

Significance Statement
Studies have suggested that estrogens may protect mice from AKI. Estrogen sulfotransferase (SULT1E1, or EST) plays an important role in estrogen homeostasis by sulfonating and deactivating estrogens, but studies of SULT1E1’s role in AKI are lacking. Using the ischemia-reperfusion model of AKI, the authors demonstrated that genetic ablation or pharmacologic inhibition of Sult1e1 can mitigate AKI in both male and female mice in a sex hormone-independent manner. A gene profiling analysis indicated that the renoprotective effect was associated with increased vitamin D receptor signaling. Liver-specific reconstitution of Sult1e1 resensitizes male Sult1e1 knockout mice to AKI, indicating that liver Sult1e1is required for ischemic AKI in males. These findings suggest that pharmacologic inhibition of SULT1E1 might represent a novel approach for clinical management of AKI.
Keywords: acute kidney injury, renal ischemia-reperfusion, estrogen sulfotransferase, kidney-liver crosstalk, calcitriol
Visual Abstract

Abstract
Background
Studies have suggested that estrogens may protect mice from AKI. Estrogen sulfotransferase (SULT1E1, or EST) plays an important role in estrogen homeostasis by sulfonating and deactivating estrogens, but studies on the role of SULT1E1 in AKI are lacking.
Methods
We used the renal ischemia-reperfusion model to investigate the role of SULT1E1 in AKI. We subjected wild-type mice, Sult1e1 knockout mice, and Sult1e1 knockout mice with liver-specific reconstitution of SULT1E1 expression to bilateral renal ischemia-reperfusion or sham surgery, either in the absence or presence of gonadectomy. We assessed relevant biochemical, histologic, and gene expression markers of kidney injury. We also used wild-type mice treated with the SULT1E1 inhibitor triclosan to determine the effect of pharmacologic inhibition of SULT1E1 on AKI.
Results
AKI induced the expression of Sult1e1 in a tissue-specific and sex-specific manner. It induced expression of Sult1e1 in the liver in both male and female mice, but Sult1e1 induction in the kidney occurred only in male mice. Genetic knockout or pharmacologic inhibition of Sult1e1 protected mice of both sexes from AKI, independent of the presence of sex hormones. Instead, a gene profiling analysis indicated that the renoprotective effect was associated with increased vitamin D receptor signaling. Liver-specific transgenic reconstitution of SULT1E1 in Sult1e1 knockout mice abolished the protection in male mice but not in female mice, indicating that Sult1e1’s effect on AKI was also tissue-specific and sex-specific.
Conclusions
SULT1E1 appears to have a novel function in the pathogenesis of AKI. Our findings suggest that inhibitors of SULT1E1 might have therapeutic utility in the clinical management of AKI.
AKI is defined as an abrupt impairment of renal function manifested by high levels of serum creatinine (1.5–1.9 times baseline or ≥0.3 mg/dl) and/or reduced urine output (0.5 ml/kg per hour for 6–12 hours).1 The AKI syndrome is common in critically ill patients and is associated with increased length of hospitalization, morbidity, CKD, and mortality.2 The etiology of AKI includes prerenal (loss of blood flow to the kidney), intrinsic (direct damage to the organ), and postrenal (obstruction of lower urinary system) causes.1−3 The clinical management of AKI is comprised of fluid support and agents to prevent life-threatening nutritional and hemodynamic changes, but no specific pharmacologic treatment for AKI has been approved.1 Understanding the pathophysiology of AKI will facilitate the development of novel strategies to manage this disease. Bilateral renal warm ischemia-reperfusion is a widely used mouse model of AKI.4
Estrogen sulfotransferase (SULT1E1, or EST) is a conjugating enzyme that belongs to the superfamily of cytosolic sulfotransferases. SULT1E1 catalyzes the transfer of a sulfur group from 3′-phosphoadenosine-5′-phosphosulfate to the hydroxyl group of its substrates. The preferred SULT1E1 substrates include endogenous and synthetic estrogens.5 Upon SULT1E1-catalyzed sulfation, estrogens have their hydrosolubility augmented, which prevents their binding to the estrogen receptor (ER).6,7 As such, SULT1E1-mediated sulfoconjugation represents a major pathway to deactivate estrogens and this enzyme plays an important role in estrogen homeostasis. The basal expression of Sult1e1 in the mouse liver is low, but its expression is highly inducible in response to local or systemic diseases, such as liver ischemia/reperfusion,8 hemorrhagic shock,9 type 2 diabetes,10 and sepsis.11 Animal studies have suggested that estrogens may protect mice from AKI.12−14 Epidemiology studies suggest that women below the age of menopause are believed to be protected because of the anti-inflammatory activities of estrogens,15,16 whereas being a man can be an independent risk factor for AKI.17,18 It has not been reported whether SULT1E1 plays a role in the pathogenesis of AKI and, if so, whether the effect of SULT1E1 on AKI is sex hormone-dependent.
Besides its renal effect, AKI has been reported to affect many distal organs, including the liver.19 In the liver, AKI may lead to increased levels of alanine aminotransferase (ALT) and aspartate aminotransferase, and impaired activity of multiple drug-metabolizing enzymes via increased inflammation, apoptosis, and oxidative stress.20−22 Most of the reported effects of AKI on drug-metabolizing enzymes are focused on the phase 1 cytochrome P450 enzymes,22 whereas the AKI effect on the expression and activity of the phase 2 conjugating enzymes is largely unknown.
In this study, we uncovered a functional interaction between AKI and SULT1E1. Specifically, AKI induced the expression of Sult1e1 in mice in a tissue- and sex-specific manner. Meanwhile, genetic ablation or pharmacologic inhibition of Sult1e1 protected mice from AKI in an estrogen- or androgen-independent manner.
METHODS
Animals
Wild-type (WT) C57BL/6 mice were purchased from the Jackson Laboratory (Bar Harbor, ME). The creations of Sult1e1 knockout (KO)23 and liver-specific liver-enriched activator protein (Lap)-EST/SULT1E1 (LE) transgenic mice11 were reported previously. The LE transgenic mice express the human EST/SULT1E1 transgene in the liver under the control of the hepatocyte-specific Lap gene promoter. The KOLE mice, which are Est KO mice with the liver-specific reconstitution of SULT1E1, were created by crossbreeding the Est KO and LE transgenic mice, as we have previously reported.24 In the KOLE mice, the Est KO allele and LE transgenic allele were independently genotyped by PCR. Mice were maintained in a temperature-controlled animal facility at the University of Pittsburgh. The use of animals complied with the guidelines established by the Institutional Animal Care and Use Committee of the University of Pittsburgh.
Ischemia-Reperfusion Model of AKI
To induce AKI, both kidneys were clamped to block blood flow for 30 minutes. After ischemia, clamps were released to start reperfusion, and mice were euthanized after 1, 2, 6, 24, or 72 hours. Blood samples were collected by cardiac puncture. Kidneys and livers were harvested for analysis. The sham surgery was a midline incision.
Histologic Analysis
The kidney and liver tissues were fixed in 10% neutral buffered formalin for 24 hours and then dehydrated, embedded in paraffin, sectioned at 4 mm, stained with periodic acid-Schiff (PAS), and counterstained with hematoxylin. Immunostaining was performed on paraffin sections. Slides were incubated overnight with the primary anti-SULT1E1 antibody (12522–1-AP) from Proteintech (Rosemont, IL), anti-CD3 antibody (MAB4841) from R&D Systems (Minneapolis, MN), anti-Cyp24a1 antibody (ab203308), anti-Ki67 antibody (ab66155) from Abcam (Cambridge, MA), or anti-neutrophil gelatinase-associated lipocalin (NGAL) antibody (MAB1857) from R&D Systems. Antibodies were diluted to 1:100 (liver) or 1:50 (kidney), and incubated in humid chambers overnight at 4°C. Specimens were then treated with fluorescence- or biotin-conjugated secondary antibodies. Terminal deoxynucleotidyl transferase-mediated digoxigenin-deoxyuridine nick-end labeling (TUNEL) staining was performed by using the In Situ Cell Death Detection Kit from Roche (Mannheim, Germany).
Serum Biochemical Analysis of Creatinine, BUN, ALT, IL-6, and 17β-E2
Creatinine levels were measured with the QuantiChrom Creatinine Assay Kit from BioAssay Systems (Hayward, CA). BUN was measured by using the QuantiChrom Urea assay kit (Catalog #DIUR-100) from BioAssay Systems. The ALT levels were analyzed with the Stanbio ALT kit from Laboratory (Boerne, TX). The concentrations of IL-6 were measured by an ELISA kit from R&D Systems. Serum levels of 17β-E2 were measured by the Center for Research in Reproduction at the University of Virginia using the ELISA (Calbiotech) kit.
Cell Cultures
Primary hepatocytes were isolated from 12-week-old male WT mice by liver perfusion.25 Hepatocytes were seeded onto type 1 collagen-coated dishes in William E medium containing 5% FBS until confluent. After 24 hours, the medium was replaced with DMEM-F12 supplemented with vehicle or IL-6 (70 ng/ml) for 24 hours before cell harvesting. Human kidney proximal tubular (HKC-8) cells were cultured in DMEM-F12 and 5% FBS until confluent. The cells were then treated with vehicle or IL-6 (35 ng/ml) for 24 hours before cell harvesting.
RNA Isolation, Quantitative Real-Time PCR, Northern Blot, and Affymetrix Microarray Analysis
Total RNA was isolated using the TRIZOL reagent from Invitrogen (Carlsbad, CA). Total RNA was treated with RNase-free DNase I, and the resultant DNA-free RNA was used to synthesize single-strand cDNA. Real-time PCR was performed on an ABI 7300 Real-Time PCR System from Applied Biosystems (Foster City, CA). The primer sequences are listed in Supplemental Table 1. Melting curve analysis was performed to determine the specificity of amplification. Gene expression was normalized to the expression of the control cyclophilin gene. Northern blot analysis using a 32P-labeled cDNA probe was performed as previously described.26 Affymetrix microarray analysis was performed at the High Throughput Genome Center at the Department of Pathology, University of Pittsburgh. Microarray data were first quantile-normalized across samples. The probe-level intensities were then mapped to gene-level expression. If multiple probes are mapped to the same gene, only the probe with the largest interquartile range will be kept. Then the top 200 upregulated and 200 downregulated genes were selected on the basis of log2 fold change. These differentially expressed genes were used as input for Ingenuity Pathway Analysis (IPA), with significant pathways defined by a false discovery rate of 5%.
Transient Transfection and Luciferase Reporter Gene Assay
The pCMX-VDR, tk-VDRE,27 and pCMX-SULT1E18 constructs were described previously. HEK293T cells were transiently transfected with pCMX-VDR and tk-VDRE plasmids, with or without the cotransfection of the pCMX-SULT1E1 plasmid, using Lipofectamine 2000 from Invitrogen. The pCMX-β-gal (β-galactosidase) plasmid was added as an internal control to monitor the transfection efficiency. After transfection, cells were treated with vehicle or calcitriol (10 nM) for 24 hours. The luciferase activity was normalized to the β-gal activity.
Statistical Analyses
All data are expressed as the mean±SD. Statistical analysis was performed using a t test or one-way ANOVA where appropriate. Differences between groups were considered statistically significant at P<0.05. Multiple comparisons were evaluated by one-way ANOVA followed by Tukey’s multiple comparison tests.
RESULTS
AKI Induces the Hepatic Expression of Sult1e1 in Both Male and Female Mice, but Induces the Renal Expression of Sult1e1 Only in Male Mice
The acute ischemic AKI model was established by clamping the renal pedicle vessels of both kidneys to block the blood flow for 30 minutes. The AKI-induced renal injury in male mice was confirmed at the biochemical, gene expression, and histologic levels (Supplemental Figure 1). We first used a microarray to determine the effect of AKI on hepatic and renal gene expression in male mice. The microarray data sets have been submitted to the NIH Gene Expression Omnibus under the records GSE138995, GSE138996, and GSE138997. The microarray results were analyzed and presented as IPA diagrams (Supplemental Figure 2) and lists of genes that were notably regulated (Supplemental Tables 2–4). The microarray results showed that Sult1e1 was the most highly induced gene in the livers of AKI mice, and the induction of hepatic Sult1e1 by AKI in both male and female mice was verified by quantitative (q)RT-PCR (Figure 1A) and northern blotting (Figure 1B). The AKI induction of hepatic Sult1e1 was also confirmed by immunofluorescence (Figure 1C). Basal expression of Sult1e1 was also detected in the kidneys of both male and female mice, but interestingly, AKI induced renal expression of Sult1e1 in male mice but not in female mice, as shown by qRT-PCR (Figure 1D) and immunofluorescence (Figure 1E).
Figure 1.
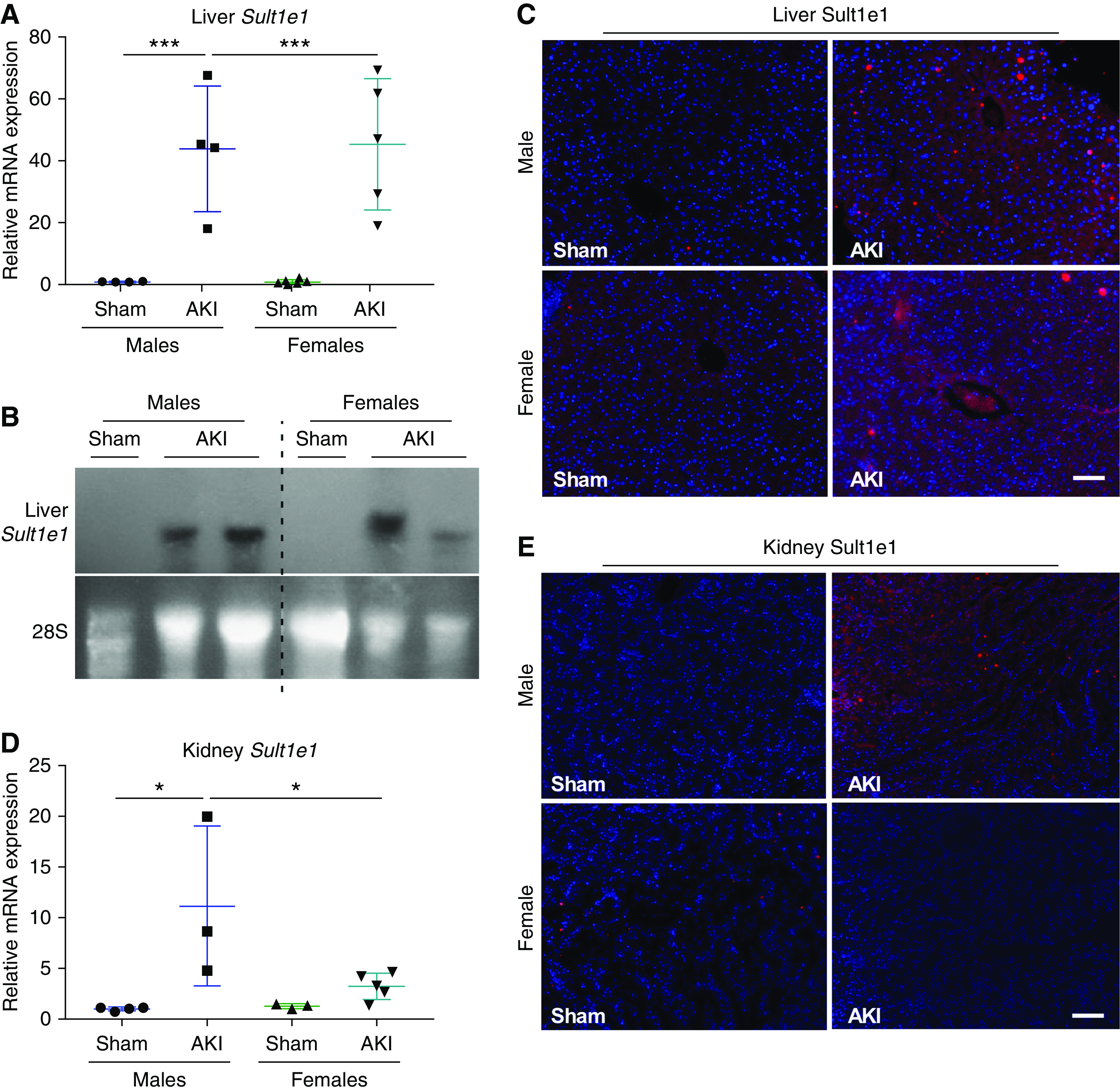
AKI induces the hepatic expression of Sult1e1 in both male and female mice, but induces the renal expression of Sult1e1 only in male mice. Mice were subject to 30-minutes of ischemic AKI or sham surgery, were euthanized, and tissues were harvested 24 hours after surgery. (A and B) Hepatic mRNA expression of Sult1e1 in male and female mice as shown by qRT-PCR (A) and northern blot analysis (B). 28S RNA was included as the loading control for northern blotting. (C) Immunofluorescence staining showed hepatic expression of Sult1e1 in male and female mice. (D and E) Renal expression of Sult1e1 in male and female mice was evaluated by qRT-PCR (D) and immunofluorescence (E). n=7 for each group. Scale bars are 50 µm. Results are presented as the mean±SD. *P<0.05; ***P<0.001, compared with the sham groups.
Inflammation is a Potential Mechanism for AKI-Responsive Induction of Sult1e1 in the Liver
The Sult1e1 gene has been reported to be positively regulated by nuclear receptors constitutive androstane receptor (CAR)28 and liver X receptor α (LXRα).29 The expression of Car was suppressed as we have previously reported,20 whereas the expression of Lxrα was not affected by AKI (Supplemental Figure 3A), suggesting that the AKI-responsive induction of hepatic Sult1e1 was nuclear receptor-independent. We have previously reported that the expression of Est/Sult1e1 can be induced by inflammation in sepsis and the Sult1e1 gene is a transcriptional target of NF-κB.11 Ischemic AKI is known to trigger local and systemic inflammatory responses,30 so we speculated that the AKI-responsive inflammation and subsequent secretion of inflammatory cytokines into the circulation may have contributed to the hepatic induction of Sult1e1. Indeed, AKI induced renal and hepatic expression of Il-6 (Supplemental Figure 3B), and increased the circulating level of Il-6 (Supplemental Figure 3C). Time course analysis showed that the induction of hepatic Il-6 and Il-1β preceded the induction of Sult1e1, in that the hepatic expression of Il-6 and Il-1β started increasing 1 hour after AKI (Supplemental Figure 3D), whereas the expression of Sult1e1 did not increase until 6 hours after AKI (Supplemental Figure 3E). The renal infiltration of cells positive for cluster of differentiation 3 (CD3), a surface marker of T cells, was increased upon AKI, consistent with the inflammatory response of the kidney (Supplemental Figure 3F). In vitro treatment with Il-6 induced the expression of Sult1e1 in primary hepatocytes (Supplemental Figure 3G) and the HKC-8 cells (Supplemental Figure 3H). Together, these results suggest that AKI may distantly induce the expression of Sult1e1 in the liver as a result of AKI-responsive inflammation, as summarized in Supplemental Figure 3I.
Genetic Ablation or Pharmacologic Inhibition of Sult1e1 Protects Mice from AKI
To determine the functional relevance of Sult1e1 and its regulation by AKI, we challenged 8-week-old male Sult1e1 KO mice with AKI or sham surgery, and the mice were euthanized for analysis 24 hours after. The Sult1e1 KO mice exhibited marked protection from AKI, including the abolishment of AKI-responsive increases in the serum levels of creatinine (Figure 2A) and BUN (Figure 2B), and attenuation of AKI-responsive induction of renal injury marker gene NGAL 2 (Ngal) at the mRNA (Figure 2C) and protein (Figure 2D) levels. At the histologic level, the Sult1e1 KO mice subjected to AKI showed fewer tubular injuries and less apoptosis, as shown by PAS staining and TUNEL assay, respectively (Figure 2E). Moreover, the AKI-responsive renal (Figure 2F) and hepatic (Figure 2G) induction of Il-6 was largely normalized in Sult1e1 KO mice, and so was the circulating level of Il-6 (Figure 2H). The renal protective effect of Sult1e1 ablation remained obvious 72 hours after the ischemic AKI, which was evidenced by lower levels of serum creatinine and BUN (Supplemental Figure 4A), decreased renal expression of Ngal (Supplemental Figure 4B), and reduced renal tubular injury and apoptosis (Supplemental Figure 4C). The protective effect of Sult1e1 KO was not sex-specific, because the female Sult1e1 KO mice were also protected from AKI, as evidenced by serum creatinine and BUN levels (Supplemental Figure 5A), renal expression of Ngal (Supplemental Figure 5B), renal histology (Supplemental Figure 5C), and renal expression of Il-6 (Supplemental Figure 5D).
Figure 2.
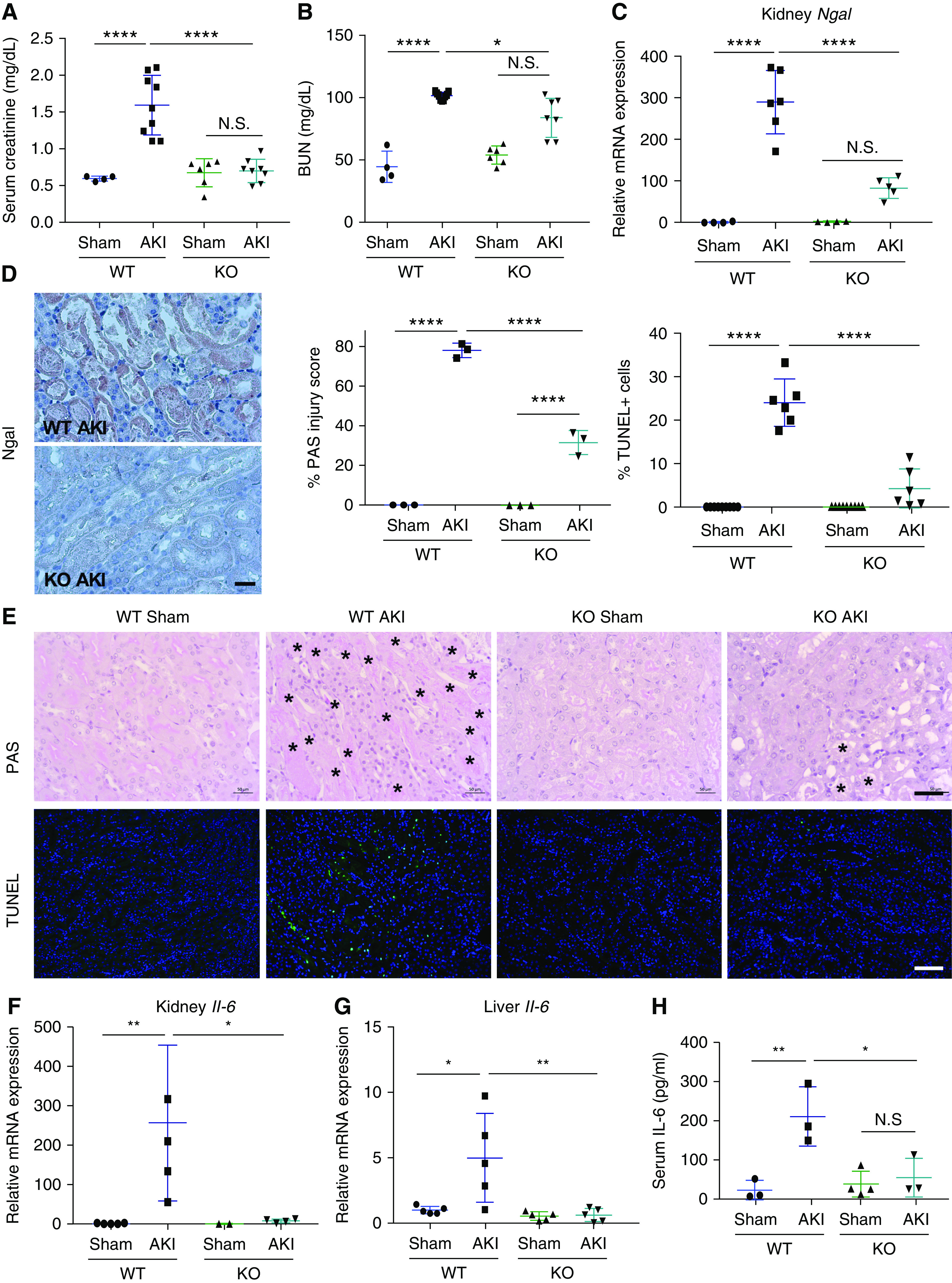
KO of Sult1e1 protects male mice from AKI. (A–H) WT and Sult1e1 KO males were subject to 30 minutes of ischemic AKI or sham surgery, and the mice were euthanized 24 hours after surgery. Shown are (A) serum level of creatinine, (B) BUN, and the (C) mRNA and (D) protein expression of renal Ngal, (E) histology as shown by PAS staining and TUNEL with the quantifications shown on the top right (with asterisks indicating tubular damage), (F and G) renal and hepatic mRNA expression of Il-6, and (H) serum level of Il-6. n=3–6 for each group. Scale bars are 50 µm. Results are presented as the mean±SD. *P<0.05; **P<0.01; ****P<0.0001; NS, statistically NS, with the comparisons labeled.
In an independent pharmacologic model of Sult1e1 inhibition, WT mice were treated with triclosan, an efficient pharmacologic inhibitor of SULT1E1,31 as outlined in Figure 3A. Consistent with results from the Sult1e1 KO mice, treatment of male mice with triclosan at 10 mg/kg or 50 mg/kg also attenuated AKI, as shown by decreased serum creatinine and BUN levels (Figure 3B), and renal expression of Ngal (Figure 3C). Triclosan-treated mice also presented improved histology (Figure 3D), and decreased renal and hepatic expression of Il-6 (Figure 3E). The 50 mg/kg dose of triclosan was also effective in attenuating AKI-responsive kidney injury in female mice (Supplemental Figure 6).
Figure 3.
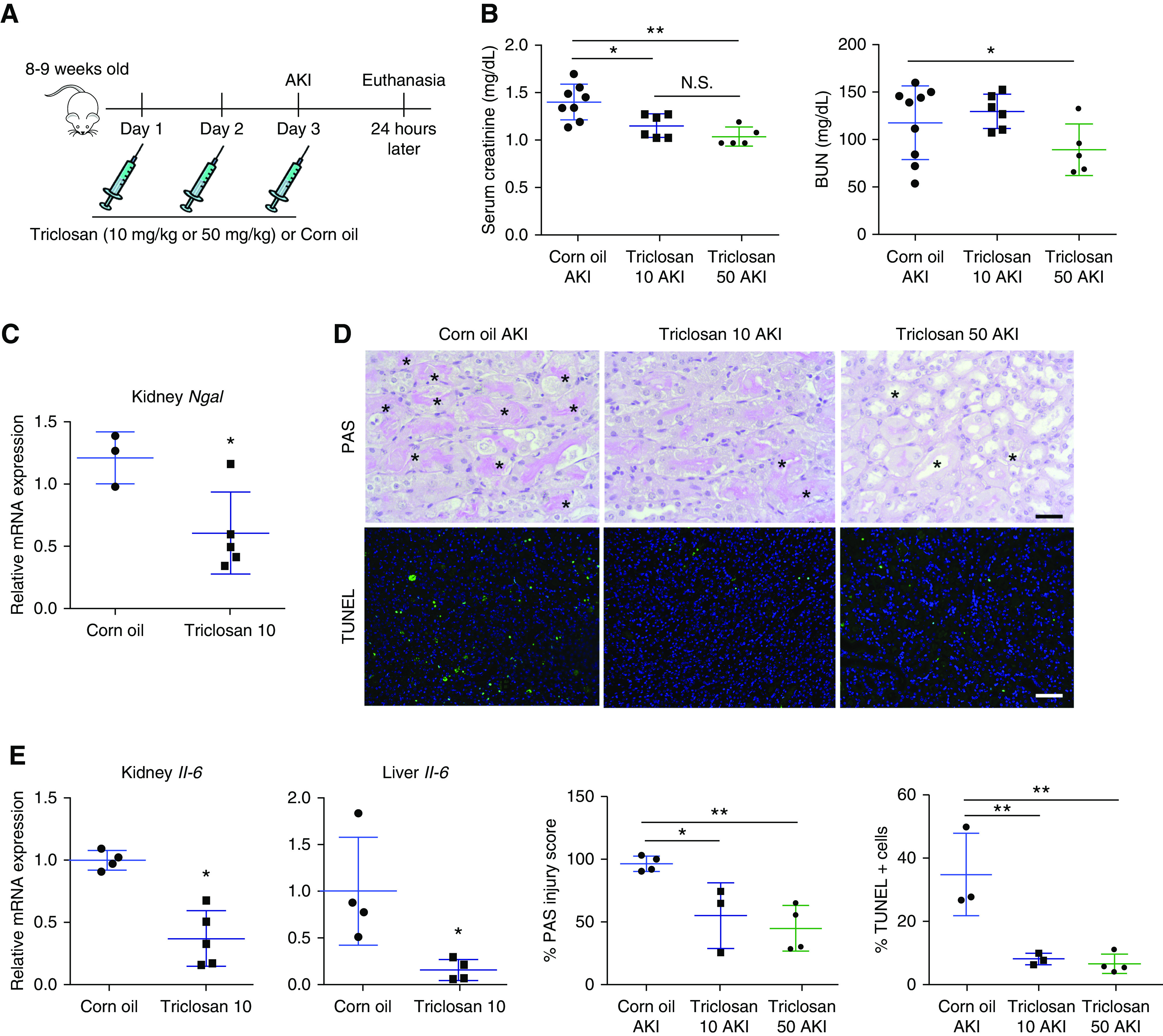
Pharmacologic inhibition of Sult1e1 protects mice from AKI. (A) Scheme of triclosan treatment. WT male mice received three doses of triclosan (10 or 50 mg/kg) or the vehicle corn oil before being subjected to 30 minutes of ischemic AKI, and the mice were euthanized 24 hours after surgery. (B–E) Shown are serum levels of (B) creatinine and BUN, (C) renal mRNA expression of Ngal, (D) histology as shown by PAS staining and TUNEL with the quantifications shown on the bottom right (with asterisks indicating tubular damage), and (E) renal and hepatic mRNA expression of Il-6. n=4–8 for each group. Scale bars are 50 µm. Results are presented as the mean±SD. *P<0.05; **P<0.01; compared with the corn oil groups or the comparisons are labeled.
The Renal Protective Effect of Sult1e1 Ablation is Estrogen- and Androgen-Independent
Because the primary function of SULT1E1 is sulfonating and deactivating estrogens,6,7 and administration of pharmacologic doses of E2 after cardiac arrest protected male mice from AKI,14 we wanted to know whether the renal protective effect of Sult1e1 ablation in male mice was estrogen-dependent. To our surprise, the serum levels of E2 (Figure 4A) were not significantly altered in male Sult1e1 KO AKI mice. The renal expression of estrogen-responsive gene Stat532 was not affected either (data not shown). Additionally, the expression of aromatase, the enzyme that converts testosterones into estrogens, was undetectable in the liver or kidney (data not shown). Castration or ovariectomy were then performed on male or female Sult1e1 KO mice to determine whether the renal protective effect of Sult1e1 ablation was androgen- or estrogen-dependent, respectively. In this experiment, castration or ovariectomy was performed on Sult1e1 KO mice 4 weeks before the AKI surgery, as outlined in Figure 4B. Both castrated Sult1e1 KO males and ovariectomized Sult1e1 KO females remained protected from AKI compared with their sham surgery counterparts, as shown by serum levels of creatinine, BUN, and histology (Figure 4, C–F). The effectiveness of the ovariectomy surgery was confirmed by decreased uterine weight as percentages of the body weight in ovariectomized mice (Figure 4G). These results suggested that the renal protective effect of Sult1e1 ablation is sex hormone-independent.
Figure 4.
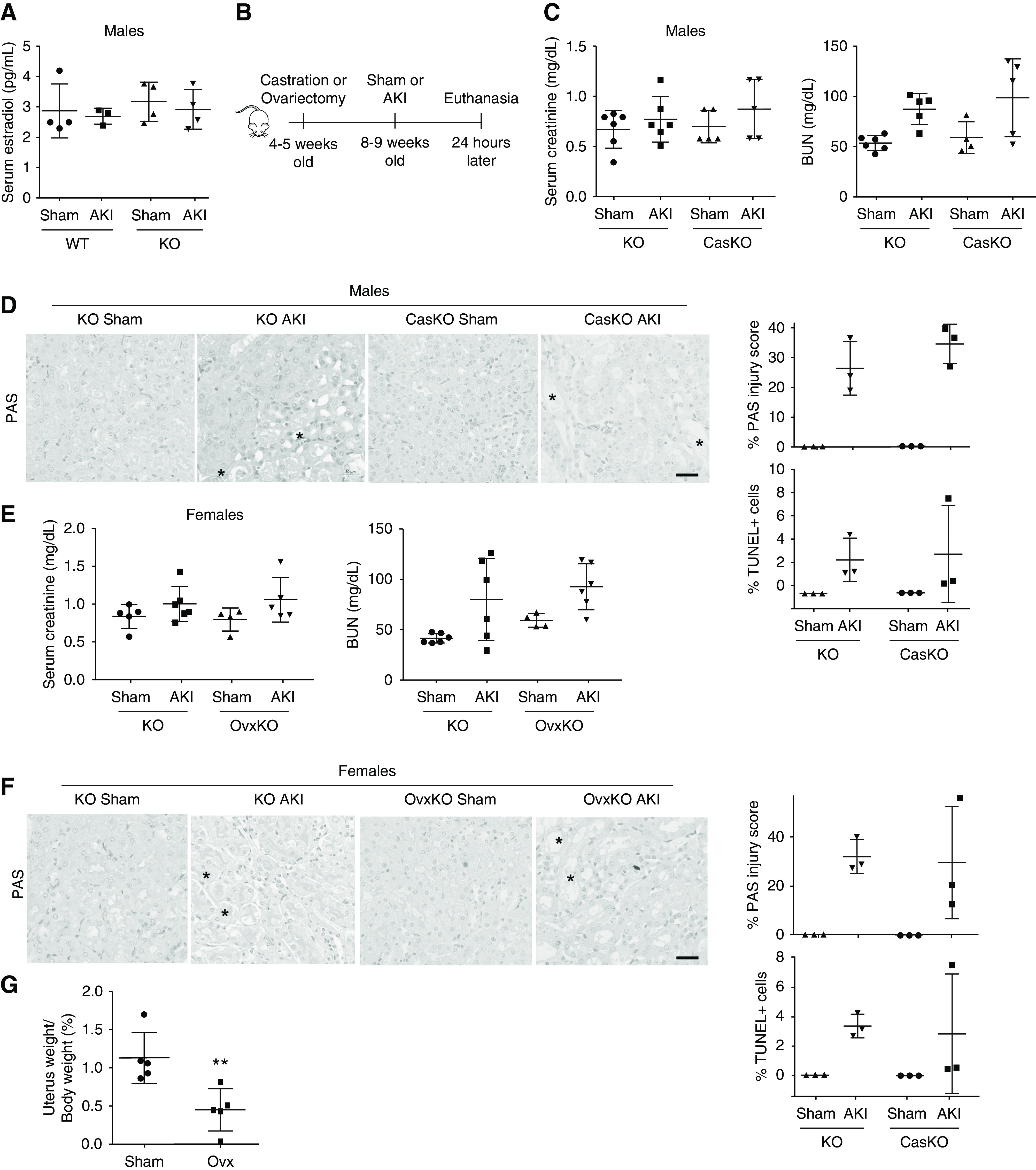
The renal protective effect of Sult1e1 ablation is estrogen- and androgen-independent. (A) Mice are the same as described in Figure 3A. Shown are the serum levels of E2. (B) Scheme of castration or ovariectomy followed by renal ischemic AKI. (C and D) Intact or castrated (CasKO) Sult1e1 KO males were subjected to sham surgery or AKI surgery. Shown are (C) serum levels of creatinine and BUN, and (D) renal histology as evaluated by PAS and TUNEL staining with their quantifications shown on the right with asterisks indicating tubular damage). (E and F) Intact or ovariectomized (OvxKO) Sult1e1 KO females were subjected to sham surgery or AKI surgery. Shown are (E) serum levels of creatinine and BUN, and (F) renal histology as evaluated by PAS and TUNEL staining with their quantifications shown on the right (with asterisks indicating tubular damage). (G) Uterine weights as percentages of the body weight in female mice subjected sham surgery or ovariectomy. n=4–6 for each group. Scale bars are 50 µm. Results are presented as the mean±SD.
Hepatic Sult1e1 is Required for AKI Renal Injury in Male, but not in Female, Mice
Knowing hepatic Sult1e1 is induced by AKI, we wanted to determine whether the hepatic expression of Sult1e1 distantly contributed to the pathogenesis of AKI. For this purpose, we reconstituted the expression of SULT1E1 tissue specifically to the liver using the KOLE mice. KOLE mice were generated by crossbreeding the Sult1e1 KO mice with the LE transgenic mice that express the human SULT1E1 transgene exclusively in the liver under the control of the Lap gene promoter,11 as outlined in Figure 5A. The KOLE mice express SULT1E1 in the liver but have a Sult1e1 KO background in other organs, including the kidney, which was verified by qRT-PCR (Figure 5B). In the male mice, restoration of liver SULT1E1 was sufficient to resensitize KOLE mice to AKI, as shown by serum levels of creatinine and BUN (Figure 5C), renal mRNA expression of Ngal (Figure 5D) and IL-6 (Figure 5E), and histology (Figure 5F). These results suggested that hepatic Sult1e1 is required for the sensitivity of male to AKI. Interestingly, the effect of hepatic reconstitution of SULT1E1 on AKI was sex-specific, because the female KOLE mice remained efficiently protected from AKI, as shown by serum levels of creatinine and BUN (Figure 5G), and histology (Figure 5H).
Figure 5.
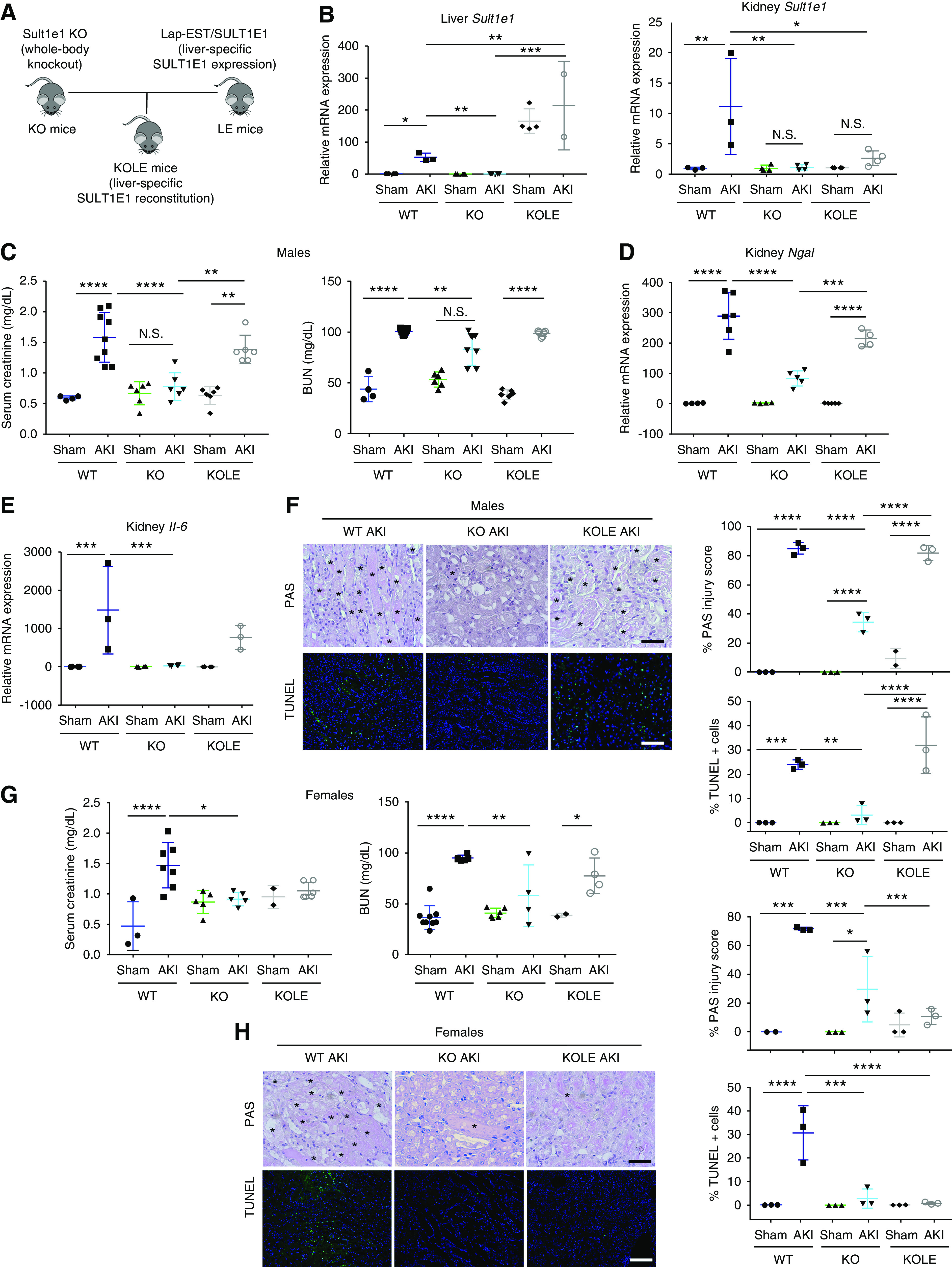
Hepatic Sult1e1 is required for AKI renal injury in male, but not in female, mice. (A) Schematic representation of the KOLE mice that were created by breeding the liver-specific LE transgene into the Sult1e1 KO background. (B) Reconstitution of SULT1E1 in the liver (left panel) and lack of reconstitution in the kidney (right panel) was confirmed by qRT-PCR. (C–F) Male mice of indicated genotypes were subjected to 30 minutes of ischemic AKI, and the mice were euthanized 24 hours after surgery. Shown are (C) serum levels of creatinine and BUN, renal mRNA expression of (D) Ngal and (E) Il-6, and (F) kidney histology as shown by PAS staining and TUNEL, with their quantifications shown on the right (with asterisks indicating tubular damage). (G and H) Experiments were the same as described in (C–F) except that female mice were used. Shown are (G) serum levels of creatinine and BUN, and (H) kidney histology as shown by PAS staining and TUNEL with their quantifications shown on the right (with asterisks indicating tubular damage). n=6 for each group. Scale bars are 50 µm. Results are presented as the mean±SD. *P<0.05, **P<0.01; ***P<0.001; ****P<0.0001; NS, statistically NS, with the comparisons labeled.
The Renal Protective Effect of Sult1e1 Ablation Is Associated with Renal Regulation of Vitamin D-Metabolizing and Cell Cycle Genes
Because the renal protective effect of Sult1e1 ablation was sex hormone-independent, we went on to determine whether the metabolism of other endogenous substrates may have been responsible for renal protection. In this effort, we performed Affymetrix microarray analysis comparing the transcriptomic profile in the kidneys of WT AKI and Sult1e1 KO AKI mice. The microarray showed altered expression of several genes involved in vitamin D metabolism and cell proliferation in the Sult1e1 KO AKI group (Supplemental Table 4). The inductions of Cyp24a1 and Ccnd1, and suppression of Fgg, were verified by qRT-PCR (Figure 6A). Cyp24a1, which encodes a vitamin D-metabolizing enzyme, is a known vitamin D receptor (VDR) target gene.33 The expression of Ccnd1, which encodes cyclin D1, has been reported upregulated at early rises of vitamin D34 and may promote cell proliferation.35 Fgg, which encodes fibrinogen, has been shown to have an inverse correlation with vitamin D levels.36 This gene expression profile suggested that the VDR signaling was enhanced in the kidneys of Sult1e1 KO mice upon AKI. Administration of the active form of vitamin D calcitriol has been shown to improve several animal models of AKI,37,38 and calcitriol is currently in clinical trials for the treatment of AKI.39 The induction of Cyp24a1 and cyclin D1 in Sult1e1 KO AKI kidneys, and the loss of these inductions in KOLE AKI kidneys, were confirmed by immunofluorescence (Figure 6, B and C), which correlated with the AKI protection and resensitization in these two genotypes, respectively. The increased renal expression of cyclin D1 was also verified by western blotting (Figure 6D). The renal immunostaining of Ki67, a cell proliferation marker, was increased in Sult1e1 KO AKI kidney and this effect was attenuated in KOLE AKI kidney (Figure 6E). Our luciferase reporter gene assay results showed that overexpression of SULT1E1 did not affect the activity of calcitriol in inducing the VDR-responsive reporter activity (Figure 6F), suggesting that calcitriol is not a direct substrate of SULT1E1.
Figure 6.
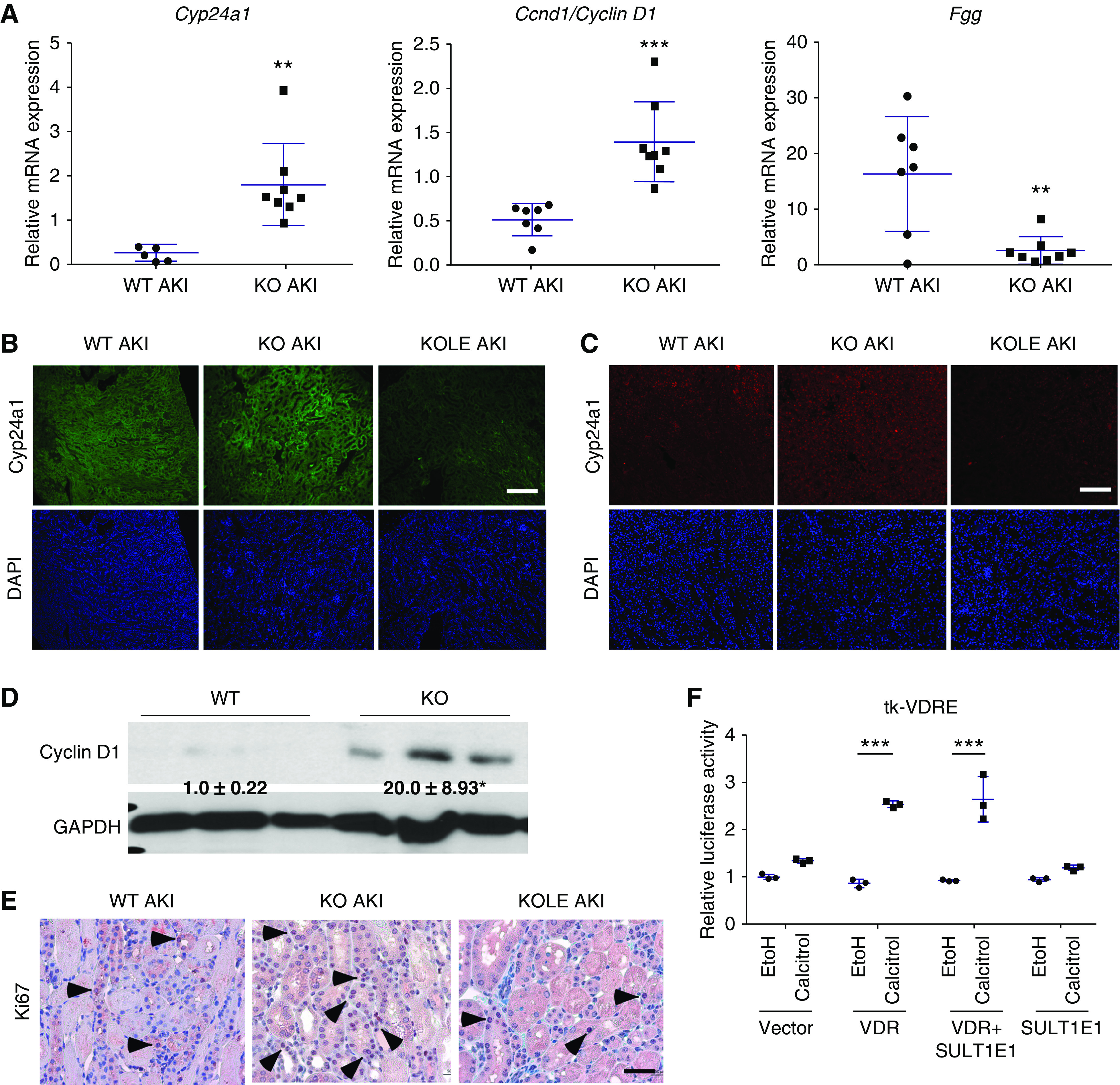
The renal protective effect of Sult1e1 ablation is associated with renal regulation of vitamin D-metabolizing and cell cycle genes. (A) Renal mRNA expression of Cyp24a1, Ccnd1, and Fgg in WT and Sult1e1 KO mice subjected to 30 minutes of ischemic AKI. (B and C) The renal expression of (B) Cyp24a1 and (C) Cyclin D1 in WT, Sult1e1 KO, and KOLE mice subjected to 30 minutes of ischemic AKI was shown by immunofluorescence. (D) The renal expression of Cyclin D1 protein in WT and Sult1e1 KO mice subjected to 30 minutes of ischemic AKI is shown by western blotting with the signal quantifications labeled. (E) Immunostaining of Ki67 in the same groups, with arrowheads indicating positive staining. (F) 293T cells were transfected with a VDR reporter gene tk-VDRE in the presence of the cotransfection of the VDR and SULT1E1 plasmids alone or in combination. Transfected cells were treated with vehicle (ethanol) or calcitriol (10 µM) for 24 hours before cell lysis and luciferase assay. n=3–8 per group. Scale bars are 50 µm. Results are presented as the mean±SD. **P<0.01; ***P<0.001; ****P<0.0001; compared with (A) WT AKI or (F) the comparisons are labeled. GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
DISCUSSION
The sex-specific effect of Sult1e1 on AKI is interesting. First, the regulation of Sult1e1 by AKI is sex-specific. AKI induced the hepatic expression of Sult1e1 in both male and female mice. However, the renal induction of Sult1e1 by AKI only occurred in males, and not in females. Interestingly, the effect of Sult1e1 ablation and reconstitution was also sex-specific. Although both male and female Sult1e1 KO mice were protected from AKI, reconstitution of SULT1E1 in the liver of male KOLE mice abolished the protective effect, whereas the female KOLE mice remained protected from AKI. Sex-specific effects of SULT1E1 ablation and/or reconstitution were also observed in our previous studies in the context of metabolic disease. We reported that Sult1e1 ablation protected female ob/ob mice from obesity and type 2 diabetes, but sensitized male ob/ob mice to metabolic syndrome.10 When the expression of SULT1E1 was reconstituted in the adipose tissue of Sult1e1-deficient ob/ob (obe) mice, the reconstitution effect was sex-specific because the adipose reconstitution of SULT1E1 improved the metabolic function of male obe mice, but had little effect on the female obe mice.40,41 Interestingly, in both AKI and metabolic disease, the effect of SULT1E1 reconstitution was uniformly obvious in males, but not in female mice. The male-specific effect of adipose reconstitution of SULT1E1 was explained to be because of high basal expression of Sult1e1 in the adipose tissue of male mice.41 The basal expression of Sult1e1 in the liver is low in both sexes, so the mechanism underlying the male-specific effect of liver constitution of SULT1E1 on AKI remains to be understood. The sex-specific effect of Sult1e1 ablation was also observed in a mouse model of liver ischemia-reperfusion-induced liver injury in that Sult1e1 ablation conferred protection to female mice, whereas the male mice were further sensitized.8 The effect of SULT1E1 reconstitution on ischemia-reperfusion-induced liver injury remains to be tested.
The tissue-specific effect of SULT1E1 on AKI is equally interesting. In male mice, although the AKI-responsive induction of Sult1e1 was observed in both the liver and kidney, our results suggested that loss of Sult1e1 in the liver, but not in the kidney, was responsible for the renoprotection, because reconstitution of SULT1E1 in the liver was sufficient to abolish the protection. The role of basal and AKI-inducible expression of renal Sult1e1 in AKI-induced kidney injury in male mice remains to be defined. In female mice, although loss of Sult1e1 in the liver was not responsible for the protection because liver reconstitution of SULT1E1 had little effect, we cannot conclude that loss of Est in the kidney accounted for the renoprotection. It will be interesting to determine whether reconstitution of SULT1E1 in the kidneys of female Sult1e1 KO mice affects the protective effect of Sult1e1 ablation. The tissue-specific effect of SULT1E1 was also observed in our previous study in the ob/ob mice. The ob/ob mice exhibited liver-specific upregulation of Sult1e1.10 Sult1e1 ablation in ob/ob (obe) males worsened the metabolic phenotype.10 Interestingly, transgenic reconstitution of SULT1E1 in the adipose tissue of male obe mice attenuated the metabolic phenotypes, including decreased local and systemic inflammation, improved insulin sensitivity, and increased energy expenditure.40,41 In contrast, reconstitution of SULT1E1 in the liver failed to improve the metabolic function of obe males.41 These results suggested that although hepatic Sult1e1 is markedly induced in ob/ob mice, it was not the loss of hepatic expression and induction of Sult1e1 that was responsible for the worsened metabolic function in obe males.
Another interesting finding of this study is the estrogen and androgen independence of the AKI protective effect of Sult1e1 ablation, because the protective effect was intact in Sult1e1 KO mice subject to ovariectomy or castration. The estrogen independence was a surprise, considering that a primary function of SULT1E1 is to regulate estrogen homeostasis, and estrogens have been suggested to be AKI protective in animals 12−14 and humans.17,18 Indeed, our previous study showed that the metabolic benefit of Sult1e1 ablation in female ob/ob mice was estrogen-dependent, because the metabolic benefit was abolished upon ovariectomy.10 The protective effect of Sult1e1 ablation on ischemia-reperfusion-induced liver injury in female mice was also estrogen-dependent.8 The androgen independence was also a surprise, because prior studies have suggested that castration ameliorates AKI, whereas testosterone administration worsens it.13 In addition, the sensitizing effect of Sult1e1 ablation on ischemia-reperfusion-induced liver injury in male mice was also androgen-dependent, because the sensitization was abolished upon castration.8 It remains to be understood why the sex hormone dependence of the SULT1E1 effect varies among disease models. Nevertheless, the sex hormone independence of the AKI protective effect of Sult1e1 ablation suggests that Sult1e1 substrates other than estrogens might have been responsible for the SULT1E1 effect on AKI.
Our gene profiling analysis suggested that increased vitamin D signaling may have contributed to the renal protective effect of Sult1e1 ablation, because the renal expression of several VDR-responsive genes, such as Cyp24a1, Ccnd1, and Fgg, was affected in Sult1e1 KO mice upon the AKI challenge. The enhanced VDR signaling may have explained the protective phenotype, because administration of calcitriol has been shown to improve several animal models of AKI,37,38 and calcitriol is currently in clinical trials for the treatment of AKI.39 Furthermore, calcitriol and VDR signaling have been reported to promote cell proliferation, especially in cancerous and damaged cells.42 We recognize that nearly all of the outcomes presented were from 24 hours, before most repair, so we cannot conclude that increased VDR signaling has promoted cellular recovery and tissue repair after AKI. Calcitriol is also known for its anti-inflammatory activity,43 which could also have contributed to the renoprotection. Because we have shown that calcitriol is not a SULT1E1 substrate, the mechanism by which Sult1e1 ablation or inhibition increases VDR signaling remains to be understood. We recognize that both VDR activation and cyclin D1 induction in AKI Sult1e1 KO mice were associations. Future studies are necessary to determine whether these two events are required for the renoprotective effect of Sult1e1 ablation.
Among the limitations, although we have identified interesting crosstalk between the liver and kidney, and shown clearly that the loss of hepatic Sult1e1 was responsible for the renoprotective effect of Sult1e1 ablation, future studies are necessary to identify the mediators released from the liver that affect kidney injury. Our microarray IPA analysis showed that the kidneys of Sult1e1 KO AKI mice had the highest positive z-score for the ataxia telangiectasia mutated (ATM) pathway (Supplemental Figure 2A). A positive z-score indicates this pathway was activated. The ATM pathway is activated in the presence of DNA damage and stimulates DNA repair, DNA recombination, and cell cycle control.44,45 Checkpoint kinase 2, the main effector of ATM kinase, is in charge of cell cycle regulation and also controls calcitriol formation.46 Future studies are necessary to determine whether ATM signaling could be a possible mediator for renoprotection. The sex-specific effect of hepatic reconstitution of SULT1E1 on Sult1e1 KO AKI mice also remains to be better understood. IPA analysis of our microarray results suggests that the livers of WT female, but not male, mice had a positive z-score for the VDR/retinoid X receptor pathway (Supplemental Figure 2, B and C), but its significance in female sensitivity to AKI remains to be defined.
In summary, we have uncovered a tissue- and sex-specific role of Sult1e1 in renal ischemic AKI. The hepatic expression of Sult1e1 is required for male animals’ sensitivity to ischemic AKI. Pharmacologic inhibition of SULT1E1 may represent a novel approach for the clinical management of AKI.
Disclosures
All authors have nothing to disclose.
Funding
This work was supported in part by National Institutes of Health grants DK117370 and ES030429 (to Dr. Xie), DK116816 (to Dr. Zhou), S10RR023461 (to Dr. Poloyac), and DK120531 (to Dr. Luo). Dr. Barbosa was supported by a Predoctoral Fellowship from the Science Without Borders Program, Government of Brazil. Dr. Xie was also supported in part by the Joseph Koslow Endowed Professorship from the University of Pittsburgh School of Pharmacy.
Supplementary Material
Acknowledgments
We thank Dr. Wen-chao Song (University of Pennsylvania) for his gift of the Sult1e1 KO mice and Junyi Li for her assistance in the analysis of tissue estrogens.
Dr. Barbosa and Dr. W. Xie designed the study. Dr. Barbosa, Dr. Choi, Dr. Zhou, Dr. W. Xie, Dr. Luo, and Dr. Y. Liu carried out experiments. Dr. Barbosa, Dr. Y. Liu, Dr. Yu, Dr. Luo, and Dr. W. Xie analyzed the data. Dr. Barbosa, Dr. Y. Liu, and Dr. W. Xie made the figures. Dr. Barbosa, Dr. Gibbs, Dr. Poloyac, Dr. Y. Liu, and Dr. W. Xie drafted and/or revised the manuscript. Dr. W. Xie received the funding. All authors approved the final version of the manuscript.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2019080767/-/DCSupplemental.
Supplemental Table 1. Sequences of qRT-PCR primers.
Supplemental Table 2. Top 200 upregulated and downregulated genes in the livers of male WT AKI mice (WT AKI versus WT sham).
Supplemental Table 3. Top 200 upregulated and downregulated genes in the livers of female WT AKI mice (WT AKI versus WT sham).
Supplemental Table 4. Top 200 upregulated and downregulated genes in the kidneys of male Sult1e1 KO AKI mice (KO AKI versus WT AKI).
References
- 1.Khwaja A: KDIGO clinical practice guidelines for acute kidney injury. Nephron Clin Pract 120: c179–c184, 2012. [DOI] [PubMed] [Google Scholar]
- 2.Chertow GM, Burdick E, Honour M, Bonventre JV, Bates DW: Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J Am Soc Nephrol 16: 3365–3370, 2005. [DOI] [PubMed] [Google Scholar]
- 3.Uchino S, Kellum JA, Bellomo R, Doig GS, Morimatsu H, Morgera S, et al. ; Beginning and Ending Supportive Therapy for the Kidney (BEST Kidney) Investigators : Acute renal failure in critically ill patients: A multinational, multicenter study. JAMA 294: 813–818, 2005. [DOI] [PubMed] [Google Scholar]
- 4.Wei Q, Dong Z: Mouse model of ischemic acute kidney injury: Technical notes and tricks. Am J Physiol Renal Physiol 303: F1487–F1494, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Falany CN, Krasnykh V, Falany JL: Bacterial expression and characterization of a cDNA for human liver estrogen sulfotransferase. J Steroid Biochem Mol Biol 52: 529–539, 1995. [DOI] [PubMed] [Google Scholar]
- 6.Driscoll WJ, Komatsu K, Strott CA: Proposed active site domain in estrogen sulfotransferase as determined by mutational analysis. Proc Natl Acad Sci U S A 92: 12328–12332, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pedersen LC, Petrotchenko E, Shevtsov S, Negishi M: Crystal structure of the human estrogen sulfotransferase-PAPS complex: Evidence for catalytic role of Ser137 in the sulfuryl transfer reaction. J Biol Chem 277: 17928–17932, 2002. [DOI] [PubMed] [Google Scholar]
- 8.Guo Y, Hu B, Huang H, Tsung A, Gaikwad NW, Xu M, et al.: Estrogen sulfotransferase is an oxidative stress-responsive gene that gender-specifically affects liver ischemia/reperfusion injury. J Biol Chem 290: 14754–14764, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xie Y, Xu M, Deng M, Li Z, Wang P, Ren S, et al.: Activation of pregnane x receptor sensitizes mice to hemorrhagic shock-induced liver injury. Hepatology 70: 995–1010, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao J, He J, Shi X, Stefanovic-Racic M, Xu M, O’Doherty RM, et al.: Sex-specific effect of estrogen sulfotransferase on mouse models of type 2 diabetes. Diabetes 61: 1543–1551, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chai X, Guo Y, Jiang M, Hu B, Li Z, Fan J, et al.: Oestrogen sulfotransferase ablation sensitizes mice to sepsis. Nat Commun 6: 7979, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maeda N, Tanaka E, Suzuki T, Okumura K, Nomura S, Miyasho T, et al.: Accurate determination of tissue steroid hormones, precursors and conjugates in adult male rat. J Biochem 153: 63–71, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kang KP, Lee JE, Lee AS, Jung YJ, Kim D, Lee S, et al.: Effect of gender differences on the regulation of renal ischemia-reperfusion-induced inflammation in mice. Mol Med Rep 9: 2061–2068, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ikeda M, Swide T, Vayl A, Lahm T, Anderson S, Hutchens MP: Estrogen administered after cardiac arrest and cardiopulmonary resuscitation ameliorates acute kidney injury in a sex- and age-specific manner. Crit Care 19: 332, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hutchens MP, Dunlap J, Hurn PD, Jarnberg PO: Renal ischemia: Does sex matter? Anesth Analg 107: 239–249, 2008. [DOI] [PubMed] [Google Scholar]
- 16.Neugarten J, Golestaneh L, Kolhe NV: Sex differences in acute kidney injury requiring dialysis. BMC Nephrol 19: 131, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hsu RK, McCulloch CE, Dudley RA, Lo LJ, Hsu CY: Temporal changes in incidence of dialysis-requiring AKI. J Am Soc Nephrol 24: 37–42, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eriksson M, Brattström O, Mårtensson J, Larsson E, Oldner A: Acute kidney injury following severe trauma: Risk factors and long-term outcome. J Trauma Acute Care Surg 79: 407–412, 2015. [DOI] [PubMed] [Google Scholar]
- 19.Grams ME, Rabb H: The distant organ effects of acute kidney injury. Kidney Int 81: 942–948, 2012. [DOI] [PubMed] [Google Scholar]
- 20.Choi Y-J, Zhou D, Barbosa ACS, Niu Y, Guan X, Xu M, et al.: Activation of constitutive androstane receptor ameliorates renal ischemia-reperfusion-induced kidney and liver injury. Mol Pharmacol 93: 239–250, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Golab F, Kadkhodaee M, Zahmatkesh M, Hedayati M, Arab H, Schuster R, et al.: Ischemic and non-ischemic acute kidney injury cause hepatic damage. Kidney Int 75: 783–792, 2009. [DOI] [PubMed] [Google Scholar]
- 22.Nolin TD, Frye RF, Matzke GR: Hepatic drug metabolism and transport in patients with kidney disease. Am J Kidney Dis 42: 906–925, 2003. [DOI] [PubMed] [Google Scholar]
- 23.Qian YM, Sun XJ, Tong MH, Li XP, Richa J, Song W-C: Targeted disruption of the mouse estrogen sulfotransferase gene reveals a role of estrogen metabolism in intracrine and paracrine estrogen regulation. Endocrinology 142: 5342–5350, 2001. [DOI] [PubMed] [Google Scholar]
- 24.Xie Y, Barbosa ACS, Xu M, Oberly PJ, Ren S, Gibbs RB, et al.: Hepatic estrogen sulfotransferase distantly sensitizes mice to hemorrhagic shock-induced acute lung injury. Endocrinology 161: bqz031, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao J, Yan J, Xu M, Ren S, Xie W: CAR suppresses hepatic gluconeogenesis by facilitating the ubiquitination and degradation of Pgc1α. Mol Endocrinol 29: 1558–1570, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou J, Zhai Y, Mu Y, Gong H, Uppal H, Toma D, et al.: A novel pregnane X receptor-mediated and sterol regulatory element-binding protein-independent lipogenic pathway. J Biol Chem 281: 15013–15020, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dwivedi PP, Hii CS, Ferrante A, Tan J, Der CJ, Omdahl JL, et al.: Role of MAP kinases in the 1,25-dihydroxyvitamin D3-induced transactivation of the rat cytochrome P450C24 (CYP24) promoter. Specific functions for ERK1/ERK2 and ERK5. J Biol Chem 277: 29643–29653, 2002. [DOI] [PubMed] [Google Scholar]
- 28.Alnouti Y, Klaassen CD: Regulation of sulfotransferase enzymes by prototypical microsomal enzyme inducers in mice. J Pharmacol Exp Ther 324: 612–621, 2008. [DOI] [PubMed] [Google Scholar]
- 29.Gong H, Guo P, Zhai Y, Zhou J, Uppal H, Jarzynka MJ, et al.: Estrogen deprivation and inhibition of breast cancer growth in vivo through activation of the orphan nuclear receptor liver X receptor. Mol Endocrinol 21: 1781–1790, 2007. [DOI] [PubMed] [Google Scholar]
- 30.Bonventre JV, Zuk A: Ischemic acute renal failure: An inflammatory disease? Kidney Int 66: 480–485, 2004. [DOI] [PubMed] [Google Scholar]
- 31.James MO, Li W, Summerlot DP, Rowland-Faux L, Wood CE: Triclosan is a potent inhibitor of estradiol and estrone sulfonation in sheep placenta. Environ Int 36: 942–949, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jelinsky SA, Harris HA, Brown EL, Flanagan K, Zhang X, Tunkey C, et al.: Global transcription profiling of estrogen activity: Estrogen receptor α regulates gene expression in the kidney. Endocrinology 144: 701–710, 2003. [DOI] [PubMed] [Google Scholar]
- 33.St-Arnaud R, Arabian A, Travers R, Barletta F, Raval-Pandya M, Chapin K, et al.: Deficient mineralization of intramembranous bone in vitamin D-24-hydroxylase-ablated mice is due to elevated 1,25-dihydroxyvitamin D and not to the absence of 24,25-dihydroxyvitamin D. Endocrinology 141: 2658–2666, 2000. [DOI] [PubMed] [Google Scholar]
- 34.Rots NY, Iavarone A, Bromleigh V, Freedman LP: Induced differentiation of U937 cells by 1,25-dihydroxyvitamin D3 involves cell cycle arrest in G1 that is preceded by a transient proliferative burst and an increase in cyclin expression. Blood 93: 2721–2729, 1999. [PubMed] [Google Scholar]
- 35.Han Y, Chen A, Umansky K-B, Oonk KA, Choi W-Y, Dickson AL, et al.: Vitamin D stimulates cardiomyocyte proliferation and controls organ size and regeneration in zebrafish. Dev Cell 48: 853–863.e5, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mellenthin L, Wallaschofski H, Grotevendt A, Völzke H, Nauck M, Hannemann A: Association between serum vitamin D concentrations and inflammatory markers in the general adult population. Metabolism 63: 1056–1062, 2014. [DOI] [PubMed] [Google Scholar]
- 37.Tan X, Wen X, Liu Y: Paricalcitol inhibits renal inflammation by promoting vitamin D receptor-mediated sequestration of NF-kappaB signaling. J Am Soc Nephrol 19: 1741–1752, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsai J-P, Lee C-J, Hsieh Y-H, Hsu B-G: Calcitriol ameliorated rhabdomyolysis induced acute renal failure in rats. Int J Clin Exp Med 10: 2430–2439, 2017 [Google Scholar]
- 39.Cameron LK, Lei K, Smith S, Doyle NL, Doyle JF, Flynn K, et al.: Vitamin D levels in critically ill patients with acute kidney injury: A protocol for a prospective cohort study (VID-AKI). BMJ Open 7: e016486, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barbosa ACS, Feng Y, Yu C, Huang M, Xie W: Estrogen sulfotransferase in the metabolism of estrogenic drugs and in the pathogenesis of diseases. Expert Opin Drug Metab Toxicol 15: 329–339, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Garbacz WG, Jiang M, Xu M, Yamauchi J, Dong HH, Xie W: Sex-and tissue-specific role of estrogen sulfotransferase in energy homeostasis and insulin sensitivity. Endocrinology 158: 4093–4104, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vuolo L, Di Somma C, Faggiano A, Colao A: Vitamin D and cancer. Front Endocrinol (Lausanne) 3: 58, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Díaz L, Noyola-Martínez N, Barrera D, Hernández G, Avila E, Halhali A, et al.: Calcitriol inhibits TNF-α-induced inflammatory cytokines in human trophoblasts. J Reprod Immunol 81: 17–24, 2009. [DOI] [PubMed] [Google Scholar]
- 44.Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, et al.: A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 268: 1749–1753, 1995. [DOI] [PubMed] [Google Scholar]
- 45.Beamish H, Williams R, Chen P, Lavin MF: Defect in multiple cell cycle checkpoints in ataxia-telangiectasia postirradiation. J Biol Chem 271: 20486–20493, 1996. [DOI] [PubMed] [Google Scholar]
- 46.Fahkri H, Zhang B, Fajol A, Hernando N, Elvira B, Mannheim JG, et al.: Checkpoint kinase Chk2 controls renal Cyp27b1 expression, calcitriol formation, and calcium-phosphate metabolism. Pflugers Arch 467: 1871–1880, 2015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.