

Abstract
Esophageal cancers comprise adenocarcinoma and squamous cell carcinoma, two distinct histologic subtypes. Both are difficult to treat and amongst the deadliest human malignancies. We describe protocols to initiate, grow, passage and characterize patient-derived organoids (PDO) of esophageal cancers as well as squamous cell carcinomas of oral/head-and-neck and anal origin. Formed rapidly (< 14 days) from a single cell suspension embedded in basement membrane matrix, esophageal cancer PDO recapitulate the histology of the original tumors. Additionally, we provide guidelines for morphological analyses and drug testing coupled with functional assessment of cell response to conventional chemotherapeutics and other pharmacological agents in concert with emerging automated imaging platforms. Predicting drug sensitivity and potential therapy resistance mechanisms in a moderate-to-high throughput manner, esophageal cancer PDO are highly translatable in personalized medicine for customized esophageal cancer treatments.
Keywords: esophageal cancer, patient-derived organoids, personalized medicine
INTRODUCTION
Esophageal cancers comprise esophageal adenocarcinoma (EAC) and esophageal squamous cell carcinoma (ESCC), two distinct histologic subtypes. Both EAC and ESCC are amongst the deadliest of all human malignancies featuring presentation at late stages, therapy resistance, early recurrence and poor prognosis (Rustgi & El-Serag, 2014). Grown rapidly ex vivo, patient-derived organoids (PDO) recapitulate the original tissue architecture of primary esophageal tumors (Kijima et al., 2019). Herein, we describe methods to generate and characterize esophageal cancer PDO in terms of growth, morphology and biology. Tissue specimens (diagnostic biopsies or surgically resected tumor tissues) are subjected to enzymatic and mechanical disruption in order to obtain single-cell suspensions, which are embedded in basement membrane matrix (Matrigel®) and cultured in the unique organoid growth media optimized for distinct histologic tumor types (i.e. adenocarcinoma vs. squamous cell carcinoma). The medium is replaced every other day. Following 14 days of culture, the resulting primary PDO are passaged, cryopreserved or harvested for morphological and functional analyses (Fig. 1).
Figure 1. Workflow of PDO generation and characterizationc.
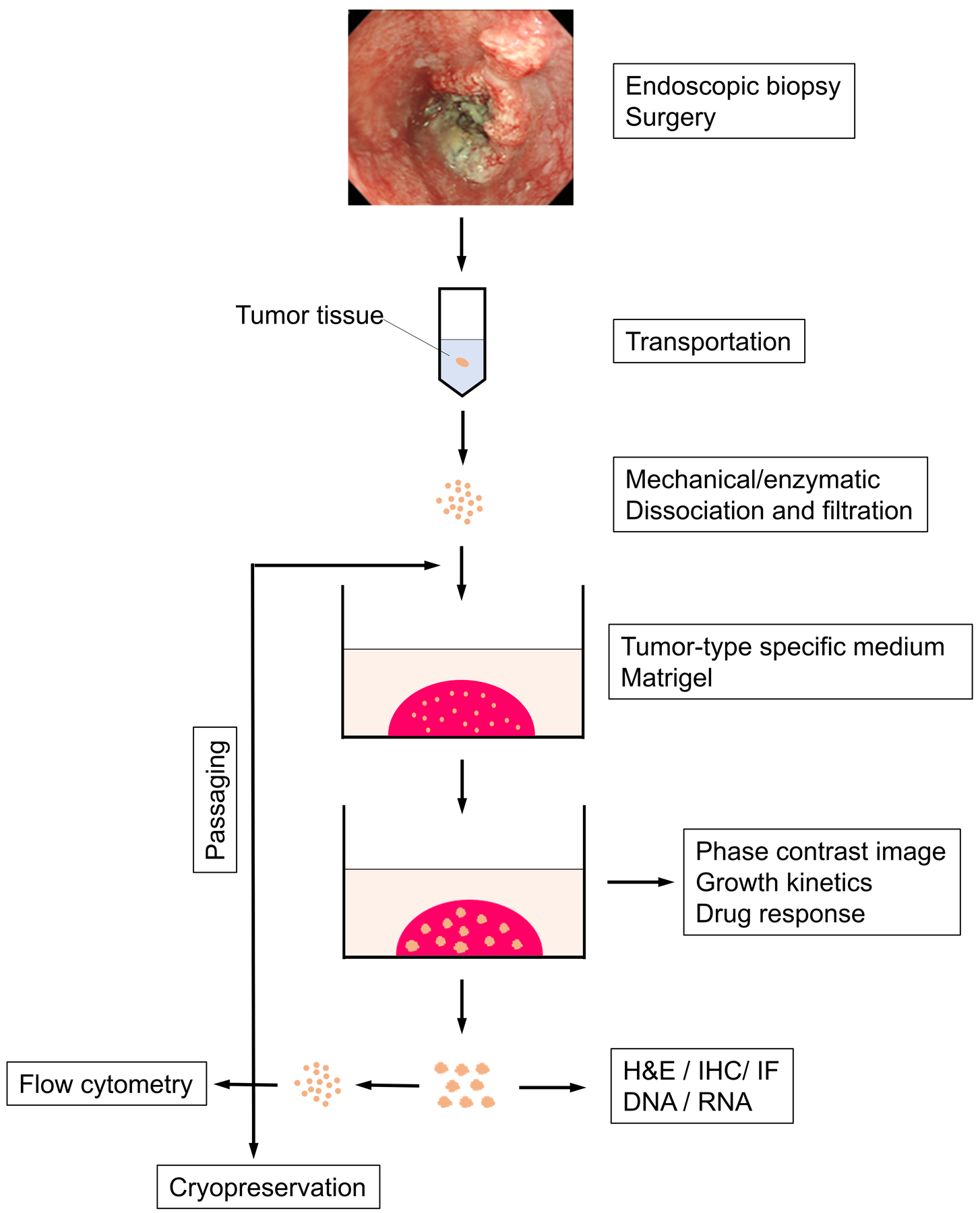
An esophageal tumor fragment, procured via either endoscopy or surgery, is dissociated and filtered into single cell suspension. Cells are seeded into Matrigel and grown with tumor type-specific organoid culture medium. Resulting PDO are processed for subculture or cryopreservation, and subjected to morphological and functional assays coupled with pharmacological drug treatments.
The harvested organoids can be subjected to a variety of morphological and functional assays including, but not limited to, immunohistochemistry, immunofluorescence, Western blotting, flow cytometry, quantitative polymerase chain reaction and RNA-sequencing (bulk and single-cell). The conditioned media from organoid cultures can be used for enzyme-linked immunosorbent assays. Passaged organoids can be tested for conventional and experimental therapeutics in a moderate-to-high throughput manner. Drug treatment of 3D organoids with variable concentrations of therapeutic agents determines their half maximal inhibitory concentration (IC50). Analysis of surviving cells provides insights into the potential drug resistance mechanisms.
Taken together, these methods provide a comprehensive experimental platform to study the molecular mechanisms underlying esophageal cancer cell propagation and drug responses.
STRATEGIC PLANNING
Studies need to be carried out as part of Institutional Review Board (IRB)-approved protocols with HIPAA compliance. Esophageal tumor specimens are procured from patients who consented to research biopsies during diagnostic upper endoscopy or surgery (e.g. esophagectomy or endoscopic mucosal resection) by expert gastroenterologists or surgeons at appropriate clinical care facilities with well-trained staff including clinical coordinators. Laboratory personnel should receive laboratory safety training about infectious agents such as human papilloma virus (HPV), hepatitis viruses (HBV, HCV) and human immunodeficiency virus (HIV) that may be potentially present in the patient materials. IRB protocols should be carefully designed to permit investigators access to corresponding patient clinical information such as age, gender, medical history, endoscopic findings, pathology details, therapy received, and patient outcomes.
Fresh tumor specimens need to be transported on wet-ice to a research lab for tissue processing and initiation of 3D organoids culture. Laboratory personnel should be notified in advance to schedule organoid culture. To maximize viable cell yield, tissue pieces should be processed as soon as possible. For overnight shipping, samples should be placed in polypropylene tubes (15-mL) filled with Basal Medium containing Penicillin, Streptomycin, Gentamicin and Amphotericin B to prevent cell culture contamination.
BASIC PROTOCOL 1
Generation of esophageal cancer PDO
A tissue specimen is obtained via diagnostic biopsy or surgery (esophagectomy or endoscopic mucosal resection) and dissociated by enzymatic digestion (Dispase and Trypsin), and embedded into a single-cell suspension in Matrigel® matrix. PDO are grown in tumor type-specific organoid medium at 37°C under a controlled atmosphere with 5% CO2 and 95% relative humidity, resulting in formation of spherical 3D structures representative of the original tumor.
Materials
All reused surgical tools (forceps and dissecting scissors) should be properly cleansed and autoclaved prior to use. Cell culture plasticware and glassware should be sterile and disposable. Water used to dissolve or dilute reagents should be ultrapure (e.g. Milli-Q®) and sterile. Standard equipment and tools for cell culture and cell biology (CO2 incubator, tissue culture hoods, liquid nitrogen cell storage tank, vacuum aspirator/collection system, centrifuges, electronic pipettors, etc.) are needed. While alternatives are available from multiple vendors, we will list key items we use routinely in our laboratories.
CO2 incubator (e.g. Heracell 150i CO2 incubator, Thermo Fisher Scientific)
Eppendorf ThermoMixer C (Thermo Fisher Scientific, cat. No. 14–285-562PM)
Spectrafuge benchtop mini-centrifuge (Spectrafuge, Labnet, cat. No. C1301) to spin 1.7-mL tubes quickly at room temperature
Eppendorf Refrigerated Centrifuge (Eppendorf 5424R, Thermo Fisher Scientific, cat. No. 5404000014) to spin 1.7-mL tubes in a controlled manner (i.e. time and relative centrifugal force)
Sorvall Centrifuge (Sorvall ST 16R, Thermo Fisher Scientific, cat. No. 75004380) Tx-400 Rotor with round buckets (Thermo Fisher Scientific, cat. No. 75003655) for 15-mL conical tubes and 5-mL round-bottom tubes for flow cytometry
ISOTEMP 220 Water bath (37°C, Thermo Fisher Scientific, cat. No. 15–462-20Q)
Countess™ II Automated Cell Counter (Invitrogen, cat. No. AMQAX1000)
Serological pipettor (pipette controller, e.g. PORTABLE PIPET-AID® W/110V charger) (Drummond Scientific, cat. No. 4–000-100)
P1000, P200, and P20 pipettor (Thermo Fisher Scientific, Cat. No. FA10006MTG, FA10005MTG, and FA10004MTG)
Forceps (VWR, Cat. No.82027–386)
Dissecting scissors with 30-mm cutting edge (VWR, Cat. No.25870–002)
60-mm cell culture dish (Thermo Fisher Scientific, cat. No. 13–690-081)
24-well plate (Thermo Fisher Scientific, cat. No. 12–556-006)
50-mL conical sterile polypropylene centrifuge tube (Thermo Fisher Scientific, cat. No. 12–565-270)
15-mL conical sterile polypropylene centrifuge tube (Thermo Fisher Scientific, cat. No. 14–959-53A)
1.7-mL microcentrifuge tube (DOT Scientific, cat. No. RN1700-GMT)
10-mL disposable plastic pipette (Thermo Fisher Scientific, cat. No. 170356)
1250-μL barrier pipette tips (GeneMate, cat. No. P-1237–1250)
200-μL barrier pipette tips (GeneMate, cat. No. P1237–200)
20-μL barrier pipette tips (GeneMate, cat. No. P1237–20)
100-μm cell strainer (Corning, cat. No. 431752)
1-mL syringe with a rubber plunger (BD Slip-Tip Tuberculin Syringe without needle, cat. No. 309659)
Countess™ cell counting chamber slide (Thermo Fisher Scientific, cat. No. C10228)
Basal Medium (Table 1)
Tumor type-specific organoid medium for ESCC (Table 2)
Tumor type-specific organoid medium for EAC (Table 3)
-
Matrigel® matrix (Corning, cat. No. 354234, stored at −20°C)
Dispense into 1.7-mL tubes (1 mL each) and store at −20°C. Thaw on ice for at least 1h or leave at 4°C for 2–3 h prior to use.
Hanks’ Balanced Salt Solution (HBSS) (Thermo Fisher Scientific, cat. No. 14175079)
50 U/mL Dispase (Corning, cat. No. 354235)
Store undiluted aliquots (1 mL) at −20°C. Thaw in water bath at 37°C for 1 min prior to use.
-
250 μg/mL Fungizone (Amphotericin B, Thermo Fisher Scientific, cat. No.15290018)
Store undiluted aliquots (1 mL) at −20°C. Thaw in water bath at 37°C for 1 min prior to use.
-
200 mg/mL Collagenase IV (optional) (Thermo Fisher Scientific, cat. No. 17104019)
Dissolve 1 g Collagenase IV into 5 mL HBSS to make a 200 mg/mL stock solution, dispense into 1 mL aliquots and store at −20°C. Thaw in water bath at 37°C for 1 min prior to use.
-
10 mM Y-27632 (optional) (ROCK1 inhibitor, Selleck Chemicals, cat. No. S1049)
Reconstituted Y-27632 2 HCl in DPBS, stored in aliquots at −20°C. Dissolve 1 mg or Y-27632 (MW=320.26) into 320 μL DPBS, dispense into 50 μL aliquots and store at −20°C. Thaw at room temperature prior to use.
-
HBSS-DF: HBSS containing Dispase and Fungizone
Dilute 50 U/mL Dispase in HBSS at 1:4 and add Fungizone to the final concentration of 0.5 μg/mL. To prepare a working solution for 2 tumor samples, add 400 μL 50U/mL Dispase and 4 μL 250 μg/mL Fungizone into 1.6 mL HBSS in 15-mL conical sterile polypropylene centrifuge tube.
-
HBSS-DFCY (optional): HBSS-DF supplemented with Collagenase IV and Y-27632
Add Collagenase IV and Y-27632 into HBSS-DF to the final concentration of 20 mg/mL and 10 μM, respectively. To prepare a working solution for 2 tumor sample, add 200 μL Collagenase IV (200 mg/mL) and 2 μL Y-27632 (10 mM) into 1.8 mL HBSS-DF in 15-mL conical sterile polypropylene centrifuge tube.
Dulbecco’s Phosphate-Buffered Saline (DPBS) (Thermo Fisher Scientific, cat. No. 14190250)
-
DNase I (optional) (Sigma-Aldrich, cat. No. 10104159001)
Reconstituted to 50 U/mL in sterile DPBS and stored in aliquots at −20°C.
0.25% Trypsin-EDTA (Invitrogen, cat. No. 25200056) stored at 4°C until use.
-
Soybean Trypsin Inhibitor (STI) (Sigma-Aldrich, cat. No. T9128).
Dissolve 250 mg STI in 1,000 mL DPBS (250 mg/L) and filter-sterile via a 1,000-mL filter cup (e.g. Nalgene™ Rapid-Flow™ Sterile Disposable Filter Unit with PES Membrane) (Thermo Fisher Scientific, cat. No. 167–0045), and dispense aliquots into 50-mL polypropylene tubes to be stored at 4°C.
0.4% Trypan Blue (Thermo Fisher Scientific, cat. No. T10282)
Tissue specimen kept at 4°C (or on wet-ice) in a 15-mL polypropylene tube containing Basal Medium (Table 1)
Table 1.
Basal medium
| Reagent | volume | Final concentration |
|---|---|---|
| Advanced DMEM/F12 | 500 mL | |
| GlutaMAX (100x) | 5 mL | 1x |
| HEPES (1 M) | 5 mL | 10 mM |
| Antibiotic-Antimycotic (100x) | 5 mL | 1x |
| Gentamicin (50 mg/mL) | 50 μL | 5 μg/ml |
Table 2.
ESCC Organoid Medium (50 mL)
| Reagent | Volume | Final concentration |
|---|---|---|
| Basal Medium | 47 mL | |
| RN conditioned medium | 1 mL | 2% |
| N-2 (100x) | 500 μL | 1x |
| B-27 (50x) | 1 mL | 1x |
| NAC (0.5 M) | 100 μL | 1 mM |
| EGF (500 ng/μL) | 5 μL | 50 ng/mL |
| Y-27632 (10 mM)* | 50 μL | 10 μM§ |
| Gentamicin (50 mg/mL) | 5 μL | 10 μg/ml |
| Antibiotic-Antimycotic (x100) | 500 μL | 1x |
, only needed when establishing during day 0-day 2
, Note that Basal Medium contains 5 μM Gentamicin before this supplementation.
Table 3.
EAC Organoid Medium (50 mL)
| Reagent | Volume | Final concentration |
|---|---|---|
| Basal Medium | 24 mL | |
| WRN Conditioned Medium | 24 mL | 50% |
| N-2 (100x) | 500 μL | 1x |
| B-27 (50x) | 1 mL | 1x |
| NAC (0.5 M) | 100 μL | 1 mM |
| CHIR99021 (5 mM) | 5 μL | 0.5 μM |
| EGF (500 ng/μL) | 25 μL | 250 ng/mL |
| A83–01 (5 mM) | 5 μL | 0.5 μM |
| SB202190 (10 mM) | 5 μL | 1 μM |
| Gastrin (1 mM) | 5 μL | 0.1 μM |
| Nicotinamide (1 M) | 1 mL | 20 mM |
| Y-27632 (10 mM) | 50 μL | 10 μM |
| Gentamicin (50 mg/mL) | 5 μL | 10 μM§ |
| Antibiotic-Antimycotic (x100) | 500 μL | 1x |
| FGF-10* (100 μg/mL) | 50 μL |
, Note that Basal Medium contains 5 μM Gentamicin before this supplementation.
, only added when establishing primary cultures and recovering from frozen stocks.
Dissociation of human esophageal biopsies to a single cell suspension
Transfer a tumor tissue with sterile forceps into a 60-mm cell culture dish.
Mince the tissue to smaller fragments (< 1 mm) with sterile dissecting scissors
Transfer minced tissue fragments into a 1.7-mL tube containing 1 mL HBSS-DF.
-
Incubate for 10 min at 37°C with simultaneous mixing at 800 rpm in Thermomixer C.
Optional: Collagenase IV and Y-27632 may be added into HBSS-DF (HBSS-DFCY) with an extended incubation time period for ~45 min to increase single cell yields.
Spin down quickly (~10 sec on Spectrafuge) at room temperature.
Discard the supernatant by a single-channel P1000 pipettor with a 1,250-μL tip.
-
Resuspend the pellet with 1 mL 0.25% trypsin-EDTA and incubate for 10 min at 37°C with simultaneous mixing at 800 rpm in Thermomixer C.
Optional: DNase I may be added in order to degrade DNA released from broken cells to minimize cell aggregates. Add 10 μL of 50 U/mL DNase I into 1 mL of 0.25% trypsin-EDTA to the final concentration of 0.5 U/mL.
Filtrate trypsinized tissue fragments (~1 mL) from step 7 over a 100-μm strainer into a 50-mL tube containing 8 mL STI.
Optional: Residual tissue fragments may be pelleted by spinning down quickly (~10 sec on Spectrafuge) at room temperature. Steps 7 and 8 may be repeated to increase single cell yields.
Remove a rubber plunger from a 1-mL syringe to use the rubber part of the plunger from a 1-mL syringe to force through the remaining tissue fragments over the strainer.
Take ~8 mL of the filtrate (cell suspension) out from the 50-mL tube using a 10-mL pipette. Use this filtrate to wash further the strainer into the identical 50-mL tube. Repeat this three times.
Transfer the filtrate into a 15-mL tube and centrifuge at 188 × g (1,000 rpm on Sorvall ST 16R) for 5 min at 4°C.
Discard the supernatant first by aspiration leaving the last ~1 mL which should be removed using a P1000 pipettor with a 1,250-μL tip so as not to disturb the pellet.
Resuspend the cell pellet in 1 mL Basal Medium with gentle pipetting.
Take 10 μL of the cell suspension from step 13 and mix with 10 μL 0.4% Trypan Blue (to stain dead cells) and load onto a cell counting chamber slide.
Determine cell density and cell viability (via Trypan Blue exclusion test) with Countess™ II Automated Cell Counter.
PDO initiation and growth in 24-well plates
In general, 2×104 cells will be seeded per well to initiate PDO in 24-well plates.
Thaw and keep Matrigel® on ice.
Pre-warm a 24-well plate at 37°C.
-
Use 6 wells to generate sufficient number of organoids for both initial propagation in a subsequent passage (3 wells) and morphological analysis (3 wells).
Prepare 1.4×105 cells (=7 wells × 2×104 cells/well for 6 wells plus an extra well). Take the necessary volume (mL) of cell suspension according to the following formula:
1.4×105 [cell number needed] ÷ [cell density (/mL) from step 15]
Transfer 1.4×105 cells into a 1.7-mL tube
Centrifuge at 500 × g (2,300 rpm on Eppendorf 5424R) for 3 min at room temperature to pellet cells.
Resuspend cells in 350 μL ice-cold Matrigel (50 μL/well × ([6+1] wells).
Dispense 50 μL each of cells-suspended Matrigel into each well of 24-well plate.
Place the 24-well plate in a CO2 incubator (37°C) for 30 min to allow the Matrigel to solidify.
Add into each well 500 μL tumor-type specific organoid medium (Tables 2 and 3) for ESCC or EAC according to clinical diagnosis and pathology report of the original tumor.
Refresh the organoid medium every 2–3 d. To remove spent media, use a P1000 pipettor or aspirate very carefully so as not to disturb the Matrigel.
Monitor contamination and organoid growth under a phase-contrast microscope (See Basic Protocol 3)
Grow organoids for up to 10–14 d to be passaged or harvested. If spherical structures (organoids) emerge but grow slowly, extend the culture period by additional 7–14 d.
BASIC PROTOCOL 2
Propagation and cryopreservation of esophageal cancer PDO
Once established, esophageal cancer cells can be isolated from the primary PDO by enzymatic dissociation to seed subsequent passage (i.e. sub-culture) in order to propagate further for histological analyses in Basic Protocol 4 and flow cytometry in Basic Protocol 5. PDO may be sub-cultured in 96-well plates for experiments such as drug treatment in Basic Protocol 6. Additionally, isolated esophageal cancer cells can be cryopreserved for long-term storage.
Materials
All cell culture equipment, centrifuges, plasticware, pipettors, reagents are the same as in Basic Protocol 1 unless otherwise listed below.
96-well plate (Thermo Fisher Scientific, cat. No. 12–556-008)
5-mL Falcon round-bottom tube with a 35-μm cell strainer cap (BD Bioscience, cat. No. 352235)
Fetal Bovine Serum (FBS) (HyClone, cat. No. SH30071.03), stored at −20°C.
Dimethyl sulfoxide (DMSO) (Sigma-Aldrich, cat. No. D4540)
-
Freezing medium
Mix 9.0 mL FBS and 1.0 mL DMSO in a 15-mL tube and store at −20°C. Do not repeat freeze-thawing cycles more than two times.
Nalgene general long-term storage cryogenic tubes (Thermo Fisher Scientific, cat. No. 03–337-7D)
Cool cell container (Corning, cat. No. 432002)
Growing PDO from Basic Protocol 1.
Disintegration of mature PDO to prepare a single cell suspension for a subsequent sub-culture
PDO growing in 3 wells (Basic Protocol 1) will be used for sub-culture.
Remove culture media carefully using a P1000 pipettor with a 1,250-μL tip so as not to disturb the Matrigel containing growing mature organoids in each well. Aspiration is not recommended at this stage because Matrigel becomes increasingly fragile as PDO grow.
Add 500 μL of cold DPBS into each well and disrupt the Matrigel mechanically into small fragments by pipetting up and down 3 times through a 1,250-μL pipette tip.
Combine PDO-containing Matrigel from 3 wells and transfer into a 1.7-mL tube.
Centrifuge at 500 × g (2,300 rpm on Eppendorf 5424R) for 3 min at room temperature to pellet the Matrigel fragments.
Discard the supernatant using a P1000 pipettor.
-
Resuspend the Matrigel fragments with 1 mL Trypsin-EDTA
Optional: if desired, supplement with DNase I as described in Basic Protocol 1, step 7.
Incubate for 10 min at 37°C with simultaneous mixing at 800 rpm in Thermomixer C.
To disintegrate organoid structures further, add 3–4 strokes of pipetting using a P1000 pipettor through a 1,250-μL pipette tip.
Filter the cell suspension from step 8 over a 35-μm cell strainer cap into a 5-mL Falcon round-bottom tube containing 3 mL STI.
Centrifuge at 188 × g (1,000 rpm on Sorvall ST 16R) for 5 min at 4°C.
Remove the supernatant by aspiration.
Resuspend the cell pellet in 1,000 μL Basal Medium.
Determine cell density and viability as in Basic Protocol 1-Steps 14–15.
Passaging PDO in 24-well plates
To passage PDO in 24-well plates, generally, seed 2×104 live cells per well in accordance with Basic Protocol 1 steps 16–27.
assaging PDO in 96-well plates
In general, 2–5×103 cells per well will be seeded in 96-well plates.
Thaw Matrigel as in Basic Protocol 1-Step 16 and keep on ice until use.
Pre-warm a 96-well plate at 37°C.
-
To seed 2×103 cells per well for treatment with a drug (e.g. 5FU) at various concentrations (e.g. 10−3, 10−2, 10−1, 1, 10, 100, 1000 μM) and vehicle (e.g. DMSO for 5FU) in triplicate, prepare 6×104 cells (=30 wells × 2×103 cells/well for 24 wells (3 wells × 8 for 5FU and DMSO) and plus extra 6 wells). Take a necessary volume (mL) of cell suspension according to the following formula:
6×104 [cell number needed] ÷ [cell density (/mL) from Basic Protocol 2 step 13]
Transfer 6×104 cells into a 1.7-mL tube
Centrifuge at 500 × g (2,300 rpm on Eppendorf 5424R) for 3 min at room temperature to pellet cells.
Resuspend cells in 150 μL ice-cold Matrigel (5 μL/well × [30 wells]).
Dispense 5 μL of cells-suspended in Matrigel into each well of 96-well plate.
Place the 96-well plate in a CO2 incubator (37°C) for 30 min to allow the Matrigel to solidify.
Dispense 100 μL tumor-type specific organoid medium into each well (Tables 2 and 3) for ESCC or EAC according to clinical diagnosis and pathology report of the original tumor.
Refresh the organoid medium every 2–3 d. Remove spent media by aspiration. Use a P200 pipettor to replenish with 100 μL medium.
Monitor contamination and organoid growth under a phase-contrast microscope (See Basic Protocol 3)
Grow organoids for up to 7–8 d until they reach 70–100 μm in dimeter for experiments (e.g. drug treatment, Basic Protocol 6).
Cryopreservation
To make three vials of frozen stock, take 3×105 live cells from Basic Protocol 2-Step 12. Determine the necessary volume of the cell suspension based on the viable cell count in step 13 and transfer the cells into a 1.7-mL tube.
Centrifuge at 500 × g (2,300 rpm on Eppendorf 5424R) at room temperature for 3 min.
Remove the supernatant using a P1000 pipettor with a 1,250-μL tip.
Resuspend the cell pellets in 1 mL freezing medium (to the final density at 3×105/mL).
Dispense 330 μL each of aliquots into three fresh cryogenic vials.
Add 670 μL fresh freezing medium into each cryogenic vial.
Freeze each vial in a freezing container at −80°C overnight.
Transfer the frozen vials into a liquid nitrogen cell storage tank.
Recovery after cryopreservation
Thaw Matrigel as in Basic Protocol 1-step 16 and keep on ice.
Pre-warm a 24-well plate at 37°C.
Thaw the cryogenic vial (step 34) in a 37°C-water-bath for 30–45 sec.
Transfer the cell suspension into a 1.7-mL tube.
Spin at 500 × g (2,300 rpm on Eppendorf 5424R) at room temperature for 3 min and remove the supernatant using a P1000 pipettor.
Resuspend the cell pellet in 1 mL DPBS.
Spin at 500 × g (2,300 rpm on Eppendorf 5424R) at room temperature for 3 min and remove the supernatant using a P1000 pipettor.
Resuspend the cell pellet in 1 mL Basal Medium, determine cell density and viability as in Basic Protocol 1-Steps 13–15.
Spin at 500 × g (2,300 rpm on Eppendorf 5424R) at room temperature for 3 min and remove the supernatant using a P1000 pipettor.
-
Resuspend cells in Matrigel and seed cells (2×104 cells/well) and proceed with organoid culture as described in Basic Protocol 1-Steps 17–27.
Optional: for EAC PDO, supplementation of 100 ng/mL FGF-10 (Table 3) in the first week dramatically increases recovery after cryopreservation.
BASIC PROTOCOL 3
Imaged-based monitoring of organoid size and growth kinetics
To evaluate organoid growth, 3D organoid structures are monitored during culture, and their size is documented by a conventional phase-contrast inverted microscope or high-throughput cell imaging instruments (e.g. Celigo Image Cytometer, Nexcelom Bioscience) with the capacity of automated multi-well rapid imaging and quantitative analysis of organoid size, number and structure.
Materials
All cell culture equipment, centrifuges, plasticware, pipettors, reagents are the same as in Basic Protocol 1 and 2 unless otherwise listed below.
Inverted microscope with imaging capacity such as Nikon Eclipse E600 microscope (Nikon, Tokyo, Japan) with a camera, EVOS FL Cell Imaging System (Thermo Fisher scientific) or Celigo imaging cytometer (Nexcelom Bioscience)
ImageJ software (NIH)
GraphPad 7.0 (Prism) or SigmaPlot (Systat) software to generate a growth curve.
Evaluating PDO growth
Grow organoids in 24-well or 96-well plates (Basic Protocol 1 and 2). Use at least three (3) wells per condition as biological replicates.
Acquire phase-contrast images (Fig. 2) manually via a conventional microscopy with a camera. Use ImageJ software to measure the diameter of at least 5 organoids per well. Alternatively, Celigo imaging cytometer can not only acquire phase-contrast images but calculate mean organoid area for all organoids imaged. Such assays can be performed in conjunction with drug treatment (Fig. 3) (Basic Protocol 6).
Repeat step 2 every other day.
Generate growth curves by plotting organoid diameter or area at each time point.
Figure 2. ESCC and EAC PDO morphological characteristicsc.
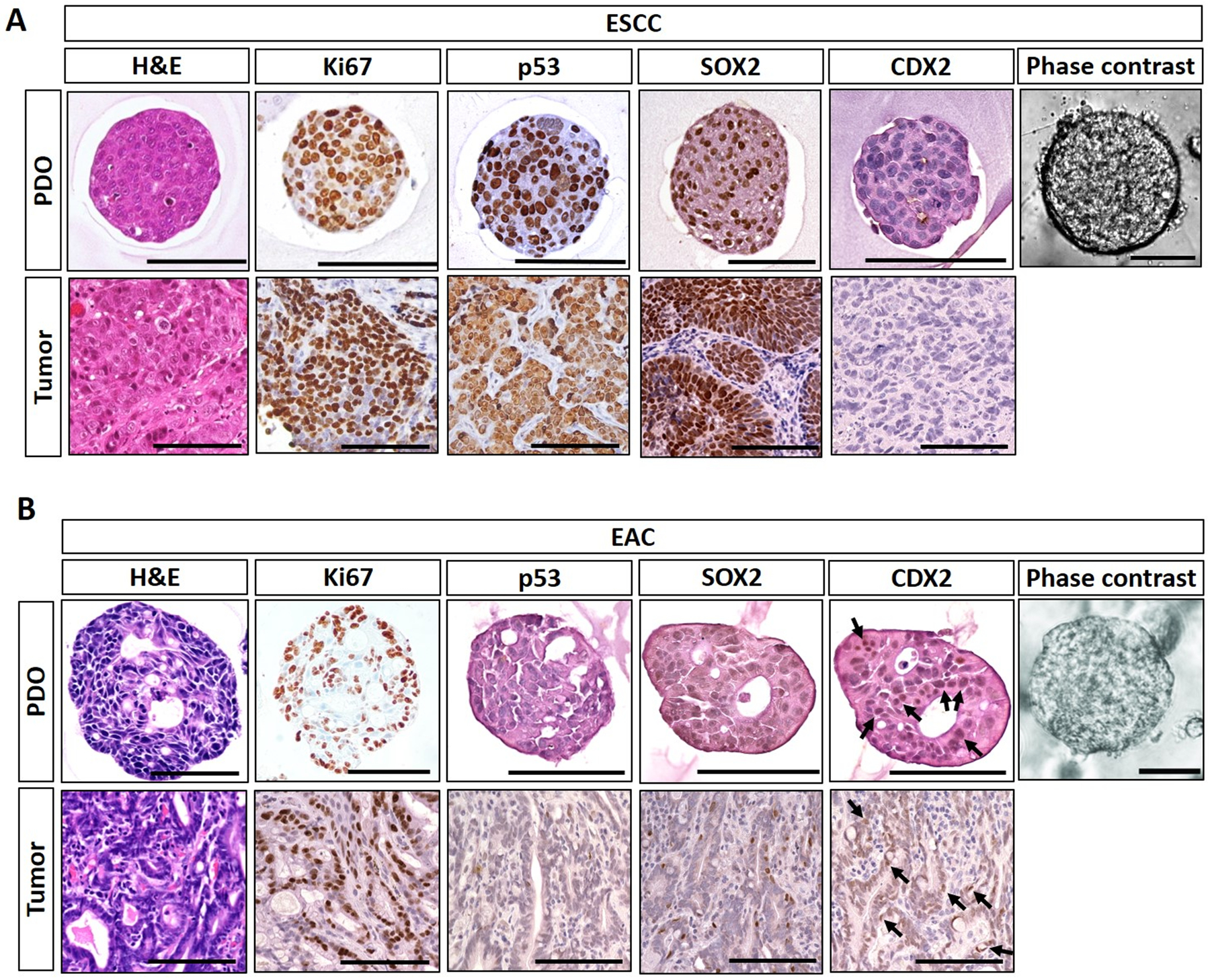
Representative ESCC (A) and EAC (B) PDO images under phase contrast microscope and histologic characterization of PDO as well as corresponding original primary tumor tissues by Hematoxylin-Eosin (H&E) staining and immunohistochemistry. ESCC PDO comprise poorly differentiated squamous cell carcinoma cells featuring increased cell proliferation (Ki67), stabilization of tumor suppressor TP53 protein, and overexpression of SOX2, an oncogene essential in ESCC. EAC PDO feature high chromatin density along with focal luminal formations reminiscent of glandular structures compatible with adenocarcinoma as corroborated by nuclear expression (arrows) of caudal type homeobox 2 protein (CDX2). Note that TP53 was negative in the representative EAC POD and original primary tumor. Cancer cells within PDO recapitulate those in original tumors. Scale bar, 100 μm.
Figure 3. IC50 curve from drug-treated PDO.
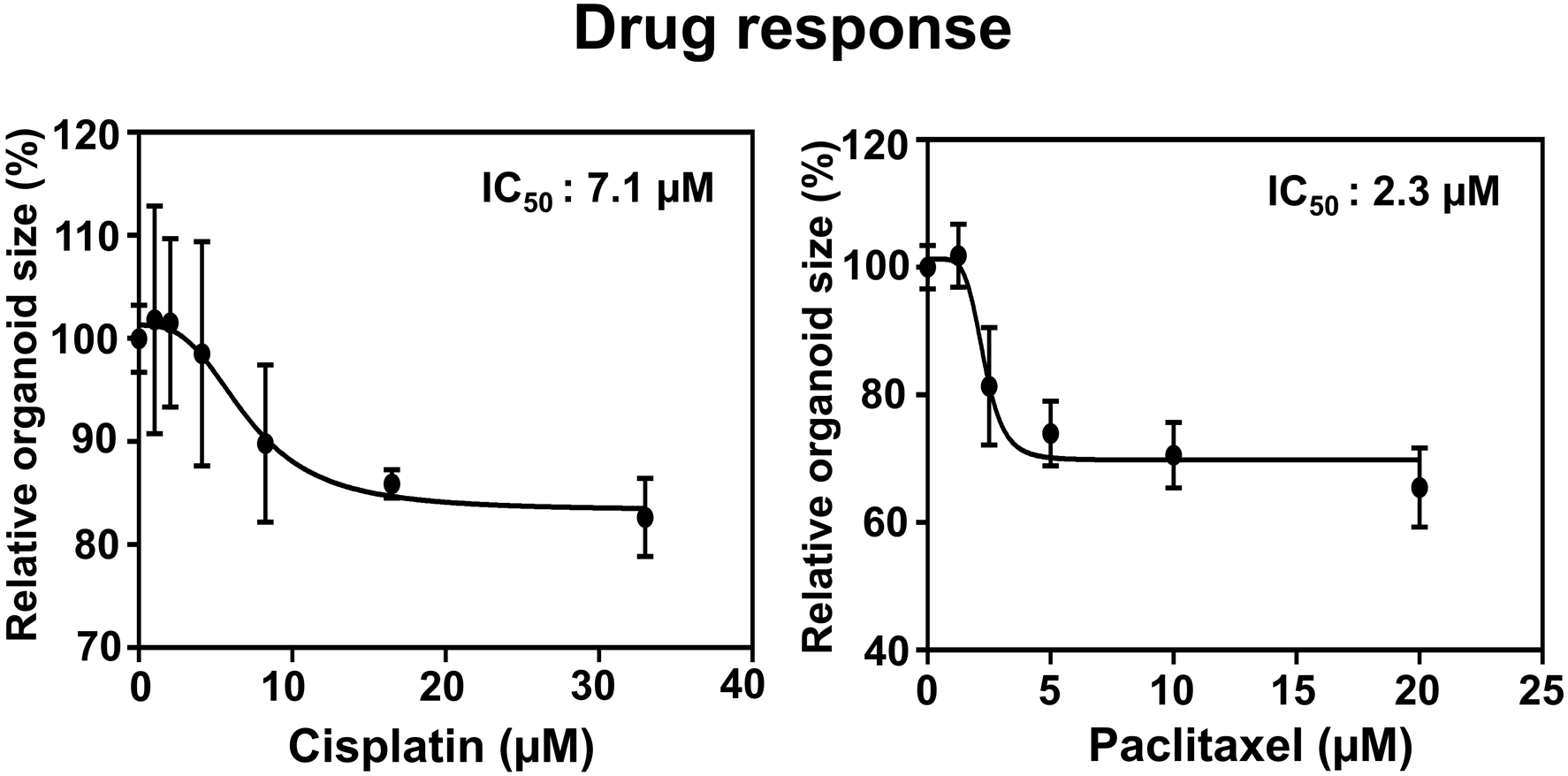
EAC PDO size were evaluated by Celigo Imaging Cytometer measuring the mean organoid size following 72 h-exposure to Cisplatin and Paclitaxel at indicated final concentrations. Organoid size was normalized by vehicle-treated control as 100%. IC50 for Cisplatin and Paclitaxel was determined as 7.1 and 2.3 μM (with R squares of 0.5874 and 0.8652), respectively.
BASIC PROTOCOL 4
Harvesting esophageal cancer PDO for histological analyses
Histological evaluation is an essential step in ensuring that PDO recapitulate the original tumor morphologically. Herein, we provide a protocol for fixation and embedding of PDO in paraffin, allowing for a long-term storage and histological analyses including hematoxylin-eosin staining, immunohistochemistry and immunofluorescence.
Materials
All cell culture equipment, centrifuges, plasticware, pipettors, reagents are the same as in Basic Protocol 1 and 2 unless otherwise listed below.
-
HBSS-D: HBSS containing Dispase
Dilute 50 U/mL Dispase in HBSS at 1:4. To prepare a working solution that is sufficient to embed organoids grown in 3 wells, add 300 μL 50U/mL Dispase into 1200 μL HBSS.
Dissecting scissors with 30-mm cutting edge (VWR, Cat. No.25870–002)
-
Embedding tip (Fig. 4A): a 200-μL pipette tip modified to have a wide opening
Use clean dissecting scissors and cut off ~9 mm the pointed end of 200-μL tips to make their opening wider.
-
Embedding bottom-less barrel (Fig. 4A): The pipettor connecting part of 200-μL pipette tip will be utilized as an embedding cylinder (a bottom-less barrel).
Use clean dissecting scissors to cut out the proximal ~4 mm of the cylinder part (i.e. the broader open end) of 200-μL tips).
Parafilm M wrapping film (Thermo Fisher Scientific, cat. No. S37440)
Microcentrifuge tube rack (5×16 holes/rack) (Southern Labware, cat. No. 0061)
-
Embedding rack
Wrap a microcentrifuge tube rack with Parafilm M over the top (Fig. 4B)
Kimble KIMAX griffin 150-mL beaker (Thermo Fisher Scientific, cat. No. 02–539J)
Bacto-agar (Becton Dickinson, cat. No. 214010)
Gelatin (Fisher Scientific, cat. No. G7–500)
-
Embedding gel (2% Bacto-Agar-2.5% Gelatin)
To prepare 50 mL of 2% (weight/volume) Bacto-Agar-2.5% (weight/volume) gelatin gel, resuspend 1 g of Bacto-agar and 1.25 g of gelatin in the total 50 mL of water in a 150-mL beaker. Swirl the suspension and let it sit at room temperature for 30–60 min. Autoclave at 121°C for 20 min and dispense into 5 mL aliquots in 15-mL conical tubes.
Fisher-brand tissue path IV tissue cassettes (Thermo Fisher Scientific, cat. No. 22–272416)
Thermo Scientific™ Shandon™ Sponge (rectangular sponges to hold specimen in cassette, 25×31mm) (Thermo Fisher Scientific, cat. No. 84–53)
Paraformaldehyde (PFA), powder, 95% (Sigma-Aldrich, cat. No. 158127–500G)
Ethanol 200 Proof (EtOH, Thermo Fisher Scientific, cat. No. 22–032-601)
Figure 4. Tools used for embedding of paraformaldehyde-fixed PDO.
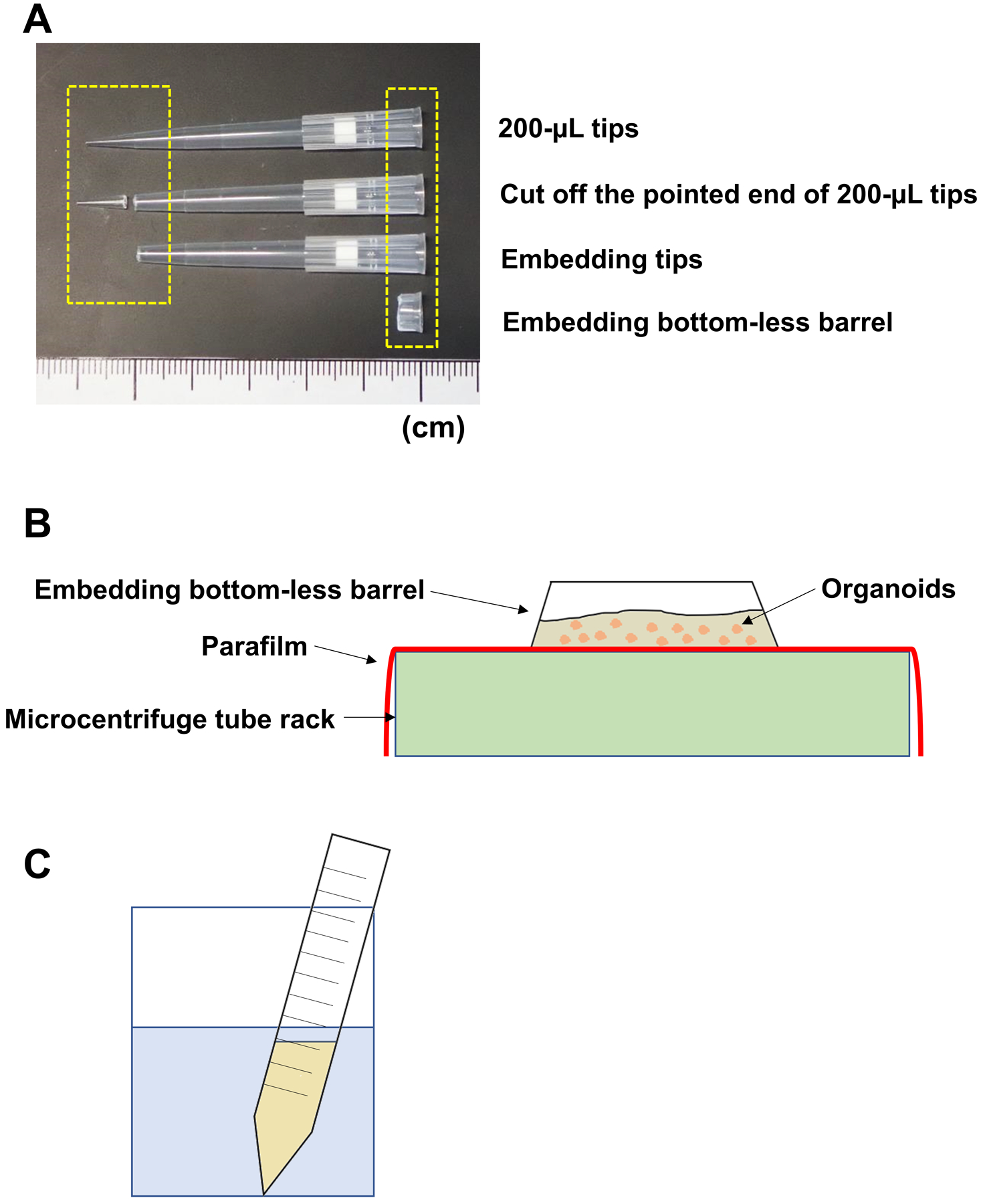
200-μL pipet tips are modified to make embedding tips and embedding bottom-less barrels (A). A polypropylene 1.7-mL tube rack, covered by a sheet of Parafilm M, is used as a scaffold to place embedding bottom-less barrel where fixed organoids will be cast in along with embedding gel (pre-heated Bacto-agar) (B). To liquefy embedding gel, an aliquot of 5-mL Bacto-agar will be microwaved for 1 min in a 150-mL beaker containing 100-mL water.
Fixation of 3D organoid cultures
PDO growing in 3 wells (Basic Protocol 1) will be used for histological analyses.
Remove culture media carefully as in Basic Protocol 2-Step 1.
Add 500 μL of cold DPBS into each well, dislodge and disrupt the Matrigel mechanically by pipetting up and down 3–4 times with a P1000 pipettor with a 1,250-μL tip.
Combine the fragmented Matrigel from 3 wells and transfer into a 1.7-mL tube.
Centrifuge at 500 × g (2,300 rpm on Eppendorf 5424R) at room temperature for 3 min to pellet the Matrigel fragments.
Remove the supernatant by P1000 with a 1,250-μL tip.
Add 1 mL DPBS to dissociate the pellet by pipetting up and down 3–4 times with a P1000 pipettor through a 1,250-μL tip.
Centrifuge at 500 × g (2,300 rpm on Eppendorf 5424R) at room temperature for 3 min to pellet the Matrigel fragments.
Remove the supernatant by P1000 with a 1,250-μL tip.
Resuspend the pellet in 500 μL 4% PFA using a P1000 pipettor with a 1,250-μL tip.
Incubate at 4°C > 2–6 hours or overnight.
Centrifuge at 500 × g (2,300 rpm on Eppendorf 5424R) at room temperature for 3 min.
Remove the supernatant by P1000 with a 1,250-μL tip.
Add 1 mL of DPBS to wash the pellet by pipetting up and down 3–4 times with a P1000 pipettor through a 1,250-μL tip.
Centrifuge at 500 × g (2,300 rpm on Eppendorf 5424R) at room temperature for 3 min.
Remove the supernatant carefully by P1000 with a 1,250-μL tip.
Remove as much as possible the remnant trace volume of the supernatant by P200 with a 200-μL tip.
Proceed to embedding. Alternatively, the cell suspension (step 14) may be stored at 4°C for up to one week prior to embedding.
Embedding and preparation of paraffin tissue blocks
Place embedding gel (5 ml in a 15-mL tube) in a 150-mL beaker containing ~100 mL water (Fig. 4C). Remove/loosen the cap! This is important for safety to use microwave in the following step 20.
Microwave at the highest power level for 1 min until the water starts boiling.
Confirm that the embedding gel has been liquefied and leave for 2–3 min.
-
Resuspend the organoid pellet in 50 μL liquefied embedding gel using an embedding tip on P200 pipettor (Fig. 4A). Transfer immediately and cast into an embedding bottom-less barrel placed on the Parafilm-covered embedding rack (Fig. 4B).
Note: The organoid pellet should be minimally dispersed in embedding gel upon pipetting. This will maximize the number of individual organoid structures visible per resulting paraffin section on a glass microscope slide. To this end, detach the packed organoid pellet from the bottom of the 1.7-mL tube (Basic Protocol 4-step 17) using a toothpick or a regular 10-μL tip (without being attached to a pipettor). Then, add 50 μL embedding gel and transfer the minimally disturbed organoid pellet into the embedding barrel using an embedding tip on P200 pipettor.
Transfer the embedding rack to 4°C and let the gel solidify for at least 30 min.
Use a fresh embedding tip to gently push out the solidified gel containing embedded organoids onto to sponge placed within a tissue cassette.
Place the tissue cassette into 70% ethanol and store at 4°C until embedding in paraffin via routine histological processing to prepare paraffin blocks.
BASIC PROTOCOL 5
PDO content analysis by flow cytometry
Single cell-derived PDO recapitulate intratumoral cell heterogeneity. Besides morphology (Basic Protocol 4), PDO content can be characterized by flow cytometry (e.g. cell surface markers). Such analysis can be done in conjunction with pharmacological treatments to explore unique signaling pathways or therapy resistance mechanisms associated with unique cell populations within PDO. Fluorescence-labeled antibodies, dyes and probes can be utilized to detect a variety of cellular antigens and molecular targets. We describe a protocol to determine cell surface CD44 expression as an example of this approach. CD44 is a glycoprotein implicated in the pathogenesis of esophageal cancer (Kinugasa et al., 2015; Natsuizaka et al., 2017; Whelan, Chandramouleeswaran, et al., 2017). Note that antibody titers, selection of fluorochromes, and other assay conditions are variable, requiring optimization for each molecule of interest.
Materials
All cell culture equipment, centrifuges, plasticware, pipettors, reagents are the same as in Basic Protocol 1 and 2 unless otherwise listed below.
Vortex-Genie2 (Scientific Industries, cat. No. SI-T236)
Aluminum Foil Roll (Thermo Fisher Scientific, cat. No. 01–213-105)
FACSCalibur or LSR II cytometers (BD Biosciences)
FlowJo software (Tree Star)
MilliporeSigma™ Steriflip™ Sterile Disposable Vacuum Filter Units (50-mL tube with a filter with 0.22-μm pore) (Fisher Scientific, cat. No. SE1M179M6)
Fluorescence-activated cell sorting (FACS) buffer: DPBS supplemented with 1% bovine serum albumin (BSA). Dissolve 0.5 g BSA in 50 mL DPBS, filter-sterilize and store at 4°C.
APC mouse anti-human CD44 (clone G44–26, BD Biosciences, cat. No. 559942)
-
4’,6-Diamidino-2-Phenylindole, Dihydrochloride (DAPI), FluoroPure grade (Thermo Fisher Scientific, cat. No. D21490)
Dissolve 10 mg DAPI into 2 mL water (5 mg/ml), and dilute further to 0.05 mg/mL (50 μg/mL) as a stock solution, dispense into 1 mL aliquots and store at −20°C. Thaw in water bath at 37°C for 1 min prior to use. Protect from light.
Flow cytometry analysis of PDO
Dissociate organoids grown in a 24-well plate to prepare a single cell suspension and determine cell density as described in Basic Protocol 2-Steps 1–12.
Transfer 2×105-1×106 cells to a 5-mL Falcon round-bottom tube.
Add 4 mL FACS buffer and centrifuge at 188 × g (1,000 rpm on Sorvall ST 16R) at 4°C for 5 min.
Discard the supernatant by aspiration.
Add 100 μl of FACS buffer containing 5 μl conjugated anti-CD44 antibody in cell suspension (1:20, pre-optimized titer) in 5-mL Falcon round-bottom tube.
Mix vigorously by vortex at an analog speed setting of 8 (max speed) for a few sec.
Incubate the reaction tubes on ice for 30 min (optimized for anti-CD44 antibody binding), protected from light by covering with aluminum foil.
Add 4 ml FACS buffer to wash cells by centrifugation at 188 × g (1,000 rpm on Sorvall ST 16R) at 4°C for 5 min.
Discard the supernatant by aspiration and resuspend cells in 500 μl FACS buffer containing 1 μl DAPI, a DNA staining dye to determine dead cells, and mix well as in Step 6.
Analyze the DAPI-negative cells for CD44 expression using a flow cytometer and FlowJo software (Whelan, Chandramouleeswaran, et al., 2017). To define cells with CD44 expression, a gate set in unstained cells (control) is applied.
BASIC PROTOCOL 6
Evaluation of drug response with determination of the half-inhibitory concentration (IC50)
One of the major goals in PDO translation is to serve as a potential guide to assist clinical decision making by physicians and surgeons in personalized/precision medicine where customized therapeutics are provided following molecular characterization of cancer cells in the original tumors. To this end, PDO need to be tested for multiple drugs in standard of care and molecularly-targeted agents (e.g. small molecule inhibitors and antibodies) in a time-sensitive manner. Drug treatment of PDO can be performed in 96-well plates containing established PDO with a broad range of drug concentrations. PDO response to drugs can be evaluated via numerous cell viability assays based upon cellular functions (e.g. ATP production and other mitochondrial activities such as formazan formation in the WST-1 reagent) and cell membrane integrity (e.g. membrane-permeating fluorescent dyes such as Calcein-AM). We describe utilization of the CellTiter-Glo® 3D cell viability assay.
Materials
96-well plate with mature PDO grown for 7–8 d until they reach ~70–100 μm in diameter (Basic Protocol 2). Note that white plates such as Nunc™ Nunclon Multi-Dishes, 96 well Plate with Lid, Color White (Thermo Fisher Scientific, cat. No. 165306) are recommended for luminescence-based CellTiter-Glo® 3D cell viability assay to prevent a signal interference from neighboring wells.
5-Fluorouracil (5-FU) (Sigma-Aldrich, cat. No. F6627): Make a stock solution by dissolving 65 mg 5-FU (MW = 230) in 1mL DMSO to the final concentration of 65 mg/mL (500 mM) and store at −20°C. Prepare tumor type-specific organoid media containing 5-FU at a range of final concentrations (10−3, 10−2, 10−1, 1, 10, 100, 1000 μM) by serial-dilution. Note that 100 μL is needed per well.
Cisplatin (Santa Cruz Biotechnology, cat. No. sc-200896): Prepare fresh working solution by dissolving 0.5 mg of Cisplatin (MW = 300) in 1 mL of 0.9% NaCl to the final concentration of 0.5 mg/mL (1.67 mM). Prepare tumor type-specific organoid media containing Cisplatin at a range of final concentrations (1.0, 2.05, 4.1, 8.25, 16.5, 33.0 μM) by serial-dilution. Note that 100 μL is needed per well.
Paclitaxel (Selleck chemicals, cat. No. S1150): Make a stock solution by dissolving 1 mg of Cisplatin (MW = 854) in 117 μL of DMSO to the final concentration of 10 mM and store at −20°C. Prepare tumor type-specific organoid media containing Paclitaxel at a range of final concentrations (1.25, 2.5, 5.0, 10.0, 20.0 μM) by serial-dilution. Note that 100 μL is needed per well.
CellTiter-Glo 3D cell viability assay (G9681, Promega): Thaw CellTiter-Glo 3D reagent at 4°C overnight. Equilibrate the CellTiter-Glo 3D reagent to room temperature prior to use. Mix 50 μL CellTiter-Glo 3D regent and 50 μL Basal Medium (1:1) (100 μL per well).
The Belly Dancer Shaker (IBI Scientific, cat. No. Z768502–1EA)
Microplate Reader for detecting luminescence (e.g. GloMax® Discover Microplate Reader Promega, Cat. No. GM3000)
GraphPad Prism 7.0
Drug treatment
Remove the spent culture medium by aspiration.
Dispense 100 μL of organoid medium containing drugs per well at the range of final concentrations to be tested.
Incubate organoids with drugs for 72 h.
Cell viability assay
Remove drug-containing medium from each well by aspiration and replace with 100 μL volume of a 1:1 cocktail of CellTiter-Glo 3D and Basal Medium. Add the cocktail into 3 wells without organoids to measure the background luminescence level.
Mix the contents vigorously using the Berry Dancer shaker at speed 4 for 5 min to induce cell lysis.
Incubate the plate at room temperature for 25 min.
Measure the luminescence by GloMax-Multi+ Microplate Multimode Reader.
Generate dose response curves using GraphPad Prism 7.0, the least squares fit (ordinary) with a variable slope (four parameters).
REAGENTS AND SOLUTIONS
Support Protocol 1: Preparation of basal medium and tumor type-specific organoid media
Mix the following reagents to generate basal, as well as tumor-type specific organoid media listed in Tables 1–3.
Materials
Advanced DMEM/F12 (Thermo Fisher Scientific, cat. No. 12634028)
GlutaMAX supplement, 100x (Thermo Fisher Scientific, cat. No. 35050061)
HEPES, 1 M (pH 7.2–7.5) (Thermo Fisher Scientific, cat. No. 15630080)
Antibiotic-Antimycotic, 100x (Thermo Fisher Scientific, cat. No. 15240062)
Gentamicin, 50 mg/mL (Thermo Fisher Scientific, cat. No. 15750060)
N-2 Supplement, 100x (Thermo Fisher Scientific, cat. No. 17502048)
B-27 supplement, 50x (Thermo Fisher Scientific, cat. No. 17504044)
N-Acetylcysteine (NAC), 0.5M (Sigma-Aldrich, cat. No. A9165), reconstituted in DPBS, filter-sterilized and stored in aliquots at −20°C.
CHIR99021, 5 mM (Cayman Chemical, cat. No. 13122), reconstituted in DMSO, stored in aliquots at −20°C.
Recombinant human epidermal growth factor (EGF), 500 ng/μL (Peprotech, cat. No. AF-100–15), reconstituted in Basal Medium, stored in aliquots at −20°C.
A83–01, 5 mM (Cayman Chemical, cat. No. 9001799), reconstituted in DMSO, stored in aliquots at −20°C.
SB202190, 10 mM (Selleck Chemicals, cat. No. S1077), reconstituted in DMSO, stored in aliquots at −20°C.
Gastrin, 1 mM (Sigma-Aldrich, cat. No. G9145), reconstituted in sterile 0.1% NaOH, stored in aliquots at −20°C.
Nicotinamide, 1M (Sigma-Aldrich, cat. No. N0636), reconstituted in DPBS, filter-sterilized and stored in aliquots at −20°C.
Y-27632, 50mM (Selleck Chemicals, cat. No. S1049), reconstituted in DMSO, stored in aliquots at −20°C.
FGF-10, 100 μg/mL (Peprotech, Cat. No. 100–26), reconstituted in Basal medium, stored in aliquots at −20°C.
L-WRN cell-conditioned medium expressing Wnt-3A, R-Spondin1 and Noggin (WRN), stored at −20°C.
L-WRN cells are available from ATCC (cat. No. CRL-3276). Produce WRN in accordance with a detailed protocol (Miyoshi & Stappenbeck, 2013).
HEK293T-conditioned medium expressing R-Spondin1 and Noggin (RN) (See Support Protocol 2), stored at −20°C.
SUPPORT PROTOCOL 2
To generate conditioned medium containing high RN activity, high-titer lentivirus expressing RN can be produced by transient transfection of HEK293T cells. The resulting high-titer virus-containing HEK293T cell conditioned medium is used to infect HEK293T cells to produce RN that will be harvested as a conditioned medium from virus-infected HEK293T cells.
Preparation of conditioned media
Organoid culture media require developmental niche factors (i.e. WNR or NR). Such factors can be harvested as cell culture conditioned media (Miyoshi & Stappenbeck, 2013), providing a more affordable alternative to commercially available recombinant proteins. Herein, we describe a protocol to produce RN in HEK293T cells via lentivirus-mediated transduction of R-spondin1 and Noggin. The produced RN can be validated in murine small-intestinal organoid formation assays (Sato et al., 2011).
Materials
HEK239T cells (ATCC, cat. No. CRL-3216)
Dulbecco’s Modified Eagle’s Medium (DMEM) (Corning, cat No. MT10013CV)
Fetal bovine serum (FBS) (HyClone, cat. No. SH3007003)
Penicillin (100 U/mL)-Streptomycin (100 μg/mL) (Thermo Fisher Scientific, cat. No. 15140122)
HEK 293T medium (Table 4), stored at 4°C
Lipofectamine 2000 reagent (life Sciences, cat. No.11668019), stored at 4°C
Opti-MEM reduced serum medium (Thermo Fisher Scientific, cat. No. 31985070), stored at 4°C
Polybrene (EMD Millipore, cat. No. TR-1003-G)
Puromycin (GoldBio, cat. No. P-600–100)
pCMVR8.74, 2nd generation lentiviral packaging plasmid (Addgene, cat. No. 22036)
VSV.G (Addgene, cat. No. 14888), mammalian expression plasmid to express VSV-G envelope
pG-N+RIP (unpublished, Rustgi Lab), lentiviral vector expressing RSpondin1 and Noggin.
T175 tissue culture flasks (Fisher Scientific, cat. No. 159910)
100-mm tissue culture dish (Fisher Scientific, cat. No. 130182)
150-mm tissue culture dish (Fisher Scientific, cat. No. 130183)
Rapid-flow vacuum filter unit (Fisher Scientific, cat. No. 566–0020)
50-mL conical tube (Fisher Scientific, cat. No. 12–565-270)
10-mL syringe (BD, cat. No. 0309604)
PES Syringe filter (0.45μm, 30mm, Sterile, CellTreat Scientific Products, cat. No. 229749)
Bleach (6% sodium hypochlorite, Clorox)
Table 4.
HEK293T medium
| Reagent | Volume | Final concentration |
|---|---|---|
| DMEM | 500 mL | |
| FBS | 50 mL | 10% |
| Penicillin-Streptomycin (100x) | 5 mL | 1x |
Preparation of RN conditioned medium
Lentivirus preparation
CAUTION
Biosafety level (BL)-2 or enhanced BL-2 is appropriate to perform lentivirus production and infection experiments. The produced virus, albeit replication incompetent, can infect human cells. Treat plasticware such as pipettes, syringe discs, glass Pasteur pipettes, suction lines with Bleach to kill the infectious virus in every step following transfection.
-
Grow HEK293T cells in HEK 293T medium at 5% CO2 and at 37°C under >95% humidity. Wash cells with DPBS, trypsinize and count cells via standard cell culture procedures.
Note that a mechanical force easily detaches HEK 293T cells from plastic plates, and thus it is important to handle cells gently during DPBS-wash and trypsinization. It takes less than 30 seconds for cells to detach from plates at room temperature when trypsinized. Washing once with DPBS before adding trypsin-EDTA is necessary because FBS in HEK 293T medium may block trypsin activity.
Seed 6×106 HEK293T cells in a 100 mm dish and grow for 48–72 h to 80–90% confluency in the following day.
Mix 40 μL of Lipofectamine 2000 in 360 μL of Opti-MEM.
Incubate at room temperature for 5min.
Add DNA (10 μg pG-N+RIP, 6.5 μg pCMVdR8.74, 3.5 μg VSV.G) in 400 μL of Opti-MEM.
Combine Opti-MEM containing Lipofectamine 2000 (step 3) and Opti-MEM containing DNA (step 5).
Incubate at room temperature for 30 min.
Add 5ml Opti-MEM into the Opti-MEM-Lipofectamine-DNA cocktail.
Remove spent media from HEK293T cell culture by aspiration and replace by the Lipofectamine-DNA-Opti-MEM mix.
Incubate at 37°C for 4 h.
Remove the medium and replace with 7ml fresh HEK293T medium.
Incubate cells at 5% CO2 and at 37°C under >95% humidity for48–72 h.
Harvest the virus at 48 h as conditioned medium and replenish the HEK293T medium.
Filter the conditioned medium using 0.45-μm syringe filter. Best used immediately, or the virus can be stored at −80°C for one month.
Harvest the virus at 72 h as conditioned medium and replenish the culture with HEK293T medium.
Filter the conditioned medium using 0.45-μm syringe filter. Best used immediately, or the virus can be stored at −80°C for one month.
Lentivirus-mediated transduction of HEK293T cells to produce RN conditioned medium
Plate HEK293T cells in two 100 mm dishes to ~ 80–90% the following day (one dish will be transfected, and the other will be used as control for Puromycin selection).
Replace the culture medium with 7 ml virus supplemented with Polybrene (3.5 μL of 10mg/mL stock). Incubate at 37°C for 4 h.
Add 3ml of HEK293T medium and incubate at 37°C for 4 h.
Replace virus-containing HEK293T medium from step 13 (or step 15).
Incubate for 24h.
Add Puromycin to the final concentration of 2 μg/ml and continue until cells from the control plate without virus infection are all dead.
Wash cells with DPBS and trypsinize the cells.
Count cells and seed 5×106 cells into as many 150 mm dishes as possible in HEK293T medium without Puromycin.
Collect the media 24, 48 and 72 hours later (store at −80°C until the last harvest).
Combine all conditioned media harvested and filter-sterilize.
Dispense into 50-ml tubes and keep at −80°C.
After thawing a 50-mL tube, dispense into 1 mL aliquots and store at −20°C.
Test RN conditioned media in murine intestinal organoid formation assays by comparing dilutions from 1–25% conditioned media to media containing defined recombinant growth factors and chose a final concentration based upon similar growth rates and morphology over the course of 7 days (Sato et al., 2011).
COMMENTARY
Background Information
Esophageal cancer is the deadliest of all human cancers owing to late stage presentation and diagnosis, therapy resistance and/or early recurrence (Rustgi & El-Serag, 2014). Esophageal cancer comprises squamous cell carcinoma (ESCC) and adenocarcinoma (EAC), two major histologic subtypes. ESCC accounts for about 90% of esophageal cancers worldwide. The incidence of EAC has been rising in Western countries at an alarming rate. Intratumoral cancer cell heterogeneity contributes to therapy resistance in these cancers, and prediction of therapeutic outcomes in individual patients remains elusive.
The nomenclature of 3D organoids is somewhat controversial as they do not typically contain non-epithelial cellular components existing in the original tissues or organs [for historic review see (Moreira et al., 2018; Whelan, Muir, & Nakagawa, 2018)]. Amongst 3D cell culture systems, for example, organotypic 3D culture reconstitutes epithelia on top of the sub-epithelial stromal compartment comprising tissue-specific fibroblasts, type I collagen and basement membrane matrix (Matrigel) (Kalabis et al., 2012). Organotypic 3D culture has been utilized to model esophageal epithelial squamous-cell differentiation (Ohashi et al., 2010), ESCC development (Naganuma et al., 2012), and ESCC invasive tumor front (Grugan et al., 2010; Okawa et al., 2007). Organotypic 3D culture requires monolayer cultures of epithelial cells (normal or neoplastic) and fibroblasts prior to 3D reconstitution. By contrasts, 3D organoids are directly generated with cells isolated from starting tissue materials (e.g. endoscopic biopsies and surgically resected tumors).
Successful recapitulation of normal and malignant intestinal epithelial structures (i.e. intestinal organoids aka enteroids) has inspired similar attempts to grow and characterize epithelial structures from a variety of organs and tumor types using a defined set of growth factors, pharmacological agents and Matrigel (Sato et al., 2009; Sen et al., 2019). In recent years, PDO have been generated from multiple tumor types (Boj et al., 2015; Broutier et al., 2016; Driehuis, Kolders, et al., 2019; Gao et al., 2014; Huang et al., 2015; Hubert et al., 2016; Kijima et al., 2019; Lee et al., 2018; Li et al., 2018; Pauli et al., 2017; Sachs et al., 2018; Saito et al., 2019; Sato et al., 2011; Tanaka et al., 2018; Taneja, 2015; van de Wetering et al., 2015; Vlachogiannis et al., 2018; Walsh, Castellanos, Nagathihalli, Merchant, & Skala, 2016) as a more powerful tool in personalized/precision medicine, compared to other platforms such as patient-derived xenograft tumors. Indeed, PDO allow for rapid drug screening and prediction of therapy response in concert with automated imaging microscopies and other high-throughput multi-well screening tools. Furthermore, in conjunction with DNA- and RNA-sequencing approaches, PDO provide structural and functional insights into gastrointestinal oncology and more broadly into tumor biology.
We were the first to describe generation of PDO from oral/head-and-neck and esophageal squamous cell carcinomas (SCCs; OSCC, HNSCC and ESCC) to evaluate therapy response and interrogate therapy resistance mechanisms (Kijima et al., 2019). We have further optimized organoid culture conditions to support the growth of PDO from broader SCCs (OSCC, HNSCC, anal SCC) as well as EAC according to the above-described protocols. These methods may be applied to cancers of other organs, such as the uterus and the lung where both squamous cell carcinomas and adenocarcinomas develop (Lin et al., 2017).
Critical Parameters, Understanding Results and Troubleshooting
Key parameters in successful PDO generation and characterization (Fig. 1) include quality of starting materials, enzymatic digestion and mechanical dissociation and straining (i.e. cell viability and cellular stress during transportation and tissue dissociation), as well as control of bacterial and fungal contamination. Medium components and Matrigel are essential reagents. Daily observation, monitoring of growth kinetics, as well as appropriate training in cell culture and other relevant techniques (e.g. histology, flow cytometry) will ensure successful generation of organoid lines. Employment of quality control reporter cell lines, standard molecular markers, functional and morphological analyses of 3D organoid products are key for ensuring that generated organoid lines are viable and recapitulate the original tumor specimen.
Starting materials:
Freshly isolated biopsies and surgical specimens serve best for successful PDO generation. After procurement, these clinical materials should be protected against ischemic conditions and thus processed as soon as possible to sustain cell viability. Surgically resected tumors often contain necrotic tissue that should be removed. A higher cell viability in the starting materials is helpful to gain an increased yield in the primary 3D organoid products, permitting more diverse subsequent phenotypic analyses (e.g. genetic profiling, morphology, flow cytometry, and drug response), cryopreservation and subculture. Due to the obstructive nature of esophageal tumors (esophageal stricture, dysphagia, food impactions), primary cultures of esophageal cancer specimens are at high risk of fungal and/or bacterial contamination. Addition of fungicides and antibiotics to all solutions and extensive rinsing of the tissue with large volumes of DPBS reduces this risk. Tissue samples should be placed in chilled DPBS or an organ transplant preserving solution (University of Wisconsin solution aka Belzer UW® Cold Storage Solution, Bridge To Life, cat. No. BUW-005), the latter for overnight shipping of tissues from a collection site. In our hands, freezing of specimens has diminished organoid formation rate.
Sample processing:
Enzymatic and mechanical dissociation of the tissue and the use of cell strainer are necessary; however, such processes can cause isolation-related stress. In a single cell suspension, ROCK inhibitor Y-27632 is utilized to minimize cell death associated with loss of cell-matrix or cell-cell contact (anoikis). One may argue that organoid formation rate may be improved by starting with cell aggregates instead of a well-dissociated single-cell suspension; however, this does not appear to be the case in our extensive experience.
Medium components, growth conditions and quality control cell lines:
Certain components of the organoid culture media, particularly the recombinant proteins in the L-WRN or RN conditioned media, have a short half-life. We recommend storing complete organoid growth media for no longer than two weeks. Growth factors and agents included in organoid media have been optimized; however, it remains elusive whether each of them is necessary, sufficient, or even beneficial for all esophageal cancer organoids from different patients. ESCC PDO tend to grow more successfully from patients with poorly-differentiated ESCC, and those showing therapy resistance (Kijima et al., 2019). It is possible that current organoid media may be selective for a subset of cancer cells (e.g. cancer stem cells characterized by high CD44 expression) within individual tumors.
Advanced DMEM/F12 is often used as a base medium to grow 3D organoids; however, media and their constitutes vary from study to study. To generate 3D organoids from human primary SCCs of oral (OSCC)/head-and-neck (HNSCC), esophagus (ESCC) and anal origin, we have omitted Wnt3A, A83–01, Nicotinamide and SB202190 from the medium we have originally described (Kijima et al., 2019). Removal of these factors did not affect formation and growth of primary or passaged 3D organoids in our ongoing practice (unpublished observations). This medium composition works well for murine ESCC 3D organoids (Natsuizaka et al., 2017) except that species-specific recombinant EGF (i.e. human EGF for human organoids) is utilized. Although patient-derived ESCC 3D organoids have not been described elsewhere, 3D organoids have been generated from OSCC/HNSCC patients (Driehuis, Kolders, et al., 2019; Driehuis, Spelier, et al., 2019; Tanaka et al., 2018). Driehuis et al. utilize advanced DMEM/F12 where they supplement CHIR99021 (GSK3β inhibitor), FGF2, FGF10, Prostaglandin E, and forskolin that we do not use. While their success rate (~60%) to form HNSCC 3D organoid is comparable with ours (~70%), their medium did not significantly improve our PDO growth efficiency (unpublished observations). Tanaka et al. utilized a human embryonic stem cell culture medium supplemented with bFGF; however, their success rate (30%) was lower than ours. Li et al. are the first to describe EAC 3D organoids from multiple patients (Li et al., 2018). Utilizing advanced DMEM/F12, their medium does not contain CHIR99021, Gastrin, Y-27632 and the N-2 supplement we use based on the principle set by Sato et al. to grow 3D organoids of intestinal cell types (Sato et al., 2011). The success rate by Li et al. is reported 31% while ours is ~80%. Of note, their condition did not support 3D organoids from Barrett’s esophagus (i.e. intestinal metaplasia), a histologic precursor of EAC.
Amongst commonly utilized growth supplements, we suggest that EGF, NAC, N2 and B27 are necessary although both N2 and B27 supplements include unknown concentrations of factors (insulin, transferrin, and selenite) that are redundantly present in the advanced DMEM/F12 base medium as well. A83–01 would be beneficial in most cases as we find useful to grow EAC, but not ESCC, 3D organoids. Given lot-to-lot variability in Matrigel, growth factors, antioxidants and other agents in media, we have utilized extensively characterized esophageal cancer cell lines (e.g. TE11, OE-33) (Kijima et al., 2019) to evaluate organoid formation in each medium tested for quality control purposes. Additionally, newly prepared L-WRN and RN can be tested in standard enteroid culture conditions. We use cell culture incubators set at 5% CO2, 37°C, >95% humidity to grow PDO. Growth conditions such as low oxygen tension (i.e. hypoxia) (Fujii et al., 2016) and air-liquid interface (Li, Ootani, & Kuo, 2016) remain yet to be validated for esophageal cancer PDO.
Growing cancer cells in primary culture meets often a common technical issue of concurrent growth of non-cancer cells. In Matrigel droplet, non-epithelial cells (e.g. fibroblasts and immune cells) do not form 3D structures; however, normal epithelial or pre-cancerous cells may form 3D organoids along with ESCC (and OSCC/HNSCC) cells (Kijima et al., 2019). In our EAC 3D organoid culture conditions, normal esophageal (squamous epithelial cell) organoids do not grow; however, 3D structures compatible with Barrett’s esophagus (i.e. intestinal metaplasia) may grow concurrently (unpublished observations). The size and morphology (phase contrast images and H&E staining) of organoids will help us to distinguish non-cancerous 3D structures from cancerous structures. Additionally, normal organoids do not grow continuously beyond 14 days in culture. Drug sensitivity to chemotherapeutic agents may be overestimated when the proportion of non-cancer 3D organoids is significant in primary 3D organoid culture. It is important to follow 3D organoid structures that remain viable following exposure to chemotherapy agents. If necessary, drugs should be tested on passaged secondary 3D organoids where cancerous organoids become predominant over non-cancerous organoids (Kijima et al., 2019). Of note, non-neoplastic human esophageal 3D organoids grow better and display a more exquisite proliferation-differentiation gradient in media with distinct compositions (Kasagi et al., 2018) with detailed protocols provided in our complementary manuscript (Nakagawa et al., Current Protocols Stem Cell Biology, In press). Outgrowth of stromal fibroblasts is a cumbersome issue in primary monolayer culture. We do occasionally observe growth of fibroblasts on the plastic surface in our organoid culture plates; however, fibroblasts are not transferred into subsequent passages because they remain adhered on the plastic surface when we harvest organoids in Matrigel without utilizing trypsin (Basic Protocol 2 steps 2–3).
Cell numbers to be seeded and growth kinetics:
Given a relatively low organoid formation rate (0.01%−1%) anticipated from human tissue materials, 20,000 viable cells are typically seeded per well in 24-well plates to initiate organoid cultures yielding 20–200 PDO per well. When passaged, organoid formation rate is anticipated to improve by 10-fold, permitting seeding of a smaller number of cells (e.g. 2,000 per well in 24-well plates; and 200 per well in 96-well plates). It should be noted that these live cells are usually seeded along with co-existing dead cells. Dead cells exude enzymes that degrade extracellular matrix components in Matrigel. Thus, seeding more cells does not necessarily increase the organoid formation rate. Trypan blue exclusion test detects dead cells, but not necessarily dying cells, overestimating the number of live cells seeded. Thus, high cell viability in the original tumor is crucial for a successful primary PDO culture, reinforcing the appropriate preservation, transportation and processing of the starting materials.
Live cancer cells also secrete matrix degrading enzymes such as matrix metalloproteases. As the organoids grow, the Matrigel may become loose and break apart. To preserve the Matrigel structure, media should be added gently and only along the wall of the well. If the Matrigel breaks, the organoids may be pelleted without trypsinization and re-embedded in fresh Matrigel. When PDO are initiated successfully, spherical structures emerge within 4–7 days and continue to grow to be passaged by day 11–14. We do not determine routinely doubling time of cancer cells within 3D organoids for a practical reason to save as many organoids-containing wells for morphological characterization and drug-testing. If necessary, we can determine the average number of live cells present in each 3D structure. By harvesting exponentially growing organoids, we have estimated doubling time of esophageal cancer PDO at approximately 24–36 h with each organoid starting from a single cell in suspension. This estimate ignores the initial lag phase, thereby raising the possibility of underestimation. The mature PDO may reach 100–250 μm in diameter. Overgrown PDO tend to contain an internal necrotic core as detected by H&E staining and should be avoided. The high number of metabolically active cells lowers the media pH which can be monitored by medium color turning to yellow a day after medium change.
Contaminations:
Clinical esophageal tumor samples are vulnerable to bacterial and fungal (yeast and mold) contaminations, as noted in the above “starting materials” section. It is recommended to have tissue culture incubators devoted for the use of primary organoid cultures. Fungicides and antibiotics may fail to prevent contamination. Bacteria and yeast grow rapidly, typically overnight. The suspected culture should be quarantined and may be treated with higher concentration (50 μg/ml) of gentamycin. Fungus-contaminated plates should be discarded immediately. Fungal contamination remains the biggest hindering factor in generation of organoids from esophageal cancer.
Molecular and functional characteristics of esophageal cancer PDO:
Analyses of PDO by standard assays such as histology (hematoxylin and eosin staining), immunohistochemistry and flow cytometry reveal unique attributes of esophageal cancer cells constituting individual organoid structures grown in each well (Fig. 2). Histology can define the degree of atypia and differentiation grade of cancer cells. While normal organoids display a well-organized differentiation gradient reminiscent of the stratified squamous-cell differentiation in the normal esophageal epithelium (Kasagi et al., 2018), cancerous organoids display a various degree of atypia and abnormal differentiation with increased proliferation, which can be documented by immunohistochemistry and immunofluorescence for markers such as Ki-67 (Kijima et al., 2019). ESCC cells may display upregulation of SOX2 (Dotto & Rustgi, 2016; Watanabe et al., 2014), while EAC cells may express CDX2 (Lord et al., 2005). Upregulation of dysfunctional tumor suppressor TP53 protein is common in both ESCC and EAC via mutant TP53 stabilization (Dotto & Rustgi, 2016; Fisher et al., 2017); however, some tumors display TP53 downregulation via other mechanisms (e.g. epigenetic silencing). It should be noted that such molecular changes in PDO recapitulate those in the original tumors (Fig. 2). Additionally, these marker expression patterns in PDO are stable between passages. Flow cytometry may reveal a subset of cancer cells with elevated expression of CD44, which may have properties of tumor initiating cells or cancer stem (stem-like) cells (Natsuizaka et al., 2017; Whelan, Chandramouleeswaran, et al., 2017). When isolated by fluorescence-activated cell sorting (FACS), such cells can be further propagated (Basic Protocol 2) to assess their organoid formation capability, and may show an increased organoid formation capability when passaged in subsequent organoid culture. Flow cytometry may also detect unique cellular functions and processes such as autophagy (Kijima et al., 2019; Whelan, Merves, et al., 2017) and epithelial-to-mesenchymal transition (EMT) (Karakasheva et al., 2018; Kinugasa et al., 2015; Natsuizaka et al., 2017), which may contribute to therapy resistance mechanisms. Additionally, PDO may be lysed for conventional gene expression analyses (e.g. immunoblotting, quantitative reverse-transcription polymerase chain reaction, RNA-sequencing) or single cell-RNA sequencing, and genetic and epigenetic profiling (e.g. whole exome sequencing). Such approach has been increasingly crucial for not only molecular subtyping of tumors but validation of the fidelity of PDO through a head-to-head comparison of molecular and functional phenotypes (e.g. specific tissue markers and drug response) and genomic alterations between original tumors and the resulting PDO.
Success rates:
Generation of primary organoids within the 14 days qualifies as success. Success rate of PDO generation is 60% (n=25) for ESCC and 80% (n=6) for EAC. Primary EAC PDO are readily passaged ≥15 times. ESCC PDO are harder to passage than EAC PDO as ~10% of primary ESCC PDO can be passaged ≥5 times. This presents the foremost limitation for PDO generated from squamous cell carcinomas. Interestingly, our PDO culture conditions are permissive for 3D organoid generation from HNSCC and ESCC cell lines (100%, n=5) and HNSCC/ESCC patient-derived xenograft tumors (PDX) (80%, n=5). The 3D organoids from these established cell lines and PDX are easily passaged in our PDO culture medium (unpublished observations). Genetic profiling of these cell line/PDX-derived 3D organoids is currently underway, comparing to primary PDO, with a hope to identify signaling pathways or genetic factors that PDO to be passaged more successfully.
Time Considerations
It is recommended that the tissue sample be processed as soon as possible after procurement from the patient. Single-cell suspension is typically generated in 1–2 hours, followed by solidification of Matrigel domes for 30 min. The organoids are ready for passage and/or harvest in up to 14 days (roughly 10 days for ESCC and 14 days for EAC). Preparation of cultures and drug treatment for IC50 determination takes 8–11 days.
Significance Statement.
Generated from endoscopic biopsies or surgically resected tumors, patient-derived organoids (PDO) recapitulate cancer cell heterogeneity within tumors. PDO represent a highly translatable platform for personalized medicine. Grown within 14 days, PDO allow rapid evaluation of therapeutic effects of drugs, both standard of care and molecularly-targeted therapies. PDO serve as an experimental platform to study genetic and environmental factors, as well as signaling pathways, in tumor development and progression that may be variable from patient to patient. Herein we describe extensive protocols to generate and characterize esophageal cancer PDO.
ACKNOWLEDGEMENT
This study was supported by the following NIH Grants: P01CA098101 (HN, KT, KAW, VG, AJK, AB, JAD, AKR), U54CA163004 (TK, JTG, HN, GWF, GGG, JQ, JA, AKR, TCW), R01DK114436 and R01AA026297 (HN), P30DK050306, P30CA013696, P30ES013508 University of Pennsylvania Center of Excellence in Environmental Toxicology (RCA, HN and AKR), K01DK103953 and R01DK121159 (KAW), K08DK106444 and R03DK118310 (ABM), K01DK100485, R03DK114463 (KEH), R01DK113144, R01DK100342, DK120650 (JQ), R01DE02680(AJY) and the American Cancer Society RP-10-033-01-CCE (AKR). VG holds the tier 2 Canada Research Chair in Gastrointestinal Stem Cell Biology. This study was also supported by the Grant-in-Aid for challenging Exploratory Research, Grant in Aid for Scientific Research B and Grant in Aid for Scientific Research C from the Ministry of Education, Culture, Sports, Science and Technology of Japan (17H04285 to SN). We thank the University of Pennsylvania NIH/NIDDK Center for Molecular Studies in Digestive and Liver Diseases and the CHOP Epithelial Biology Center of Excellence. We are grateful for the Herbert Irving Comprehensive Cancer Center Biostatistics, Genomics and High Throughput Screening and Molecular Pathology Shared Resources.
LITERATURE CITED
- Boj SF, Hwang CI, Baker LA, Chio II, Engle DD, Corbo V, … Tuveson DA (2015). Organoid models of human and mouse ductal pancreatic cancer. Cell, 160(1–2), 324–338. doi: 10.1016/j.cell.2014.12.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broutier L, Andersson-Rolf A, Hindley CJ, Boj SF, Clevers H, Koo BK, & Huch M (2016). Culture and establishment of self-renewing human and mouse adult liver and pancreas 3D organoids and their genetic manipulation. Nat Protoc, 11(9), 1724–1743. doi: 10.1038/nprot.2016.097 [DOI] [PubMed] [Google Scholar]
- Dotto GP, & Rustgi AK (2016). Squamous Cell Cancers: A Unified Perspective on Biology and Genetics. Cancer Cell, 29(5), 622–637. doi: 10.1016/j.ccell.2016.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driehuis E, Kolders S, Spelier S, Lohmussaar K, Willems SM, Devriese LA, … Clevers H (2019). Oral Mucosal Organoids as a Potential Platform for Personalized Cancer Therapy. Cancer Discov, 9(7), 852–871. doi: 10.1158/2159-8290.CD-18-1522 [DOI] [PubMed] [Google Scholar]
- Driehuis E, Spelier S, Beltran Hernandez I, de Bree R, S MW, Clevers H, & Oliveira S (2019). Patient-Derived Head and Neck Cancer Organoids Recapitulate EGFR Expression Levels of Respective Tissues and Are Responsive to EGFR-Targeted Photodynamic Therapy. J Clin Med, 8(11). doi: 10.3390/jcm8111880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher OM, Lord SJ, Falkenback D, Clemons NJ, Eslick GD, & Lord RV (2017). The prognostic value of TP53 mutations in oesophageal adenocarcinoma: a systematic review and meta-analysis. Gut, 66(3), 399–410. doi: 10.1136/gutjnl-2015-310888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii M, Shimokawa M, Date S, Takano A, Matano M, Nanki K, … Sato T (2016). A Colorectal Tumor Organoid Library Demonstrates Progressive Loss of Niche Factor Requirements during Tumorigenesis. Cell Stem Cell, 18(6), 827–838. doi: 10.1016/j.stem.2016.04.003 [DOI] [PubMed] [Google Scholar]
- Gao D, Vela I, Sboner A, Iaquinta PJ, Karthaus WR, Gopalan A, … Chen Y (2014). Organoid cultures derived from patients with advanced prostate cancer. Cell, 159(1), 176–187. doi: 10.1016/j.cell.2014.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grugan KD, Miller CG, Yao Y, Michaylira CZ, Ohashi S, Klein-Szanto AJ, … Rustgi AK (2010). Fibroblast-secreted hepatocyte growth factor plays a functional role in esophageal squamous cell carcinoma invasion. Proc Natl Acad Sci U S A, 107(24), 11026–11031. doi: 10.1073/pnas.0914295107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Holtzinger A, Jagan I, BeGora M, Lohse I, Ngai N, … Muthuswamy SK (2015). Ductal pancreatic cancer modeling and drug screening using human pluripotent stem cell- and patient-derived tumor organoids. Nat Med, 21(11), 1364–1371. doi: 10.1038/nm.3973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubert CG, Rivera M, Spangler LC, Wu Q, Mack SC, Prager BC, … Rich JN (2016). A Three-Dimensional Organoid Culture System Derived from Human Glioblastomas Recapitulates the Hypoxic Gradients and Cancer Stem Cell Heterogeneity of Tumors Found In Vivo. Cancer Res, 76(8), 2465–2477. doi: 10.1158/0008-5472.CAN-15-2402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalabis J, Wong GS, Vega ME, Natsuizaka M, Robertson ES, Herlyn M, … Rustgi AK (2012). Isolation and characterization of mouse and human esophageal epithelial cells in 3D organotypic culture. Nat Protoc, 7(2), 235–246. doi: 10.1038/nprot.2011.437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakasheva TA, Lin EW, Tang Q, Qiao E, Waldron TJ, Soni M, … Rustgi AK (2018). IL-6 Mediates Cross-Talk between Tumor Cells and Activated Fibroblasts in the Tumor Microenvironment. Cancer Res, 78(17), 4957–4970. doi: 10.1158/0008-5472.CAN-17-2268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasagi Y, Chandramouleeswaran PM, Whelan KA, Tanaka K, Giroux V, Sharma M, … Nakagawa H (2018). The Esophageal Organoid System Reveals Functional Interplay Between Notch and Cytokines in Reactive Epithelial Changes. Cell Mol Gastroenterol Hepatol, 5(3), 333–352. doi: 10.1016/j.jcmgh.2017.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kijima T, Nakagawa H, Shimonosono M, Chandramouleeswaran PM, Hara T, Sahu V, … Natsugoe S (2019). Three-Dimensional Organoids Reveal Therapy Resistance of Esophageal and Oropharyngeal Squamous Cell Carcinoma Cells. Cell Mol Gastroenterol Hepatol, 7(1), 73–91. doi: 10.1016/j.jcmgh.2018.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinugasa H, Whelan KA, Tanaka K, Natsuizaka M, Long A, Guo A, … Nakagawa H (2015). Mitochondrial SOD2 regulates epithelial-mesenchymal transition and cell populations defined by differential CD44 expression. Oncogene, 34(41), 5229–5239. doi: 10.1038/onc.2014.449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Hu W, Matulay JT, Silva MV, Owczarek TB, Kim K, … Shen MM (2018). Tumor Evolution and Drug Response in Patient-Derived Organoid Models of Bladder Cancer. Cell, 173(2), 515–528 e517. doi: 10.1016/j.cell.2018.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Francies HE, Secrier M, Perner J, Miremadi A, Galeano-Dalmau N, … Garnett MJ (2018). Organoid cultures recapitulate esophageal adenocarcinoma heterogeneity providing a model for clonality studies and precision therapeutics. Nat Commun, 9(1), 2983. doi: 10.1038/s41467-018-05190-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Ootani A, & Kuo C (2016). An Air-Liquid Interface Culture System for 3D Organoid Culture of Diverse Primary Gastrointestinal Tissues. Methods Mol Biol, 1422, 33–40. doi: 10.1007/978-1-4939-3603-8_4 [DOI] [PubMed] [Google Scholar]
- Lin EW, Karakasheva TA, Lee DJ, Lee JS, Long Q, Bass AJ, … Rustgi AK (2017). Comparative transcriptomes of adenocarcinomas and squamous cell carcinomas reveal molecular similarities that span classical anatomic boundaries. PLoS Genet, 13(8), e1006938. doi: 10.1371/journal.pgen.1006938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord RV, Brabender J, Wickramasinghe K, DeMeester SR, Holscher A, Schneider PM, … DeMeester TR (2005). Increased CDX2 and decreased PITX1 homeobox gene expression in Barrett’s esophagus and Barrett’s-associated adenocarcinoma. Surgery, 138(5), 924–931. doi: 10.1016/j.surg.2005.05.007 [DOI] [PubMed] [Google Scholar]
- Miyoshi H, & Stappenbeck TS (2013). In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat Protoc, 8(12), 2471–2482. doi: 10.1038/nprot.2013.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira L, Bakir B, Chatterji P, Dantes Z, Reichert M, & Rustgi AK (2018). Pancreas 3D Organoids: Current and Future Aspects as a Research Platform for Personalized Medicine in Pancreatic Cancer. Cell Mol Gastroenterol Hepatol, 5(3), 289–298. doi: 10.1016/j.jcmgh.2017.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naganuma S, Whelan KA, Natsuizaka M, Kagawa S, Kinugasa H, Chang S, … Nakagawa H (2012). Notch receptor inhibition reveals the importance of cyclin D1 and Wnt signaling in invasive esophageal squamous cell carcinoma. Am J Cancer Res, 2(4), 459–475. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/22860235 [PMC free article] [PubMed] [Google Scholar]
- Natsuizaka M, Whelan KA, Kagawa S, Tanaka K, Giroux V, Chandramouleeswaran PM, … Nakagawa H (2017). Interplay between Notch1 and Notch3 promotes EMT and tumor initiation in squamous cell carcinoma. Nat Commun, 8(1), 1758. doi: 10.1038/s41467-017-01500-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi S, Natsuizaka M, Yashiro-Ohtani Y, Kalman RA, Nakagawa M, Wu L, … Nakagawa H (2010). NOTCH1 and NOTCH3 coordinate esophageal squamous differentiation through a CSL-dependent transcriptional network. Gastroenterology, 139(6), 2113–2123. doi: 10.1053/j.gastro.2010.08.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okawa T, Michaylira CZ, Kalabis J, Stairs DB, Nakagawa H, Andl CD, … Rustgi AK (2007). The functional interplay between EGFR overexpression, hTERT activation, and p53 mutation in esophageal epithelial cells with activation of stromal fibroblasts induces tumor development, invasion, and differentiation. Genes Dev, 21(21), 2788–2803. doi: 10.1101/gad.1544507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauli C, Hopkins BD, Prandi D, Shaw R, Fedrizzi T, Sboner A, … Rubin MA (2017). Personalized In Vitro and In Vivo Cancer Models to Guide Precision Medicine. Cancer Discov, 7(5), 462–477. doi: 10.1158/2159-8290.CD-16-1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rustgi AK, & El-Serag HB (2014). Esophageal carcinoma. N Engl J Med, 371(26), 2499–2509. doi: 10.1056/NEJMra1314530 [DOI] [PubMed] [Google Scholar]
- Sachs N, de Ligt J, Kopper O, Gogola E, Bounova G, Weeber F, … Clevers H (2018). A Living Biobank of Breast Cancer Organoids Captures Disease Heterogeneity. Cell, 172(1–2), 373–386 e310. doi: 10.1016/j.cell.2017.11.010 [DOI] [PubMed] [Google Scholar]
- Saito Y, Muramatsu T, Kanai Y, Ojima H, Sukeda A, Hiraoka N, … Saito H (2019). Establishment of Patient-Derived Organoids and Drug Screening for Biliary Tract Carcinoma. Cell Rep, 27(4), 1265–1276 e1264. doi: 10.1016/j.celrep.2019.03.088 [DOI] [PubMed] [Google Scholar]
- Sato T, Stange DE, Ferrante M, Vries RG, Van Es JH, Van den Brink S, … Clevers H (2011). Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology, 141(5), 1762–1772. doi: 10.1053/j.gastro.2011.07.050 [DOI] [PubMed] [Google Scholar]
- Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, … Clevers H (2009). Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature, 459(7244), 262–265. doi: 10.1038/nature07935 [DOI] [PubMed] [Google Scholar]
- Sen M, Hahn F, Black TA, DeMarshall M, Porter W, Snowden E, … Carpenter EL (2019). Correction: Flow based single cell analysis of the immune landscape distinguishes Barrett’s esophagus from adjacent normal tissue. Oncotarget, 10(49), 5119. doi: 10.18632/oncotarget.27161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka N, Osman AA, Takahashi Y, Lindemann A, Patel AA, Zhao M, … Myers JN (2018). Head and neck cancer organoids established by modification of the CTOS method can be used to predict in vivo drug sensitivity. Oral Oncol, 87, 49–57. doi: 10.1016/j.oraloncology.2018.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taneja SS (2015). Re: Organoid cultures derived from patients with advanced prostate cancer. J Urol, 193(4), 1442–1443. doi: 10.1016/j.juro.2015.01.059 [DOI] [PubMed] [Google Scholar]
- van de Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, Pronk A, … Clevers H (2015). Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell, 161(4), 933–945. doi: 10.1016/j.cell.2015.03.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlachogiannis G, Hedayat S, Vatsiou A, Jamin Y, Fernandez-Mateos J, Khan K, … Valeri N (2018). Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science, 359(6378), 920–926. doi: 10.1126/science.aao2774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh AJ, Castellanos JA, Nagathihalli NS, Merchant NB, & Skala MC (2016). Optical Imaging of Drug-Induced Metabolism Changes in Murine and Human Pancreatic Cancer Organoids Reveals Heterogeneous Drug Response. Pancreas, 45(6), 863–869. doi: 10.1097/MPA.0000000000000543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H, Ma Q, Peng S, Adelmant G, Swain D, Song W, … Bass AJ (2014). SOX2 and p63 colocalize at genetic loci in squamous cell carcinomas. J Clin Invest, 124(4), 1636–1645. doi: 10.1172/JCI71545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan KA, Chandramouleeswaran PM, Tanaka K, Natsuizaka M, Guha M, Srinivasan S, … Nakagawa H (2017). Autophagy supports generation of cells with high CD44 expression via modulation of oxidative stress and Parkin-mediated mitochondrial clearance. Oncogene, 36(34), 4843–4858. doi: 10.1038/onc.2017.102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan KA, Merves JF, Giroux V, Tanaka K, Guo A, Chandramouleeswaran PM, … Nakagawa H (2017). Autophagy mediates epithelial cytoprotection in eosinophilic oesophagitis. Gut, 66(7), 1197–1207. doi: 10.1136/gutjnl-2015-310341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan KA, Muir AB, & Nakagawa H (2018). Esophageal 3D Culture Systems as Modeling Tools in Esophageal Epithelial Pathobiology and Personalized Medicine. Cell Mol Gastroenterol Hepatol, 5(4), 461–478. doi: 10.1016/j.jcmgh.2018.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
KEY REFERENCE (optional)
Kijima et al. describe generation and drug testing upon PDO from ESCC and head-and-neck squamous cell carcinoma clinical samples (Kijima et al., 2019).