

Significance Statement
Neutrophil gelatinase–associated lipocalin (NGAL) is produced by injured renal cells and by neutrophils that are central to ANCA-associated vasculitis. The authors show that circulating and urinary NGAL is not only a marker for ANCA-induced necrotizing crescentic GN, but also that neutrophil NGAL is involved mechanistically in ANCA-associated vasculitis. They demonstrate that ANCA-activated neutrophils release NGAL, and that chimeric mice deficient in neutrophil-derived NGAL develop accelerated myeloperoxidase-ANCA–induced crescentic GN, with increased renal CD4+ T cells—particularly T helper 17 (TH17) cells—acting as mediators of the accelerated phenotype. They also demonstrated that iron siderophore–loaded NGAL suppresses TH17 polarization. Their findings indicate that bone marrow–derived NGAL, presumably from neutrophils, protects from ANCA-induced necrotizing and crescentic GN by downregulating TH17 immunity.
Keywords: ANCA glomerulonephritis, ngal, TH17, neutrophil
Visual Abstract

Abstract
Background
Neutrophil gelatinase–associated lipocalin (NGAL) is a diagnostic marker of intrinsic kidney injury produced by damaged renal cells and by neutrophils. ANCA-associated vasculitis features necrotizing crescentic GN (NCGN), and ANCA-activated neutrophils contribute to NCGN. Whether NGAL plays a mechanistic role in ANCA-associated vasculitis is unknown.
Methods
We measured NGAL in patients with ANCA-associated vasculitis and mice with anti-myeloperoxidase (anti-MPO) antibody–induced NCGN. We compared kidney histology, neutrophil functions, T cell proliferation and polarization, renal infiltrating cells, and cytokines in wild-type and NGAL-deficient chimeric mice with anti-MPO antibody–induced NCGN. To assess the role of TH17 immunity, we transplanted irradiated MPO-immunized MPO-deficient mice with bone marrow from either wild-type or NGAL-deficient mice; we also transplanted irradiated MPO-immunized MPO/IL-17A double-deficient mice with bone marrow from either IL-17A–deficient or NGAL/IL-17A double-deficient mice.
Results
Mice and patients with active ANCA-associated vasculitis demonstrated strongly increased serum and urinary NGAL levels. ANCA-stimulated neutrophils released NGAL. Mice with NGAL-deficient bone marrow developed worsened MPO-ANCA–induced NCGN. Intrinsic neutrophil functions were similar in NGAL-deficient and wild-type neutrophils, whereas T cell immunity was increased in chimeric mice with NGAL-deficient neutrophils with more renal infiltrating TH17 cells. NGAL-expressing neutrophils and CD3+ T cells were in close proximity in kidney and spleen. CD4+ T cells showed no intrinsic difference in proliferation and polarization in vitro, whereas iron siderophore–loaded NGAL suppressed TH17 polarization. We found significantly attenuated NCGN in IL-17A–deficient chimeras compared with MPO-deficient mice receiving wild-type bone marrow, as well as in NGAL/IL-17A–deficient chimeras compared with NGAL-deficient chimeras.
Conclusions
Our findings support that bone marrow–derived, presumably neutrophil, NGAL protects from ANCA-induced NCGN by downregulating TH17 immunity.
ANCAs to either myeloperoxidase (MPO) or proteinase 3 (PR3) are found in patients with small-vessel vasculitis.1,2 ANCA-associated vasculitis (AAV) is a group of systemic autoimmune diseases characterized by necrotizing vasculitis with organ damage, including necrotizing and crescentic GN (NCGN). ANCA binding to their respective antigens, exclusively expressed by neutrophils and monocytes, activates these myeloid cells that in turn damage the vasculature.3 Neutrophil serine proteases, chemokines, reactive oxygen species (ROS), complement, and extracellular DNA traps are examples of inflammatory mediators, which act in concert to promote endothelial injury.4−7 ANCA-activated neutrophils and monocytes interact with each other and with adaptive immune cells. However, these interactions need further characterization.
Neutrophil gelatinase–associated lipocalin (NGAL), also known as lipocalin-2 (LCN2), siderocalin, or 24p3 is a member of the lipocalin family that consists of several secreted proteins functioning as carriers for small hydrophobic molecules. NGAL is a 25-kDa glycoprotein that was first purified from, and therefore named after, neutrophils. It is stored in mature granules and released during cell activation.8 Originally, NGAL was thought to inhibit bacterial growth by sequestering iron.9,10 However, NGAL was subsequently shown to be expressed by other cell types where it controls several physiologic cellular functions, including apoptosis, differentiation, migration, and proliferation.11
Over the last decade, NGAL has received ample attention from nephrologists because it is rapidly induced and released by damaged kidney cells, particularly in the thick ascending limb of the loop of Henle and collecting duct.12 Several clinical and animal studies established NGAL as a noninvasive, early biomarker for AKI. For example, we showed that NGAL allows diagnostic and prognostic stratification in the emergency department and helps to distinguish prerenal from intrinsic AKI.13 Whether NGAL is more than a diagnostic marker and participates mechanistically in kidney damage is not clear. Only a few mechanistic renal studies have been performed. One study showed protective NGAL effects in urinary tract infections.14 Conflicting results were reported for murine nephrotoxic nephritis with either protective15 or deleterious effects.16 More recently, recombinant NGAL provided protection in a mouse model of kidney transplantation.17
We studied NGAL in ANCA-induced NCGN using patient material and a murine MPO-ANCA disease model. We generated NGAL−/− chimeric mice and NGAL−/−/IL-17A−/− double-deficient mice and focused specifically on neutrophil-derived NGAL because neutrophil is an NGAL source and ANCA-activated neutrophils cause vascular inflammation, leading to vasculitis and NCGN. Our data indicate that bone marrow (BM) cell–derived NGAL is mechanistically involved in ANCA-induced NCGN and provides renoprotection by downregulating the T cell response, particularly T helper 17 (TH17) cells.
Methods
Materials
Complete and incomplete Freund Adjuvants, liberase, DNAse, FBS, zymosan, SOD, cytochalasin B, TNF-α, N-formyl-methionyl-leucyl-phenylalanine (Fmlp), and PMA were from Sigma-Aldrich (Steinheim, Germany); urine dipsticks were from Roche Diagnostics (Indianapolis, IN); and RPMI 1640 medium and PBS without magnesium and calcium ions were from Biochrom GmbH (Berlin, Germany). The ovalbumin (ova) peptide323–339 (ISQAVHAAHAEINEAGR) was produced by Rudolf Volkmer-Engert in house.
Human Samples
Urine and serum were collected from patients with either active AAV or in remission, non-ANCA patients with AKI, patients with SLE, and from healthy controls (Supplemental Table 1). All samples were stored at −80°C. Collection and analysis of human samples were approved by the local ethics committee (EA3/011/08, EA3/011/06, EA2/103/17, and EA1/034/10).
Neutrophil and IgG Isolation
Blood from healthy human donors was obtained after approval by Charité and after written informed consent was obtained. Neutrophils were isolated by density gradient centrifugation as described previously.5 Human neutrophil preparations contained <10% other cells, such as basophils and eosinophils, and we use the term neutrophils throughout the manuscript. Isolation of murine neutrophils from the BM of wild-type (WT) and NGAL−/− mice was performed as previously described.18 IgG from murine and human serum was isolated as described previously.5
Murine Neutrophil Functions
Degranulation
Isolated neutrophils were primed for 15 minutes with 5 ng/ml murine TNF-α and further stimulated for 1 hour with either 150 μg/ml polyclonal anti-BSA or anti-MPO antibodies. Degranulation was assessed by measuring PR3-specific proteolytic activity in cell supernatants using the FRET substrate 2-Abz-VAD-norV-ADYQ-EDA-Dnp (Biosynthan, Berlin, Germany) prepared as described previously.19 In brief, 100 μl of neutrophil supernatants were incubated with FRET substrate (20 μM) and fluorescence (excitation 320 nm, emission 420 nm) was recorded over 45 minutes using a SpectraMax M5 fluorescence plate reader (Molecular Devices, Sunnyvale, CA). The corresponding Vmax for each condition was reported.
ROS Production
Extracellular ROS production was measured using the ferricytochrome C reduction assay as described before.20 Neutrophils were pretreated with 5 mg/ml cytochalasin B for 15 minutes. Cells (0.75×106) were then pretreated with SOD (300 U/ml) for 15 minutes and primed with 5 ng/ml TNF-α for 15 minutes before activation with either 150 μg/ml antibody (anti-BSA or anti-MPO), 25 ng/ml PMA, or 100 µg/ml zymosan. Samples were incubated in 96-well plates at 37°C and the absorption of samples was scanned repetitively at 550 nm using a Microplate Autoreader (Molecular Devices, Munich, Germany). Results are reported as nmol superoxide/0.75×106 neutrophils per 45 min.
Intracellular ROS production was measured using the dihydrorhodamine oxidation assay. Prewarmed neutrophils were loaded with 1 μM rhodamine 123 (Sigma-Aldrich) for 10 minutes at 37°C. Cells were then primed and activated with stimuli described for extracellular ROS production. After 30 minutes, the reaction was stopped with ice-cold PBS/1% BSA and samples were analyzed using a FACS Calibur (Becton Dickinson, Heidelberg, Germany). The mean fluorescent intensity is reported.
Neutrophil Extracellular Trap Formation
Isolated neutrophils were stained with Sytox green (Invitrogen), primed for 15 minutes with 5 ng/ml murine TNF-α, and stimulated with either 150 μg/ml anti-BSA or polyclonal anti-MPO antibodies. After 3 hours, neutrophil extracellular traps (NETs) were observed by microscopy.
Phagocytosis
Isolated neutrophils were incubated for 30 minutes with 1 μm fluorescent yellow-green beads (Sigma-Aldrich) and phagocytosis (FITC+ neutrophils) was assessed by flow cytometry.
Apoptosis
Isolated neutrophils were incubated in RPMI 1640 medium/10% FBS overnight and apoptosis was determined using Annexin V labeling and flow cytometry.
Migration
Migration was tested in fibronectin-coated transwells (3 μm pore size, 6.5 mm diameter; Corning, Amsterdam, Netherlands). Isolated neutrophils were seeded at the density of 1.5×106 and stimulated with either 50 ng/ml colony-stimulating factor 2 or 400 ng/ml CXCL1 (R&D Systems) applied to the lower well. After 3 hours, the total number of transmigrated cells was quantified by MPO assay as described previously.19
Animal Experiments
Mice were kept under specific pathogen-free conditions at the Max Delbrück Center for Molecular Medicine (Berlin, Germany) animal facility. MPO−/− and MPO−/−/IL-17A−/− (generated by crossing MPO−/− mice with IL-17A−/− mice) mice were immunized intraperitoneally with murine MPO in CFA, boosted intraperitoneally after 4 weeks with murine MPO in incomplete Freund's adjuvant, sublethally irradiated, and subsequently transplanted intravenously with BM cells (1.5×107) from either C57/Bl6 WT (The Jackson Laboratory) or NGAL−/− (kindly provided by Dr. Akira, Osaka University, Osaka, Japan) mice.9 MPO−/−/IL-17A−/− double-deficient mice were transplanted with BM cells from IL-17A−/− mice or with NGAL−/−/IL-17A−/− double-deficient mice that were generated by crossing NGAL−/− and IL-17A−/− mice. All mice were on Bl6 background. Purification of murine MPO from WEHI-3 cells, immunization, and irradiation protocols for MPO−/− were performed as previously described.21 Animal experiments were approved by the local authorities (Landesamt für Gesundheit und Soziales, Berlin, Germany) and followed the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines.22
Histologic Examination of Renal Damage
Renal tissues were collected at the time of euthanasia in cold PBS, fixed in 10% formalin overnight, and embedded in paraffin using routine procedures. Sections were stained with Periodic acid–Schiff and all glomeruli on each kidney section assessed by light microscopy using an Axio Imager M2 microscope (Zeiss, Jena, Germany). Glomerular crescents and necrosis were expressed as the mean percentage of glomeruli with crescents and necrosis in each animal and scored in a blinded manner. When indicated, paraffin sections were stained with polyclonal antibody (rabbit) directed again human CD3 (Dako, Glostrup, Denmark) in combination with the EnVision+System-HRP Kit (Dako).
Functional Measurement of Renal Damage
Mice were transferred to metabolic cages with free water and food access 1 day before they were euthanized. Urine was collected over 16 hours. Urine was screened by dipstick for proteinuria, erythrocyturia, and leukocyturia. Results were expressed on a scale of zero (none) to four (severe) for erythrocyturia, and of zero to three for proteinuria and leukocyturia. Urine albumin concentration was determined by ELISA (Bethyl Laboratories, Inc., Montgomery, TX).
Isolation of Renal and Splenic Leukocytes
Previously described methods for the isolation of murine renal leukocytes were used.23 In brief, kidney biopsies were minced with scissors and digested for 50 minutes at 37°C with 0.2 mg/ml liberase and 100 U/ml DNAse in PBS without magnesium and calcium ions (PBS). Cell suspensions were washed with PBS, filtered through 70-μm meshes, resuspended in PBS/1% FBS, and assessed by flow cytometry. Spleens were collected, minced, and filtered through 70-μm meshes. Erythrocytes were lysed using the Red Blood Cell Lysing Buffer Hybri-Max (Sigma). After washing, splenic cells were resuspended in buffer for further experiments.
Flow Cytometry
The following fluorochrome-conjugated antibodies were used to identify renal myeloid cells by flow cytometry: CD45 (30F11; BD Bioscience, Heidelberg, Germany); and CD11b (M1/70), Ly6C (HK1.4), and Ly6G (1A8), all from Biolegend (San Diego, CA). Anti-mouse CD16/32 antibody (Biolegend) was used to block Fc receptors. Dead cells were excluded using the LIVE/DEAD Fixable Near-IR Dead Cell Stain Kit (Thermo Fisher Scientific, Rockford, IL).
To identify T cells and TH subsets, renal cell suspensions were stimulated for 4 hours with phorbol 12-myristate 13-acetate (50 ng/ml; Sigma) and ionomycin (1 μg/ml; Calbiochem-Merck, Darmstadt, Germany) in the presence of 1× Brefeldin A (Biolegend) in X-VIVO10 medium (Biozym, Hessisch Oldendorf, Germany). After washing, cells were stained with the following antibodies: anti-CD45 (30F11) from BD Biosciences, and anti-CD3 (17A2), anti-CD4 (GK1.5), and anti-CD8 (53-6-7) from Biolegend. After fixation with 4% paraformaldehyde, cells were permeabilized with 0.1% nonidet P-40 (NP-40) for 20 minutes and intracellular staining was performed with anti–IL-17A (TC11-18H10.1) and anti-IFNγ (XMG12) from Biolegend.
All samples were acquired using an FACS CANTO II flow cytometer with BD FACSDiva software version 6.1.2 (BD Biosciences). Data were analyzed using FlowJo software version 10 (Treestar, Ashland, OR).
CD4+ T Cell Proliferation and Polarization
CD4+ T cells were isolated from spleens of WT and NGAL−/− mice using the CD4+ T Cell Isolation Kit (Miltenyi Biotec, Bergisch Gladbach, Germany), following the manufacturer’s instructions. For proliferation assays, isolated CD4+ T cells or splenocytes (all immune cells) were stained with carboxyfluorescein succinimidyl ester (Thermo Fisher Scientific), washed, and seeded in a 96-well plate (2.5×105 cells/well) with Dynabeads Mouse T-Activator CD3/CD28 (Thermo Fisher Scientific) at a bead/cell ratio of 1:1. Murine recombinant NGAL (Biolegend) or iron siderophore–loaded NGAL was added as indicated. After 3 days in culture, cells were labeled with anti-CD3 and anti-CD4 antibodies and cell proliferation was analyzed by flow cytometry. For polarization assays, isolated CD4+ T cells or splenocytes were seeded in a 96-well plate (2.5×105 cells/well) with Dynabeads Mouse T-Activator CD3/CD28 (Thermo Fisher Scientific) at a bead/cell ratio of 1:1. Cells were cultured in RPMI 1640 medium with 10% FBS supplemented with cytokines (IL-1β, IL-6, IL-23, TGFβ) and antibodies (anti-IFNγ, anti–IL-2 and anti–IL-4) from the CytoBox TH17 Kit (Miltenyi Biotec) following the manufacturer’s instructions. After 7 days in culture, cells were labeled with anti-CD45, anti-CD3, anti-CD4, anti–IL-17A, and anti-IFNγ antibodies and TH cell subsets were analyzed by flow cytometry.
Antigen-Specific Proliferation and Polarization Assay
For antigen-specific assays, naive CD4+ T cells were isolated from the spleen of OT-II mice with the CD4+ T Cell Isolation Kit (Miltenyi Biotec) and anti-CD62L microbeads (Miltenyi Biotec). Antigen-presenting cells (APCs) were isolated from spleens of WT and NGAL−/− mice by depleting T cells with anti-CD90 beads and LS columns from Miltenyi Biotech. For cell proliferation, naive carboxyfluorescein succinimidyl ester– labeled CD4+ T cells and APCs were cocultured at a cell ratio of 1:4 in absence or presence of either 0.6 or 0.12 μM ova peptide323–339 (ISQAVHAAHAEINEAGR) in RPMI 1640 medium. After 3 days in culture, proliferation of CD4+ T cells was assessed by flow cytometry. For the polarization assay, unlabeled naive CD4+ T cells and APCs were cocultured at a cell ratio of 1:4 in absence or presence of 0.6 μM ova peptide323–339 in RPMI 1640 medium with 10% FBS supplemented with polarization cytokines and antibodies as above. After 7 days in culture, TH17 polarization was assessed by flow cytometry.
Quantitative RT-PCR
Whole-kidney RNA was extracted with the RNeasy Mini Kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions and reverse transcription was performed with The SuperScript III One-Step RT-PCR System (Thermo Fisher Scientific). Renal Il-1β and CCL20 gene expression was determined by quantitative RT-PCR using the QuantStudio 3 instrument (Thermo Fisher Scientific) and primers synthetized by BioTeZ Berlin-Buch GmbH (Berlin, Germany).
ELISA
Renal tissues were dissociated in Precellys Ceramic-Kit 1.4/2.8 mm (VWR, Darmstadt, Germany) with 500 μl buffer containing 50 mM Tris/hydrochloride, pH 7.5, 150 mM sodium chloride, 200 mM PMSF, 1% NP-40, 0.1% SDS, 0.5% deoxycholate, and supplemented with complete protease (Roche) and phosphatase (Roche) inhibitor cocktails. Tissue lysates were centrifuged and supernatants were collected and stored at −80°C. Mouse IL-1β/IL-1F2 Quantikine ELISA and mouse CCL20/MIP-3α DuoSet ELISA Kits (both R&D Systems) were used to quantify IL-1β and CCL20 levels in these renal extracts. Mouse IL-17 and mouse IFNγ Quantikine ELISA Kits (R&D Systems) were used to quantify IL-17A and IFNγ levels in supernatants from splenocytes stimulated with phorbol 12-myristate 13-acetate (50 ng/ml) and ionomycin (1 μg/ml) for 4 hours in the presence of 1× Brefeldin A. NGAL concentration in murine and human serum and urine as well as in supernatants from stimulated neutrophils was determined with the NGAL (LCN2) ELISA Kit from Dianova (Hamburg, Germany). For the neutrophil stimulation experiments, 1×106 cells were treated with the indicated stimuli.
Immunoblotting
Monocytes and neutrophils were sorted from human blood and murine BM by flow cytometry. Spleens from WT and NGAL−/− mice were collected and CD4+ T cells were isolated as described above and cultured with or without Dynabeads Mouse T-Activator CD3/CD28 (Thermo Fisher Scientific) at a bead/cell ratio of 1:1 for 3 days. Cells and organs were lysed in buffer containing 50 mM Tris/hydrochloride, pH 7.5, 150 mM sodium chloride, 200 mM PMSF, 1% NP-40, 0.1% SDS, 0.5% deoxycholate, and supplemented with complete protease and phosphatase inhibitor cocktails. Protein lysates were resolved by SDS-PAGE and transferred onto nitrocellulose membranes. Membranes were probed with the following antibodies: mouse anti-human NGAL (Dianova), rabbit anti–β-actin (Cell Signaling Technology, Frankfurt am Main, Germany), and rat anti-mouse LCN2/NGAL (R&D Systems). Horseradish peroxidase–labeled donkey anti-rabbit IgG (GE Healthcare, Little Chalfont, United Kingdom), rabbit anti-mouse IgG (Dako), and rabbit anti-rat IgG (Dako) were used as secondary antibodies. Thermo Scientific SuperSignal West Dura Chemiluminescent Substrate (Thermo Fisher Scientific) was used for detection.
Preparation of Recombinant NGAL
Murine NGAL was expressed, purified, and complexed with ferric enterobactin as previously described.17 Briefly, murine NGAL without the signal peptide (NP_032517) was expressed and purified as a glutathione S-transferase fusion protein in Escherichia coli BL21 according to manufacturer’s instructions (GE Healthcare, Vienna, Austria). Following cleavage with thrombin protease, NGAL was purified to >99% purity by chromatography on CIM-SO3 (BIA Separations, Ljubljana, Slovenia). To produce the recombinant NGAL-siderophore-iron complex (iron siderophore–loaded NGAL) the recombinant NGAL protein was incubated for 4 hours at 4°C with an equimolar amount of ferric enterobactin (EMC Microcollections, Tübingen, Germany).
Immunofluorescence Staining
Tissue was dissected into blocks of 1–3 μm thickness, dehydrated by graded ethanol series into xylene, and embedded in paraffin. Tissue blocks were sectioned at 4 μm. Sections were deparaffinized in xylene and rehydrated in a graded ethanol series. Heat-induced epitope retrieval was performed for 6 minutes in citrate buffer (pH 6) in a pressure cooker. Skim milk (5%) in PBS was used as a blocking buffer for 30 minutes at room temperature. Sections were incubated overnight at 4°C in 5% skim milk/PBS containing primary antibodies (goat anti-NGAL and rabbit anti-CD3). Subsequently, sections were fluorescence labeled with secondary antibodies (anti-goat and anti-rabbit; Jackson ImmunoResearch) for 1 hour at room temperature. Samples were mounted in 1:9 PBS/glycerol and examined in a fluorescence microscope. Images were acquired using a Zeiss Axio Imager Z2 equipped with ×20/numeric aperture 1.3 and ×63/ numeric aperture 1.4 objectives. The sections were illuminated at 488 nm and 543 nm wavelengths to excite green and red fluorescence dyes, respectively.
Statistical Analysis
Results are given as means±SEM. Comparisons were made using ANOVA with post hoc analysis by GraphPad Prism5 software. Differences were considered significant at P<0.05.
Results
Serum and Urine NGAL Is Increased in Patients with Active AAV and in a Murine AAV Disease Model
Damaged nephrons strongly upregulate NGAL, providing an established marker for intrinsic kidney damage. We measured serum and urine NGAL in patients with active and inactive AAV and controls. Patients with active AAV displayed strongly upregulated serum and urine NGAL levels compared with patients in remission, to healthy controls, and to patients with lupus nephritis (Supplemental Figure 1A). Increased NGAL levels in active AAV were similar to those found in patients with non-AAV–related AKI. In addition, we tested serum and urine from mice with experimental anti-MPO antibody–induced NCGN. We observed significantly increased serum and urine NGAL, indicating that our murine AAV model reflects the human NGAL kinetics (Figure 1A).
Figure 1.
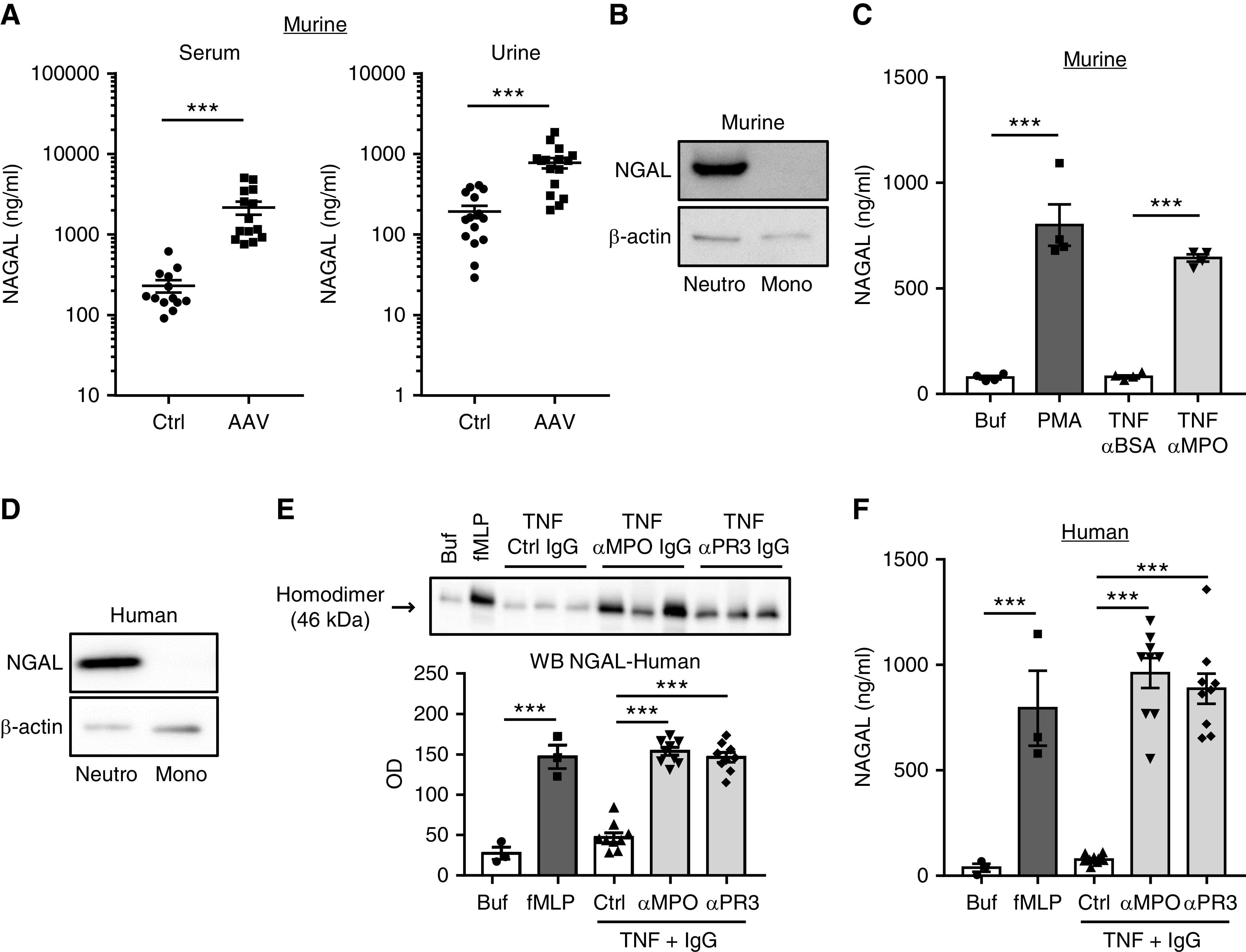
NGAL is increased in mice with ANCA-induced GN and is released by ANCA-activated murine and human neutrophils. (A) Circulating and urinary NGAL levels detected by ELISA were increased in mice with anti-MPO–induced NCGN (AAV) compared with control mice (Ctrl). (B) Representative immunoblots show strong NGAL protein expression in murine neutrophils (Neutro), but not in monocytes (Mono) that were isolated by cell sorting. Actin served as loading control. (C) NGAL protein by ELISA is increased in supernatants of murine neutrophils primed with TNFα and stimulated with 125 µg/ml anti-MPO IgG. Stimulation with murine anti-BSA IgG and 25 ng/ml PMA, respectively, served as controls. (D) Representative immunoblots show strong NGAL protein expression in human neutrophils, but not in monocytes that were purified by cell sorting. Actin served as loading control. (E) TNFα-primed human neutrophils were stimulated with three different human MPO-ANCA, PR3-ANCA, or control IgG preparations (125 µg/ml), respectively. Treatment with either 10−6 fMLP or buffer (Buf) served as controls. ANCA caused a strong release of neutrophil-specific NGAL 46-kDa homodimers into the supernatant by immunoblotting. A representative immunoblotting experiment is depicted together with the corresponding statistics of all experiments. (F) NGAL by ELISA is increased in supernatants of ANCA-stimulated human neutrophils treated as in (E). ***P>0.001.
Neutrophils provide a nonrenal NGAL source that may contribute to increased NGAL levels in active AAV. Because ANCA activate both neutrophils and monocytes, we analyzed NGAL in both cell types separately. Immunoblotting indicates that human and murine neutrophils, but not monocytes, harbored NGAL protein (Figure 1, B and D). Next, we tested whether murine BM neutrophils released NGAL in response to murine polyclonal anti-MPO IgG and found a strong NGAL release that was not observed with control anti-BSA IgG (Figure 1C). Similarly, human blood neutrophils released NGAL from intracellular granules into the culture medium after stimulation with PR3- or MPO-ANCA. Immunoblotting detected the 46-kDa homodimer that is neutrophil specific and was used for quantification (Figure 1E).24 The data were independently confirmed by ELISA (Figure 1F). In contrast, IgG from patients with lupus did not stimulate NGAL release (Supplemental Figure 1B).
Collectively, these data demonstrated increased NGAL in blood and urine of patients with active AAV and mice with ANCA-induced NCGN and that neutrophils release NGAL when activated by ANCA. Monocytes do not express NGAL, indicating neutrophils are the major NGAL source within the BM-derived myeloid cell compartment. Whether neutrophil-derived NGAL plays any causative role in NCGN is not clear.
ANCA-Induced GN Is Aggravated in NGAL−/− Chimeric Mice
Next, we used our murine MPO-ANCA mouse model21 to test the hypothesis that neutrophil-derived NGAL contributes to NCGN. MPO-deficient mice were immunized with murine MPO, irradiated, and transplanted with BM cells from either WT or NGAL-deficient (NGAL−/−) animals to generate MPO−/−/WT mice and MPO−/−/NGAL−/− chimeric mice, respectively. At 8 weeks after transplantation, mice were euthanized and urine, serum, and kidneys were analyzed (Figure 2A). Microscopy showed that mice from both groups developed NCGN. However, quantification indicated that NGAL−/− chimeric mice developed significantly more glomerular crescents and necrotic lesions compared with WT mice (Figure 2, B and C). In line with these histologic findings, we found aggravated proteinuria and erythrocyturia by dipstick analysis and albuminuria by ELISA in NGAL−/− chimeric mice (Figure 2, D and E). Interestingly, serum and urine NGAL levels were similar in both groups, suggesting the quantitative contribution of activated neutrophils to the NGAL increase was rather small (Figure 2F). Nevertheless, our results indicate that neutrophil-derived NGAL provided renoprotection, because mice with NGAL-deficient BM developed more severe NCGN. No difference in anti-MPO titers was detected between the two groups, indicating humoral immunity was not affected by NGAL (Figure 2G).
Figure 2.
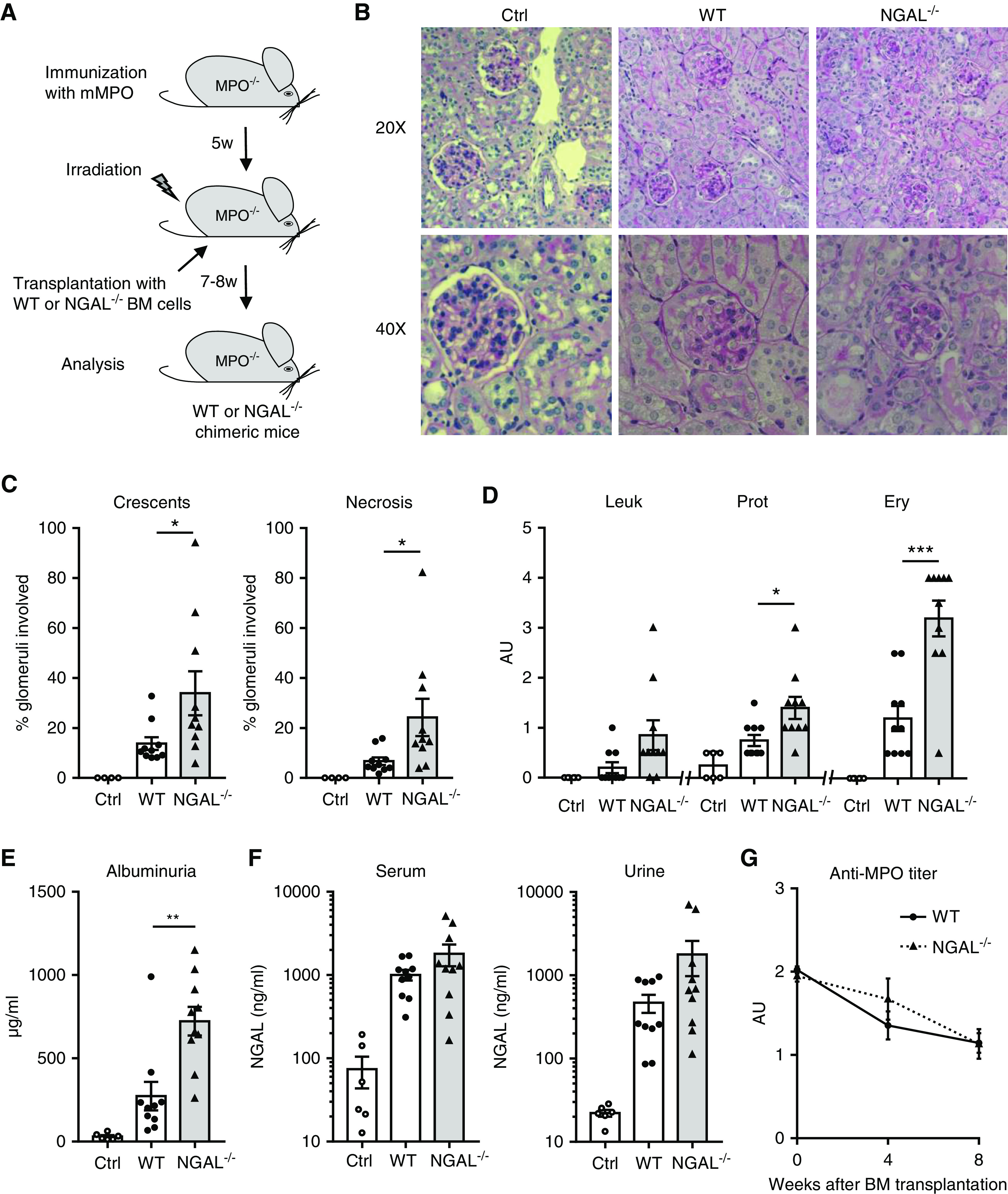
NGAL−/− chimeric mice develop more severe anti-MPO–induced NCGN, but show similar NGAL levels in serum and urine compared with WT mice. (A) Experimental scheme depicting the induction of NCGN in MPO-deficient (MPO−/−) mice immunized with murine MPO (mMPO) that were subsequently irradiated and transplanted with either WT or NGAL-deficient (NGAL−/−) BM cells. All chimeric mice were analyzed 7–8 weeks after transplantation. (B) NGAL−/− chimeric mice developed more severe NCGN than WT chimeric mice with (C) higher percentage of crescents and necrosis. Shown are representative images of kidney sections stained with Periodic acid–Schiff at low (20×) and high (40×) magnification and quantitative analyses of glomeruli with crescents and necrosis. (D) Urine analysis reveals more erythrocyturia (Ery) and proteinuria (Prot) in NGAL−/− chimeric mice by dipstick and (E) higher albuminuria by ELISA, whereas (F) serum and urinary NGAL levels were similar in WT and NGAL−/− chimeric mice by ELISA. (G) Anti-MPO titer determined by ELISA was similar in WT and NGAL−/− chimeric mice. Ctrl, control; Leuk, leukocyturia. *P<0.05; **P<0.01; ***P<0.001.
NGAL-Deficient Neutrophils Do Not Differ in Several Functional Responses
The importance of neutrophils for anti-MPO antibody–induced NCGN was shown previously.25 Thus, a potential explanation for the accelerated disease phenotype was that NGAL functions as a brake for neutrophil responses. We studied neutrophil functions from NGAL−/− and WT mice and observed no difference in intra- and extracellular ROS generation to anti-MPO IgG (Figure 3, A and B), spontaneous apoptosis (Figure 3C), phagocytosis (Figure 3D), chemotactic migration toward colony-stimulating factor 2 and CXCL1 (Figure 3E), and anti-MPO IgG-induced NET generation (Figure 3F), respectively. The only difference was a slightly increased PR3 degranulation response to anti-MPO IgG in NGAL−/− neutrophils (Figure 3G). Moreover, white blood cell numbers and leukocyte subgroups did not differ between unchallenged WT and NGAL−/− mice (Supplemental Figure 2). These data suggest that neutrophil NGAL does not protect from ANCA-induced NCGN by downregulating intrinsic neutrophil effector functions but rather by controlling effector cells other than neutrophils.
Figure 3.
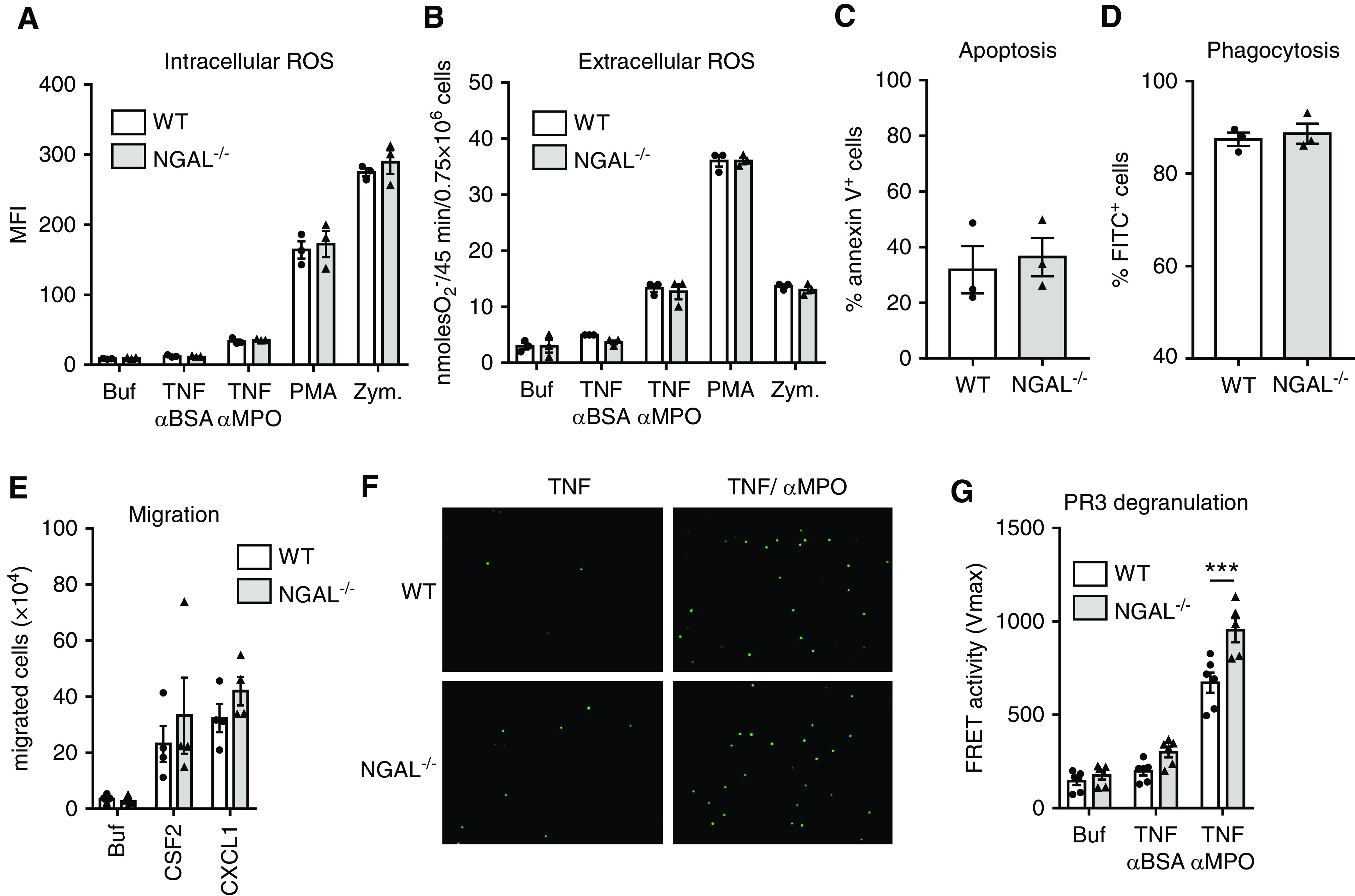
ANCA-related neutrophil functions are similar in NGAL−/− and WT neutrophils. (A and B) Murine WT or NGAL−/− neutrophils were isolated from the BM and stimulated with buffer (Buf), anti-BSA IgG, anti-MPO IgG, PMA, or zymosan (Zym). (A) Intracellular ROS generation was measured by dihydrorhodamine 123 staining and flow cytometry. (B) Extracellular ROS generation was detected by ferricytochrome reduction assay. (C) Neutrophils were incubated overnight in culture medium and spontaneous apoptosis was measured using Annexin V staining and flow cytometry. (D) Neutrophils were incubated with FITC-labeled particles and phagocytosis (FITC+ neutrophils) was analyzed by flow cytometry. (E) Chemotactic migration toward colony-stimulating factor 2 (CSF2, 50 ng/ml) and CXCL1 (400 ng/ml) was assessed. (F) Neutrophils were stimulated with either TNF or TNF/anti-MPO IgG and NET generation was detected with Sytox green staining. Representative images of three independent experiments are depicted at low magnification (20×). (G) Degranulation was measured by PR3 FRET assay in culture supernatants of WT and NGAL−/− neutrophils stimulated with TNF/anti-MPO IgG. Results are reported as Vmax. Buffer and anti-BSA IgG were used as controls. MFI, mean fluorescence intensity; O2−, superoxide. ***P<0.001.
NGAL−/− Chimeric Mice Show Differences in CD4+ T Cell Responses that Predominantly Affect TH17 Cells
To identify the protective mediators used by BM cell–derived NGAL, we analyzed kidney-infiltrating inflammatory cells by flow cytometry. In agreement with the in vitro neutrophil migration data above, we observed similar renal neutrophil and classic monocyte influx in NGAL−/− chimeric and WT mice (Figure 4A). In contrast, we detected strongly increased renal CD4+ T cells, mainly of the TH1 subtype, and an even more pronounced increase of the TH17 subtype (Figure 4B). Using microscopy, the strongest T cell infiltrates were observed in the glomerular and periglomerular compartment of the NGAL−/− chimeric mice (Figure 4C). When we analyzed kidneys from the anti-MPO antibody–challenged mice for CCL20 and IL1β, we found that CCL20, an important chemokine that attracts TH17 cells, was increased in kidneys from NGAL−/− chimeric mice (Figure 4D).
Figure 4.
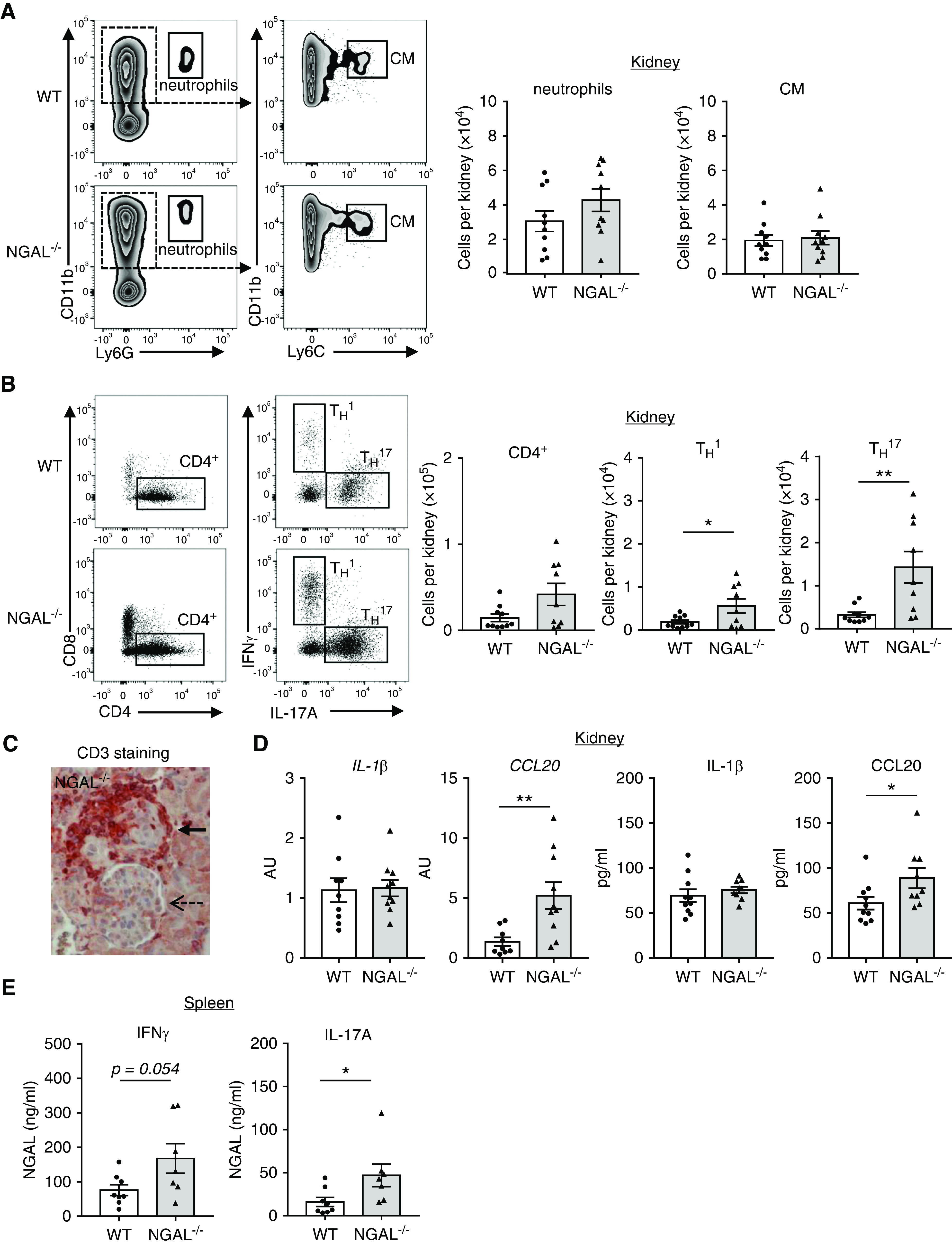
NGAL−/− chimeric mice show similar renal myeloid cell, but stronger TH17 cell influx and increased renal CCL20 expression. MPO−/− mice immunized with murine MPO were irradiated and transplanted with either WT or NGAL−/− BM cells as described in Figure 2A. All chimeric mice were analyzed 7–8 weeks after transplantation. (A) Representative FACS plot with gating strategy and corresponding quantification of renal CD45+CD11b+Ly6G+ neutrophils (neutro) and CD45+CD11b+Ly6Chi classic monocytes (CM). (B) Representative FACS plots showing IL-17A and IFNγ expression by renal CD4+ T cells after PMA/ionomycin stimulation ex vivo. Quantification reveals increased renal total CD4+, CD4+ TH1 (IL-17A−IFNγ+) and CD4+ TH17 (IL-17A+IFNγ−) T cells in NGAL−/− compared with WT chimeric mice. (C) Immunohistochemistry of kidney sections stained for the T cell marker CD3 shows that most of the CD3+ T cells were detected within and around affected glomeruli (bold arrow), but not in nonaffected glomeruli (dotted arrow). (D) Quantification of renal CCL20 and IL1β mRNA by quantitative PCR with HPRT used as reference gene and renal CCL20 and IL1β protein by ELISA. Both renal CCL20 mRNA and protein were increased in mice transplanted with NGAL−/− BM. (E) Splenic cells from WT and NGAL−/− chimeric mice were stimulated with PMA/ionomycin ex vivo and IFNγ and IL-17A was determined in the supernatants by ELISA. Cells isolated from mice transplanted with NGAL−/− BM produced more IFNγ and IL-17A compared with WT BM-transplanted mice. *P<0.05; **P<0.01.
The spleen provides an immune organ with complex interactions between T cells, APCs, and neutrophils. Therefore, we studied splenocytes from anti-MPO antibody–challenged mice and found that cells from NGAL−/− chimeric mice produced significantly more IL-17A protein compared with cells from WT mice (Figure 4E). Together, these findings support the notion that BM cell–derived NGAL, most likely neutrophil NGAL, downregulates CD4+ T cells, particularly TH17 cells. Whether this NGAL effect is direct or indirect is unclear.
Additional staining demonstrated close proximity of NGAL and CD3+ T cells in both kidneys and spleens of mice with anti-MPO–induced vasculitis, making intimate interactions possible (Figure 5, A and B).
Figure 5.

NGAL localize close to T cells in kidney and spleen of mice with AAV. Immunofluorescence reveals the close proximity of NGAL (green) and T cells (CD3; red) in kidneys glomerulus and spleen pulpa from mice with anti-MPO induced NCGN. Boxed areas in 20× objective magnification panels were sequentially enlarged to 63× objective magnification. Bars indicate magnification.
Iron Siderophore–Loaded NGAL Controls CD4+ T Cell Polarization toward TH17 T Cells in Splenocyte Culture
We observed that neither resting nor CD3/CD28 antibody–treated CD4+ T cells express NGAL, but encounter NGAL in the spleen, most likely from neutrophils (Figure 6A). Because CD4+ T cells were shown to express the 24p3 NGAL receptor,26 we treated WT and NGAL−/− CD4+ T cells with recombinant NGAL. Flow cytometry showed that the proliferation rate of CD3/CD28 antibody–treated CD4+ T cells from WT and NGAL−/− chimeric mice (Figure 6B) and the polarization toward TH17 cells (Figure 6C) were similar both in the absence and presence of recombinant NGAL. CD4+ T cells in splenocyte cultures did not display a difference either (Figure 6D). However, these experiments cannot exclude differences in antigen-specific CD4+ T cell proliferation and TH17 polarization. Such studies are not feasible with MPO as an antigen. Therefore, we used OT-II transgenic mice that express an OVA-specific T cell receptor together with the CD4 coreceptor instead. We detected similar antigen-specific CD4+ T cell proliferation (Supplemental Figure 3A) and TH17 polarization (Supplemental Figure 3B), independently of whether these T cells were coincubated with APCs from WT or from NGAL−/− mice.
Figure 6.
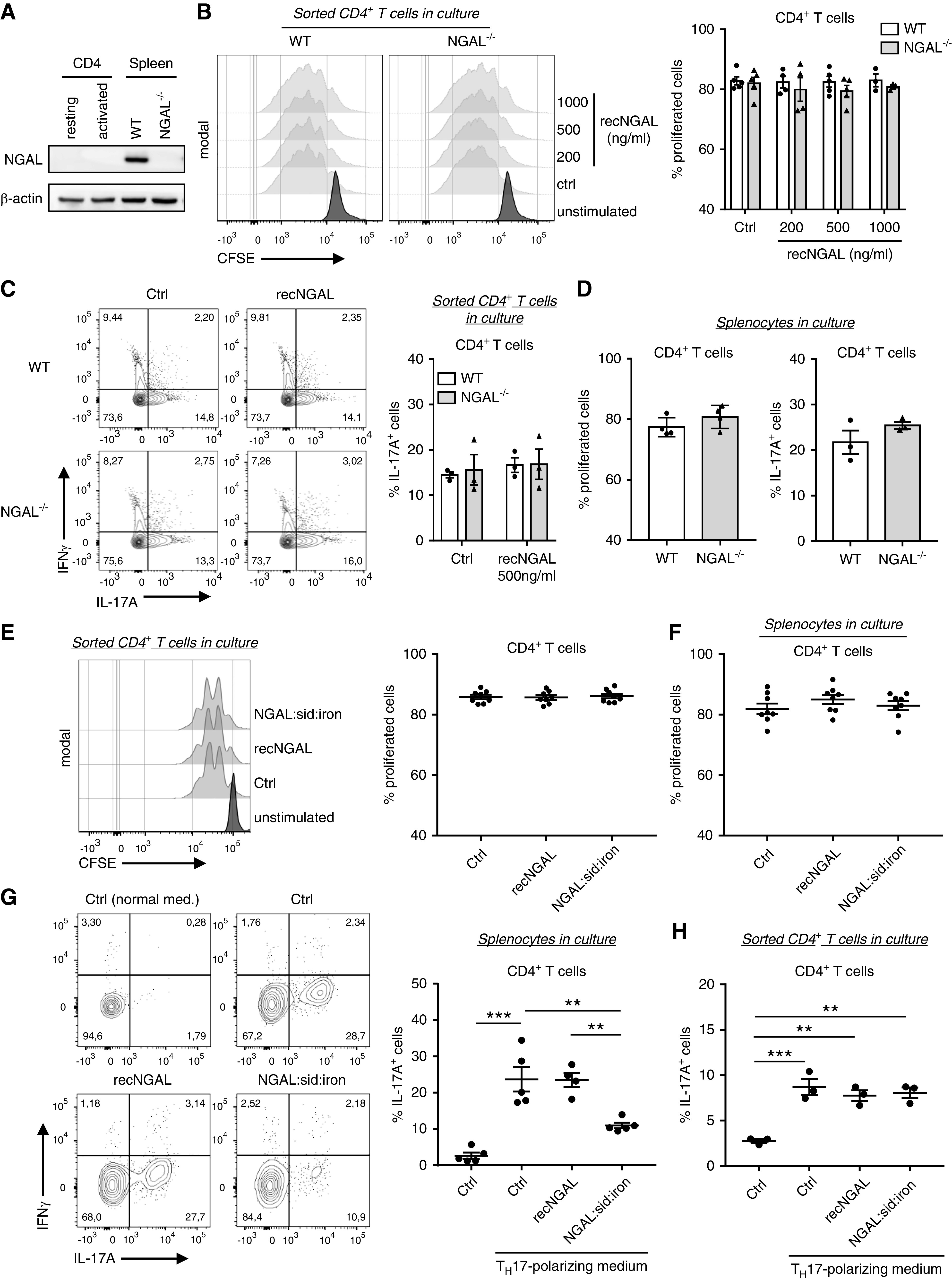
Isolated WT and NGAL−/− CD4+ T cells show similar proliferation and TH17 polarization whereas siderophore iron–loaded NGAL inhibited TH17 polarization in splenocyte cultures. (A) Representative Western blot of NGAL and β-actin (loading control) expression in resting and CD3/CD28 Dynabeads-activated (activated) CD4+ cells isolated from WT mice spleens and in total splenocytes from WT and NGAL−/− mice. (B) CD4+ T cells isolated from spleens of WT and NGAL−/− mice were labeled with carboxyfluorescein succinimidyl ester (CFSE) and incubated with CD3/CD28 dynabeads in the absence or presence of the indicated recombinant murine NGAL concentrations. After 72 hours in culture, proliferation was assessed by flow cytometry. Representative histograms and the corresponding percentages of proliferated cells are shown. (C) Isolated CD4+ T cells from spleens of WT and NGAL−/− mice were incubated with CD3/CD28 dynabeads in TH17 polarization medium in absence or presence of 500 ng/ml recombinant murine NGAL for 6 days and analyzed by flow cytometry. Representative histograms and corresponding percentages of TH17 T cells (IL-17A+IFNγ −) are shown. (D) Proliferation and polarization assays were performed with total splenocytes from WT and NGAL−/− mice in culture. Proliferation and polarization of WT and NGAL−/− CD4+ T cells were found to be similar. (E) Proliferation of CD4+ T cells isolated from spleens of WT −/− mice was measured in the absence or presence of 1 μg/ml of either normal NGAL or iron siderophore–loaded recombinant murine NGAL. Representative histograms and the corresponding percentages of proliferated cells are shown. (F) Total splenocytes from WT mice were prepared and cultured as in (E). Proliferation of gated WT CD4+ T cells were similar in all conditions after 72 hours in culture. (G) Polarization assay of splenocytes from WT mice were performed as in (B) in absence or presence of 1 μg/ml of either normal NGAL or iron siderophore–loaded NGAL. Iron siderophore–loaded NGAL significantly reduced TH17 polarization compared with control conditions and normal NGAL, respectively. Representative histograms and corresponding percentages of TH17 cells (IL-17A+IFNγ−) are shown. (H) TH17 polarization assay under the same conditions as in (G) was performed with CD4+ T cells isolated from spleens of WT mice. Iron siderophore–loaded NGAL had no direct effect on CD4+ T cells during TH17 polarization. Percentages of TH17 T cells (IL-17A+IFNγ−) are shown. Ctrl, control; recNGAL, recombinant NGAL; sid, siderophore. **P<0.01; ***P<0.001.
Next, we questioned whether iron siderophore–loaded NGAL, the biologically active state of NGAL, affects T cells. We detected no difference in proliferation of either isolated CD4+ T cell or CD4+ T cells in splenocyte cultures (Figure 6, E and F). However, iron siderophore–loaded NGAL profoundly inhibited splenocyte TH17 polarization (Figure 6G), which was not observed with isolated CD4+ T cells (Figure 6H).
In summary, our data strongly suggest that biologically active iron siderophore–loaded NGAL suppresses TH17 polarization, thereby providing a negative feedback loop in ANCA-induced vasculitis.
IL-17A Deficiency Protects from Aggravated anti-MPO NCGN in NGAL −/− Mice
Finally, we tested whether increased TH17 T cells and IL-17A are merely markers or rather pathogenic drivers of increased kidney damage in NGAL−/− chimeric mice. To study these possibilities, we bred two additional mouse lines, namely MPO−/−/IL-17A−/− mice as BM recipients and NGAL−/−/IL-17A−/− mice as BM donors. We subsequently compared three groups of murine MPO-immunized MPO−/− mice: MPO−/− transplanted with WT BM (WT group), MPO−/− transplanted with NGAL−/− BM (NGAL−/− group), and MPO−/−/IL-17A−/− mice transplanted with NGAL−/−/IL-17A−/− BM (NGAL−/−/IL-17A−/− group) (Figure 7A). We confirmed in this second set of independent experiments that NGAL−/− chimeric mice displayed aggravated NCGN compared with WT mice (Figure 7B). In addition, we found that MPO−/−/IL-17A−/− mice that received NGAL−/−/IL-17A−/− BM were protected from anti-MPO antibody–induced glomerular crescents and necrosis (Figure 7B). These mice also developed significantly less erythrocyturia and a trend toward less albuminuria compared with NGAL−/− chimeric mice (Figure 7, C and D). Interestingly, and in agreement with the data in Figure 2F, circulating and urinary NGAL levels were similar between the groups despite quantitative differences in renal damage (Figure 7E). By flow cytometry, renal influx of neutrophils, classic monocytes, and CD4+ T cells was reduced in the protected NGAL−/−/IL-17A−/− mice as a further indicator of strong interactions between innate and adaptive immunity (Figure 7F).
Figure 7.
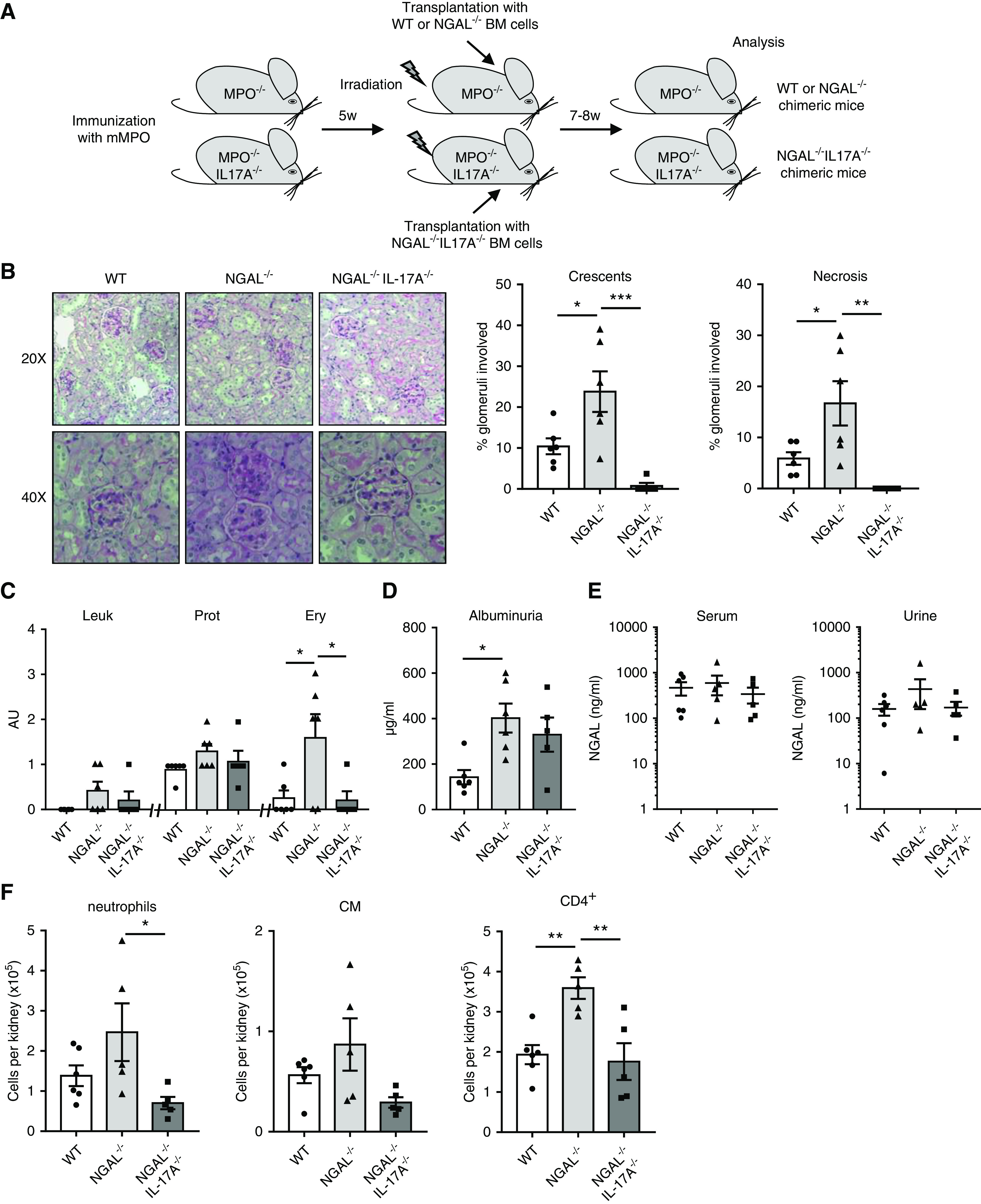
IL-17A gene deficiency rescues NGAL−/− chimeric mice from aggravated NCGN. (A) Experimental scheme for the induction of anti-MPO induced NCGN in WT, NGAL−/−, and IL-17A−/−/MPO−/− chimeric mice. MPO-immunized MPO−/− mice were irradiated and transplanted with either WT or NGAL-deficient (NGAL−/−) BM. Murine MPO-immunized IL-17A−/−/MPO−/− mice were irradiated and transplanted with NGAL−/−/IL-17A−/− BM. All chimeric mice were analyzed 7–8 weeks after transplantation. (B) Representative image of kidney sections stained with Periodic acid–Schiff at low (20×) and high (40×) magnification. Glomeruli with crescents and necrosis were increased in NGAL−/− chimeric mice compared with WT chimeric mice and reduced in the NGAL−/−/IL-17A−/− chimeric mice. (C) Leukocyturia (Leuk), proteinuria (Prot), and erythrocyturia (Ery) were measured in urine by dipstick. (D) Albuminuria was quantified by ELISA. (E) Serum and urinary NGAL levels were measured by ELISA. (F) Renal influx of neutrophils, classic monocytes (CM), and CD4+ T cells was increased in NGAL−/− chimeric mice compared with WT mice and reduced in the NGAL−/−/IL-17A−/− mice. Immune cells were identified using specific markers by flow cytometry. Total number of neutrophils, CM, and CD4+ T cells per kidney are shown. *P<0.05; **P<0.01; ***P<0.001.
Additionally, when anti-MPO disease was induced in immunized MPO−/− mice transplanted with WT BM (WT group) and MPO−/−/IL-17A−/− transplanted with IL-17A−/− BM (IL-17A−/− group), we observed less kidney damage with reduced glomerular crescents and necrosis, erythrocyturia, and albuminuria in the latter group (Supplemental Figure 4, A–E).
Together, these findings strongly support a central pathogenic role of TH17 cells in inducing ANCA GN and an important role of NGAL in regulating TH17 immunity.
Discussion
Our major novel finding is that NGAL is not only a marker of AKI, including in AAV, but also that neutrophil NGAL in particular has a protective role in AAV. NGAL deficiency in BM-derived cells resulted in accelerated MPO-ANCA–induced NCGN in a murine NGAL-chimeric disease model. In addition, we show for the first time that TH17 immunity provides an important disease mechanism in AAV and that neutrophil NGAL exerted renoprotection by reducing TH17 immunity. The protective neutrophil NGAL effect required rather complex interaction between innate and adaptive immunity because NGAL per se did not directly affect neutrophil functions, CD4+ T cell proliferation, and TH17 polarization. However, iron siderophore–loaded NGAL inhibited TH17 polarization in splenocytes—an effect not seen in isolated CD4+ T cells. Possible therapeutic interventions regarding these interactions need to be elucidated in future studies.
NGAL is a 25-kDa glycoprotein of the lipocalin family with high affinity for siderophores. NGAL has a major role in infection through iron chelation, thereby preventing bacterial growth.9,14 In addition, NGAL was implicated in cancer and in metabolic and inflammatory diseases.
In the kidney, NGAL is freely filtered and almost completely reabsorbed by megalin-dependent endocytosis in the proximal tubule. In AKI, NGAL mRNA expression is strongly upregulated, particularly in the thick ascending limb of the loop of Henle and A-intercalated cells of the collecting duct.12,14 Urinary NGAL is now an established early biomarker of intrinsic AKI. We now show that urinary (and serum) NGAL is increased in patients with active AAV, similar to patients with non-ANCA AKI, and that these levels returned to normal during remission. Our murine MPO-ANCA NCGN disease model recapitulates the NGAL increase. However, although neutrophils harbor and release NGAL after ANCA stimulation, the NGAL increase was not diminished in mice that were deficient in BM cell–derived NGAL, but still capable of upregulating renal NGAL. This finding suggests that the quantitative neutrophil contribution to the total NGAL increase was rather small. We did not detect NGAL in monocytes, suggesting that the neutrophil is the major NGAL source in the BM-derived myeloid cell compartment.
An important hypothesis of our study was that neutrophil NGAL was mechanistically involved in AAV. Indeed, our data establish a protective role of neutrophil NGAL in MPO-ANCA NCGN. WT mice had less severe NCGN compared with NGAL−/− chimeric mice that lacked neutrophil NGAL but had similar serum and urinary NGAL levels. This finding suggested that renal NGAL was of minor mechanistic importance for protection from NCGN and that the neutrophil itself or a NGAL-dependent neutrophil interaction with other cells provided the protective effect. We did not find fundamental differences in intrinsic neutrophil functions with relevance to AAV. We detected slightly less degranulation, but doubt that this finding would explain the phenotype. We then turned toward CD4+ T cells because these cells, particularly the TH17 subset, were significantly upregulated in kidneys from NGAL−/− chimeric mice with accelerated NCGN. Increased IL-17A was also detected in spleens of NGAL−/− chimeric mice with MPO-ANCA NCGN, showing that neutrophil NGAL-dependent interactions with the adaptive immunity also occur in other organs. We found that CD4+ T cells did not express NGAL but, because they harbor the NGAL receptor,26 they are technically responsive to NGAL. However, in vitro, we did not find differences between WT and NGAL−/− mice in proliferation or TH17 polarization, neither intrinsically nor when cells were incubated with recombinant NGAL. The same result was obtained when we used T cells from OT-II transgenic mice incubated with antigen-specific APCs from WT and NGAL−/− mice. However, iron siderophore–loaded NGAL had a profound inhibitory effect on splenocyte TH17 polarization in vitro. Because inhibition was only observed in splenocytes but not in isolated T cells, the role of iron siderophore–loaded NGAL in controlling TH17 immunity is complex requiring additional players. However, the possibility of such complex interactions is supported by the close proximity of NGAL-positive neutrophils and T cells in kidneys and spleens.
Only a few mechanistic NGAL studies have been performed in immunologic renal diseases. Ashraf et al.17 reported recently that recombinant NGAL prevented renal damage in a mouse allogeneic kidney transplantation model by a yet unidentified mechanism. In a nephrotoxic nephritis model, conflicting data were reported. Complete NGAL deficiency developed more severe GN15 with increased markers of cell death. T cells were not analyzed and could be the mediators of the accelerated disease. The opposite result was observed in another study with attenuated kidney disease in NGAL-deficient mice and exacerbation of the disease by exogenous NGAL.16
We show that NGAL/IL-17A double-deficient mice were rescued from the accelerated MPO-ANCA NCGN. First, these data establish that TH17 cells are indeed the pathogenic disease driver. The fact that TH17 promote autoimmune crescentic GN was previously shown in a nephrotoxic nephritis model.27−29 However, the nephrotoxic nephritis model differs mechanistically from ANCA-induced NCGN because, in the former, a foreign antiserum is injected, with deposits at the glomerular basement membrane subsequently inducing a secondary immune reaction. In contrast, AAV GN is induced by ANCA-activated neutrophils and monocytes. Therefore, our data advance the understanding of the specific role of TH17 cells in the development of ANCA-induced GN. Second, the data underscores the fact that complex interactions between neutrophils and adaptive immunity occur in AAV. Such interactions were also reported in a T cell–dependent experimental autoimmune encephalomyelitis model where NGAL downregulated cerebral inflammation.30 An explanation was not provided, but others found that purified NGAL increased regulatory T cells in vitro, thereby suggesting a possible direct link.31 Our data indicate that the regulation of T cell immunity by neutrophil NGAL is rather indirect. NGAL-dependent modulation of APCs promoting TH1 immunity has been suggested,32 whereas others26 observed suppressive T cell effects. We found also suppressive T cell effects and did not detect NGAL-dependent APC modulation in an ova-specific OT-II transgenic mouse model. However, we cannot exclude that MPO-specific TH17 immunity is regulated differently, compared with the ova model. The CCR6-directed TH17 cell chemokine CCL2027,33 was upregulated in kidneys from NGAL−/− chimeric mice with MPO-ANCA NCGN and possibly provides a link between neutrophils and increased TH17 immunity. Neutrophils, but also other myeloid cells as well as kidneys cells, were shown to produce CCL20.34,35
In summary, we established BM cell–derived, most likely neutrophil NGAL as an important immune modulator in AAV by keeping TH17 immunity in check. Neutrophil NGAL protects from kidney inflammation and injury in ANCA-induced NCGN. Further studies are needed to understand the precise mechanisms of how neutrophil NGAL regulates renal TH17-mediated inflammation.
Disclosures
All authors have nothing to disclose.
Funding
This work was supported by Deutsche Forschungsgemeinschaft grants SCHR 771/8-1 and SCHR 771/6-1 (to A. Schreiber) and grant 394046635–SFB 1365 (to R. Kettritz and A. Schreiber), and Experimental and Clinical Research Center grants (to R. Kettritz and A. Schreiber).
Supplementary Material
Acknowledgments
We thank Sylvia Lucke, Susanne Rolle, and Tanja Filipowski for excellent technical assistance, and Hubert G. Schwelberger (Department of Visceral, Transplant and Thoracic Surgery, Medical University Innsbruck, Innsbruck, Austria) for preparation of recombinant NGAL. We thank Dr. Shizuo Akira (WPI Immunology Frontier Research Center, Osaka University, Japan) for kindly providing the LCN2−/− mice.
R. Kettritz, A. Rousselle, and A. Schreiber designed the study; M. Ashraf, J. Bontscho, U. Jerke, J. Klocke, S. Popovic, A. Rousselle, A. Schreiber, V. Siffrin, and K. Schmidt-Ott carried out experiments; S. Bachmann, J. Bontscho, U. Jerke, A. Rousselle, A. Schreiber, and V. Siffrin analyzed the data; S. Bachmann, A. Rousselle, and A. Schreiber made the figures; and all authors drafted and/or revised the paper and approved the final version of the manuscript.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2019090879/-/DCSupplemental.
Supplemental Figure 1. NGAL is increased in serum and urine of patients with ANCA-induced GN and NGAL is released by ANCA-activated neutrophils.
Supplemental Table 1. Demographic and clinical features of patients with AAV, AKI, SLE and healthy controls.
Supplemental Figure 2. Wildtype and NGAL−/− mice have a similar hematological profile.
Supplemental Figure 3. Antigen-presenting cells from NGAL−/− mice have normal functions.
Supplemental Figure 4. IL-17A genetic deletion protect mice from anti-MPO induced NCGN.
References
- 1.Falk RJ, Jennette JC: Anti-neutrophil cytoplasmic autoantibodies with specificity for myeloperoxidase in patients with systemic vasculitis and idiopathic necrotizing and crescentic glomerulonephritis. N Engl J Med 318: 1651–1657, 1988. [DOI] [PubMed] [Google Scholar]
- 2.van der Woude FJ: Anticytoplasmic antibodies in Wegener’s granulomatosis. Lancet 2: 48, 1985. [DOI] [PubMed] [Google Scholar]
- 3.Schreiber A, Choi M: The role of neutrophils in causing antineutrophil cytoplasmic autoantibody-associated vasculitis. Curr Opin Hematol 22: 60–66, 2015. [DOI] [PubMed] [Google Scholar]
- 4.Schreiber A, Rousselle A, Becker JU, von Mässenhausen A, Linkermann A, Kettritz R: Necroptosis controls NET generation and mediates complement activation, endothelial damage, and autoimmune vasculitis. Proc Natl Acad Sci U S A 114: E9618–E9625, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schreiber A, Pham CTN, Hu Y, Schneider W, Luft FC, Kettritz R: Neutrophil serine proteases promote IL-1β generation and injury in necrotizing crescentic glomerulonephritis. J Am Soc Nephrol 23: 470–482, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schreiber A, Luft FC, Kettritz R: Phagocyte NADPH oxidase restrains the inflammasome in ANCA-induced GN. J Am Soc Nephrol 26: 411–424, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jerke U, Hernandez DP, Beaudette P, Korkmaz B, Dittmar G, Kettritz R: Neutrophil serine proteases exert proteolytic activity on endothelial cells. Kidney Int 88: 764–775, 2015. [DOI] [PubMed] [Google Scholar]
- 8.Kjeldsen L, Johnsen AH, Sengeløv H, Borregaard N: Isolation and primary structure of NGAL, a novel protein associated with human neutrophil gelatinase. J Biol Chem 268: 10425–10432, 1993. [PubMed] [Google Scholar]
- 9.Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, Strong RK, et al.: Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature 432: 917–921, 2004. [DOI] [PubMed] [Google Scholar]
- 10.Goetz DH, Holmes MA, Borregaard N, Bluhm ME, Raymond KN, Strong RK: The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol Cell 10: 1033–1043, 2002. [DOI] [PubMed] [Google Scholar]
- 11.Buonafine M, Martinez-Martinez E, Jaisser F: More than a simple biomarker: The role of NGAL in cardiovascular and renal diseases. Clin Sci (Lond) 132: 909–923, 2018. [DOI] [PubMed] [Google Scholar]
- 12.Paragas N, Qiu A, Zhang Q, Samstein B, Deng S-X, Schmidt-Ott KM, et al.: The Ngal reporter mouse detects the response of the kidney to injury in real time. Nat Med 17: 216–222, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nickolas TL, Schmidt-Ott KM, Canetta P, Forster C, Singer E, Sise M, et al.: Diagnostic and prognostic stratification in the emergency department using urinary biomarkers of nephron damage: A multicenter prospective cohort study. J Am Coll Cardiol 59: 246–255, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paragas N, Kulkarni R, Werth M, Schmidt-Ott KM, Forster C, Deng R, et al.: α-Intercalated cells defend the urinary system from bacterial infection. J Clin Invest 124: 2963–2976, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eller K, Schroll A, Banas M, Kirsch AH, Huber JM, Nairz M, et al.: Lipocalin-2 expressed in innate immune cells is an endogenous inhibitor of inflammation in murine nephrotoxic serum nephritis. PLoS One 8: e67693, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pawar RD, Pitashny M, Gindea S, Tieng AT, Levine B, Goilav B, et al.: Neutrophil gelatinase-associated lipocalin is instrumental in the pathogenesis of antibody-mediated nephritis in mice. Arthritis Rheum 64: 1620–1631, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ashraf MI, Schwelberger HG, Brendel KA, Feurle J, Andrassy J, Kotsch K, et al.: Exogenous lipocalin 2 ameliorates acute rejection in a mouse model of renal transplantation. Am J Transplant 16: 808–820, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schreiber A, Rolle S, Peripelittchenko L, Rademann J, Schneider W, Luft FC, et al.: Phosphoinositol 3-kinase-gamma mediates antineutrophil cytoplasmic autoantibody-induced glomerulonephritis. Kidney Int 77: 118–128, 2010. [DOI] [PubMed] [Google Scholar]
- 19.Jerke U, Marino SF, Daumke O, Kettritz R: Characterization of the CD177 interaction with the ANCA antigen proteinase 3. Sci Rep 7: 43328, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schreiber A, Xiao H, Jennette JC, Schneider W, Luft FC, Kettritz R: C5a receptor mediates neutrophil activation and ANCA-induced glomerulonephritis. J Am Soc Nephrol 20: 289–298, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schreiber A, Xiao H, Falk RJ, Jennette JC: Bone marrow-derived cells are sufficient and necessary targets to mediate glomerulonephritis and vasculitis induced by anti-myeloperoxidase antibodies. J Am Soc Nephrol 17: 3355–3364, 2006. [DOI] [PubMed] [Google Scholar]
- 22.Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG: Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. PLoS Biol 8: e1000412, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rousselle A, Kettritz R, Schreiber A: Monocytes promote crescent formation in anti-myeloperoxidase antibody-induced glomerulonephritis. Am J Pathol 187: 1908–1915, 2017. [DOI] [PubMed] [Google Scholar]
- 24.Cai L, Rubin J, Han W, Venge P, Xu S: The origin of multiple molecular forms in urine of HNL/NGAL. Clin J Am Soc Nephrol 5: 2229–2235, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiao H, Heeringa P, Liu Z, Huugen D, Hu P, Maeda N, et al.: The role of neutrophils in the induction of glomerulonephritis by anti-myeloperoxidase antibodies. Am J Pathol 167: 39–45, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nam Y, Kim J-H, Seo M, Kim J-H, Jin M, Jeon S, et al.: Lipocalin-2 protein deficiency ameliorates experimental autoimmune encephalomyelitis: The pathogenic role of lipocalin-2 in the central nervous system and peripheral lymphoid tissues. J Biol Chem 289: 16773–16789, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Turner J-E, Paust H-J, Steinmetz OM, Peters A, Riedel J-H, Erhardt A, et al.: CCR6 recruits regulatory T cells and Th17 cells to the kidney in glomerulonephritis. J Am Soc Nephrol 21: 974–985, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steinmetz OM, Summers SA, Gan P-Y, Semple T, Holdsworth SR, Kitching AR: The Th17-defining transcription factor RORγt promotes glomerulonephritis. J Am Soc Nephrol 22: 472–483, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Disteldorf EM, Krebs CF, Paust H-J, Turner J-E, Nouailles G, Tittel A, et al.: CXCL5 drives neutrophil recruitment in TH17-mediated GN. J Am Soc Nephrol 26: 55–66, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berard JL, Zarruk JG, Arbour N, Prat A, Yong VW, Jacques FH, et al.: Lipocalin 2 is a novel immune mediator of experimental autoimmune encephalomyelitis pathogenesis and is modulated in multiple sclerosis. Glia 60: 1145–1159, 2012. [DOI] [PubMed] [Google Scholar]
- 31.La Manna G, Ghinatti G, Tazzari PL, Alviano F, Ricci F, Capelli I, et al.: Neutrophil gelatinase-associated lipocalin increases HLA-G(+)/FoxP3(+) T-regulatory cell population in an in vitro model of PBMC. PLoS One 9: e89497, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Floderer M, Prchal-Murphy M, Vizzardelli C: Dendritic cell-secreted lipocalin2 induces CD8+ T-cell apoptosis, contributes to T-cell priming and leads to a TH1 phenotype. PLoS One 9: e101881, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krebs CF, Paust H-J, Krohn S, Koyro T, Brix SR, Riedel J-H, et al.: Autoimmune renal disease is exacerbated by S1P-Receptor-1-Dependent intestinal Th17 cell migration to the kidney. Immunity 45: 1078–1092, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.González-Guerrero C, Morgado-Pascual JL, Cannata-Ortiz P, Ramos-Barron MA, Gómez-Alamillo C, Arias M, et al.: CCL20 blockade increases the severity of nephrotoxic folic acid-induced acute kidney injury. J Pathol 246: 191–204, 2018. [DOI] [PubMed] [Google Scholar]
- 35.Scapini P, Laudanna C, Pinardi C, Allavena P, Mantovani A, Sozzani S, et al.: Neutrophils produce biologically active macrophage inflammatory protein-3alpha (MIP-3alpha)/CCL20 and MIP-3beta/CCL19. Eur J Immunol 31: 1981–1988, 2001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.