we utilized NMR spectroscopy. The complex was initially examined through chemical shift perturbation (CSP) experiments. Sequential 1H,15N-HSQC spectra were collected on 15N-AT-BD while titrating in either nc-DNA or SELEX-DNA. Both substrates led to substantial CSPs indicating binding (Supplementary Figure S2). Both titrations were in fast-to-intermediate exchange on the NMR timescale consistent with micromolar affinity. Notably, peaks corresponding to the BD and the AT-hook shifted concomitantly (i.e. in response to the same concentration of DNA), suggesting a multivalent contribution from both the BD and AT-hook to DNA binding, consistent with our previous observations[17].
Backbone resonances were assigned using standard triple resonance experiments (BMRB 28048, Supplementary Figure S3A). Plotting the normalized CSPs against residue number for both titrations reveals the residues that are directly and indirectly involved in binding to nc-DNA or SELEX-DNA (Figure 2A). In both titrations, we observed significant perturbations in residues corresponding to both the AT-hook and the BD. However, SELEX-DNA induced additional significant CSPs not seen with ncDNA. These include residues Lys1450, Leu1451, Ser1452, and Asn1454 . SELEX-DNA also led to substantially larger perturbations compared to nc-DNA for residues in the AT-hook (Lys1438, Lys1439, Gln1440 and Lys1442) and residues in the BD (Ser1490, Lys1492, Ile1501, Arg1502, Lys1509, His1517, Tyr1519 and Arg1520) (Figure 2A, bottom). Mapping the CSPs onto the previously solved structure, reveals a binding pocket which is formed by αA and the ZA loop for both nc-DNA and SELEX-DNA, with additional contributions by the very N-terminal end of αZ and the AB loops for SELEX-DNA (Figure 2B). This binding pocket is lined with basic residues that form a larger basic patch on the BD surface (Supplemental Figure S3B), consistent with what we previously observed[17]. Together, this data suggests a more extensive binding pocket and a more stable complex with SELEX-DNA compared to nc-DNA.
Figure 2. NMR reveals the molecular basis of enhanced binding to SELEX-DNA.

(A) Plots of normalized CSPs (Δδ) for AT-BD with SELEX-DNA (top), nc-DNA (middle), and the difference between the two (ΔΔδ, bottom). Residues perturbed more than the average plus two standard deviations (denoted by the gray line) are labeled. Where the average was calculated after trimming the top 10% of perturbations. “P” indicates a proline, “*” indicates an unassigned residue and “****” represent a signal that broadened beyond detection upon titration with ligand. The AT-BD secondary structure architecture is depicted on top of the plots for reference. (B) Residues in the AT-BD with significant CSPs upon binding to the SELEX-DNA (top) or nc-DNA (bottom) are plotted onto a cartoon representation of the previously solved structure of the BD (PDB ID 3UVD), with the AT-hook drawn in (a small break is left between the structure and the drawn-in segment).
While changes in the magnitude of CSPs between the two substrates can indicate stabilization of the complex if along the same trajectory, differences in the trajectory of CSPs indicate a unique bound state. Differences in the trajectory of CSPs between SELEX-DNA and nc-DNA were observed in several residues in the AT-hook, linker, and the BD indicating a unique binding mode at these residues. These included residues Lys1439, Lys1441, Arg1443, Lys1450 and Leu1451 in the AT-hook and linker and residues Thr1459, Tyr1498, and Tyr1519 in the BD (Figure 3).
Figure 3. NMR reveals unique binding to SELEX-DNA.

1H,15N-HSQC spectral overlays are shown for AT-BD residues saturated with either SELEX-DNA (red) or nc-DNA (salmon). Shown are resonances for residues which show a unique trajectory of CSP between the two substrates, indicating a unique binding mode. This was observed for residues in the bromodomain (Thr1459, Tyr1498, Tyr1519, Ser1490) the AT-hook (Lys1439, Lys1441, Lys1443) and the linker (Lys1450, Lys1451).
A similar analysis was carried out for the BD alone. Spectra of the 15N-BD construct saturated with nc-DNA or SELEX-DNA were overlaid. Chemical shift perturbations were generally smaller in magnitude for the BD alone as compared to the BD in the context of the AT-BD construct except at the C-terminal end (Supplementary FigureS4). This is consistent with the weaker binding seen in the EMSAs (Supplementary FigureS1). As was seen with the AT-BD, larger CSPs were induced in the BD resonances by SELEX-DNA (Supplementary Figure S5 ). However, in contrast to the AT-BD there were no perceptible changes in trajectories of CSPs between nc-DNA or SELEX-DNA (Supplementary Figure S5 ). Analysis of the CSPs as a function of substrate DNA concentration reveals that the BD alone binds ~10× weaker than the AT-BD (Supplementary Figure S5C), confirming that both the AT-hook and BD are needed for robust binding.
Together the CSP experiments suggest that both the AT-hook and BD contribute to the preference for SELEX-DNA. The AT-BD binds more robustly to both the nc-DNA and SELEX-DNA than the BD alone. However, t he increased magnitude of CSPs indicates that both the BD alone and AT-BD form a more stable complex with SELEX-DNA compared to nc-DNA. In addition, the additional CSPs and altered trajectories indicate that the AT-BD adopts a unique structure in binding to SELEX-DNA as compared to nc-DNA.
The AT-hook and BD span the minor and major grooves of SELEX-DNA
Having defined the DNA-binding interface of the protein, we sought to identify the protein-binding interface on DNA, also using NMR spectroscopy. Resonances for SELEX-DNA non-exchangeable (H2, H5, H6/H8, H1’, H2', H2") and exchangeable (iminos) protons were assigned (Supplementary Figure S6 ). Initial collection of 1D-proton spectra of SELEX-DNA upon titration of increasing amounts of AT-BD revealed CSPs indicating binding (Figure 4A, Supplementary Figure S7A). Notably, the most substantial CSPs were seen for the imino protons of A6, T7, T8, A9 and A10, which are in the core of the SELEX determined sequence.
Figure 4. The AT-BD spans the major and minor groove.

(A) CSPs (Δδ) of the imino resonances of the SELEX-DNA saturated with AT-BD. “*” indicates an unassigned or not visible resonance. (B) Table of Intermolecular NOEs observed between the 13C-AT-BD and 12C-SELEX-DNA. (C) Schematic of AT-hook and BD contacts with SELEX-DNA (strands 1 and 2 labeled) in the major (left) and minor (right) grooves. Contacts are highlighted by red lines. The core SELEX determined sequence is in bold. (D) structure of an AT base pair with hydrogens forming NOEs labeled, as well as the major and minor groove sides. (E) Representative NOE cross-peaks between AT-BD and SELEX-DNA indicate that these protons are <6Å from each other in space.
We then measured intermolecular NOEs (Nuclear Overhauser Enhancements) to detect proton-proton distances of <6 Å between the AT-BD and SELEX-DNA via an isotope edited/filtered approach using 13C/15N-labeled AT-BD and unlabeled SELEX-DNA. In this experiment, the intermolecular NOE cross-peaks between 13C-H protons of AT-BD and 12C-H protons of DNA were detected. Side chain assignments of the AT-BD were made by tracking the chemical shift in a 1H,13C-HMQC upon titration of SELEX-DNA (Supplementary Figure S7B).
Several intermolecular NOEs were observed between the AT-hook and adenines in SELEX-DNA sequence (Figure 4 and Supplementary Figure S7C). Specifically, NOEs were seen between the Hδ proton of Arg1443 in the AT-hook and the H2 protons of both 1A6 and 2A9 in SELEX-DNA, where 1 and 2 refer to the forward and reverse strand respectively (Figure 4B,C,E). Similarly, we observed NOEs between Arg1445 Hδ proton of the AT-hook and the H2 protons of 1A9 and 2A8. Consistent with this, we also observed NOEs between Arg1443 Hδ protons and HI’ protons of both 1T7 and 2T10 and between Arg1445 Hδ protons the HI’ protons of 1A9 and 2A8 (Figure 4B,C and Supplementary Figure S7C). In B-form DNA, H1’ protons and adenine H2 protons are most easily accessible through the minor groove, indicating that the AT-hook is inserting into the minor groove (Figure 4D) [19, 48]. Notably, in 1H,13C-HMQC spectra of AT-BD single peaks are observed for the Hδ protons of Arg1443 and Arg1445, but upon titration of SELEX-DNA these peaks split into two. This suggests a fixed position of these arginines upon binding to DNA (Supplementary Figure S7B). This is consistent with a canonical AT-hook binding mode, in which the narrowed minor groove of AT-rich DNA is favored. Indeed, evidence of minor groove narrowing in the apo SELEX-DNA is supported by an inter-strand NOE observed in the homonuclear NOESY spectrum between 2T7 H1’ and 1A10 H2 protons (Supplementary Figure S6 F) [48,49].
In addition, NOEs were observed between the BD and SELEX-DNA (Figure 4 and Supplementary Figure S7 C). These NOE cross-peaks were observed to the methyl groups of 2T5, 2T6, and 2T7, indicating that the BD is interacting in the major groove (Figure 4B,C,E and Supplementary Figure S7 C). The corresponding protein chemical shifts for these NOE cross-peaks are consistent with Hδ/Hε protons of two tyrosine residues. Unfortunately, these tyrosines were not observed in either the HCCONH nor the CCONH experiments, and thus they could not be definitely assigned. However, only two tyrosines experienced significant CSPs in the 15N-HSQC upon titration of SELEX-DNA into AT-BD, and only three tyrosines are in the binding pocket. These are Tyr1497 and Tyr1498 in the ZA loop and Tyr1519 at the end of the αA helix (see Figure 2). This strongly suggests that two of these three tyrosine residues are making contact with the SELEX-DNA major groove, and thus at least one must be a Tyr in the ZA-loop. Though these tyrosines could not be assigned, given the positioning of the AT-hook, one tyrosine should be adjacent to 2T6 and 2T7, and another should be adjacent to 2T5. An additional NOE was seen between a tyrosine Hδ proton and the H6 protons of 2T6 and 2T7 (Figure 4B,C and Supplementary Figure S7 C).
Together, the NOEs reveal that the AT-hook and BD span the minor and major grooves. The NOEs are consistent with the Arg-Gly-Arg (Arg1443, Gly1444, Arg1445) of the AT-hook inserting into the minor groove of the narrowed AT-rich segment. In addition, the NOEs between the BD and DNA would be consistent with two aromatic/methyl interactions between tyrosines and thymines, a common major groove mode of binding. At least one of these interactions is being formed with the ZA loop with an additional interaction in the ZA loop or αA helix. The NOEs are consistent with the binding pocket identified by CSPs (Figure 2). The CSPs suggest further electrostatic interactions between basic residues in both the AT-hook and BD and SELEX-DNA. This is supported by the detection of several additional NOEs that would be consistent with Lys/DNA interactions, however substantial spectral overlap precluded our ability to resolve these.
Molecular modeling reveals the importance of the linker in AT-BD binding
To better understand how the AT-BD docks and binds SELEX-DNA, a hierarchy of computational methods were used to model the complex. Initial complexes of AT-BD with SELEX-DNA were obtained using the data-driven docking program HADDOCK[50], which is a flexible docking approach that imposes distance restraints based on experimentally defined binding interfaces. Unambiguous interaction restraints on the AT-BD were assigned based on CSP analysis (see Figure 2), while no restraints were imposed on the DNA. The structures with the top-ten highest HADDOCK Z-score were selected for further analysis (see Supplementary Figure S8). These poses were used as starting structures for triplicate all-atom molecular dynamics (MD) simulations of 100 ns in length.
Several binding modes of the AT-BD along the SELEX-DNA were obtained in the initial ten docked structures (see Supplementary Figure S8), and the observed binding modes observed at the end of the 100 ns simulations were similar to their starting conformations. Of these, five had the AT-hook bound to the SELEX-DNA minor groove and the BD in proximity of the major groove consistent with the NOE data. In the other conformations the AT-hook was bound to the DNA major groove with ocA in either the major or minor groove, or the AT-hook bound in the minor groove, but inconsistent with the experimental data. Notably, these were also predicted to be the least favorable based on docking score. As such, these five binding modes were discarded from further analysis, reducing the number of binding conformations from ten to five.
MD simulations were rerun on the remaining five conformations using a multi-step restraint-enabled refinement approach, followed by 500 ns of unrestrained MD simulations (Supplementary Figure S9 A). In this step the restraints were based on experimental NOEs. At the end of these runs, three of the simulations had the AT-hook consistently bound in the DNA minor groove while in the other two it was associated in the major groove. The two binding modes with the AT-hook bound to the major groove were discarded and further analysis was performed on the three states with the AT-hook bound to the minor groove. Notably, two of the three are not fully consistent with the NMR data. In binding mode 2 the AT-hook is associated with, but not inserted into the minor groove, while in binding mode 3 it is positioned in a reverse manner in the minor groove (Supplementary Figure S9C,D ). The position of the AT-hook and BD in binding mode 1 however, is overall consistent with the experimental data (Figure 5A,C).
Figure 5. Molecular model of the AT-BD/SELEX-DNA complex.

(A) Model of AT-BD/SELEX-DNA complex. The AT-hook is highlighted in blue, the αA in deep sky blue, the ZA loop in ice blue and the linker in cyan. The DNA and the rest of the BRG1 BD are in gray. In this binding mode (mode 1) the AT-hook is positioned in the minor groove and the ZA loop the major groove, consistent with NOE data. (B) A zoom in of the linker shows a significant bend induced at in the polypeptide backbone at Pro1456. (C) A zoom in shows the AT-hook arginines (1443 and 1445) inserted into the minor groove. Carbons are colored in cyan, oxygens in red and nitrogens in blue. (D) The minimum distance between AT-BD elements and SELEX-DNA. (E) The number of hydrogen bonds between AT-BD elements and SELEX-DNA in the percentage of frames during the 500 ns simulations.
The overall stability of the binding modes was assessed by calculating the root mean square deviation (RMSD) of AT-BD relative to SELEX-DNA. The average RMSD for all three binding modes was ≤ 4 Å during the simulations, with the exception of binding mode 2 in which there was an increase in the final 50 ns that corresponded to a slight twisting of αA relative to the DNA (Supplementary Figure S9 B). However, only in binding mode 1 are both the AT-hook and BD associated stably with the DNA throughout the simulation.
To further investigate the binding mode, minimum distances between the DNA and AT-BD elements were calculated for each binding mode. Our results show that the minimum distance between the AT-hook and DNA during the 500 ns simulation (as measured between any protein heavy atom and any DNA heavy atom) is stable at 1.76 ± 0.08 Å for all three binding modes. For the BD, in binding mode 1, the distance between αA and the DNA is stable at 1.81± 0.13 Å and between the ZA loop and DNA is stable at 1.79 ± 0.09 Å over the course of the 500 ns simulations (Figure 5D). In binding modes 2 and 3 the BD is far more dynamic with the DNA/αA distance fluctuating from 1.58 Å to 5.03 Å, and the DNA/ZA loop distance fluctuating from 1.46 Å to 9.55 Å (Supplementary Figure S9 C,D). DNA base-pair parameters for both DNA free in solution and bound to the AT-BD were calculated to quantify the effects of binding on the DNA. Results showed little differences in the base-pairs parameters in all simulations, suggesting that AT-BD binding does not change the geometry of the double helix (results not shown).
Contact analysis showed that AT-hook and BD made significant contacts with the DNA in all three simulations. The highest contact frequency is observed for Lys1442, Arg1443, Gly1444 and Ala1448 in the AT-hook consistent with NMR CSPs. Analysis of hydrogen bonding between the DNA and residues in the AT-hook, αA and ZA loop demonstrate that the AT-hook remains stably bound to the DNA. In contrast, the BD interactions are more transient (Figure 5E and Supplementary Figure S9 C,D). For binding mode 1 the average number of hydrogen bonds formed between the DNA and AT-hook is the highest, 5.60 ± 1.76, whereas otA and the ZA loop formed fewer hydrogen bonds at 2.76 ± 1.26 and 2.72 ± 1.46 respectively. Similar trends are seen for binding modes 2 and 3 (Supplementary Figure S9 C,D).
Notably, the model indicates that the composition of the linker between the AT-hook and BD is critical for spanning the major and minor grooves. From the AT-hook, the linker is seen to wrap around one strand of the SELEX-DNA allowing the BD ZA loop to insert into the major groove. The conformation of Pro1456 in the linker adopts a unique conformation (Figure 5B and Supplementary Figure S9 E) and provides a critical turn in the polypeptide necessary for both the AT-hook and BD to associate with both the major and minor grooves. This is consistent with the CSP analysis, in which the linker resonances were uniquely shifted for SELEX-DNA.
Together, simulation results show that the AT-hook and the BD can bind to the DNA in different locations. Though only one of these conformations is fully consistent with the experimental data, the others may be representative of minor populated states. Both minimum distance calculations between the DNA and AT-BD, and hydrogen bond analysis show that the complex is driven by interactions of the AT-hook, αA and ZA loop with DNA. The AT-hook interaction is the most stable while the BD interaction is more transient. Finally, the simulations reveal that the linker is critical in mediating the multivalent interaction of the AT-hook and BD with SELEX-DNA.
Cancer mutations alter the mode of AT-BD DNA binding
It is estimated that 20% of human tumors present a mutation in at least one of the subunits of BAF[8]. As seen in the cBio Cancer Genomics Portal several point and truncation mutations have been identified in the DNA binding pocket of the BRG1 AT-BD[51,52] in a broad range of cancers. We investigated six of these mutations to determine the effect on DNA binding: R1445W in the AT-hook, P1456L in the linker, R1502H and P1504L in the ZA loop, and R1515H in the αA helix. In addition, we examined R1520H in the AB loop. To investigate global DNA association, EMSAs were carried out with the 147 bp Widom 601 DNA naked or formed into unmodified nucleosome core particles (NCPs) and each of the six mutant AT-BD constructs (Supplementary Figure S10). Notably, the 601 sequence includes four AT-rich regions. Analysis of the fraction bound by way of quantitating the remaining unbound state, revealed that mutants P1456L (linker) and R1502H (ZA loop) have abrogated binding . Notably, almost all mutants show some change in the laddering pattern observed upon binding to the DNA or NCPs, with the exception of P1504L. Similar results were seen for both naked and NCP DNA. Together, this indicates that excepting P1504L, all mutants tested have some effect on the mode of AT-BD DNA binding.
We further investigated P1456L (linker) and R1502H (ZA loop) using EMSA, NMR spectroscopy, and BLI. Binding affinities for SELEX-DNA were determined by BLI and yielded only small changes as compared to wild type (WT): Kd=1.2 μM±0.1 for P1456L, Kd=2.4±0.2 μM for R1502H, Kd= 1.8 μM±0.2 WT (see Supplementary Figure S11A). In addition, competitive EMSAs indicate that the mutants still have moderate preference for SELEX-DNA (Supplementary Figure S11B). However, changes seen in the EMSAs with the 601 DNA (Supplementary Figure S10 ) suggest that the disruption in binding mode leads to more substantial defects on longer pieces of DNA. To assess if either mutation affected complex formation, 1H,15N-HSQC spectra were collected on both mutants. Comparing the mutant spectra to WT revealed that both mutants retain a similar overall fold (Supplementary Figure S12 ). To investigate differences in DNA binding by the mutants as compared to WT, sequential 1H,15N-HSQC spectra were collected on 15N-labeled mutants upon addition of increasing amounts of SELEX-DNA. Both mutants show differences in CSPs around the mutation as expected. However, for both mutants, differences are also seen in the magnitude of CSPs throughout the AT-hook and BD (Figure 6 and Supplementary Figure S13) indicating a change in the mode of DNA binding. Notably, these were most pronounced for the linker mutation, which is consistent with the model that predicts that the linker composition is important in allowing the AT-hook and BD to span the major and minor grooves.
Figure 6. Cancer mutations alter the mode of DNA binding.
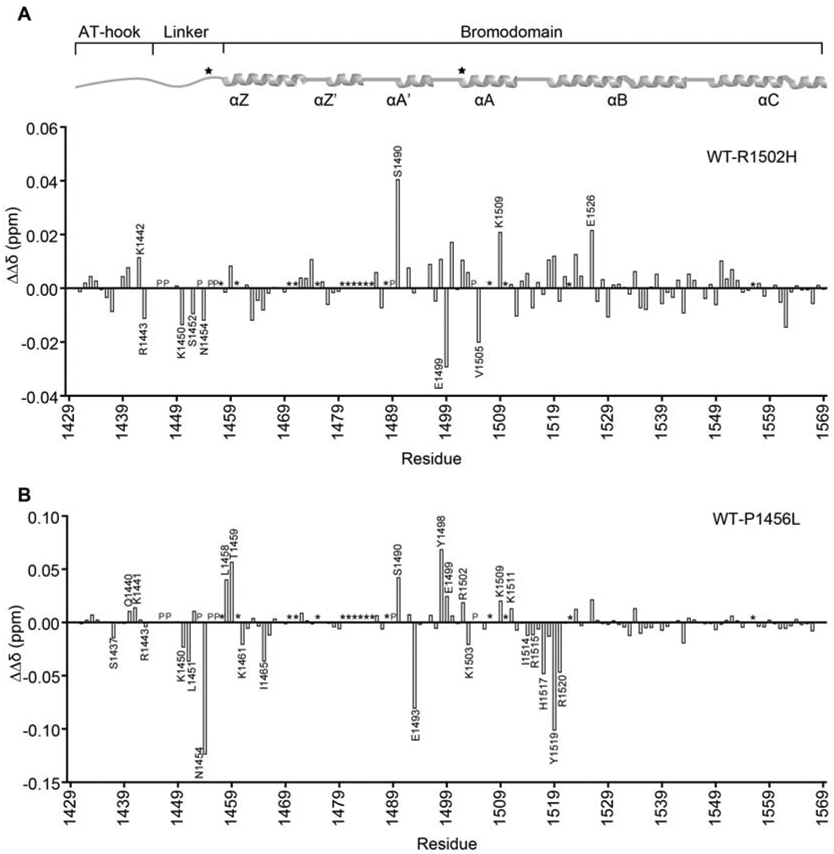
Differences in the magnitude of CSPs (ΔΔδ) for binding of AT-BD to SELEX-DNA were computed between wild-type (WT) and the R1502H (A) or P1456L (B) mutants. Residues which show substantially different binding are marked. “P” denotes proline and “*” denotes unassigned residues. The AT-BD secondary structure architecture is depicted on top of the plots for reference with mutation sites marked with a star.
Discussion
Here we have identified a preferred consensus sequence for the AT-hook and bromodomain of BRG1 and have determined the molecular basis for DNA binding and selectivity. We show that the AT-hook and bromodomain span the minor and major grooves of the preferred sequence, with the AT-hook inserted into the minor groove and the bromodomain ZA loop inserting into the major groove. This binding mode is reminiscent of HOX proteins, in which a homeodomain inserts into the major groove and the nearby disordered “N-terminal arm”, often arginine rich, associates in the adjacent minor groove[53]. Sequence selectivity appears to arise largely from shape recognition of the DNA by the AT-hook with additional base specific contacts by the BD. In particular, the narrowing of the minor groove characteristic of AT-rich sequences leads to electrostatic focusing, which is known to enhance the affinity of arginines for DNA [26,54]. Thus, the AT-hook has preference for this shape. In addition, two base specific contacts were detected between tyrosines in the BD and thymines in the major groove that help stabilize interaction with the consensus sequence. Notably, the linker composition appears critical for this binding conformation. Indeed mutation of the linker leads to changes in both the AT-hook and BD binding.
AT-hooks are known drivers of the interaction of HMG proteins with DNA [55]. They have also been found in other transcription factors such as ELF3, in which an AT-hook adjacent to the ETS specificity domain is required for function[56]. For BRG1 the AT-hook and bromodomain appear to be structurally and functionally coupled and act in a composite manner to associate specifically with DNA. Here the AT-hook is not simply an accessory motif, neither is it solely defining DNA binding. Notably, there is substantial variation in the target sequence of AT-hooks, likely arising not only from differences in motif sequence, but also from the surrounding protein context.
AT-hooks are found in a number of chromatin associated proteins. Of the 61 human bromodomain-containing proteins, we found that 14 also contain at least one canonical GRP containing AT-hook. Of these, the bromodomain of ASH1L is predicted to bind nucleic acid[17]. In addition, several other reader domains (e.g. chromodomains and PWWP domains) that have been identified to bind nucleic acid contain adjacent canonical AT-hooks, AT-like-hooks, extended AT-hooks, or extensions rich in basic residues [57]. Thus, this multivalent mode of interaction may be common. However, additional studies would need to be done to determine the sequence selectivity of these motifs and the molecular basis of binding.
Though we have shown that the AT-BD can associate with both linear DNA and DNA formed into the nucleosome particle, we do not yet know the effect of nucleosome structure on the mode of DNA binding. For most transcription factors nucleosome wrapping substantially inhibits association with DNA both by inhibiting association and enhancing dissociation [58,59]. However, for others, binding to nucleosomes is as efficient as naked DNA, and some even prefer nucleosomes [60,61] It is not fully clear how this is achieved mechanistically but it has recently been shown that this is due to decreased dissociation rates[62] and for some may include stabilizing contacts with the histones[63]. Our previous work revealed that concomitant binding of DNA and H3K14ac histone tails by the BD is possible, suggesting that the AT-BD may participate in dual histone/DNA binding at acetylated nucleosomes[17]. A recent cryo-EM structure of the BAF complex bound to a nucleosome does not resolve the AT-BD. However, based on the position of BRG1 in this complex, the AT-BD would be positioned near the entry-exit DNA, and notably near the H3 tail[64]. This suggests that at least part of the time the AT-BD is associating with linear linker DNA, though it does not preclude additional interactions with nucleosomal DNA.
Further work is also necessary to reveal how post-translational modification of the AT-BD may regulate the interaction with DNA. BRG1 is known to be posttranslationally modified, including in and around the AT-BD region. This includes phosphorylation and acetylation. Phosphorylation would likely inhibit interaction with DNA, whereas acetylation may both inhibit interaction, as well as regulate AT-BD function through intramolecular interaction with the BD [65-67].
The results presented here provide insight into the mechanisms of DNA binding of this composite domain and lay the groundwork for determining its role in BAF function. In addition, they provide insight into how cancer mutations may alter this function. It remains to be determined exactly how the AT-BD DNA binding activity contributes to the overall function of the BAF complex. Chromatin remodeling activity is known to be affected by remodeling complex composition and chromatin status[68,69]. The latter includes histone modifications, and DNA sequence. The CHD1 remodeling complex as well as the yeast RSC complex (BAF homologue) are known to be stimulated by AT-rich DNA[69,70]. In the case of CHD1 this is due in part to an AT-hook. Though the sequence preference is not substantial these subtle differences lead to changes in the release of the nucleosome leading to unique positioning by each remodeling complex and thus orchestrated chromatin structural changes. Thus, the activity of the BRG1 AT-BD likely plays a role in recognizing the underlying DNA sequence, fine-tuning the affinity of the BAF complex for nucleosomes. Such fine-tuning would not require high sequence selectivity, which rather might be detrimental to global chromatin remodeling function. This fine-tuning could be further regulated by recognition of acetylated histone tails by the BD. In addition to nucleosome affinity, AT-BD DNA binding may play a role direct role in the chromatin remodeling , through regulation of ATPase or translocase activity. These regulatory activities may be more important at some regions of chromatin than others leading to the context dependent effects.
Supplementary Material
Highlights.
The BRG1 AT-BD bind multivalently to DNA
The BRG1 AT-BD is selective for AATTAAAT containing sequences
The BRG1 AT-BD span the minor and major grooves of DNA
Acknowledgements
We would like to thank Lokesh Gakhar for his assistance in experimental design and data analysis, Chris Ptak, Madeline Shea, Adina M. Kilpatrick, and Ryan Mhaling for many thoughtful discussions of data.
Funding
This project was supported by The Holden Comprehensive Cancer Center at The University of Iowa and its National Cancer Institute Award P30CA086862. Work in the Musselman Lab is funded by the National Institutes of Health (GM128705) and the National Science Foundation (CAREER-1452411). J.C.S and M.N.H received funding through the interdisciplinary Institutional Training Grant in Pharmacological Sciences (T32 GM067795). Work in the Wereszczynski group is funded by the National Institutes of Health (1R35GM119647) and used the Extreme Science and Engineering Discovery Environment, which is supported by the National Science Foundation (ACI-1053575). Work in the Pufall Lab is funded by the National Science Foundation (CAREER-1552862).
Footnotes
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ, Crystal structure of the nucleosome core particle at 2.8 Å resolution, Nature. 389 (1997) 251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- [2].Wu JI, Lessard J, Crabtree GR, Understanding the Words of Chromatin Regulation, Cell. 136 (2009) 200–206. doi: 10.1016/j.cell.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Alpsoy A, Dykhuizen EC, Glioma tumor suppressor candidate region gene 1 (GLTSCR1) and its paralog GLTSCR1-like form SWI/SNF chromatin remodeling subcomplexes, J Biol Chem. 293 (2018) 3892–3903. doi: 10.1074/jbc.ra117.001065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Gatchalian J, Malik S, Ho J, Lee D-S, Kelso TWR, Shokhirev MN, Dixon JR, Hargreaves DC, A non-canonical BRD9-containing BAF chromatin remodeling complex regulates naive pluripotency in mouse embryonic stem cells., Nature Communications. 9 (2018) 5139. doi: 10.1038/s41467-018-07528-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Alfert A, Moreno N, Kerl K, The BAF complex in development and disease, Epigenet Chromatin. 12 (2019) 19. doi: 10.1186/s13072-019-0264-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Narlikar GJ, Sundaramoorthy R, Owen-Hughes T, Mechanisms and functions of ATP-dependent chromatin-remodeling enzymes., Cell. 154 (2013) 490–503. doi: 10.1016/j.cell.2013.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hargreaves DC, Crabtree GR, ATP-dependent chromatin remodeling: genetics, genomics and mechanisms., Cell Research. 21 (2011) 396–420. doi: 10.1038/cr.2011.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kadoch C, Crabtree GR, Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics., Sci Adv. 1 (2015) el500447. doi: 10.1126/sciadv.1500447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ho L, Crabtree GR, Chromatin remodelling during development : Article : Nature, Nature. (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Helming KC, Wang X, Roberts CWM, Vulnerabilities of mutant SWI/SNF complexes in cancer., Cancer Cell. 26 (2014) 309–317. doi: 10.1016/j.ccr.2014.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kadam S, Emerson BM, Transcriptional Specificity of Human SWI/SNF BRG1 and BRM Chromatin Remodeling Complexes, Molecular Cell. 11 (2003) 377–389. doi: 10.1016/sl097-2765(03)00034-0. [DOI] [PubMed] [Google Scholar]
- [12].Flowers S, Nagl NG, Beck GR, Moran E, Antagonistic roles for BRM and BRG1 SWI/SNF complexes in differentiation., J Biological Chem. 284 (2009) 10067–75. doi: 10.1074/jbc.m808782200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bultman S, Gebuhr T, Yee D, Mantia CL, Nicholson J, Gilliam A, Randazzo F, Metzger D, Chambon P, Crabtree G, Magnuson T, A Brg1 null mutation in the mouse reveals functional differences among mammalian SWI/SNF complexes., Molecular Cell. 6 (2000) 1287–1295. [DOI] [PubMed] [Google Scholar]
- [14].Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert J-P, Barsyte-Lovejoy D, Felletar I, Volkmer R, Muller S, Pawson T, Gingras A-C, Arrowsmith CH, Knapp S, Histone recognition and large-scale structural analysis of the human bromodomain family., Cell. 149 (2012) 214–231. doi: 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Singh M, Popowicz GM, Krajewski M, Holak TA, Structural Ramification for Acetyl-Lysine Recognition by the Bromodomain of Human BRG1 Protein, a Central ATPase of the SWI/SNF Remodeling Complex, ChemBioChem. 8 (2007) 1308–1316. doi: 10.1002/cbic.200600562. [DOI] [PubMed] [Google Scholar]
- [16].Shen W, Xu C, Huang W, Zhang J, Carlson JE, Tu X, Wu J, Shi Y, Solution Structure of Human Brg1 Bromodomain and Its Specific Binding to Acetylated Histone Tails †,‡, Biochemistry. 46 (2007) 2100–2110. doi: 10.1021/bi0611208. [DOI] [PubMed] [Google Scholar]
- [17].Morrison EA, Sanchez JC, Ronan JL, Farrell DP, Varzavand K, Johnson JK, Gu BX, Crabtree GR, Musselman CA, DNA binding drives the association of BRGl/hBRM bromodomains with nucleosomes., Nature Communications. 8 (2017) 16080. doi: 10.1038/ncomms16080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Miller TCR, Simon B, Rybin V, Grötsch H, Curtet S, Khochbin S, Carlomagno T, Müller CW, A bromodomain-DNA interaction facilitates acetylation-dependent bivalent nucleosome recognition by the BET protein BRDT., Nature Communications. 7 (2016) 13855. doi: 10.1038/ncomms13855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Rahnamoun H, Lee J, Sun Z, Lu H, Ramsey KM, Komives EA, Lauberth SM, RNAs interact with BRD4 to promote enhanced chromatin engagement and transcription activation., Nature Structural & Molecular Biology. 25 (2018) 687–697. doi: 10.1038/s41594-018-0102-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Philpott M, Rogers CM, Yapp C, Wells C, Lambert J-P, Strain-Damerell C, Burgess-Brown NA, Gingras A-C, Knapp S, Müller S, Assessing cellular efficacy of bromodomain inhibitors using fluorescence recovery after photobleaching., Epigenetics & Chromatin. 7 (2014) 14. doi: 10.1186/1756-8935-7-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Fedorov O, Castex J, Tallant C, Owen DR, Martin S, Aldeghi M, Monteiro O, Filippakopoulos P, Picaud S, Trzupek JD, Gerstenberger BS, Bountra C, Willmann D, Wells C, Philpott M, Rogers C, Biggin PC, Brennan PE, Bunnage ME, Schule R, Gunther T, Knapp S, Muller S, Selective targeting of the BRG/PB1 bromodomains impairs embryonic and trophoblast stem cell maintenance., Science Advances. 1 (2015) e1500723. doi: 10.1126/sciadv.1500723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Trotter KW, Fan H-Y, Ivey ML, Kingston RE, Archer TK, The HSA Domain of BRG1 Mediates Critical Interactions Required for Glucocorticoid Receptor-Dependent Transcriptional Activation In Vivo, Mol Cell Biol. 28 (2007) 1413–1426. doi: 10.1128/mcb.01301-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Vangamudi B, Paul TA, Shah PK, Kost-Alimova M, Nottebaum L, Shi X, Zhan Y, Leo E, Mahadeshwar HS, Protopopov A, Futreal A, Tieu TN, Peoples M, Heffernan TP, Marszalek JR, Toniatti C, Petrocchi A, Verhelle D, Owen DR, Draetta G, Jones P, Palmer WS, Sharma S, Andersen JN, The SMARCA2/4 ATPase Domain Surpasses the Bromodomain as a Drug Target in SWI/SNF-Mutant Cancers: Insights from cDNA Rescue and PFI-3 Inhibitor Studies., Cancer Research. 75 (2015) 3865–3878. doi: 10.1158/0008-5472.can-14-3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Reeves R, Nissen MS, The A T-DNA-binding domain of mammalian high mobility group I chromosomal proteins. A novel peptide motif for recognizing DNA structure., J Biological Chem. 265 (1990) 8573–82. [PubMed] [Google Scholar]
- [25].Filarsky M, Zillner K, Araya I, Villar-Garea A, Merkl R, Langst G, Nemeth A, The extended AT-hook is a novel RNA binding motif, RNA Biology. 12 (2015) 864–876. doi: 10.1080/15476286.2015.1060394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Rohs R, West SM, Sosinsky A, Liu P, Mann RS, Honig B, The role of DNA shape in protein-DNA recognition, Nature. 461 (2009) 1248–1253. doi: 10.1038/nature08473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Reeves R, Structure and Function of the HMGI(Y) Family of Architectural Transcription Factors, Environ Health Persp. 108 (2000) 803–809. doi: 10.1289/ehp.00108s5803. [DOI] [PubMed] [Google Scholar]
- [28].Aravind L, Landsman D, AT-hook motifs identified in a wide variety of DNA-binding proteins., Nucleic Acids Research. 26 (1998) 4413–4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zhang L, Martini GD, Rube HT, Kribelbauer JF, Rastogi C, FitzPatrick VD, Houtman JC, Bussemaker HJ, Pufall MA, SelexGLM differentiates androgen and glucocorticoid receptor DNA-binding preference over an extended binding site., Genome Research. 28 (2018) 111–121. doi: 10.1101/gr.222844.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Delaglio F, Grzesiek S, Vuister G, Zhu G, Pfeifer J, Bax A, NMRPipe: A multidimensional spectral processing system based on UNIX pipes, Journal of Biomolecular NMR. 6 (1995). doi: 10.1007/bf00197809. [DOI] [PubMed] [Google Scholar]
- [31].Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas M, Ulrich EL, Markley JL, Ionides J, Laue ED, The CCPN data model for NMR spectroscopy: development of a software pipeline., Proteins. 59 (2005) 687–696. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- [32].Burgering M, Boelens R, Kaptein R, Observation of intersubunit NOEs in a dimeric P22 Mnt repressor mutant by a time-shared [15N, 13C] double half-filter technique, J Biomol Nmr. 3 (1993) 709–714. doi: 10.1007/bf00198373. [DOI] [Google Scholar]
- [33].Šali A, Blundell TL, Comparative Protein Modelling by Satisfaction of Spatial Restraints, J Mol Biol. 234 (1993) 779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- [34].van Dijk M, Bonvin AMJJ, 3D-DART: a DNA structure modelling server., Nucleic Acids Res. 37 (2009) W235–9. doi: 10.1093/nar/gkp287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].van Zundert GCP, Rodrigues JPGLM, Trellet M, Schmitz C, Kastritis PL, Karaca E, Melquiond ASJ, van Dijk M, de Vries SJ, Bonvin AMJJ, The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes., Journal of Molecular Biology. 428 (2016) 720–725. doi: 10.1016/j.jmb.2015.09.014. [DOI] [PubMed] [Google Scholar]
- [36].Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E, GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers, Softwarex. 1-2 (2015) 19–25. doi: 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- [37].Lindorff-Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO, Shaw DE, Improved side-chain torsion potentials for the Amber ff99SB protein force field., Proteins. 78 (2010) 1950–8. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Galindo-Murillo R, Robertson JC, Zgarbová M, Šponer J, Otyepka M, Jurecka P, Cheatham TE, Assessing the Current State of Amber Force Field Modifications for DNA., J Chem Theory Comput. 12 (2016) 4114–27. doi: 10.1021/acs.jctc.6b00186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Dans PD, Ivani I, Hospital A, Portella G, González C, Orozco M, How accurate are accurate force-fields for B-DNA?, Nucleic Acids Res. 45 (2017) 4217–4230. doi: 10.1093/nar/gkw1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML, Comparison of simple potential functions for simulating liquid water, The Journal of Chemical Physics. 79 (1983) 926–935. doi: 10.1016/0009-2614(80)85344-9. [DOI] [Google Scholar]
- [41].Parrinello M, Rahman A, Polymorphic transitions in single crystals: A new molecular dynamics method, J Appl Phys. 52 (1981) 7182–7190. doi: 10.1063/1.328693. [DOI] [Google Scholar]
- [42].Darden T, York D, Pedersen L, Particle mesh Ewald: An N ·log( N ) method for Ewald sums in large systems, J Chem Phys. 98 (1993) 10089–10092. doi: 10.1063/1.464397. [DOI] [Google Scholar]
- [43].Humphrey W, Dalke A, Schulten K, VMD: Visual molecular dynamics, J Mol Graphics. 14 (1996) 33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- [44].Lu X, Olson WK, 3DNA: a software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures, Nucleic Acids Res. 31 (2003) 5108–5121. doi: 10.1093/nar/gkg680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kumar R, Grubmuller H, do_x3dna: a tool to analyze structural fluctuations of dsDNA or dsRNA from molecular dynamics simulations., Bioinform Oxf Engl. 31 (2015) 2583–5. doi: 10.1093/bioinformatics/btv190. [DOI] [PubMed] [Google Scholar]
- [46].Riley TR, Slattery M, Abe N, Rastogi C, Liu D, Mann RS, Bussemaker HJ, SELEX-seq: a method for characterizing the complete repertoire of binding site preferences for transcription factor complexes, Methods in Molecular Biology (Clifton, N.J.). 1196 (2014) 255–278. doi: 10.1007/978-1-4939-1242-l_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Foat BC, Morozov AV, Bussemaker HJ, Statistical mechanical modeling of genome-wide transcription factor occupancy data by MatrixREDUCE, Bioinformatics. 22 (2006) e141–el49. doi: 10.1093/bioinformatics/btl223. [DOI] [PubMed] [Google Scholar]
- [48].Sarma RH, Sarma MH, Dai L, Umemoto K, GC rich DNA oligonucleotides with narrow minor groove width, Febs Lett. 418 (1997) 76–82. doi: 10.1016/s0014-5793(97)01351-3. [DOI] [PubMed] [Google Scholar]
- [49].Searle MS, Embrey KJ, Sequence-specific interaction of Hoechst 33258 with the minor groove of an adenine-tract DNA duplex studied in solution by 1H NMR spectroscopy., Nucleic Acids Res. 18 (1990) 3753–62. doi: 10.1093/nar/18.13.3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Dominguez C, Boelens R, Bonvin AMJJ, HADDOCK: A Protein-Protein Docking Approach Based on Biochemical or Biophysical Information, Journal of the American Chemical Society. 125 (2003) 1731–1737. doi: 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- [51].Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data, Cancer Discovery. (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N, Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal., Science Signaling. 6 (2013) pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Laughon A, DNA binding specificity of homeodomains, Biochemistry-Us. 30 (1991) 11357–11367. doi: 10.1021/bi00112a001. [DOI] [PubMed] [Google Scholar]
- [54].Joshi R, Passner JM, Rohs R, Jain R, Sosinsky A, Crickmore MA, Jacob V, Aggarwal AK, Honig B, Mann RS, Functional Specificity of a Hox Protein Mediated by the Recognition of Minor Groove Structure, Cell. 131 (2007) 530–543. doi: 10.1016/j.cell.2007.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Reeves R, Structure and Function of the HMGI(Y) Family of Architectural Transcription Factors, Environmental Health Perspectives. 108 (2000) 803. doi: 10.2307/3454310. [DOI] [PubMed] [Google Scholar]
- [56].Kopp JL, Wilder PJ, Desler M, Kinarsky L, Rizzino A, Different Domains of the Transcription Factor ELF3 Are Required in a Promoter-specific Manner and Multiple Domains Control Its Binding to DNA, J Biol Chem. 282 (2007) 3027–3041. doi: 10.1074/jbc.m609907200. [DOI] [PubMed] [Google Scholar]
- [57].Weaver T, Morrison E, Musselman C, Reading More than Histones: The Prevalence of Nucleic Acid Binding among Reader Domains, Molecules. 23 (2018) 2614–25. doi: 10.3390/molecules23102614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Turner B, Chromatin: Structure and Function, third edition, Alan Wolffe, Chromosome Res. 8 (2000) 457–458. doi: 10.1023/a:1009223202157. [DOI] [Google Scholar]
- [59].Luo Y, North JA, Rose SD, Poirier MG, Nucleosomes accelerate transcription factor dissociation., Nucleic Acids Research. 42 (2014) 3017–3027. doi: 10.1093/nar/gkt1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Zhu F, Farnung L, Kaasinen E, Sahu B, Yin Y, Wei B, Dodonova SO, Nitta KR, Morgunova E, Taipale M, Cramer P, Taipale J, The interaction landscape between transcription factors and the nucleosome, Nature. 562 (2018) 76–81. doi: 10.1038/s41586-018-0549-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Garcia MF, Moore CD, Schulz KN, Alberto O, Donague G, Harrison MM, Zhu H, Zaret KS, Structural Features of Transcription Factors Associating with Nucleosome Binding., Mol Cell. 75 (2019) 921–932.e6. doi: 10.1016/j.molcel.2019.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Donovan BT, Chen H, Jipa C, Bai L, Poirier MG, Dissociation rate compensation mechanism for budding yeast pioneer transcription factors., ELife. 8 (2019). doi: 10.7554/elife.43008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Zaret KS, Mango SE, Pioneer transcription factors, chromatin dynamics, and cell fate control., Curr Opin Genet Dev. 37 (2016) 76–81. doi: 10.1016/j.gde.2015.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].He S, Wu Z, Tian Y, Yu Z, Yu J, Wang X, Li J, Liu B, Xu Y, Structure of nucleosome-bound human BAF complex., Sci New York N Y. 367 (2020) 875–881. doi: 10.1126/science.aaz9761. [DOI] [PubMed] [Google Scholar]
- [65].Bourachot B, Growth inhibition by the mammalian SWI-SNF subunit Brm is regulated by acetylation, The EMBO Journal. 22 (2003) 6505–6515. doi: 10.1093/emboj/cdg621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M, Lysine acetylation targets protein complexes and co-regulates major cellular functions., Science. 325 (2009) 834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- [67].Muchardt C, Reyes JC, Bourachot B, Leguoy E, Yaniv M, The hbrm and BRG-1 proteins, components of the human SNF/SWI complex, are phosphorylated and excluded from the condensed chromosomes during mitosis., Embo J. 15 (1996) 3394–3402. doi: 10.1002/j.1460-2075.1996.tb00705.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Rippe K, Schrader A, Riede P, Strohner R, Lehmann E, Längst G, DNA sequence- and conformation-directed positioning of nucleosomes by chromatin-remodeling complexes., Proceedings of the National Academy of Sciences of the United States of America. 104 (2007) 15635–15640. doi: 10.1073/pnas.0702430104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Lorch Y, Maier-Davis B, Kornberg RD, Role of DNA sequence in chromatin remodeling and the formation of nucleosome-free regions., Gene Dev. 28 (2014) 2492–7. doi: 10.1101/gad.250704.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Stokes DG, Perry RP, DNA-binding and chromatin localization properties of CHD1., Molecular and Cellular Biology. 15 (1995) 2745–2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.