

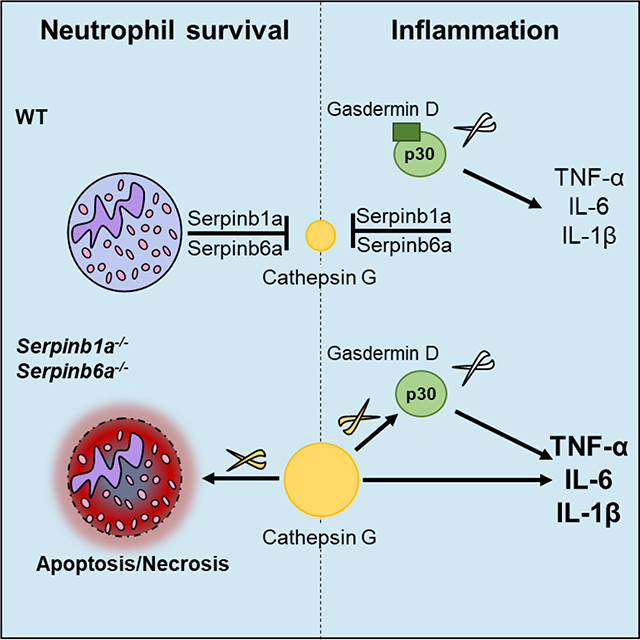
Summary
Neutrophil granule serine proteases contribute to immune responses through cleavage of microbial toxins and structural proteins. They induce tissue damage and modulate inflammation if levels exceed their inhibitors. Here, we show that the intracellular protease inhibitors Serpinb1a and Serpinb6a contribute to monocyte and neutrophil survival in steady state and inflammatory settings by inhibiting cathepsin G (CatG). Importantly, we found that CatG efficiently cleaved gasdermin D (GSDMD) to generate the signature N-terminal domain GSDMD-p30 known to induce pyroptosis. Yet, GSDMD deletion did not neutrophil survival in Sb1a.Sb6a−/− mice. Furthermore, Sb1a.Sb6a−/− mice released high levels of pro-inflammatory cytokines upon endotoxin challenge in vivo in a CatG-dependent manner. Canonical inflammasome activation in Sb1a.Sb6a−/− macrophages showed increased IL-1β release that was dependent on CatG and GSDMD. Together, our findings demonstrate that cytosolic serpins expressed in myeloid cells prevent cell death and regulate inflammatory responses by inhibiting CatG and alternative activation of GSDMD.
Graphical Abstract
Introduction
Regulated forms of cell death are essential for the development of multicellular organisms and for their immune responses. Apoptosis, the most studied form of regulated cell death (RCD), is triggered by intrinsic or extrinsic cues transmitted to signalling routes converging on the proteolytic activation of executioner caspases. These apoptotic caspases cleave multiple protein targets in the nucleus, the cytoplasm and the cytoskeleton but do not directly compromise the integrity of the plasma membrane (Taylor et al., 2008). Apoptosis therefore proceeds slowly and without alarming neighbouring cells; indeed, the removal of apoptotic bodies by phagocytes induces the release of anti-inflammatory signals (Ravichandran, 2011). Until recently, RCD was synonymous with apoptosis but it is now recognized that RCD may also involve necrosis resulting from distinct molecular pathways that have been principally defined as necroptosis and pyroptosis (Bliss-Moreau et al., 2017; Wallach et al., 2016). Such pathways may have evolved to trigger inflammation in response to potentially concealed infections and cytosolic microbes (Jorgensen et al., 2017; Miao et al., 2010). Necroptosis occurs in many cell types and can be triggered by tumour necrosis factor-α (TNF-α) as well as other stimuli converging on receptor-interacting protein kinase-3 (RIPK3)(Cho et al., 2009; He et al., 2009; Zhang et al., 2009), which phosphorylates the pseudo-kinase mixed-lineage kinase-like protein (MLKL). Phosphorylated MLKL oligomerizes, leading to cell death (Murphy et al., 2013). Pyroptosis has been principally described in monocytes and macrophages and is elicited by inflammatory caspases, caspase-1/4/5/11 (Miao et al., 2010). This form of RCD is molecularly defined by the specific, limited cleavage of gasdermin D (GSDMD) to release its N-terminal domain GSDMD-p30 (He et al., 2015; Kayagaki et al., 2015; Shi et al., 2015). GSDMD-p30 assembles to form pores at the plasma membrane leading to cell lysis (Aglietti et al., 2016; Ding et al., 2016; Liu et al., 2016).
Neutrophils are major effectors of the immune response to infection and drive inflammatory reactions. Neutrophil serine proteases (NSPs) (e.g., neutrophil elastase (NE), cathepsin G (CatG), proteinase-3 (PR3) and NSP4) contribute to these functions in multiple ways. NSPs are found in specialized secretory lysosomes that fuse with phagosomes or are released into the extracellular milieu upon degranulation. NSPs have antimicrobial functions through defined proteolysis of microbial toxins and structural proteins (Belaaouaj et al., 2000; Tkalcevic et al., 2000; Weinrauch et al., 2002). Importantly, NSPs promote inflammation through cleavage-mediated activation or inhibition of cytokines, chemokines, opsonins and receptors (Clancy et al., 2018; Henry et al., 2016; Kessenbrock et al., 2008; Lefrancais et al., 2012; Padrines et al., 1994; Raptis et al., 2005). NSPs have been associated with neutrophil death but the mechanisms are not fully elucidated and appear different for each NSP (Benarafa and Simon, 2017). Most notably, heterozygous mutations in the gene for NE, ELANE, are the most common cause of severe congenital neutropenia and the only known cause of cyclic neutropenia (Makaryan et al., 2015). NE is expressed at high levels at the promyelocyte stage and mutant proteins induce cell death and an apparent maturation block at the promyelocyte stage. How the various NE mutants induce cell death is not defined, but it may be due to the unfolded protein response or to mislocalized mutant NE because NE proteolytic activity is not required (Tidwell et al., 2014). By contrast, CatG and PR3 can directly activate apoptotic pro-caspase-7 and pro-caspase-3, respectively (Loison et al., 2014; Zhou and Salvesen, 1997). NSPs are also expressed in a subset of monocytes (Kargi et al., 1990). Recent evidence suggests that NSP-expressing monocytes derive from a neutrophil-like common ancestor (Yanez et al., 2017), but the function of NSPs in monocyte homeostasis is unknown.
Secreted endogenous inhibitors regulate the activity of NSPs in plasma (e.g. α1-antitrypsin) and mucosal surfaces (e.g. secretory leukocyte protease inhibitor, SLPI). Inhibitors of the clade B serpin family are cytosolic and expressed in monocytes and neutrophils (Remold-O’Donnell et al., 1989; Scott et al., 1999). Among the latter, Serpinb1 is found at high levels in the cytosol of neutrophils and monocytes and inhibits all NSPs except NSP4 (Benarafa et al., 2011; Cooley et al., 2001; Perera et al., 2012). Mice deficient in the mouse ortholog Serpinb1a (Sb1a−/−) have reduced neutrophil numbers, show increased mortality and produce high levels of inflammatory cytokines upon lung infection (Benarafa et al., 2011; Benarafa et al., 2007; Gong et al., 2011). Serpinb6 is a related clade B serpin, also expressed in monocytes and neutrophils, which inhibits CatG but not NE or PR3 (Scott et al., 1999). Notably, deficiency in the mouse ortholog, Serpinb6a (Sb6a−/−), was not associated with defects in leukocyte numbers possibly because Serpinb1a levels are higher in these animals (Scarff et al., 2004). In this study, we first investigated the effects of combined deficiency of Sb1a and Sb6a (Sb1a.Sb6a−/−) on leukocyte homeostasis and the effect on RCD pathways of monocytes and neutrophils in vitro. We discovered that the two serpins collaborate to protect myeloid cells through blockade of cell-specific, CatG-dependent RCD. We found that CatG efficiently cleaves GSDMD to generate GSDMD-p30, but deletion GSDMD did not rescue CatG-mediated neutrophil death in vivo and in vitro. In contrast, we demonstrated that Sb1a.Sb6a−/− mice released more IL-1β, TNF-α and IL-6 in a CatG- and GSDMD-dependent manner in in vivo models of inflammation and following classical inflammasome activation in macrophages in vitro.
Results
Serpinb1a and Serpinb6a are survival factors for neutrophils in vivo
To address the function of Sb1a and Sb6a in myeloid cell homeostasis, we analysed the leukocyte subsets from single and double-deficient 6-week-old mice in steady state. Sb1a.Sb6a−/− mice showed a severe reduction in absolute numbers and proportion of neutrophils in the bone marrow (Figure 1a, S1a,b). The neutropenia of Sb1a.Sb6a−/− mice was more severe than that observed in Sb1a−/− mice; and Sb6a−/− mice had normal proportions and numbers of neutrophils (Figure S1b) (Benarafa et al., 2011; Scarff et al., 2004). Neutrophil counts in blood were significantly reduced only in Sb1a.Sb6a−/− mice compared to WT mice (Figure S1c). Sb1a and Sb6a are both inhibitors of CatG (Benarafa et al., 2002; Scott et al., 1999) and CatG deletion rescues the neutropenia of Sb1a−/− mice (Baumann et al., 2013). Thus, we generated mice lacking both serpins and CatG and found that deletion of CatG was indeed sufficient to restore normal neutrophil numbers in bone marrow and blood in steady state (Figure 1a, Figure S1c).
Figure 1. Sb1a.Sb6a−/− mice present a CatG-dependent neutropenia in steady state and inflammation.

(a) Analysis of bone marrow neutrophils of 6 week-old female mice, n=10–11/genotype. (b) Percentage and absolute counts of neutrophils and macrophages in peritoneal lavage fluid 4 hours after injection of 0.5mg zymosan/mouse, n=8–16/genotype of 6 week-old female and male mice. (a,b) Scatter plots show data for individual mice from 4 independent experiments, horizontal lines indicate mean ± SEM. Data were analysed by Mann-Whitney test (**** P<0.0001; ***P<0.001; **P<0.01; *P<0.05). (c) Representative cytospins of peritoneal cells, quantification of zymosan containing cells and of number of zymosan particles per neutrophil. Data from male and female mice n=5–9/genotype from 4 independent experiments and analysed by one-way ANOVA. Left panel, percentage of zymosan positive cells for individual mice, bars indicate mean ± SEM. Right panel, numbers of zymosan particles per neutrophil (40 neutrophils/mouse from 5–9 mice/genotype) for all mice per genotype, data shown as box and whiskers. Scale bars are 15μm. See also Figure S1 and Tables S1, S2.
Neutrophils are rapidly mobilized from the bone marrow in response to infection and have a high phagocytic capacity. To evaluate the relevance of the neutropenia of Sb1a.Sb6a−/− mice on neutrophil recruitment and efficient clearance of fungal particles, we injected opsonized zymosan intraperitoneally. We observed a reduced number of peritoneal neutrophils in Sb1a.Sb6a−/− mice 4h after zymosan injection (Figure 1b). Furthermore, the removal of zymosan was impaired as extracellular particles were still visible in cytospins of peritoneal washes of Sb1a.Sb6a−/− mice, there were higher numbers of neutrophils containing yeast particles, and more zymosan per phagocyte was observed compared to WT mice (Figure 1c). Defective neutrophil recruitment and impaired clearance of zymosan particles were fully rescued in CatG.Sb1a.Sb6a−/− mice (Figure 1b–c). Thus, control of CatG by Sb1a and Sb6a is crucial to maintain neutrophil survival in steady state and to support efficient innate immune responses.
Apoptotic and necrotic pathways in neutrophils are accelerated by CatG
Loss of granule integrity can result in the cytosolic release of proteases, which trigger RCD pathways unless opposed by endogenous protease inhibitors (Baumann et al., 2013; Bird et al., 2014; Bird et al., 1998; Luke et al., 2007). Cell death of bone marrow neutrophils was therefore evaluated in vitro following treatment with the lysosomotropic compound L-leucyl-L-leucine-methyl-ester (LLME), which is assembled in membranolytic metabolites by the transferase activity of the cysteine protease dipeptidyl-peptidase I (DPPI) to release granule contents into the cytosol (Thiele and Lipsky, 1990). Reflecting the in vivo phenotype, Sb1a.Sb6a−/− neutrophils were highly sensitive to LLME-induced cell death (Figure 2a, S2a). Sb1a.Sb6a−/− neutrophils died significantly faster than Sb1a−/− neutrophils (1h) after LLME treatment, and few neutrophils remained alive after 2 and 4h in both genotypes. Sb6a−/− neutrophils showed an intermediate phenotype between Sb1a−/− and WT neutrophils (Figure S2b). Live cell microscopy recordings of bone marrow cells treated with LLME indicated that Sb1a−/− and Sb1a.Sb6a−/− cells initiate an accelerated death process measurable 30 minutes after LLME treatment (Figure S2c,d). LLME-induced cell death was not significantly altered by treatment with the broad caspase inhibitor QVD-OPh alone or in combination with the RIPK1 inhibitor necrostatin-1 (Figure S2b). In contrast, LLME-induced cell death was fully prevented in CatG.Sb1a.Sb6a−/− neutrophils, where CatG.Sb1a.Sb6a−/− neutrophils exhibited significantly improved survival compared to Sb1a.Sb6a−/− (Figure 2a, S2e).
Figure 2. CatG-dependent necrosis is executed following induction of multiple RCD pathways in Sb1a.Sb6a−/− neutrophils.

(a) Survival of neutrophils treated with 100μM LLME in the presence or absence of 50μM Q-VD-OPh for 4 hours. (b) Spontaneous apoptosis and secondary necrosis of neutrophils cultured in vitro with or without 50μM Q-VD-OPh. (c) Neutrophil survival following stimulation with TNF-α (100ng/ml) and actinomycin D (200ng/ml) with or without 50μM Q-VD-OPh and 20μM necrostatin-1 over 48 hours. Viability of neutrophils was assessed by flow cytometry using Annexin V-APC and 7-AAD. Cells were from 6–12 week-old female and male mice. Data are shown as mean ± SEM of 4–8 independent experiments and were analyzed by two-way ANOVA. See also Figures S1–S4.
Neutrophils have a limited lifespan in vitro due to spontaneous apoptosis mediated principally by the intrinsic mitochondrial apoptotic pathway culminating with activation of apoptotic caspase-3 and −7 (Geering and Simon, 2011). Apoptotic neutrophils then rapidly proceed to secondary necrosis. Cultured Sb1a.Sb6a−/− neutrophils showed increased necrosis in the presence of the caspase inhibitor (Figure 2b, S3a). This defect was dependent on CatG since CatG.Sb1a.Sb6a−/− neutrophil spontaneous death in vitro was indistinguishable from that of WT neutrophils. Percentages of apoptotic cells, measured by annexin V staining, were not significantly different between cells of different genotypes (Figure 2b, S3a).
Neutrophils are sensitive to death receptor stimulation, which leads to apoptosis or necroptosis depending on caspase-8 activity and X-linked inhibitor of apoptosis (XIAP) (Wang et al., 2018; Wicki et al., 2016). Neutrophils were stimulated with TNF-α for 24 and 48h leading to a massive induction of cell death that was indistinguishable between genotypes. Caspase inhibition with Q-VD-OPh considerably reduced TNF-induced neutrophil death in all genotypes. Yet, Sb1a.Sb6a−/− neutrophils showed increased necrosis and reduced apoptosis in a CatG-dependent manner (Figure 2c, S3b). Necroptosis induced by TNF-α depends on the interaction of RIPK1 and RIPK3, which is allowed by RIPK1 autophosphorylation (Cho et al., 2009). Inhibition of RIPK1 with necrostatin-1 in presence of Q-VD-OPh reduced neutrophil necrosis in all genotypes but did not abrogate the increased necrosis observed in Sb1a.Sb6a−/− neutrophils (Figure 2c, S3b).
Reactive oxygen species (ROS) produced by the NADPH-oxidase contribute to neutrophil death in inflammatory conditions (von Gunten et al., 2005). To explore the role of ROS, we evaluated leukocyte subsets of mice lacking the critical p47phox subunit of the NADPH oxidase (Huang et al., 2000). We found that both Ncf1.Sb1a−/− and Sb1a−/− mice presented similarly reduced neutrophil numbers in the bone marrow compared to WT and Ncf−/− mice in steady state (Figure S3c,d). Granzymes and caspases can induce ROS-mediated cell death by cleaving mitochondrial complex I subunit NDUFS1 (Martinvalet et al., 2008). Scavenging of mitochondrial ROS with MitoQ did not significantly alter LLME-induced and TNF-induced neutrophil death (Figure S3e,f). We found that CatG did not cleave the respiratory chain components NDUFS1 and NDUFS3 in Sb1a.Sb6a−/− neutrophils (Figure S3g). Overall, these data indicate that Sb1a and Sb6a inhibit CatG-mediated apoptotic and necrotic death induced by multiple stimuli such as survival factor withdrawal, death receptor stimulation and loss of granule integrity. Yet, neutrophil death was partly blocked by caspase inhibition and was independent of RIPK1 and ROS.
Compensatory effects of Sb1a and Sb6a in monocyte survival
Beyond neutrophils, Sb1a.Sb6a−/− mice presented significantly reduced monocyte numbers and percentage in bone marrow (Figure S1a, S4a). The monocyte defect was largely resolved in the bone marrow of CatG.Sb1a.Sb6a−/− mice, which showed monocyte numbers and percentage similar to WT and significantly higher percentage than Sb1a.Sb6a−/− mice. Sb1a−/− and Sb6a−/− single knockout mice had normal monocyte numbers and percentage in bone marrow, as previously reported (Benarafa, 2011; Scarff et al., 2004)(Figure S4a). Differences in blood monocytes were variable and showed a subtle downward trend for Sb6a−/− and Sb1a.Sb6a−/− mice (Figure S4b). No difference between the genotypes were observed in other major blood leukocyte subsets and erythrocytes (Tables S1, S2). Monocytes were generally less sensitive to LLME than neutrophils and only Sb1a.Sb6a/- monocytes showed a significant increase in necrotic cell death following LLME treatment compared to WT (Figure S4c,d). Cell death in Sb1a−/− and Sb6a−/− monocytes treated with LLME were at intermediate levels between WT and Sb1a.Sb6a−/− (Figure S4d). In Sb1a.Sb6a−/− mice, deletion of CatG only partly corrected the accelerated monocyte death after LLME treatment (Figure S4e). As in neutrophils, the in vivo and in vitro findings suggest that Sb1a and Sb6a additively, although modestly, contribute to monocyte survival.
CatG directly cleaves GSDMD into a stable N-terminal domain GSDMD-p30
Because CatG induces a regulated form of necrosis with fast kinetics, we hypothesized that CatG might process GSDMD to induce cell lysis. We indeed found that purified human CatG cleaved both human and mouse GSDMD to form a stable characteristic GSMDp30 fragment virtually indistinguishable from that generated by recombinant mouse caspase-11 (Figure 3a,b, S5a,b). Under the conditions used, purified human NE and PR3 failed to produce a stable GSDMD-p30 fragment at low nM concentrations and had a largely degrading activity on human and mouse GSDMD at higher concentrations (Figure 3c,d, S5c,d). Pre-treatment of the proteases with the caspase inhibitor QVD-OPh blocked the cleavage of GSDMD by caspase-11 but not by CatG, ruling out indirect activation of caspases in THP-1 and transfected HEK cell lysates (Figure S5a). Conversely, CatG inhibitor I (CatG-Inh) had no effect on caspase-11 activity, while effectively inhibiting CatG (Figure S5a–d). We found that cleavage of mouse GSDMD by high concentrations of NE was inhibited by CatG-Inh, suggesting that the observed cleavage may not be caused by NE but by residual CatG activity in the purified preparation of NE from human sputum (Figure S5c).
Figure 3. CatG cleaves GSDMD to generate GSDMD-p30 N-terminal fragment.

Immunoblots of (a) HEK cells, transfected with mouse Flag-GSDMD plasmid, (b) THP-1 lysates (c) HEK cells, transfected with mouse Flag-GSDMD plasmid and (d) HEK cells, transfected with human Flag-GSDMD plasmid. Cells were lysed without protease inhibitors, incubated (12.5–25μg total proteins) with indicated concentrations of proteases (nM) for 1h at 37°C and were resolved on SDS-PAGE and immunoblotted for GSDMD (b) or Flag (a,c,d). See also Figure S5.
Purified recombinant mouse GSDMD with a C-terminal His-tag (rGSDMD) and active site titrated CatG were used to determine the second order rate constant for CatG cleavage of GSDMD (Figure S5e). The kcat/KM value was 1.09×106 M−1sec−1, which is one or two order of magnitude greater than what we previously reported for caspase-1 (<105 M−1sec−1) and caspase-11 (<104 M-1sec−1), respectively (Gonzalez Ramirez et al., 2018). The p20 C-terminal fragment generated by CatG was excised and subjected to Edman degradation to reveal that CatG cleaved GSDMD at Leu-274 (Figure S5e,f). Substitution of Leu-274 for Ala (L274A) or for Gly (L274G) substantially reduced the cleavage of GSDMD by CatG (Figure S5g). Incomplete abrogation of CatG cleavage of the L274 mutants suggests that alternative, less preferred, cleavage sites for CatG may exist in the linker region between the N- and C-terminal regions when high protease concentrations are used (Figure S5f). Together, these finding demonstrate that GSDMD is a preferred substrate of CatG, which cleaves GSDMD only two residues upstream of the caspase cleavage site at Asp-276 (Kayagaki et al., 2015; Shi et al., 2015).
Sb1a.Sb6a−/− neutropenia is not due to pyroptosis mediated by GSDMD
We then ruled out a role for pyroptotic caspases in the steady state neutropenia due to Sb1a deficiency. Deletion of both mouse inflammatory caspases (Casp1 and Casp11) did not rescue the bone marrow neutropenia in Casp1/11.Sb1a−/− mice (Figure S6a,b). Furthermore, we observed no difference in cell death kinetics after granule permeabilization of Casp1/11.Sb1a−/− neutrophils compared to Sb1a−/− neutrophils treated with LLME (Figure S6c). To address whether Gsdmd is required for neutrophil death in vivo, we generated Gsdmd knockout mice by CRISPR/Cas9 targeting in Sb1a.Sb6a−/− zygotes. Five mutant alleles were identified and each was bred to homozygosity (Figure S7). We found that Gsdmd.Sb1a.Sb6a−/− had reduced neutrophils in the bone marrow similarly as Sb1a.Sb6a−/− mice, while Gsdmd−/− neutrophil numbers were the same as in WT mice in steady state (Figure 4a). Furthermore, granule permeabilization with LLME induced identical kinetics of necrosis in Gsdmd.Sb1a.Sb6a−/− as in Sb1a.Sb6a−/− neutrophils (Figure 4b, S8a). Likewise, spontaneous apoptosis was not altered in neutrophils lacking Gsdmd in WT or Sb1a.Sb6a−/− backgrounds (Figure 4c; S8b). We also found that TNF-mediated death pathways were consistently increased in Gsdmd.Sb1a.Sb6a−/− (as in Sb1a.Sb6a−/−) compared to Gsdmd−/− and WT neutrophils (Figure 4d; S8c). Deletion of Gsdmd appears to reduce monocyte numbers in the bone marrow compared to WT mice, but no further decrease in monocyte numbers and percentage was observed in Gsdmd.Sb1a.Sb6a−/− compared to Sb1a.Sb6a−/− bone marrow (Figure S8d). Moreover, LLME-induced death was similarly enhanced in Gsdmd.Sb1a.Sb6a−/− and Sb1a.Sb6a−/− monocytes and no difference was observed between WT and Gsdmd−/− monocytes in this assay (Figure S8e). Neutrophil and monocyte percentage in blood of Gsdmd−/− and Gsdmd.Sb1a.Sb6a−/− were similar to each other and intermediate between WT and Sb1a.Sb6a−/− (Figure S8f). Higher absolute counts of neutrophils in blood in Gsdmd.Sb1a.Sb6a−/− and Gsdmd−/− compared to Sb1a.Sb6a−/− mice does not reflect a specific rescue but is rather due to elevated total white blood counts (WBC) in Gsdmd−/− and Gsdmd.Sb1a.Sb6a−/− compared to other genotypes (Figure S8f, Table S2). Overall, our findings demonstrate that Gsdmd is dispensable for the death pathways mediated by CatG and regulated by Sb1a and Sb6a in vivo and in vitro.
Figure 4. GSDMD does not mediate neutrophil death in Sb1a.Sb6a−/− mice.

(a) Leukocyte subset analysis of bone marrow neutrophils of 6 week-old male and female mice, n=9–16/genotype from 6 independent experiments. Scatter plots show data for individual mice, horizontal lines indicate mean ± SEM. Data were analysed by Mann-Whitney test (**** P<0.0001; ***P<0.001). (b) Survival of neutrophils treated with 100μM LLME in the presence or absence of 50μM Q-VD-OPh for 4 hours. (c) Spontaneous apoptosis and secondary necrosis of neutrophils cultured in vitro with or without 50μM Q-VD-OPh. (d) Neutrophil survival following stimulation with TNF-α (100ng/ml) and actinomycin D (200ng/ml) with or without 50μM Q-VD-OPh and 20μM necrostatin-1 over 48 hours. Viability was assessed by flow cytometry using Annexin V-APC and 7-AAD. Cells were from 6–12 week-old female and male mice. Data are shown as mean ± SEM of 3–6 independent experiments and were analyzed by two-way ANOVA. See also Figure S6–S8.
Sb1a and Sb6a regulate endotoxin-mediated inflammation in a CatG- and GSDMD-dependent manner
Activation of GSDMD by inflammatory caspases and assembly of GSDMD-p30 at the plasma membrane contribute in part to the release of mature IL-1β and Gsdmd−/− mice are largely protected against endotoxemic shock (Kayagaki et al., 2015; Shi et al., 2015). Conversely, we have previously shown that Sb1a−/− mice release increased levels of inflammatory cytokines in association with failed clearance of Pseudomonas aeruginosa infection (Benarafa et al., 2007). To test the physiological relevance of the cleavage of GSDMD by CatG, we measured the early systemic cytokine response to intraperitoneal injection of a sub-lethal dose of LPS. We found that Sb1a.Sb6a/- mice had significantly higher levels of TNF-α at 2h after injection and increased IL-6 and IL-1 β at 6h compared to WT mice (Figure 5a). Importantly, increased systemic inflammation was dependent on CatG, as cytokine levels in CatG.Sb1a.Sb6a−/− mirrored those of WT mice (Figure 5a). As expected, GSDMD was essential for the detection of IL-1β but, importantly, deletion of GSDMD also reduced TNF-α levels (2h) but not IL-6 levels (6h) in Gsdmd.Sb1a.Sb6a−/− mice (Figure 5a). Similarly, in a lung inflammation model induced by intranasal instillation of LPS, Sb1a.Sb6a−/− mice showed a significant increase in TNF-α and IL-6 in bronchoalveolar lavage (BAL) 14h after instillation and this increase was dependent on both CatG and GSDMD (Figure 5b). At this time point, IL-1β levels in BAL were very low to undetectable in all genotypes.
Figure 5. Sb1a.Sb6a−/− mice exhibit CatG-dependent enhanced pro-inflammatory responses upon LPS challenge.

(a) Systemic cytokine release in plasma after intraperitoneal injection of LPS. Data are from male and female 6 week-old mice n=5–15/genotype from 7 independent experiments and analysed by two-way ANOVA. (b) Local cytokine release in BAL after intranasal LPS instillation. Data are from female 6 week-old mice n=5–6/genotype from 3 independent experiments and analysed by one-way ANOVA. (c) IL-1β released by BMDMs primed with LPS and stimulated with nigericin and ATP for 3 hours and 18 hours. BMDMs were from 6–12 week-old female and male mice. Data are from n=10–14/genotype from 9 independent experiments and analysed by one-way ANOVA at each time point. All data are shown as mean ± SEM with individual scatter plots. See also Figure S8.
To evaluate direct effects of the cytosolic serpins on IL-1β release, we finally investigated the effects of canonical inflammasome activation in bone marrow-derived macrophages (BMDMs). We found that IL-1β release by Sb1a.Sb6a−/− BMDMs was significantly higher than by WT primed BMDMs stimulated with nigericin or ATP at 3 and 18h. Furthermore, this effect was again completely dependent on both CatG and GSDMD (Figure 5c). In all conditions, we did not observe any significant difference in cell death (LDH release) between genotypes (Figure S8g). Taken together, our data indicate that Sb1a and Sb6a regulate inflammatory responses through the regulation of CatG and in part through prevention of GSDMD processing in macrophages.
Discussion
Serpinb1 and Serpinb6 are ancient clade B serpin genes and are conserved in all vertebrates (Benarafa and Remold-O’Donnell, 2005; Kaiserman and Bird, 2005). In this study, we found that both mouse orthologs Serpinb1a and Serpinb6a are survival factors of neutrophils and monocytes. In neutrophils, cell death was increased in single serpin knockout mice and it was significantly more severe in Sb1a.Sb6a−/− neutrophils. In monocytes, each serpin compensated for the absence of the other and reduced survival of monocytes was observed only in double knockout Sb1a.Sb6a−/− mice. Both serpins have very fast inhibitory second order rate constants for CatG, 107 mol/L−1s−1 for Serpinb6 and 2×106 mol/L−1s−1 for Serpinb1 (Cooley et al., 2001; Scott et al., 1999). Since CatG deletion rescues the neutrophil defect of Sb1a−/− neutrophils (Baumann et al., 2013), we anticipated and demonstrated that CatG is essential in inducing cell death in Sb1a.Sb6a−/− neutrophils in homeostatic conditions in vivo. Moreover, granule permeabilization-induced cell death was reduced in CatG.Sb1a.Sb6a−/− neutrophils, indicating that the CatG/serpin axis is critical in this neutrophil RCD pathway. In monocytes, CatG deletion also rescued monocyte numbers in mice lacking both serpins.
Increased spontaneous death and TNF-induced death of Sb1a.S6a−/− neutrophils in vitro also highlighted the contribution of the two serpins in regulating CatG in these RCD pathways. Spontaneous neutrophil apoptosis is largely driven by the intrinsic apoptotic pathway and can be significantly delayed by sustained expression of anti-apoptotic BCL-2 family proteins such as Mcl-1 and A1 and by apoptotic caspase inhibitors (Akgul et al., 2001). TNF-α induces apoptosis through the activation of RIPK1, p38 MAPK, PI3K, and generation of ROS by the NADPH oxidase triggering caspase-3 cleavage (Geering and Simon, 2011). Caspase inhibition shifts the apoptotic pathway to necroptosis (Wallach et al., 2016). Here, caspase and RIPK1 inhibition improved survival of Sb1a.S6a−/− neutrophils in spontaneous and TNF-induced apoptosis, respectively. Yet, Sb1a.S6a−/− neutrophils showed more necrotic cells at late time points in the presence of these inhibitors. Therefore, while apoptotic caspases and RIPK1-RIPK3-MLKL are the principal drivers of these RCD pathways, CatG significantly and independently contributes to the acceleration of the dying process towards necrosis (cell lysis). Indeed, granule/lysosomal permeabilization is a late event during TNF-induced death or after NLRP3 inflammasome activation; and this process is associated with cleavage of mitochondrial complex 1 proteins and triggered or enhanced by mitochondrial ROS (Heid et al., 2013; Huai et al., 2013; Oberle et al., 2010). In neutrophils, the prime source of ROS is the NADPH oxidase but we found that deletion of the essential p47phox subunit in Ncf1.Sb1a−/− mice did not rescue neutrophil survival in vivo and in vitro. Furthermore, CatG did not cleave NDUFS1 and NDUFS3, which are essential for the production of ROS by mitochondria, and were shown to be proteolytically inactivated by caspases and granzymes to induce apoptosis (Huai et al., 2013; Jacquemin et al., 2015; Martinvalet et al., 2008). The ROS scavenger MitoQ did not alter cell death mediated by LLME and TNF-α in neutrophils in vitro, indicating that CatG-mediated RCD can largely proceed independently of ROS.
Live cell imaging experiments demonstrated that LLME-treated Sb1a−/− and Sb1a.Sb6a−/− neutrophils proceeded rapidly through necrotic death with cell membrane blebbing, swelling and rupture suggestive of pyroptosis (Liu and Lieberman, 2017). We found that human CatG directly cleaves human and mouse GSDMD to generate an N-terminal discreet cleavage product. The cleavage site of mouse GSDMD was identified at Leu-274, only two residues upstream of the conserved Asp-276 cleaved by caspase-1/11. This is reminiscent of the activation of caspase-7 by CatG, which is also two residues upstream of the canonical Asp site (Zhou and Salvesen, 1997). Furthermore, the kinetics of cleavage indicate that GSDMD is a preferred substrate of CatG. A recent study reported that excess recombinant NE cleaved GSDMD and that this cleavage of GSDMD induced neutrophil death. They also reported that Gsdmd−/− mice were less effective in clearing intraperitoneally injected E. coli (Kambara et al., 2018). In our hands, human purified NE did not cleave neither human nor mouse GSDMD into a stable GSDMD-p30 fragment. CatG inhibitor I inhibited the cleavage of GSDMD with high NE concentrations indicating the presence of residual CatG in the purified NE preparation. Furthermore, we found that deletion of caspase-1/11 and, more critically, deletion of GSDMD had no effect on the neutropenia of Sb1a−/− and Sb1a.Sb6a−/− mice. In addition, caspase-1/11 and GSDMD did not directly contribute to cell death induced by granule permeabilisation. We previously showed that NE-deficient neutrophils were equally sensitive to granule permeabilization-induced death as WT neutrophils (Baumann et al., 2013). Serpinb1a also inhibits PR3 (Benarafa et al., 2002), which was shown to activate caspase-3 leading spontaneous apoptosis (Loison et al., 2014). We have shown here that PR3 can cleave GSDMD but generates several fragments that are rapidly degraded and therefore PR3 may disarm GSDMD-dependent pyroptosis. Release of PR3 together with CatG may in part explain why GSDMD is not involved in cell death in neutrophils. Alternatively, similarly to caspase-3, CatG may have multiple target proteins leading to cell death in addition to caspase-7 and GSDMD and only combined disruption of multiple pathways may restore neutrophil survival in absence of Serpinb1 and Serpinb6.
Importantly, our study revealed that cytosolic serpins regulate inflammatory cytokine responses and this effect was dependent of CatG and GSDMD. We found increased IL-1β release from Sb1a.Sb6a−/− activated macrophages and high levels of IL-6 and TNF-α following local and systemic injection of LPS. The increased cytokine levels in vitro and in vivo were all dependent on CatG and GSDMD suggesting that the serpins likely regulate release of this inflammatory cytokine in part through inhibition of processing of GSDMD by CatG. Higher levels of IL-1β and danger signals released from necrotic cells in Sb1a.Sb6a−/− mice may in turn lead to the sustained production of TNF-α and IL-6. IL-1β is produced in the cytosol as a biologically inactive pro-form that is activated by cleavage of an N-terminal propeptide. NSPs can process several pro-forms of the IL-1 family members, including IL-1β, IL-33, IL-36α, IL-36β and Il-36γ (Clancy et al., 2018; Hazuda et al., 1990; Henry et al., 2016; Lefrancais et al., 2012; Macleod et al., 2016). While inflammatory caspases have a predominant role in IL-1β processing in activated myeloid cells, NSPs substantially contribute to enhancing the responses in vivo (Adkison et al., 2002; Kono et al., 2012). NSPs are thought to contribute to inflammation in part through processing of IL-1 family members after they are released from necrotic cells as an unprocessed pro-forms. Indeed, the reported cleavage sites by NSPs on IL-1 cytokines are located N-terminally of the Asp residues cleaved by caspases. Our study suggests an additional pathway where CatG promotes the release of IL-1β cells via GSDMD activation. Thus, more pro-IL-1β may be processed and released by activated myeloid cells when cytosolic serpins are downregulated or overwhelmed by proteases leading to enhanced release via GSDMD processing by CatG. This may explain in part previous observations of increased release of IL-1β in lungs of Sb1a−/− mice infected with Pseudomonas aeruginosa or influenza A virus (Benarafa et al., 2007; Gong et al., 2011). Whether this process is occurring with other IL-1 family cytokines and in other cells than macrophages is currently under study. As we were resubmitting this revised manuscript, a study reported a non-covalent interaction between C-terminal domain of Serpinb1 and the CARD domain of inflammatory caspases that was proposed to prevent spontaneous caspase activation (Choi et al., 2019). They show that Sb1a−/− mice are more susceptible to a lethal dose of LPS and to bacterial infection. These data are in agreement with our current data and our previous studies showing that clearance of P. aeruginosa is reduced in Sb1a−/− mice. However, we have also shown that clearance of P. aeruginosa can be rescued by increasing neutrophil numbers following enhanced myelopoiesis with G-CSF treatment (Basilico et al., 2016). In contrast to recent report of Choi and colleagues (Choi et al., 2019), we did not observe increased spontaneous release of IL-1β when macrophages were incubated with LPS for 5h in absence of nigericin or ATP (Figure S8h). Thus, without contradicting a potential direct interaction between caspases and Sb1a shown by these authors, our data presented here demonstrate a more conventional and straightforward mechanism dependent on CatG inhibition by Serpinb1a and Serpinb6a leading to inflammation via GSDMD processing.
In summary, our findings indicate that Serpinb1a and Serpinb6a are key survival factors in neutrophils and monocytes. They protect neutrophils from multiple death pathways through inhibition of CatG, which activates both executors of apoptosis and of pyroptosis: caspase-7 and GSDMD, respectively. Yet, our data indicate that CatG does not rely exclusively on any of the known RCD pathways. By contrast, GSDMD is a critical target of CatG-mediated death of monocytes in steady state in vivo but not after granule permeabilization. Finally, we demonstrate that Serpinb1a and Serpinb6a critically regulate IL-1β release and systemic inflammation by regulating myeloid cell necrosis and an alternative activation of GSDMD.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Charaf Benarafa (charaf.benarafa@vetsuisse.unibe.ch)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse husbandry
All animal studies were approved by the Cantonal Veterinary Office of Bern and conducted in accordance with the Swiss federal legislation on animal welfare. Mice were kept in SPF facilities, in individually ventilated cages (Tecniplast, blue line), with 12/12 light/dark cycle, autoclaved acidified water, autoclaved cages including food, bedding and environmental enrichment. Age and gender of mice is indicated for each in vivo or ex vivo model described below as well as in figure legends.
Previously described mouse lines
All mice were in C57BL/6J background or backcrossed for at least 10 generations. Sb1a−/− (Serpinb1atm1.1Cben) and Sb6a−/− (Serpinb6atm1.1Pib) mice were generated previously (Benarafa et al., 2007; Scarff et al., 2003). CatG−/− (Ctsgtm1Ley) mice were provided by Christine Pham (Washington University, St. Louis) (MacIvor et al., 1999), Ncf1−/− (Ncf1m1J) mice were from The Jackson Laboratory (Huang et al., 2000). Casp1/11−/− (B6J.129Casp1tm1Flv) mice (Kuida et al., 1995) were provided by Jens Stein. We confirmed by PCR and sequencing that the Casp1/11−/− mice (but not any other strain) carried the Casp129 5-nt deletion in Casp11 described previously (Kayagaki et al., 2011).
Newly generated mouse lines
Sb1a.Sb6a−/− mice were generated by mating compound heterozygous F1 mice. The two genes are separated by 1.2 Mb (0.25cM) on mouse chromosome 13 and we observed 5 crossover events from 378 F2 pups, of which only one crossover event had two deleted alleles on the same chromosome (Sb1a+/−.Sb6a−/−) and the others had two wild-type alleles on the same chromosome (Sb1a+/.Sb6a+/+ or Sb1a+/−.Sb6a+/+). Gsdmd−/− mice were generated at the transgenic unit of the Theodor Kocher Institute, University of Bern by microinjection of wild-type and Sb1a.Sb6a−/− zygotes with ribonucleoprotein (RNP) complexes as described (Aida et al., 2015) and detailed hereafter. Recombinant Cas9 nuclease, tracrRNA, crRNA and nuclease-free duplex buffer were from IDT. Target-specific crRNA sequence used was AGCATCCTGGCATTCCGAG, which was previously shown to successfully target Gsdmd in mouse zygotes (Shi et al., 2015). To prepare RNPs, tracrRNA and crRNA stock solutions were reconstituted at 10μM concentrations in nuclease-free duplex buffer and stored at-20°C. tracrRNA and crRNA were mixed (4.5μl each) and incubated at 95°C for 2 minutes, then cooled down at room temperature for at least 15 minutes for annealing of the duplex. EmbryoMax buffer (Millipore) (20.5μl) was added to the RNA duplex at room temperature, mixed by pipetting twice. Cas9 protein (0.5μl) was added to the RNA duplex and mixed again by pipetting. The RNP was incubated at 37°C for 20 minutes, centrifuged at 18’000g for 10 min at 4°C and placed on ice until microinjection into the male pronucleus of zygotes on the same day. After overnight incubation at 37°C, live 2-cell stage embryos were transferred into the infundibulum of pseudopregnant CD1 females using standard protocols. Founders were positively screened by PCR and T7 endonuclease assay. Mosaic founders were crossed with C57BL/6J or Sb1a.Sb6a−/− and mutated alleles in F1 mice were identified by DNA sequencing (Figure S7). F1 mice with identical mutations were intercrossed to generate Gsdmd−/−, Gsdmd.Sb1a.Sb6a−/− mice. F2 progeny of four distinct Gsdmd−/− mouse lines and their littermates were studied.
Mouse cohorts
For each experimental set up involving in vivo experimentation or isolated cells, gender and age of the mice is indicated in METHODS DETAILS below and in figure legends. For in vivo experiments and determination of leukocyte numbers in blood and bone marrow, animals were used at 6 weeks of age and data was pooled and analysed using mice from both sex, except for the model of lung instillation of LPS, where only females were used as indicated below. For assays using isolated primary mouse leukocytes, mice were used between 6–12 weeks of age. Data was pooled from both male and female mice from all genotypes. Figure legends indicate the numbers of individual mice per genotype (n) and explicitly mention the number of independent experiments that we have performed.
Cell lines
Human embryonic kidney (HEK-293) cells were cultured in DMEM supplemented with 10% heat-inactivated FBS and 1% penicillin/streptomycin and maintained at 37°C in 5% CO2. Human monocytic cell line THP-1 cells were cultured in RPMI supplemented with 10% heat-inactivated FBS and 1% penicillin/streptomycin and maintained at 37°C in 5% CO2. In cell death assays, primary mouse bone marrow leukocytes were cultured in DMEM supplemented with 1% FBS and 1% penicillin/streptomycin at 1×106 cells/ml. Primary mouse bone marrow-derived macrophages (BMDMs) were generated by culturing bone marrow leukocytes in RPMI supplemented with 10% FBS, 1% penicillin/streptomycin and 10ng/ml M-CSF (PeproTech) for 7 days. Each preparation of primary cell cultures were from 1–2 sex- and age-matched 6–12 week-old male and female mice of each genotype.
METHOD DETAILS
Hematology and flow cytometry
Erythrocyte (RBC), platelet (PLT) and total white blood cell (WBC) counts were determined in whole blood collected in EDTA of 6 week-old female and male mice using a VetABC hematology analyser. Blood and bone marrow leukocyte subsets percentages and cell death kinetics of bone marrow cells were determined using a 4-color FACS Calibur (BD Biosciences) using single cell suspensions blocked with anti-CD16/CD32 (clone 2.4G2) and stained for 30–40 min on ice with fluorescently labeled antibodies (1:200; BioLegend, BD Biosciences) as previously (Benarafa et al., 2011). Analysis was performed using FlowJo gating within nucleated live CD45+ cells as neutrophils (CD11b+/Ly6G+), monocytes (CD11b+/CD115+), B cells (CD45R/B220+), eosinophils (SSChigh/SiglecF+), NK cells (NK1.1+/CD3neg) and T cells (CD3+). Apoptosis and necrosis of neutrophils and monocytes in vitro was determined by Annexin V-allophycocyanin and 7aminoactinomycin D (7AAD) labelling for 15min at room temperature prior to FACS analysis. Flow cytometry sorting of neutrophils (CD11b+/Ly6G+) was performed on single-cell suspensions of bone marrow leukocytes using a FACS Aria II sorter (BD Biosciences) at the flow cytometry core facility of the University of Bern.
Zymosan induced peritonitis model
Groups of 2–3 female or male 6 week-old mice per genotype were injected intraperitoneally with 0.5mg opsonized zymosan (1mg/ml). The experiment was repeated 4 times. Peritoneal cells were collected with 5ml PBS supplemented with 1% FBS 4 hours after injection. Cells were counted manually in a Neubauer chamber. Relative percentages of live leukocyte subsets were determined by flow cytometry, analysis was performed using FlowJo gating within nucleated live CD45+ cells as neutrophils (CD11b+/Ly6G+) and macrophages (CD11b+/CD115+). Phagocytosis of zymosan particles was determined by microscopy analysis of cytospins (Shandon) stained with Differential Quik stain (Electron Microscopy Science). One hundred cells/mouse were counted to calculate the percentage of zymosan positive cells and 40 zymosan positive neutrophils/mouse were further evaluated for the number of particles per phagocyte.
Systemic and lung LPS challenge in vivo
Systemic inflammation was induced in groups of male and female mice by intraperitoneal injection of 10μg/g LPS from P. aeruginosa (Sigma, L8643) diluted in 0.9% NaCl. EDTA plasma was collected 2 or 6 hours after injection. Local lung inflammation was induced in groups of anesthetized female mice (intraperitoneal ketamine/xylazine 100mg/kg, 10mg/kg, respectively) by intranasal instillation of LPS (10μg/mouse in 20μl). Bronchoalveolar lavage (BAL) was collected with 3× 1ml PBS supplemented with 1% FBS 14 hours after injection. TNF-α, IL-6 and IL-1β in plasma and BAL were measured by ELISA (eBioscience).
Cell death assays
In cell death induction assays, leukocytes (1×106 cells/ml) were incubated at indicated concentrations with L-leucyl-L-leucine methyl ester (LLME) (G-2550; Bachem), Q-VD-OPh (ApexBio), necrostatin-1 (Enzo LifeScience), TNF-α (Promokine) and actinomycin (PeproTech). MitoQ was a gift from Michael Murphy. Viability was assessed by flow cytometry as described above. Live cell images were acquired on a Zeiss Axio Observer of total bone marrow cells of female and male 6–12 week-old mice stained for 30–40 min on ice with fluorescently labeled antibody for neutrophils (Ly6G+) and DAPI to visualize dead cells. Cells were kept at 37°C following administration of 100μM LLME. Life cell images were taken every 30sec over a time frame of 120min. Live cell images over time were analysed using dot quantification feature of IMARIS quantifying the number of Ly-6G+ neutrophils and DAPI+ dead cells over time.
IL-1β release by BMDMs
BMDMs (2×106 cells/ml) were primed with 100ng/ml ultra-pure LPS (InvivoGen) for 5 hours followed by stimulation with 5mM ATP or 5μM nigericin (InvivoGen) for 3 and 18 hours in RPMI supplemented with 10% FBS, 1% penicillin/streptomycin and 10ng/ml M-CSF (PeproTech). Mature IL-1β in culture medium was measured by ELISA (eBioscience).
Plasmids and transfection
pFlag-CMV-4 containing N-terminal Flag-tagged mouse GSDMD was a gift from Judy Lieberman (Addgene plasmid # 80950)(Liu et al., 2016). Human GSDMD cDNA was synthetized and cloned into pcDNA3.1(+)-N-DYK (Flag) using BamHI/EcoRI (GenScript). Alanine substitution mutants were generated using Q5 site-directed mutagenesis kit (NEB). WT and mutant Flag-GSDMD plasmids were purified (Macherey-Nagel) and verified by DNA sequencing (Microsynth). Transient transfection of HEK cells with Flag-Gsdmd plasmids was performed using TurboFect (ThermoFisher Scientific) following the manufacturer’s procedure. In brief, 2×105 HEK cells were seeded in a 6-well plate followed by transfection with 6μl TurboFect and 4μg plasmid DNA. HEK cells were grown without selection pressure until 90% confluency after transfection to allow high protein yield during lysis.
Proteolysis of cell lysates
HEK and THP-1 cells were lysed in 0.1% Triton X-100 lysis buffer without protease inhibitors followed by 2× 30sec sonication (Soniprep 150 plus, MSE). Lysates were clarified at 10’000g for 10min at 4°C and supernatant was collected. Total protein concentration in lysates was determined by the BCA assay (ThermoFisher Scientific). Cell lysates (12.5 μg HEK; 25μg THP-1) were incubated for 1h at 37°C with recombinant caspase-11 (175nM final concentration) (Enzo LifeScience, BML-SE155–5000) or with purified human CatG, NE or PR3 (Athens Research Technologies) at indicated concentrations ranging from 4–136nM. Where indicated, Q-VD-OPh (1mM)(ApexBio) or cathepsin G inhibitor I (CatGInh)(1mM)(Calbiochem, 429676–93-7) were preincubated for 5 min at 37°C before the protease treatment. Proteolysis was stopped by adding final concentration of 1x Laemmli Buffer with DTT (25mM) to the reaction and incubated for 5 min at 95°C prior to loading on SDS-PAGE.
Western Blot
Lysates were resolved by SDS-PAGE under reducing conditions using Tris-Glycine buffer. After transfer on nitrocellulose, blocking was performed using 0.5% skimmed milk and blots were probed with anti-Flag M2 (Sigma, F1804), rabbit anti-GSDMD (Sigma, G7422; Novus, NBP-33244) antibodies. Blots were stripped (Restore Western Blot Stripping Buffer, ThermoFisher, 21059) and reprobed with anti-β-actin antibody (Abcam, ab8227). Cleavage of NDUFS3 and NDUFS1 was performed on sorted bone marrow neutrophils, which were preincubated in PBS with protease inhibitor cocktail (Roche) for 5 min at 37°C before lysis in 1% NP-40 lysis buffer. Blots were probed with monoclonal mouse anti-NDUFS1 (Santa Cruz, E-8, sc-271510) and monoclonal mouse-anti NDUFS3 (Invitrogen, 459130) antibodies, both gifts from Denis Martinvalet.
Recombinant mouse GSDMD (rGSDMD)
The sequence encoding mouse full length GSDMD was purchased from Integrated DNA Technologies and cloned into pET29b(+) (Novagen) containing a C-terminal His tag and transformed into BL21 (DE3) competent E. coli. Expression was induced with 0.2mM isopropyl β-D-1-thiogalactopyranoside (IPTG) for 4 hours at 25°C shaking at 250 RPM. The GSDMD cell pellets were resuspended in 50mM HEPES, 100 mM NaCl, pH 8.0 and lysed via sonication. Cell lysates were centrifuged at 4°C for 30 minutes at 29,000 × g, supernatants were filtered through a 0.22μm filter (Millipore) and the soluble fractions were applied to a 1ml Ni-chelating sepharose resin (GE Healthcare Life Sciences) in a chromatography column. The rGSDMDbound resin was washed in 50mM HEPES, 500mM NaCl, pH 8.0 and then eluted in 50mM HEPES, 100mM NaCl, pH 8.0 with stepwise increments of imidazole from 12.5 – 100 mM. rGSDMD purity was analyzed by 4–12% Bis-Tris SDS/PAGE gel (Novex Life Technologies) stained with Instant Blue (Expedeon).
Cleavage of rGSDMD and N-terminal sequencing
To determine the active concentration of CatG (Athens Research Technologies), a serial dilution series of AcAAPF-OPh2 was incubated with CatG for 30 minutes at 37°C. The fluorogenic substrate Suc-AAPF-AMC was added to a final concentration of 100μM in 100μl of assay buffer (100mM Tris-HCL pH 7.5 500mM NaCl) and enzymatic activity was measured. Velocities were plotted against inhibitor concentration allowing the calculation of active enzyme concentration (Denault and Salvesen, 2003). Active site titrated CatG was subjected to two-fold series dilutions and incubated separately for 30 minutes at 37°C with 2μM of GSDMD in 100mM Tris-HCL pH 7.5 500mM NaCl in a final assay volume of 20μl. Reactions were terminated by heating at 95°C with 10μl of 3x SDS loading buffer to give a final concentration of 1x loading buffer. Samples were analyzed on 4–12% Bis-Tris SDS-PAGE gels by Instant Blue staining. The gels were then scanned and imported to Image Studio (LI-COR Biosciences) for protein band intensity quantification. Band intensity values were plotted against enzyme concentration and IC50 values were determined via GraphPad Prism. Those values were then used to determine kcat/KM using the following equation kcat/KM = ln2/(E½ * t). Here, kcat/KM is the second order rate constant for substrate hydrolysis, E½ is the concentration of protease for which half the substrate is consumed, E is the concentration of enzyme, and t is the incubation time (Pop et al., 2008). Protein sample was resolved by SDS/PAGE and transferred to an Immobilon-P membrane (Millipore, Bedford, MA, USA) by electroblotting. The membrane was briefly stained with Coomassie Brilliant Blue R-250, destained and washed with water. The appropriate band was excised and sequenced by Edman degradation with a ABI Procise at UC Davis Proteomics Core Facility.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis and graphs were generated using Prism 8.0 (GraphPad, San Diego, CA). As indicated in figure legends, independent experiments were performed and data was pooled. Where applicable data was shown for each replicate or mouse using scatter plots and horizontal lines or bars showed mean ± SEM. Box and whiskers showed median, interquartile range, 5–95 percentiles and individual values above 95th percentile. For analysis, non-parametric tests were used to analyse data from in vivo and ex vivo studies. Experiments were analysed by Mann-Whitney test, one-way or two-way ANOVA with Tukey post-test as appropriate and as indicated in figure legends. P<0.05 was considered statistically significant (**** P<0.0001; ***P<0.001; **P<0.01; *P<0.05).
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-mouse Ly-6G APC/Cy7 | Biolegend | Clone 1A8, Cat.#127624 |
| anti-mouse CD115 (CSF-1R) PE | Biolegend | Clone AFS98, Cat.#135506 |
| anti-mouse/human CD11b PE/Cy7 | Biolegend | Clone M1/70, Cat.#101216 |
| anti-mouse CD11c APC | Biolegend | Clone N418, Cat.#117310 |
| anti-mouse CD45R (B220) Pacific Blue | Biolegend | Clone RA3-6B2, Cat.#103227 |
| anti-mouse Ly6C Pacific Blue | Biolegend | Clone HK1.4, Cat.#128014 |
| anti-mouse CD3 PE/Cy7 | Biolegend | Clone 17A2, Cat.#100220 |
| anti-mouse CD4 APC | Biolegend | Clone GK1.5, Cat.#100412 |
| anti-mouse CD8a APC/Cy7 | Biolegend | Clone 53-6.7, Cat.#100714 |
| anti-mouse NK1.1 APC | Biolegend | Clone PK136, Cat.#108710 |
| Annexin V APC | Biolegend | Cat.#640941 |
| 7-AAD | Biolegend | Cat.#420404 |
| Anti-mouse CD16/CD32 purified | Biolegend | Clone 2.4G2, Cat.#101302 |
| anti-mouse Siglec F PE | BD Bioscience | Clone E50-2440, Cat.#562068 |
| anti-mouse CD45 V500 | BD Bioscience | Clone 30-F11, Cat.#561487 |
| Mouse Anti-mouse NDUFS3 | Invitrogen | Cat.#459130 |
| Mouse Anti-mouse NDUFS1 | Santa Cruz | Clone E-8, Cat.#sc-271510 |
| Rabbit anti-mouse Actin | Abcam | Cat.#ab8227 |
| Rabbit anti-human GSDMD | Sigma | Cat.#G7422 |
| Rabbit anti-mouse GSDMD | Novus | Cat.#NBP-33244 |
| Mouse anti-Flag M2 | Sigma | Cat.#F1804 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| L-leucyl-L-leucine methyl ester | Bachem | Cat.#G-2550 |
| Q-VD-OPh hydrate | ApexBio | Cat.#A1901 |
| Necrostatin-1 | Enzo Life Science | Cat.#BML-AP309-0100 |
| Mouse TNF-α | Promokine | Cat.#D-63720 |
| Actinomycin D | Promokine | Cat.#PK-CA577-1036-5MG |
| MitoQ | In house | Provided by Michael Murphy |
| Recombinant caspase-11 | Enzo Life Science | Cat.#BML-SE155-5000 |
| Purified human Cathepsin G | Athens Research Technologies | Cat.#16-14-030107 |
| Purified human Elastase | Athens Research Technologies | Cat.#16-14-051200 |
| Purified human Proteinase-3 | Athens Research Technologies | Cat.#16-14-161820 |
| Cathepsin G Inhibitor | Calbiochem | Cat.#429676-93-7 |
| Suc-AAPF-AMC | Bachem | Cat. #4012873.0050 |
| Immobilon-P membrane | Millipore | |
| Zymosan A from S. cerevisiae | Sigma | Cat.#ZA250 |
| LPS from P. aeuroginosa | Sigma | Cat.#L8643 |
| Mouse recombinant M-CSF | PeproTech | Cat.#315-02 |
| Ultra Pure LPS from E. coli | InvivoGen | Cat.#tlrl-3pelps |
| Nigericin | InvivoGen | Cat.#tlrl-nig |
| ATP | InvivoGen | Cat.#tlrl-atpl |
| Alt-R™ S.p. Cas9 Nuclease 3NLS | Integrated DNA Technologies (IDT) | Lot.#289717 |
| 5nmol Alt-R™ CRISPR tracrRNA | Integrated DNA Technologies (IDT) | Lot.#275957 |
| Nuclease Free Duplex Buffer | Integrated DNA Technologies (IDT) | Lot.#271987 |
| Restore™ Western Blot Stripping Buffer | ThermoFisher Scientific | Cat. #21059 |
| TurboFect Transfection Reagent | ThermoFisher Scientific | Cat. #R0531 |
| Commercial Assays | ||
| pFlag-CM-4 mouse GSDMD plasmid | Addgene, Li et al., 2016 | Cat.#80950 |
| pcDNA3.1 human GSDMD plasmid | GenScript, this paper | N/A |
| pET29b(+) | Novagen (Sigma) | Cat.#71463-M |
| Mouse IL-1β ELISA | Thermo Fisher Scientific (eBioscience) | Cat.#88-7013-88 |
| Mouse IL-6 ELISA | Thermo Fisher Scientific (eBioscience) | Cat.#88-7064-88 |
| Mouse TNF-α ELISA | Thermo Fisher Scientific (eBioscience) | Cat.#88-7324-88 |
| Differential Quik Stain Kit (modified Giemsa) | Electron Microscopy Science | Cat.#26096-50 |
| Experimental Models: Cell Lines | ||
| Human monocyte THP-1 | ATCC | Cat.#ATCC TIB-202, gift from Alfred Walz |
| Human embryonic kidney HEK-293 | ATCC | Cat.#ATCC CRL-1573 |
| Experimental Models: Organisms/Strains | ||
| Serpinb1atm1.1Cben | Benarafa et al., 2007 | N/A |
| Serpinb6atm1.1Pib | Scarff et al., 2003 | N/A |
| Sb1a.Sb6a−/− | In house | N/A |
| Ctsgtm1Ley | MacIvor et al., 1999 | N/A |
| CatG.Sb1a.Sb6a−/− | In house | N/A |
| B6(Cg)-Ncf1m1J/J | JAX Labs, Huang et al., 2000 | Cat.# 004742 |
| B6J.129-Casp1tm1Flv | Kuida et al., 1995 | N/A |
| Gsdmdem1, em4, em5, em6 | In house | N/A |
| Gsdmdem1, em4, em5, em6.Sb1a.Sb6a−/− | In house | N/A |
| Software and Algorithms | ||
| Prism 8 (version 8.0) | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Image Studio 4.0 | LI-COR | https://www.licor.com/bio/image-studio/ |
| Imaris Software | Oxford Instruments | https://imaris.oxinst.com/ |
| Other | ||
Acknowledgements
We thank Judy Lieberman, Denis Martinvalet, Michael Murphy and Jens Stein for reagents. We thank Ruth Lyck for assistance with live cell imaging. We thank Albert Witt and Urban Deutsch for the RNP microinjection. This work was supported by grants from the Swiss National Science Foundation to CB (310030-149790, 310030-173137) and the Novartis Foundation for medical-biological research.
Footnotes
Declaration of interests
The authors declare that the data supporting the findings of this study are available within the paper and its supplementary information files. The authors have no competing interest to declare.
References
- Adkison AM, Raptis SZ, Kelley DG, and Pham CT (2002). Dipeptidyl peptidase I activates neutrophil-derived serine proteases and regulates the development of acute experimental arthritis. J Clin Invest 109, 363–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, Ciferri C, Dixit VM, and Dueber EC (2016). GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci U S A 113, 78587863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aida T, Chiyo K, Usami T, Ishikubo H, Imahashi R, Wada Y, Tanaka KF, Sakuma T, Yamamoto T, and Tanaka K (2015). Cloning-free CRISPR/Cas system facilitates functional cassette knock-in in mice. Genome Biol 16, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akgul C, Moulding DA, and Edwards SW (2001). Molecular control of neutrophil apoptosis. FEBS Lett 487, 318–322. [DOI] [PubMed] [Google Scholar]
- Basilico P, Cremona TP, Oevermann A, Piersigilli A, and Benarafa C (2016). Increased Myeloid Cell Production and Lung Bacterial Clearance in Mice Exposed to Cigarette Smoke. Am J Respir Cell Mol Biol 54, 424–435. [DOI] [PubMed] [Google Scholar]
- Baumann M, Pham CT, and Benarafa C (2013). SerpinB1 is critical for neutrophil survival through cell-autonomous inhibition of cathepsin G. Blood 121, 3900–3907, S3901–3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belaaouaj A, Kim KS, and Shapiro SD (2000). Degradation of outer membrane protein A in Escherichia coli killing by neutrophil elastase. Science 289, 1185–1188. [DOI] [PubMed] [Google Scholar]
- Benarafa C (2011). The SerpinB1 knockout mouse a model for studying neutrophil protease regulation in homeostasis and inflammation. Methods Enzymol 499, 135–148. [DOI] [PubMed] [Google Scholar]
- Benarafa C, Cooley J, Zeng W, Bird PI, and Remold-O’Donnell E (2002). Characterization of four murine homologs of the human ov-serpin monocyte neutrophil elastase inhibitor MNEI (SERPINB1). J Biol Chem 277, 42028–42033. [DOI] [PubMed] [Google Scholar]
- Benarafa C, LeCuyer TE, Baumann M, Stolley JM, Cremona TP, and Remold-O’Donnell E (2011). SerpinB1 protects the mature neutrophil reserve in the bone marrow. J Leukoc Biol 90, 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benarafa C, Priebe GP, and Remold-O’Donnell E (2007). The neutrophil serine protease inhibitor serpinb1 preserves lung defense functions in Pseudomonas aeruginosa infection. J Exp Med 204, 1901–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benarafa C, and Remold-O’Donnell E (2005). The ovalbumin serpins revisited: perspective from the chicken genome of clade B serpin evolution in vertebrates. Proc Natl Acad Sci U S A 102, 1136711372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benarafa C, and Simon HU (2017). Role of granule proteases in the life and death of neutrophils. Biochem Biophys Res Commun 482, 473–481. [DOI] [PubMed] [Google Scholar]
- Bird CH, Christensen ME, Mangan MS, Prakash MD, Sedelies KA, Smyth MJ, Harper I, Waterhouse NJ, and Bird PI (2014). The granzyme B-Serpinb9 axis controls the fate of lymphocytes after lysosomal stress. Cell Death Differ 21, 876–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird CH, Sutton VR, Sun J, Hirst CE, Novak A, Kumar S, Trapani JA, and Bird PI (1998). Selective regulation of apoptosis: the cytotoxic lymphocyte serpin proteinase inhibitor 9 protects against granzyme B-mediated apoptosis without perturbing the Fas cell death pathway. Mol Cell Biol 18, 6387–6398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss-Moreau M, Chen AA, D’Cruz AA, and Croker BA (2017). A motive for killing: effector functions of regulated lytic cell death. Immunol Cell Biol 95, 146151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, and Chan FK (2009). Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137, 1112–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YJ, Kim S, Choi Y, Nielsen TB, Yan J, Lu A, Ruan J, Lee HR, Wu H, Spellberg B, et al. (2019). SERPINB1-mediated checkpoint of inflammatory caspase activation. Nat Immunol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy DM, Sullivan GP, Moran HBT, Henry CM, Reeves EP, McElvaney NG, Lavelle EC, and Martin SJ (2018). Extracellular Neutrophil Proteases Are Efficient Regulators of IL-1, IL-33, and IL-36 Cytokine Activity but Poor Effectors of Microbial Killing. Cell Rep 22, 2937–2950. [DOI] [PubMed] [Google Scholar]
- Cooley J, Takayama TK, Shapiro SD, Schechter NM, and Remold-O’Donnell E (2001). The serpin MNEI inhibits elastase-like and chymotrypsin-like serine proteases through efficient reactions at two active sites. Biochemisstry 40, 15762–15770. [DOI] [PubMed] [Google Scholar]
- Denault JB, and Salvesen GS (2003). Expression, purification, and characterization of caspases. Curr Protoc Protein Sci Chapter 21, Unit 21 13. [DOI] [PubMed] [Google Scholar]
- Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, Sun H, Wang DC, and Shao F (2016). Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535, 111–116. [DOI] [PubMed] [Google Scholar]
- Geering B, and Simon HU (2011). Peculiarities of cell death mechanisms in neutrophils. Cell Death Differ 18, 1457–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong D, Farley K, White M, Hartshorn KL, Benarafa C, and Remold-O’Donnell E (2011). Critical role of serpinB1 in regulating inflammatory responses in pulmonary influenza infection. J Infect Dis 204, 592–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez Ramirez ML, Poreba M, Snipas SJ, Groborz K, Drag M, and Salvesen GS (2018). Extensive peptide and natural protein substrate screens reveal that mouse caspase-11 has much narrower substrate specificity than caspase-1. J Biol Chem. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazuda DJ, Strickler J, Kueppers F, Simon PL, and Young PR (1990). Processing of precursor interleukin 1 beta and inflammatory disease. J Biol Chem 265, 6318–6322. [PubMed] [Google Scholar]
- He S, Wang L, Miao L, Wang T, Du F, Zhao L, and Wang X (2009). Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 137, 1100–1111. [DOI] [PubMed] [Google Scholar]
- He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, Yang ZH, Zhong CQ, and Han J (2015). Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res 25, 1285–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heid ME, Keyel PA, Kamga C, Shiva S, Watkins SC, and Salter RD (2013). Mitochondrial reactive oxygen species induces NLRP3-dependent lysosomal damage and inflammasome activation. J Immunol 191, 5230–5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry CM, Sullivan GP, Clancy DM, Afonina IS, Kulms D, and Martin SJ (2016). Neutrophil-Derived Proteases Escalate Inflammation through Activation of IL-36 Family Cytokines. Cell Rep 14, 708–722. [DOI] [PubMed] [Google Scholar]
- Huai J, Vogtle FN, Jockel L, Li Y, Kiefer T, Ricci JE, and Borner C (2013). TNFalpha-induced lysosomal membrane permeability is downstream of MOMP and triggered by caspase-mediated NDUFS1 cleavage and ROS formation. J Cell Sci 126, 4015–4025. [DOI] [PubMed] [Google Scholar]
- Huang CK, Zhan L, Hannigan MO, Ai Y, and Leto TL (2000). P47(phox)-deficient NADPH oxidase defect in neutrophils of diabetic mouse strains, C57BL/6J-m db/db and db/+. J Leukoc Biol 67, 210–215. [DOI] [PubMed] [Google Scholar]
- Jacquemin G, Margiotta D, Kasahara A, Bassoy EY, Walch M, Thiery J, Lieberman J, and Martinvalet D (2015). Granzyme B-induced mitochondrial ROS are required for apoptosis. Cell Death Differ 22, 862–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen I, Rayamajhi M, and Miao EA (2017). Programmed cell death as a defence against infection. Nat Rev Immunol 17, 151–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiserman D, and Bird PI (2005). Analysis of vertebrate genomes suggests a new model for clade B serpin evolution. BMC Genomics 6, 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kambara H, Liu F, Zhang X, Liu P, Bajrami B, Teng Y, Zhao L, Zhou S, Yu H, Zhou W, et al. (2018). Gasdermin D Exerts Anti-inflammatory Effects by Promoting Neutrophil Death. Cell Rep 22, 2924–2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kargi HA, Campbell EJ, and Kuhn C 3rd (1990). Elastase and cathepsin G of human monocytes: heterogeneity and subcellular localization to peroxidase-positive granules. J Histochem Cytochem 38, 1179–1186. [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, et al. (2015). Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671. [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, et al. (2011). Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121. [DOI] [PubMed] [Google Scholar]
- Kessenbrock K, Frohlich L, Sixt M, Lammermann T, Pfister H, Bateman A, Belaaouaj A, Ring J, Ollert M, Fassler R, et al. (2008). Proteinase 3 and neutrophil elastase enhance inflammation in mice by inactivating antiinflammatory progranulin. J Clin Invest 118, 2438–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono H, Orlowski GM, Patel Z, and Rock KL (2012). The IL-1-dependent sterile inflammatory response has a substantial caspase-1-independent component that requires cathepsin C. J Immunol 189, 3734–3740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS, and Flavell RA (1995). Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science 267, 2000–2003. [DOI] [PubMed] [Google Scholar]
- Lefrancais E, Roga S, Gautier V, Gonzalez-dePeredo A, Monsarrat B, Girard JP, and Cayrol C (2012). IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proc Natl Acad Sci U S A 109, 1673–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, and Lieberman J (2017). A Mechanistic Understanding of Pyroptosis: The Fiery Death Triggered by Invasive Infection. Adv Immunol 135, 81–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, and Lieberman J (2016). Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loison F, Zhu H, Karatepe K, Kasorn A, Liu P, Ye K, Zhou J, Cao S, Gong H, Jenne DE, et al. (2014). Proteinase 3-dependent caspase-3 cleavage modulates neutrophil death and inflammation. J Clin Invest 124, 4445–4458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luke CJ, Pak SC, Askew YS, Naviglia TL, Askew DJ, Nobar SM, Vetica AC, Long OS, Watkins SC, Stolz DB, et al. (2007). An intracellular serpin regulates necrosis by inhibiting the induction and sequelae of lysosomal injury. Cell 130, 1108–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacIvor DM, Shapiro SD, Pham CT, Belaaouaj A, Abraham SN, and Ley TJ (1999). Normal neutrophil function in cathepsin G-deficient mice. Blood 94, 4282–4293. [PubMed] [Google Scholar]
- Macleod T, Doble R, McGonagle D, Wasson CW, Alase A, Stacey M, and Wittmann M (2016). Neutrophil Elastase-mediated proteolysis activates the anti-inflammatory cytokine IL-36 Receptor antagonist. Sci Rep 6, 24880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makaryan V, Zeidler C, Bolyard AA, Skokowa J, Rodger E, Kelley ML, Boxer LA, Bonilla MA, Newburger PE, Shimamura A, et al. (2015). The diversity of mutations and clinical outcomes for ELANE-associated neutropenia. Curr Opin Hematol 22, 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinvalet D, Dykxhoorn DM, Ferrini R, and Lieberman J (2008). Granzyme A cleaves a mitochondrial complex I protein to initiate caspase-independent cell death. Cell 133, 681–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, Warren SE, Wewers MD, and Aderem A (2010). Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol 11, 1136–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, Lewis R, Lalaoui N, Metcalf D, Webb AI, et al. (2013). The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39, 443–453. [DOI] [PubMed] [Google Scholar]
- Oberle C, Huai J, Reinheckel T, Tacke M, Rassner M, Ekert PG, Buellesbach J, and Borner C (2010). Lysosomal membrane permeabilization and cathepsin release is a Bax/Bak-dependent, amplifying event of apoptosis in fibroblasts and monocytes. Cell Death Differ 17, 1167–1178. [DOI] [PubMed] [Google Scholar]
- Padrines M, Wolf M, Walz A, and Baggiolini M (1994). Interleukin-8 processing by neutrophil elastase, cathepsin G and proteinase-3. FEBS Lett 352, 231–235. [DOI] [PubMed] [Google Scholar]
- Perera NC, Schilling O, Kittel H, Back W, Kremmer E, and Jenne DE (2012). NSP4, an elastase-related protease in human neutrophils with arginine specificity. Proc Natl Acad Sci U S A 109, 6229–6234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pop C, Salvesen GS, and Scott FL (2008). Caspase assays: identifying caspase activity and substrates in vitro and in vivo. Methods Enzymol 446, 351–367. [DOI] [PubMed] [Google Scholar]
- Raptis SZ, Shapiro SD, Simmons PM, Cheng AM, and Pham CT (2005). Serine protease cathepsin G regulates adhesion-dependent neutrophil effector functions by modulating integrin clustering. Immunity 22, 679–691. [DOI] [PubMed] [Google Scholar]
- Ravichandran KS (2011). Beginnings of a good apoptotic meal: the find-me and eat-me signaling pathways. Immunity 35, 445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remold-O’Donnell E, Nixon JC, and Rose RM (1989). Elastase inhibitor. Characterization of the human elastase inhibitor molecule associated with monocytes, macrophages, and neutrophils. J Exp Med 169, 1071–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarff KL, Ung KS, Nandurkar H, Crack PJ, Bird CH, and Bird PI (2004). Targeted disruption of SPI3/Serpinb6 does not result in developmental or growth defects, leukocyte dysfunction, or susceptibility to stroke. Mol Cell Biol 24, 4075–4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarff KL, Ung KS, Sun J, and Bird PI (2003). A retained selection cassette increases reporter gene expression without affecting tissue distribution in SPI3 knockout/GFP knock-in mice. Genesis 36, 149–157. [DOI] [PubMed] [Google Scholar]
- Scott FL, Hirst CE, Sun J, Bird CH, Bottomley SP, and Bird PI (1999). The intracellular serpin proteinase inhibitor 6 is expressed in monocytes and granulocytes and is a potent inhibitor of the azurophilic granule protease, cathepsin G. Blood 93, 2089–2097. [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, and Shao F (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. [DOI] [PubMed] [Google Scholar]
- Taylor RC, Cullen SP, and Martin SJ (2008). Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol 9, 231–241. [DOI] [PubMed] [Google Scholar]
- Thiele DL, and Lipsky PE (1990). Mechanism of L-leucyl-L-leucine methyl ester-mediated killing of cytotoxic lymphocytes: dependence on a lysosomal thiol protease, dipeptidyl peptidase I, that is enriched in these cells. Proc Natl Acad Sci U S A 87, 83–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidwell T, Wechsler J, Nayak RC, Trump L, Salipante SJ, Cheng JC, Donadieu J, Glaubach T, Corey SJ, Grimes HL, et al. (2014). Neutropenia-associated ELANE mutations disrupting translation initiation produce novel neutrophil elastase isoforms. Blood 123, 562–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkalcevic J, Novelli M, Phylactides M, Iredale JP, Segal AW, and Roes J (2000). Impaired immunity and enhanced resistance to endotoxin in the absence of neutrophil elastase and cathepsin G. Immunity 12, 201–210. [DOI] [PubMed] [Google Scholar]
- von Gunten S, Yousefi S, Seitz M, Jakob SM, Schaffner T, Seger R, Takala J, Villiger PM, and Simon HU (2005). Siglec-9 transduces apoptotic and nonapoptotic death signals into neutrophils depending on the proinflammatory cytokine environment. Blood 106, 1423–1431. [DOI] [PubMed] [Google Scholar]
- Wallach D, Kang TB, Dillon CP, and Green DR (2016). Programmed necrosis in inflammation: Toward identification of the effector molecules. Science 352, aaf2154. [DOI] [PubMed] [Google Scholar]
- Wang X, Yousefi S, and Simon HU (2018). Necroptosis and neutrophil-associated disorders. Cell Death Dis 9, 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinrauch Y, Drujan D, Shapiro SD, Weiss J, and Zychlinsky A (2002). Neutrophil elastase targets virulence factors of enterobacteria. Nature 417, 91–94. [DOI] [PubMed] [Google Scholar]
- Wicki S, Gurzeler U, Wei-Lynn Wong W, Jost PJ, Bachmann D, and Kaufmann T (2016). Loss of XIAP facilitates switch to TNFalpha-induced necroptosis in mouse neutrophils. Cell Death Dis 7, e2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanez A, Coetzee SG, Olsson A, Muench DE, Berman BP, Hazelett DJ, Salomonis N, Grimes HL, and Goodridge HS (2017). Granulocyte-Monocyte Progenitors and Monocyte-Dendritic Cell Progenitors Independently Produce Functionally Distinct Monocytes. Immunity 47, 890902 e894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, and Han J (2009). RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325, 332–336. [DOI] [PubMed] [Google Scholar]
- Zhou Q, and Salvesen GS (1997). Activation of procaspase-7 by serine proteases includes a non-canonical specificity. Biochem J 324 ( Pt 2), 361–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.