

Significance Statement
Although genetic mutations in α-actinin-4 (ACTN4) are linked with proteinuric glomerulosclerosis in humans, the effect of post-translational modifications is unknown. The authors show that ACTN4—an actin crosslinking cytoskeletal protein—is phosphorylated at serine 159 (S159) in podocytes. Compared with wild-type ACTN4, phosphomimetic ACTN4 protein demonstrated increased binding affinity to F-actin, and phosphomimetic mouse podocytes exhibited more spatially correlated F-actin alignment and a higher rate of detachment under mechanical stress compared with controls. Phosphomimetic Actn4 mice developed proteinuria and glomerulosclerosis after subtotal nephrectomy. These biochemical, cellular, and renal effects are similar to those seen in mutant ACTN4-mediated proteinuric glomerulosclerosis. High extracellular glucose and TGF-β levels stimulate ACTN4 phosphorylation. These findings suggest that, in addition to genetic mutations, increased phosphorylation of ACTN4 may mediate podocyte injury and kidney disease.
Keywords: chronic kidney disease, focal segmental glomerulosclerosis, glomerular disease, cytoskeleton, podocyte, proteinuria
Visual Abstract

Abstract
Background
Genetic mutations in α-actinin-4 (ACTN4)—an important actin crosslinking cytoskeletal protein that provides structural support for kidney podocytes—have been linked to proteinuric glomerulosclerosis in humans. However, the effect of post-translational modifications of ACTN4 on podocyte integrity and kidney function is not known.
Methods
Using mass spectrometry, we found that ACTN4 is phosphorylated at serine (S) 159 in human podocytes. We used phosphomimetic and nonphosphorylatable ACTN4 to comprehensively study the effects of this phosphorylation in vitro and in vivo. We conducted x-ray crystallography, F-actin binding and bundling assays, and immunofluorescence staining to evaluate F-actin alignment. Microfluidic organ-on-a-chip technology was used to assess for detachment of podocytes simultaneously exposed to fluid flow and cyclic strain. We then used CRISPR/Cas9 to generate mouse models and assessed for renal injury by measuring albuminuria and examining kidney histology. We also performed targeted mass spectrometry to determine whether high extracellular glucose or TGF-β levels increase phosphorylation of ACTN4.
Results
Compared with the wild type ACTN4, phosphomimetic ACTN4 demonstrated increased binding and bundling activity with F-actin in vitro. Phosphomimetic Actn4 mouse podocytes exhibited more spatially correlated F-actin alignment and a higher rate of detachment under mechanical stress. Phosphomimetic Actn4 mice developed proteinuria and glomerulosclerosis after subtotal nephrectomy. Moreover, we found that exposure to high extracellular glucose or TGF-β stimulates phosphorylation of ACTN4 at S159 in podocytes.
Conclusions
These findings suggest that increased phosphorylation of ACTN4 at S159 leads to biochemical, cellular, and renal pathology that is similar to pathology resulting from human disease–causing mutations in ACTN4. ACTN4 may mediate podocyte injury as a consequence of both genetic mutations and signaling events that modulate phosphorylation.
Podocytes are essential to maintaining the glomerular filtration barrier. To carry out their function, podocytes rely on an intricate actin-based cytoskeleton to maintain their structural integrity against the mechanical stresses resulting from pulsatile blood flow and glomerular filtration.1−4 Importantly, loss of normal podocyte foot process architecture and podocyte detachment are seminal events signifying the progression of CKD.5−8 Mutations in several key actin cytoskeletal proteins have been shown to cause familial forms of FSGS, providing support for the theory that dysfunction of the cytoskeleton represents a common disease pathway.9−17
α-Actinins (ACTNs) are essential cytoskeleton proteins that crosslink actin filaments and provide structural support for multiple cell types, including platelets (ACTN1),18 cardiac muscle (ACTN2),19 skeletal muscle (ACTN3),20 and kidney podocytes (ACTN4).9 Mutations in the actin crosslinking protein ACTN4 cause a highly penetrant autosomal dominant form of FSGS.9 All of the disease-causing ACTN4 mutations identified to date reside within the actin-binding domain (ABD) of the encoded ACTN4 protein and increase the binding affinity of ACTN4 to filamentous actin (F-actin).9,21,22 At the protein level, the F-actin network formed by crosslinking with mutant ACTN4 is more brittle than F-actin networks crosslinked with wild-type (WT) ACTN4, with a lower threshold for breaking.23 Homozygous mutant Actn4K256E/K256E podocytes (a mouse K256E mutation is homologous to the FSGS-causing K255E mutation in humans; we use lowercase Actn4 to refer to the mouse protein and uppercase ACTN4 to refer to the human protein) fail to recover their baseline contraction after stretch and develop irreparable disruptions in their actin cytoskeletons, leading to a more brittle podocyte compared with WT podocytes.24 Moreover, mutant Actn4K256E/K256E mice develop albuminuria and FSGS, similar to human renal phenotype.24−26 These findings have revealed that mutations contribute to disease via altering the ACTN4–F-actin interaction, leading to a disrupted cytoskeleton and impaired podocyte that is vulnerable to the mechanical stresses it constantly experiences in the kidney.
Although genetic mutations in ACTN4 have been linked to podocyte vulnerability and proteinuric glomerulosclerosis, the effect of post-translational modifications of ACTN4 on podocyte and kidney function is not known. In this study, we searched for sites of ACTN4 modification in cultured human podocytes. Using mass spectrometry, we found that ACTN4 was phosphorylated at serine 159 (S159), located within its ABD, the domain that contains all known disease-causing mutations. Increased phosphorylation at this site leads to similar effects as does disease-causing mutant ACTN4 when examined at a biochemical, cellular, or whole-animal level. We further found that both high extracellular glucose and TGF-β stimulate phosphorylation of ACTN4 in podocytes. Altogether, our findings demonstrate a new mechanism by which a post-translational modification to ACTN4 recapitulates similar podocyte vulnerability as do dominantlyacting ACTN4 point mutations.
Methods
Human Podocytes Culture
Immortalized human podocyte cells were cultured in complete RPMI medium (Thermo Fisher Scientific) supplemented with 10% FBS, Antibiotic-Antimycotic Solution (Corning), and ITS Liquid Media Supplement (100×; Sigma-Aldrich) as described previously.27 Immortalized human podocyte cells were cultured under 33°C to propagate and then switched to 37°C for 14 days of differentiation before any treatment. At both temperatures, these cells were fed with fresh medium every 2–3 days. To study the effect of high glucose, cells were cultured in base RPMI medium (serum free, ITS free, and antibiotic-antimycotic free) containing 4.5 g/L glucose (2 g/L glucose in premixed base medium to which we supplemented an additional 2.5 g/L glucose [Sigma-Aldrich]) for 72 hours. For osmolarity control, cells were cultured in base RPMI medium containing 2.5 g/L D-mannose (Sigma-Aldrich) for 72 hours. To study the effect of TGF-β (R&D SYSTEMS), cells were cultured in base RPMI medium either with TGF-β (20 ng/ml) or without TGF-β.
Mass Spectrometry
Podocytes were lysed using NP40 lysis buffer (Boston BioProducts) containing 6 M potassium iodide. Total protein extract was quantified by the BCA method, and cell lysates were fractionated using SDS-PAGE and visualized with colloidal blue staining (Invitrogen). Targeted protein bands (100±20 kD) were excised, digested with trypsin (Promega), and extracted following in-gel digestion protocol described previously with slight modification.28,29 Peptides were analyzed by nanoflow reversed-phase HPLC (SCIEX) connected to a Q Exactive mass spectrometer (Thermo Fisher Scientific). Nonphosphopeptide FAIQDISVEETSAKheavy and phosphor-peptide FAIQDI(pS)VEETSAKheavy were synthesized, and the C-terminal lysine (K) of each peptide was isotopically labeled with 13C6 and 15N2 heavy isotopes (New England Peptide). These isotopically labeled peptides were used as internal standards for quantification. Nonphosphopeptide FAIQDISVEETSAKlight and phosphorylated peptide FAIQDI(pS)VEETSAKlight refer to endogenous peptides derived from each sample. Each biologic sample was coinjected with 30 fmol of FAIQDISVEETSAKheavy and 30 fmol of phosphorylated peptide FAIQDI(pS)VEETSAKheavy. Each sample was separated at a flow rate of 1000 nl/min with a linear 40 minutes of gradient from 98% solvent A (0.1% formic acid in water) to 30% solvent B (100% acetonitrile and 0.1% formic acid) followed by a linear 5 minutes of gradient from 30% solvent B to 35% solvent B. Parallel reaction monitoring mass spectrometric method was used for the identification and quantification of the above described four targeted peptides.30,31 Mass spectrums were searched by Mascot v.2.6.1 software (Matrix Science, London, United Kingdom) against the human proteome database containing 42,353 protein sequences. Mascot search criteria included (1) mass tolerance of 10 ppm; (2) fragment mass tolerance of 0.6 D; (3) fixed modification: carbamidomethyl (C); (4) variable modifications: labels: 13C(6)15N(2) (K); Phospho (STY); and (5) cleavage specificity: trypsin, with up to two missed cleavages allowed. Ions identification scores above 20 were used as identification cutoff from the Mascot search. Xcalibur (Thermo Fisher Scientific) software was used to quantify the intensity of targeted peptides as described previously.29
Protein Expression
A DNA fragment encoding the WT actin binding domain (ABD) of ACTN4 (amino acids 47–271) linked with an N-terminal tobacco etch virus protease site was synthesized as gBlocks Gene Fragments (Integrated DNA Technologies) and cloned into the pET-28a vector (Millipore Sigma). The plasmids encoding the ABDs of S159D ACTN4 and S159A ACTN4 were generated by site-directed mutagenesis. Bl21-CodonPlus (DE3)-RILP–competent Escherichia coli cells (Agilent Technology) were used for transformation. Protein expression was induced by the addition of 1 mM isopropyl β-d-1-thiogalactopyranoside for 4 hours at 37°C. The cell pellets were lysed in lysis buffer containing 50 mM Tris/HCl (pH 8.0), 200 mM NaCl, 100 mM Imidazole (pH 8.0), and 2 mM β-Mercaptoethanol supplemented with protease inhibitor tablet (Millipore Sigma). The ABDs of ACTN4 proteins were purified by Ni-NTA chromatography (Qiagen).
Full-length WT ACTN4 was subcloned into the bacterial expression vector pET-28a as described previously.22 Other ACTN4 expression plasmids were generated by site-directed mutagenesis. The plasmid was transformed into BL21-CodonPlus (DE3)-RILP Competent Cells (Agilent Technology), and the expression was induced for 3 hours at 37°C by 1 mM isopropyl β-d-1-thiogalactopyranoside. The cell pellet was lysed in B-PER buffer (Thermo Fisher Scientific) containing Protease Inhibitor Cocktail Tablet (Sigma-Aldrich). ACTN4 was purified using a Cobalt purification kit (Thermo Fisher Scientific). Purified protein was concentrated by Amicon Ultra 0.5-ml centrifugal filters (Millipore).
X-Ray Crystallography
The Ni-NTA–purified WT and S159D ACTN4 proteins were incubated with in-house generated tobacco etch virus protease to remove His tags. These proteins were further purified by ion exchange and size exclusion chromatography using UNO Q12 (Biorad) and Superdex 75 (GE Healthcare) columns, respectively. WT and S159D ACTN4 proteins were concentrated to approximately 14 mg/ml using Amicon Ultra centrifugal filters (Millipore Sigma) in a final buffer of 20 mM Tris-HCl (pH 8.0), 150 mM NaCl, and 1 mM dithiothreitol (DTT). Initial crystals were obtained by the sitting drop vapor diffusion method with a well solution containing 100 mM Imidazole (pH 7.4), 50 mM NaCl, 1 mM EDTA, 5% (vol/vol) glycerol, and 18% (wt/vol) polyethylene glycol 5000 monomethyl-ether. The glycerol concentration was raised to 20% for crystal freezing prior to x-ray data collection. Datasets were collected at the APS beamline 17-ID (S159D) and the ESRF beamline ID30B (WT) using Dectris Pilatus3 6M (Baden, Switzerland) detectors. Processing and scaling were performed in HKL2000 (HKL Research, Inc.).32 The structures were solved by molecular replacement in Molrep33 using the coordinates of the ACTN4-K255E mutant structure (PDB ID code 2R0O)34 as the search model. Analysis of the Matthews coefficient indicated one protein molecule per crystallographic asymmetric unit in each case.35 Isotropic refinement with TLS parameters (WT) and anisotropic refinement (S159D) was carried out with REFMAC5.36 The structure building and the root-mean-square deviation were calculated using COOT.37 The geometry of the final refined models (Table 1) was assessed using Molprobity.38 There were no outliers in the Ramachandran plots.39 Superposition of the Cα positions of ABD of WT (PDB ID code 6O31) and S159D ACTN4 (PDB ID code 6OA6) was generated with PovScript+40 and ray traced with Povray (http://www.povray.com). Electrostatic maps were generated using PyMol APBS plug-in.41
Table 1.
Structure determination and refinement statistics
| Parameter | WT | S159D |
| PDB identification code | 6O31 | 6OA6 |
| Unit cell, Åa | a=b=38.1, c=303.2 | a=45.6, b=61.7, c=89.5 |
| Resolution range, Åa | 40.03–1.51 (1.56–1.51) | 50.83–1.37 (1.39–1.37) |
| Wavelength, Å | 0.9762 | 1.0719 |
| Space group | P41212 | P212121 |
| Observed reflections | 326,833 | 309,288 |
| Unique reflections | 36,924 | 52,586 |
| Completeness, % | 99.9 (99.9) | 97.7 (92.2) |
| Redundancy | 8.9 (8.9) | 5.9 (3.1) |
| Rsym, %b | 9.4 (75.5) | 13.0 (69.2) |
| Rpim, %c | 3.3 (26.1) | 5.4 (41.6) |
| CC1/2 | 0.989 (0.779) | 0.968 (0.665) |
| Overall <I/σ(I)> | 25.3 (2.3) | 13.1 (1.4) |
| Rcryst/Rfree, %d | 20.8/ 25.4 | 13.4/17.2 |
| Ramachandran plot | ||
| Favored/allowed/outliers, % | 97.0/3.0/0.0 | 99.0/1.0/0.0 |
| Bond lengths, Åe | 0.013 | 0.009 |
| Bond angles, °e | 1.825 | 1.534 |
PDB, protein data bank; CC1/2, Pearson correlation coefficient between two random half datasets.
Values in parentheses are for the highest-resolution shell.
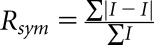 .
.
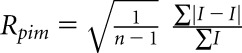 , where I is the observed integrated intensity, <I> is the average integrated intensity obtained from multiple measurements, and the summation is over all observed reflections.
, where I is the observed integrated intensity, <I> is the average integrated intensity obtained from multiple measurements, and the summation is over all observed reflections.
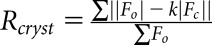 . Fo and Fc are the observed and calculated structure factors, respectively, and k is a scaling factor. The summation is over all measurements. Rfree is calculated as Rcryst using 5% of the reflections chosen randomly and omitted from the refinement calculations.
. Fo and Fc are the observed and calculated structure factors, respectively, and k is a scaling factor. The summation is over all measurements. Rfree is calculated as Rcryst using 5% of the reflections chosen randomly and omitted from the refinement calculations.
Bond lengths and angles are root-mean-square deviations from ideal values. Differences in side chain rotamers were attributed to distinct crystal packing effects.
F-Actin Spin-Down Assay and F-Actin Bundling Assays
The F-actin spin-down assay and F-actin bundling assay were performed using the Actin-Binding Protein Biochem Kit (Cytoskeleton) according to the manufacturer’s protocol. Specifically, for the spin-down assay, 3 μM ABD of WT, S159A, and S159D ACTN4 proteins was separately incubated with 18.4 μM F-actin at room temperature for 30 minutes and then, centrifuged at 24°C at 150,000×g for 1.5 hours. For the F-actin bundling assay, 0.3 μM full-length WT, S159A, and S159D ACTN4 proteins were separately incubated with 3 μM F-actin, incubated at room temperature for 30 minutes, and then, centrifuged at 24°C at 14,000×g for 1 hour. Proteins in the supernatants and pellets after centrifuge were solubilized in equal amounts of SDS sample buffer, boiled, and subjected to 4%–20% SDS-PAGE gel (Biorad).
Mouse Podocyte Isolation and Culture
Primary mouse podocytes were isolated from male mice aged between 4 and 6 weeks old using previously described methods.24,42 Briefly, both kidneys were removed; minced; and sequentially pressed through 100-, 70-, and 40-μm cell strainers. The isolated glomeruli were seeded on collagen I (Advanced Biomatrix)–coated plates. After 4 days of podocyte growth, podocytes were passaged and used within 24 days since the day of isolation; podocytes from passages 2 and 3 were used for experiments. The method yields primary podocytes with 90% purity, confirmed via staining with the podocyte-specific marker Wilms tumor 1 (WT-1) on day 24 after isolation. Primary mouse podocytes were cultured in complete RPMI medium, and they were fed with fresh medium every 2–3 days.27
Immunofluorescence Staining
Mouse podocytes were seeded on the collagen I–coated coverslip (Thermo Fisher Scientific) in complete RPMI medium for 8 hours (Figure 4). They were then fixed in 4% paraformaldehyde, permeabilized in 0.1% Triton X-100, and blocked with BlockAid Blocking Solution (Thermo Fisher Scientific). Podocytes were stained with primary antibody vinculin (Abcam) followed by coincubation of secondary antibody Alexa Fluor 647 and rhodamine phalloidin staining (Thermo Fisher Scientific). Nuclei were stained with Hoechst 33342 (Thermo Fisher Scientific). Confocal images were acquired using a Zeiss LSM 880 confocal system equipped with a plan-Apochromat 63×/1.40 oil objective lens. Z-stack images were recorded at slice intervals of 0.5 μm.
Figure 4.

Phosphomimetic Actn4S160D/S160D podocytes demonstrate altered F-actin alignment. (A) Representative 63× images of F-actin within WT, Actn4S160A/S160A, and Actn4S160D/S160D podocytes grown on collagen-coated coverslips. Light yellow lines indicate the averaged angles of F-actin vectors within a region of 1.9×1.9 μm2. Scale bar, 20 μm. (B) Quantification of the distribution of F-actin alignment across each group of podocytes. This quantification was calculated by the autocorrelation function C(r) (y axis), which is a measure of the spatial correlation of the F-actin alignment over increasing distances (r) across the cell (x axis). Data are plotted as mean C(r) ± SEM. Solid curves were fit to these mean data within each group of podocytes. (C) Quantification of autocorrelation length for each group of podocytes. Each dot represents one podocyte’s autocorrelation length (see Methods and Supplemental Figure 1 for calculation). The higher the autocorrelation length, the more spatially correlated the F-actin across the cell. Box plots represent median and IQR for WT (n=22), Actn4S160A/S160A (n=24), and Actn4S160D/S160D (n=18) podocytes. Data are pooled from three independent experiments. *Significant difference from the WT ( P<0.05, t test).
Measurement of F-Actin Orientation Autocorrelation
F-actin orientation was quantified by previously described methods.24 Briefly, confocal images of F-actin of mouse podocytes were analyzed using ImageJ plug-in OrientationJ Vector field.43 Local actin orientation θ is calculated by the intensity in a region of 1.9×1.9 μm2. Spatial autocorrelation C(r) was calculated using the following equation: C(r)=<cos2(θi−θj(r))>, where 0<r<30 μm.44 j represents all regions within the circle radius r from the center region I; ⟨ ⟩ indicates calculating the average value. The autocorrelation curve was fitted using the curve_fit function under the scipy.optimize module in Scipy (version 1.2.1), and the equation of ae−r/b+c was used to fit the curve (a, b, and c were the parameters to be fitted). The x-axis intercept of the tangent line for the fitted autocorrelation curve at r=0 defined the autocorrelation length for that specific podocyte (Supplemental Figure 1).
Podocyte Detachment Assay Using Microfluidic Culture
Two-channel organ-on-a-chip microfluidic culture devices made of polydimethylsiloxane (Ellsworth Adhesives) containing a top channel (1×1×16.7 mm), a bottom channel (1×0.2×16.7 mm), and two lateral vacuum chambers were produced as previously described,45 except that the upper and lower channels were separated by a solid 50-μm-thick membrane rather than one with pores. The chips were washed with 70% ethanol, activated using 0.5 mg/ml Sulfo-SANPAH solution (ProteoChem) under an ultraviolet lamp (Nailstar) for 20 minutes, rinsed with cold PBS, and coated overnight with 0.1 mg/ml collagen I (Advanced Biomatrix). Primary podocytes (2800 cells) were seeded in the bottom channel of the device by inversion for 2 hours in complete RPMI medium and then flipped, allowing podocytes to attach for an additional 16 hours before initiating experiments. These chips were transferred to the Zeiss Axio Observer system equipped with a 5×/0.16 phase objective lens as well as heat and CO2 modules to maintain the temperature at 37°C and CO2 at 5% throughout the entire imaging period. Phase-contrast images were taken every 5 minutes for 48 hours. The bottom channel was continuously perfused with the complete RPMI media using an Ismatec IPC-N digital peristaltic pump (Cole-Parmer) at a flow rate of 85.8 μl/min (shear stress =1.5 dyn/cm2).46 The cyclic strain was simultaneously applied continuously by using a programmable vacuum regulator system built in house to apply cyclic suction to the side chambers, thereby deforming the lateral walls and the membrane with attached cells. This system consists of a vacuum regulator ITV0091–2BL (SMC Corporation of America) that was electronically controlled by an Arduino Uno with an MAX517 digital to analog converter; a sinusoidal vacuum profile producing 10% cyclic strain47 at 0.1 Hz was used in these studies. Inline Bubble Traps (Precigenome LLC) were used to prevent bubble formation. At the end of each experiment, phase-contrast images were reviewed to manually count the number of podocytes that detached and the number that remained adhered.
Animal Models
Actn4S160D/S160D and Actn4S160A/S160A mice were developed at the Beth Israel Deaconess Medical Center (BIDMC) transgenic core using the RNA-guided CRISPR nickase Cas9 approach as described previously.24 The genotypes of the founder mice were verified by Sanger sequencing. A mutant male mouse was crossed with an FVB/NJ female (Jackson Laboratory). From the offspring of the backcross, heterozygous female and male mice were intercrossed to generate homozygous Actn4S160D/S160D and Actn4S160A/S160A and their WT littermates. Mice were genotyped using custom-designed S160D and S160A Taqman SNP analysis assays: guide RNA 1: GATGGCAAATCTGAGGATGATGG; guide RNA 2: TCTCTGTGGAAGGTAAGACATGG. ssDNA for the generation of Actn4S160D/S160D mice was ATGACCCTGGGAATGATCTGGACCATCATCCTCAGATTCGCGATCCAGGACATCGACGTGGAAGGTAAGACATGGCAGAGAGTACCTCT. ssDNA for the generation of Actn4S160A/S160A mice was ATGACCCTGGGAATGATCTGGACCATCATCCTCAGATTCGCGATCCAGGCTATCGACGTGGAAGGTAAGACATGGCAGAGAGTACCTCT (bold and underlined letters indicate the DNA codon change).
Subtotal Nephrectomy and Measurement of Urine Albumin-Creatinine Ratio
Subtotal nephrectomy was performed at the Duke O’Brien Center on male mice at a median age of 23 weeks using previously described procedures.48 Urine samples were collected using a metabolic cage.49 Urine albumin was quantified byELISA according to the manufacturer’s protocol (Bethyl Laboratories Inc.). Urine creatinine was quantified by mass spectrometry.50
Kidney Section Staining
Mice kidneys were formalin fixed and paraffin embedded using routine protocols. Some of the 5-μm kidney sections were stained with periodic acid–Schiff and evaluated by a renal pathologist using light microscopy. The pathologist was blinded to the genotype of the kidney sections.
Statistics
Statistical analyses were performed using R.51 Mann–Whitney U tests52 or t tests were used to evaluate differences between two groups. Fisher exact test was used to assess for the difference in the proportion of sclerosed glomeruli between two groups using fisher.test() function in R. Fisher exact test was also used to assess for the difference in proportions of podocytes that detached between Actn4S160D/S160D and WT podocytes. All figures were generated by Plotly (https://plot.ly) unless otherwise specified. A P value of 0.05 was considered statistically significant.
Study Approval
All animal procedures were approved by the BIDMC Animal Care and Use Committee. Subtotal nephrectomy was performed at the Association for Assessment and Accreditation of Laboratory Animal Care–accredited animal facility at Durham Veterans Affairs under National Institutes of Health guidelines.
Results
ACTN Is Phosphorylated at S159 in Human Podocytes
We used mass spectrometry to determine whether there are any post-translational modifications to ACTN4 in immortalized human podocytes cultured in complete RPMI medium. The only post-translational modification we detected in these cells was phosphorylation of ACTN peptide 153FAIQDIpSVEETSAK166 at S159 site (Figure 1A). The presence of y(8)-98 product ion in the Tandem mass spectrometry (MS2)spectrum of the peptide (153FAIQDISVEETSAK166) precursor ion at m/z 809.3752 (z=2) confirms the site of phosphorylation at S159 (y8 position) (Figure 1A). S159 is located in a critical linker region between the CH1 and CH2 domains within the ABD of ACTN4 (Figure 1B). This S159 is evolutionarily conserved among ACTN orthologs from frogs to humans, suggesting the functional importance of this site (Figure 1C). Of note, the peptide containing S159 is shared across ACTN1, ACTN2, ACTN3, and ACTN4. Our assay does not distinguish which ACTN is phosphorylated. However, ACTN4 is the predominant ACTN expressed in podocytes.9 Moreover, because several genetic mutations in the ABD of ACTN4 have been found to cause human FSGS,9,21,22 we sought out to determine the relevance of phosphorylation of ACTN4 at S159 to kidney function.
Figure 1.
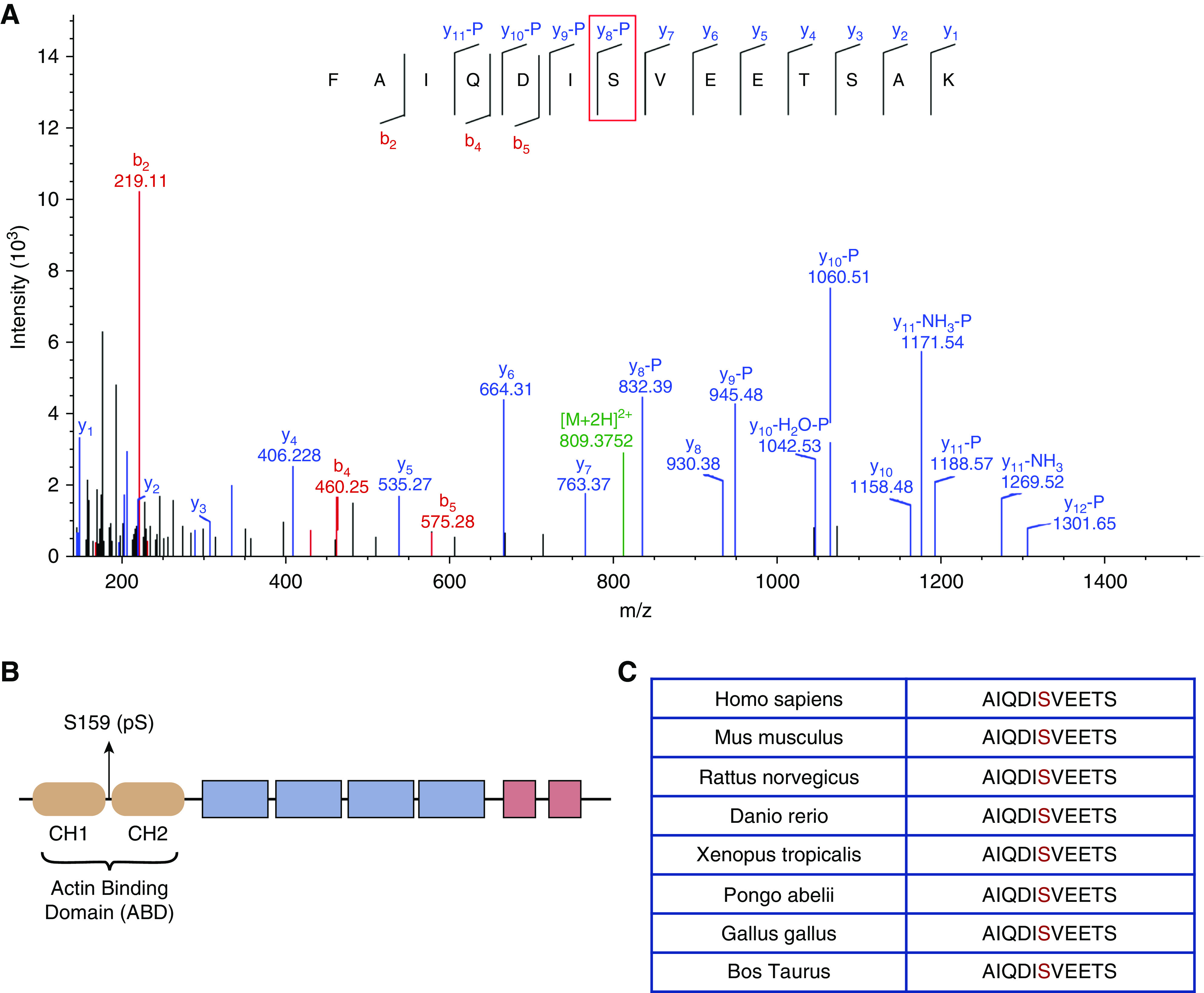
Phosphorylation of ACTN4 is detected at conserved. S159. (A) The representative tandem mass spectrometry (MS2)spectrum of the phosphopeptide (153FAIQDISVEETSAK166) showing S159 phosphorylation. The MS2 spectrum of the peptide at m/z=809.3752 (z=2) is shown in green in (A). Detection of specific y (blue) and b (red) fragment ions allowed identification of the peptide sequence 153FAIQDISVEETSAK166 and assignment of phosphorylation site to S159. Specifically, the presence of the y(8)-98 fragment ion confirms the site of phosphorylation at S159. (B) Functional domains of the human ACTN4 protein. The ABD consists of CH1 and CH2 domains. S159 is located in the linker region (amino acids 157–164) between CH1 and CH2 domain. (C) The S159 phosphorylation site (±5 amino acids from S159) is evolutionarily conserved across species.
Phosphomimetic S159D ACTN4 Changes the Charge at S159 Site without Changing Its Conformation
Because the kinase(s) that phosphorylates ACTN4 at S159 is unknown, we used a phosphomimetic serine (S) to aspartic acid (D) substitution (S159D) to study the effect of phosphorylation at ACTN4 S159, with WT ACTN4 and a nonphosphorylatable form (S159A) serving as controls.53 Previous experiments have shown that such phosphomimetic proteins behave similarly to kinase-phosphorylated proteins, supporting phosphomimetic models as valid surrogates for phosphorylated proteins.54,55
To examine the effect of S159 phosphorylation on the structure of ACTN4, we crystallized and solved the three-dimensional structures of the ABDs of WT and S159D ACTN4 (Table 1). Although the two proteins crystallized in different space groups (Table 1), the Cα atoms of the WT and S159D structures were superimposed with a root-mean-square deviation of 0.613 Å, indicating no significant difference between the two structures (Figure 2A). However, electrostatic maps revealed significant changes in surface negative charge in the region of the S159D substitution (Figure 2C) in comparison with the WT (Figure 2B). Overall, these findings suggest that the phosphomimetic S159D mimics the charge effect of phosphorylation while not disrupting the structural integrity of the protein.
Figure 2.
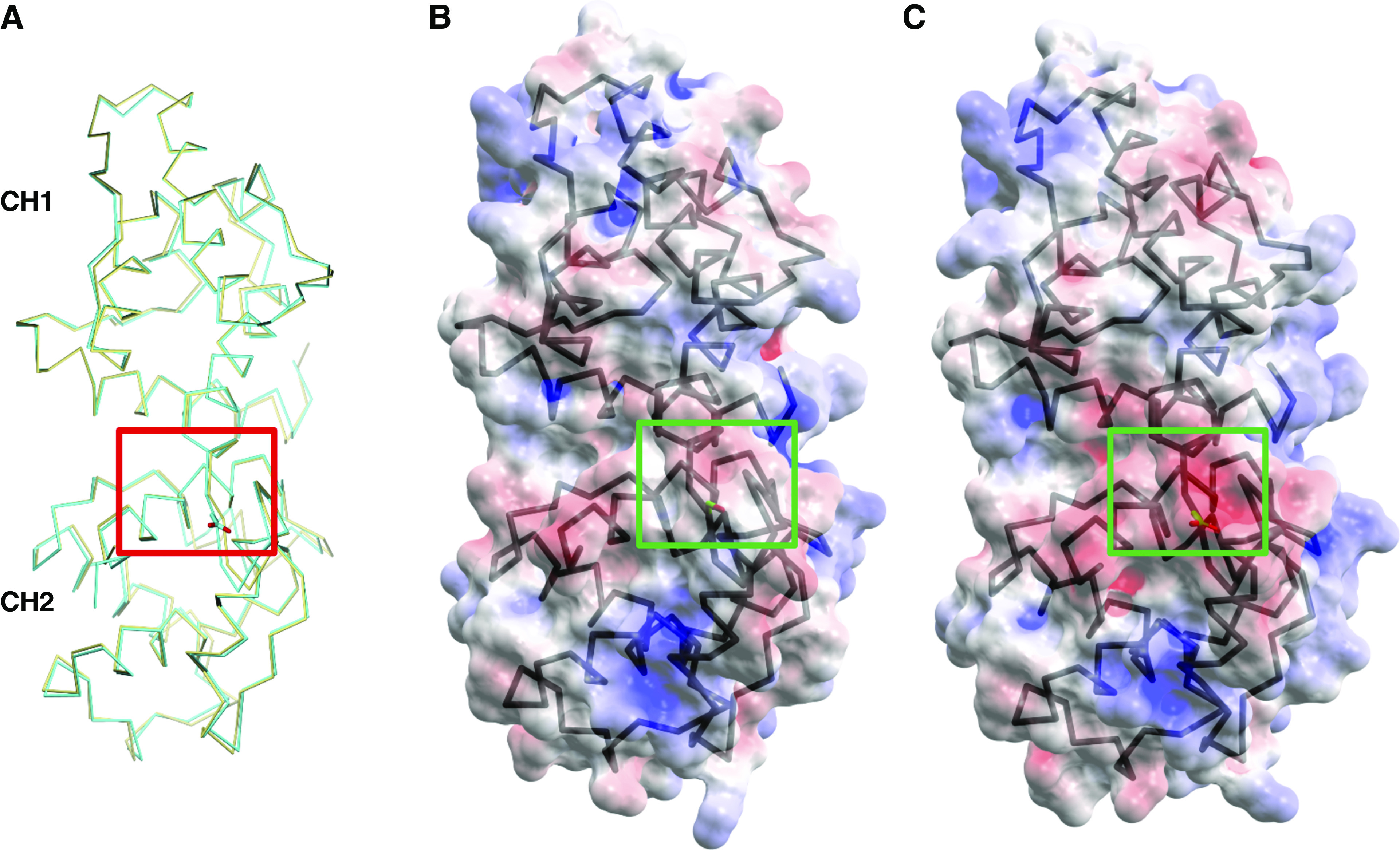
Phosphomimetic S159D ACTN4 does not change the conformation of its ABD. (A) Superposition of the Cα positions of the ABD of WT (khaki) and phosphomimetic S159D (cyan) ACTN4. The red box highlights the side chain of aspartate (D) at position 159. The CH1 domain is shown at the bottom, and the CH2 domain is shown at the top. Semitransparent electrostatic surface representation of (B) WT ACTN4 ABD and (C) phosphomimetic S159D ACTN4 ABD; both are colored according to electrostatic potential (red [acidic, −5 kBT], white [neutral, 0 kBT], and blue [basic, 5 kBT]). Cα positions of the ABD of (B) WT and (C) phosphomimetic S159D ACTN4 are indicated by the black lines. The red box highlights negative charge (represented by red shading) conferred by D159 substitution in (C), which is different from more neutral charge conferred by S159 in (B). Different side chain orientations can be attributed to distinct crystal packing effects.
Phosphomimetic S159D Increases F-Actin Binding Affinity and Bundling Activity
A major role of ACTN4 is to bundle F-actin.56 All of the disease-causing ACTN4 mutations identified to date reside within the ABD of the encoded protein and increase the binding affinity of ACTN4 to F-actin.9,21 This increased binding leads to increased F-actin bundling activity.22 Because the phosphorylation site of interest (S159) is within the ABD of ACTN4, we sought to determine whether similar increases in binding affinity and bundling activity occurred as a result of phosphorylation at this site. The ACTN4 ABD and F-actin binding assay is illustrated in Figure 3A. These ABDs were used to assess binding affinity to F-actin. We incubated F-actin with three different groups of the ACTN4 ABDs: WT, S159A, and S159D. After subjecting the mixtures to high-speed ultracentrifugation, the amount of pellet-total (P/T) ratio of F-actin and ACTN4 ABD was quantified. Because the majority of the F-actin should be present in the pellet after high-speed ultracentrifugation, the P/T ratio of F-actin should not change across groups, as confirmed in our study (Figure 3B). For the ABD of ACTN4, we found that P/T ratio of S159D ACTN4 was significantly higher than that of WT ACTN4 (P= 0.03) (Figure 3C). Thus, S159D ACTN4 exhibits increased F-actin binding affinity compared with WT ACTN4. There was no difference in the P/T ratio between the ABD of WT and S159A ACTN4, reflecting similar binding affinity of the ABD of WT and S159A to F-actin.
Figure 3.
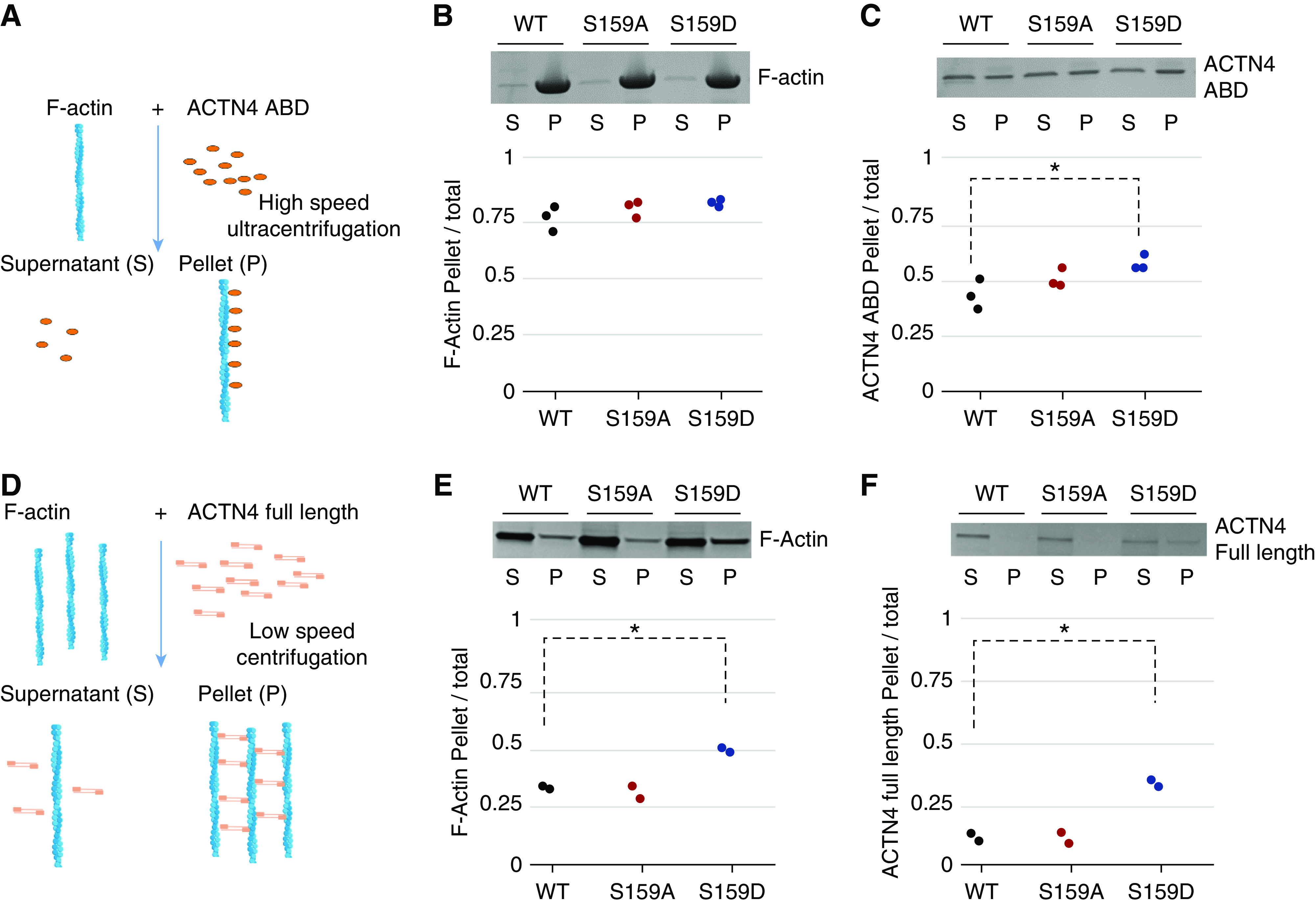
Phosphomimetic S159D ACTN4 demonstrates increased F-actin binding affinity and F-actin bundling activity. (A) Illustration of F-actin binding assay. F-actin was incubated with the ABD of WT, S159A, and S159D ACTN4. These ABDs were used to assess binding affinity to F-actin. After high-speed ultracentrifugation, the pellet (P) contains ACTN4 ABD bound to F-actin, and supernatant (S) contains unbound ACTN4 ABD. The higher the binding affinity, the higher the ratio of ACTN4 ABD in the P to total amount. (B) Representative image of Coomassie Blue–stained SDS-PAGE gel of F-actin (one of three independent experiments). Bands correspond to the amount of F-actin in S and P after ultracentrifugation. Dot plots show ratios of the amount of F-actin in the P to the total amount of F-actin. (C) Representative image of Coomassie Blue–stained SDS-PAGE gel of ACTN4 ABD (one of three independent experiments). Bands correspond to the amount of ACTN4 ABD in S and P after ultracentrifugation. Dot plots show the ratio of the amount of ACTN4 ABD in the P to the total amount of ACTN4 ABD. (D) Illustration of bundling assay. F-actin was incubated with full-length WT, S159A, and S159D ACTN4 proteins. Full-length ACTN4 protein forms antiparallel homodimers with an ABD at either end, enabling an assessment of actin bundling. After low-speed centrifugation, the P contains F-actin bundled by full-length ACTN4, and the S contains unbundled F-actin and full-length ACTN4. The higher the bundling activity, the higher the ratio of full-length ACTN4-bundled F-actin in the P to total amount. (E) Representative image of Coomassie Blue–stained SDS-PAGE gel of full-length ACTN4 (one of two independent experiments). Bands correspond to the amount of F-actin in S and P after centrifugation. Dot plots show ratios of the amount of F-actin in the P to the total amount of F-actin. (F) Representative image of Coomassie Blue–stained SDS-PAGE gel of F-actin (one of two independent experiments). Bands correspond to the amount of full-length ACTN4 in S and P after centrifugation. Dot plots show ratios of the amount of full-length ACTN4 protein in the P to total amount of full-length ACTN4 protein. *Significant difference from the WT ( P<0.05, t test).
Because binding affinity between the ABD of ACTN4 and F-actin is expected to correlate with ACTN4/F-actin bundling activity, we determined if the full-length S159D ACTN4 bundled more F-actin than did WT ACTN4 (Figure 3D). Full-length ACTN4 protein forms antiparallel homodimers with an ABD at either end, enabling an assessment of actin bundling. We incubated F-actin with full-length ACTN4—WT, S159A, or S159D. After subjecting the mixtures to low-speed ultracentrifugation, the amount of P/T ratio of F-actin and full-length ACTN4 was quantified. For F-actin, we found that more bundled F-actin was present in the pellet when F-actin was incubated with S159D ACTN4 than when F-actin was incubated with WT ACTN4 ( P=0.008) (Figure 3E). We also observed that more full-length S159D ACTN4 was present in association with bundled F-actin compared with full-length WT ACTN4 ( P=0.04) (Figure 3F). Again, there was no difference in the P/T ratio between the full-length of WT and S159A ACTN4, reflecting similar bundling activity between full-length WT and S159A ACTN4. Altogether, similar to disease-causing mutant K255E ACTN4, phosphomimetic S159D ACTN4 exhibits both increased F-actin binding affinity and bundling activity compared with WT ACTN4.
Phosphomimetic Actn4S160D/S160D Podocytes Demonstrate Altered F-Actin Alignment and an Increased Rate of Detachment under Mechanical Stress
To test whether the above in vitro abnormalities associated with phosphomimetic ACTN4 translate into cellular abnormalities, we used CRISPR/Cas9 technology to generate phosphomimetic S160D Actn4 and nonphosphorylatable S160A Actn4 knock-in mouse models (S160 in mice is homologous to S159 in humans). We isolated primary mouse podocytes with a purity of around 90% from these models (Supplemental Figure 2). Using primary cells has the benefit of allowing the study of phosphomimetic, nonphosphorylatable, and WT Actn4 under the control of the cell’s endogenous regulatory machinery rather than an overexpression system. We have observed in prior work that podocytes isolated from disease-causing mutant Actn4K256E/K256E mice demonstrate more spatially oriented F-actin alignment, correlating with a more brittle podocyte.24
To assess whether ACTN4 phosphorylation at S159 changes F-actin alignment, we quantified the F-actin alignment in phalloidin-stained podocytes using the autocorrelation function [C(r)] (Figure 4, A and B, Supplemental Figure 1).24,44 All cells used in this experiment also stained positive for Actn4, confirming that they were podocytes (Supplemental Figure 3). Actn4S160D/S160D podocytes cultured in complete RPMI medium (see Methods) demonstrated significantly increased median autocorrelation length (median=23.6 μm; interquartile range [IQR], 16.3–27.7 μm) compared with WT podocytes (median=16.7 μm; IQR, 13.2–20.9 μm; P=0.03), indicating more spatially correlated F-actin in Actn4S160D/S160D podocytes (Figure 4C). In turn, WT showed significantly increased median autocorrelation length than Actn4S160A/S160A (median=13.7 μm; IQR, 10.1–18.4 μm; P=0.04) (Figure 4C). This result suggests that WT Actn4 in podocytes is partially phosphorylated in complete RPMI medium.
We used mechanically actuatable microfluidic organ-on-a-chip culture devices45 to assess whether Actn4S160D/S160D podocytes detach at a higher rate than WT podocytes under simultaneous fluid shear stress and cyclic strain. Simultaneous exposure to these forces mimics the mechanical stress experienced by podocytes in the glomerulus.4,57 In three independent experiments, Actn4S160D/S160D podocytes detached at a higher rate than WT podocytes after 48 hours of simultaneous cyclic mechanical deformation (10% strain) and fluid shear stress (1.5 dyn/cm2) (Figure 5, B and C and Supplemental Figure 4). Actn4S160D/S/160D podocytes demonstrated nearly a threefold higher rate of detachment (28 of 154; 18.2%) than WT podocytes (12 of 170; 7.1%; P= 0.004). Taken together, these results indicate that phosphorylation of Actn4 at S160, as mimicked by Actn4S160D/S160D, leads to more correlated F-actin alignment and higher rates of podocyte detachment, which is similar to the effect resulting from the disease-causing mutant Actn4.24
Figure 5.
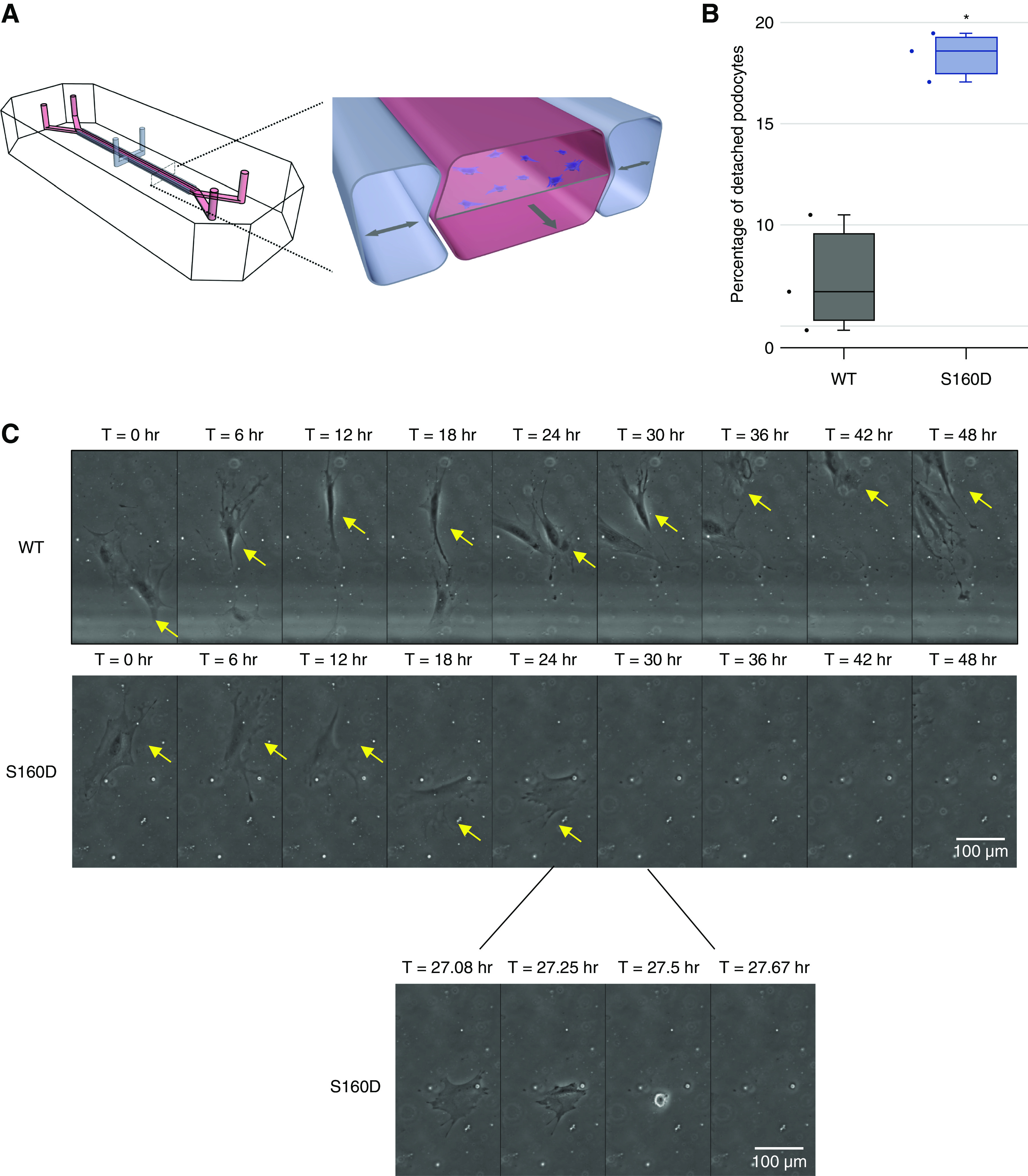
Phosphomimetic Actn4S160D/S160D podocytes demonstrate increased rate of detachment rate under simultaneous exposure to fluid shear stress and cyclic strain. (A) Schematic of organ-on-a-chip microfluidic culture device used for podocyte detachment assay. Cross-section shows top and bottom channels with central membrane on which podocytes are seeded (in the bottom channel). Culture medium is continuously perfused through the bottom channel to subject podocytes to fluid shear stress while cyclic suction is applied to lateral vacuum chambers to apply cyclic strain to the side walls and central membrane with adherent cells. (B) Detachment rates of WT and Actn4S160D/S160D podocytes after 48 hours of exposure to simultaneous mechanical strain and fluid shear stress. * P=0.004 compared with the WT (Fisher exact test of pooled proportions from three independent experiments). (C) Representative series of images depicting podocytes responding to simultaneous strain and shear stress in the mechanically actuatable microfluidic culture devices (each yellow arrow indicates one adherent podocyte). WT podocytes (top panel) remained adherent to the extracellular matrix (ECM)-coated central membrane even after 48 hours of exposure to simultaneous strain and shear stress, whereas Actn4S160D/S160D podocytes (middle panel) detached between T=24 hours and T=30 hours (bottom panel; a series of images between T=24 and 30 hours to more precisely show when Actn4S160D/S160D podocyte detaches). See videos that show the continuous stream of images depicting Actn4S160D/S160D podocyte detachment and WT podocyte adherence in response to 48 hours of simultaneous fluid shear stress and cyclic strain (Supplemental Figure 4). Scale bar, 100 µm.
Phosphomimetic Actn4 S160D/S160D Mice Develop Albuminuria and FSGS after Subtotal Nephrectomy
We next sought to assess whether these biochemical and cellular abnormalities associated with phosphomimetic ACTN4 that we observed in vitro translate into in vivo pathology. Specifically, we wanted to determine whether increased phosphorylation (via Actn4S160D/S160D mice) or absent phosphorylation (via Actn4S160A/S160A mice) leads to abnormal podocyte responses to glomerular hypertension and hyperfiltration—mechanical stress induced by subtotal nephrectomy.48,58 To assess renal injury, we measured albuminuria via albumin-creatinine ratio (micrograms per milligram) at 1 week before nephrectomy and again at 2, 3, and 7 weeks after subtotal nephrectomy (Figure 6A).
Figure 6.
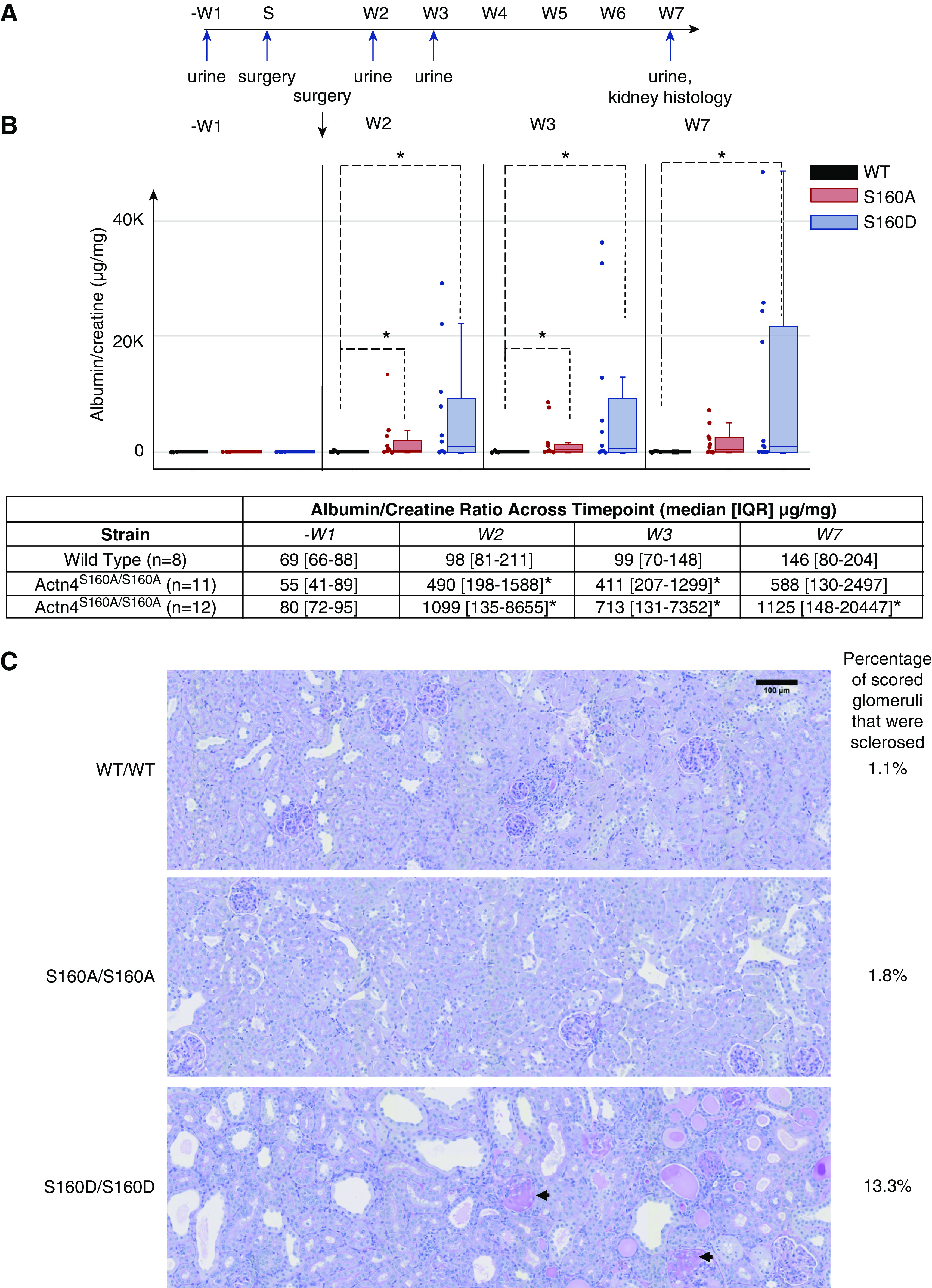
Phosphomimetic S159D mice develop albuminuria and FSGS after subtotal nephrectomy (A) Timeline of urine collection and kidney tissue collection in relation to the day of subtotal nephrectomy. S, surgery; W, week postsurgery; −W1, 1 week prior to surgery. (B) Urine was collected from male WT (n=7), Actn4S160A/S160A (n=12), and Actn4S160D/S160D mice (n=12) at −W1, W2, W3, and W7. Albumin/creatinine ratio (micrograms per milligram) at each timepoint by genotype was quantified. *Significant difference from WT (P<0.05, Mann–Whitney U test). (C) Representative kidney sections from all groups of mice stained with periodic acid–Schiff and imaged at ×20 magnification. Kidney tissues were collected and sectioned at W7. Arrows indicate representative sclerosed glomeruli from Actn4S160D/S160D mice. Scale bar, 100 µm.
We found no significant differences in albuminuria between all groups before subtotal nephrectomy (Figure 6B). By contrast, Actn4S160D/S160D mice demonstrated significantly increased albuminuria compared with WT mice at weeks 2, 3, and 7 after subtotal nephrectomy. Actn4S160A/S160A mice also developed increased albuminuria compared with WT mice, although to lesser degrees, across weeks 2, 3, and 7 after subtotal nephrectomy. At the end of the seventh week after nephrectomy, kidney sections were examined histologically (Figure 6C). A significantly higher proportion of sclerosed glomeruli was found in Actn4S160D/S160D mice (13.3%) compared with the proportion of sclerosed glomeruli found in WT (1.1%; P<0.001) and the proportion in Actn4S160A/S160A mice (1.8%; P<0.001) (Figure 6C). These results suggest that increased phosphorylation of Actn4 at S160, as mimicked by Actn4S160D/S160D, leads to albuminuria and glomerulosclerosis after subtotal nephrectomy. Of note, preventing phosphorylation of Actn4 at S160 (mimicked by Actn4S160A/S160A) also leads to albuminuria (Figure 6B) after subtotal nephrectomy but does not lead to histologically observable FSGS (Figure 6C). These findings suggest that regulation of phosphorylation in vivo is necessary for the cytoskeleton in a healthy podocyte to withstand mechanical forces (supported by the evolutionary conservation of S159) (Figure 1C), whereas pathologically elevated levels of phosphorylation lead to podocyte vulnerability and glomerulosclerosis.
High Extracellular Glucose and TGF-β Stimulate Phosphorylation of ACTN at S159
Our studies demonstrated that phosphomimetic S159D ACTN4 mimics the biochemical and cellular changes and renal pathology seen in patients with FSGS-causing mutant K255E ACTN4. Having observed that increased ACTN4 S159 phosphorylation—as mimicked by an S159D substitution—can cause renal pathology, we sought to identify upstream signaling pathways that lead to increased phosphorylation at ACTN S159. Because high extracellular glucose and TGF-β can both lead to kidney disease and podocyte dysfunction,59−62 we examined whether these stimuli lead to increased ACTN phosphorylation at S159.
We developed an assay using mass spectrometry that quantitatively measures changes in phosphorylation. We used this assay to assess changes in phosphorylation at ACTN S159 in human podocytes cultured in either high glucose (4.5 g/L) or mannitol control base RPMI medium (see Methods). We also used the assay to assess changes in phosphorylation in human podocytes with or without TGF-β treatment (20 ng/ml). After 3 days, mean S159 phosphorylation was nearly twofold higher in the high glucose–treated podocytes in comparison with the mannitol-treated control podocytes (P=0.001) (Figure 7D). Similarly, after 3 days, mean S159 phosphorylation was nearly twofold higher in the TGF-β–treated podocytes in comparison with untreated podocytes (P=0.001) (Figure 7E). These results suggest that high extracellular glucose and TGF-β, stimuli associated with podocyte injury, increase ACTN phosphorylation at S159.
Figure 7.
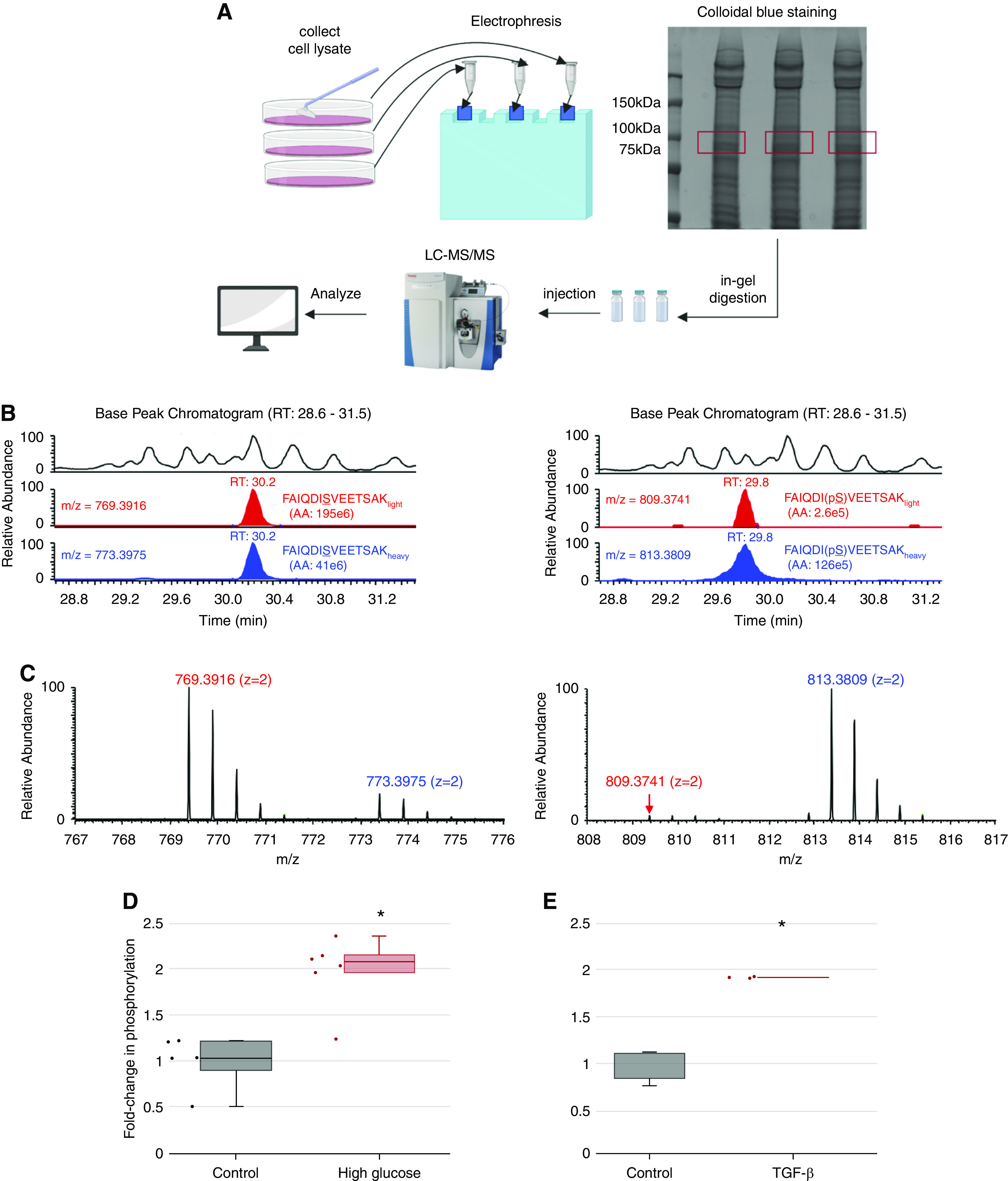
High extracellular glucose and TGF-β stimulate ACTN phosphorylation at S159. (A) Workflow for identification and quantification of peptide phosphorylation by mass spectrometry. Proteins from podocyte lysates were separated by SDS-PAGE and visualized by colloidal blue staining. Prior to mass spectrometry, the gel slice corresponding to the target protein (highlighted in red; around 105 kD) was excised, digested with trypsin, and extracted following in-gel digestion protocol. Tryptic peptides were analyzed by liquid chromatography tandem mass spectrometry (LC-MS/MS). (B) Identification and quantification of target peptide by mass spectrometry. Left panel (from top to bottom) shows the representative zoomed base peak chromatogram of the full mass spectrum, extracted ion chromatogram (XIC) of FAIQDISVEETSAKlight (red), and XIC of FAIQDISVEETSAKheavy (blue) with matching retention times at 30.2 minutes. Right panel (from top to bottom) shows the representative zoomed base peak chromatogram of the full mass spectrum, XIC of FAIQDI(pS)VEETSAKlight (red), and XIC of FAIQDI159(pS)VEETSAKheavy (blue) with matching retention times at 29.8 minutes. RT, room temperature. (C) Mass spectrum of nonphosphopeptide and phosphopeptide. (Left panel) Mass spectra of doubly charged nonphosphopeptide FAIQDISVEETSAKlight and FAIQDISVEETSAKheavy are shown at m/z 769.3916 (z=2) and 773.3975 (z=2), respectively. (Right panel) Mass spectra of doubly charged phosphopeptide FAIQDI(pS)VEETSAKlight and FAIQDI159(pS)VEETSAKheavy are shown at m/z 809.3741(z=2) and 813.3809 (z=2), respectively. (D) Effect of glucose on ACTN phosphorylation at S159. Samples were derived from human podocytes cultured in either high glucose medium (4.5 g/L) or mannitol control base RPMI medium. The phosphorylation level for each sample was calculated using the following equation: 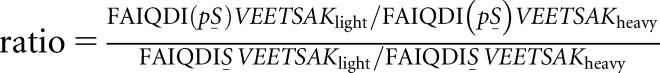 . The numerator of this ratio reflects the amount of phosphopeptide, and the denominator reflects the amount of nonphosphopeptide. The equation, therefore, calculates the ratio of phosphopeptide to nonphosphopeptide for each sample. The average ratio of phosphopeptide to nonphosphopeptide was calculated for the control group, and all results were normalized by this value to represent the fold change of phosphorylation (y axis) in response to high glucose treatment. *Significant difference from control (P<0.01, t test). (E) Effect of TGF-β treatment on ACTN phosphorylation at S159. Samples were derived from human podocytes cultured in base RPMI medium with or without TGF-β treatment (20 ng/ml). *Significant difference from control (P<0.01, t test).
. The numerator of this ratio reflects the amount of phosphopeptide, and the denominator reflects the amount of nonphosphopeptide. The equation, therefore, calculates the ratio of phosphopeptide to nonphosphopeptide for each sample. The average ratio of phosphopeptide to nonphosphopeptide was calculated for the control group, and all results were normalized by this value to represent the fold change of phosphorylation (y axis) in response to high glucose treatment. *Significant difference from control (P<0.01, t test). (E) Effect of TGF-β treatment on ACTN phosphorylation at S159. Samples were derived from human podocytes cultured in base RPMI medium with or without TGF-β treatment (20 ng/ml). *Significant difference from control (P<0.01, t test).
Discussion
Point mutations in the ABD of ACTN4 cause a podocyte-mediated form of proteinuric glomerulosclerosis by increasing the strength of the interaction between ACTN4 and F-actin. The altered interaction renders the podocyte vulnerable to mechanical stress.24 This study shows that a post-translational modification to the ABD of ACTN4—increased phosphorylation at S159—leads to similar alterations in its interaction with F-actin that also correlate with podocyte vulnerability and proteinuric kidney disease in mice. Phosphomimetic S159D ACTN4 demonstrated increased F-actin binding and bundling activity compared with WT, and Actn4S160D/S160D podocytes harbored more correlated F-actin alignment and demonstrated higher rates of substrate detachment in response to mechanical stress. Phosphomimetic Actn4S160D/S160D mice developed albuminuria and glomerulosclerosis after subtotal nephrectomy. Moreover, we found that high extracellular glucose and TGF-β stimulate phosphorylation of ACTN at S159. Our findings demonstrate that ACTN4 may modulate kidney function and podocyte response to stress via a nongenetic modification. They also reinforce that enhanced F-actin binding is a common mediator of ACTN4-associated kidney disease.
Other phosphorylation sites have been detected in ACTN4 across cell types (including podocytes), and 12 of these sites reside within the ABD of ACTN4.63 For example, Y265 phosphorylation has been detected in a variety of tissues. Phosphomimetic Y265E ACTN4 showed increased F-actin binding affinity and predominantly located to the perinuclear actin network.64 At the N terminus of the ACTN4 (N-terminal to the ABD), tyrosine 4 (Y4) and Y31 were found to be phosphorylated by the EGF receptor in fibroblasts.65 A dually phosphomimetic ACTN4 Y4E/Y31E showed decreased F-actin binding affinity.65,66 Although the above-mentioned phosphorylation events were associated with cellular changes, we did not detect phosphorylation at these sites in human podocytes. We, therefore, focused on S159 because (1) it is located in the same functional domain (ABD) as the FSGS-causing mutation K255E; (2) it is highly conserved across species; and (3) when S159 phosphorylation is mimicked, we observe pathologic consequences to the kidney.
All known disease-causing mutations of ACTN4 enhance ACTN4’s binding affinity to F-actin.56 Our findings show that phosphorylation of ACTN4 also enhances binding affinity. Two main theories have been proposed to explain the mechanism underlying this increased affinity. One theory posits that mutations change the ABD conformation to increase the accessibility of its F-actin binding site to F-actin.67,68 The other theory suggests that mutations alter protein charge, but not conformation, to enhance ACTN4’s binding affinity to F-actin.34 We found that phosphorylation of S159 (mimicked by S159D) within the ABD does not change the structure of the ACTN4 protein by itself but does change a localized region of surface charge from neutral to negative. However, we cannot rule out the possibility that dynamic conformational changes occur when ACTN4 actively binds to F-actin in a cellular environment. Resolving the structure of ACTN4 crosslinked with F-actin could provide more mechanistic insights into normal and abnormal interactions between these two key proteins in the podocyte cytoskeleton.
The increased binding affinity resulting from phosphorylation was associated with more correlated F-actin alignment in podocytes and increased rates of podocyte detachment. In our prior work, more correlated F-actin alignment was seen in disease-causing mutant podocytes. This altered alignment was associated with brittle podocytes that, when faced with mechanical stress, demonstrated breakages in their cytoskeletons, failure of contraction, and increased rates of detachment.24,57 In the current work, the more correlated F-actin alignment caused by phosphomimetic S159D was also associated with podocyte dysfunction in response to mechanical stress—manifested by increased rates of detachment under simultaneous fluid shear stress and cyclic strain as well as albuminuria and glomerulosclerosis in mice subjected to subtotal nephrectomy. These findings suggest that, like the disease-causing mutation K255E, increased phosphorylation of ACTN4 at S159 alters F-actin alignment to compromise the cytoskeleton, rendering the podocyte vulnerable to the mechanical stresses it experiences in vivo.
We found that high glucose and TGF-β stimulate phosphorylation of ACTN in WT podocytes. These stimuli are known injurious markers in common forms of CKD. Podocytes are subjected to high extracellular glucose in diabetic nephropathy, and glucose may injure podocytes and other renal cells through proapoptotic and proinflammatory pathways.59,62 TGF-β has been implicated in both diabetic and hypertensive nephropathy, and it is thought to cause cellular hypertrophy, proliferation, and apoptosis.60,61 Our results open the possibility that high glucose and TGF-β may also compromise WT podocyte integrity through affecting its cytoskeleton by stimulating the phosphorylation of ACTN4. These findings call for further investigation into the role of ACTN4 and its regulation in more common, nongenetic forms of CKD.
Identification of the kinases and phosphatases that regulate ACTN4 S159 phosphorylation will allow for more direct and dynamic studies of how phosphorylation regulates ACTN4–F-actin binding. Although phosphomimetic models such as S159D have been validated to mimic the effects of true phosphorylation,53,55 these models represent an “all-or-none” phenomenon. Indeed, the mild albuminuria associated with the S159A nonphosphorylatable mouse model may suggest that the ability to regulate ACTN4–F-actin binding by phosphorylation is physiologically important. A physiologic state of phosphorylation (static or dynamic) is likely necessary for healthy podocyte cytoskeleton homeostasis. Knowledge of the relevant kinase(s) and phosphatase(s) will enable a more detailed mechanistic study of dose-dependent alterations of the podocyte cytoskeleton resulting from varying degrees of phosphorylation.
In conclusion, our study shows that ACTN4 phosphorylation at S159 regulates the interaction between ACTN4 and F-actin. Increased phosphorylation of ACTN4 leads to biochemical, cellular, and whole-animal pathology that is similar to mutant ACTN4-mediated FSGS. Although genetic mutations and their role in podocyte-mediated kidney disease have been well described, our findings suggest that alteration of ACTN4’s function by post-translational modification may also be a mediator of podocyte injury.
Disclosures
Dr. Alper reports a grant and consultation fees from QUEST Diagnostics, consultation fees from the Broad Institute of Harvard and Massachusetts Institute of Technology, from the Medical University of Vienna, and from the Swiss National Science Foundation, all unrelated to this submitted work. Dr. Ingber reports personal fees and equity holdings from Emulate Inc., grants from Astrazeneca and Fulcrum, personal fees from Roche, and multiple patents licensed to Emulate Inc. Dr. Muntel reports personal fees from Biognosys AG outside the submitted work. Dr. Novak reports multiple patents licensed to Emulate, Inc. Dr. Pollak reports patents related to APOL1, owns equity in Apolo1bio, and receives research funding and has consulted for Vertex, unrelated to the submitted work. Dr. Schlondorff and Dr. Pollak are named as inventors on a patent for INF2 mutation analysis in FSGS, unrelated to the submitted work. All remaining authors have nothing to disclose.
Funding
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases grants K01 DK114329 (to Dr. Feng), T32 DK007199 (to Dr. Feng), P30 DK096493 (to Dr. Gurley), and R37DK059588 (to Dr. Pollak). It was also supported by funding from the Wyss Institute for Biologically Inspired Engineering at Harvard University (to Dr. Ingber).
Supplementary Material
Acknowledgments
We thank Lay-Hong Ang for technical assistance with the confocal microscope. We acknowledge the staff at beamlines ID30B [European Synchrotron Radiation Facility (ESRF), Grenoble] and 17-ID [Advanced Photon Source (APS), Chicago] for assistance during data collection. We thank Lei Jin for generating the illustration shown in Figure 5A. Figure 3A and D and Figure 7A were generated using BioRender. We also thank Clark DuMontier for editing the manuscript.
Dr. Feng and Dr. Pollak conceptualized the study; Dr. Ahmed, Dr. Alper, Dr. Birrane, Dr. Ding, Dr. Feng, Dr. Ferrante, Dr. Gurley, Dr. Ingber, Dr. Kumar, Ms. Marquez, Dr. Muntel, Mr. Ng, Dr. Novak, Dr. Schlondorff, Dr. Steen, and Dr. Wang were responsible for methodology; Dr. Birrane, Dr. Feng, Dr. Gurley, and Dr. Stillman were responsible for investigation; Dr. Ding, Dr. Feng, Dr. Ferrante, Dr. Kumar, and Dr. Wang were responsible for visualization; Dr. Feng and Dr. Pollak wrote the original draft; Dr. Ahmed, Dr. Alper, Dr. Birrane, Dr. Ding, Dr. Feng, Dr. Ferrante, Dr. Gurley, Dr. Ingber, Dr. Kumar, Ms. Marquez, Dr. Muntel, Mr. Ng, Dr. Novak, Dr. Pollak, Dr. Schlondorff, Dr. Steen, Dr. Stillman, and Dr. Wang reviewed and edited the writing; Dr. Feng, Dr. Ingber, and Dr. Pollak were responsible for funding acquisition; Dr. Feng, Dr. Gurley, Dr. Ingber, Dr. Pollak, and Dr. Steen were responsible for resources; and Dr. Feng, Dr. Ingber, Dr. Pollak, and Dr. Steen were responsible for supervision.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2019101032/-/DCSupplemental.
Supplemental Figure 1. Quantification of autocorrelation length for a representative podocyte (related to Figure 4C).
Supplemental Figure 2. Representative immunofluorescence image of Wilms tumor 1 (WT-1) (podocyte-specific marker) used to assess the purity of mouse podocyte isolation (related to Figures 4 and 5).
Supplemental Figure 3. Representative immunofluorescence images of F-actin and Actn4 (related to Figure 4A).
Supplemental Figure 4. Supplemental videos showing continuous stream of images depicting Actn4S160D/S160D (right panel) podocyte detachment and WT (left panel) podocyte adherence in response to 48 hours of simultaneous fluid shear stress and cyclic strain (related to Figure 5C).
References
- 1.Welsh GI, Saleem MA: The podocyte cytoskeleton--key to a functioning glomerulus in health and disease. Nat Rev Nephrol 8: 14–21, 2011. [DOI] [PubMed] [Google Scholar]
- 2.Saleem MA, Zavadil J, Bailly M, McGee K, Witherden IR, Pavenstadt H, et al.: The molecular and functional phenotype of glomerular podocytes reveals key features of contractile smooth muscle cells. Am J Physiol Renal Physiol 295: F959–F970, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Neal CR, Crook H, Bell E, Harper SJ, Bates DO: Three-dimensional reconstruction of glomeruli by electron microscopy reveals a distinct restrictive urinary subpodocyte space. J Am Soc Nephrol 16: 1223–1235, 2005. [DOI] [PubMed] [Google Scholar]
- 4.Kriz W, Lemley KV: A potential role for mechanical forces in the detachment of podocytes and the progression of CKD. J Am Soc Nephrol 26: 258–269, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jefferson JA, Shankland SJ: The pathogenesis of focal segmental glomerulosclerosis. Adv Chronic Kidney Dis 21: 408–416, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kriz W, Shirato I, Nagata M, LeHir M, Lemley KV: The podocyte’s response to stress: The enigma of foot process effacement. Am J Physiol Renal Physiol 304: F333–F347, 2013. [DOI] [PubMed] [Google Scholar]
- 7.Chen YM, Liapis H: Focal segmental glomerulosclerosis: Molecular genetics and targeted therapies. BMC Nephrol 16: 101, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fogo AB: Causes and pathogenesis of focal segmental glomerulosclerosis. Nat Rev Nephrol 11: 76–87, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaplan JM, Kim SH, North KN, Rennke H, Correia LA, Tong HQ, et al.: Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat Genet 24: 251–256, 2000. [DOI] [PubMed] [Google Scholar]
- 10.Brown EJ, Schlöndorff JS, Becker DJ, Tsukaguchi H, Tonna SJ, Uscinski AL, et al.: Mutations in the formin gene INF2 cause focal segmental glomerulosclerosis [published correction appears in Nat Genet 42: 361, 2010]. Nat Genet 42: 72–76, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mele C, Iatropoulos P, Donadelli R, Calabria A, Maranta R, Cassis P, et al. ; PodoNet Consortium : MYO1E mutations and childhood familial focal segmental glomerulosclerosis. N Engl J Med 365: 295–306, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heath KE, Campos-Barros A, Toren A, Rozenfeld-Granot G, Carlsson LE, Savige J, et al.: Nonmuscle myosin heavy chain IIA mutations define a spectrum of autosomal dominant macrothrombocytopenias: May-Hegglin anomaly and Fechtner, Sebastian, Epstein, and Alport-like syndromes. Am J Hum Genet 69: 1033–1045, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Akilesh S, Suleiman H, Yu H, Stander MC, Lavin P, Gbadegesin R, et al.: Arhgap24 inactivates Rac1 in mouse podocytes, and a mutant form is associated with familial focal segmental glomerulosclerosis. J Clin Invest 121: 4127–4137, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gupta IR, Baldwin C, Auguste D, Ha KC, El Andalousi J, Fahiminiya S, et al.: ARHGDIA: A novel gene implicated in nephrotic syndrome. J Med Genet 50: 330–338, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gbadegesin RA, Hall G, Adeyemo A, Hanke N, Tossidou I, Burchette J, et al.: Mutations in the gene that encodes the F-actin binding protein anillin cause FSGS. J Am Soc Nephrol 25: 1991–2002, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gee HY, Zhang F, Ashraf S, Kohl S, Sadowski CE, Vega-Warner V, et al.: KANK deficiency leads to podocyte dysfunction and nephrotic syndrome. J Clin Invest 125: 2375–2384, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schell C, Huber TB: The evolving complexity of the podocyte cytoskeleton. J Am Soc Nephrol 28: 3166–3174, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kunishima S, Okuno Y, Yoshida K, Shiraishi Y, Sanada M, Muramatsu H, et al.: ACTN1 mutations cause congenital macrothrombocytopenia. Am J Hum Genet 92: 431–438, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chiu C, Bagnall RD, Ingles J, Yeates L, Kennerson M, Donald JA, et al.: Mutations in alpha-actinin-2 cause hypertrophic cardiomyopathy: A genome-wide analysis. J Am Coll Cardiol 55: 1127–1135, 2010. [DOI] [PubMed] [Google Scholar]
- 20.North KN, Yang N, Wattanasirichaigoon D, Mills M, Easteal S, Beggs AH: A common nonsense mutation results in alpha-actinin-3 deficiency in the general population. Nat Genet 21: 353–354, 1999. [DOI] [PubMed] [Google Scholar]
- 21.Weins A, Kenlan P, Herbert S, Le TC, Villegas I, Kaplan BS, et al.: Mutational and Biological Analysis of alpha-actinin-4 in focal segmental glomerulosclerosis. J Am Soc Nephrol 16: 3694–3701, 2005. [DOI] [PubMed] [Google Scholar]
- 22.Feng D, Steinke JM, Krishnan R, Birrane G, Pollak MR: Functional validation of an alpha-actinin-4 mutation as a potential cause of an aggressive presentation of adolescent focal segmental glomerulosclerosis: Implications for genetic testing. PLoS One 11: e0167467, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yao NY, Becker DJ, Broedersz CP, Depken M, Mackintosh FC, Pollak MR, et al.: Nonlinear viscoelasticity of actin transiently cross-linked with mutant α-actinin-4. J Mol Biol 411: 1062–1071, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feng D, Notbohm J, Benjamin A, He S, Wang M, Ang LH, et al.: Disease-causing mutation in α-actinin-4 promotes podocyte detachment through maladaptation to periodic stretch. Proc Natl Acad Sci U S A 115: 1517–1522, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yao J, Le TC, Kos CH, Henderson JM, Allen PG, Denker BM, et al.: Alpha-actinin-4-mediated FSGS: An inherited kidney disease caused by an aggregated and rapidly degraded cytoskeletal protein. PLoS Biol 2: e167, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Henderson JM, Al-Waheeb S, Weins A, Dandapani SV, Pollak MR: Mice with altered alpha-actinin-4 expression have distinct morphologic patterns of glomerular disease. Kidney Int 73: 741–750, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saleem MA, O’Hare MJ, Reiser J, Coward RJ, Inward CD, Farren T, et al.: A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J Am Soc Nephrol 13: 630–638, 2002. [DOI] [PubMed] [Google Scholar]
- 28.Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M: In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc 1: 2856–2860, 2006. [DOI] [PubMed] [Google Scholar]
- 29.Kumar M, Joseph SR, Augsburg M, Bogdanova A, Drechsel D, Vastenhouw NL, et al.: MS western, a method of multiplexed absolute protein quantification is a practical alternative to western blotting. Mol Cell Proteomics 17: 384–396, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bourmaud A, Gallien S, Domon B: Parallel reaction monitoring using quadrupole-Orbitrap mass spectrometer: Principle and applications. Proteomics 16: 2146–2159, 2016. [DOI] [PubMed] [Google Scholar]
- 31.Gallien S, Duriez E, Crone C, Kellmann M, Moehring T, Domon B: Targeted proteomic quantification on quadrupole-orbitrap mass spectrometer. Mol Cell Proteomics 11: 1709–1723, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Otwinowski Z, Minor W: [20] Processing of X-ray diffraction data collected in oscillation mode. Meth. Enzymol 276: 307–326 10.1016/S0076-6879(97)76066-X [DOI] [PubMed] [Google Scholar]
- 33.Vagin A, Teplyakov A: Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr 66[Pt 1]: 22–25, 2010. 10.1107/S0907444909042589 [DOI] [PubMed] [Google Scholar]
- 34.Lee SH, Weins A, Hayes DB, Pollak MR, Dominguez R: Crystal structure of the actin-binding domain of alpha-actinin-4 Lys255Glu mutant implicated in focal segmental glomerulosclerosis. J Mol Biol 376: 317–324, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kantardjieff KA, Rupp B: Matthews coefficient probabilities: Improved estimates for unit cell contents of proteins, DNA, and protein-nucleic acid complex crystals. Protein science: a publication of the Protein Society, 12: 1865–1871, 2003 [DOI] [PMC free article] [PubMed]
- 36.Murshudov GN, Vagin AA, Dodson EJ: Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr 53[Pt 3]: 240–255, 1997. 10.1107/S0907444996012255 [DOI] [PubMed] [Google Scholar]
- 37.Emsley P, Lohkamp B, Scott WG, Cowtan K: Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr 66[Pt 4]: 486–501, 2010. 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, et al.: MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr 66[Pt 1]: 12–21, 2010. 10.1107/S0907444909042073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.RAMACHANDRAN GN, RAMAKRISHNAN C, SASISEKHARAN V: Stereochemistry of polypeptide chain configurations. J. Mol. Biol 7: 95–99, 1963. 10.1016/s0022-2836(63)80023-6 [DOI] [PubMed] [Google Scholar]
- 40.Fenn TD, Ringe D, Petsko GA: POVScript+: A program for model and data visualization using persistence of vision ray-tracing. J Appl Cryst 36: 944–947, 2003 [Google Scholar]
- 41.Jurrus E, Engel D, Star K, Monson K, Brandi J, Felberg LE, Brookes DH, Wilson L, Chen J, Liles K, Chun M, Li P, Gohara DW, Dolinsky T, Konecny R, Koes DR, Nielsen JE, Head-Gordon T, Geng W, Krasny R, Wei GW, Holst MJ, McCammon JA, Baker NA: Improvements to the APBS biomolecular solvation software suite. Protein Science: A publication of the Protein Society, 27: 112–128, 2018 [DOI] [PMC free article] [PubMed]
- 42.Mundel P, Reiser J, Zúñiga Mejía Borja A, Pavenstädt H, Davidson GR, Kriz W, et al.: Rearrangements of the cytoskeleton and cell contacts induce process formation during differentiation of conditionally immortalized mouse podocyte cell lines. Exp Cell Res 236: 248–258, 1997. [DOI] [PubMed] [Google Scholar]
- 43.Püspöki Z, Storath M, Sage D, Unser M: Transforms and operators for directional bioimage analysis: A survey. Adv Anat Embryol Cell Biol 219: 69–93, 2016. [DOI] [PubMed] [Google Scholar]
- 44.Gupta M, Sarangi BR, Deschamps J, Nematbakhsh Y, Callan-Jones A, Margadant F, et al.: Adaptive rheology and ordering of cell cytoskeleton govern matrix rigidity sensing. Nat Commun 6: 7525, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Novak R, Didier M, Calamari E, Ng CF, Choe Y, Clauson SL, et al.: Scalable fabrication of stretchable, dual channel, microfluidic organ chips [published online ahead of print October 20, 2018]. J Vis Exp doi:10.3791/58151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Srivastava T, Celsi GE, Sharma M, Dai H, McCarthy ET, Ruiz M, et al.: Fluid flow shear stress over podocytes is increased in the solitary kidney. Nephrol Dial Transplant 29: 65–72, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ferrell N, Sandoval RM, Bian A, Campos-Bilderback SB, Molitoris BA, Fissell WH: Shear stress is normalized in glomerular capillaries following ⅚ nephrectomy. Am J Physiol Renal Physiol 308: F588–F593, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Salzler HR, Griffiths R, Ruiz P, Chi L, Frey C, Marchuk DA, et al.: Hypertension and albuminuria in chronic kidney disease mapped to a mouse chromosome 11 locus. Kidney Int 72: 1226–1232, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chang JH, Gurley SB: Assessment of diabetic nephropathy in the Akita mouse. Methods Mol Biol 933: 17–29, 2012. [DOI] [PubMed] [Google Scholar]
- 50.Young S, Struys E, Wood T: Quantification of creatine and guanidinoacetate using GC-MS and LC-MS/MS for the detection of cerebral creatine deficiency syndromes. Curr Protoc Hum Genet Chapter 17: Unit 17.3, 2007 [DOI] [PubMed] [Google Scholar]
- 51.R Core Team : R: A Language and Environment for Statistical Computing, Vienna, Austria, R Foundation for Statistical Computing, 2016 [Google Scholar]
- 52.Mann HB, Whitney DR: On a test of whether one of two random variables is stochastically larger than the other. Ann Math Stat 18: 50–60, 1947 [Google Scholar]
- 53.Sieracki NA, Komarova YA: Studying Cell Signal Transduction with Biomimetic Point Mutations, Chicago, University of Illinois, 2013 [Google Scholar]
- 54.Singh SA, Winter D, Kirchner M, Chauhan R, Ahmed S, Ozlu N, et al.: Co-regulation proteomics reveals substrates and mechanisms of APC/C-dependent degradation. EMBO J 33: 385–399, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kristensen AS, Jenkins MA, Banke TG, Schousboe A, Makino Y, Johnson RC, et al.: Mechanism of Ca2+/calmodulin-dependent kinase II regulation of AMPA receptor gating. Nat Neurosci 14: 727–735, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Feng D, DuMontier C, Pollak MR: The role of alpha-actinin-4 in human kidney disease. Cell Biosci 5: 44, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Feng D, DuMontier C, Pollak MR: Mechanical challenges and cytoskeletal impairments in focal segmental glomerulosclerosis. Am J Physiol Renal Physiol 314: F921–F925, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nagata M, Kriz W: Glomerular damage after uninephrectomy in young rats. II. Mechanical stress on podocytes as a pathway to sclerosis. Kidney Int 42: 148–160, 1992. [DOI] [PubMed] [Google Scholar]
- 59.Lewko B, Stepinski J: Hyperglycemia and mechanical stress: Targeting the renal podocyte. J Cell Physiol 221: 288–295, 2009. [DOI] [PubMed] [Google Scholar]
- 60.Schnaper HW, Jandeska S, Runyan CE, Hubchak SC, Basu RK, Curley JF, et al.: TGF-beta signal transduction in chronic kidney disease. Front Biosci 14: 2448–2465, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lavoie P, Robitaille G, Agharazii M, Ledbetter S, Lebel M, Larivière R: Neutralization of transforming growth factor-beta attenuates hypertension and prevents renal injury in uremic rats. J Hypertens 23: 1895–1903, 2005. [DOI] [PubMed] [Google Scholar]
- 62.Toth-Manikowski S, Atta MG: Diabetic kidney disease: Pathophysiology and therapeutic targets. J Diabetes Res 2015: 697010, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hornbeck PV, Kornhauser JM, Tkachev S, Zhang B, Skrzypek E, Murray B, et al.: PhosphoSitePlus: A comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res 40: D261–D270, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Feng Y, Ngu H, Alford SK, Ward M, Yin F, Longmore GD: α-Actinin1 and 4 tyrosine phosphorylation is critical for stress fiber establishment, maintenance and focal adhesion maturation. Exp Cell Res 319: 1124–1135, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shao H, Wu C, Wells A: Phosphorylation of alpha-actinin 4 upon epidermal growth factor exposure regulates its interaction with actin. J Biol Chem 285: 2591–2600, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Travers T, Shao H, Joughin BA, Lauffenburger DA, Wells A, Camacho CJ: Tandem phosphorylation within an intrinsically disordered region regulates ACTN4 function. Sci Signal 8: ra51, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Weins A, Schlondorff JS, Nakamura F, Denker BM, Hartwig JH, Stossel TP, et al.: Disease-associated mutant alpha-actinin-4 reveals a mechanism for regulating its F-actin-binding affinity. Proc Natl Acad Sci U S A 104: 16080–16085, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Galkin VE, Orlova A, Salmazo A, Djinovic-Carugo K, Egelman EH: Opening of tandem calponin homology domains regulates their affinity for F-actin. Nat Struct Mol Biol 17: 614–616, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.