

Abstract
We report eight cases of a distinctive, previously undescribed renal cell carcinoma associated with somatic mutations in the neurofibromin 2 (NF2) gene. All patients were adults, ranging from 51 to 78 years of age and of cases of known gender six of seven were males. The carcinomas were predominantly unencapsulated, and all had a rounded, nodular interface with the native kidney. The neoplasms were all solid with papillary architecture evident in most cases (7/8), while one was only tubular. All cases were biphasic, characterized by larger and smaller carcinoma cells. The smaller cells clustered around basement membrane material similar to the characteristic pattern of the t(6;11) renal cell carcinoma associated with TFEB gene fusions. In 6 of 8 carcinomas, branching nodules of small cells clustered around basement membrane material within larger acini yielding a distinctive glomeruloid pattern. In 6 of 8 carcinomas, the small cells were focally spindle-shaped and unassociated with basement membrane material. The stroma was sclerotic in all eight carcinomas, and all eight contained psammoma bodies that were abundant in two. In some carcinomas, focal or predominant areas had a less distinctive appearance; two had areas that resembled clear cell renal cell carcinoma, two had high-grade eosinophilic areas, while one had branching tubular architecture that resembled mucinous tubular and spindle cell carcinoma. Two carcinomas demonstrated cellular necrosis. While we have minimal clinical follow-up, one case presented with distant metastasis, progressed, and resulted in patient death. While NF2 mutations may be found in other established renal cell carcinoma subtypes (often as secondary genetic alterations), they are potentially the genetic driver of this distinctive entity.
Keywords: Renal Neoplasm, NF2; Merlin, Renal Cell Carcinoma, Mutation
INTRODUCTION
Perhaps more than any other cancer, distinctive morphologic features of renal cell carcinomas (RCCs) correlate with specific genetic alterations. For example, prominent nucleoli with a perinucleolar halo correlate with fumarate hydratase deficiency,1, eosinophilic cytoplasmic inclusions correlate with succinate dehydrogenase (SDH) deficiency2, basophilic cytoplasmic precipitates correlate with tuberous sclerosis complex (TSC) gene mutations3–5, and the constellation of clear cells with papillary architecture, voluminous cytoplasm and psammoma bodies correlate with translocations involving chromosome Xp11 resulting in gene fusions involving the TFE3 transcription factor gene6,7.
We report herein eight cases of a morphologically distinctive, previously undescribed renal cell carcinoma that is associated with somatic mutations in the neurofibromin 2 (NF2) gene. As NF2 has previously been implicated in renal neoplasia, we review these publications to put our findings into context.
METHODS
IRB Approval
This study was approved by the Institutional Review Boards at our institutions.
Cases
The eight cases reported herein were retrieved from the files of three authors (PA, VER, JNE) during a review of unclassified primary renal carcinomas. Seven of the eight cases were reviewed as consultations for renal tumor classification, while the eighth (Case #3) was an in house case reviewed during routine sign out at one of our institutions. Cases were submitted with a variety of differential diagnoses; these included papillary renal cell carcinoma type 2 (2 cases), MiTF family translocation carcinoma (2 cases), and ectopic/metastatic sex cord stromal tumor (2 cases).
Immunohistochemistry
Immunohistochemistry with HMB45, cathepsin K, cytokeratins AE1/3 and Cam5.2, epithelial membrane antigen (EMA), PAX8, S100 protein, melan A, desmin, SF-1, GATA3, smooth muscle actin, and TFE3 was performed as previously described8,9. Immunohistochemistry for YAP1 was performed using the Abcam antibody #52771 at 1:200 dilution. Immunohistochemistry for NF2 was performed using the Abcam antibody #217016 at 1:500 dilution.
DNA and RNA sequencing
DNA and RNA sequencing were performed by the Genomics and Molecular Pathology Core at UT Southwestern Medical Center on all cases except case #3. Briefly, tumor hematoxylin and eosin slides were examined and marked by 2 pathologists (P.A. and D.N.P.) for subsequent macro-dissection, nucleic acid isolation, and molecular testing. Areas enriched with tumor were then scraped from adjacent 5-μm thick formalin-fixed paraffin embedded (FFPE) sections. Adjacent normal tissue was separately isolated and processed for select cases when available. Extraction and purification were performed using Qiagen Allprep kits (Qiagen, Germantown, MD). Libraries were prepared using KAPA Hyperplus kits (Roche Sequencing and Life Science Kapa Biosystems, Wilmington, MA) with genomic regions of interest captured by custom DNA probes covering all exons of over 1425 cancer-related genes. The libraries were sequenced to an average unique read depth of >600X (95% of exons at or above 100X) using Sequencing by Synthesis (SBS) paired-end cluster generation on the Illumina NextSeq 550 platform (Illumina Inc., San Diego, CA). Sequence reads were aligned to reference genome GRCh38 and subsequent analyses were performed using custom germline, somatic, and mRNA bioinformatics pipelines run on the UTSW Bio-High Performance Computer cluster and optimized for detection of single nucleotide variants, indels, and known gene fusions. Minor allele frequency limit of detection for the assay: single nucleotide variants - 5%; indels and known gene fusions - 10%. Variants in exons with less than 100X coverage are not reported.
Somatic variants were identified on the basis of their variant allele frequencies (VAF) and relative absence in matched normal tissue (if available) and/or presence at frequencies below 0.5% in databases of germline variants (dbSNP, gnomAD). Variants were classified according to the Association for Molecular Pathology/American Society for Clinical Oncology/College of American Pathologists guidelines10, and all variant calls were inspected using Integrated Genomics Viewer version 2.3.4 (IGV; Broad Institute, MIT Harvard, Cambridge, MA) prior to reporting.
Copy number variants (CNV) were detected using CNVKit and an internally derived panel of normal FFPE tissue samples.
Case #3 was evaluated using MSK-Impact (Integrated Mutational Profiling of Actionable Cancer Targets), a targeted ultra-deep next generation sequencing platform designed to capture all exons and selected introns of over 400 oncogenes, tumor suppressor genes, and members of targetable pathways11.
Case #1 was additionally subjected to RNA sequencing using a different technique. RNA was extracted from formalin-fixed paraffin-embedded (FFPE) tissue using Amsbio’s ExpressArt FFPE Clear RNA Ready kit (Amsbio LLC, Cambridge, MA). Fragment length was assessed with an RNA 6000 chip on an Agilent Bioanalyzer (Agilent Technologies, Santa Clara, CA). RNA-sequencing libraries were prepared using 20 to 100 ng total RNA with the TruSight RNA Fusion Panel (Illumina, San Diego, CA). Each sample was subjected to targeted RNA sequencing on an Illumina MiSeq at 8 samples per flow cell (∼3 million reads per sample). All reads were independently aligned with STAR (version 2.3) and BowTie2 against the human reference genome (hg19) for Manta-Fusion and TopHat-Fusion analysis, respectively.
Photomicrographs
Composite images were compiled using Adode Photoshop CC.
RESULTS
Clinical History and Gross/Radiologic Appearance
The clinical and pathologic features of the cases are summarized in Tables 1 and 2. Patient ages ranged from 51 to 78 years. There were six males, one female, and one patient of unknown gender. One neoplasm (case 2) arose in a native atrophic kidney 21 years after that patient had received a renal transplant for end stage renal disease. In two other cases, the surrounding kidney demonstrated simple cysts. Another neoplasm arose in the setting of renal adrenal fusion. Of the seven patients for whom the procedure was specified, five underwent partial nephrectomy while two underwent radical nephrectomy. Clinical follow-up was limited. However, patient 3 presented with bone metastasis, and immediately after partial nephrectomy was found to have lung metastasis. The patient died of disease two months later.
Table 1:
Clinical, Pathologic, and Genetic Features of Cases
| Case | Age/Sex | Tumor Size/Stage | IHC+ | IHC- | Comment | NF2 Variant | Other Molecular Findings |
|---|---|---|---|---|---|---|---|
| 1 | 78/M | 4.2cm; pT1bNX; | HNF-1 beta, PAX8, CK7, EMA (focal) | WT-1, Melan A, cathepsin k-, GATA3, SF-1, inhibin; FH, SDHB intact | TFE3/B FISH -; died of complications 1 month after operation | p.Tyr66* | Chr loss: 1p, 6, 22q; Chr gain: 16, 20. XPO1 p.Asn735Lys; POLQ p.Phe944Val; SPTBN1 p.Trp1893*; EXT2 p.Tyr421*; NPM1 p.Asp165_Glu169del; NCOA1 p.Met476Ile; AUTS2 p.Gly216Arg; ARID2 p.Arg314Ser; LMO7 p.Pro715Ser; CYLD p.Pro78Thr; MIB1 p.Ser408Asn; CSNK1G2 c.187+1G>T |
| 2 | 60/U | 7cm; pT2NX | PAX8, EMA,CK7 | WT-1, TFE3,cathepsin k, TFEB, HMB45, Melan A chromogranin, synaptophysin, GATA3, SF-1, inhibin | TFE3/B FISH -; ESRD; tumor in native kidney 21 years after transplant | p.Asn293fs | Chr loss: 1p, 6, 22q; Chr gain: 16, 20. WNT4 p.Val97Ile; RAD54L p.Arg202Cys; MRE11A p.Asp495His; ERBB2 p.Asp880Tyr; PI4KA p.Pro514Leu |
| 3 | 69/M | 4cm; pT1cNXM1 (bone metastasis) | CK7, racemase, HNF-1 beta | TFE3, HMB45, Melan A, cathepsin K, AR,ER, CA-IX, GATA3 | Lung metastasis; died of disease 2 months later | p.Gln111* | Chr loss: 9; Chr gain: 20. GLI1 p.Gly1097fs; RB1 p.Glu539_Thr543del; TERT promoter variant g.1295228C>T |
| 4 | 53/F | 2.5cm; pT1NX | GATA3, WT-1, cathepsin K, inhibin, SF-1 | TFE3/B FISH -; h/o bilateral ovarian cystectomy | c.600–1G>A | Chr loss: 1p, 4, 6, 21q, 22q; Chr gain: 1, 7, 20. ARID1A p.Pro1619fs; CDK6 p.Thr198fs; RAD50 p.Gln479Arg; MLLT6 p.Ser608Tyr; SUGP2 p.Ala75Ser; THBS1 p.Arg458Gly; LRIG3 p.Asp56Glu; PLCG2 p.Glu567Lys; KMT2B p.Gly100Asp |
|
| 5 | 73/M | 3cm L partial nephrectomy variegated pink orange multilobulated | CK7 | CA-IX, cathepsin K, GATA3, WT-1, inhibin, SF-1 -; FH, SDHB intact | TFE3/B FISH- | p.Lys364* | Chr loss: 1, 6, 9q, 15q, 19q; Chr gain: 9p, 16. CHD6 p.Asn2152Ser; NUP98 p.Ser103*; SAMD9 p.Phe801fs; MIB1 c.2211+1G>A; CIITA p.Gly174Arg; MECOM p.Glu81Gln; FLT4 p.Trp1233Cys; ABL2 p.Asn152fs; ASPSCR1 p.Asp217fs; HIST1H2AG p.Pro81Arg; FBXW7 p.Arg505His; KDM1A p.Val400Ile; SQSTM1 p.Cys27Ser |
| 6 | 71/M | 1.5cm; pT3NX (perirenal fat and vascular invasion) | PAX8, CK7 | Cathepsin k, GATA3, WT-1, inhibin, SF-1-; FH intact |
Concurrent clear cell RCC; TFE3/B FISH-, | p.Leu316fs | Chr loss: 1p, 6, 9, 15q; Chr gain: 16, 17q, 20. SMARCA4 c.3546+3A>T; FANCD2 p.Ser352Leu; BCL2 p.Leu185Met; PAK5 p.Ala120Val; PPP2R1B p.His661Leu; NOTCH2 p.Phe2091Ser; PICALM p.Val183Gly; HIP1 c.2891–2A>G; AKAP9 p.Glu2697Gln; STAT1 p.Ala531Thr; DLEC1 p.Gln1740Arg; PRICKLE1 p.Pro786Thr; LAMA5 p.Asn479Ser |
| 7 | 52/M | 1cm; pTXNX | PAX8 | Chromogranin, Synaptophysin, cathepsin K, Melan A, WT-1, inhibin, SF-1, calretinin, GATA3 | Renal-adrenal fusion; concurrent renal cyst |
p.Arg516_Lys523delinsGln | Chr loss: 14q, 19q, 22q; Chr gain: 2p, 5, 13q, 20. PPP2R2B p.Arg129fs; RPL22 p.Lys13fs; BICC1 p.Val254Leu; WDFY3 c.1591+4A>C; TFEB p.Arg4Pro; FZD3 p.Gly595Ala; TNC p.Phe1196Tyr |
| 8 | 51/M | 1.5cm; pT1NX | Concurrent renal cyst | p.Lys130fs | Chr loss: 1p, 3, 6, 11, 14q, 19, 22q; Chr gain: 20. ERCC5 p.Leu844*; CLTC p.Asp662fs; WHSC1 c.1674+6dupT; SNX29 p.Lys513Ile; CNTN1 c.2185–4T>A; SGK1 p.Lys338Glu; ITGA7 p.Glu120Val; GNAI1 p.Arg142Ile; TPR p.Glu1594Asp; RASGRF1 p.Asn1075Tyr; LIFR p.Met773Leu |
U=unknown gender; FH=fumarate hydratase; SDHB=succinate dehydrogenase subunit B; Chr=chromosome; IHC=immunohistochemistry
Table 2:
Morphologic Features of Cases
| Case | Slides | Capsule | Vaguely Nodular | Biphasic | Basement Membrane Nodules | Small Cell Spindling | Papillary | Glomeruloid Bodies | Sclerotic Stroma | Psammoma Bodies | Other |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 3 | −/+ | + | + | + | + | + | + | + | + | |
| 2 | 3 | - | + | + | + | + | + | + | + | + | necrosis |
| 3 | 1 | na | + | + | + | + | + | + | + | + | |
| 4 | 2 | -* | + | + | + | + | + | + | + | +++ | necrosis |
| 5 | 3 | + | + | + | Minimal | + | + | - | + | +++ | Clear cells, pink cells, MTSC like |
| 6 | 2 | −/+ * | + | Minimal | + | Minimal | + | + | + | + | Pink cells, entraps tubules |
| 7 | 4 | −/+ | + | + | ++ | - | - | - | + | +++ | All tubular |
| 8 | 2 | - | + | + | ++ | - | + | + | + | + | Clear cells, entraps tubules |
na-not evaluable;
-prominent peritumoral chronic inflammation; MTSC-mucinous tubular and spindle cell carcinoma
Gross Pathology
Tumor sizes ranged from 1 to 7cm. All tumors were grossly solid. In six cases in which the neoplasm’s color was described, the color was either white or tan in five, while the other was described as variegated, orange and pink (Figure 1). The texture was described as firm in all four cases in which it was commented upon. All tumors were organ confined at diagnosis with the exception of case 3 (which presented with distant metastasis) and case 6 (which invaded perirenal fat).
Figure 1:
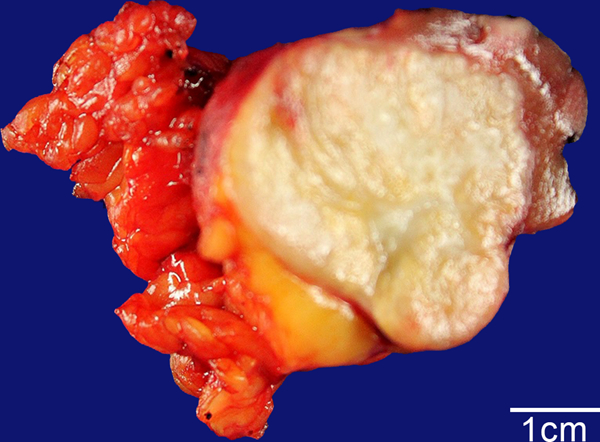
Biphasic Hyalinizing Psammomatous RCC case #3. The 4 cm neoplasm has a white, fibrous appearance, which contrasts with the yellow perinephric fat lobules to the left and the maroon-red native renal parenchyma to the upper right.
Microscopic Pathology and Immunoprofile.
All the neoplasms shared distinctive morphologic features and therefore they are described together (Figures 2–9). All neoplasms had a rounded, nodular appearance at low power. Among seven cases in which the interface with the native kidney was sampled, three were unencapsulated, three were partially unencapsulated, while one was completely encapsulated (Figures 2A, 3A, 4A, 5A, 7A, 8A, 9A). In two cases, the unencapsulated neoplasm entrapped native renal tubules (Figure 9E).
Figure 2:
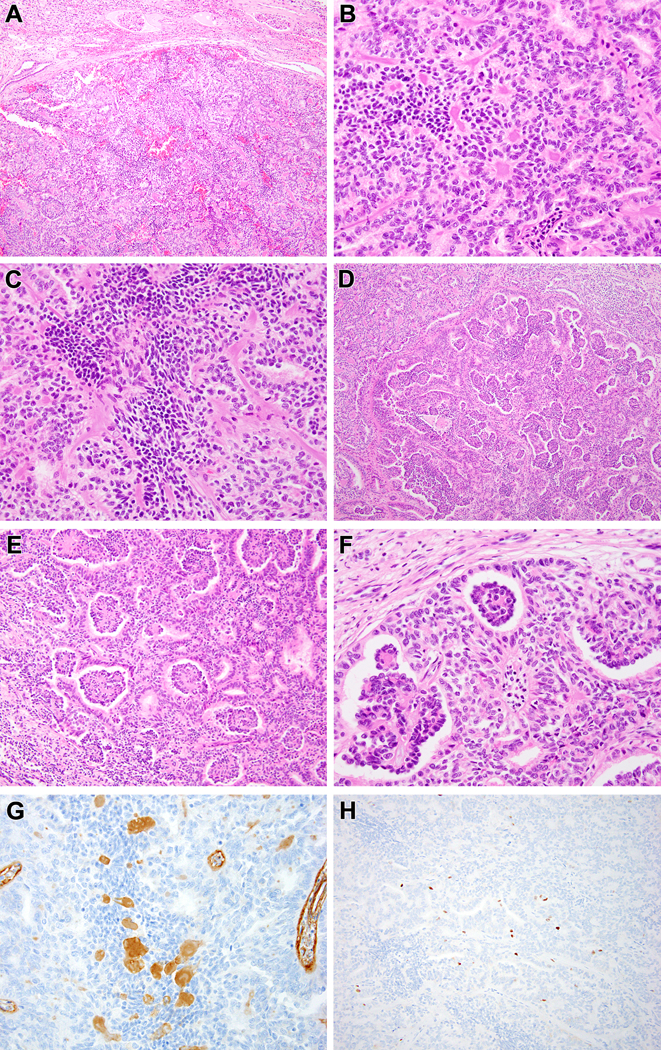
Biphasic Hyalinizing Psammomatous RCC case #1. The neoplasm is unencapsulated and has a rounded, nodular border with the native kidney at the top of the figure (A). The biphasic pattern is evident, as smaller cells with condensed chromatin cluster around hyalinized material while larger cells with vesicular chromatin form tubules and larger acini (B). The smaller cells also form solid spindle cell foci unassociated with basement membrane material (C). The small cells on papillae associated with basement membrane branch within larger acini, resulting in a glomeruloid pattern (D-F). The hyalinized material labels with type IV collagen, consistent with basement membrane material (G). The neoplasm has a low proliferative index as measured by Ki67 immunohistochemistry, particularly in the smaller cells (H).
Figure 9:
Biphasic Hyalinizing Psammomatous RCC case #8. At low power, the neoplasm has a solid and papillary architecture, and has an unencapsulated border with the native kidney to the right (A). The neoplasm has a biphasic appearance, with solid, tubular and papillary areas to the right and a nested clear cell area to the left (B). The solid papillary areas demonstrate the typical biphasic appearance with smaller cells clustered on basement membrane material and larger cells at the periphery (C). The solid clear cell areas are indistinguishable from clear cell RCC (D). Other areas of this neoplasm demonstrated sclerotic and desmoplastic stroma, as cords and tubules of neoplastic cells permeate amongst renal native tubules and glomeruli (E). In other areas, the neoplastic cells form linear cords in a desmoplastic and sclerotic stroma (F).
Figure 3:
Biphasic Hyalinizing Psammomatous RCC case #2. The neoplasm has a rounded, nodular border with the native kidney (A). At intermediate power, one can appreciate the biphasic nature of the neoplasm, with smaller cells clustered around the basement membrane material within larger acini formed by larger epithelioid cells (B). Branching clusters of smaller cells associated with basement membrane material within larger acini produced the striking glomeruloid appearance (C). This neoplasm demonstrated central tumor necrosis as shown to the left of the figure (D). Cytokeratin 7 preferentially labels the larger cells forming the larger acini (E). Epithelial membrane antigen (EMA) preferentially labels the smaller cells in the glomeruloid bodies (F).
Figure 4:
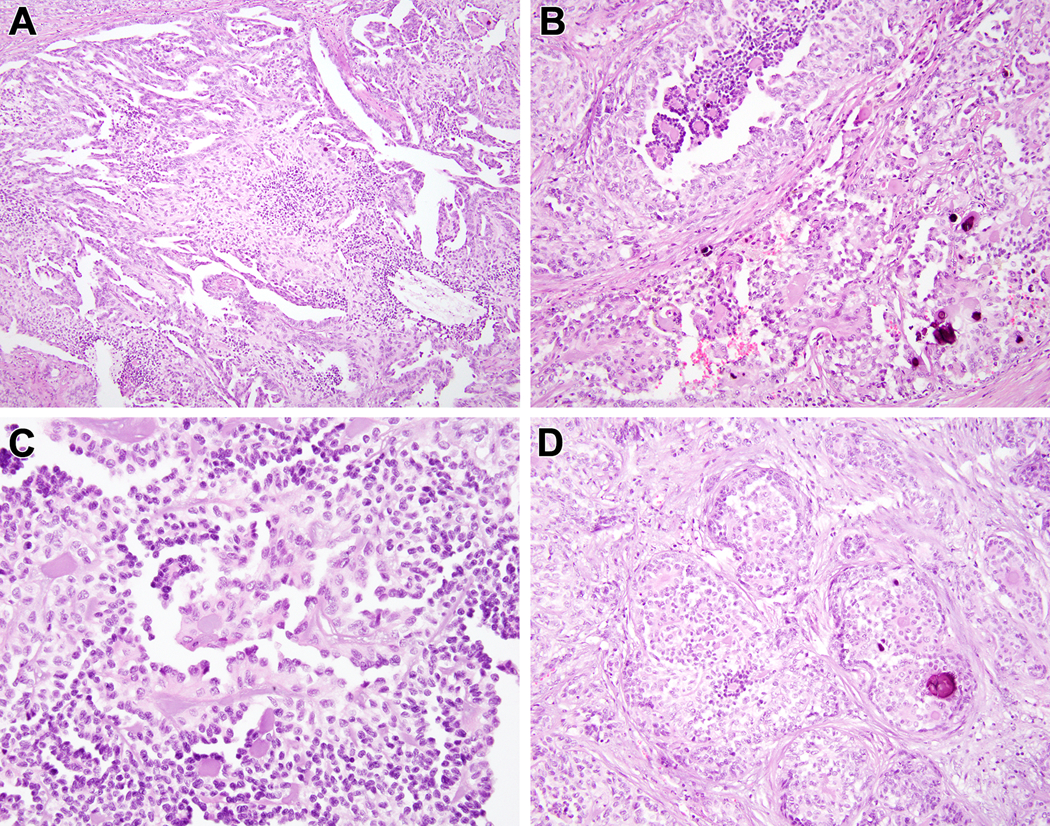
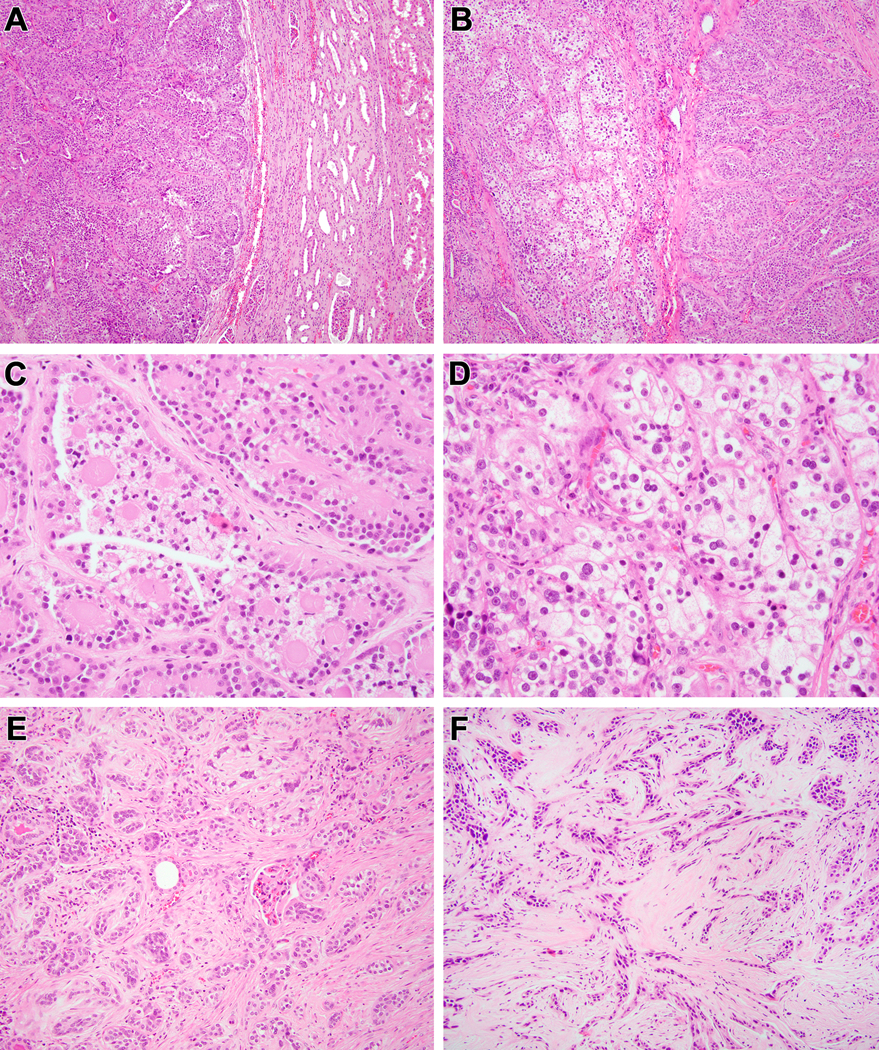
Biphasic Hyalinizing Psammomatous RCC case #3. The neoplasm is unencapsulated; note the native renal parenchyma at the upper left. The neoplasm has a striking biphasic appearance, with solid clusters of larger epithelioid clear cells and smaller cells forming branching papillae (A). At higher power, one can appreciate the smaller cells with condensed chromatin clustered around basement membrane material, the larger cells with vesicular chromatin, as well as extensive psammomatous calcification (B) (C). In other areas, the neoplasm has more fibrotic stroma and the architecture is more solid with epithelioid cells surrounding smaller cells around hyalinized material and psammomatous calcifications (D).
Figure 5:
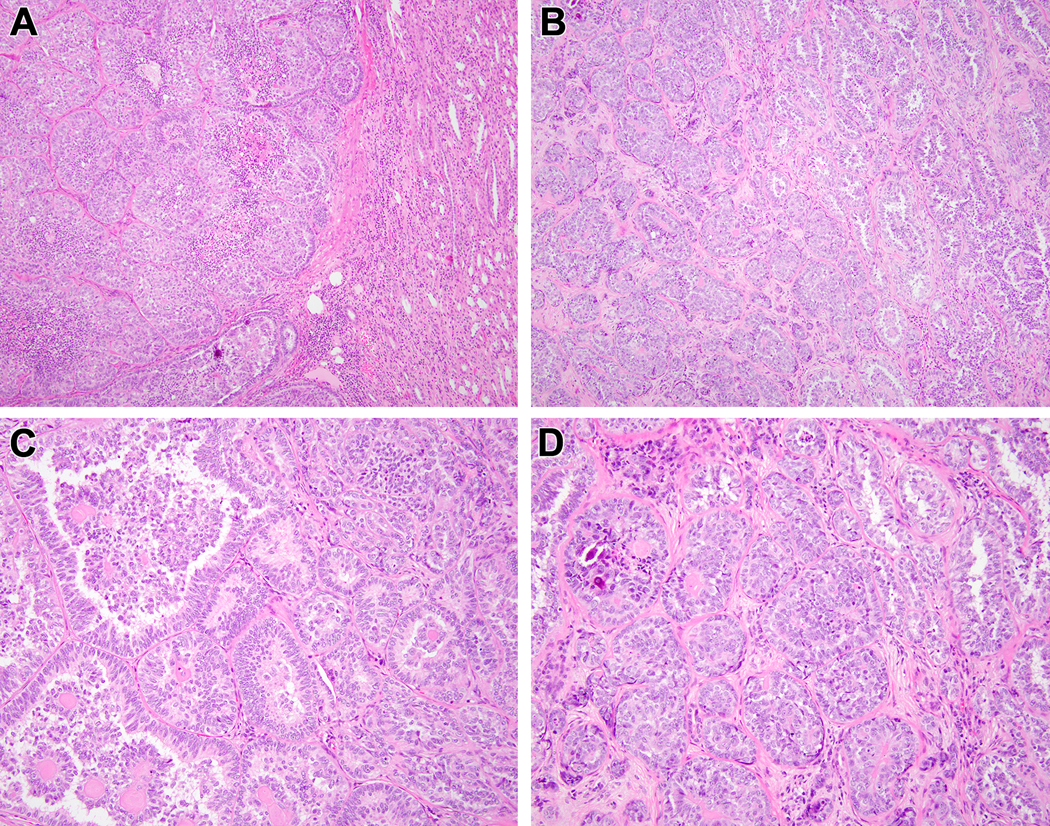
Biphasic Hyalinizing Psammomatous RCC case #4. At lower power, one can appreciate the unencapsulated nature of the neoplasm that borders the native kidney at the right (A). At intermediate power, one can appreciate both the tubulopapillary pattern at the right and the solid tubular pattern at the left (B). The tubulopapillary pattern demonstrates smaller cells clustered around basement membrane material in larger acini, yielding a glomeruloid pattern (C). The more solid areas feature sclerotic stroma in which there are tubules, more cribriform structures clustered around basement membrane material, and psammomatous calcification (D).
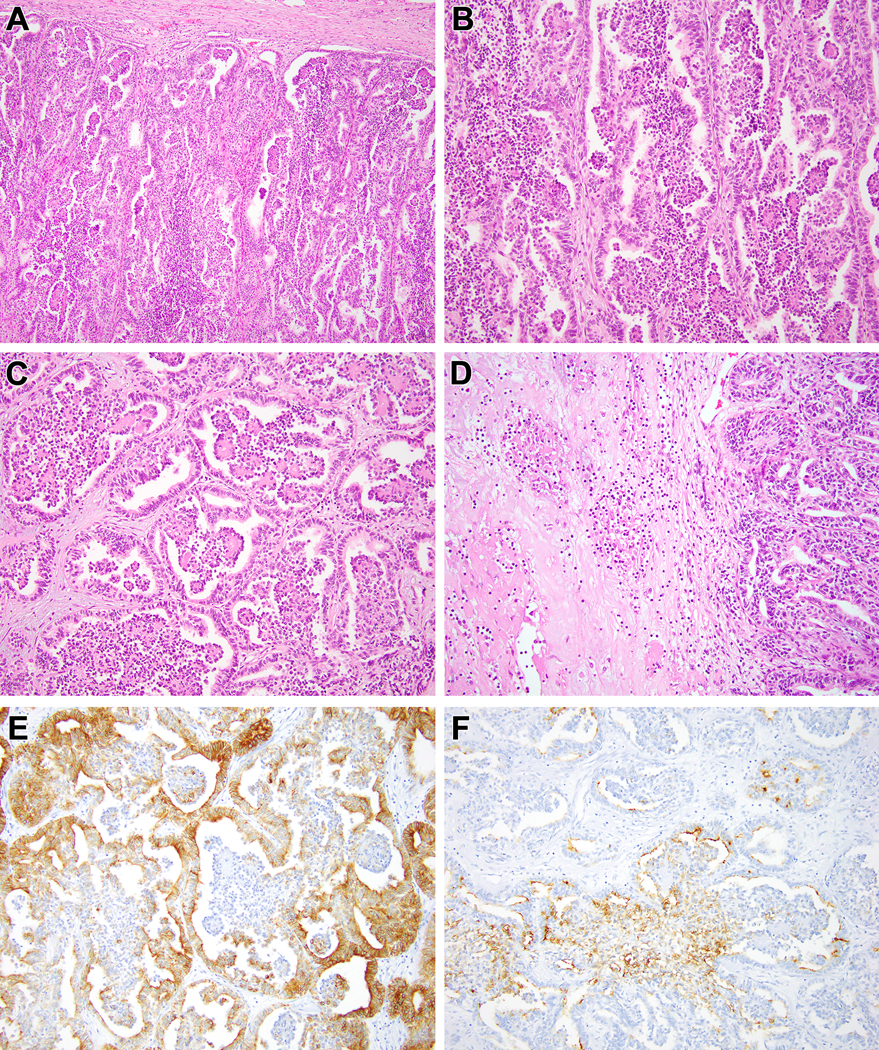
Figure 7:
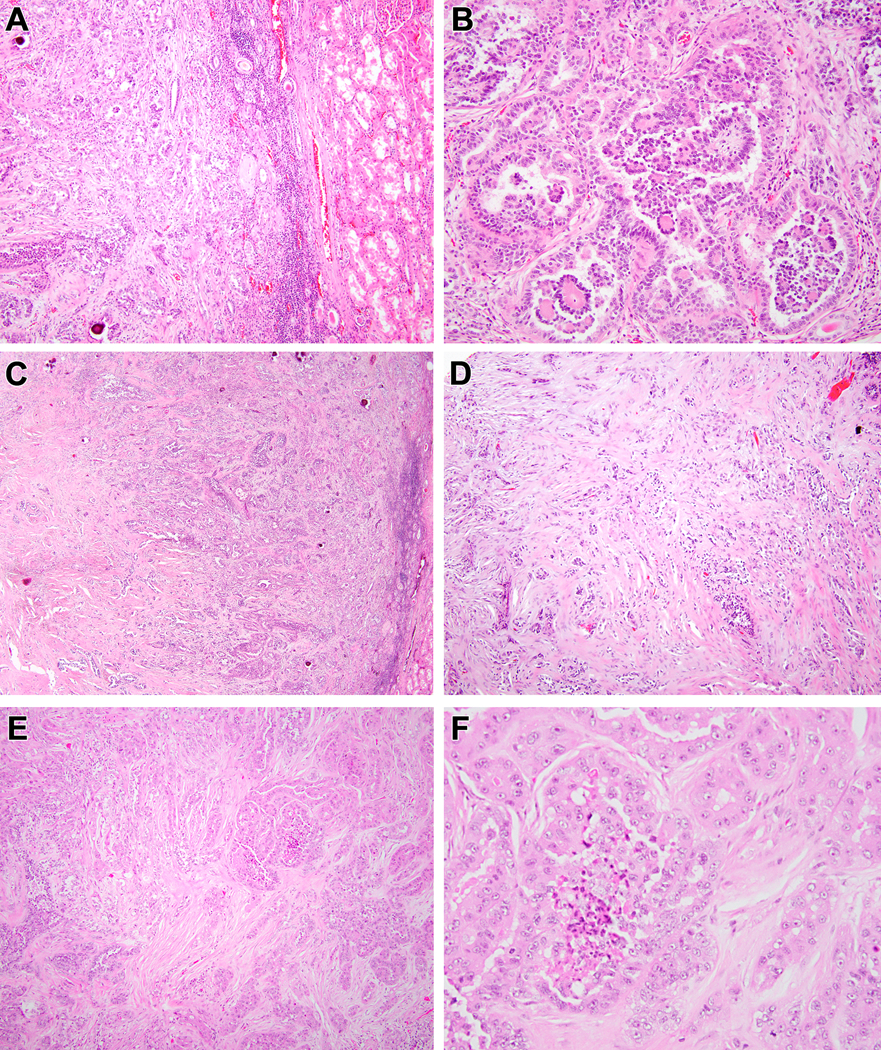
Biphasic Hyalinizing Psammomatous RCC case #6. The neoplasm demonstrated an unencapsulated border with the native kidney to the right (A). The majority of the neoplasm demonstrates the typical biphasic appearance with small cells forming glomeruloid bodies within larger acini lined by larger cells (B). Centrally, the neoplasm demonstrates extensive sclerosis (C). Neoplastic epithelium within this sclerotic and desmoplastic stroma has a cord-like and tubular appearance that raises the differential diagnosis of collecting duct carcinoma (D). The more typical biphasic areas (left bottom) merge with areas having more prominent eosinophilic cytoplasm (upper right) (E). The neoplastic cells in the latter areas have abundant eosinophilic cytoplasm, vesicular chromatin and prominent nucleoli and are associated with necrosis (F).
Figure 8:
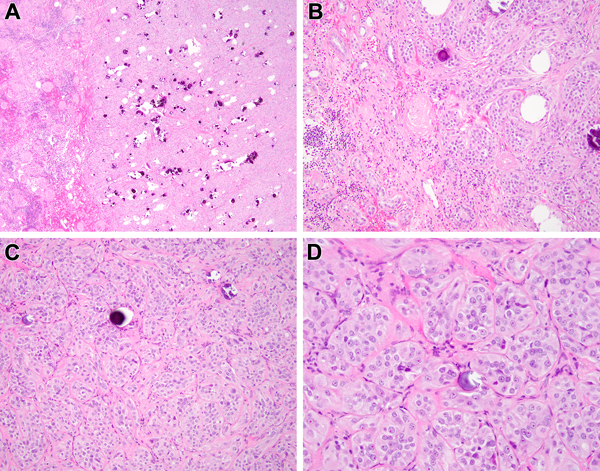
Biphasic Hyalinizing Psammomatous RCC case #7. This neoplasm has an unencapsulated border with the native kidney. The dominant appearance is that of a solid tubular lesion with extensive psammomatous calcifications (A). The neoplasm intermingles amongst native renal elements at its border to the left (B). At higher power, one can appreciate solid nests of neoplastic cells with vesicular chromatin, prominent nucleoli and abundant eosinophilic cytoplasm forming solid nests, frequently associated with psammomatous calcification (C, D).
All neoplasms were predominantly solid, though papillary architecture was evident in seven cases. The other case had only solid and tubular architecture (Figure 8A).
All neoplasms had a characteristic biphasic appearance, with both larger epithelial cells with vesicular chromatin and well-developed pale cytoplasm and smaller cells with darker condensed chromatin and minimal cytoplasm. In all cases, the smaller cells clustered around hyaline nodules similar to the Call-Exner bodies of adult granulosa cell tumor and the small cell population typical of the t(6;11) RCC12,13 (Figures 2B-C, 3A-B, 4B-C, 7B, 9C). This appearance was prominent in seven cases and only focal in one. In six cases, the small cells surrounding hyaline material and capillaries branched within dilated acini of larger cells, forming distinctive structures vaguely resembling glomeruli (“glomeruloid bodies”) (Figures 2D-F, 3C, 5C). In five cases, the smaller cells were focally spindle-shaped and formed solid foci that extended away from the hyaline nodules (Figure 2C). All eight cases contained psammoma bodies that were abundant in two.
While these neoplasms were solid and cellular in most areas, all at least focally demonstrated hyalinized sclerotic stroma. In two cases, neoplastic epithelium was compressed within more desmoplastic stroma, forming cords or trabeculae (Figure 7D, 9F). Two cases had focal clear cell areas that suggested clear cell RCC (Figures 6B, 9D), while two had focal areas with prominent eosinophilic cytoplasm and round nuclei with prominent nucleoli (Figures 7E,7F, 8C,8D). One case had focal branching architecture similar to that seen in mucinous tubular and spindle cell carcinoma (Figures 6C, 6D). Two neoplasms demonstrated necrosis (Figures 3D, 7F). Mitotic figures were typically sparse, with fewer than one per 10 high power fields in all cases except case 1 (2 per 10 high power fields) and case 4 (3 per 10 high power fields).
Figure 6:
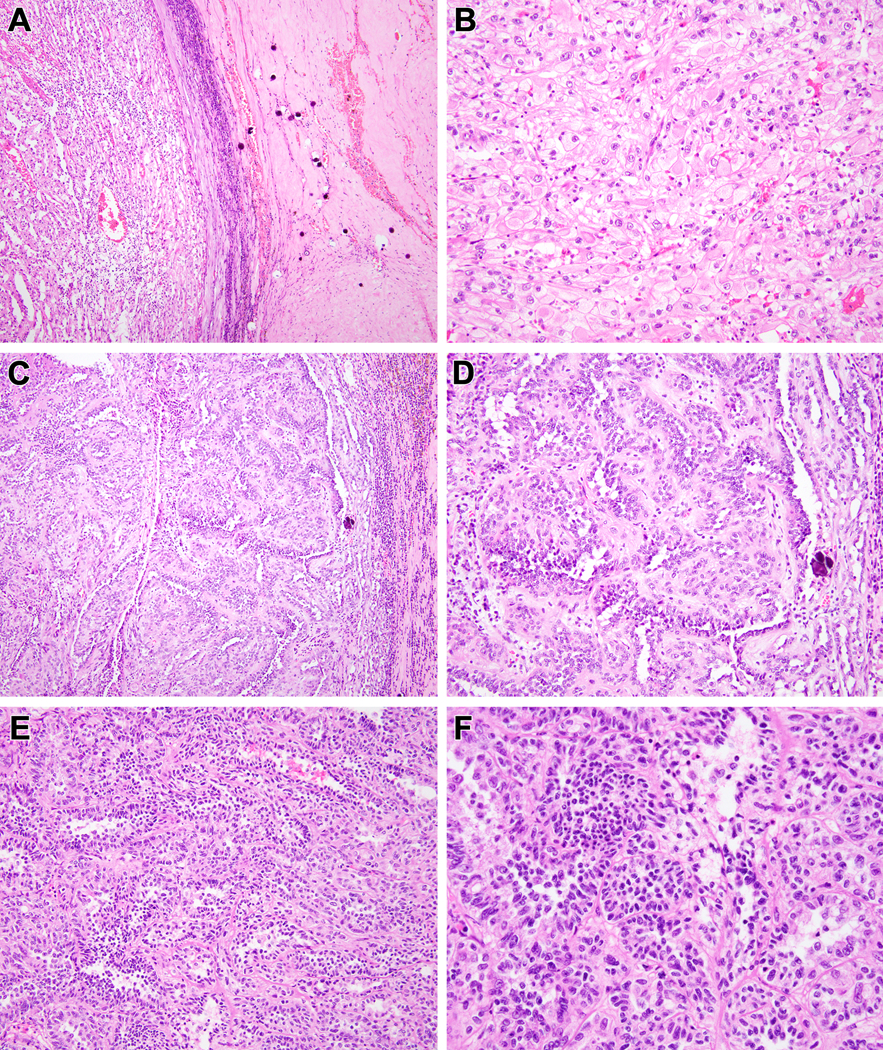
Biphasic Hyalinizing Psammomatous RCC case #5. Much of this neoplasm had the appearance of an unclassifiable RCC. At low power, the neoplasm demonstrates hyalinization with prominent psammomatous calcifications to the right, and a clear cell appearance to the left (A). Higher power view of the clear cell area reveals a nondescript solid clear cell proliferation that would be difficult to distinguish from high-grade conventional clear cell RCC (B). Other areas of the neoplasm demonstrates anastomosing tubular pattern in myxoid stroma that is reminiscent of mucinous tubular and spindle cell carcinoma (note the psammomatous calcification to the right of both Figures) (C, D). In other areas, one can appreciate tubular architecture and biphasic cytology (E). Higher power view of these areas reveals larger epithelioid cells with open chromatin and eosinophilic cytoplasm and smaller cells with condensed chromatin and minimal basophilic cytoplasm (F).
The immunohistochemical profiles were not specific. By immunohistochemistry, all cases tested were positive for PAX8 (4/4), CK7 (5/5), HNF1-beta (2/2), and EMA (2/2). In case 2, CK7 preferentially labelled the larger cells while EMA preferentially labeled the smaller cells (Figures 3E, F). All cases tested were negative for GATA3 (0/7), cathepsin K (0/7), Melan A (0/4), inhibin (0/6), SF-1 (0/6), and WT-1 (0/6). SDHB (2/2 cases) and fumarate hydratase (3/3 cases) were intact. The hyaline material labeled for type 4 collagen in the one case with available material for this stain, consistent with duplicated basement membrane material as seen in the t(6;11) RCC (Figure 2G). All five cases tested were negative for TFE3 and TFEB rearrangements by break apart FISH14,15. One other case was negative for TFE3 overexpression and one of the cases that was FISH negative was also negative for TFE3 and TFEB overexpression by immunohistochemistry (IHC)13,16.
The neoplasms generally demonstrated low Ki-67 indices. In three cases, the Ki-67 index was less than 5% (Figure 2H), in three it was approximately 5%, while in one (case 3, which had the highest mitotic index) it was 10%. Notably, the Ki-67 index was not increased in the small cell component, and in cases 1 and 4 specifically appeared to be decreased in it. We attempted YAP1 staining as a marker of Hippo pathway activation; however, high background staining in the native kidney was found, possibly due to the age of the unstained slides available. Nonetheless, three of 8 cases demonstrated nuclear and cytoplasmic labeling; in two of these the labeling was stronger in the tumor than the surrounding native renal tubules (Supplementary Figure 1). Four neoplasms demonstrated cytoplasmic staining which was similar to that of the surrounding native renal tubules, while neither the neoplasm nor the surrounding kidney stained in one case.
RNA sequencing
No fusions were identified in the eight cases studied.
DNA Mutational Analysis
All cases (8/8) harbored pathogenic or likely pathogenic variants in the NF2 gene (Figure 10). Most variants were frameshift (3/8) or truncating (3/8) mutations. Other NF2 alterations included an in-frame deletion of 7 amino acids (case 7) and a splice acceptor variant (case 4). The in-frame deletion occurs in a highly conserved C-terminal region of the merlin protein and includes the functionally significant serine-518 residue, the phosphorylation of which modulates merlin’s ability to form intramolecular association between residues in the N-terminal and C-terminal domains17. The splice acceptor variant (c.600–1G>A) is likely pathogenic having been previously reported as a germline mutation in a young patient with bilateral vestibular schwannomas and meningioma18. Most of the NF2 variants were confirmed as somatically acquired (cases 1, 3, 4, 6–8). The two cases (cases 2 and 5) that did not have matched normal tissue sequenced had either a frameshift or truncating stop mutation occurring at allelic frequencies that closely approximated tumor purity estimates (73% and 81%, respectively).
Figure 10:
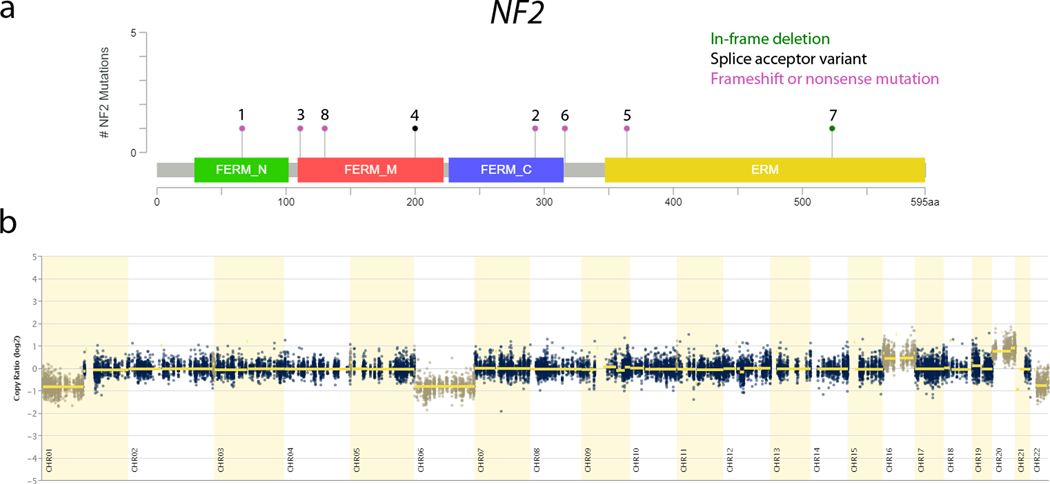
Recurrent molecular findings in biphasic hyalinizing psammomatous renal cell carcinoma (BHP RCC). a) Lollipop plot of the NF2 gene (ENST00000338641, NM_000268) showing the distribution of alterations identified. Each mutation is plotted with the corresponding case number. In-frame deletion – green; splice acceptor variant – black; frameshift or nonsense mutation – magenta; b) CNV scatter plot of Case 2 showing broad copy number alterations in chromosomes 1p, 6, 16, 20, and 22q.
While there were no other recurrently altered genes, variants in recurrently altered cellular pathways were noted in many cases (DNA repair –ERCC5, FANCD2, MRE11A, POLQ, RAD50, RAD54L, WHSC1; chromatin remodeling – ARID1A, ARID2, AUTS2, CHD6, KMT2B, NCOA1, SMARCA4; cell cycle and differentiation – CHD6, CYLD, GLI1, PPP2R1B, PPP2R2B, RB1; cell proliferation, survival, and apoptosis – BCL2, MIB1, NPM1, SGK1, SAMD9).
Multiple broad chromosomal copy number variants were also present. The most frequent chromosomal alterations included gain of chromosome 20 (7/8), loss of chromosome 6 (6/8), loss of chromosome 1p (6/8), and loss of chromosome 22q (the location of NF2) (5/8). Gain of chromosome 16 was seen in 4/8 cases, while loss of 9q and 19q was seen in 3/8 cases each.
DISCUSSION
We report eight cases of a distinctive, previously undescribed renal cell carcinoma associated with somatic NF2 mutations. Key morphologic features included a biphasic appearance with larger and smaller carcinoma cells clustered around basement membrane material, spindling of smaller cells, papillary architecture, glomeruloid bodies, sclerotic stroma (likely contributing to the firm white gross appearance), and psammoma bodies. We refer to this tumor as biphasic hyalinizing psammomatous renal cell carcinoma, or BHP RCC for short.
The neurofibromin 2 (NF2) gene encodes merlin, a protein that suppresses multiple receptor-dependent mitogenic signaling pathways that promote tumor growth. Merlin inhibits cellular proliferation in response to cell-cell contact. Merlin blocks cell growth by binding and disrupting CD44 function, inhibiting the oncogenic effects of Ras, inhibiting phosphate inositide 3-kinase (PI3K)/AKT pathway activity, negatively regulating the Hippo developmental pathway, and blocking mTORC1 pathway signaling. The latter presents a potential therapeutic target for neoplasms harboring NF2 inactivation. Inactivation of NF2 has been described in many tumor types19,20. Patients with neurofibromatosis type 2 syndrome are not known to develop renal cell carcinomas at increased frequency, though a distinctive benign sclerosing peritubular lesion that does not resemble the neoplasms described in this report has been described21–23. Five of our 8 NF2 mutant RCC demonstrated loss of chromosome 22q where NF2 resides, consistent with biallelic inactivation of this tumor suppressor gene. We suspect that other mechanisms of inactivation of the second allele (such as methylation or copy-neutral loss of heterozygosity which our assays would not detect) may be at play in the remaining cases. We were unable to more convincingly demonstrate functional effects of the NF2 mutations in our cohort of cases. We attempted to demonstrate downstream Hippo pathway activation in these neoplasms using immunohistochemistry for YAP1, a downstream target of this pathway. While all but one case labeled for YAP1, we also noted labeling for native renal tubules at similar levels in most cases, precluding a definitive assessment of Hippo pathway activation. Similarly, we noted loss of labeling for NF2 protein by immunohistochemistry in the only case we tested (case 1), but the absence of labeling in native renal tubules precluded our using this as evidence of NF2 protein loss (data not shown). Both of these proteins are difficult to detect reliably by immunohistochemistry in formalin-fixed, paraffin-embedded tissue. Hence, the functional consequences of the NF2 mutations found in our cases remain unproven, though the absence of other consistent genetic alterations along with the pathogenic or likely pathogenic nature of the NF2 mutations found suggest that they are potentially the drivers of this cancer.
The role of the NF2 gene in renal neoplasia is not well defined. The Cancer Genome Atlas (TCGA) sequencing studies have shown that the NF2 gene is infrequently mutated in the common subtypes of renal cell carcinoma. Approximately 1.7% of clear cell RCCs (6 out of 354) published by the TCGA group have alterations in NF2 (one nonsense mutation, one missense mutation, four splice site mutations) (Table 3). Three of these are genetically completely consistent with clear cell RCC, as 2 of these 6 NF2 altered cases have concurrent VHL missense mutations (one L135F, one D121Y), while the third case has a deep chromosomal deletion involving the VHL locus on 3p2524. Review of the online images of these three clear cell carcinomas from the TCGA database with both 3p alterations and NF2 mutations (TCGA-CJ-4901, TCGA-CJ-4638, and TCGA-B8–4153) reveals typical morphology for clear cell RCC without any unusual features (Supplementary Figure 2). Therefore, we believe that the NF2 alterations most likely represent secondary, progression-related changes in neoplasms driven by VHL loss. Along these lines, these NF2-mutated clear cell RCC have a significantly worse prognosis relative to NF2 wild-type clear cell RCC (log rank test p value 4.9e-5), suggesting a significant biologic role for NF2 loss in neoplastic progression. NF2 mutations were reported in slightly more than 3.2% of papillary RCCs in the TCGA database (9 of 283)(Table 4). However, two of the nine papillary RCC illustrated in that database (TCGA-EV-5902 and TCGA-SX-A7SL) show biphasic morphology and hyaline nodules that overlaps with the neoplasms described herein, and we suspect that these two cases may represent examples of BHP RCC (Supplementary Figure 3). Case TCGA-Y8-A896 shows some morphologic features that suggest this BHP RCC, but is not definitive. The other papillary RCC in this cohort do not. 25 More recently, mutations in NF2 (22% of cases) and phosphatase non-receptor type 14 (PTPN14) (31% of cases) resulting in inactivation of the Hippo pathway have been identified as the primary driving genetic alteration in mucinous tubular and spindle cell carcinoma (MTSC) 26,27. These results are particularly interesting given the MTSC-like areas in case #5 of this series. Another study found mutations of NF2 in 29% of cases classified as collecting duct carcinomas, though these cases were not illustrated28. Finally, NF2 mutations and Hippo pathway activation have recently been associated with sarcomatoid transformation of RCC29.
Table 3:
NF2 Mutated Clear Cell Renal Cell Carcinoma in TCGA PanCancer Atlas Database
| Case | NF2 Variant | Age/Sex | Tumor Size/Stage | Morphology | Outcome | Other Notable Genetic Anomalies |
|---|---|---|---|---|---|---|
| TCGA-CJ-4901–01 | R341* | 47/M | 14cm/pT3b | Clear cell | Alive 47months | VHL, MLH1, BAP1 deletions; loss of 3p: partial gain of 5q |
| TCGA-CJ-4638–01 | X172_splice | 46/M | 15cm/pT3aN1M1 | Clear cell | DOD 14 months | VHL, BAP1 mutations; CDKN2a deletion; partial loss of 3p: partial gain of 5q |
| TCGA-B8–4153-01 | D494N | 74/M | 5cm/pT3a | Clear cell | Alive 25 months | VHL, ARID1B mutations; PBRM1 deletion; partial loss of 3p |
| TCGA-B8–5098-01 | X200_splice | 53/F | 4cm/pT1 | Unclassified, rhabdoid features | Died 52 months (ESRD) | CDK12, NF1, TP53, MLH1, APC, EP400, SMARCA4 mutations: partial loss of 3p |
| TCGA-B0–5084-01 | X121_splice | 33/M | 8cm/pT3N1M1 | Unclassified, possibly clear cell | DOD 7 months | NSD1, NOTCH1 mutations |
| TCGA-B0–4698-01 | X81_splice | 75/M | 10.5cm/pT4 | Unclassified with rhabdoid features | DOD 1 month | KRAS,TP53, ARID3A, EP300 mutations |
ESRD=End Stage Renal Disease
Table 4:
NF2 Mutated Papillary Renal Cell Carcinoma in TCGA PanCancer Atlas Database
| Case | NF2 Variant | Age/Sex | Tumor Size/Stage | Morphology | Outcome | Other Notable Genetic Anomalies |
|---|---|---|---|---|---|---|
| TCGA-BQ-5875 | Y132* | 68/F | 13cm/pT3a | Oncocytic, reverse polarity | Alive 36 months | SETD2, STAG2 mutations; probable 16q and 17q gain; 22q loss |
| TCGA-EV-5902 | R359Kfs*11 | 58/M | 3cm/pT1N0 | Biphasic, hyaline nodules | Alive 19 months | Probable 5, 16, 20 gain |
| TCGA-SX-A7SL | G151Vfs*23 | 7/M | 4cm/pT1 | Biphasic hyaline nodules | Alive 27 months | Probable 16,17q,20 gain; 6 loss |
| TCGA-Y8-A896 | D277Gfs*20 | 62/M | 6cm/pT3aNX | Unclassified but suggestive of biphasic morphology | Alive 18 months | Probable 7 gain; 6, 9, 15q, 22q loss |
| TCGA-G7–6797 | X200_splice | 46/M | 2.6cm (multifocal)/pT1a | Solid papillary NOS | Alive 25 months | BAP1, MSH2, FANCD2, BCL11B mutations; probable 16, 20 gain and 1, 6p, 22q loss; |
| TCGA-BQ-5877 | K171* | 60/M | 3cm/pT3aN1M1 | Papillary NOS | DOD 8 months | SETD2, BAP1 mutation; CDKN2A/B deletion; probable 12 gain and 1p,3p,11,18, 22q loss |
| TCGA-GL-7966 | L295Rfs*14 | 28/F | 6.5cm/pT3N1 | Tubulopapillary NOS | Alive 3 months | FH and B2M nonsense mutations; probable partial 6p gain and 1p, 4, partial 5p, 10, 13q, 15q, 18,22q loss |
| TCGA-P4-A5EA | X270_splice | 54/F | 8cm/pT3N1 | Papillary NOS | DOD 6 months | CREBBP, MAP3K1, RASA1, BCOR nonsense or frameshift mutations |
| TCGA-B1-A47M | P134H | 79/M | 4.5cm, pT3a | Papillary NOS | Alive 21 months | SMARCB1, NIFE2L2, ERRFI1, TET1 mutations; probable 7 gain |
NOS-not otherwise specified; DOD=dead of disease
We suspect that the role of NF2 in renal neoplasia is similar to that of the tuberous sclerosis genes TSC1 and TSC2. First, both NF2 and TSC1/TSC2 genes may be inactivated as secondary events in established subtypes of renal cell carcinoma, such as the clear cell carcinomas with established VHL gene inactivation noted above. In the case of NF2, an association with more aggressive clinical course appears likely. Second, in less common distinctive neoplasms, TSC1/TSC2 or NF2 can be the primary driver mutation, and these neoplasms have a distinctive morphology. TSC1/TSC2 mutations are the key underlying genetic alteration in eosinophilic solid and cystic renal cell carcinoma (ESC RCC)3–5, and TSC2 or mTOR mutations have now been described in a lesion characterized as “eosinophilic and vacuolated RCC” or “high grade oncocytic tumor” which overlaps morphologically with ESC RCC30,31. Similarly, NF2 mutations appear to be the genetic driver of MTSC and may be the driver of distinctive BHP RCC reported herein, which as case #5 of this series shows may overlap morphologically. Third, some cases of high grade RCC which are currently considered unclassified are driven by NF2 or TSC1/2 alterations. It is possible that careful examination of some of these cases may reveal clues as to origin from a distinctive lower-grade lesion such as ESC RCC or BHP RCC. For example, the high-grade eosinophilic morphology of cases #5 and #6 and clear cell areas of cases #5 and 8 of the BHP RCC in this series contributed to their initially being considered unclassifiable, as the distinctive features of these neoplasms were only focally or incompletely developed. Along these lines, in a landmark study of unclassified renal cell carcinomas, Chen et al. found somatic NF2 mutations in approximately 18% and mTOR/TSC/PTEN pathway alterations in approximately 21% of high grade unclassified renal cell carcinomas32, and noted that the NF2 mutated RCC were more aggressive clinically. While these mostly high-grade neoplasms did not have a hallmark, distinctive morphology, Chen et al. did note that some of the NF2-mutated neoplasms were nodular.
The differential diagnosis for BHP RCC is broad as reflected in the submitted consultation differential diagnoses. The glomeruloid architecture seen in most cases raises the differential diagnosis of papillary renal cell carcinoma, which was the submitted diagnosis in several of these cases. As noted above, nine cases included within the TCGA cohort of papillary renal cell carcinoma harbor NF2 mutations, and the provided images of two of them demonstrate significant overlap with the BHP RCC cases reported herein. We suspect that NF2 mutated BHP RCC comprise a small but distinct genetic subset of lesions having the papillary renal cell carcinoma type 2 phenotype33. Since a high-grade papillary pattern is a recognized phenotype of collecting duct carcinoma, it is also possible that some BHP RCC may have been grouped into the latter category; this might explain reports in NF2 mutations in a subset of collecting duct carcinomas28. The biphasic cytology and frequent presence of hyaline basement membrane nodules raised the differential diagnosis of MiTF family translocation carcinoma, particularly those harboring the t(6;11) (p21;q12) translocation resulting in a MALAT1-TFEB gene fusion. While MiTF family translocation RCC have a broad morphologic spectrum as documented previously, the absence of immunoreactivity for cathepsin K and absence of rearrangements for TFE3/TFEB in these cases help exclude that diagnosis. Moreover, among 14 cases of renal cell carcinomas with TFE3 or TFEB rearrangements present in the TCGA database of clear cell and papillary RCC, none demonstrates NF2 gene mutations (www.cbioportal.org) 24,25. The primitive appearance of the small cells and their spindle-shapes raise the possibility of adult Wilms tumor or metanephric neoplasia. The absence of WT1 immunoreactivity, absence of blastemal elements and open, non- primitive chromatin of the larger cells excludes adult Wilms tumor. Gonadal sex cord stromal tumors such as gonadoblastoma or granulosa cell tumor were considered given the clustering of smaller cells around basement membrane material, resembling Call-Exner bodies. As previously noted, such structures are also hallmarks of the TFEB-rearranged renal carcinomas. The absence of immunoreactivity for inhibin and SF-1, along with the absence of clinical evidence of similar lesions in the gonads of these patients excludes this possibility. The variably tubulopapillary and glomeruloid architecture, hyaline nodules, and HNF1-beta immunoreactivity raise the possibility of a mesonephric adenocarcinoma34. The latter have not been described in the kidney, which is of metanephric origin. Moreover, mesonephric adenocarcinomas are associated with GATA3 immunoreactivity and Kras mutations which distinguish them immunohistochemically and genetically from the BHP RCC35.
We have minimal clinical follow-up in these cases. However, the primitive cytology, along with the cellular necrosis suggested that they have malignant potential, which was confirmed by the hematogenous metastases and lethal clinical course of case 3. However, the low Ki-67 indices and low mitotic rates found suggest that these are low-grade carcinomas.
In summary, we report a novel, morphologically distinctive subtype of renal cell carcinoma associated with NF2 mutations. The latter suggests the potential for targeted therapy, such as small molecular inhibitors of the downstream target protein YAP1, in cases that behave aggressively36.
Supplementary Material
Supplemental Figure 1: YAP1 immunohistochemistry reveals strong nuclear and cytoplasmic labeling in BHP RCC#5. Note that the stroma is appropriately negative.
{kind=link}
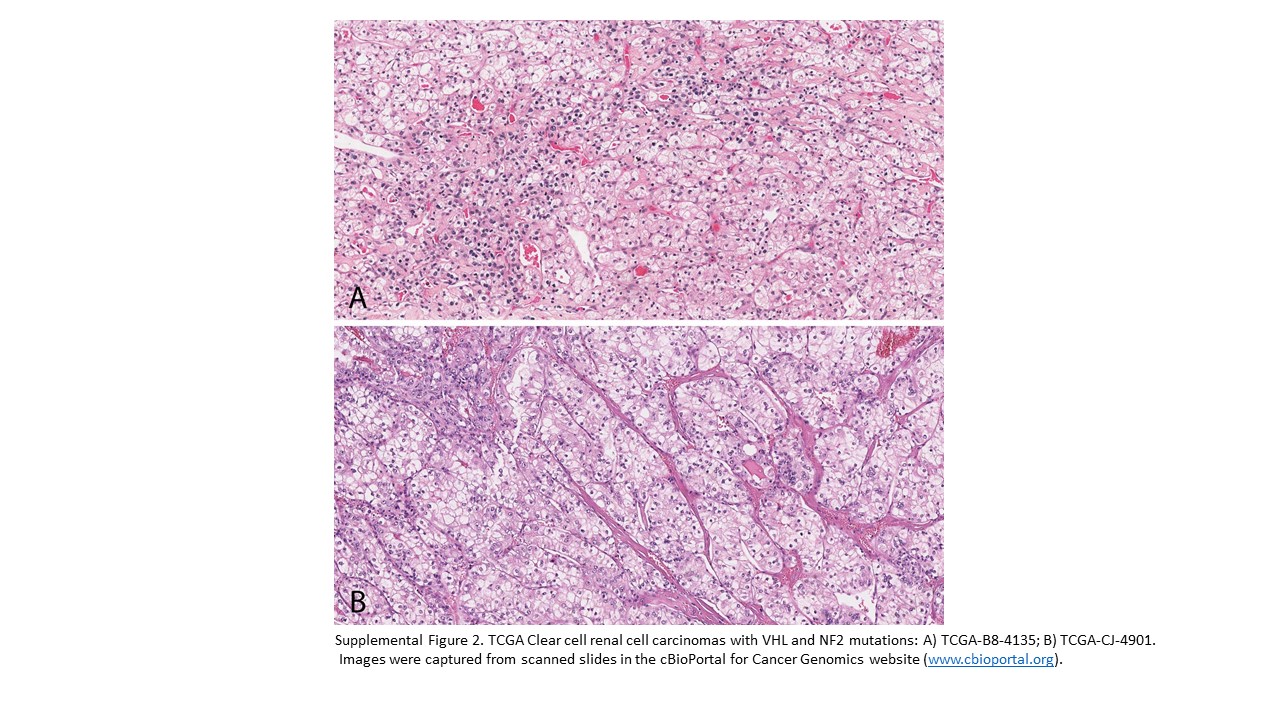
Supplemental Figure 2. TCGA Clear cell renal cell carcinomas with VHL and NF2 mutations: A) TCGA-B8–4135; B) TCGA-CJ-4901. Images were captured from scanned slides in the cBioPortal for Cancer Genomics website (www.cbioportal.org)
Supplemental Figure 3. TCGA Papillary renal cell carcinomas with NF2 mutations similar to cases of BHP RCC: A) TCGA-EV-5902; B) TCGA-SX-A7SL. Images were captured from scanned slides in the cBioPortal for Cancer Genomics website (www.cbioportal.org)
{kind=link}
Acknowledgement:
We thank Norman Barker MA, MS, RBP for expert photographic assistance.
Disclosures: Supported in part by: Dahan Translocation Carcinoma Fund (PA) and Joey’s Wings (PA)
REFERENCES
- 1.Sanz-Ortega J, Vocke C, Stratton P, et al. Morphologic and molecular characteristics of uterine leiomyomas in hereditary leiomyomatosis and renal cancer (HLRCC) syndrome. Am J Surg Pathol. 2013;37:74–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gill AJ, Hes O, Papathomas T, et al. Succinate dehydrogenase (SDH)-deficient renal carcinoma: a morphologically distinct entity: a clinicopathologic series of 36 tumors from 27 patients. Am J Surg Pathol. 2014;38:1588–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palsgrove DN, Li Y, Pratilas CA, et al. Eosinophilic Solid and Cystic (ESC) Renal Cell Carcinomas harbor TSC mutations: Molecular analysis supports an expanding clinicopathologic spectrum. Am J Surg Pathol 2018;42:1166–1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mehra R, Vas P, Cao X,et al. Somatic Bi-allelic loss of TSC genes in Eosinophilic Solid and Cystic Renal Cell Carcinoma. Eur Urol 2018;74:483–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parilla M, Kadri S, Patil SA et al. Are Sporadic Eosinophilic Solid and Cystic Renal Cell Carcinomas Characterized by Somatic Tuberous Sclerosis Gene Mutations? Am J Surg Pathol. 2018;42:911–917 [DOI] [PubMed] [Google Scholar]
- 6.Argani P, Antonescu CR, Illei PB, et al. Primary renal neoplasms with the ASPL-TFE3 gene fusion of alveolar soft part sarcoma: A distinctive tumor entity previously included among renal cell carcinomas of children and adolescents. Am J Pathol 2001;159:179–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Argani P, Antonescu CR, Couturier J, et al. PRCC-TFE3 renal carcinomas: Morphologic, immunohistochemical, ultrastructural, and molecular analysis of an entity associated with the t(X;1)(p11.2;q21). Am J Surg Pathol 2002;26:1553–66. [DOI] [PubMed] [Google Scholar]
- 8.Argani P, Zhong M, Reuter VE, et al. TFE3-Fusion Variant Analysis Defines Specific Clinicopathologic Associations Among Xp11 Translocation Cancers. Am J Surg Pathol. 2016;40:723–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Argani P, Zhang L, Reuter VE, et al. RBM10-TFE3 Renal Cell Carcinoma: A Potential Diagnostic Pitfall Due to Cryptic Intrachromosomal Xp11.2 In version Resulting in False-negative TFE3 FISH. Am J Surg Pathol. 2017;41:655–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li MM, Datto M, Duncavage EJ, et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn. 2017;19:4–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zehir A, Benayed R, Shah RH, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23:703–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Argani P, Hawkins A, Griffin CA, et al. A distinctive pediatric renal neoplasm characterized by epithelioid morphology, basement membrane production, focal HMB45 immunoreactivity, and t(6;11)(p21.1;q12) chromosome translocation. Am J Pathol 2001;158:2089–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Argani P, Laé M, Hutchinson B, et al. Renal carcinomas with the t(6;11)(p21;q12): clinicopathologic features and demonstration of the specific alpha-TFEB gene fusion by immunohistochemistry, RT-PCR, and DNA PCR. Am J Surg Pathol. 2005;29:230–40. [DOI] [PubMed] [Google Scholar]
- 14.Argani P, Yonescu R, Morsberger L, et al. Molecular confirmation of t(6;11)(p21;q12) renal cell carcinoma in archival paraffin-embedded material using a break-apart TFEB FISH assay expands its clinicopathologic spectrum. Am J Surg Pathol 2012;36:1516–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Green WM, Yonescu R, Morsberger L, et al. Utilization of a TFE3 break-apart FISH assay in a renal tumor consultation service. Am J Surg Pathol 2013;37: 1150–1163. [DOI] [PubMed] [Google Scholar]
- 16.Argani P, Lal P, Hutchinson B, et al. Aberrant nuclear immunoreactivity for TFE3 in neoplasms with TFE3 gene fusions: A sensitive and specific immunohistochemical assay. Am J Surg Pathol 2003;27:750–61. [DOI] [PubMed] [Google Scholar]
- 17.Rong R, Surace EI, Haipek CA, et al. Serine 518 phosphorylation modulates merlin intramolecular association and binding to critical effectors important for NF2 growth suppression. Oncogene. 2004;23:8447–54. [DOI] [PubMed] [Google Scholar]
- 18.Kim H-J, Seo GH, Kim YM, et al. Genetic and clinical characteristics of Korean patients with neurofibromatosis type 2. J Genet Med 2017;14:56–61 [Google Scholar]
- 19.Petrilli AM, Fernandez-Valle C. Role of Merlin/NF2 inactivation in tumor biology. Oncogene 2016; 35:537–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harvey KF, Zhang X, Thomas DM. The Hippo pathway and human cancer. Nat Rev Cancer. 2013. April;13(4):246–57. [DOI] [PubMed] [Google Scholar]
- 21.Gokden N, Jamal SE, Gokden M, Lamps LW. Renal sclerosing peritubular nodules in a patient with neurofibromatosis type 2: a case report with immunohistochemical and electron microscopic studies. Hum Pathol 2009; 40:1650–1654. [DOI] [PubMed] [Google Scholar]
- 22.Morita T, Kimura K, Kakita A. Renal sclerosing peritubular nodules in a patient with neurofibromatosis type 2: A case report with immunohistochemical and electron microscopic studies. Hum Pathol 2010; 41:1051–2. [DOI] [PubMed] [Google Scholar]
- 23.Thomas S, Pahoff C, McClymont K, Parnham A. Renal sclerosing peritubular nodule-how rare is it? Clin Kidney 2013; 6:426–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013; 499:43–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cancer Genome Atlas Research Network. Comprehensive Molecular Characterization of Papillary Renal-Cell Carcinoma N Engl J Med. 2016;374:135–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mehra R, Vats P, Cieslik M, et al. Biallelic Alteration and Dysregulation of the Hippo Pathway in Mucinous Tubular and Spindle Cell Carcinoma of the Kidney. Cancer Discov. 2016;6:1258–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ged Y, Chen YB, Knezevic A, et al. Mucinous tubular and spindle-cell carcinoma of the kidney: clinical features, genomic profiles, and treatment outcomes. Clin Genito Cancer 2019; 17:268–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pal SK, Choueiri TK, Wang K, et al. Characterization of Clinical Cases of Collecting Duct Carcinoma of the Kidney Assessed by Comprehensive Genomic Profiling. Eur Urol. 2016;70:516–21 [DOI] [PubMed] [Google Scholar]
- 29.Malouf GG, Flippot R, Dong Y, et al. Molecular characterization of sarcomatoid clear cell renal cell carcinoma unveils new candidate oncogenic drivers. Sci Rep. 2020;10:701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen YB, Mirsadraei L, Jayakumaran G, et al. Somatic Mutations of TSC2 or MTOR Characterize a Morphologically Distinct Subset of Sporadic Renal Cell Carcinoma With Eosinophilic and Vacuolated Cytoplasm. Am J Surg Pathol. 2019. 43:121–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.He H, Trpkov K, Martinek P, et al. “High-grade oncocytic renal tumor”: morphologic, immunohistochemical, and molecular genetic study of 14 cases. Virchows Arch. 2018;473:725–738 [DOI] [PubMed] [Google Scholar]
- 32.Chen YB, Xu J, Skanderup AJ, et al. Molecular analysis of aggressive renal cell carcinoma with unclassified histology reveals distinct subsets. Nat Commun 2016; 7:13131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Delahunt B, Eble JN. Papillary renal cell carcinoma: a clinicopathologic and immunohistochemical study of 105 tumors. Mod Pathol 1997;10:537–44. [PubMed] [Google Scholar]
- 34.Howitt BE, Nucci MR. Mesonephric proliferations of the female genital tract. Pathology. 2018;50:141–150. [DOI] [PubMed] [Google Scholar]
- 35.Mirkovic J, Sholl LM, Garcia E, et al. Targeted genomic profiling reveals recurrent KRAS mutations and gain of chromosome 1q in mesonephric carcinomas of the female genital tract. Mod Pathol. 2015;28:1504–14. [DOI] [PubMed] [Google Scholar]
- 36.Crawford JJ, Bronner SM, Zbieg JR. Hippo pathway inhibition by blocking the YAP/TAZ-TEAD interface: a patent review. Expert Opin Ther Pat. 2018;28:867–873 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: YAP1 immunohistochemistry reveals strong nuclear and cytoplasmic labeling in BHP RCC#5. Note that the stroma is appropriately negative.
Supplemental Figure 2. TCGA Clear cell renal cell carcinomas with VHL and NF2 mutations: A) TCGA-B8–4135; B) TCGA-CJ-4901. Images were captured from scanned slides in the cBioPortal for Cancer Genomics website (www.cbioportal.org)
Supplemental Figure 3. TCGA Papillary renal cell carcinomas with NF2 mutations similar to cases of BHP RCC: A) TCGA-EV-5902; B) TCGA-SX-A7SL. Images were captured from scanned slides in the cBioPortal for Cancer Genomics website (www.cbioportal.org)