

Significance Statement
CKD is associated with increased oxidative stress that correlates with the occurrence of cardiovascular events. Oxidative stress induces modifications that particularly affect circulating lipoproteins such as HDL that exhibit atheroprotective properties in vitro. However, information about the antithrombotic properties of HDL in CKD is lacking. The authors demonstrate that HDL from a CKD rabbit model and patients on hemodialysis exhibited an impaired ability to inhibit platelet aggregation, suggesting that properties of altered HDL may contribute to the increased cardiovascular risk in this patient population. They also describe the putative role of carbonylation by 4-hydroxynonenal adduction in these properties. This study provides important insights into the potential implication of HDL modifications in atherothrombosis and cardiovascular morbidity and mortality among patients on dialysis.
Keywords: chronic kidney disease, platelets, cardiovascular disease, dyslipidemia, thrombosis, dialysis
Visual Abstract

Abstract
Background
CKD is associated with increased oxidative stress that correlates with occurrence of cardiovascular events. Modifications induced by increased oxidative stress particularly affect circulating lipoproteins such as HDL that exhibit antiatheromatous and antithrombotic properties in vitro.
Methods
To explore the specific role of oxidative modifications of HDL in CKD and their effect on the platelet-targeting antiaggregant properties of HDL, we used a CKD (5/6 nephrectomy) rabbit model. For ex vivo assessment of the antiaggregant properties of HDL, we collected blood samples from 15 healthy volunteers, 25 patients on hemodialysis, and 20 on peritoneal dialysis. We analyzed malondialdehyde, 4-hydroxynonenal (HNE), and 4-hydroxy-2-hexenal protein adduct levels. Platelet aggregation and activation were assessed by aggregometry, thromboxane B2 assay, or FACS. We modified HDL from controls by incubating it overnight at 37°C with 100 µM of HNE.
Results
HDL from CKD rabbits and patients on hemodialysis had HNE adducts. The percentage of platelet aggregation or activation induced by collagen was significantly higher when platelets were incubated with HDL from CKD rabbit and hemodialysis groups than with HDL from the control group. In both rabbits and humans, platelet aggregation and activation were significantly higher in the presence of HNE-modified HDL than with HDL from their respective controls. Incubation of platelets with a blocking antibody directed against CD36 or with a pharmacologic inhibitor of SRC kinases restored the antiaggregative phenotype in the presence of HDL from CKD rabbits, patients on hemodialysis and peritoneal dialysis, and HNE-modified HDL.
Conclusions
HDL from CKD rabbits and patients on hemodialysis exhibited an impaired ability to inhibit platelet aggregation, suggesting that altered HDL properties may contribute to the increased cardiovascular risk in this population.
CKD is recognized as a major cardiovascular risk factor.1–3 It is involved in the onset of cardiovascular events such as myocardial infarction, peripheral arterial disease, and cerebral ischemia.1 Cardiovascular mortality remains the major cause of death in patients on hemodialysis (HD) and peritoneal dialysis (PD) despite the constant improvement of RRT.4 CKD is associated with increased oxidative stress that is correlated with the occurrence of cardiovascular events.5–7 This stress is associated with the accumulation of many uremic toxins,8 some of which have recognized cardiovascular effects.9–12 Modifications induced by increased oxidative stress particularly affect circulating lipoproteins such as HDL that exhibit antiatheromatous and antithrombotic properties in vitro.
There are many mechanisms that lead to lipoprotein modification, both enzymatic and nonenzymatic, and these affect different lipoprotein sites.13 Oxidized and carbamylated LDL are present at higher concentrations in patients with CKD compared with patients who are healthy5,14 and play a major role in the onset and aggravation of atherosclerotic lesions.14,15 The concentration of oxidized and carbamylated LDL are, however, lowered by statins.16–18 HDL is considered to be antiatherogenic as a result of its antiaggregant, anti-inflammatory, and antiapoptotic properties,19 as well as its ability to induce cholesterol efflux from macrophages,20 a mechanism known to be atheroprotective.21 HDL is not affected by conventional hypolipidemic strategies, and functional studies in CKD have found impaired biologic effects, including decreased capacity of macrophage cholesterol efflux22–24 and antioxidation,24 as well as impaired endothelial protection.25 These disorders become more significant with CKD progression.25 In parallel, HDL oxidation has been related to the onset of cardiovascular events in patients with CKD.26 These functional modifications could partly explain the failure of statins to reduce the cardiovascular risks associated with HD.27,28
Platelets are the target of antiaggregant treatments that have been shown to be beneficial in reducing cardiovascular events in at-risk populations. Oxidized LDL of patients who are obese and diabetic has shown some proaggregating properties.29,30 In CKD, carbamylated LDL exhibits proaggregant properties through binding with lectin-like oxidized LDL receptor 1,31 and oxidized LDL through binding to the CD36 receptor.32 Oxidation of HDL leads to a loss of antiaggregation properties in some populations,33 although this observation was not found in type 2 diabetes,34 where oxidized HDL has shown antiaggregant properties through a Scavenger receptor class B type 1 (SR-B1)–mediated pathway.35 However, to the best of our knowledge, there is no published data regarding the effect of CKD on the antiaggregant properties of HDL.
In this study, we aimed to explore the specific role of CKD in the oxidation profile of HDL and the effects of HDL modification on platelet aggregation in a rabbit model of CKD. We then aimed to assess, ex vivo, the antiaggregant properties of HDL isolated from patients on HD and PD.
Methods
Subjects and Ethics Statement
Patients were sampled at the Lyon teaching hospitals. Control patients were healthy volunteers for a living kidney donation, hospitalized for their predonation laboratory and clinical workup. Patients on HD were sampled within the HD unit of the Edouard Herriot Hospital (Lyon, France) before the midweek session. Patients on PD were sampled in the PD unit of the Association pour l’Utilisation du Rein Artificiel (AURAL; Lyon, France). Inclusion criteria were participants aged ≥18 years who were undergoing HD or PD for >6 months. Exclusion criteria were the presence of diabetes mellitus, ongoing inflammatory disease, liver cirrhosis, recent cardiovascular event (<3 months; myocardial infarction, stroke, acute peripheral artery occlusion), uncontrolled anemia, coagulopathy, and body mass index >35 kg/m2. The study was conducted in accordance with the Declaration of Helsinki and was approved by the institutional review board (Comité de protection des personnes Lyon Sud Est IV, Centre Léon Berard, reference, L16-57). A written informed consent was obtained from all subjects. Blood samples were obtained by venipuncture on EDTA-coated tubes. Blood samples were centrifuged at 3500 × g for 10 minutes to isolate plasma and were then stored at −80°C until use (Supplemental Methods).
Animal Procedures
All experiments were performed under the authorization number 69-266-0501 and agreed with the guidelines laid down by the French Ministry of Agriculture (number 2013-118) and the European Union Council Directive for the protection of animals used for scientific purposes of September 22, 2010 (2010/63UE). Adult male white New Zealand rabbits (CEGAVssc, Saint Mars d’Egrenne, France) were housed in individual cages at constant ambient temperature (21–23°C) and humidity (45%–50%) with a 12-hour light cycle. All animals had free access to tap water. After a 7-day period of acclimation, rabbits were randomized to either the 5/6 nephrectomy group or the control group. Nephrectomy was performed as described by Gotloib et al.36 Briefly, rabbits were anesthetized using an intramuscular injection of ketamine (50 mg/kg), xylazine (5 mg/kg), and acepromazine (0.5 mg/kg; Centravet, Lapalisse, France). The rabbit was placed in the right lateral decubitus position. Local anesthesia was performed by a subcutaneous injection of xylocaine 2% (2 ml) at the incision site. The left kidney was externalized and the perirenal adipose tissue was gently dissected. The two poles of the kidney were electrocoagulated using an electric needle to produce a two-third reduction of the left kidney mass. At 1 week after surgery, a unilateral right nephrectomy was performed. The kidney pedicle was carefully sectioned between the kidney and the ligature. The control animals underwent the same procedures (general anesthesia, skin, and muscle incisions) followed by a simple kidney mobilization. All rabbits were given buprenorphine (0.05 mg/kg subcutaneously, two times a day) for 2 days to prevent postsurgical pain. Food intake and body weight were measured on a daily and twice-weekly basis, respectively (Supplemental Methods).
Measurement of GFR
The GFR in rabbits was measured through the kinetics of decrease of plasma concentration of iohexol as described in Florens et al.37 Rabbits were anesthetized with an intravenous injection of 27.5 mg/kg of sodium pentobarbital (Centravet). A PE-50 catheter was introduced in the left jugular vein for the injections and a PE-60 catheter was inserted in the left carotid artery for the blood sampling. A 1-ml bolus of iohexol (Omnipaque 300; GE Healthcare, Chicago, IL) was performed and the timer was started after flushing the residue left in the jugular catheter with a saline solution. Blood was sampled at 5, 15, 30, 45, 60, 120, and 180 minutes on lithium-heparin–coated tubes. The plasma was aliquoted after centrifugation at 5900 × g for 2 minutes and stored at −20°C. The serum iohexol concentration was measured by HPLC as previously described38 and GFR was calculated using a bicompartmental model equation.
Euthanasia, Necropsy, and Renal Histology
At the end of the GFR measurement, rabbits were deeply anesthetized with an overdose of sodium pentobarbital (70 mg/kg, intravenously). Blood was removed by cardiac puncture and placed in EDTA-coated tubes. After a centrifugation at 1250 × g for 10 minutes, plasma was aliquoted and stored at −80°C. Urine was obtained via a direct bladder puncture and stored at −20°C. Kidneys were removed, weighed, and stored in buffered formalin 10% (w/v) for histologic examination. Kidneys were dehydrated using ethanol, embedded in paraffin, and sliced. Hematoxylin Erythrosine Saffron and Sirius Red stainings were performed.
Isolation of Lipoproteins from the Plasma
Lipoproteins were separated from plasma by stepwise potassium bromide (KBr) density gradient ultracentrifugation as described by Havel et al.39 Briefly, plasma was fractionated in the Beckman ultracentrifuge with a TLA 100.3 Rotor (Beckman, Brea, CA). A first centrifugation at 100,000 × g for 3 hours 30 minutes at 15°C was performed to remove the top layer corresponding to VLDL and chylomicrons. Then, the plasma density was adjusted to 1.063 g/ml with KBr (M=119.01 g/mol). After a second centrifugation at 100,000 × g for 5 hours at 4°C, the orange ring, corresponding to LDL, was collected. Finally, plasma density was adjusted to 1.21 g/ml with KBr and, after centrifugation at 100,000 × g for 6 hours 30 minutes at 4°C, the orange ring corresponding to HDL was collected. For the isolation of all of the lipoproteins together, a single ultracentrifugation was performed at 100,000 × g for 6 hours 30 minutes at 4°C after an adjustment of plasma density to 1.21 g/ml with KBr.
For platelet aggregation and the copper-induced HDL oxidation assay, freshly isolated HDL samples were used to prevent HDL ultrastructure modification due to freezing.40 After ultracentrifugation, lipoproteins were extensively dialyzed against PBS with 1 mM EDTA for 3 hours, twice at room temperature and then overnight at 4°C. A last dialysis without EDTA was performed just before the platelet aggregation and copper-induced oxidation. HDL samples were stored at 4°C for a maximum of 48 hours before the experiments.
Biochemistry
Serum creatinine measurement was performed using the Siemens enzymatic method (on the Dimension Vista System; Siemens Healthcare, Erlangen, Germany). Urea was measured using the urease test (Vista 1500). Cholesterol and triacylglycerol levels were measured using enzymatic kits (Biomerieux, Marcy l’Etoile, France). HDL concentration was measured with an enzymatic kit (Abcam, Paris, France). Malondialdehyde (MDA) was measured by HPLC coupled to ultraviolet-visible detection (Diode Array Detector) as described by Grotto et al.41 Antioxidant activity (AOA) of the plasma was measured as described by Koracevic et al.42 Proteinuria was measured using the Bradford protein assay (BioRad, Marne-la-Coquette, France) using BSA as a standard.
Lipoprotein Assays
MDA concentration in HDL was determined by HPLC according to the method described by Therasse and Lemonnier.43 Anti–4-hydroxynonenal (HNE)-Michael adduct (reference 393207) and anti–4-hydroxy-2-hexenal (HHE)-Michael adduct (reference NOF-N213730-EX) antibodies were obtained from Calbiochem (San Diego, CA) and Cosmobio (Tokyo, Japan), respectively. A total of 50 μg HDL was loaded directly onto a nitrocellulose membrane using the Bio-Dot apparatus (BioRad). Following saturation with 5% BSA, membranes were probed overnight with primary antibodies: anti-HHE-Michael adducts or anti-HNE-Michael adducts. After incubation with horseradish peroxidase–coupled secondary antibodies, membranes were processed for chemiluminescence (ECL Plus; GE Healthcare) and quantitated by densitometry using ImageJ software (National Institutes of Health, Bethesda, MD). 8-Isoprostane was measured using an immunoassay (Bertin Pharma, Montigny Le Bretonneux, France). HNE was synthetized as described by Soulère et al.44 and was diluted in DMSO. HNE was added to HDL solutions to final concentrations of 1, 10, 50, or 100 µM. After a 16 hour (overnight) incubation at 37°C in a water bath, HDL samples were dialyzed three times against PBS to remove the free fraction of HNE.
Platelet Aggregation, Activation, and Intracellular Pathway Assays
Blood was collected at the regional blood center from healthy volunteers who had not ingested any aspirin or any other nonsteroidal anti-inflammatory drug in the previous 10 days. Platelets were prepared for the assays as described by Lagarde et al.45 The platelet function test was carried out according to the Born turbidimetric method.46 Platelet aggregation was measured in isolated platelets in a dual-channel aggregometer (Chrono-log; Coulter, Margency, France). Platelet suspensions were preincubated for 5 minutes at 37°C in the presence or absence of lipoproteins (0.025 mg of proteins/ml for rabbit, 0.050 mg/ml for human) and stimulated with threshold concentrations of collagen (75±9 ng/ml) while being continuously stirred at 1000 rpm. The threshold concentration of collagen was defined as the concentration of collagen that induced a 50% increase in light transmission. The extent of platelet aggregation was expressed in terms of percentage of change in light transmission 4 minutes after the addition of collagen. Blocking of CD36 or SRB1 receptor was achieved by preincubation with 10 µl of an anti-CD36 (Ab-CD36) or anti-SRB1 antibodies (dilution 1:500; Abcam) for 10 minutes at 37°C before the incubation with HDL. Inhibition of SRC kinases was achieved by the preincubation with Naphthyl PP1 (final concentration 1 µM; Santa Cruz Biothechnologies, Dallas, TX) for 10 minutes at 37°C before the incubation with HDL. Aggregation values from lipoprotein assays were expressed as a percentage of the maximum aggregation induced by the collagen alone (considered as 100%). Levels of thromboxane B2 (TxB2) was measured using an immunoassay expressed as a percentage of the maximum level induced by the collagen alone (considered as 100%; Cayman Chemical, Ann Arbor, MI). After preincubation with HDL and antibodies or pharmacologic inhibitor, platelets were activated with collagen and then fixed in 5% paraformaldehyde and stained with anti-CD62/P-selectin (Thermo Fisher Scientific, San Jose, CA) according to the manufacturer’s protocol and analyzed by flow cytometry.47
Copper-Induced HDL Oxidation
Aliquots of freshly dialyzed HDL (50 µg proteins) were oxidized in the presence of 2.5 µM of copper sulphate within a day after the last dialysis. The oxidation was monitored by measuring the formation of conjugated dienes at 234 nm.48
Mass Spectrometry
Samples were reduced, alkylated, and digested with trypsin at 37°C overnight. They were desalted with spin column C18, dried, and then analyzed in triplicate using an UltiMate 3000 RSLCnano (Thermo Fisher Scientific) coupled online with a Q-Orbitrap (Q Exactive HF; Thermo Fisher Scientific). Briefly, peptides were separated on a C18 nanocolumn with a linear gradient of acetonitrile and analyzed in a Top 20 HCD (higher collision dissociation) data-dependent mass spectrometer. Data were processed by database searching using SequestHT (Thermo Fisher Scientific) with Proteome Discoverer 2.2 software (Thermo Fisher Scientific) against a human Swissprot database and quantified with a label-free quantitation approach. Precursor and fragment mass tolerance were set at 10 ppm and 0.02 Da, respectively. Trypsin was set as enzyme, and up to two missed cleavages were allowed. Oxidation (M), acetylation (protein N-terminus), and HNE (+156.115 Da on lysine, K or histidine, H) were set as variable modification. Peptides were filtered with false discovery rate at 1%. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD013301 (DOI, 10.6019/PXD013301; username, reviewer26456@ebi.ac.uk; password, oMD4gyDT).
Molecular Modeling
To study the accessibility of residues, the structure of truncated human apo A-I (Protein Data Bank [PDB] code 1av1) was downloaded from the PDB database.49 All observations were performed using PyMOL software by representing the α-helices and histidine and lysine residues (colored in blue) in CPK coloring to facilitate the study. The same method was achieved with the HDL model which was download from the PDB database.50
Statistical Analysis
Data were expressed as median±interquartile range. All analyses were performed using GraphPad Prism version 6.0 (GraphPad software, La Jolla, CA). Simple comparisons were made using the Mann–Whitney U test. Multiple comparisons were made with Kruskal–Wallis test and, whenever appropriate, Dunn tests. Differences were considered as significant at P<0.05.
Results
General Characteristics of the CKD Rabbit Model
Rabbits developed a significant renal insufficiency in the CKD group; there was a significant difference (P<0.001) in the median GFR between the control group and the CKD group (−58%; Table 1). CKD rabbits exhibited increased levels of lipid peroxidation in the plasma as evidenced by the tenfold, significant increase in MDA concentration (P<0.001; Table 1). Median AOA of the plasma was significantly (P<0.05) lower in the CKD group compared with control (−15%; Table 1). Histologic examination found a diffuse fibrosis in the parenchyma in the CKD group as well as glomerulomegaly (Supplemental Figure 1).
Table 1.
General characteristics of sham-operated (control) and 5/6 nephrectomized rabbits
| Characteristics | Control (N=9) | CKD (N=8) | P Value |
|---|---|---|---|
| Biometry | |||
| Body weight (kg) | 3.5 (3.2–3.9) | 3.1 (3.0–3.3) | 0.04 |
| Body weight gain (kg) | 1.0 (0.9–1.2) | 0.7 (0.6–0.8) | 0.001 |
| Kidney weight (g) | 19 (16–22) | 14 (12–16) | 0.008 |
| Kidney weight (g/kg body wt) | 5 (4–7) | 5 (4–5) | 0.21 |
| Biologic features | |||
| Urea (mmol/L) | 6.8 (5.6–8.4) | 12.8 (12.6–19.2) | <0.001 |
| Creatinine (µmol/L) | 57 (52–75) | 193 (154–270) | <0.001 |
| Bicarbonate (mmol/L) | 20.1 (16.8–24.0) | 21.0 (14.6–22.7) | 0.70 |
| Glucose (mmol/L) | 5.6 (4.8–6.9) | 7.4 (7.1–8.1) | 0.04 |
| Total cholesterol (mg/dl) | 47 (37–68) | 109 (73–198) | <0.001 |
| HDL cholesterol (mg/dl) | 29 (18–37) | 56 (35–85) | 0.04 |
| HDL-C/TC ratio | 0.66 (0.33–0.85) | 0.49 (0.35–0.53) | 0.27 |
| Triacylglycerols (mg/dl) | 270 (228–677) | 316 (165–719) | 0.84 |
| Protidemia (g/L) | 59 (48–69) | 39 (32–49) | 0.02 |
| Proteinuria (g/mmol creatinine) | 617 (60–1287) | 19 (9–56) | 0.20 |
| Iohexol clearance | |||
| GFR (ml/min per kg) | 4.3 (4.1–4.8) | 2.2 (1.8–2.5) | 0.008 |
| Oxidative stress markers (plasma) | |||
| MDA (µmol/L) | 0.8 (0.5–1.2) | 2.3 (1.2–3.0) | 0.009 |
| AOA (mmol/L) | 1.0 (0.8–1.1) | 0.8 (0.7–0.9) | 0.02 |
Data are expressed as median (interquartile range). P<0.05 was considered as significant by Mann–Whitney test. Creatinine, ×0.011 for mg/dl; urea, ×2.8 for mg/dl. HDL-C, HDL cholesterol; TC, total cholesterol.
HDL Oxidation Level and Oxidizability in CKD Rabbits
HDL from CKD rabbits had significantly higher levels of HNE-Michael adducts on proteins (P<0.05; Figure 1A). In contrast, HHE adducts on proteins were not significantly higher in the HDL of the CKD group (Figure 1A). MDA concentration was significantly higher in the HDL of the CKD group (Figure 1B), whereas the 8-isoprostane concentration was not significantly different (Figure 1C). Despite a nonsignificant difference in the concentration of tocopherols (Figure 1D), HDL lipids from CKD rabbits were more prone to oxidize than HDL from control animals; there was a significant difference from 70 minutes until the end of the experiment (P<0.05; Figure 1E).
Figure 1.
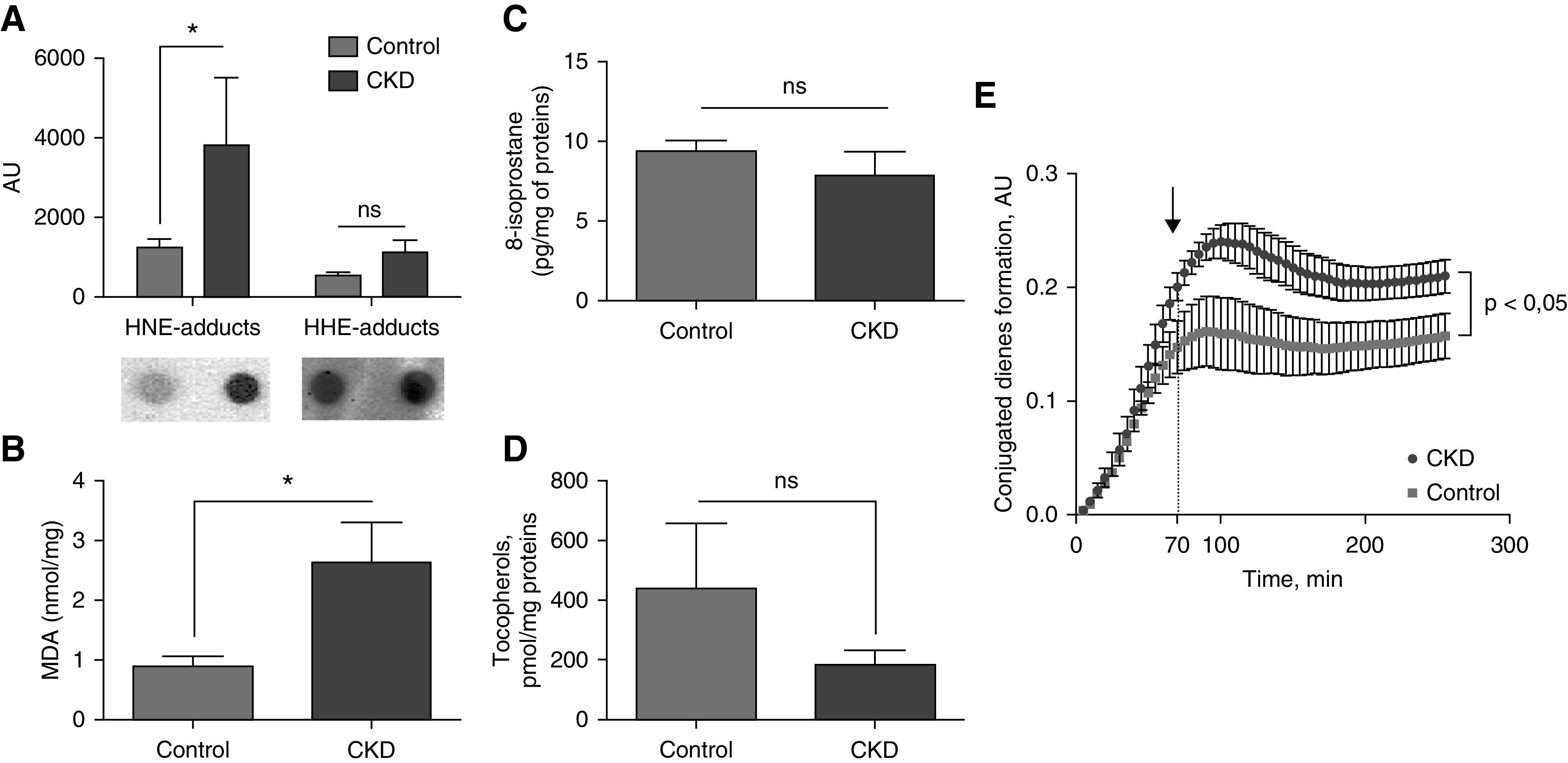
Oxidation of HDL particles from CKD rabbits. HNE adducts were increased in HDL from (A) CKD rabbits as well as (B) MDA concentration. No difference was observed with (C) 8-isoprostane and (D) tocopherol concentrations. (E) HDL from CKD rabbits were more prone to oxidation after a copper-induced oxidation as described. n=8 and 9, CKD and control, respectively; *P<0.05, Mann–Whitney test. Data are expressed as median and interquartile range.
Platelet Aggregation Induced by HDL from Rabbits
When human platelets were incubated with HDL from the CKD group, the median aggregation was 75% as compared with collagen alone; when incubated with HDL from the control group, it was 30% (P<0.05 as compared with incubation with HDL from the CKD group; Figure 2A). Because HNE adducts were increased in CKD HDL, we incubated control rabbit HDL with 100 µM HNE; median platelet aggregation in the presence of HNE-modified HDL was 85% (P<0.05 as compared with incubation with HDL from the control group; Figure 2A). Preincubation of platelets with Ab-CD36 restored the antiaggregant effect of the HDL from CKD rabbits and HNE-modified HDL (median, 25% and 22%, respectively; Figure 2, A and C). Preincubation of platelets with Ab-CD36 did not change significantly the aggregation induced by collagen (median, 95% versus 100%; NS).
Figure 2.
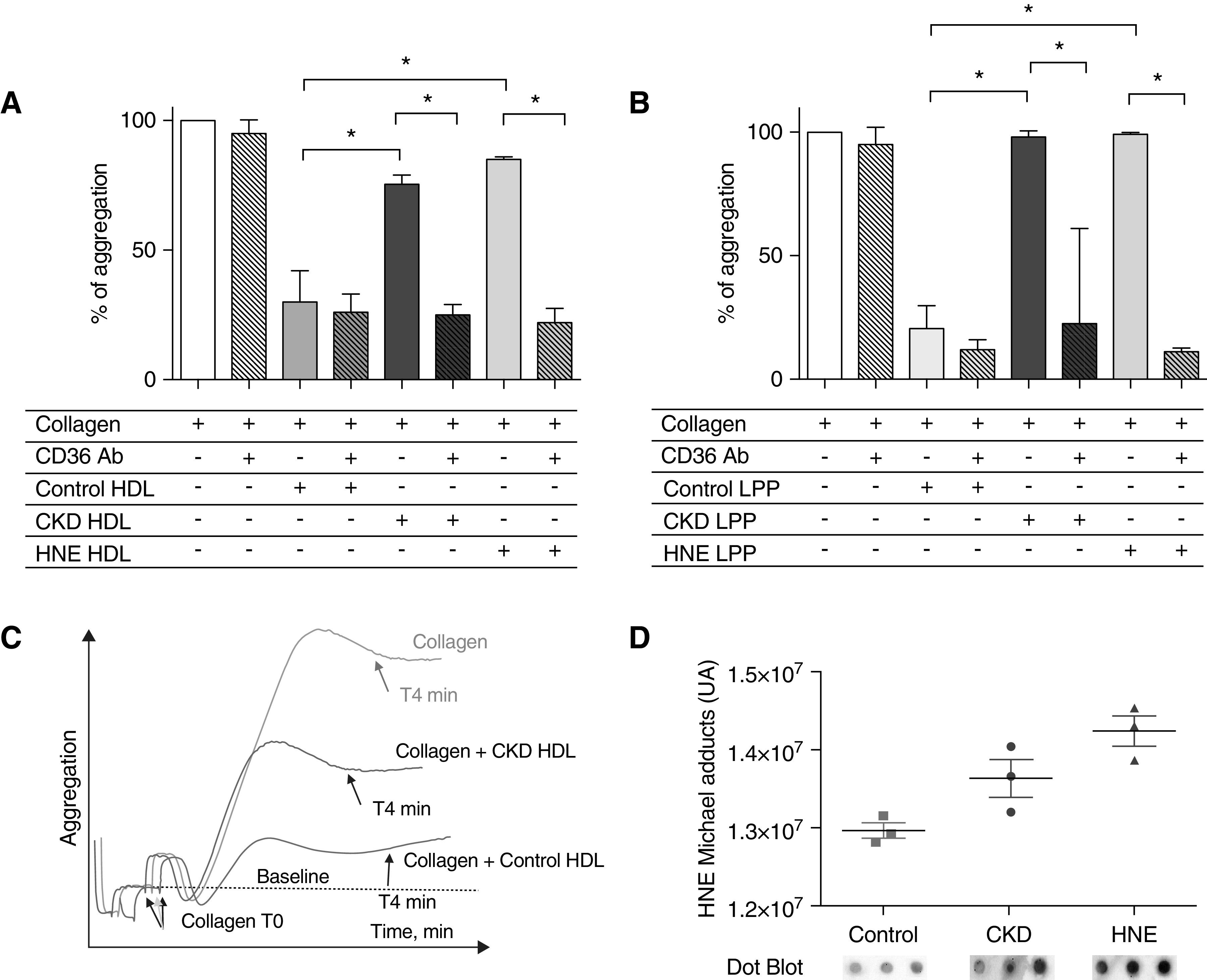
CKD is responsible for impaired antiaggregant properties of HDL in rabbits. (A) CKD HDL exhibited blunted antiaggregative properties compared with control HDL. Control HDL modified by an incubation overnight with HNE (HNE HDL) solution exhibited similar blunted properties to CKD HDL compared with control HDL. Preincubation with Ab-CD36 restored the CKD and HNE HDL antiaggregant properties like control HDL. (B) Typical aggregation curves obtained with collagen, control HDL, and CKD HDL. Collagen aggregation level is considered as 100%. The same results were observed with a lipoprotein mix containing triglyceride-rich lipoproteins (VLDL and chylomicrons), LDL, and HDL (LPP) from control and CKD rabbits. CKD LPP exhibited blunted antiaggregative properties compared with control LPP. (C) Control LPP modified by an incubation overnight with HNE solution (HNE LPP) exhibited similar properties to CKD LPP. Preincubation with Ab-CD36 restored the CKD and HNE LPP antiaggregant properties like control LPP. (D) HNE HDL had increased the level of HNE adducts compared with control HDL. n=8 and 9, CKD and control, respectively. *P<0.05, Mann–Whitney test. Data are expressed as median and interquartile range.
To investigate the effect of total lipoproteins (HDL, LDL, and triglyceride-rich lipoproteins; LPP) of CKD animals, platelets were incubated with a lipoprotein mix resulting from a single ultracentrifugation. The median aggregation in the CKD LPP group was 98% versus 21% in the control group (P<0.05; Figure 2B). Platelet aggregation in the presence of HNE-modified LPP was 99% (P<0.05 compared with control LPP group). Preincubation of platelets with Ab-CD36 restored the antiaggregant effect of the LPP from CKD rabbits and HNE-modified LPP (median, 23% and 11%, respectively; Figure 2B). HNE-adduct levels in HNE HDL and LPP were higher than in control using an immunoblot (Figure 2D).
Carbonylation of HDL from Patients on HD
Nine controls were compared with nine nondiabetic patients on HD, the main characteristics of whom are presented in Supplemental Table 1. Patients on HD had significantly lower total cholesterol, LDL cholesterol, and HDL cholesterol than the controls (P<0.05). There was a higher level of HNE adducts in HDL from patients on HD than in controls (Figure 3A). To map sites on proteins susceptible to carbonylation, HDL from control participants and patients on HD were digested with trypsin; digested peptides were separated by liquid chromatography, and peptides were analyzed for carbonylation with tandem mass spectrometry. We then expressed the ratio of amount of HNE in the HD patient sample versus the mean amount of control patients. A higher ratio (greater than one) of HNE-Michael adduct was detected on 48 amino acids (lysine or histidine) from eight constitutive proteins of HDL from patients on HD (Tables 2 and 3). The majority were located on apo A1 and concerned various important functional sites of the protein (Figure 3B, Supplemental Figure 2). Lys 250, located in the C terminal part of apo A1, was the most frequently modified site in patients on HD (5.3-fold increase compared with controls, P<0.01; Figure 3C).
Figure 3.
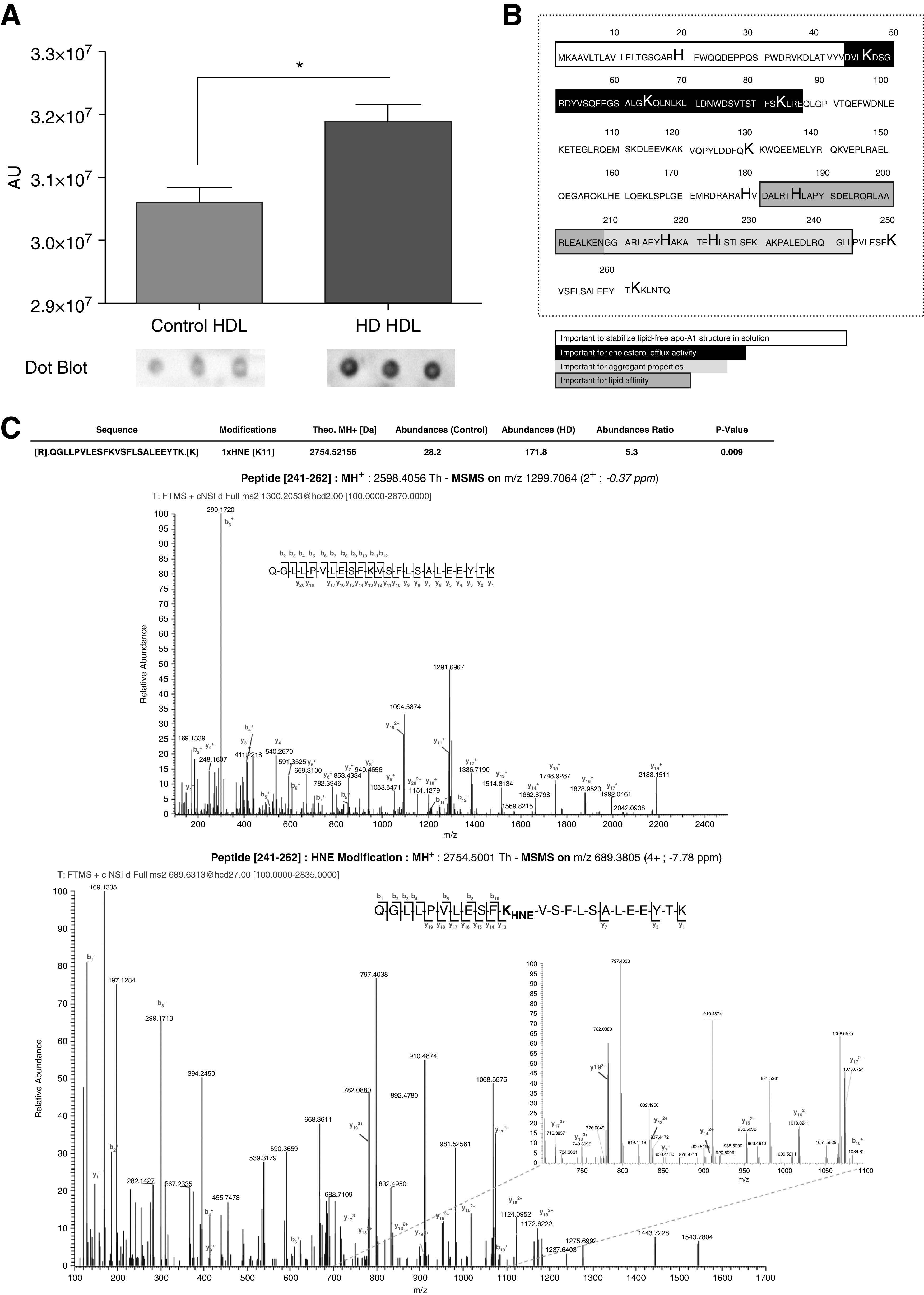
HNE adducts are increased in HDL from patients on HD compared with controls. (A) Immunoblotting (dot blot) of HNE-Michael adducts were performed as described in the Methods section. Identification of increased HNE-Michael adduct on lysine and histidine residues of HDL from patients on HD by liquid chromatography–tandem mass spectrometry assay (MSMS). HDL was reduced, alkylated, and digested with trypsin. (B) Sequence of apo A1 (accession number, P02647-1) indicating the position of the adducts (bold characters) and the sequences of interest (highlighted and framed). Typical MSMS spectra from unmodified peptide (upper spectrum) and modified peptide (lower spectrum). (C) The abundance of HNE adduct of Lys 250 in HD HDL was 5.3-fold higher than in controls (P<0.01). n=9 and 9, HD and control HDL, respectively. * P<0.05, Mann–Whitney test. Data are expressed as median and interquartile range.
Table 2.
General characteristics of controls and patients on HD and PD
| Characteristics | Control (N=15) | HD (N=25) | PD (N=20) |
|---|---|---|---|
| General characteristics | |||
| Age, yr | 46 (36–58) | 56 (43–73) | 67 (56–84)a,b |
| Gender, % male | 56 | 43 | 56 |
| BMI, kg/m2 | 26 (24–28) | 24 (20–26) | 25 (23–28) |
| Comorbidities | |||
| HT, n (%) | 3 (21) | 17 (74) | 19 (95) |
| Stroke, n (%) | 0 (0) | 2 (9) | 1 (5) |
| CHD, n (%) | 0 (0) | 5 (22) | 3 (15) |
| Cardiopathy, n (%) | 0 | 9 (39) | 8 (40) |
| PVD, n (%) | 0 | 4 (17) | 3 (15) |
| Therapies, n | |||
| Statins | 0 | 6 | 5 |
| PI | 0 | 5 | 6 |
| RASi | 1 | 3 | 11 |
| β-Blockers | 1 | 5 | 4 |
| CCB | 1 | 1 | 6 |
| Biologic parameters | |||
| Urea, mmol/L | 5.4 (4.4–7.5) | 17.1 (13.3–19.8)a | 18.8 (16.1–22.6)a |
| Creatinine, µmol/L | 73 (69–85) | 702 (506–862)a | 575 (422–758)a |
| mGFR, ml/min per 1.73 m2 | 94 (86–96) | — | — |
| Total cholesterol, mg/dl | 208 (189–237) | 149 (113–180)a | 209 (155–232)b |
| LDL cholesterol, mg/dl | 139 (105–158) | 71 (54–103)a | 125 (81–159)b |
| HDL cholesterol, mg/dl | 59 (53–66) | 41 (30–47)a | 45 (36–62)a |
| Triacylglycerols, mg/dl | 91 (64–116) | 119 (88–173)a | 149 (118–207)a |
| CRP, mg/L | 1.9 (0.5–3.5) | 3.3 (1.9–8.0) | 3.7 (1.7–5.8) |
Data are expressed as median (interquartile range). eGFR measured by the CKD Epidemiology Collaboration formula. BMI, body mass index; HT, hypertension; CHD, coronary heart disease; PVD, peripheral vascular disease; PI, platelet inhibitor; RASi, renin-angiotensin system inhibitor; CCB, calcium-channel blocker; mGFR, measured GFR by iohexol clearance; CRP, C-reactive protein.
P<0.05 versus controls.
P<0.05 versus HD, Mann–Whitney U test.
Table 3.
List of HNE-adducted amino acids in HDL constitutive proteins from patients on HD
| Protein Name | Protein Label | %a | Site of Adduction | Abundance Ratio (HD 1–9, respectively)b | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| apo B100 | APOB | 0.04 | K3689 | 0.293 | 0.825 | 0.468 | 4.099 | 8.148 | 1.488 | 1.626 | 3.606 | 0.322 |
| K4498 | — | — | — | — | 4.421 | — | — | 2.339 | — | |||
| Serum albumin | ALB | 1.15 | H152 | 2.646 | 5.05 | 0.373 | — | 0.036 | 0.072 | 1.263 | 0.874 | 6.837 |
| K160 or H152 | 2.956 | 6.049 | 0.229 | — | 0.295 | — | 0.718 | 0.405 | 12.994 | |||
| K347 | — | 1.452 | — | 0.879 | — | 0.582 | 1.767 | 2.442 | 1.966 | |||
| H170 | 1.463 | — | — | 0.919 | — | 0.178 | 0.957 | — | — | |||
| K581 | 0.861 | 1.148 | — | — | — | — | 0.668 | 1.944 | 2.204 | |||
| K198 | 1.715 | — | — | — | — | 2.864 | — | — | 1.28 | |||
| apo A1 | APOA1 | 4.50 | H20 | 0.516 | 0.863 | 1.131 | 2.298 | 1.414 | 1.284 | 1.133 | 1.109 | 0.556 |
| H179 | 2.448 | 2.756 | 5.133 | 1.117 | 0.995 | 1.306 | 0.316 | 0.802 | 0.853 | |||
| H186 | 2.646 | 2.508 | 4.401 | 0.613 | 0.806 | 0.93 | 0.168 | 1.221 | 1.767 | |||
| H223 | 0.456 | 0.549 | 1.471 | 0.101 | 0.125 | 0.188 | — | 0.136 | — | |||
| H217 | 1.342 | 0.939 | 3.216 | 0.039 | 0.175 | 0.266 | — | 0.14 | — | |||
| K36 | 0.89 | 1.362 | 1.181 | 1.64 | 0.996 | 1.36 | 1.873 | 1.863 | 1.128 | |||
| K250 | 1.423 | 3.189 | 0.548 | 4.329 | 31.087 | 1.035 | 3.154 | 30.549 | 2.522 | |||
| K130 | 0.332 | 1.236 | 0.856 | — | — | — | 1.018 | — | 0.728 | |||
| K47 | 0.596 | 1.345 | 1.237 | 2.083 | 0.861 | 1.211 | 2.596 | 2.217 | 1.557 | |||
| K83 | 0.393 | — | 0.943 | 1.004 | 0.559 | 0.79 | 2.181 | 2.642 | 2.131 | |||
| K64 | 0.571 | 1.139 | 1.079 | 1.643 | 1.119 | 1.543 | 1.403 | 1.616 | 0.935 | |||
| K262 | — | 10.052 | — | 0.757 | 7.534 | — | 0.442 | 42.042 | — | |||
| apo(a) | LPA | 0.02 | K215 | — | 100 | — | — | — | — | — | 100 | — |
| apo A2 | APOA2 | 1 | K26 | — | 1.983 | — | 0.134 | — | — | 0.963 | 0.29 | 0.689 |
| Keratin, type I, cytoskeletal 10 | KRT10 | 0.17 | K362 | 2.598 | 3.076 | 3.419 | 1.44 | 0.596 | 1.462 | 0.778 | 2.66 | 1.595 |
| Trypsin-3 | PRSS3 | 0.32 | H153 | 1.292 | 0.7 | 1.056 | 1.332 | 0.827 | 5.646 | 0.972 | 0.914 | 0.906 |
| UDP-glucose:glycoprotein | UGGT2 | 0.06 | H1388 | 1.46 | 1.385 | 3.62 | 3.525 | 4.433 | 2.838 | 5.1 | 11.999 | 1.877 |
| glucosyltransferase 2 | ||||||||||||
K, lysine; —, not found; H, histidine; UDP, uridine diphosphate.
Percentage of potential sites of adduction regarding all of the amino acids of the protein sequence.
Abundance ratio was calculated against the mean of the nine controls.
In Silico Modeling of HNE Adduction onto apo A1
To gain insights into the accessibility of histidine and lysine residues of apo A1, a molecular modeling study was achieved with the lipid-free structure of the truncated protein (Figure 4A).49 Histidine and lysine residues mentioned in the primary sequence of amino acids—namely K83, K130, H179, H186, H216, H223, K226, K250, and K262—were examined and were found to be readily accessible and susceptible to react with HNE (not available for the H20, K47, and K64 residues in the truncated protein). Therefore, the modification of these residues may occur in a random event on free apo A1. Careful examination of the model of HDL proposed by Wu et al. (Figure 4B)50 led to the following observations: some histidine and lysine residues are not involved in interactions with lipids and are located at the external part of HDL. These residues that can react with HNE may not affect the binding of apo A-I with lipids but with other proteins involved in HDL metabolism (see Figure 3B, residues K47, K83, H216, K250 and K262); in contrast, some histidine and lysine residues (residues K64, K130, H179, H186, and H223) interact tightly with lipids in the internal side of the complex. In this case, it can be proposed that these modifications may alter the ability of apo A-I to bind lipids. Overall, this molecular modeling study shows that the reaction of lysine and histidine residues leading to structural modifications may occur randomly but may be critical for lipid and protein binding, depending on the residue which is modified.
Figure 4.

Structure of truncated human apo A-I (pdb code 1av1) revealed several accessible sites for post-translational modifications by 4-HNE. Four molecules are associated with their hydrophobic faces to form an antiparallel four-helix bundle. (A) Lysine and histidine residues possibly modified by HNE are colored in cyan with CPK representation. (B) Model of HDL:phospholipids (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) are colored in yellow and cholesterol in magenta. Lysine and histidine residues possibly modified by HNE are colored in cyan with CPK coloring representation (pdb code 3k2s).
Platelet Activation and Aggregation Phenotype of HDL from Patients on HD and PD and Effects of Carbonylation
Because the HD procedure per se can lead to enhanced oxidative and carbonylated stress, we included patients on PD to avoid any effects resulting from bio-incompatibility of HD devices. A total of 15 healthy controls were compared with 25 nondiabetic patients on HD and 20 patients on PD, the main characteristics of whom are presented in Table 2. Patients on HD had significantly lower total cholesterol, LDL cholesterol, and HDL cholesterol than the controls (P<0.05). Platelet activation was measured with the production of TxB2 and expressed as a percentage of the amount found in platelets activated with collagen. When human platelets were incubated with HDL from the HD group, the median activation was 147% as compared with collagen alone; when incubated with HDL from the control group it was 75% (P<0.05 as compared incubation with HDL from the HD group; Figure 5A). When human platelets were incubated with HDL from the PD group, the median activation was 221% as compared with collagen alone (P<0.05 as compared with incubation with HDL from the control group, P=NS as compared with incubation with HDL from the HD group; Figure 5A). Because HNE adducts were increased in HD HDL, we incubated five healthy donors’ HDL with 100 µM HNE; median platelet activation in the presence of HNE-modified HDL was 123% as compared with collagen alone, when incubated with HDL from the control group it was 29% (P<0.05 as compared with incubation with HDL from the healthy donor group; Figure 5B).
Figure 5.
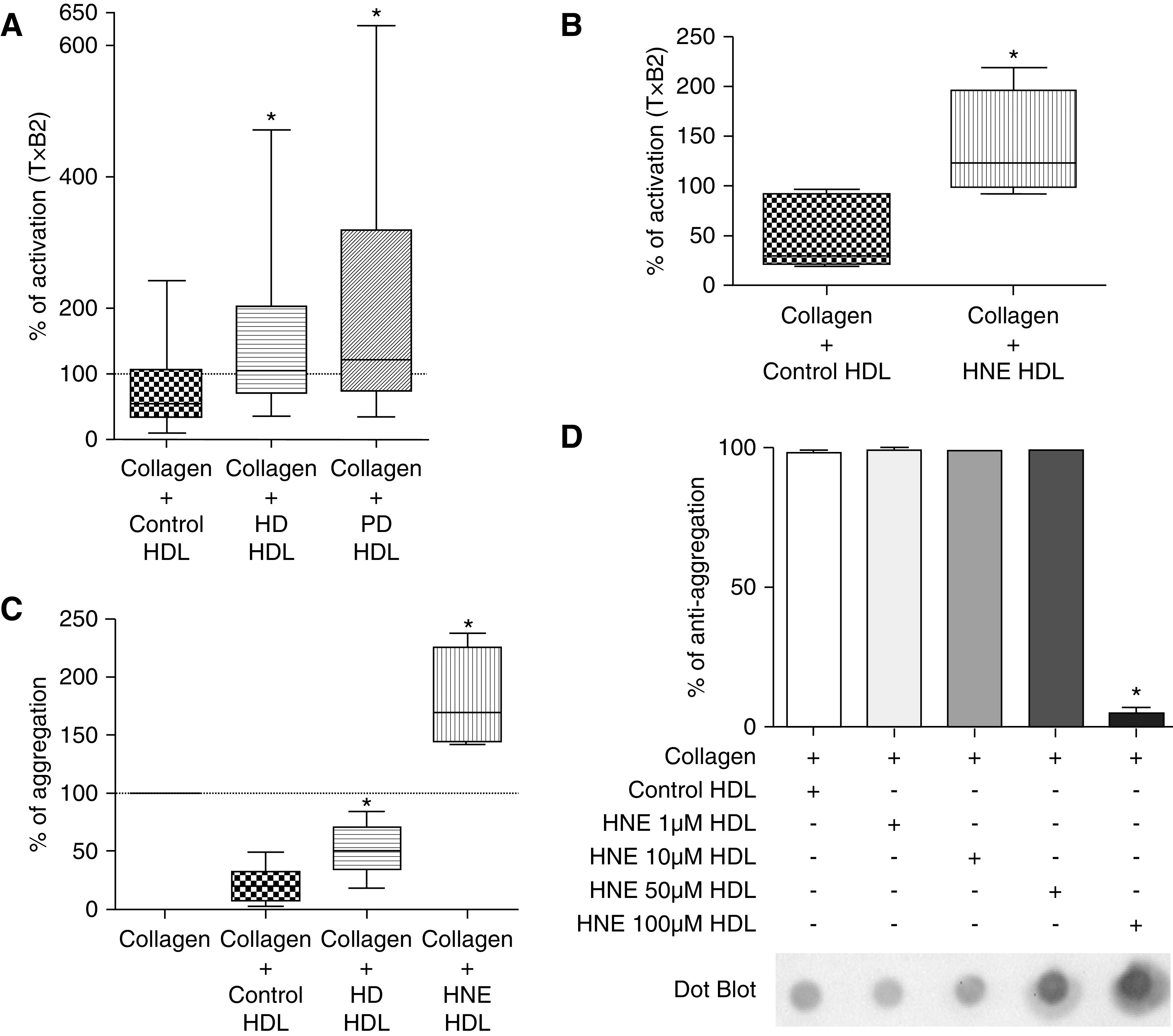
HDL from patients on PD and HD exhibited impaired antiaggregant properties. (A) Regarding levels of TxB2, HD and PD HDL triggered overactivation of platelets compared with control HDL (n=15 for control, n=25 for HD group, and n=20 for PD group). (B) The same observations were found for healthy volunteers’ HDL modified by an incubation overnight with HNE (HNE HDL, n=5 for each group). (C) HD, PD, and HNE HDL exhibited blunted antiaggregant properties compared with control HDL (n=9 for control and HD groups, n=5 for HNE group). (D) We found a threshold effect of HNE modification on HDL because only a 100 µM solution of HNE permitted the significant alteration of the antiaggregant properties of control HDL (n=4 for each group). *P<0.05, Mann–Whitney test and Kruskal–Wallis test (in D). Data are expressed as median and interquartile range.
As we performed aggregation assays in rabbits, we chose nine control and nine patients on HD (characteristics in Supplemental Table 1) and performed platelet aggregation assays. When human platelets were incubated with HDL from the HD group, the median aggregation was 50% as compared with collagen alone; when incubated with HDL from the control group it was 19% (P<0.05 as compared with incubation with HDL from the HD group; Figure 5C). We found a good correlation between percentage of activation with TxB2 assay and percentage of aggregation (r2=0.39, P<0.05; Supplemental Figure 3). Because HNE adducts were increased in HD HDL, we incubated healthy donors HDL with 100 µM HNE; median platelet aggregation in the presence of HNE-modified HDL was 170% (P<0.05 as compared with incubation with HDL from the control group; Figure 5C).
Because there was an interindividual variability of platelet response to HD HDL, we tested several concentrations of HNE for the modification of control HDL from healthy donors (1, 10, 50, and 100 µM). A threshold effect was found because only the 100 µM solution of HNE led to a significant alteration of the antiaggregant properties of HDL (median, 5% at 100 µM versus 98%, 99%, and 99% at 1, 10, and 50 µM, respectively; P<0.05; Figure 5D). Immunoblotting found a dose-dependent increase of HNE adducts in HDL incubated with HNE (1–100 µM; Supplemental Figure 4).The median levels of HNE adducts in HNE-modified HDL were similar to those of patients on HD and PD (see Supplemental Figure 5).
Activation Is Mediated by the Phosphorylation of c-Jun N-Terminal Kinase through a CD36 and SRC Kinase Pathway
To get insight into the potential pathways involved in the effects described herein, the effect of CD36, SRB1 blockade, and SRC kinase pharmacologic inhibitor were tested. Preincubation of platelets with Ab-CD36 significantly decreased their activation as measured by TxB2 levels when incubated with HDL from control, HD, PD, and HNE-modified HDL (median, 39%, 41%, 22%, and 49%, respectively; P<0.05, as compared with incubation without the antibody; Figure 6A). P-selectin expression was also lowered by the preincubation of platelets with Ab-CD36 for HDL from control, HD, PD, and HNE-modified HDL (median, −43%, −30%, −42%, and −27%, respectively; P<0.05 as compared with incubation without the antibody; Figure 6B). Preincubation of platelets with Ab-CD36 restored the antiaggregant effect of the HDL from HNE-modified HDL (median, 12%; P<0.05 as compared with incubation without the antibody; Supplemental Figure 6). Preincubation of platelets with Ab-CD36 did not significantly change the aggregation induced by collagen (median, 107% versus 100%; NS; Supplemental Figure 6).
Figure 6.

The impaired antiaggregant properties of HDL in patients on PD and HD and healthy volunteers’ HDL modified by an incubation overnight with HNE (HNE HDL) is mediated by CD36 binding and SRC kinase–mediated pathways. (A) Regarding levels of TxB2, the preincubation of platelets with an antibody against CD36 receptor (Ab anti-CD36) significantly lowered the activation of platelets exposed with control (CTL), HD, PD, and HNE compared with incubation without the antibody. The preincubation of platelets with an antibody against SRB1 receptor (Ab anti-SRB1) significantly increased the activation of platelets exposed with CTL, PD, and HNE compared with incubation without the antibody. The preincubation of platelets with a pharmacologic inhibitor of SRC kinases (Naphthyl PP1) significantly lowered the activation of platelets exposed to CTL, HD, PD, and HNE compared to incubation without the inhibitor. (B) In a FACS assay, activation of platelets were determined by the expression of P-selectin. The preincubation of platelets with Ab-CD36 and Naphthyl PP1 significantly decreased the activation of platelets exposed to CTL, HD, PD, and HNE HDL compared with the samples without the antibody/inhibitor. The preincubation of platelets with Ab anti-SRB1 significantly increased the activation of platelets exposed to CTL, HD, PD, and HNE HDL compared with the samples without the antibody/inhibitor. n=5 for each group. *P<0.05 compared with control HDL without blocker; $P<0.05 compared with HD HDL without blocker; †P<0.05 compared with PD HDL without blocker; *P<0.05 compared with HNE HDL without blocker; Mann–Whitney test. Without blocker, without either Ab-CD36, Ab anti-SRB1, or Naphthyl PP1.
Preincubation of platelets with an anti-SRB1 antibody significantly increased their activation as measured by TxB2 levels when incubated with HDL from control, HD, PD, and HNE-modified HDL (median, 385%, 178%, 288%, and 758%, respectively; P<0.05 as compared with incubation without the antibody; Figure 6A). P-selectin expression was also increased by the preincubation of platelets with an anti-SRB1 antibody for HDL from control, HD, PD, and HNE-modified HDL (median: +35%, +93%, +54%, and +88%, respectively; P<0.05 as compared with incubation without the antibody; Figure 6B). Preincubation of platelets with anti-SRB1 did not significantly alter the aggregation induced by collagen (data not shown).
Preincubation of platelets with Naphthyl PP1, a potent pharmacologic inhibitor of SRC kinases, decreased their activation as measured by TxB2 levels when incubated with HDL from control, HD, PD, and HNE-modified HDL (median, 22%, 46%, 24%, and 22%, respectively; P<0.05 as compared with incubation without the inhibitor; Figure 6A). P-selectin expression was also decreased by the preincubation of platelets with Naphthyl PP1 for HDL from control, HD, PD, and HNE-modified HDL (median, −48%, −48%, −53%, and −14%, respectively; P<0.05 as compared with incubation without the inhibitor; Figure 6B).
We confirmed that intraplatelet phosphorylation of c-Jun N-terminal kinase (JNK) was reduced by blocking of CD36 and SRC kinases, whereas blocking SRB1 increased its phosphorylation (data not shown).
Discussion
This study confirms that HDL isolated from patients with CKD exhibit impaired biologic functions that may participate in the onset of cardiovascular disease in this population. The work emphasizes the effect of carbonylation from long-chain n-6 fatty acids (HNE adducts) on the antiaggregative properties of HDL. This adduction onto the protein component of the HDL particle was responsible for a deep and significant alteration of the antiaggregant properties of HDL in CKD. Combined with all of the other impaired functions of HDL, this could be a potent contributor to the increased cardiovascular morbidity and mortality in CKD3 and the failure of statins in patients in HD.27,28 Moreover, a significant part of this pathologic phenotype was mediated by a CD36-dependent pathway because its blockade restored normal antiaggregant properties. Furthermore, CKD per se was responsible for a large part of this carbonylation because these adducts were obtained in a rabbit CKD model without other cardiovascular risk factors.
The rabbit model is of interest for cardiovascular research (e.g., atherosclerosis, lipid metabolism) because these animals have a lipoprotein metabolism much closer to humans than mice or rats due to the expression of the cholesteryl ester transfer protein gene. The CKD rabbits, despite having a lower body weight, developed a metabolic disturbance because their plasma cholesterol and glucose levels were significantly higher. The median measured GFR reduction in CKD animals corresponded approximately to stage 3 CKD in humans, and results of oxidative stress assays (MDA and AOA) were consistent with values observed in a human CKD population,5 which supports the appropriateness of the model. However, a natural history of 3 weeks is unlikely to lead to the same modifications as several years of interaction between cardiovascular risk factors and uremia in humans. Nevertheless, we found an increase in HNE adducts and the impaired antiaggregant properties of HDL in patients on HD and PD, rendering the animal findings relevant.
The results of this study suggest that CKD per se may trigger a strong oxidation of HDL particles. Interestingly, HNE adducts were both increased in the HDL of CKD rabbits and patients on HD and PD. This can be due to a loss of antioxidant capacity of HDL particles in CKD because HDL from CKD rabbits were more sensitive to copper-induced oxidation. This is in accordance with data from Holzer et al.24 who reported in patients on PD and HD a profound modification of HDL structure, in particular a lower concentration of paraoxonase. Moreover, the short natural history of CKD in the model used here suggests that these modifications occur during the early stages of the disease. These results are consistent with that reported by Shroff et al.25 and Kaseda et al.51 in pediatric patients with CKD, who can be considered as close to the model used because of a short natural history of CKD and a lower number of CKD-associated comorbidities.
The results regarding platelet activation and aggregation are consistent with the altered properties of HDL in CKD; we observed a significant increase in the activation of platelets incubated with HDL from patients on HD and PD and a twofold reduction in antiaggregation with HDL from patients on HD and CKD rabbits, suggesting a blunted antithrombotic profile of uremic HDL. To exclude that the blunted antiaggregant properties in patients on HD did not only result from the HD procedure per se, we confirmed this profile in patients on PD. This strengthened the role of CKD and especially ESKD in the pathologic properties of HDL. This is consistent with the increase in the incidence of thrombotic events in CKD, especially in patients on HD27,28,52,53 and with a higher platelet reactivity in CKD.54–57 However, we cannot totally rule out the role of age as a confounding factor in the different aggregation behavior of HDL because patients on PD were older than the patients on HD and controls. The pathophysiologic pathways of the platelet aggregation profile in CKD has only been studied for LDL; Holy et al.31 found a proaggregant profile of LDL in CKD that was mostly explained by carbamoylation of these proteins. Moreover, this study highlighted the role of the lectin-like oxidized LDL receptor 1 in the onset of this profile; a receptor mainly involved in LDL-mediated pathophysiologic pathways. The proagreggant profile of oxidized LDL has been described by Chen et al.32 to be mediated by CD36 and this receptor is also known to bind heavily modified HDL58,59 and oxidized albumin.60 The results of this study suggest a strong implication in the blunted antiaggregant profile of the CD36-mediated pathway because blocking CD36 restored the antiaggregant properties of CKD and HNE-modified HDLs. Furthermore, this study suggests the involvement of SRC kinase–mediated pathways in the platelet activation process in response to CKD and HNE-modified HDLs because the use of a specific pharmacologic inhibitor (Naphthyl PP1) also restored the antiaggregant properties of these HDLs. Finally, we confirmed that the binding of HDL onto the CD36 receptor resulted in a significant increase in JNK phosphorylation as demonstrated by Chen et al.32 for oxidized LDL. A schematic summary of these pathways is summed up in Figure 7.
Figure 7.

HDL from patients with CKD and rabbit (CKD HDL) exhibited blunted antiaggregant properties by interaction with CD36 receptor. Carbonylation by HNE of proteins from HDL was found to be part of this pathologic behavior. Activation of the platelets in response to this binding was through a SRC kinase–mediated pathway. SRC kinase activation resulted with an increase of phosphorylated JNK (pJNK). Native and CKD HDL apo A1 binds SRB1 and inhibits platelet activation.
The study also suggests that there is a threshold effect of HNE adduction on aggregative properties of HDL because the incubation of control HDL with HNE in concentrations <100 µM did not modify the antiaggregant profile of HDL. This could be explained by the multiple receptors that are involved in the aggregant properties of HDL. Indeed, native and mildly oxidized HDL binds ApoER2 and SRB1 scavenger receptor, inducing an intracellular downregulation of platelet activation pathways. Conversely, heavily oxidized HDL binds CD36 and platelet activation is upregulated.58,61 Thus, we can hypothesize that the adduction induced by the incubation with HNE at a concentration <100 µM caused light or mild modifications of the protein component of HDL insufficient to bind CD36. HNE adducts to the protein component of HDL particles were associated with proaggregative properties of HDL. Interestingly, the mass spectrometry assay specifically identified such HNE adducts on lysines and histidines from eight constitutive proteins of HDL and especially on apo A1 of patients on HD. This apo is widely responsible for the antiaggregant properties of HDL33 and several modifications were located on key parts of the protein involved in cholesterol efflux, platelet aggregation, and lipid binding.
Post-translational modifications of proteins represent a wide spectrum of potentially toxic modifications such as carbamoylation, generations of advanced glycation end products, chlorination, or nitration. For example, carbamoylation of LDL particles was responsible for proaggregant properties of platelets in patients with CKD.31 Despite a strong effect of the carbonylation by HNE described herein, we cannot exclude that other post-translational modifications could also contribute to the proaggregant properties of HDL. However, further studies should be dedicated to explore the specific role of other kinds of post-translational modification of lipopoproteins. To conclude, we describe herein for the first time the impaired antiaggregant properties of HDL in CKD. To improve cardiovascular outcomes in CKD, future studies need to focus on HDL quality as well as quantity.
Disclosures
All authors have nothing to disclose.
Funding
Dr. Florens was supported by a Agence Régionale de Santé de Rhône Alpes (ARS) grant “Année Recherche,” Hospices Civils de Lyon (University Hospital of Lyon), and by Société Française Néphrologie Dialyse et Transplantation (French Society of Nephrology) grant “IRCT – Dialyse.” The authors acknowledge the financial support from ITMO Cancer AVIESAN (Alliance Nationale pour les Sciences de la Vie et de la Santé; National Alliance for Life Sciences and Health) within the framework of the cancer plan for the founding of the Orbitrap mass spectrometer.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge Philip Robinson (Department of Clinical Research and Innovation, University Hospital of Lyon) for help in manuscript preparation. The authors acknowledge D. Yi and E. Hoibian (CarMeN Laboratory) for their help during the experiments. The authors acknowledge Dr. M. Rabeyrin (Department of Pathology, E. Herriot Hospital, Lyon, France) for preparation of the histological samples and Dr A. Varennes (Department of Biochemistry, E. Herriot Hospital, Lyon, France) for urea and creatinine measurements.
Dr. Florens, Dr. Juillard, Dr. Moulin, and Dr. Soulage conceptualized and designed the study; Dr. Barba, Dr. Boulet, Dr. Calzada, Dr. Florens, Dr. Guillot, Dr. Lemoine, Ms. Roux, and Dr. Soulage performed the experiments; Dr. Delolme and Dr. Page performed the mass spectrometry analysis; Dr. Florens and Dr. Soulage analyzed the data and wrote the manuscript; Dr. Guebre-Egziabher edited the manuscript; and Dr. Boulet, Dr. Calzada, Dr. Florens, Dr. Guebre-Egziabher, Dr. Juillard, Dr. Laville, Dr. Lemoine, Dr. Moulin, Dr. Poux, and Dr. Soulage revised the manuscript. Dr. Florens is the guarantor of this work and, as such, had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2019111205/-/DCSupplemental.
Supplemental Table 1. General characteristics of hemodialysis and control patients for aggregation assay.
Supplemental Figure 1. Histological examinations of rabbit kidneys.
Supplemental Figure 2. Examples of modified amino-acid MS/MS spectra.
Supplemental Figure 3. Spearman correlation of TxB2 assay and aggregation assay.
Supplemental Figure 4. Spearman correlation of the amount of 4-HNE adducts on in-vitro modified HDL.
Supplemental Figure 5. Immunoblotting of 4-HNE adducts median levels of HNE-modified HDL, HD, PD and control HDL.
Supplemental Figure 6. Aggregation levels of HNE-modified HDL versus control and CD36 blockade effect.
References
- 1.Go AS, Chertow GM, Fan D, McCulloch CE, Hsu C-Y: Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med 351: 1296–1305, 2004. [DOI] [PubMed] [Google Scholar]
- 2.Schiffrin EL, Lipman ML, Mann JFE: Chronic kidney disease: Effects on the cardiovascular system. Circulation 116: 85–97, 2007. [DOI] [PubMed] [Google Scholar]
- 3.Tonelli M, Muntner P, Lloyd A, Manns BJ, Klarenbach S, Pannu N, et al.: Alberta Kidney Disease Network: Risk of coronary events in people with chronic kidney disease compared with those with diabetes: A population-level cohort study. Lancet 380: 807–814, 2012. [DOI] [PubMed] [Google Scholar]
- 4.Maduell F, Moreso F, Pons M, Ramos R, Mora-Macià J, Carreras J, et al.; ESHOL Study Group: High-efficiency postdilution online hemodiafiltration reduces all-cause mortality in hemodialysis patients [published correction appears in J Am Soc Nephrol 25: 1130, 2014]. J Am Soc Nephrol 24: 487–497, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuchta A, Pacanis A, Kortas-Stempak B, Cwiklińska A, Ziętkiewicz M, Renke M, et al.: Estimation of oxidative stress markers in chronic kidney disease. Kidney Blood Press Res 34: 12–19, 2011. [DOI] [PubMed] [Google Scholar]
- 6.Dounousi E, Papavasiliou E, Makedou A, Ioannou K, Katopodis KP, Tselepis A, et al.: Oxidative stress is progressively enhanced with advancing stages of CKD. Am J Kidney Dis 48: 752–760, 2006. [DOI] [PubMed] [Google Scholar]
- 7.Aveles PR, Criminácio CR, Gonçalves S, Bignelli AT, Claro LM, Siqueira SS, et al.: Association between biomarkers of carbonyl stress with increased systemic inflammatory response in different stages of chronic kidney disease and after renal transplantation. Nephron Clin Pract 116: c294–c299, 2010. [DOI] [PubMed] [Google Scholar]
- 8.Vanholder R, Massy Z, Argiles A, Spasovski G, Verbeke F, Lameire N; European Uremic Toxin Work Group : Chronic kidney disease as cause of cardiovascular morbidity and mortality. Nephrol Dial Transplant 20: 1048–1056, 2005. [DOI] [PubMed] [Google Scholar]
- 9.Dou L, Sallée M, Cerini C, Poitevin S, Gondouin B, Jourde-Chiche N, et al.: The cardiovascular effect of the uremic solute indole-3 acetic acid. J Am Soc Nephrol 26: 876–887, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ito S, Osaka M, Edamatsu T, Itoh Y, Yoshida M: Crucial role of the aryl hydrocarbon receptor (AhR) in indoxyl sulfate-induced vascular inflammation. J Atheroscler Thromb 23: 960–975, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin C-J, Liu H-L, Pan C-F, Chuang C-K, Jayakumar T, Wang T-J, et al.: Indoxyl sulfate predicts cardiovascular disease and renal function deterioration in advanced chronic kidney disease. Arch Med Res 43: 451–456, 2012. [DOI] [PubMed] [Google Scholar]
- 12.Velasquez MT, Ramezani A, Manal A, Raj DS: Trimethylamine N-oxide: The good, the bad and the unknown. Toxins (Basel) 8: E326, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levitan I, Volkov S, Subbaiah PV: Oxidized LDL: Diversity, patterns of recognition, and pathophysiology. Antioxid Redox Signal 13: 39–75, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Apostolov EO, Ray D, Savenka AV, Shah SV, Basnakian AG: Chronic uremia stimulates LDL carbamylation and atherosclerosis. J Am Soc Nephrol 21: 1852–1857, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Apostolov EO, Ok E, Burns S, Nawaz S, Savenka A, Shah S, et al.: Carbamylated-oxidized LDL: Proatherosclerotic effects on endothelial cells and macrophages. J Atheroscler Thromb 20: 878–892, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van den Akker JM, Bredie SJH, Diepenveen SHA, van Tits LJH, Stalenhoef AFH, van Leusen R: Atorvastatin and simvastatin in patients on hemodialysis: Effects on lipoproteins, C-reactive protein and in vivo oxidized LDL. J Nephrol 16: 238–244, 2003. [PubMed] [Google Scholar]
- 17.Tsirpanlis G, Boufidou F, Manganas S, Chantzis K, Bleta A, Stamatelou K, et al.: Treatment with fluvastatin rapidly modulates, via different pathways, and in dependence on the baseline level, inflammation in hemodialysis patients. Blood Purif 22: 518–524, 2004. [DOI] [PubMed] [Google Scholar]
- 18.Baigent C, Landray MJ, Reith C, Emberson J, Wheeler DC, Tomson C, et al.; SHARP Investigators: The effects of lowering LDL cholesterol with simvastatin plus ezetimibe in patients with chronic kidney disease (Study of Heart and Renal Protection): A randomised placebo-controlled trial. Lancet 377: 2181–2192, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Navab M, Reddy ST, Van Lenten BJ, Fogelman AM: HDL and cardiovascular disease: atherogenic and atheroprotective mechanisms. Nat Rev Cardiol 8: 222–232, 2011. [DOI] [PubMed] [Google Scholar]
- 20.Khera AV, Cuchel M, de la Llera-Moya M, Rodrigues A, Burke MF, Jafri K, et al.: Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med 364: 127–135, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rosenson RS, Brewer HB Jr., Davidson WS, Fayad ZA, Fuster V, Goldstein J, et al.: Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation 125: 1905–1919, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holzer M, Birner-Gruenberger R, Stojakovic T, El-Gamal D, Binder V, Wadsack C, et al.: Uremia alters HDL composition and function. J Am Soc Nephrol 22: 1631–1641, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamamoto S, Yancey PG, Ikizler TA, Jerome WG, Kaseda R, Cox B, et al.: Dysfunctional high-density lipoprotein in patients on chronic hemodialysis. J Am Coll Cardiol 60: 2372–2379, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holzer M, Schilcher G, Curcic S, Trieb M, Ljubojevic S, Stojakovic T, et al.: Dialysis modalities and HDL composition and function. J Am Soc Nephrol 26: 2267–2276, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shroff R, Speer T, Colin S, Charakida M, Zewinger S, Staels B, et al.: HDL in children with CKD promotes endothelial dysfunction and an abnormal vascular phenotype. J Am Soc Nephrol 25: 2658–2668, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Honda H, Ueda M, Kojima S, Mashiba S, Michihata T, Takahashi K, et al.: Oxidized high-density lipoprotein as a risk factor for cardiovascular events in prevalent hemodialysis patients. Atherosclerosis 220: 493–501, 2012. [DOI] [PubMed] [Google Scholar]
- 27.Wanner C, Krane V, März W, Olschewski M, Mann JFE, Ruf G, et al.; German Diabetes and Dialysis Study Investigators: Atorvastatin in patients with type 2 diabetes mellitus undergoing hemodialysis [published correction appears in N Engl J Med 353: 1640, 2005]. N Engl J Med 353: 238–248, 2005. [DOI] [PubMed] [Google Scholar]
- 28.Fellström BC, Jardine AG, Schmieder RE, Holdaas H, Bannister K, Beutler J, et al.; AURORA Study Group: Rosuvastatin and cardiovascular events in patients undergoing hemodialysis [published correction appears in N Engl J Med 362: 1450, 2010]. N Engl J Med 360: 1395–1407, 2009. [DOI] [PubMed] [Google Scholar]
- 29.Calzada C, Coulon L, Halimi D, Le Coquil E, Pruneta-Deloche V, Moulin P, et al.: In vitro glycoxidized low-density lipoproteins and low-density lipoproteins isolated from type 2 diabetic patients activate platelets via p38 mitogen-activated protein kinase. J Clin Endocrinol Metab 92: 1961–1964, 2007. [DOI] [PubMed] [Google Scholar]
- 30.Colas R, Sassolas A, Guichardant M, Cugnet-Anceau C, Moret M, Moulin P, et al.: LDL from obese patients with the metabolic syndrome show increased lipid peroxidation and activate platelets. Diabetologia 54: 2931–2940, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Holy EW, Akhmedov A, Speer T, Camici GG, Zewinger S, Bonetti N, et al.: Carbamylated low-density lipoproteins induce a prothrombotic state via LOX-1: Impact on arterial thrombus formation in vivo. J Am Coll Cardiol 68: 1664–1676, 2016. [DOI] [PubMed] [Google Scholar]
- 32.Chen K, Febbraio M, Li W, Silverstein RL: A specific CD36-dependent signaling pathway is required for platelet activation by oxidized low-density lipoprotein. Circ Res 102: 1512–1519, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nofer J-R, Brodde MF, Kehrel BE: High-density lipoproteins, platelets and the pathogenesis of atherosclerosis. Clin Exp Pharmacol Physiol 37: 726–735, 2010. [DOI] [PubMed] [Google Scholar]
- 34.Calzada C, Véricel E, Colas R, Guillot N, El Khoury G, Drai J, et al.: Inhibitory effects of in vivo oxidized high-density lipoproteins on platelet aggregation: Evidence from patients with abetalipoproteinemia. FASEB J 27: 2855–2861, 2013. [DOI] [PubMed] [Google Scholar]
- 35.Valiyaveettil M, Kar N, Ashraf MZ, Byzova TV, Febbraio M, Podrez EA: Oxidized high-density lipoprotein inhibits platelet activation and aggregation via scavenger receptor BI. Blood 111: 1962–1971, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gotloib L, Crassweller P, Rodella H, Oreopoulos DG, Zellerman G, Ogilvie R, et al.: Experimental model for studies of continuous peritoneal’dialysis in uremic rabbits. Nephron 31: 254–259, 1982. [DOI] [PubMed] [Google Scholar]
- 37.Florens N, Lemoine S, Pelletier CC, Rabeyrin M, Juillard L, Soulage CO: Adenine rich diet is not a surrogate of 5/6 nephrectomy in rabbits. Nephron 135: 307–314, 2017. [DOI] [PubMed] [Google Scholar]
- 38.Soman RS, Zahir H, Akhlaghi F: Development and validation of an HPLC-UV method for determination of iohexol in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci 816: 339–343, 2005. [DOI] [PubMed] [Google Scholar]
- 39.Havel RJ, Eder HA, Bragdon JH: The distribution and chemical composition of ultracentrifugally separated lipoproteins in human serum. J Clin Invest 34: 1345–1353, 1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holzer M, Kern S, Trieb M, Trakaki A, Marsche G: HDL structure and function is profoundly affected when stored frozen in the absence of cryoprotectants. J Lipid Res 58: 2220–2228, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grotto D, Santa Maria LD, Boeira S, Valentini J, Charão MF, Moro AM, et al.: Rapid quantification of malondialdehyde in plasma by high performance liquid chromatography-visible detection. J Pharm Biomed Anal 43: 619–624, 2007. [DOI] [PubMed] [Google Scholar]
- 42.Koracevic D, Koracevic G, Djordjevic V, Andrejevic S, Cosic V: Method for the measurement of antioxidant activity in human fluids. J Clin Pathol 54: 356–361, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Therasse J, Lemonnier F: Determination of plasma lipoperoxides by high-performance liquid chromatography. J Chromatogr A 413: 237–241, 1987. [DOI] [PubMed] [Google Scholar]
- 44.Soulère L, Queneau Y, Doutheau A: An expeditious synthesis of 4-hydroxy-2E-nonenal (4-HNE), its dimethyl acetal and of related compounds. Chem Phys Lipids 150: 239–243, 2007. [DOI] [PubMed] [Google Scholar]
- 45.Lagarde M, Bryon PA, Guichardant M, Dechavanne M: A simple and efficient method for platelet isolation from their plasma. Thromb Res 17: 581–588, 1980. [DOI] [PubMed] [Google Scholar]
- 46.Born GV: Aggregation of blood platelets by adenosine diphosphate and its reversal. Nature 194: 927–929, 1962. [DOI] [PubMed] [Google Scholar]
- 47.Skinner SC, Diaw M, Pialoux V, Mbaye MN, Mury P, Lopez P, et al.: Increased prevalence of type 2 diabetes-related complications in combined type 2 diabetes and sickle cell trait. Diabetes Care 41: 2595–2602, 2018. [DOI] [PubMed] [Google Scholar]
- 48.Esterbauer H, Gebicki J, Puhl H, Jürgens G: The role of lipid peroxidation and antioxidants in oxidative modification of LDL. Free Radic Biol Med 13: 341–390, 1992. [DOI] [PubMed] [Google Scholar]
- 49.Borhani DW, Rogers DP, Engler JA, Brouillette CG: Crystal structure of truncated human apolipoprotein A-I suggests a lipid-bound conformation. Proc Natl Acad Sci U S A 94: 12291–12296, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu Z, Gogonea V, Lee X, Wagner MA, Li X-M, Huang Y, et al.: Double superhelix model of high density lipoprotein. J Biol Chem 284: 36605–36619, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kaseda R, Jabs K, Hunley TE, Jones D, Bian A, Allen RM, et al.: Dysfunctional high-density lipoproteins in children with chronic kidney disease. Metabolism 64: 263–273, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kawamura M, Fijimoto S, Hisanaga S, Yamamoto Y, Eto T: Incidence, outcome, and risk factors of cerebrovascular events in patients undergoing maintenance hemodialysis. Am J Kidney Dis 31: 991–996, 1998. [DOI] [PubMed] [Google Scholar]
- 53.Konishi A, Shinke T, Otake H, Nakatani D, Nakagawa M, Inoue T, et al.: Impact of hemodialysis on local vessel healing and thrombus formation after drug-eluting stent implantation. J Cardiol 64: 25–31, 2014. [DOI] [PubMed] [Google Scholar]
- 54.Yang K, Du C, Wang X, Li F, Xu Y, Wang S, et al.: Indoxyl sulfate induces platelet hyperactivity and contributes to chronic kidney disease-associated thrombosis in mice. Blood 129: 2667–2679, 2017. [DOI] [PubMed] [Google Scholar]
- 55.Jalal D, Chonchol M, Targher G: Disorders of hemostasis associated with chronic kidney disease. Semin Thromb Hemost 36: 34–40, 2010 [DOI] [PubMed] [Google Scholar]
- 56.Nishi T, Ariyoshi N, Nakayama T, Fujimoto Y, Sugimoto K, Wakabayashi S, et al.: Impact of chronic kidney disease on platelet inhibition of clopidogrel and prasugrel in Japanese patients. J Cardiol 69: 752–755, 2017. [DOI] [PubMed] [Google Scholar]
- 57.Vazzana N, Santilli F, Lattanzio S, Liani M, Giacci L, Del Rosso G, et al.: Determinants of thromboxane biosynthesis in patients with moderate to severe chronic kidney disease. Eur J Intern Med 33: 74–80, 2016. [DOI] [PubMed] [Google Scholar]
- 58.van der Stoep M, Korporaal SJA, Van Eck M: High-density lipoprotein as a modulator of platelet and coagulation responses. Cardiovasc Res 103: 362–371, 2014. [DOI] [PubMed] [Google Scholar]
- 59.Park YM: CD36, a scavenger receptor implicated in atherosclerosis. Exp Mol Med 46: e99, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pasterk L, Lemesch S, Leber B, Trieb M, Curcic S, Stadlbauer V, et al.: Oxidized plasma albumin promotes platelet-endothelial crosstalk and endothelial tissue factor expression. Sci Rep 6: 22104, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Assinger A, Koller F, Schmid W, Zellner M, Babeluk R, Koller E, et al.: Specific binding of hypochlorite-oxidized HDL to platelet CD36 triggers proinflammatory and procoagulant effects. Atherosclerosis 212: 153–160, 2010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.