

Abstract
Reduced anionic flavin adenine dinucleotide (FADH−) is the critical cofactor in DNA photolyase (PL) for the repair of cyclobutane pyrimidine dimers (CPD) in UV-damaged DNA. The initial step involves photoinduced electron transfer from *FADH− to the CPD. The adenine (Ade) moiety is nearly stacked with the flavin ring, an unusual conformation compared to other FAD-dependent proteins. The role of this proximity has not been unequivocally elucidated. Some studies suggest that Ade is a radical intermediate, but others conclude that Ade modulates the electron transfer rate constant (kET) through superexchange. No study has succeeded in removing or modifying this Ade to test these hypotheses. Here, FAD analogs containing either an ethano- or etheno-bridged Ade between the AN1 and AN6 atoms (e-FAD and ε-FAD, respectively) were used to reconstitute apo-PL, giving e-PL and ε-PL respectively. The reconstitution yield of e-PL was very poor, suggesting that the hydrophobicity of the ethano group prevented its uptake, while ε-PL showed 50% reconstitution yield. The substrate binding constants for ε-PL and rPL were identical. ε-PL showed a 15% higher steady-state repair yield compared to FAD-reconstituted photolyase (rPL). The acceleration of repair in ε-PL is discussed in terms of an ε-Ade radical intermediate vs superexchange mechanism.
INTRODUCTION
It has been well established that DNA is damaged by UV light (1,2). The predominant UV lesion is the cyclobutylpyrimidine dimer or CPD, with a second pyrimidine dimer, the (6-4) photolesion, also formed in significant yield (3). This damage, if not repaired, could lead to termination or errors in replication leading to mutations. Consequently, DNA repair proteins have evolved so that the cell can continue its normal activities. In humans, nucleotide excision repair (NER) enzymes excise these UV lesions (4). In bacteria, archaea and many eukaryotes, CPDs can be repaired without excision by an enzyme called DNA photolyase (5), a flavoprotein that utilizes flavin adenine dinucleotide as the catalytic chromophore (6) (see Scheme 1, top). In its fully reduced anionic form, FADH− absorbs blue-green light, resulting in ultrafast photoinduced electron transfer to the CPD (bound in a light-independent step) to affect repair within 2 nanoseconds (7). It is interesting to note that there is a separate photolyase for the repair of the 6–4 photolesion (8) due to structural and chemical differences between the 6–4 photodimer and the CPD. However, both photolyases employ FAD and share significant sequence and structural homology (9).
Scheme 1.
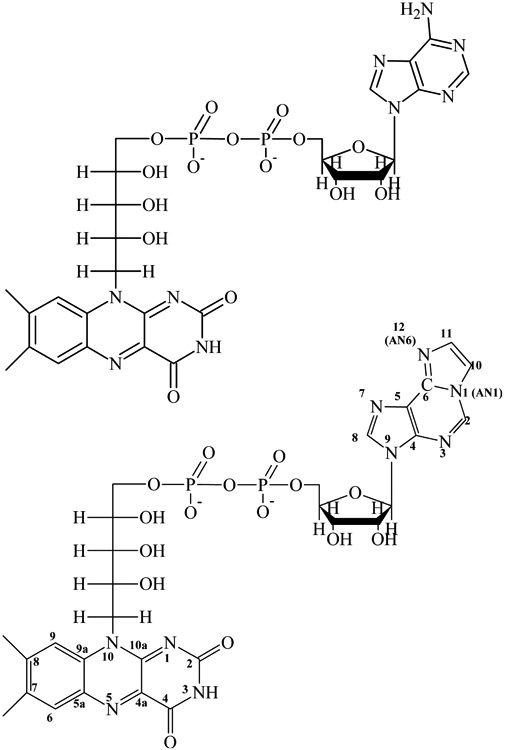
Structures of FAD and etheno-FAD (ε-FAD), showing the differences in the adenine and ε-Ade structures. Numbering of the adenines is based on the purine system (77).
Since the discovery and elucidation of photolyase structure and function, there has been a steady stream of reports about other flavoproteins that use blue light for different photobiological processes. Some of these proteins, such as the phototropins (10), use flavin mononucleotide (FMN) as the light absorbing cofactor. Many of these undergo reversible covalent photochemistry as part of their catalytic or signaling cycle (11). Others use FAD (e.g. AppA (12), one of many BLUF domains (13,14), that regulates of the synthesis of photosynthetic proteins).
An intriguing difference between these proteins and the photolyases (and their cryptochrome (CRY) homolog (15)) is the conformation of the FAD in the flavin binding pocket. In photolyase (16) and CRY (15,17), the Ade moiety is nearly stacked on the flavin (or isoalloxazine) ring system in a “U” conformation (see Fig. 1, left. From Anacystis nidulans ITEZ.PDB (18), chain C). Anacystis nidulans PL was chosen because it is the best characterized protein:substrate crystal structure available. In this view, the Ade is center stage surrounded by a chorus of water molecules engaged in hydrogen bonding. In all other known FAD-binding proteins, the ribose-phosphate chain connecting the flavin to the adenosine is significantly extended (19) and the two heterocycles are far from each other.
Figure 1.

The active site of DNA photolyase without (left, ITEZ.PDB (18), chain C) and with substrate (right, ITEZ.PDB (18), chain A). The adenine and CPD are shown as sticks. The isoalloxazine ring is shown in wire form. The conserved water molecules are shown in red and white balls and sticks with hydrogen bonds shown as blue lines. Glu283 and Asn349, which make contacts with the CPD and/or Ade. In the PL:CPD complex, Ade is in vdW contact with the CPD. The ribityl-phosphate linker has been removed for clarity.
Since the elucidation of the first crystal structure of Escherichia coli PL (16), there has been much speculation over whether the Ade molecule is part of the electron transfer pathway to the CPD. The debate centers over whether photoinduced electron transfer (PET) from the flavin proceeds through an adenine intermediate followed by a second ET to the CPD: *FlH−/Ade/CPD→FlH•/Ade•−/CPD→FlH•/Ade/CPD•−, or whether excitation of the isoalloxazine results in direct ET: *FlH−/Ade/CPD→FlH•/Ade/CPD•−. In this latter case, the Ade may or may not participate in the reaction through superexchange (20). In superexchange, the electron transfer rate is increased through the participation of unoccupied orbitals of the bridging molecule (Ade) where the coupling is inversely proportional to the energy difference between the donor SOMO (singly occupied molecular orbital) and the bridge LUMO (lowest unoccupied molecular orbital). The rate constant for electron transfer via superexchange can be written as (20-22):
| (1) |
where the superexchange interaction, Vexch, takes into account the interaction energies between the donor and bridge states, VD-B, the interaction energy between the bridge and the acceptor, VB-A, and the energy difference between the donor and the bridge states, ΔED-B, given as:
| (2) |
An obvious test of this hypothesis would be to reconstitute apo-PL with FMN. Frustratingly, no successful experiments incorporating FMN or other flavin derivatives lacking the Ade nucleotide have been reported (23,24).
In 2004, the crystal structure of the A. nidulans PL:CPD complex became available (18), showing that the Ade moiety, and not the isoalloxazine ring, is in vdW contact with the CPD (see Fig. 1, ITEZ.PDB (18), chain A). The CPD is bound to PL (in a light-independent step (25)), and resulting complex has the CPD flipped out of the helix (26,27) at an optimal distance to the FAD (~3Å from the Ade) for efficient electron transfer (18,28,29). Conserved water molecules that hydrogen bonded to the Ade of the FAD are displaced by the CPD upon substrate binding. These stabilizing water interactions are replaced by CPD contacts with the AN6 amino group through the carbonyl groups of the thymine dimer. Additionally, the CPD is stabilized by contacts with Glu283 and Asn349, but there are no direct protein contacts with the Ade ring, only those mediated through water.
If Ade is a real intermediate in the electron transfer pathway, then the formation of the Ade radical anion (Ade•−) should be measurable by transient absorption (TA) spectroscopy by an increased absorbance in the near-UV and visible regions (30,31). However, the CPD radical anion is also expected to have absorbance in this part of the spectrum (32), making a unequivocal identification of either species difficult. If the role of Ade is that of a virtual intermediate via superexchange (33), then no spectral signature of this interaction would be obvious. EPR methods, however, have suggested that the adenosine is part of the PET pathway (34).
The photoinduced electron transfer rate constant for this reaction has been studied by several groups and the lifetimes lie within the 30–300 ps range depending on the experimental approach and source of PL used (7,35-40). Although oxidation of the flavin has been observed supporting the electron transfer mechanism, there is still no direct evidence for either an adenosine radical anion or a CPD radical anion, even though these species should have spectral signatures in the visible region of the spectrum. In 2003, MacFarlane and Stanley employed ultrafast blue pump/UV probe spectroscopy and detected the direct repair of the CPD substrate through a sequential bond-breaking mechanism (39). In the absence of substrate they observed that in fitting the decay of *FADH− in PL and comparing it to *FADH− in buffer no new states were required. Indeed, the cofactor in the protein has a longer lifetime than in solution (41). However, given the single wavelength probe, this study could not determine whether the Ade•− was formed in substrate-loaded enzyme. The lack of a spectroscopic signature of the radical Ade anion was been confirmed by more complete experiments in 2005 by Kao et al. (38) and in 2011 by Brettel (42) and Zhong (40,43). In 2012, more detailed and extensive blue/UV studies (40) by Zhong’s group did not detect an adenosine radical intermediate, but through the use of different substrates, their results lend stronger support for the role of superexchange for adenosine in PET reaction.
There have been several computational/theoretical studies in which the role of Ade in modulating electron transfer efficiency has been the focus, but these studies have yielded often contradictory results (29,33,44-46). We consider these contributions in more detail in the Discussion.
In this study, we have used adenine-modified FADs (1-N6-etheno-FAD (47,48) and 1-N6-ethano-FAD (49)) to reconstitute apo-PL from E. coli and compared their properties to apo-PL reconstituted with FAD (rPL). Our hypothesis is that the incorporation of a bridged Ade molecule lowers the Ade LUMO and therefore increases ΔED-B and both VD-B and VB-A in the superexchange mechanism. We therefore expect an increase in the rate of electron transfer.
MATERIALS AND METHODS
Luria and Terrific broth (LB and TB, respectively) were made using the recipe from Sambrook et al. (50). FAD (MP Biomedical, 95% purity) and chloroacetaldehyde (Sigma) were used as received. The buffer components were made from molecular biology grade (or better) reagents. Buffers were made from distilled/deionized water. Buffer T consists of 50 mm potassium phosphate, 100 mm potassium chloride, 0.1 mm ethylenediaminetetraacetate, disodium salt (EDTA), 10 mm 2-mercaptoethanol, pH 7.5. Buffer Apo-A is 50 mm potassium phosphate, pH 7.0, containing 1.7 m ammonium sulfate, 0.5 mm EDTA, 10 mm DTT and 20% glycerol. Buffer Apo-B is Apo-A saturated with KBr, pH 3.5. Buffer Apo-C is 100 mm potassium phosphate, pH 7.0, with 0.5 mm EDTA, 10 mm DTT and 50% ethylene glycol (EG). Storage buffer is 50 mm Tris, pH 7.4, with 50 mm NaCI, 1.0 mm EDTA, 10 mm DTT and 50% glycerol. All liquids were filtered using a 0.22 μm filter.
Overexpression and purification of photolyase.
Escherichia coli DNA photolyase was overexpressed using the MS09 (51) cell line which contains the phrB gene, both generous gifts of Prof. Aziz Sancar. Purification was accomplished using the protocol reported by Gindt et al. (52). Briefly, MS09 cells were grown in LB or TB. Overexpression of PL was induced using 2 mm IPTG when the cell density reached 0.6–1.0 OD. The cells were grown for 4–6 h after induction, harvested, homogenized in lysis buffer, and stored at −80°C until further use.
The harvested cells were lysed by incubation with lysozyme (~1 mg g−1 cells) with trace DNase, RNase and protease inhibitors with stirring at 4°C, followed by ultrasonication. The lysed cells were centrifuged at 20 000 g, and the cell debris discarded. PL was precipitated from the supernatant by the addition of ammonium sulfate to 65% saturation. The ammonium sulfate pellet was recovered by centrifugation at 20 000 g and stored at −20°C as necessary. The pellet was dissolved in binding buffer, desalted using size exclusion desalting columns (Bio-Rad PDG-10) and loaded onto a blue sepharose column (GE) for purification. The column was rinsed with loading buffer until the A280 reached a minimum, at which point the protein was eluted in a step gradient using a high salt buffer (2M KCl). The blue fraction protein showed a clear semi-quinone flavin absorption from 400 to 680 nm and MTHF absorption at 390 nm. This fraction was further purified on a heparin sepharose column (GE) using a linear gradient to ~90% purity as estimated by SDS-PAGE (data not shown). Purified PL was desalted as described above and buffer exchanged into storage buffer, mixed with glycerol to 50% and flash-frozen at −80°C until needed.
Preparation of apophotolyase.
Apophotolyase (apo-PL) was prepared from the holoenzyme using the protocol of Jorns et al. (23). Heparin sepharose-purified photolyase was buffer exchanged into buffer Apo-A and loaded onto a pre-equilibrated phenyl-sepharose column (GE). This was followed by a rinse with Apo-B low-pH buffer until all the chromophores were removed, as judged by absorption spectroscopy on the column fractions. The column was re-equilibrated with Apo-A, which allows the protein to refold to its native conformation. Finally, the apoprotein was eluted from the column using buffer Apo-C.
Reconstitution of apo-PL with FAD, ε-FAD and e-FAD.
Three different flavins were used in the reconstitution of apo-PL, flavin adenine dinucleotide and the adenine-modified ethano- and etheno-FAD (Scheme 1). ε-FAD was synthesized using the protocol of Barrio et al. (48,49,53,54). The purification and characterization of ε-FAD are described in more detail in the Supporting Information. e-FAD was a generous gift of the late Andreas Bürkmann (University of Konstanz).
Reconstitution of apo-PL with these flavins was carried out according the protocol of Jorns et al. (23). Typically, 7 μm apo-PL was buffer exchanged into buffer Apo-C. The solution was slowly stirred with ~200 μm flavin ([Fl]:[Apo-PL]~30) and incubated at 4°C for 24–48 h. This mixture was concentrated using a Vivaspin 20 concentrator (10 or 30 kD MWCO) and buffer exchanged into buffer Apo-A. This protein was loaded onto a HiTrap phenyl-sepharose column, which was pre-equilibrated with buffer Apo-A. The column was washed with 3–4 column volumes of buffer Apo-A, and the reconstituted photolyase was eluted with buffer Apo-C. Purity was ascertained by SDS-PAGE analysis and by UV/vis spectroscopy.
A modification of this procedure proved to be useful. Instead of the phenyl-sepharose purification following reconstitution, the crude reconstitution mixture was loaded onto a Bio-Rad P-10 size exclusion column equilibrated in either buffer Apo-C with various amounts of EG or Buffer T with various amounts of glycerol. The protein was eluted by adding a second volume of the buffer, while free cofactor and other small buffer components were held up in the column. Protein obtained in this way was usually very pure, as judged by UV/vis spectroscopy.
Preparation of thymidine dimer substrates.
HPLC-purified dT5 (“5-mer”) and an “11-mer” oligo (5′-GCAAGTTGGAG-3′) were purchased (IDT, Inc.) and used to prepare the thymidine dimer substrates. The protocol of Banerjee et al. (55) was used for preparation of CPD from the 5-mer. The 11-mer CPD-containing oligo was made using the protocols of Kemmink et al. (56), Mu et al. (57) and Nakayama et al. (58). Details of substrate preparation and HPLC purification are described in the Supporting Information.
The 11-mer duplex was prepared by annealing the 11-mer strand containing a thymidine dimer with the complementary sequence designated “3-2AP”, 5′-CTCCAACTTGC-3′, where A denotes the fluorescent adenosine analog 2-aminopurine (59,60). The 11-mer and the 3′-2AP strands were mixed in a 1.4:1 mole ratio (60) in a total volume of 50 μL. The sample was heated in a ~80°C water bath for 10 min and cooled to room temperature on the laboratory bench for at least 60 min.
Determination of the substrate binding constant using steady-state fluorescence spectroscopy.
In previous studies, we showed that the fluorescence emission from 2-aminopurine is a reporter for CPD base flipping in duplex DNA when bound by PL (27,60-62). This property was exploited to determine the substrate binding constant, KA, for both rPL and ε-PL. Samples consisted of 250 nm 2AP-containing dsDNA with varying concentrations (50 nm–2 μm) of the reconstituted photolyases in buffer T. Correction of the spectra for tryptophan emission from the protein was achieved by subtracting the emission measured from an identical protein sample without substrate.
Steady-state fluorescence emission of PL-CPD/3′-2AP complexes was obtained using a Horiba Jobin Yvon Fluoromax-2 fluorimeter. A 0.4 × 1.0 cm quartz cuvette was used with excitation along the longer dimension and emission collected at 90° through the shorter dimension. The cuvette was held at 15°C using a chilled water recirculator. The excitation wavelength was fixed at 317 nm, and the emission was measured from 335 to 601 nm. The excitation band pass was 8 nm, and the emission band pass was 12 nm. The fluorescence was collected with a 2 nm step size and an integration time of 0.5 s per point. Spectra were corrected for excitation intensity fluctuations in real time.
Repair assay.
PL was purged with argon for 30 min. in a sealed cuvette (0.4 × 1.0 cm or 1.0 × 1.0 cm) and immersed in ice water in a Pyrex beaker to remove infrared and ultraviolet light during photoreduction. Photoreduction was achieved by loosely focusing the filtered output of a 150W Xe arc lamp (380 nm long pass) onto the cuvette. Photoreduction times were approximately 30 min. Substrate was then added to the cuvette in an argon-purged glove bag using an air-tight syringe purged with argon. Absorption spectra were taken to follow the photoreduction process using an HP8452A spectrophotometer.
The reaction mixture containing photoreduced PL and 5-mer substrate (1:40 mole ratio PL:CPD) was irradiated at 365 nm with a 4 W Spectroline hand lamp 4 cm above the cuvette. Note that the ε-Ade molecule has no absorption at this wavelength. Conversely, the S0→S2 transition in reduced flavin has its maximal absorption near 360 nm (63). Repair was tracked by an increase in absorbance at 266 nm due to the breaking of the cyclobutane C5-C5′ and C6-C6′ bonds with restoration of the double bonds in the thymines.
The change in absorption, ΔA = A(t)–A(0), was converted to a change in concentration, Δc = ΔA/(lΔε266), where is calculated from the extinction coefficients given by Johns et al. (64). The extinction coefficient of the undamaged T-T at 265 nm was and that of the dimeric CPD was . The fraction of repaired CPD is given by fCPD = [CPD(t)]repaired/[CPD(0)].
Absorption/Fluorescence spectroscopy.
Samples of the flavins or protein with or without substrate were prepared in 50 mm Tris, pH 9.0 with 1 mm EDTA as the sacrificial electron donor. Ar-purged solutions were irradiated with light for ~15 min. from a blue LED array peaked around 450 nm (Phillips). An HP 8452 diode-array UV–Vis spectrophotometer was used with a bandpass of 2 nm.
Chromophore excitation and emission spectra were obtained on anoxic samples in a septum-sealed cuvette using a PTI Q25 fluorescence spectrometer. The cuvette was thermostatted to 15°C. Further protocols for protein reconstitution, CPD preparation and supplemental graphs S1 and S2 can be found online in the Supporting Information section.
RESULTS
For reference, the spectra of the oxidized cofactors used in this study are shown in Fig. 2a along with FMN, Ade and ε-Ade for comparison. Each molecule was dissolved in 0.1 m potassium phosphate buffer, pH 6.8. The spectrum of FAD is well known, and the ε-FAD spectrum has also been described previously (48). In aqueous solution, the S0→S1 (S01) and S02 transitions (450 and 374 nm, respectively) of FAD are almost identical to FMN (446 and 374 nm). This is true for ε-FAD as well (452 and 374 nm). The peak of the S03 transition for FMN, FAD is 268 nm, and ε-FAD is 268 nm but here the similarity ends. FMN has its S04 transition at 224 nm, while FAD shows only a shoulder.
Figure 2.

(a) Absorption spectra of FAD (–––), FMN (–––) and ε-FAD (–––). Also shown are Ade (●) and ε-Ade (●) for comparison. The inset shows the region from 290 to 340 nm where the flavin molecules have a minimum in their absorbance, while the ε-Ade group has roughly 3 times the extinction at ca. 305 nm. (b) Photoreduction of ε-FAD→ε-FADH− does not reduce the ε-Ade molecule, as shown by the unchanged spectral band around 305 nm. FADH− is also shown for comparison.
The ε-Ade group has increased extinction at ~235 nm due to the overlap of its S03 and S04 bands and two overlapping transitions and 275 nm (S02) and 305 nm (S01) (65). The inset spectrum shows that ε-FAD has more than double the extinction of either FAD or FMN around 305 nm due to the ε-Ade moiety (65). ε-FAD has its S04 maximum at 224 nm (65). Both FAD and ε-FAD undergo intramolecular stacking in polar solvents (53,66), and the lower than expected extinction at 235 nm in ε-FAD probably reflects hypochromism due to the overlap of the flavin and ε-Ade transition dipole moments (65).
The reduced cofactors are shown in Fig. 2b. The spectrum of reduced ε-FADH− has not been reported, to our knowledge. The most important difference is the increase in extinction around 300 nm, suggesting that photoreduction only affects the flavin ring.
Reconstitution of apo-photolyase with FAD, ε-FAD and e-FAD
Although various flavin-modified FADs have been used for reconstitution of apo-PL (23,24,67), this is the first time, to our knowledge, that an adenine-modified FAD has been incorporated into the active site of this protein. Apo-PL was reconstituted successfully with either FAD or ε-FAD but less effectively with e-FAD (data not shown). Reconstitution efficiencies were calculated based on the ratio of the number of moles of reconstituted PL recovered to the initial number of moles of apo-PL used for reconstitution. The reconstitution yield was explored by varying the ratio of flavin to apo-PL, ranging from about 15–60. No trend could be discerned, suggesting that a 15-fold excess of flavin was sufficient for maximal reconstitution. Reconstitution times were also varied from 24 to 48 h with no change in outcome. The yield of rPL using FAD was 51 ± 37% based on four experiments. Using ε-FAD, the reconstitution yield was 43 ± 32% based on ten separate experiments, which is equivalent to FAD reconstitution within experimental error. Reconstitution of apo-PL with e-FAD was very poor (≤13%). Incorporation of the more hydrophobic e-FAD was so inefficient that it could not be made in sufficient quantities to study further.
The spectra of the reconstituted photolyases are shown in Fig. 3, including a magnified view of the two lowest electronic transitions. Maxima for the reconstituted proteins in the UV are the result of both cofactor and protein (tryptophan) absorption. The concentrations of rPL and ε-PL were estimated using ε444 = 11 200 m−1 cm−1 for both flavins (68). The wavelength maxima for the flavins in the visible to near-UV (S0n for n = 1–3) were identical within experimental error and blue-shifted from the FAD(aq) spectrum. The prominent vibronic structure of the S01 and S02 transitions in FAD-reconstituted apo-PL, also observed by Jorns’ (23) and Sancar’s (24) groups, suggests that both flavins are tightly bound in the FAD-binding pocket. The slightly less resolved ε-PL vibronic structure is probably due to a small loss of binding energy due to fewer hydrogen bonds to the ε-Ade.
Figure 3.

Absorption spectrum of FAD (rPL) and ε-FAD (ε-PL) reconstituted photolyase. The spectra were taken in buffer Apo-A (which includes 1.7 m ammonium sulfate) and normalized to 11 200 m−1 cm−1 at 444 nm. The flavin vibronic features are clearly seen in the S0→S2 band around 444 nm. Inset: The S0→S1,2 bands of ε-PL show a small degree of broadening compared to the rPL spectrum. This suggests that the modified cofactor is not as rigidly bound as the FAD.
Escherichia coli apo-PL has an extinction of 100 100 m−1 cm−1 at 280 nm (23,24,67). Upon reconstitution with FAD, both the Ade and flavin moieties contribute to the extinction at 280 nm (ε280 = 21 200 m−1 cm−1) (67). Thus, rPL has an extinction of about 121 300 m−1 cm−1 giving a calculated ratio of A280/A444 = 11 for pure reconstituted apo-PL. The measured value is ε280 = 117 581 m−1 cm−1 with an A280/A444 = 10.5, well within our experimental error. In some cases, the purity ratio varied from ~11 to 20, suggesting a purity of 55–100%. The major contaminant was assumed to be apo-PL. As apo-PL does not bind the substrate (24), further purification was not deemed necessary for either repair or binding assays.
FAD has a lower extinction at 280 nm (20 201 m−1 cm−1) than ε-FAD (21 918 m−1 cm−1) because the ε-Ade molecule has higher extinction than Ade at this wavelength (65,69). This gives a calculated extinction for ε-PL of ε280 = 122 018 m−1 cm−1. Perfectly reconstituted ε-PL would have a calculated ratio of A280/A444 = 10.9, compared to the value of 9.8 found here. Thus, reconstituted ε-PL was estimated to be > 95% pure. It should be noted that these high purity ratios were most often achieved by size exclusion purification of the reconstitution mixture. Lower purity was found when purifying by phenyl-sepharose chromatography.
Both rPL and ε-PL can be stored at −80°C in 50% glycerol/buffer and used as needed. After 6 months of storage, ε-PL exhibited a small loss of vibronic structure compared to rPL suggesting that it is slightly less stable under these conditions. Interestingly, buffer exchange using a size exclusion column into buffer T containing no glycerol leads to dissociation of ε-FAD from ε-PL, while FAD is retained in rPL (data not shown). This suggests that the ε-FAD cofactor is less tightly bound than the native FAD.
ε-PL efficiently repairs CPDs
The repair activities of the reduced PLs were assayed using a dT5 substrate containing an average of one CPD per oligo. The change in concentration of the repaired substrate with irradiation time is shown in Fig. S1 for both rPL and ε-PL. These data were converted to the fraction of CPDs repaired and plotted vs irradiation time as shown in Fig. 4. The error bars represent the standard deviation from the mean, determined from three replicates.
Figure 4.

The fraction of repaired T5 substrate with irradiation time for rPL and ε-PL. The error bars represent the standard error from the mean derived from three replicates. The repair yield of ε-PL is about 15% higher than rPL.
As repair proceeds the slopes of the curves decrease, reflecting the depletion of substrate with concomitantly slower bimolecular kinetics. Repair activity saturates between 60 and 70% of total substrate. This is consistent with what has been reported for substrates consisting of 4–6 nucleotides (70,71). Interestingly, ε-PL repairs CPDs with about 15% higher yield than rPL at all times within the error bars of the experiment. This result could be due to a difference in substrate binding, which is now addressed.
Binding constant determination
Because the adenine is in vdW contact with the substrate (18), it is important to compare the binding constants of the two PLs to find out whether the etheno group affects substrate binding. KA, the association constant for substrate with protein, was measured in both reconstituted proteins using a fluorescently tagged substrate molecule. We have shown previously that the fluorescence change of a 2-aminopurine (2AP) base placed complementary to the CPD could be used to assay for binding (27,60). When the 2AP in this duplex is excited, its emission is well quenched. However, when bound to PL, the CPD is base-flipped into the protein (18,27) and base stacking surrounding the 2AP decreases. This leads to an eight-fold increased fluorescence quantum yield relative to the unbound base-stacked state (60). If the flavin cofactor of the protein is maintained in the oxidized state (aerobic conditions), then no repair occurs during the assay, affording a quantitative interpretation of the degree of substrate binding.
Figure S2 shows the fluorescence emission intensity as a function of substrate binding (and base flipping) with λex:λem at 317 nm: 335–600 nm, respectively. Varying concentrations of rPL and ε-PL were incubated with a fixed concentration (250 nm) of a 2AP-containing CPD-dsDNA (PL:CPD/3′-2AP). At each protein concentration, the emission spectrum of reconstituted PL at that concentration was subtracted from the PL:duplex spectrum to remove contributions from protein emission and Raman scattering. Tryptophan has significant emission in this spectral range, and E. coli PL has 15 Trp residues. Even though Trp has a very low extinction coefficient at 317 nm, at least 50 times lower than 2AP, the sheer number of Trp residues requires the background subtraction procedure. A similar rationale applies to tyrosine or tyrosinate (E. coli PL has 14 Tyr residues). While Tyr is modestly fluorescent, tyrosinate has a much higher quantum yield. However, the extinction coefficient for tyrosinate at 317 nm is about 25 m−1 cm−1. Thus excitation of a single tyrosinate in the presence of 2AP (ε317 ~ 4500 m−1 cm−1 (59)) would be improbable.
The competition for excitation light between ε-Ade (or flavin) and 2AP in the ε-PL case is more troublesome. As shown in Fig. 2, ε-FAD has an extinction of ε317~4360 m−1 cm−1, with εAde contributing about 2100 m−1 cm−1 to this value. Thus, 2AP is the more probable absorber. Energy transfer from 2AP*→εAde or vice versa is probably unlikely as the emission of either chromophore is so red-shifted that the overlap with the acceptor absorption spectrum would be negligible. Finally, direct excitation of the εAde moiety could lead to energy transfer to the flavin. Their proximity suggests that this process cannot be ruled out. However, excitation transfer from εAde to the flavin ultimately results in emission quenching as has been described by Barrio et al. (53).
To address these concerns, we have constructed and analyzed binding curves by integrating the emission over several wavelength regions to reflect the emission of the 2AP, εAde and flavin chromophores, as well as an integration of the emission from 343 to 453 nm, reflecting the majority of all emission from the sample. In the latter case, no significant emission is observed after the subtraction of the protein emission spectrum, suggesting that energy transfer to the flavin is a small concern. The 2AP and εAde integrated intensities have identical concentration dependencies, again suggesting that emission is dominated by 2AP*, while no significant dependence of flavin emission with protein concentration was observed (data not shown).
The corrected integrated emission of IPL:CPD-IPL as a function of protein concentration at 250 nm substrate was fitted to a simple hyperbolic function (72):
| (3) |
where A is the amplitude of the curve, KD is the dissociation constant for the enzyme-substrate complex (see Fig. 5). The association constant KA is obtained from 1/KD. These constants are tabulated in Table 1 for the corresponding wavelength ranges discussed above.
Figure 5.

Plots of integrated fluorescence emission vs. protein concentration against a fixed concentration of substrate (250 nm). Protein emission sans substrate was subtracted at each concentration point. The emission data were then integrated from 359 to 387 nm to exclude emission from the ε-Ade chromophore. KA for rPL was (3.9 ± 1.4) × 106 m−1 and that for ε-PL was (4.0 ± 1.5) × 106 m−1, as obtained from a fit to a hyperbolic function (solid lines).
Table 1.
KA (±σ) from fits to binding curves (×10−6 m−1).
| Δλ (nm)/KA | rPL:CPD | ε-PL:CPD |
|---|---|---|
| 359–387 | 3.9 (1.4) | 4.0 (1.5) |
| 343–453 | 3.7 (1.5) | 4.7 (0.8) |
| 411–439 | 3.9 (2.3) | 6.8 (1.4) |
Association constants for rPL are fairly constant over all integration windows at around KA = 3.9 × 106 m−1, showing that the protein subtraction procedure is removing all but 2AP* emission. In contrast, the ε-PL:CPD association constant is smallest when the emission is integrated over 359–387 nm where 2AP* emission is expected to dominate and grows when the window is centered over the ε-Ade emission range (411–439 nm). Integration of a broader wavelength range appears to reduce the KA for ε-PL:CPD somewhat, suggesting that a lower value of KA is probably more precise. This result suggests that the ε-Ade quantum yield is slightly higher in the presence of the CPD. A simple explanation for this hypothesis is that excitation of 2AP leads to energy transfer to ε-Ade, generating more εAde emission at 420 nm than the protein without substrate (2AP*+ε-Ade→2AP+ε-Ade*→2AP+ε-Ade+hν420 nm). Resonance energy transfer would be possible if the emission spectrum of 2AP* overlaps with the absorption spectrum of ε-Ade. As 2AP* has emission down to 325 nm where ε-Ade absorbs (65,73), this appears possible, assuming the orientation factor (κ2) is nonzero. When integrated over the 2AP* range, KA = (4.0 ± 1.5) × 106 m−1 for ε-PL:CPD, essentially identical with the corresponding KA = (3.9 ± 1.4) × 106 m−1 for rPL:CPD.
DISCUSSION
Reconstitution of apophotolyase with adenine-modified FADs
Although flavin-modified FADs have been used for reconstitution of apo-PL in previous studies (23,24), this is the first time, to our knowledge, that an adenine-modified FAD has been successfully incorporated into this protein. Apophotolyase was successfully reconstituted with oxidized FAD and ε-FAD with about equal yield. Reconstitution with sterically similar ethano-adenine-modified FAD was less efficient by a factor of 3–4. ε-PL binds an 11-mer CPD-containing duplex with the same affinity as rPL, but the repair yield of ε-PL is slightly better than rPL. To understand this outcome, it is useful to examine the contacts made between the FAD adenine and amino acid groups and/or conserved water molecules and to see how these contacts change upon substitution of ε-Ade for Ade.
The FAD in E. coli photolyase bridges α-helical domains I and II (16). Amino acids 234–288 in α-helix I provide for six of the eight interactions with the diphosphate group. Other interactions between the α-helix II and the riboflavin moiety stabilize the cofactor (16). That ε-FAD is tightly bound is shown by its clear vibronic structure, and it appears that the isoalloxazine ring maintains similar interactions with α-helix I and II inside the protein active site. However, the loss of ε-FAD from ε-PL in glycerol-free buffer suggests that polar contacts between the protein and the Ade moiety are lost when the Ade AN6-AN1 atoms are bridged by the etheno group. This bridge eliminates the possibility of hydrogen bonding at the AN6 amino group and potentially blocks stabilizing interactions involving the AN1 lone pair. It is likely that the sudden decrease in viscosity during the exchange causes the protein to expand somewhat. As the adenine is closer to the protein surface than the flavin, its greater solvent exposure and weaker contacts may facilitate cofactor dissociation.
This additional conformational flexibility may translate into closer contact between the ε-Ade and CPD. This may be a static or dynamic phenomenon, but the electron transfer coupling is exponentially sensitive to distance. What is interesting here is that contacts to the flavin ring are probably unperturbed by the ε-Ade, so that the isoalloxazine-CPD distance, ~ 7Å, would be unchanged if the Ade molecule was modified to ε-Ade.
Interestingly, there are no direct amino acid contacts with the adenine ring. In Ade, the AN1, AN3, and AN7 lone pairs are hydrogen bonded to water molecules that are additionally stabilized by the contacts with the ribityl tail and the Asn349 and Glu283 residues (A. nidulans) (18) which do not have direct contacts with the adenine. The AN6 hydrogens are also involved in hydrogen bonds with at least two and likely three water molecules in the absence of substrate (see Fig. 1, left panel). The protonation state of ε-Ade might play a role in binding, but given that the pKA of this N-H bond is 3.9 (47,53,74) ε-Ade is likely to be neutral.
The AN6 amino group is also important in stabilizing the substrate, hydrogen bonding to both C4 = O groups on the base-flipped CPD either directly or through a conserved water molecule. The Glu and Asn residues now make direct contacts with the CPD at the N3-H and C4 = O sites. This direct involvement displaces three water molecules (Fig. 1, right panel).
ε-FAD in PL cannot make the same kinds of contacts. To illustrate this, see Fig. 6. This picture was constructed using the “Build Structure” module of Chimera (75) v. 1.5.3 (UCSF) based on the 1TEZ crystal structure of A. nidulans PL. Chain A of 1TEZ shows the bound CPD, while chain C lacks substrate. No minimization was carried out on the ε-Ade group, but the bond lengths were adjusted based on the crystal structure of 1, N6-ethenoadenine by Jaskólski (76). The etheno group subsumes the AN6 H-bond donors which are clearly important in stabilizing the CPD and may be part of the electron transport pathway. The fact that their loss does not change the substrate binding constant suggests that these interactions are significantly less important than the protein:DNA backbone contacts (the Glu and Asn contacts make contacts directly to the CPD) and are preserved in the presence of the ε-Ade. However, as pointed out by others (76,77), the AN6 nitrogen atom can act as a hydrogen bond acceptor.
Figure 6.

Left. Cartoon of ε-FAD structure inside of apo-PL constructed using Chimera (UCSF (75)). Water molecules that are no longer hydrogen bonded to adenine are shown in yellow and are probably lost due to steric clashes from the ε-Ade group. Right: Chimera cartoon of how the ε-Ade group eliminates important contacts with the CPD, while increasing orbital overlap with the flavin ring.
Without a crystal structure, it remains unknown whether this acceptor site is used to stabilize the cofactor and/or CPD to any great extent. What appears to be true is that the etheno bridge extension of the adenine does not clash with either the CPD or amino acid residues. Nonetheless, water molecules that are conserved in the adenine-containing structure clearly clash with the bridging group (cf. Fig. 1 left and Fig. 6 left, in yellow). We come back to the contribution of the bridging group to electron transfer below.
Binding constant measurements
The binding constant (KA) for the formation of PL:CPD complex was calculated from a hyperbolic fit to the data obtained from an assay based on fluorescence enhancement due to CPD base flipping (27,60). The KA for rPL was (3.9 ± 1.4) × 106 m−1. Using a gel-shift assay Sancar et al. reported, the KA = 2.6. × 108 m−1 for E. coli photolyase (semiquinone form) with a 43-mer containing a single T< >T (78) and KA = 3.6 × 108 m−1 for rPL with a 48-mer with a single T< >T (79). Sancar et al. reported KA = (5.7 ± 1.7) × 107 m−1 for wild-type E. coli PL (semiquinone form) bound to UV-damaged pBR322 plasmid in a buffer solution with an ionic strength of 125 mm. More recently, Gindt et al. used isothermal titration calorimetry (ITC) and measured the KA for the wild-type photolyase and oxidized photolyase to be (1.45 ± 0.05) × 105 m−1 and (1.11 ± 0.07) × 105 m−1 with UV-damaged ss-dT10, respectively (80). It has to be mentioned that the concentration of PL and CPD used by Sancar et al. was in the nm range while that of Gindt et al. was in the μm (PL) to mm (CPD) range. Our results, using a completely different approach than either the Sancar or Gindt groups, were obtained with protein and CPD concentrations about two orders of magnitude different from both groups.
The significantly higher KA values reported by Sancar et al. can be attributed to tighter binding associated with longer substrate (70), but it is not clear why the KA value from Gindt’s study, using a relatively long 10-mer substrate, would yield a value ~3 orders of magnitude lower than Sancar et al. The 11-mer duplex substrate with the fluorescent reporter base used in the present study suggests that substrate binding may have a sequence dependence that has not been elucidated.
Repair efficiencies of rPL and ε-PL
Both reconstituted photolyases repair thymidine dimer substrate under conditions of saturating substrate and blue light, as shown in Fig. 4. Interestingly, ε-PL repairs CPDs at a slightly higher rate. The repair quantum yield for PL is about Φrepair=0.82 (15). From ultrafast measurements, the quantum yield for the ultrafast fast electron transfer reaction is ΦET = kET/(kET+kNET) = 0.87 (38) (NET = not electron transfer). Given that the substrate binding constants for rPL and ε-PL are essentially equal, the 15% increase in overall repair yield for the ε-PL construct appears to be due to either an increase in the forward electron transfer rate constant, kET, or a decrease in either the recombination rate of the radical pair or some internal conversion process in competition with forward electron transfer. These constants can be represented by kNET. While there is some debate about the initial electron transfer rate constant (7,35,38,39,42,43), a value of 1010 s−1 is reasonable. Based on this rate constant, kNET = 1.8 × 109 s−1. A 15% increase in Φrepair = kET/(kET+kNET) based solely on an increase in the forward electron transfer rate constant due to the ε-Ade moiety would give = 8.8 × 1010 s−1. This gives a lifetime of about 11 ps, easily distinguishable from the 100 ps lifetime of native PL. Ultrafast spectroscopic measurements of the ε-PL:CPD complex are currently underway to test this supposition.
ε-PL repairs a larger fraction of CPDs than rPL. This difference in repair yield is not due to differences in substrate affinity or repair rate. It may be due to the greater conformational freedom of ε-PL in the binding cavity, as evidenced by the more poorly resolved vibronic structure of ε-FAD in PL. A 10% increase in kET can be obtained if ε-FAD can get closer to the CPD by 0.1–0.2 Ẫ. However, it is also possible that the lower relative energy of ε-Ade compared to Ade has opened a new pathway for repair that of two-step hopping using ε-Ade as a real intermediate.
PET between *FlH− and the CPD: Is Ade a real intermediate or superexchange bridge?
Evidence for CT in PLRED has not yet been demonstrated. In 2005, Kao et al. (2) saw no spectroscopic evidence for Ade•−, even with subpicosecond time resolution when probing in the visible region. As noted above, the two very recent TA studies of Brettel (42) and Zhong (43) confirm this assignment. However, in 2008, Li and Glusac (81) saw a difference of repair rate constants for bimolecular collisions between a CPD substrate and *FADH− vs *FMNH− giving convincing evidence that PET to the Ade was important for these model systems, although this may be due to adenine:dimer alignment effects rather than changes in the electron transfer pathway due to the adenine.
For Ade to be a real intermediate on the electron transfer pathway, the free energy for the electron transfer reaction must be negative. The sequential mechanism can be outlined as *FlH−: Ade: CPD→FlH•: Ade•−: CPD→FlH•: Ade: CPD•−. Thermodynamically, if either forward ET reaction is endergonic then charge separation will be too slow to compete with deactivation channels such as internal conversion. The same arguments can be made if Ade is replaced by ε-Ade. However, even though the reduction potential of the CPD is −1.96V vs NHE (82), a coexistence of one-step (superexchange) and two-step (hopping) mechanisms may be possible (83). An examination of the reduction potentials of the reaction partners is a reasonable first step in addressing this question. From these, the free energy of photoinduced electron transfer can be computed as (84):
| (4) |
where is the reduction potential of the excited state donor (*PLRED) and is the reduction potential of the acceptor (Ade or ε-Ade).
Measurements of have been made by two groups. Gindt’s group obtained = 0.081 V and 0.016 V vs NHE for PLRED with and without CPD (85). Brettel’s group measured = −0.039 ± 0.005 V for E. coli PL (86). An estimate of E00 is obtained by taking an average over the range from 2.48 to 2.58 V used by Zhong’s group (40).
The ground state reduction potential of adenine is available from Seidel et al. (87) or Crespo-Hernandez et al. (88), respectively, as −2.52 ± 0.05 and −2.71 V vs NHE (in DMF). Using −2.52V – 0.05V = −2.57V leads to the result that the Ade•− is not formed whether substrate is bound or not. However, the lower limit, −2.52 + 0.05 = −2.47V, gives the result that Ade•− can be formed, but only in the absence of the CPD, a result that runs counter to transient absorption measurements (39,40). This suggests that superexchange must be operative. However, the reduction potential of Ade is given in DMF, and it is likely that is lower (less negative) in the low dielectric environment of photolyase. Additionally, as sequential electron transfer is faster than superexchange for small differences in donor-bridge energies (22), we would expect a two-step hopping mechanism to be plausible so long as it is thermodynamically allowed.
The ground state reduction potential of ε-Ade in aqueous solvent is about −1.1 V vs NHE as measured by polarography (89). Kelly and Barton estimated ε-Ade* (excited state) reduction potential at 1.4 V vs NHE (90). Assuming an E00 = 28 324 cm−1 (3.53 eV), the ground state reduction potential would be 1.4 V–3.5 V = −2.1 V, in significant disagreement with the polaro-graphic measurement. Given the large uncertainties in this estimate, the reduction in ε-Ade by *FlH− is quite possible.
Computational studies (33,34,44,45) disagree that superexchange is operative, although the details of this interaction differ depending on the specific approach. The Stuchebrukhov group used the extended Hückel method to calculate the electronic couplings between the *FADH− donor and the CPD acceptor. Molecular dynamics/docking calculations were used to optimize conformation of the CPD relative to the flavin so that the electron transfer matrix element matched experimental values of the electron transfer rate constant (35). Their analysis predicted that the CPD would lie 3Å from the flavin ring and that the adenine ring must act to support superexchange between the reduced anionic flavin and the CPD. An important result was that the acceptor wavefunction is highly localized on the C4 = O4 carbonyl group of the 3′-T of the CPD, very close to the adenine. In particular, the AN6 nitrogen atom of the adenine and its hydrogen bonds to 3′-C = O groups on the CPD modulate the electron transfer matrix element significantly. The prediction was made that modulation of the aromaticity of the adenine should lead to changes in the electron transfer rate constant and quantum yield of repair. Our work is the first test of this hypothesis.
The work of Prytkova et al. (45) used extensive molecular dynamics simulations with the ten lowest excited singlet states of the flavin computed using TDDFT/BHandHLYP and solvation by PL amino acid residues and conserved water molecules. These results showed that only the isoalloxazine C8 methyl group was important for electron transfer to the CPD. They would predict that no changes in rate or yield should occur if the adenine is modified. However, they do acknowledge that low probability structural fluctuations could open up secondary superexchange pathways between the Ade and CPD. It is certainly possible that substitution of the ε-Ade group not only makes the superexchange coupling stronger between Ade and *FlH− but by its increased size improves overlap between the LUMOs of the ε-Ade and either the CPD or the flavin. A more precise understanding will be afforded by crystal structure studies of the ε-PL protein, if and when they become available.
Acocela et al. (44) predicted that the Ade NH2 group is all that is necessary to promote efficient ET from the flavin to the CPD through an electrostatic perturbation of the electron. Clearly, this is not the case based on the repair yields for ε-PL in our study, as the NH2 moiety is subsumed into the etheno bridge which will have completely different partial charges. The polarization of the AN6 nitrogen bridged with the etheno group will be much smaller than the original amino group. This will reduce any electrostatic perturbation and should lead to a reduction in the electron transfer rate. Interestingly, MacFarlane and Stanley (28) introduced a similar idea regarding the influence of the dipolar electric field of the CPD itself on the driving force of the excited state flavin, which has been supported by the work of Gindt (85) and Schelvis (91,92).
What is the role of the FAD adenine in DNA photolyase?
Simply put, the red-shifted absorption spectrum of ε-Ade compared to Ade suggests that the LUMO of ε-Ade is lower than the corresponding LUMO of Ade. If electron transfer does involve Ade as a real intermediate (for which there is no evidence yet), then the driving force to ε-Ade should be larger and the repair rate should increase. If, instead, Ade acts as a superexchange bridge, then the lower LUMO of ε-Ade could enhance the rate of superexchange-mediated ET, as the superexchange coupling goes as the inverse of the energy difference between donor and bridge states. Temperature-dependent studies could differentiate between these two scenarios, as superexchange has a weaker temperature dependence than hopping (22). Given what has been shown here, the ε-PL system offers the possibility of a direct proof of superexchange in the photolyase system. Definitive confirmation of this using ultrafast laser spectroscopy on the ε-PL:CPD complex is underway in our laboratory.
CONCLUSIONS
The main aim of this work was to provide an experimental system for studying the role of adenine in the FAD of DNA photolyase. Toward this end, an adenine-modified FAD, ε-FAD, was synthesized and reconstituted successfully into apophotolyase. The reconstituted (FAD and ε-FAD) photolyases both repair thymidine dimers in double-stranded 11-bp DNA. Substrate binding efficiencies of these PLs were found to be about the same. ε-PL gives a distinctly higher quantum yield of repair under steady-state conditions. The lower reduction potential of ε-Ade suggests that two-step hopping may dominate. However, without a direct measurement of the electron transfer rate constant, and observation of a radical anion intermediate of ε-Ade, the role of ε-Ade (and Ade) in DNA repair by photolyase remains elusive.
Supplementary Material
Figure S1. Typical repair assays of Escherichia coli rPL and εPL with a 5-mer substrate containing one T<>T.
Figure S2. Plot of fluorescence emission intensity (arb. units) at various concentrations of rPL (top) and εPL (bottom).
Acknowledgements
We wish to thank Dr. Charlie Debrosse, Department of Chemistry, Temple University, for help with measuring and interpreting NMR data. We are grateful to Prof. Yvonne Gindt and Prof. Hans Schelvis (Montclair State Univ.), and Prof. David Beratan (Duke Univ.) for very useful discussions. K.M. was supported by a HHMI Undergraduate Summer Research Fellowship. G.K. and R.J.S. are grateful for support from the NSF Molecular Biosciences Division (MCB-0347087). M.N and R.J.S. are thankful for a Temple University Bridge Grant. V.R.S. was supported by NSF grant (CHE-0847855) which also supported, in part, M.N. and K.J. G.K. and M.N acknowledges support from Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation (Grant # TG-CHE140112).
Footnotes
This article is part of the Special Issue highlighting Dr. Aziz Sancar’s outstanding contributions to various aspects of the repair of DNA photodamage in honor of his recent Nobel Prize in Chemistry.
SUPPORTING INFORMATION
Additional Supporting Information may be found in the online version of this article:
REFERENCES
- 1.Taylor J-S (1994) Unraveling the molecular pathway from sunlight to skin cancer. Acc. Chem. Res 27, 76–82. [Google Scholar]
- 2.Kao Y-T, Saxena C, He T-F, Guo L, Wang L, Sancar A and Zhong D (2008) Ultrafast dynamics of flavins in five redox states. J. Am. Chem. Soc 130, 13132–13139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.LeClerc JE, Borden A and Lawrence CW (1991) The thymine-thymine pyrimidine-pyrimidone (6-4) ultraviolet light photoproduct is highly mutagenic and specifically induces 3’ thymine-to-cytosine transitions in Escherichia coli. Proc. Natl Acad. Sci. USA 88, 9685–9689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sancar A, Franklin KA and Sancar GB (1984) Escherichia coli DNA photolyase stimulates uvrABC excision nuclease in vitro. Proc. Natl Acad. Sci. USA 81, 7397–7401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Minato S and Werbin H (1971) Spectral properties of the chromophoric material associated with the deoxyribonucleic acid photoreactivating enzyme isolated from baker’s yeast. Biochemistry 10, 4503–4508. [DOI] [PubMed] [Google Scholar]
- 6.Sancar GB, Smith FW and Sancar A (1983) Identification and amplification of the E. coli phr gene product. Nucleic Acids Res 11, 6667–6678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim ST, Heelis PF, Okamura T, Hirata Y, Mataga N and Sancar A (1991) Determination of rates and yields of interchromophore (folate → flavin) energy transfer and intermolecular (flavin → DNA) electron transfer in Escherichia coli photolyase by time-resolved fluorescence and absorption spectroscopy. Biochemistry 30, 11262–11270. [DOI] [PubMed] [Google Scholar]
- 8.Todo T, Takemori H, Ryo H, Ihara M, Matsunaga T, Nikaido O, Sato K and Nomura T (1993) A new photoreactivating enzyme that specifically repairs ultraviolet light-induced (6-4)photoproducts. Nature (London) 361, 371–374. [DOI] [PubMed] [Google Scholar]
- 9.Kanai S, Kikuno R, Toh H, Ryo H and Todo T (1997) Molecular evolution of the photolyase-blue-light photoreceptor family. J. Mol. Evol 45, 535–548. [DOI] [PubMed] [Google Scholar]
- 10.Briggs WR, Tseng T-S, Cho H-Y, Swartz TE, Sullivan S, Bogomolni RA, Kaiserli E and Christie JM (2007) Phototropins and their LOV domains: versatile plant blue-light receptors. J. Integr. Plant Biol 49, 4–10. [Google Scholar]
- 11.Kennis JTM, Crosson S, Gauden M, van Stokkum IHM, Moffat K and van Grondelle R (2003) Primary reactions of the LOV2 domain of phototropin, a plant blue-light photoreceptor. Biochemistry 42, 3385–3392. [DOI] [PubMed] [Google Scholar]
- 12.Kraft BJ, Masuda S, Kikuchi J, Dragnea V, Tollin G, Zaleski JM and Bauer CE (2003) Spectroscopic and mutational analysis of the blue-light photoreceptor AppA: a novel photocycle involving flavin stacking with an aromatic amino acid. Biochemistry 42, 6726–6734. [DOI] [PubMed] [Google Scholar]
- 13.Gomelsky M and Kaplan S (1998) AppA, a redox regulator of photosystem formation in Rhodobacter sphaeroides 2.4.1, is a flavoprotein - Identification of a novel FAD binding domain. J. Biol. Chem 273, 35319–35325. [DOI] [PubMed] [Google Scholar]
- 14.Masuda S and Bauer CE (2002) AppA is a blue light photoreceptor that antirepresses photosynthesis gene expression in Rhodobacter sphaeroides. Cell 110, 613–623. [DOI] [PubMed] [Google Scholar]
- 15.Sancar A (2003) Structure and function of DNA photolyase and cryptochrome blue-light photoreceptors. Chem. Rev 103, 2203–2237. [DOI] [PubMed] [Google Scholar]
- 16.Park H-W, Kim S-T, Sancar A and Deisenhofer J (1995) Crystal structure of DNA photolyase from Escherichia coli. Science 268, 1866–1872. [DOI] [PubMed] [Google Scholar]
- 17.Huang Y, Baxter R, Smith BS, Partch CL, Colbert CL and Deisenhofer J (2006) Crystal structure of cryptochrome 3 from Arabidopsis thaliana and its implications for photolyase activity. Proc. Natl Acad. Sci. USA 103, 17701–17706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mees A, Klar T, Gnau P, Hennecke U, Eker APM, Carell T and Essen L-O (2004) Crystal structure of a photolyase bound to a CPD-like DNA lesion after in situ repair. Science (Washington, DC, U.S.) 306, 1789–1793. [DOI] [PubMed] [Google Scholar]
- 19.Dym O and Eisenberg D (2001) Sequence-structure analysis of FAD-containing proteins. Protein Sci. 10, 1712–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marcus RA (1987) Superexchange versus an intermediate bacteriochlorophyll ion (BChl−) mechanism in reaction centers of photosynthetic bacteria. Chem. Phys. Lett 133, 471–477. [Google Scholar]
- 21.Davis WB, Ratner MA and Wasielewski MR (2001) Conformational gating of long distance electron transfer through wire-like bridges in donor-bridge-acceptor molecules. J. Am. Chem. Soc 123, 7877–7886. [DOI] [PubMed] [Google Scholar]
- 22.Paulson BP, Miller JR, Gan W-X and Closs G (2005) Superexchange and sequential mechanisms in charge transfer with a mediating state between the donor and acceptor. J. Am. Chem. Soc 127, 4860–4868. [DOI] [PubMed] [Google Scholar]
- 23.Jorns MS, Wang B, Jordan SP and Chanderkar LP (1990) Chromophore function and interaction in Escherichia coli DNA photolyase: reconstitution of the apoenzyme with pterin and/or flavin derivatives. Biochemistry 29, 552–561. [DOI] [PubMed] [Google Scholar]
- 24.Payne G, Wills M, Walsh C and Sancar A (1990) Reconstitution of Escherichia coli photolyase with flavins and flavin analogs. Biochemistry 29, 5706–5711. [DOI] [PubMed] [Google Scholar]
- 25.Sancar GB and Sancar A (1987) Structure and function of DNA photolyases. Trends Biochem. Sci 12(7), 259–261. [Google Scholar]
- 26.Berg BJV and Sancar GB (1998) Evidence for dinucleotide flipping by DNA photolyase. J. Biol. Chem 273, 20276–20284. [DOI] [PubMed] [Google Scholar]
- 27.Christine KS, IV MacFarlane AW, Yang K and Stanley RJ (2002) Cyclobutylpyrimidine dimer base flipping by DNA photolyase. J. Biol. Chem 277, 38339–38344. [DOI] [PubMed] [Google Scholar]
- 28.MacFarlane AW IV and Stanley RJ (2001) Evidence of powerful substrate electric fields in DNA photolyase: implications for thymidine dimer repair. Biochemistry 40, 15203–15214. [DOI] [PubMed] [Google Scholar]
- 29.Medvedev D and Stuchebrukhov AA (2001) DNA repair mechanism by photolyase: electron transfer path from the photolyase catalytic cofactor FADH-to DNA thymine dimer. J. Theor. Biol 210, 237–248. [DOI] [PubMed] [Google Scholar]
- 30.Steenken S (1989) Purine bases, nucleosides, and nucleotides: aqueous solution redox chemistry and transformation reactions of their radical cations and e- and OH adducts. Chem. Rev 89, 503–520. [Google Scholar]
- 31.Candeias LP and Steenken S (1992) Electron adducts of adenine nucleosides and nucleotides in aqueous-solution - protonation At 2 carbon sites (C2 And C8) and intramolecular and intermolecular catalysis by phosphate. J. Phys. Chem 96, 937–944. [Google Scholar]
- 32.Yeh SR and Falvey DE (1991) Model studies of DNA photorepair: radical anion cleavage of thymine dimers probed by nanosecond laser spectroscopy. J. Am. Chem. Soc 113, 8557–8558. [Google Scholar]
- 33.Antony J, Medvedev DM and Stuchebrukhov AA (2000) Theoretical study of electron transfer between the photolyase catalytic cofactor FADH- AND DNA thymine dimer. J. Am. Chem. Soc 122, 1057–1065. [Google Scholar]
- 34.Weber S, Moebius K, Richter G and Kay CWM (2001) The electronic structure of the flavin cofactor in DNA photolyase. J. Am. Chem. Soc 123, 3790–3798. [DOI] [PubMed] [Google Scholar]
- 35.Langenbacher T, Zhao X, Bieser G, Heelis PF, Sancar A and Michel-Beyerle ME (1997) Substrate and temperature dependence of DNA photolyase repair activity examined with ultrafast spectroscopy. J. Am. Chem. Soc 119, 10532–10536. [Google Scholar]
- 36.Heelis PF, Okamura T and Sancar A (1990) Excited-state properties of Escherichia coli DNA photolyase in the picosecond to millisecond time scale. Biochemistry 29, 5694–5698. [DOI] [PubMed] [Google Scholar]
- 37.Okamura T, Sancar A, Heelis PF, Begley TP, Hirata Y and Mataga N (1991) Picosecond laser photolysis studies on the photorepair of pyrimidine dimers by DNA photolyase. 1. Laser photolysis of photolyase -2-deoxyuridine dinucleotide photodimer complex. J. Am. Chem. Soc 113, 3143–3145. [Google Scholar]
- 38.Kao YT, Saxena C, Wang LJ, Sancar A and Zhong DP (2005) Direct observation of thymine dimer repair in DNA by photolyase. Proc. Natl Acad. Sci. USA 102, 16128–16132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.MacFarlane AW IV and Stanley RJ (2003) Cis-syn thymidine dimer repair by DNA photolyase in real time. Biochemistry 42, 8558–8568. [DOI] [PubMed] [Google Scholar]
- 40.Liu ZY, Guo XM, Tan C, Li J, Kao YT, Wang LJ, Sancar A and Zhong DP (2012) Electron tunneling pathways and role of adenine in repair of cyclobutane pyrimidine dimer by DNA photolyase. J. Am. Chem. Soc 134, 8104–8114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Enescu M, Lindqvist L and Soep B (1998) Excited-state dynamics of fully reduced flavins and flavoenzymes studied at subpicosecond time resolution. Photochem. Photobiol 68, 150–156. [DOI] [PubMed] [Google Scholar]
- 42.Thiagarajan V, Byrdin M, Eker APM, Muller P and Brettel K (2011) Kinetics of cyclobutane thymine dimer splitting by DNA photolyase directly monitored in the UV. Proc. Natl Acad. Sci. USA 108, 9402–9407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu Z, Tan C, Guo X, Kao Y-T, Li J, Wang L, Sancar A and Zhong D (2011) Dynamics and mechanism of cyclobutane pyrimidine dimer repair by DNA photolyase. Proc. Natl Acad. Sci 108, 14831–14836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Acocella A, Jones GA and Zerbetto F (2010) What is adenine doing in photolyase? J. Phys. Chem. B 114, 4101–4106. [DOI] [PubMed] [Google Scholar]
- 45.Prytkova TR, Beratan DN and Skourtis SS (2007) Photoselected electron transfer pathways in DNA photolyase. Proc. Natl Acad. Sci. USA 104, 802–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harrison CB, O’Neil LL and Wiest O (2005) Computational studies of DNA photolyase. J. Phys. Chem. A 109, 7001–7012. [DOI] [PubMed] [Google Scholar]
- 47.Penzer GR (1973) The solution conformation and some spectroscopic properties of 1, N6-ethenoadenosine monophosphate, a fluorescent analogue of AMP. Eur. J. Biochem 34, 297–305. [DOI] [PubMed] [Google Scholar]
- 48.Harvey RA (1980) Flavin 1,N6-ethenoadenine dinucleotide In Methods Enzymol, Vol. 66 (Edited by Donald LDW and McCormick B), pp. 290–294. Academic Press, New York. [DOI] [PubMed] [Google Scholar]
- 49.Buckmann AF, Wray V and Stocker A (1997) Synthesis of N6-(2-aminoethyl)-FAD, N6-(6-carboxyhexyl)-FAD, and related compounds. Methods Enzymol. 280, 360–374. [DOI] [PubMed] [Google Scholar]
- 50.Sambrook J, Fritsch EF and Maniatis T (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Woodbury, NY. [Google Scholar]
- 51.Sancar GB, Smith FW, Lorence MC, Rupert CS and Sancar A (1984) Sequences of the Escherichia coli photolyase gene and protein. J. Biol. Chem 259, 6033–6038. [PubMed] [Google Scholar]
- 52.Gindt YM, Vollenbroek E, Westphal K, Sackett H, Sancar A and Babcock GT (1999) Origin of the transient electron paramagnetic resonance signals in DNA photolyase. Biochemistry 38, 3857–3866. [DOI] [PubMed] [Google Scholar]
- 53.Barrio JR, Tolman GL, Leonard NJ, Spencer RD and Weber G (1973) Flavin 1, N6-ethenoadenine dinucleotide: dynamic and static quenching of fluorescence. Proc. Natl Acad. Sci. USA 70, 941–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Harvey RA and Damle S (1972) A fluorescent modification of flavin adenine dinucleotide. FEBS Lett. 26, 341–343. [DOI] [PubMed] [Google Scholar]
- 55.Banerjee SK, Christensen RB, Lawrence CW and LeClerc JE (1988) Frequency and spectrum of mutations produced by a single cis-syn thymine-thymine cyclobutane dimer in a single-stranded vector. Proc. Natl Acad. Sci. USA 85, 8141–8145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kemmink J, Boelens R, Koning TMG, Kaptein R, Van der Marel GA and Van Boom JH (1987) Conformational changes in the oligonucleotide duplex d(GCGTTGCG).d(CGCAACGC) induced by formation of a cis-syn thymine dimer. A two-dimensional NMR study. Eur. J. Biochem 162, 37–39. [DOI] [PubMed] [Google Scholar]
- 57.Mu W, Han Q, Luo Z and Wang Y (2006) Production of cis-syn thymine-thymine cyclobutane dimer oligonucleotide in the presence of acetone photosensitizer. Anal. Biochem 353, 117–123. [DOI] [PubMed] [Google Scholar]
- 58.Nakayama T, Todo T, Notsu S, Nakazono M and Zaitsu K (2004) Assay method for Escherichia coli photolyase activity using single-strand cis-syn cyclobutane pyrimidine dimer DNA as substrate. Anal. Biochem 329, 263–268. [DOI] [PubMed] [Google Scholar]
- 59.Ward DC, Reich E and Stryer L (1969) Fluorescence studies of nucleotides and polynucleotides. J. Biol. Chem 244, 1228–1237. [PubMed] [Google Scholar]
- 60.Yang K and Stanley RJ (2006) Differential distortion of substrate occurs when it binds to DNA photolyase: A 2-aminopurine study. Biochemistry 45, 11239–11245. [DOI] [PubMed] [Google Scholar]
- 61.Yang K, Matsika S and Stanley RJ (2007) 6MAP, a fluorescent adenine analogue, is a probe of base flipping by DNA photolyase. J. Phys. Chem. B 111, 10615–10625. [DOI] [PubMed] [Google Scholar]
- 62.Yang K and Stanley RJ (2008) The extent of DNA deformation in DNA photolyase-substrate complexes: A solution state fluorescence study. Photochem. Photobiol. 84, 741–749. [DOI] [PubMed] [Google Scholar]
- 63.Siddiqui MSU, Kodali G and Stanley RJ (2008) The electronic transition dipole moment directions of reduced flavin in stretched poly(vinyl alcohol) films. J. Phys. Chem. B 112, 119–126. [DOI] [PubMed] [Google Scholar]
- 64.Fisher GJ and Johns HE (1976) Pyrimidine Photodimers. In Photochemistry and Photobiology of Nucleic Acids, Vol. 1 (Edited by Wang SY), pp. 226–294. Academic Press, New York, NY. [Google Scholar]
- 65.Holmen A, Albinsson B and Norden B (1994) Electronic transition dipole moments of the 1, N6-Ethenoadenine chromophore. J. Phys. Chem 98, 13460–13469. [Google Scholar]
- 66.Weber G (1950) Fluorescence of riboflavin and flavin-adenine dinucleotide. Biochem. J 47, 114–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang B and Jorns MS (1989) Reconstitution of Escherichia coli DNA photolyase with various folate derivatives. Biochemistry 28, 1148–1152. [DOI] [PubMed] [Google Scholar]
- 68.Müller F (1983) The Flavin Redox-System and Its Biological Function In Top. Curr. Chem, Vol. 108, pp. 71–107. Springer-Verlag, Berlin. [DOI] [PubMed] [Google Scholar]
- 69.Secrist JA, Weber G, Leonard NJ and Barrio JR (1972) Fluorescent modification of adenosine-containing coenzymes - biological-activities and spectroscopic properties. Biochemistry 11, 3499–000. [DOI] [PubMed] [Google Scholar]
- 70.Jorns MS, Sancar GB and Sancar A (1985) Identification of oligothymidylates as new simple substrates for E. coli DNA photolyase and their use in a rapid spectrophotometric enzyme assay. Biochemistry 24, 1856–1861. [DOI] [PubMed] [Google Scholar]
- 71.Kim ST and Sancar A (1991) Effect of base, pentose, and phosphodiester backbone structures on binding and repair of pyrimidine dimers by Escherichia coli DNA photolyase. Biochemistry 30, 8623–8630. [DOI] [PubMed] [Google Scholar]
- 72.Nelson DL and Cox M (2000) Principles of Biochemistry. Freeman WH, New York. [Google Scholar]
- 73.Holmen A, Norden B and Albinsson B (1997) Electronic transition moments of 2-aminopurine. J. Am. Chem. Soc 119, 3114–3121. [Google Scholar]
- 74.Imakubo K (1978) Effects of protonation and cooling on the fluorescence of 1, N6-ethenoadenosine. Nucleic Acids Res. 1, s357–s362. [Google Scholar]
- 75.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC and Ferrin TE (2004) UCSF chimera - A visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- 76.Jaskolski M (1982) 1, N6-Ethenoadenosine. Acta Crystallogr. B 38, 3171–3174. [Google Scholar]
- 77.Seela F, Ding P, Leonard P, Eickmeier H and Reuter H (2011) 1, N 6-Etheno-2 ‘-deoxytubercidin hemihydrate. Acta Crystallogr. C Struct. Chem 67, O111–O114. [DOI] [PubMed] [Google Scholar]
- 78.Husain I and Sancar A (1987) Binding of E. coli DNA photolyase to a defined substrate containing a single T<>T dimer. Nucleic Acids Res 15, 1109–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Payne G and Sancar A (1990) Absolute action spectrum of E-FADH2 and E-FADH2-MTHF forms of Escherichia coli DNA photolyase. Biochemistry 29, 7715–7727. [DOI] [PubMed] [Google Scholar]
- 80.Sokolowsky K, Newton M, Lucero C, Wertheim B, Freedman J, Cortazar F, Czochor J, Schelvis JPM and Gindt YM (2010) Spectroscopic and thermodynamic comparisons of Escherichia coli DNA photolyase and vibrio cholerae cryptochrome 1. J. Phys. Chem. B 114, 7121–7130. [DOI] [PubMed] [Google Scholar]
- 81.Li G, Sichula V and Glusac KD (2008) Role of adenine in thymine-dimer repair by reduced flavin-adenine dinucleotide. J. Phys. Chem. B 112, 10758–10764. [DOI] [PubMed] [Google Scholar]
- 82.Scannell MP, Fenick DJ, Yeh S-R and Falvey DE (1997) Model studies of DNA photorepair: reduction potentials of thymine and cytosine dimers measured by fluorescence quenching. J. Am. Chem. Soc 119, 1971–1977. [Google Scholar]
- 83.Lee W, Kodali G, Stanley RJ and Matsika S (2016) Coexistence of different electron-transfer mechanisms in the DNA repair process by photolyase. Chem. Eur. J 22, 11371–11381. [DOI] [PubMed] [Google Scholar]
- 84.Kavarnos GJ (1993) Fundamentals of Photoinduced Electron Transfer. VCH Publishers Inc, New York, NY. [Google Scholar]
- 85.Gindt YM, Schelvis JPM, Thoren KL and Huang TH (2005) Substrate binding modulates the reduction potential of DNA photolyase. J. Am. Chem. Soc 127, 10472–10473. [DOI] [PubMed] [Google Scholar]
- 86.Balland V, Byrdin M, Eker APM, Ahmad M and Brettel K (2009) What makes the difference between a cryptochrome and DNA photolyase? A spectroelectrochemical comparison of the flavin redox transitions J. Am. Chem. Soc 131, 426–+. [DOI] [PubMed] [Google Scholar]
- 87.Seidel CAM, Schulz A and Sauer MHM (1996) Nucleobase-specific quenching of fluorescent dyes. 1. Nucleobase one-electron redox potentials and their correlation with static and dynamic quenching efficiencies. J. Phys. Chem 100, 5541–5553. [Google Scholar]
- 88.Crespo-Hernandez CE, Close DM, Gorb L and Leszczynski J (2007) Determination of redox potentials for the Watson-Crick base pairs, DNA nucleosides, and relevant nucleoside analogues. J. Phys. Chem. B 111, 5386–5395. [DOI] [PubMed] [Google Scholar]
- 89.Bojarska E (1986) Electrochemical reduction Of e-Adenine, e-Adenosine, And e-NAD+ In Aqueous-Media. Bioelectrochem. Bioenerget 16, 287–300. [Google Scholar]
- 90.Kelley SO and Barton JK (1999) Electron transfer between bases in double helical DNA. Science 283, 375–381. [DOI] [PubMed] [Google Scholar]
- 91.Kapetanaki SM, Ramsey M, Gindt YM and Schelvis JPM (2004) Substrate electric dipole moment exerts a pH-dependent effect on electron transfer in Escherichia coli photolyase. J. Am. Chem. Soc 126, 6214–6215. [DOI] [PubMed] [Google Scholar]
- 92.Schelvis JPM, Ramsey M, Sokolova O, Tavares C, Cecala C, Connell K, Wagner S and Gindt YM (2003) Resonance raman and UV-Vis spectroscopic characterization of FADH.bul. in the complex of photolyase with UV-damaged DNA. J. Phys. Chem. B 107, 12352–12362. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Typical repair assays of Escherichia coli rPL and εPL with a 5-mer substrate containing one T<>T.
Figure S2. Plot of fluorescence emission intensity (arb. units) at various concentrations of rPL (top) and εPL (bottom).