

Abstract
How somatic mutations accumulate in normal cells is poorly understood. A comprehensive analysis of RNA-sequencing data from ~6,700 samples across 29 normal tissues reveals multiple somatic variants, demonstrating that macroscopic clones can be found in many normal tissues. We confirm that sun-exposed skin, esophagus, and lung have a higher mutation burden than other tested tissues, suggesting that environmental factors can promote somatic mosaicism. Mutation burden is associated with both age and tissue-specific cell proliferation rate, highlighting that mutations accumulate over time and number of cell divisions. Finally, we find that normal tissues harbor mutations in known cancer genes and hotspots. This study provides a broad view of macroscopic clonal expansion in human tissues, thus serving as the basis to associate clonal expansion with environmental factors, aging and risk of disease.
One Sentence Summary:
Multiple macroscopic clonal expansions are detected across normal tissues, including clones with mutations in cancer genes.
Introduction
As cells divide during life, they accumulate somatic mutations. While most of these mutations are thought to be either neutral or slightly deleterious (1), a few may increase cellular fitness and contribute to clonal expansion. This process is associated with aging as well as with diseases such as coronary heart disease (2, 3), neurological disorders (4), and cancer (5). In cancer, the accumulation of several mutations (known as “cancer drivers”) eventually may transform the cells and promote uncontrolled cellular growth. Despite work contributing to our understanding of the molecular and cellular aspects of cancer (6–14), we still only partially understand the initiation and progression of this disease. Acknowledging this gap, studies have focused on studying somatic mutations in normal human tissues and pre-cancerous lesions, aiming to identify early clonal expansions (3, 15–18). Clonal expansions detected in normal blood are enriched with mutations in several genes implicated in hematologic cancers (3, 19). Ultra-deep sequencing studies by Martincorena et al. (17, 18) in normal skin and esophagus tissues focused on 74 cancer genes and detected a high burden of low-allele frequency mutations associated with skin and esophagus squamous cell carcinoma. Despite these associations, which specific clones will eventually develop into cancer remains unclear. Collectively, these findings emphasize the need to comprehensively map and study the prevalence and size of clonal expansion across human tissues.
Results
A pipeline for detecting somatic mutations using RNA-seq data
For genomic data derived from normal tissues, we leveraged the Genotype–Tissue Expression (GTEx) project (20), a collection of data generated from over 30 normal primary tissues from hundreds of healthy individuals. These data include RNA-sequencing (RNA-seq) data of the tissues as well as whole–genome and –exome sequencing data of DNA extracted from matched blood samples (release V7), providing an opportunity to explore all genes and tissues for the existence of macroscopic clones that have expanded to a detectable level in bulk RNA-seq.
To detect somatic mutations from bulk RNA-seq data, we needed to first develop a pipeline, called RNA-MuTect, to analyze this type of data. To develop our approach for detecting somatic mutations from RNA-seq data, we initially focused on a training set of 243 tumor samples (representing 6 tumor types) from The Cancer Genome Atlas (TCGA), for which both DNA and RNA were co-isolated from the same cells (table S1). Applying our standard somatic mutation calling pipeline (that was developed for DNA) to both DNA and RNA from the tumor samples, and using the matched-normal DNA as a germline control (21), we found 5-fold more mutations in RNA than in the corresponding DNA (Figs. 1A, S1A and (22)). Moreover, 65% of the DNA-based mutations were not detected in the RNA, and 92% RNA-based mutations were not found in the DNA (22). One obvious reason for not detecting DNA-based mutations in the RNA is the insufficient sequence coverage in lowly expressed genes––indeed, in a typical RNA sample, only 55% of the transcriptome had sufficient coverage (≥95% sensitivity) to detect mutations at the median DNA allele fraction (fig. S1B). When accounting for the actual allele fractions of the DNA mutations and coverage of RNA transcripts, RNA-MuTect detected 82% of the sufficiently covered mutations (fig. S2A–D, (21, 22)).
Fig. 1: Validation of RNA-MuTect in TCGA samples.
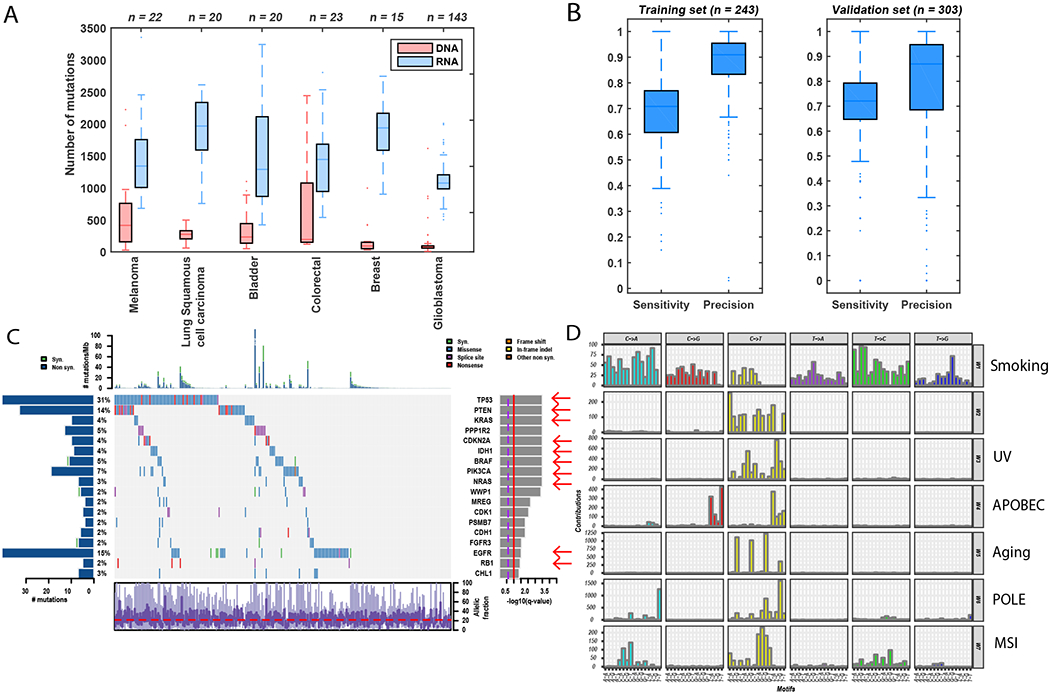
(A) Total number of mutations detected before filtering in DNA (red) and RNA (blue) across samples in each TCGA cohort. (B) Sensitivity and precision of sufficiently covered sites, across training and validation samples. Box plots show median, 25th and 75th percentiles. The whiskers extend to the most extreme data points not considered outliers, and the outliers are represented as dots. (C) Co-mutation plot with mutations across the 243 TCGA samples, overall frequencies, allele fractions, and significance levels of candidate cancer genes (Q value < 0.05) identified by applying MutSig2CV (25) on the mutations detected in the RNA. Genes marked with a red arrow were also identified as significantly mutated in the DNA. (D) Mutational signatures identified by SignatureAnalyzer (26) on the basis of mutations detected in the RNA. The mutational signatures identified are: a mixture of smoking and nucleotide-excision repair signatures (W1, combination of COSMIC signatures 4 and 5, cosine similarities of 0.7 and 0.75, respectively); UV (W3, COSMIC signature 5, cosine similarity = 0.95); APOBEC (W4, COSMIC signature 13, cosine similarity = 0.9); Aging (W5, COSMIC signature 1, cosine similarity = 0.9); POLE (W6, COSMIC signature 10, cosine similarity = 0.88), and MSI (W7, COSMIC signature 15, cosine similarity = 0.8).
Next, to address the excessive mutations detected only in the RNA, we included several key filtering steps into the RNA-MuTect pipeline (fig. S3): (i) removal of alignment errors by using two different RNA aligners; (ii) removal of sequencing errors by a site-specific error model built upon thousands of normal RNA-seq data; and (iii) removal of RNA editing sites using known databases (21). The vast majority (93%) of RNA mutations were filtered out (fig. S2E–G), reaching a median precision of 0.91 across samples (Fig. 1B), and only a median of 3 detected mutations/sample remained in the RNA alone. RNA-MuTect retained a high overall median sensitivity of 0.7 after filtering (Figs. 1B, S2E (22)), removing as few as 10% of mutations that were detected in the DNA. Of note, RNA-MuTect outperformed previous methods (23, 24) in terms of both sensitivity and precision for detecting mutations in RNA-seq (22). To evaluate the robustness of RNA-MuTect on an independent dataset, we collected a validation set of 303 TCGA samples representing 6 tumor types (5 differed from the training set, table S1). RNA-MuTect achieved high sensitivity and precision on the validation set, in agreement with the training set results (sensitivity of 0.72 and precision of 0.87, Fig. 1B).
The high overall performance of RNA-MuTect enabled us to apply our standard tools for finding drivers and mutational signatures to RNA-based mutations (21, 25, 26), which yielded very similar results to what was found in the DNA (Figs. 1C, D, S4, S5 and (22)). Our analysis did, however, identify a yet-unreported mutational signature in the RNA dominated by C>T mutations; this signature represented only 7% of the mutations, with the majority originating from a single colon cancer sample (Fig. 1D). Of these mutations, 75% were sufficiently covered but not detected in the DNA, suggesting that this signature may reflect a C>U RNA-editing process.
Notably, to obtain a conservative (i.e., higher) estimate of the false-positive rate, we considered mutations as false positives if they were detected in the RNA but not in the DNA while having sufficient coverage in the DNA. Although these mutations could in theory be true RNA-only mutations generated via RNA-specific processes (but not in the RNA-editing databases), it is more likely that they are in fact present in the DNA but at allele fractions too low to be detected, as our detection sensitivity calculations assume that the underlying allele fractions of a mutation are the same in the DNA and RNA. Although these values are often close, they can vary due to variable gene- and allele-specific expression in different cells types within the sample. One way to test this is by examining the correlation between the number of RNA-only mutations and the number of true-positive mutations detected in both RNA and DNA. We observed a high correlation (Spearman R = 0.6, P value = 4.2 × 10−30), suggesting that many of the RNA-only mutations are likely also in the DNA, since we would not expect any correlation to exist between false-positive (generated by either noise or RNA processes) and true-positive mutations. Nonetheless, we continued with our conservative approach throughout this study and considered all RNA-only mutations detected in sufficiently covered corresponding DNA loci to be false-positive mutations. Overall, we conclude that high precision analysis of somatic mutations based on RNA is achievable despite the apparent limitations in calling mutations de novo from RNA-seq data, allowing for most cancer-associated genes as well as mutational processes to be revealed from RNA-seq data.
Finally, to evaluate the performance of RNA-MuTect on normal tissues, we applied it to a set of 35 tumor-adjacent normal samples collected in TCGA, wherein DNA and RNA were co-isolated from the same sample (table S1). After ensuring the samples were not contaminated with tumor cells (27), we detected 114 DNA-based mutations, with a median allele fraction of 0.06. These mutations and their low allele fractions reflect the existence of small, yet macroscopic, clones in these samples, as expected in normal tissues. Out of only 8 mutations detected in the DNA that had sufficient sequencing coverage in the RNA to enable detection (22), 3 were indeed detected, one had evidence in 2 reads (just below our detection level), and the remaining 4 had no supporting reads in the RNA (table S2). Similarly, the 175 RNA-based mutations had an average allele fraction of 0.07; of the 86 that were sufficiently covered in the DNA, 13 mutations were detected. Overall, the number of RNA-only mutations per sample was very low (median of 1, and average of 2; table S2). As only half of the RNA-based mutations had sufficient coverage in the DNA, we conservatively estimated the total number of false-positive RNA-based mutations per sample to be between 2 to 4.
Overall, when applying RNA-MuTect to normal samples with co-extracted DNA and RNA data, we found that DNA mutations with allele fraction of >0.07 could be detected in the RNA in cases where the gene was sufficiently highly expressed (as a specific example, a mutation with an allele fraction of 0.05 required coverage by at least 124 reads in order to have >95% chance of being detected, and ~17% of a typical transcriptome from a TCGA RNA-seq sample is covered to that depth [see fig. S1B]). More importantly, RNA-MuTect detected a low number of potential false-positive calls per sample in normal tissues, consistent to what we found in our cancer samples.
Detecting somatic clonal expansions in normal tissues
After establishing RNA-MuTect’s performance on both cancer and normal samples, we sought to study somatic mutations across a comprehensive collection of normal tissues by analyzing RNA-seq data from the GTEx project (20). For a mutation to be detected in bulk-RNA extracted from a normal tissue, a macroscopic clone that harbors and expresses the somatic mutation needs to contribute a sufficient amount of RNA such that the signal can be observed over the background RNA from other cells in the sample (e.g., muscle and fat cells typically do not proliferate, thus diluting the signal from the expanding clone) (Fig. 2A). Thus, the ability to detect a somatic mutation depends on: (i) the clonal diversity of the sample, (ii) the depth of sequencing, and (iii) the expression level of the mutated gene. In the GTEx dataset, RNA was extracted from a relatively large amount of tissue material (~20 mg of tissues, estimated to represent 30,000–730,000 cells depending on tissue type (22)), limiting our ability to identify mutations present in microscopic clonal populations. We did, however, expect to detect macroscopic clones harboring mutations found in ~10% of the cells.
Fig. 2: Somatic clonal expansion in normal tissues.
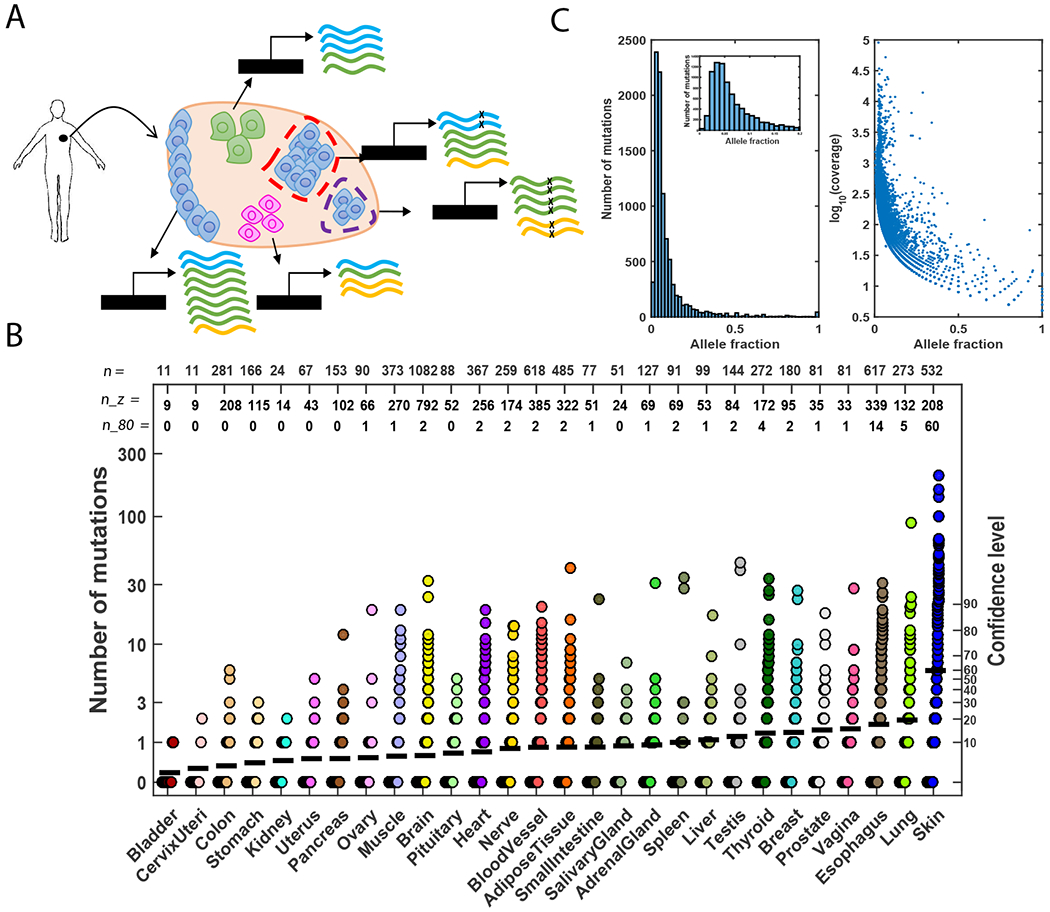
(A) An illustration of the composition of bulk RNA extracted from a normal human tissue. The biopsy consists of three different cell types that express different transcripts (marked in blue, green, and yellow) at different levels. Blue cells represent cells with a higher probability to form clones. Two clones, small and large, are shown denoted by purple- and red-dashed outlines, respectively. Mutated reads are marked with an “x”. The allele fractions of the mutations in the blue and green genes are the same (0.25; 2/8 and 4/16 reads, respectively), despite the different clone sizes. Additionally, the allele fraction of the mutation in the yellow gene is higher than the allele fractions of the mutations in the blue and green genes (0.33; 2/6 reads), despite the fact that the yellow mutation is supported by the same (or smaller) number of reads. These scenarios illustrate the challenge of identifying somatic mutations in bulk normal tissue due to a mixture of cell types and the relatively small clones. Moreover, inferring clone size is limited due to different cell types that exist in different proportions and express transcripts at different levels. (B) Number of mutations detected in the RNA-seq of all studied tissues. Each sample is represented with a circle. The black horizontal line represents the mean number of mutations in each tissue type. A confidence level from our estimation of false positives in the validation data is indicated in the right y-axis. Specifically, this confidence level is computed as the xth percentile on the number of false positive calls (RNA-only mutations in DNA-powered sites) found in the validation set. “n” values represent the total number of samples analyzed in each tissue; “n_z” values represent the number of samples in which no mutations were detected; and “n_80” values represent the number of samples in which more than 13 mutations were found (equivalent to a confidence level of 80%). (C) Left panel: Distribution of allele fraction across all samples in which somatic mutations were detected. Inset: mutations with allele fraction ≤ 0.2. Right panel: Allele fraction as a function of the log10(coverage) for all detected mutations.
Applying RNA-MuTect to 6,707 RNA-seq samples against their matched-blood DNA, which spanned 29 human tissues and 488 individuals (21), we detected 8,870 somatic mutations in 37% (2,519) of the samples, representing nearly all individuals (95%, 467/488; Fig. 2B and table S3). Applying our conservative estimate based on the TCGA data of 2–4 false positives per sample, 374 samples across 24 tissues had more than 4 mutations (within these, 106 samples across 13 tissues had >13, which is the conservative estimate at the 80th percentile of false calls). Note that mutations detected in samples with 4 or fewer mutations are not necessarily false positives; for example, some of these samples harbored known cancer driver mutations that likely increased cell fitness. The analyses described below provide evidence indicating that many of the detected mutations are somatic mutations that reflect clonal expansions in normal tissue.
Similar to what we observed from analyzing the tumor-adjacent normal samples from TCGA, the median allele fraction of the mutations in the GTEx normal tissue samples was 0.05 (Fig. 2C). Although our ability to detect low-allele fraction mutations in both DNA and RNA in GTEx samples was limited because they were extracted from adjacent, but different, samples, we were able to experimentally validate 5 of 28 mutations by deep sequencing (table S4; (21)). Consistent with the majority of mutations being passengers, like we observe in cancer, ~59% were missense mutations (fig. S6). However, we also found that a few mutations in normal tissue types matched mutations observed in their corresponding cancer types (table S5). Overall, these results support that macroscopic clonal expansion occurs across many normal tissues throughout the body.
As expected, we found a negative correlation between RNA sequencing coverage and allele fraction (Spearman R = −0.8, P value < 10−200, Fig. 2C) due to a higher sensitivity to detect low-allele fraction mutations from highly covered sites. However, after correcting for detection sensitivity (given the mutation allele fraction and the effective gene coverage (21)), we also observed a negative correlation between expression level and expected number of mutations (fig. S7). Similar findings suggest that transcription-coupled repair occurs in genes that are highly expressed in cancers (28–30).
The tissues that typically harbor the greatest number of mutations are skin, lung, and esophagus. Associations between cancer incidence in these tissues and environmental factors such as UV radiation, air pollution, smoking, and nutritional habits were previously shown (31–37). Of note, sorting the normal tissues by mutation frequency rather than by absolute number of mutations yields essentially the same order (fig. S8). Looking at tissue sub-regions, we found that sun-exposed skin had more mutations than non-exposed skin and contained the highest number of mutations overall. Similarly, esophagus mucosa, from which esophageal squamous cell carcinoma derive rather than from either gastroesophageal junction or esophagus muscularis, had the second-highest mutational burden (fig. S9). Interestingly, the only tissue with a significant difference in the number of mutations between males and females was breast (two-sided Wilcoxon P value = 2.1 × 10−5, fig. S10), reflecting the observation that breast tissue samples from males in the GTEx dataset are mainly composed of fat cells, while female breast tissue are also composed of epithelial cells.
Finally, we examined whether somatic mutations could be detected in the blood (21). Focusing on a previously defined set of 332 single nucleotide variants detected in the blood of healthy individuals (3), we identified 87 mutations in the DNA across 83 individuals (17% of the studied individuals). For each of these 83 individuals, we next tested whether the exact variant(s) was present in other solid tissues from the same individual. Only 7 mutations were found in at least one RNA sequencing read in other tissues (each in a different individual) across different tissue types (5 brain, 1 thyroid, 1 heart, table S6). This result most likely suggests that blood had been captured in the tissue samples. Previous results found an increase in the number of detected mutations above the age of 70 (3). Although the oldest person in our dataset was 70 years old, we did observe a trend (with borderline significance: one-sided Wilcoxon P value = 0.049) in which these 83 individuals were older than the rest of the cohort.
Clonal expansion increases with age and tissue-specific cell proliferation rate
Several factors can affect the number of mutations accumulated in normal tissues: (i) age, (ii) accumulated DNA damage, and (iii) a tissue’s propensity for forming macroscopic clones. All are expected to be more prominent in tissues with a higher cell proliferation rate (38, 39). To test for these associations, we examined whether the age of the individual correlated with the average number of accumulated mutations across tissues. After the age of 45 (the cohort median age), both the number of CpG>T mutations (aging signature) as well as the total number of mutations significantly increased (one-sided Wilcoxon test P value = 0.001 and P value = 2.2 × 10−4, respectively, Fig. 3A, top panels). Importantly, this significant association remained after (i) controlling for the number of tissues sequenced in each individual (table S7) and (ii) splitting all individuals to three age groups (fig. S11A–B, E). As expected, when considering the top 10 tissues with the highest level of cell proliferation (as determined by MKI67 expression, a marker of proliferation (21), table S8), this relationship became more significant for the total number of mutations (one-sided Wilcoxon test P value = 2.3 × 10−5) and remained similar for the aging mutations (one-sided Wilcoxon test P value = 0.004). In the 10 tissues with the lowest cell proliferation, no significant association with age was observed.
Fig. 3: Mutation load is associated with age and tissue-specific proliferation rate.
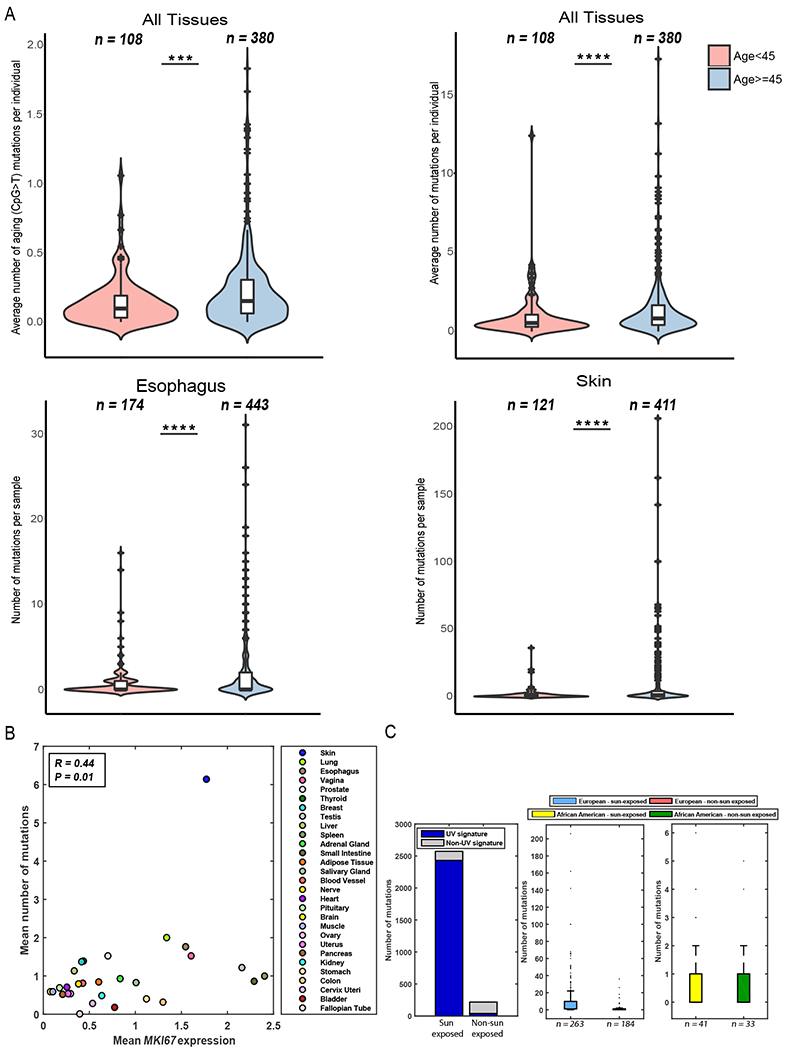
(A) Top panels: Differences in the average number of aging-related and total number of mutations before and after the age of 45 (left and right panels, respectively). Bottom panels: Differences in mutation number in esophagus and skin samples before and after the age of 45 (left and right panel, respectively). Box plots show median, 25th, and 75 percentiles in each group. Red crosses represent the outliers, and black crosses represent the mean. (B) Mean expression of the proliferation marker MKI67 vs. the average number of mutations found in each tissue. (C) Left panel: Number of mutations associated with the UV signature in sun-exposed and non–sun exposed skin samples. Middle panel: Number of mutations found in sun-exposed and non–sun exposed skin samples taken from individuals of European ancestry. Right panel: Number of mutations found in sun-exposed and non–sun exposed skin samples taken from individuals of African-American ancestry.
Next, we tested if there was a tissue-specific association with age. A significant association was detected in skin and esophagus tissues (one-sided Wilcoxon test P value = 2.1 × 10−6 and P value = 1.5 × 10−5, respectively, Fig. 3A, bottom panels). When considering sun-exposed and non–sun exposed skin separately, we found that while the number of observed mutations increased with age in both skin types, the increase was significantly greater in sun-exposed vs. non–sun exposed skin (Odds ratio = 3.29 and 1.26, Fisher’s P value = 1.7 × 10−8 and 0.68, respectively, using the number of mutations below or above the tissue’s median mutation number [med. = 2], fig. S11C). Since these samples derive from the same tissue type, and hence are expected to have a similar cell proliferation rate, this result can either suggest that (i) increased exposure to UV light and other environmental factors contributes to DNA damage as an individual ages, or (ii) the size of clones increases in both skin types with increasing age, but the clones in sun-exposed skin enable us to detect the mutations that were acquired earlier in life. Differences were also observed when testing esophagus-derived mucosa, gastroesophageal junction, and muscularis tissues (Odds ratio = 4.3, 0.87, and 4.7; Fisher’s P value = 2.6 × 10−7, 1 and 0.17, respectively; [med. = 2]; fig. S11D). The lack of association in other tissues could be due to either low cell proliferation rates or the presence of clones below our detection threshold.
We next directly examined whether cell proliferation associated with the number of accumulated mutations across tissues and indeed found a significantly higher expression level of MKI67 in tissues with a higher number of mutations (one-sided Wilcoxon test P value = 8.2 × 10−4 and P value = 1.2 × 10−4 for all primary and sub-region tissues, respectively, with an overall Spearman correlation of R = 0.44 and P value = 0.01, Figs. 3B and S11F (21)). Overall, these data suggest that both aging and exposure to mutagenic factors contribute to the number of accumulated mutations, especially in tissues with a high cell proliferation rates (38, 40).
Mutational signatures in normal tissues
In addition to the identified aging mutations (CpG>T), we examined whether and which other mutational processes were active in normal samples by applying SignatureAnalyzer (21, 26). Since most samples had a small number of mutations (fig. S12), we analyzed only the 169 samples with ≥10 mutations. SignatureAnalyzer identified a UV signature in skin samples. This UV signature is common in melanoma and has been reported in skin fibroblasts and normal skin samples (17, 37). When examining sun-exposed and non–sun exposed skin separately, the UV signature was active in 62/67 sun-exposed samples and only 1/5 non sun–exposed samples (Fig. 3C, Fisher’s exact test P value = 5.7 × 10−4). Interestingly, all the skin samples with ≥10 mutations analyzed here were from the 447 individuals of European ancestry. In contrast, none of the samples from the 74 individuals of African ancestry had more than 6 mutations (Fig. 3C), regardless of sun-exposure. Indeed, no difference in mutations was found between sun-exposed and non–sun exposed skin among African-American individuals (an average of 0.87 and 0.81 mutations, respectively; one-sided Wilcoxon P value = 0.58). Overall, skin was the only tissue that showed a significant difference between the total number of mutations detected in European vs. African-ancestry samples (one-sided Wilcoxon P value = 1.9 × 10−5, fig. S13).
Mutations in cancer genes in normal tissues
To determine whether somatic mutations in normal tissues occur in known cancer genes, we tested the frequency of non-synonymous mutations within Cancer Gene Census (CGC) genes (41). This CGC set represents genes in which mutations have been causally implicated in cancer. We found that 3% of the samples and 33% of the individuals carried at least one non-synonymous mutation in a CGC gene. Examining the tissues enriched with non-synonymous mutations (21), we identified that skin, esophagus, adipose, adrenal gland, and uterus tissues were significantly enriched with mutations in CGC genes (empirical Q value < 0.1), after controlling for both gene length and coverage (fig. S14A).
Consistent with previous findings (17, 18), the most frequently mutated cancer genes in our data were TP53 and NOTCH1 (Fig. 4A). Examining whether the number of mutations differed between samples carrying TP53 mutations and those that did not, we found that the TP53-associated samples had significantly more mutations (two-sided Wilcoxon P value = 9.2 × 10−9). To test if these TP53 mutations conferred a growth advantage to the cell, we analyzed their allele fraction level relative to all other detected mutations in the same sample (21). Indeed, the allele fractions of TP53 mutations were significantly higher than other mutations in the corresponding sample (empirical P value < 0.02; fig. S14B). Similarly, we also found that the NOTCH1-mutated cases had a significant increase in the overall number of mutations (two-sided Wilcoxon P value = 1 × 10−7) as well as a significantly higher allele fraction of the NOTCH1 mutation (empirical P value < 9.9 × 10−4, fig. S14C). These findings were independent of TP53 and NOTCH1 expression levels (fig. S14B–C). This higher allele fraction of the TP53 and NOTCH1 mutations compared to other mutations in the same samples suggests that these mutations appear early in the history (i.e., the trunk) of these clones. However, since early appearance in the trunk does not guarantee that these mutations conferred a growth advantage, we cannot rule out the possibility that these early events are the result of genetic drift; we do consider this possibility unlikely, however, since both TP53 and NOTCH1 are known cancer genes. Overall, samples carrying TP53 or NOTCH1 mutations were found only in skin and esophagus tissues (with equal proportions in each tissue, table S3), but no samples harbored mutations in both of these genes.
Fig. 4: Mutations in cancer genes across normal tissues.
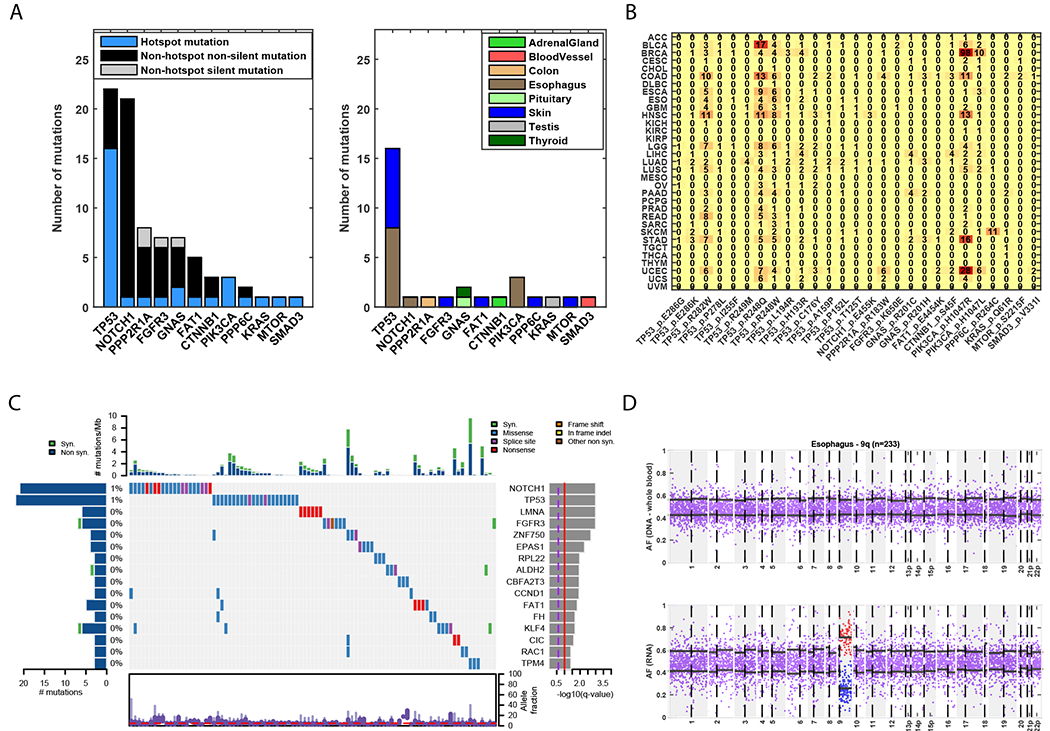
(A) Genes in which hotspot mutations were detected. Left panel: Number of hotspot mutations detected in each gene, and number of silent and non-silent mutations that are not in hotspots. Right panel: Normal tissues in which the hotspot mutations were detected. All hotspot mutations except two (FAT1 p.E4454K; FGFR3 p.K650E) were annotated as pathogenic. (B) Occurences of each hotspot mutation found in different TCGA cohorts. (C) Co-mutation plot for genes significantly mutated in a pan-normal analysis, ordered by their significance level (by MutSig2CV); data show 93 of 6707 samples with at least one mutation in these genes and the overall frequency among samples with at least one mutation. The distribution of allele fraction of mutations appears at the bottom. (D) Allelic imbalance in chromosme 9q of a normal esophagus sample. Top panel: Allele fraction of heterozygous sites based on DNA from a matched-blood sample. Bottom panel: Allele fraction of heterozygous sites based on RNA from the esophagus sample. The black horizonal lines indicate the mean allele fraction per chromosomal arm of sites with allele fraction smaller or greater than 0.5.
We next examined whether any of the ~1760 recurrently mutated sites (hotspots) in known cancer genes were observed in normal tissues (table S9, (21)). We found 30 such mutations in 8 tissues that overall included 27 hotspots in 12 genes (Fig. 4A, table S10). The gene with the greatest number of detected hotspot mutations was TP53 with 16 known hotspot mutations in both skin and esophagus samples, 14 of which were observed once in our dataset (Fig. 4A). In total, 10 of these mutations were previously reported in either (i) normal human skin, peritoneal or uterine lavage fluids taken from healthy women, or (ii) in human pluripotent stem cells (16, 17, 42, 43). Reviewing IARC TP53 database (44), we found that all of these mutations were annotated as deleterious by SIFT (45). Interestingly, although all of the mutations were annotated as loss-of-function in yeast, 3 (R248Q, R248W, R282W) were reported to have gain-of-function activities (46). R248Q knock-in mice showed an earlier onset of tumor formation and reduced lifespan, as well as an expansion of hematopoietic and mesenchymal stem cell progenitors (47). The R248W variant was involved in multiple gain-of-function activities, including promotion of cell invasion (48) and increased cell proliferation (49), among others (46). The R282W variant increased colony formation (50). We found that these 14 hotspot sites shared some tissue specificity with the corresponding primary cancerous tissue, wherein 4 skin and 5 esophagus mutations were also observed in melanoma and esophagus TCGA samples, respectively (Fig. 4B).
Among the other 14 non-TP53 hotspot mutations, all but 2 were annotated as pathogenic by FATHMM (51), and 7 were also observed in their corresponding cancer type (Fig. 4B). Three PIK3CA mutations in the p.H1047L and p.H1047R hotspots, which are common in multiple cancers (including esophageal cancer), were observed in normal esophagus mucosa samples. The p.Q61R KRAS hotspot mutation found in a normal testis sample of a 58-year-old male had been detected in a testicular germ cell cancer. The p.R183W hotspot mutation in the cell-growth regulator PPP2R1A detected in a normal colon sample here was also detected in colorectal cancer. While the β isoform (PPP2R1B) was discovered as a tumor suppressor in colon cancer cell lines and primary tumors (52), the α isoform had also been observed in a cohort of primary colon tumors (53). The hotspot mutation p.S45F in CTNNB1 (β-catenin) found in the normal adrenal gland sample of a 58-year-old female had previously been detected in adrenocortical adenomas; this hotspot was also found significantly mutated in adrenocortical tumors (10, 54, 55) that resulting in Wnt/β-catenin pathway deregulation. The hotspot mutation p.R264C in the PPP6C gene that we detected in normal skin was also observed in melanoma, wherein this gene was found to be significantly mutated (56).
To further explore whether clonal expansion observed in normal tissues was in part due to positive selection, we computed the dN/dS ratio per gene (57), taking into account the trinucleotide context and the mutational spectrum (21). We found that both CGC genes and cancer genes listed in Lawrence et al. (25) were enriched with genes exhibiting a higher rate of non-synonymous mutations (one-sided Wilcoxon P value = 3.4 × 10−4 and P value = 9.3 × 10−4, respectively). These data suggest that some of these mutations may confer a selective advantage. Of note, these results become insignificant when removing genes identified in skin and esophagus tissues––this finding could either be due to the overall low number of mutations detected in the other tissues, or alternatively suggest that clones in skin and esophagus tissues undergo positive selection, while clones in the other tissues reflect genetic drift.
To more specifically identify which of these cancer genes are significantly mutated, we performed a pan-normal analysis by applying MutSig2CV (25) to all 2519 samples in which we detected at least one mutation, restricting the test to 718 known cancer genes ((21), table S11). This analysis yielded 16 significantly mutated genes, with 99 non-silent mutations spanning 17 tissues, 90 samples, and 80 individuals (Figs. 4C and S14D). In addition to TP53, NOTCH1, and FAT1 previously reported as significantly mutated in normal skin (17), we also identified other genes such as RAC1 and ZNF750, which are significantly mutated in melanoma and esophagus squamous cell carcinoma, respectively (9, 25). Overall, our results show that cancer genes and hotspots are present in normal tissues, especially in skin and esophagus tissues.
Allelic imbalance in normal tissues
To study other somatic alterations in normal samples, we developed a method for identifying allelic imbalance across chromosome arms using RNA-seq data (21), which is similar to previous approaches used for detecting allelic imbalance (58, 59). To test our approach, we applied it to four TCGA samples for which DNA and RNA were co-extracted and showed that the vast majority of allelic imbalance events at the chromosomal arm level detected in the RNA were also found in the DNA, and vice versa (fig. S15). In addition, we found a high correlation between the allele fraction of heterozygous sites in the RNA and in the DNA (R range = 0.45–0.7; P value < 8 × 10−225, fig. S16), suggesting that approaches developed for detecting allelic imbalance in DNA can also work for RNA.
Similar to a recent concurrent study of normal esophagus DNA (18), we identified 8 esophagus mucosa samples that had an allelic imbalance in 9q (Figs. 4D and S17). Two out of the 8 samples also had a nonsense or missense mutation in NOTCH1 (hypergeometric P value = 0.02), a gene also located on 9q. The allele fraction of these mutations was relatively high (0.22 and 0.12) and at the top quintile of their corresponding samples. This might suggest that either the wild-type copy of these chromosome arms was lost, or that the mutated copy was gained. Frequent amplifications of NOTCH1 were reported in esophageal squamous cell carcinoma (8). Interestingly, 9q loss was more common in esophageal dysplasia than in esophageal squamous cell carcinoma (60). Its detection here in non-dysplastic lesions suggests that this may be an early event in the development of dysplasia. One additional sample with 9q imbalance was found to carry mutations in both TP53 and FAT1. An allelic imbalance in 22p and a mutation in NOTCH1 were also identified in an additional esophagus sample (fig. S17). Finally, we identified a testis sample with a strong allelic imbalance in 17p, with no point mutation detected (fig. S17).
Discussion
This study presents a comprehensive overview of somatic clonal expansion in human tissues. While detecting somatic mutations using RNA is limited to expressed genes, we show that RNA analysis can reveal true somatic variations after accounting for both sequencing and alignment noise; moreover, RNA-based analysis can identify both underlying mutational processes and significantly mutated genes. Taking advantage of our approach, thousands of somatic mutations were detected across all human tissues and in almost all tested individuals, including mutations at cancer hotspots and other cancer genes.
Macroscopic clonal expansion was detected in all tissues. However, a greater number of accumulated mutations were observed in sun-exposed skin, esophagus mucosa, and lung than in other tissues. All 3 of these tissues are exposed to carcinogenic environmental factors, emphasizing the contribution of extrinsic factors to the mutagenesis process. Indeed, these tissues are also among those carrying the greatest number of somatic mutations in cancer patients (29), consistent with the notion that a non-negligible proportion of the mutations observed in cancer accumulate well before disease (38). In both skin and esophagus, we observed an association between the number of mutations in normal tissue and age, suggesting a contribution of somatic mosaicism to the aging phenotype (61). The lack of association with age in normal lung tissue may be masked due to effects of other factors that are missing in our data, such as smoking or exposure to air pollution.
Beyond these intrinsic and extrinsic factors, the cellular microenvironment and tissue architecture are likely to influence the differences observed among tissues. Studies of different tumor types have shown differences in both the composition of the microenvironment as well as the transcriptional program active in each tissue (62–69). In addition, it was previously argued that tissue compartmentalization can affect the rate at which cancer mutations accumulate (70). For instance, the arrangement of the intestinal epithelium into crypts and villi is believed to limit the expansion of fitter cells (71). Overall, the complex nature of transformation from a normal to a cancer cell within different tissues is a result of the interplay among genetic and epigenetic events, tissue structure, exposure, and the tissue microenvironment. More comprehensive and dedicated data and metadata from various tissues should be collected to further study these relationships.
Compared to studies focusing on microscopic clones (17, 18), we find a significantly lower number of clonal expansions, even despite the fact that our scale is much larger and not restricted to a specific set of genes. While this result can be partially explained by our missing mutations in lowly expressed genes, it also suggests that the majority of clones remain microscopic and do not expand to a size that can currently be detected by bulk RNA-seq. In addition, while TP53 and NOTCH1 were the most mutated genes in our data with a relatively high allele fraction, their overall frequency was lower than previously observed in microscopic clones. This suggests that these gene mutations do not drive clonal growth beyond a certain size without additional genetic, epigenetic, or environmental contributions. Furthermore, it should be noted that we identified known driver genes in some clones but not in many others. This observation may suggest that these clones do not have greater fitness and are the result of genetic drift. In this study, due to the non-trivial relation between variant allele fraction in RNA-seq and clone size, we have decided not to draw any conclusions from the distribution of allele fractions on selection beyond our findings for TP53 and NOTCH1. Large scale studies analyzing DNA sequencing data are needed to better distinguish selection vs. drift in macroscopic clones in normal tissues.
The overall low rate of cancer-related events in our data (<10%) most likely reflects both our detection sensitivity and the fact that we have analyzed only a single biopsy from each tissue type in each individual. Given previous results from deep sequencing on much smaller tissue biopsies (17, 18), it is reasonable to assume that we would have detected a larger number of somatic mutations across all normal tissues if we analyzed more biopsies from any given tissue type, and if those biopsies were more enriched with epithelial cells. This implies that while these macroscopic clones have expanded to the point of detection, they would remain harmless and may not develop into cancer until––and only if––additional transforming events occur. Also, the detection of hotspots and other mutations in cancer genes across various normal human tissues emphasizes the need for identifying drivers of the disease while considering the non-pathogenic landscape of mutations. Such findings may greatly impact the selection of therapeutic and vaccination targets.
Understanding the earliest genetic events that occur in human tissues may advance our understanding of aging and cancer. Therefore, initiatives such as the Pre-Cancer Genome Atlas (72) will significantly aid in our ability to detect and treat the disease in its early stages. As all individuals in this study are deceased, we cannot determine whether the detected clones would have eventually developed into cancer upon acquisition of additional genetic or epigenetic abnormalities. Studying clonal expansion of normal samples longitudinally as they progress from normal tissue to microscopic clones and finally to macroscopic clones will shed light on which of these pre-malignant lesions has the capacity to transform into cancer; moreover, such a longitudinal study can reveal the required combinations of genetic and/or epigenetic events needed for transformation.
Supplementary Material
Brief methods.
We developed a method, called RNA-MuTect, for identifying somatic mutations using a tissue RNA sample and its matched-normal DNA. RNA-MuTect includes several filtering steps designed for RNA sequences. RNA-MuTect was validated on both cancer and normal samples from The Cancer Genome Atlas (TCGA), wherein DNA and RNA were co-extracted from the same samples. A power analysis was performed to evaluate the statistical power of observing a mutation, given the mutation allele fraction and sequence coverage at the site. MutSigCV and SignatureAnalyzer (25, 26) were applied for identifying significantly mutated genes and mutational signatures, respectively. A context dependent dN/dS analysis was performed for identifying genes with an excessive number of protein-altering mutations. We applied HaplotypeCaller and fitted a beta distribution in order to detect events of allelic imbalance at the chromosome arm level.
Acknowledgements
We would like to thank B. Ebert, A. Bass, and T. R. Golub for helpful comments on the manuscript. We would also like to thank J. Gastier-Foste and E. Zmuda for helpful information regarding TCGA samples. We thank Mendy Miller for help in editing the manuscript.
Funding: K.Y was funded by the Broad-ISF postdoctoral fellowship and the Weizmann award for Women in Science. G.G was partially funded by the GTEx LDACC (HHSN268201000029C) and the Paul C. Zamecnick, MD, Chair in Oncology at MGH.
Competing interests: G.G. receives research funds from IBM and Pharmacyclics. G.G. is an inventor on patents related to MuTect, MutSig, and ABSOLUTE.
Footnotes
Data and materials availability: All data are available in the manuscript or the supplementary materials. The code of RNA-MuTect is available on Zenodo: https://zenodo.org/record/2620062
References
- 1.McFarland CD, Korolev KS, Kryukov GV, Sunyaev SR, Mirny LA, Impact of deleterious passenger mutations on cancer progression. Proc. Natl. Acad. Sci 110, 2910–2915 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vermulst M et al. , DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat. Genet 40, 392–394 (2008). [DOI] [PubMed] [Google Scholar]
- 3.Jaiswal S et al. , Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med 371, 2488–98 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Poduri A, Evrony GD, Cai X, Walsh CA, Somatic Mutation, Genomic Variation, and Neurological Disease. Science (80-. ). 341 (2013) (available at http://science.sciencemag.org/content/341/6141/1237758.abstract). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Greaves M, Maley CC, Clonal evolution in cancer. Nature. 481, 306 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abeshouse A et al. , The Molecular Taxonomy of Primary Prostate Cancer. Cell. 163, 1011–1025 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brennan CW et al. , The somatic genomic landscape of glioblastoma. Cell. 155, 462–477 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim J et al. , Integrated genomic characterization of oesophageal carcinoma. Nature (2017), doi: 10.1038/nature20805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin D-C et al. , Genomic and molecular characterization of esophageal squamous cell carcinoma. Nat. Genet 46, 467–473 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zheng S et al. , Comprehensive Pan-Genomic Characterization of Adrenocortical Carcinoma. Cancer Cell. 29, 723–736 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burk RD et al. , Integrated genomic and molecular characterization of cervical cancer. Nature. 228 (2017), doi: 10.1038/nature21386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoadley KA et al. , Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell. 173, 291–304.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Genovese G et al. , Synthetic vulnerabilities of mesenchymal subpopulations in pancreatic cancer. Nature. 542, 362 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Knijnenburg TA et al. , Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep. 23, 239–254.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cai X et al. , Single-Cell, Genome-wide Sequencing Identifies Clonal Somatic Copy-Number Variation in the Human Brain. Cell Rep. 8, 1280–1289 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krimmel JD et al. , Ultra-deep sequencing detects ovarian cancer cells in peritoneal fluid and reveals somatic TP53 mutations in noncancerous tissues. Proc. Natl. Acad. Sci . 113, 6005–6010 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martincorena I et al. , High burden and pervasive positive selection of somatic mutations in normal human skin. Science (80-. ). 348, 880–886 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martincorena I et al. , Somatic mutant clones colonize the human esophagus with age. Science (80-. ). (2018) (available at http://science.sciencemag.org/content/early/2018/10/17/science.aau3879.abstract). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Genovese G et al. , Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N. Engl. J. Med 371, 2477–2487 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lonsdale J et al. , The Genotype-Tissue Expression (GTEx) project. Nat Genet. 45, 580–585 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Supplementary Methods.
- 22.Supplementary text.
- 23.Tang X et al. , The eSNV-detect: a computational system to identify expressed single nucleotide variants from transcriptome sequencing data. Nucleic Acids Res. 42, e172–e172 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sheng Q, Zhao S, Li C-I, Shyr Y, Guo Y, Practicability of detecting somatic point mutation from RNA high throughput sequencing data. Genomics. 107, 163–169 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lawrence MS et al. , Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 505, 495–501 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim J et al. , Somatic ERCC2 mutations are associated with a distinct genomic signature in urothelial tumors. Nat Genet. 48, 600–606 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taylor-Weiner A et al. , DeTiN: overcoming tumor-in-normal contamination. Nat. Methods 15, 531–534 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chapman MA et al. , Initial genome sequencing and analysis of multiple myeloma. Nature. 471, 467–472 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lawrence MS et al. , Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 499, 214–218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pleasance ED et al. , A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 463, 191–196 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.a Gilchrest B, Eller MS, Geller AC, Yaar M, I Nduced By U Ltraviolet. N. Engl. J. Med 340, 1341–1348 (1999). [DOI] [PubMed] [Google Scholar]
- 32.Beate P et al. , Cigarette smoking and lung cancer—relative risk estimates for the major histological types from a pooled analysis of case–control studies. Int. J. Cancer 131, 1210–1219 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kamangar F, Chow WH, Abnet CC, Dawsey SM, Environmental Causes of Esophageal Cancer. Gastroenterol. Clin. North Am 38, 27–57 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raaschou-Nielsen O et al. , Air pollution and lung cancer incidence in 17 European cohorts: prospective analyses from the European Study of Cohorts for Air Pollution Effects (ESCAPE). Lancet Oncol. 14, 813–822 (2013). [DOI] [PubMed] [Google Scholar]
- 35.Islami F et al. , High-temperature beverages and foods and esophageal cancer risk—A systematic review. Int. J. Cancer 125, 491–524 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen Y et al. , Consumption of hot beverages and foods and the risk of esophageal cancer: a meta-analysis of observational studies. BMC Cancer. 15, 449 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saini N et al. , The Impact of Environmental and Endogenous Damage on Somatic Mutation Load in Human Skin Fibroblasts. PLoS Genet. 12, 1–25 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tomasetti C, Vogelstein B, Parmigiani G, Half or more of the somatic mutations in cancers of self-renewing tissues originate prior to tumor initiation. Proc. Natl. Acad. Sci. U. S. A 110, 1999–2004 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tomasetti C, Vogelstein B, Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science (80-. ) 347, 78–81 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alexandrov LB et al. , Clock-like mutational processes in human somatic cells. Nat. Genet 47, 1402–7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Forbes SA et al. , COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 43, D805–D811 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nair N et al. , Genomic Analysis of Uterine Lavage Fluid Detects Early Endometrial Cancers and Reveals a Prevalent Landscape of Driver Mutations in Women without Histopathologic Evidence of Cancer: A Prospective Cross-Sectional Study. PLOS Med. 13, e1002206 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Merkle FT et al. , Human pluripotent stem cells recurrently acquire and expand dominant negative P53 mutations. Nature, 1–11 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Petitjean A et al. , Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum. Mutat 28, 622–629 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Kumar P, Henikoff S, Ng PC, Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc 4, 1073–1081 (2009). [DOI] [PubMed] [Google Scholar]
- 46.Muller PAJ, Vousden KH, Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell. 25, 304–317 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hanel W et al. , Two hot spot mutant p53 mouse models display differential gain of function in tumorigenesis. Cell Death Differ. 20, 898–909 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Muller PAJ et al. , Mutant p53 Drives Invasion by Promoting Integrin Recycling. Cell. 139, 1327–1341 (2009). [DOI] [PubMed] [Google Scholar]
- 49.Yan W, Chen X, Identification of GRO1 as a critical determinant for mutant p53 gain of function. J. Biol. Chem 284, 12178–12187 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Scian MJ et al. , Tumor-derived p53 mutants induce oncogenesis by transactivating growth-promoting genes. Oncogene. 23, 4430–4443 (2004). [DOI] [PubMed] [Google Scholar]
- 51.Shihab HA et al. , Predicting the Functional, Molecular, and Phenotypic Consequences of Amino Acid Substitutions using Hidden Markov Models. Hum. Mutat 34, 57–65 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang SS et al. , Alterations of the PPP2R1B gene in human lung and colon cancer. Science. 282, 284–287 (1998). [DOI] [PubMed] [Google Scholar]
- 53.Muzny DM et al. , Comprehensive molecular characterization of human colon and rectal cancer. Nature. 487, 330–337 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bonnet S et al. , Wnt/β-catenin pathway activation in adrenocortical adenomas is frequently due to somatic CTNNB1-activating mutations, which are associated with larger and nonsecreting tumors: A study in cortisol-secreting and -nonsecreting tumors. J. Clin. Endocrinol. Metab 96, 419–426 (2011). [DOI] [PubMed] [Google Scholar]
- 55.Leal LF et al. , Wnt/β-catenin pathway deregulation in childhood adrenocortical tumors. J. Clin. Endocrinol. Metab 96, 3106–3114 (2011). [DOI] [PubMed] [Google Scholar]
- 56.Hodis E et al. , A Landscape of Driver Mutations in Melanoma. Cell. 150, 251–263 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.KIMURA M, Preponderance of synonymous changes as evidence for the neutral theory of molecular evolution. Nature. 267, 275–276 (1977). [DOI] [PubMed] [Google Scholar]
- 58.González JR et al. , A fast and accurate method to detect allelic genomic imbalances underlying mosaic rearrangements using SNP array data. BMC Bioinformatics. 12, 166 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weissbein U, Schachter M, Egli D, Benvenisty N, Analysis of chromosomal aberrations and recombination by allelic bias in RNA-Seq. Nat. Commun 7, 12144 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shi ZZ et al. , Consistent and differential genetic aberrations between esophageal dysplasia and squamous cell carcinoma detected by array comparative genomic hybridization. Clin. Cancer Res 19, 5867–5878 (2013). [DOI] [PubMed] [Google Scholar]
- 61.Risques RA, Kennedy SR, Aging and the rise of somatic cancer-associated mutations in normal tissues. PLOS Genet. 14, e1007108 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lin EW, Karakasheva TA, Hicks PD, Bass AJ, Rustgi AK, The tumor microenvironment in esophageal cancer. Oncogene. 35, 5337 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sui X, Lei L, Chen L, Xie T, Li X, Inflammatory microenvironment in the initiation and progression of bladder cancer. Oncotarget. 8, 93279–93294 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Villanueva J, Herlyn M, Melanoma and the tumor microenvironment. Curr. Oncol. Rep 10, 439–446 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Quail DF, Joyce JA, The Microenvironmental Landscape of Brain Tumors. Cancer Cell. 31, 326–341 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Place AE, Jin Huh S, Polyak K, The microenvironment in breast cancer progression: biology and implications for treatment. Breast Cancer Res. 13, 227 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Soncin I et al. , The tumour microenvironment creates a niche for the self-renewal of tumour-promoting macrophages in colon adenoma. Nat. Commun 9, 582 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ghoneum A, Afify H, Salih Z, Kelly M, Said N, Role of tumor microenvironment in ovarian cancer pathobiology. Oncotarget. 9, 22832–22849 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mittal V et al. , The Microenvironment of Lung Cancer and Therapeutic Implications. Adv. Exp. Med. Biol 890, 75–110 (2016). [DOI] [PubMed] [Google Scholar]
- 70.Cairns J, Mutation selection and the natural history of cancer. Sci. Aging Knowl. Environ 2006, cp1 (2006). [Google Scholar]
- 71.Quastler H, Sherman FG, Cell population kinetics in the intestinal epithelium of the mouse. Exp. Cell Res 17, 420–438 (1959). [DOI] [PubMed] [Google Scholar]
- 72.Campbell JD et al. , The Case for a Pre-Cancer Genome Atlas (PCGA). Cancer Prev. Res 9, 119–124 (2016). [DOI] [PubMed] [Google Scholar]
- 73.Cibulskis K et al. , ContEst: estimating cross-contamination of human samples in next-generation sequencing data. Bioinformatics. 27, 2601–2602 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cibulskis K et al. , Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotech. 31, 213–219 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Costello M et al. , Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucleic Acids Res. 41, e67–e67 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ellrott K et al. , Scalable Open Science Approach for Mutation Calling of Tumor Exomes Using Multiple Genomic Pipelines. Cell Syst. 6, 271–281.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ramos AH et al. , Oncotator: Cancer Variant Annotation Tool. Hum. Mutat 36, E2423–E2429 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dobin A et al. , STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kim D, Langmead B, Salzberg SL, HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lek M et al. , Analysis of protein-coding genetic variation in 60,706 humans. Nature. 536, 285–291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kiran A, V Baranov P, DARNED: a DAtabase of RNa EDiting in humans. Bioinformatics. 26, 1772–1776 (2010). [DOI] [PubMed] [Google Scholar]
- 82.Ramaswami G, Li JB, RADAR: a rigorously annotated database of A-to-I RNA editing. Nucleic Acids Res. 42, D109–D113 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Haradhvala NJ et al. , Mutational Strand Asymmetries in Cancer Genomes Reveal Mechanisms of DNA Damage and Repair. Cell. 164, 538–549 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.DePristo MA et al. , A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 43, 491–498 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Blanc V, Davidson NO, C-to-U RNA editing: Mechanisms leading to genetic diversity. J. Biol. Chem 278, 1395–1398 (2003). [DOI] [PubMed] [Google Scholar]
- 86.Radenbaugh AJ et al. , RADIA: RNA and DNA Integrated Analysis for Somatic Mutation Detection. PLoS One. 9, e111516 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Piskol R, Ramaswami G, Li JB, Reliable Identification of Genomic Variants from RNA-Seq Data. Am. J. Hum. Genet 93, 641–651 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Network TCGA et al. , Comprehensive molecular portraits of human breast tumours. Nature. 490, 61 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.