

Summary
The processing of RNA transcripts from mammalian genes occurs close to their transcription. Here we describe a phenomenon affecting thousands of genes that we call exon-mediated activation of transcription starts (EMATS), in which the splicing of internal exons impacts promoter choice and the expression level of the gene. We observed that evolutionary gain of internal exons is associated with gain of new transcription start sites (TSS) nearby and increased gene expression. Inhibiting exon splicing reduced transcription from nearby promoters. Conversely, creation of new splice sites that enabled splicing of new exons activated transcription from cryptic promoters. The strongest effects occurred for weak promoters located proximal and upstream of efficiently spliced exons. Together, our findings support a model in which splicing recruits transcription machinery locally to influence TSS choice, and identify exon gain, loss and regulatory change as major contributors to the evolution of alternative promoters and gene expression in mammals.
Graphical Abstract
eTOC Blurb
Transcription and splicing occur in close proximity. Fiszbein et al. show that splicing of internal exons activates transcription from nearby upstream promoters, especially those that are weak or even cryptic, which they call “exon-mediated activation of transcription starts” (EMATS). A model is proposed in which splicing factors recruit transcription machinery to locally boost transcription. These findings imply that regulation of splicing can be and often is used to alter the transcriptional output of mammalian genes.
In Brief Sentence
Fiszbein et al. show that splicing of internal exons activates transcription from nearby upstream promoters, especially those that are weak or even cryptic, suggesting that regulation of splicing can be used to alter the transcriptional output of mammalian genes.
Introduction
RNA transcripts from mammalian genes are processed within seconds or minutes after their synthesis, creating opportunities for functional connections between transcription and splicing (Custódio and Carmo-Fonseca, 2016). Several links between splicing and transcription are known, and both transcription rate and chromatin structure can influence splicing outcomes (Bentley, 2014; Kornblihtt et al., 2013; Schor et al., 2013). However, more recent evidence suggests that splicing also feeds back on transcription (Braunschweig et al., 2013). Adding an intron to an intron-less gene often boosts gene expression in plants, animals, and fungi; although the mechanisms are not fully understood, impacts on transcription, nuclear export, mRNA stability, and/or translation have been noted (Furger et al., 2002; Shaul, 2017). Splicing can impact the rate of transcription elongation (Fong and Zhou, 2001), and in yeast the presence of an intron can generate a transcriptional checkpoint that is associated with pre-spliceosome formation (Chathoth et al., 2014). Furthermore, recruitment of the spliceosome complex can stimulate transcription initiation by enhancing preinitiation complex assembly (Damgaard et al., 2008), and inhibition of splicing can reduce levels of histone 3 lysine 4 trimethyl (H3K4me3), a chromatin mark associated with active transcription (Bieberstein et al., 2012).
Several components of the splicing machinery associate with RNA polymerase II (RNAPII) and other transcription machinery (Das et al., 2007; Emili et al., 2002; Kameoka et al., 2004; Morris and Greenleaf, 2000; Mortillaro et al., 1996; Neugebauer and Roth, 1997; Vincent et al., 1996). The U1 and U2 small nuclear ribonucleoprotein particles (snRNPs) associate with general transcription factors (GTFs) TFIIH/GTF2H1 (Kwek et al., 2002), TFIIF/GTF2F2 (Kameoka et al., 2004), and the carboxy-terminal domain (CTD) of RNAPII (Emili et al., 2002; Morris and Greenleaf, 2000). In addition to its role in splicing, U1 snRNP acts as a general repressor of proximal downstream premature cleavage and polyadenylation (PCPA) sites (Gunderson et al., 1998; Kaida et al., 2010). The relative abundance of U1 snRNP binding sites upstream in the antisense orientation from promoters contributes to frequent termination of antisense transcripts at PCPA sites, yielding short and unstable transcripts (Almada et al., 2013).
Alternative transcription initiation and termination sites drive a substantial portion of transcript isoform differences between human tissues (Reyes and Huber, 2018). Recent analyses of full-length mRNAs suggests that transcription starts and splicing may be coordinated (Anvar et al., 2018). However, whether exon splicing commonly impacts transcription start site (TSS) location and activity remains unknown. Here we describe a phenomenon we call “exon-mediated activation of transcription starts” (EMATS) in which the splicing of internal exons, especially those near gene 5′ ends, alters gene expression by influencing which TSSs are used, contributing to expression regulation of thousands of genes.
Results
Increased exon splicing is associated with increased gene expression and alternative TSS usage
We used a comparative approach to explore potential connections between splicing and TSS usage, examining transcript patterns in orthologous genes of mouse and rat that differed by the presence/absence of an internal exon. Previously, we identified over one thousand such exons that were not detected in RNA-seq data from diverse organs/tissues of other mammals including rat, macaque, and cow, and therefore likely arose recently in the murine lineage. We also identified a similar number of exons that are unique to the rat, as well as several hundred exons uniquely lost in mouse or in rat (Figure 1A). Most of such evolutionarily new exons are located in 5′ untranslated regions (UTRs) and are spliced in an alternative and tissue-specific fashion (Merkin et al., 2015). Comparing closely related species, we have observed that genes with evolutionarily new internal exons tend to have increased gene expression, but only in those tissues where the new exons are included in mRNAs (Figure S1A and Table S1) (Merkin et al., 2015). This trend was stronger for exons that were efficiently spliced – assessed by “percent spliced in” (PSI or ψ) values > 0.95, indicating that more than 95% of mRNAs from the gene include the exon (Figure 1B) – suggesting an association between the extent of exon splicing and level of gene expression.
Figure 1. Splicing of new exons is associated with increased gene expression and gain of TSSs.
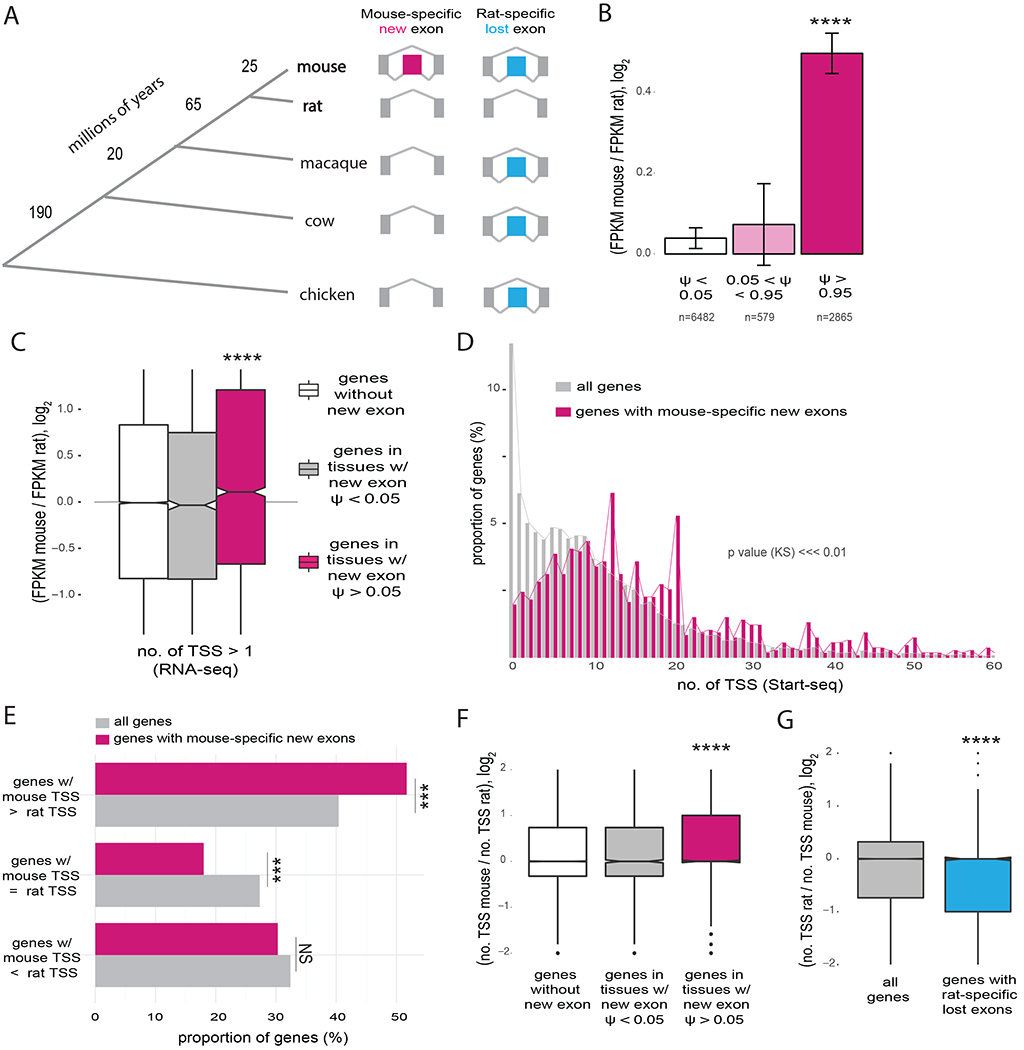
A, Phylogenetic tree representing the main species used for dating evolutionarily new exons and approximate branch lengths in millions of years. The patterns of inclusion/exclusion used to infer mouse-specific new exons (n = 1089) and rat-specific lost exons (n = 515) are shown. B, Fold change in gene expression in genes with mouse-specific exons (assessed by fragments per kilobase of exon per million mapped reads, FPKM) between mouse and rat in 9 organs, binned by ψ value of the new exon in each tissue. Number of gene-tissue pairs in each category is indicated. Mean ± SEM of displayed distributions is shown. C, Fold change in gene expression between corresponding tissues of mouse and rat in genes with multiple TSSs in mouse (no. of TSS > 1) for mouse control genes with no new exons (white), genes with mouse-specific new exons in tissues where inclusion of the new exon is not detected, PSI < 0.05 (grey), and genes with new mouse-specific exons in tissues were the exon is included, PSI > 0.05 (pink). D, Distribution of the number of TSSs per gene using Start-seq data from murine macrophages for all genes expressed in mouse and genes with mouse-specific new exons. TSS peaks located within 50 bp from each other were merged. Genes with mouse-specific new exons have increased numbers of TSSs (p < 2.2e−16 by Kolmogorov-Smirnov test). E, Proportion of genes that gained TSSs in mouse (mouse TSS > rat TSS), genes that lost TSSs in mouse (mouse TSS < rat TSS) and genes with same number of TSSs in both species (mouse TSS = rat TSS) for all genes expressed in both species and genes with mouse-specific new exons. F, Fold change in the number of TSSs used per gene between mouse and rat for 9 tissues, for mouse genes grouped as in (C). G, Ratio of number of TSSs used in rat over number used in mouse, for all genes expressed in both species (grey) and for genes with rat-specific lost exons (blue).
See also Figure S1.
Grouping genes by the number of promoters used, we observed a positive association between inclusion of new exons and gene expression for genes with multiple TSSs while this association was not observed for genes with only one TSS (Figure 1C). Furthermore, our RNA-seq data (from (Merkin et al., 2012)) showed that genes with mouse-specific new exons were far more likely to have multiple TSSs compared to all expressed genes in mouse (Figure S1B and S1C). We confirmed that genes with new mouse-specific exons are more likely to have multiple TSSs using other methods to define TSS locations, including H3K4me3 ChIP-seq peaks (Yu et al., 2015) and data from high-resolution sequencing of polymerase-associated RNA (Start-seq) (Scruggs et al., 2015) (Figure 1D, S1D, Table S2). Genes with rat-specific new exons (n = 1517) were also far more likely to have multiple TSSs than rat genes overall (Figure S1E). Furthermore, genes that gained new species-specific exons were more likely to have gained TSSs in the same species, suggesting that the evolutionary gain of an internal exon is connected to evolutionary gain of TSSs in a locus (Figure 1E and S1F).
To investigate this connection further, we examined the new exons and TSSs used by a gene across different tissues. We observed that genes containing mouse-specific exons used more TSSs than their rat orthologs (Figure S1G), and that this association was specific to mouse tissues where the new exon was included with PSI > 0.05 (Figure 1F and S1H), showing a connection between splicing and TSS usage across mammalian organs. We also observed higher PSI values for new exons in genes with multiple alternative TSSs relative to genes with a single TSS (Figure S1I). Conversely, loss of internal exons was associated with TSS loss and decreased gene expression levels (Figure 1G and S1J). Together, these observations indicate that the usage of new TSSs and the splicing of new internal exons tend to occur in the same genes, tissues, and species, suggesting an intimate connection between splicing, increased gene expression and new TSSs.
TSSs arise proximal and upstream of new exons
We observed that increased gene expression in mouse relative to rat was restricted to those genes that gained TSSs in mouse (Figure 2A and S2A), confirming a tight connection between evolution of promoters, internal exons and gene expression levels. Only 10% of genes with new TSSs gained mouse-specific new exons (Figure S2B), not different from the fraction of analyzed genes overall (Figure 2B). This directional bias suggests that the gain of species-specific new exons favors the gain of new TSSs rather than vice versa.
Figure 2. TSSs arise proximal and upstream of new exons.
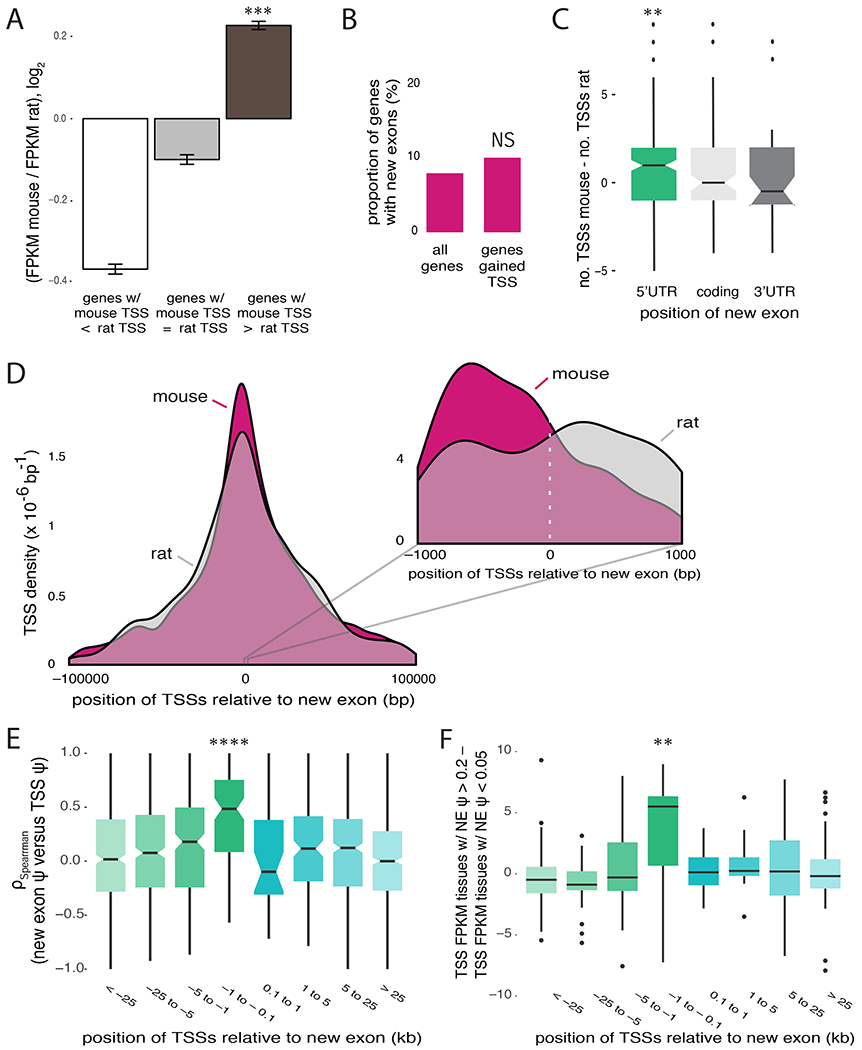
A, Fold change in gene expression between mouse and rat for genes that gained TSSs in mouse (mouse TSS > rat TSS), genes that lost TSSs in mouse (mouse TSS < rat TSS) and genes with same number of TSSs in both species (mouse TSS = rat TSS). B, Proportion of genes that gained mouse-specific new exons from all genes expressed in mouse and rat, and genes that gained new TSSs. C, Ratio between number of TSSs used in mouse and in rat for genes with mouse-specific new exons, binned by location of the exon within the gene. D, Histogram of TSS locations in mouse (pink) and rat (grey) in all 9 tissues for genes with mouse-specific new exons, centered on start of mouse new exon or homologous genomic position in rat. Inset shows zoom-in of locations within 1 kb of new exon. Distributions were smoothed with kernel density estimation. E, Spearman correlations between relative TSS usage and new exon PSI across mouse tissues, for TSSs binned by position relative to mouse-specific exon. F, Difference in expression (in units of FPKM) in mouse tissues for transcripts including TSSs in tissues where new exon is moderately or highly included (PSI > 0.2) versus tissues where new exon is excluded (PSI < 0.05), grouped by TSS location relative to new exon.
See also Figure S2.
We observed a positional effect in which the increase in the number of TSS per gene was associated predominantly with new exons located in 5′ UTRs (Figure 2C). We examined the distribution of the locations of all mouse TSSs relative to the locations of mouse-specific new exons (Figure S2C, Table S3), and compared it to the distribution of rat TSSs relative to sites homologous to mouse-specific exons. This comparison showed an enrichment of TSSs in mouse within a few kilobases (kb) upstream of new exons (Figure 2D). Thus, evolutionary gain of new internal exons was specifically associated with gain of proximal, upstream TSSs.
We then asked about the relationship between splicing levels and usage of alternative TSSs within the same gene. Considering relative TSS usage (representing the fraction of transcripts from a gene that derive from a given TSS) we found that use of the most proximal upstream TSS (designated TSS −1) was positively correlated with new exon inclusion, especially for TSSs located within about 1 kb upstream of the new exon (Figure 2E and S2D). Furthermore, absolute expression of transcripts from nearby TSSs increased specifically in tissues where new exons were included at moderate or high levels (Figure 2F). These observations suggest a positive influence of splicing on nearby transcription.
Manipulation of exon splicing impacts upstream transcription initiation
To directly test whether splicing impacts nearby transcription, we chose two candidate mouse genes, Gper1 (G protein-coupled estrogen receptor 1), and Tsku (Tsukushi, small leucine rich proteoglycan). These genes both have widespread, moderate expression and contain a mouse-specific 5′ UTR internal exon whose splicing is positively correlated with the expression of the gene across mouse tissues (Spearman ρ = 0.64 and 0.57, respectively; Figure 3A and 3B left panels). When cultured mouse fibroblasts were treated with morpholino antisense oligonucleotides (MO) targeting splice sites of the new exons in these genes, exon inclusion decreased by about 4-fold in both Gper1 (Figure 3A) and Tsku (Figure 3B). Moreover, gene expression levels of these two genes were depressed to a similar extent (Figure S3A), consistent with a positive effect of exon inclusion on gene expression. We observed similar levels of repression when assaying metabolically labeled nascent RNA (Figure 3A and 3B) as with total mRNA (Figure S3A), indicating that the effect is primarily at the level of transcription rather than mRNA stability.
Figure 3. Manipulation of exon splicing impacts upstream transcription initiation.
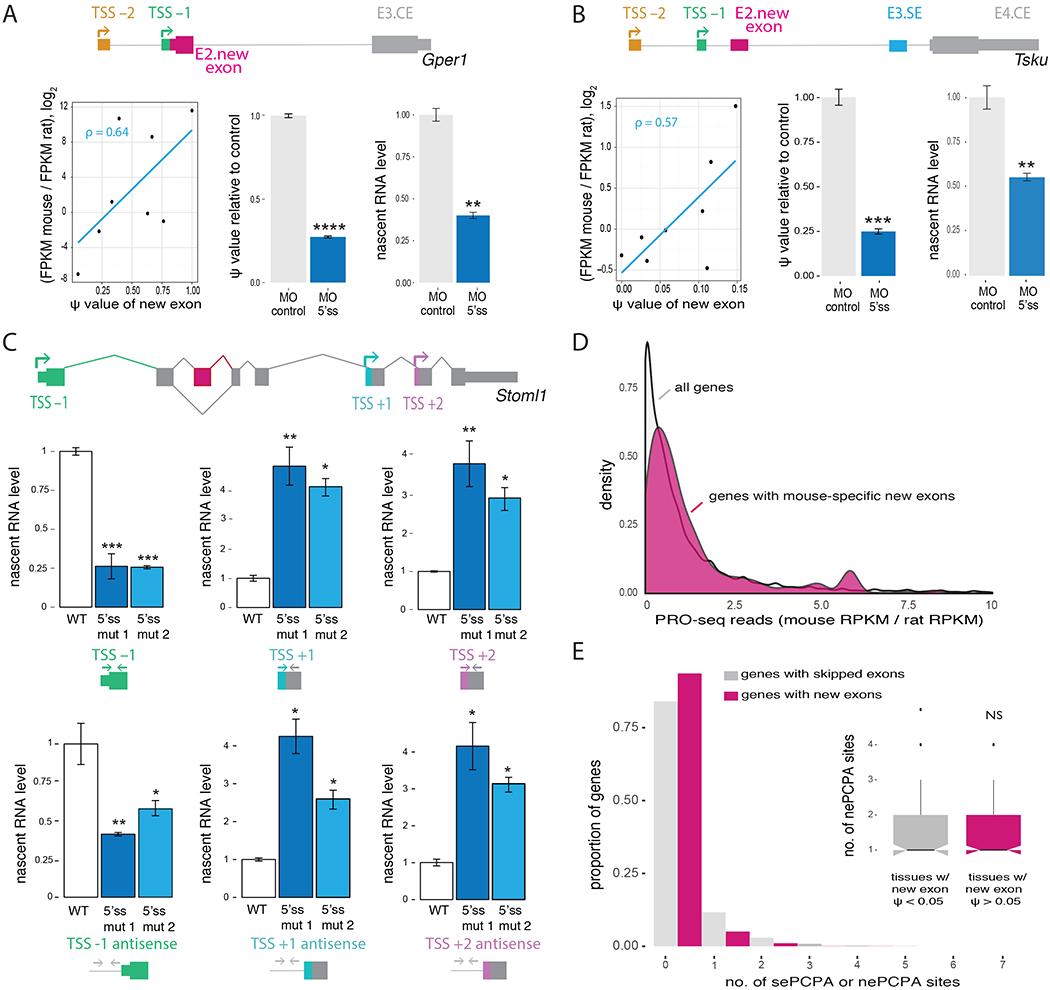
A, (Left) Relationship between fold change in gene expression between mouse and rat and new exon PSI value across 8 tissues for Gper1 gene. (Right) qRT-PCR analysis of fold change in new exon PSI value (middle) and gene expression (right) in nascent RNA metabolically labeled for 10 minutes with 5-ethynyl uridine, following treatment of NIH3T3 cells with MO targeting new exon 5′ splice site relative to control treatment. Mean ± SEM of displayed distributions, n=3 biological replicates. B, As in (A) for mouse Tsku gene. C, Fold change in nascent sense (top) and antisense (bottom) RNA levels of Stoml1 in CAD cells measured by qRT-PCR of RNA metabolically labeled for 10 minutes with 5-ethynyl uridine and normalized using housekeeping genes Gapdh, Hprt and Hspcb. Wild type cells in white and CRISPR-Cas cells with mutations in the 5′ splice site of the new exon in blue. Mean ± SEM of displayed distributions, n=3 independent experiments. A schematic diagram of Stoml1 exon-intron organization is shown at top. D, Fold change in nascent RNA expression between mouse and rat for all genes expressed in both species (grey) and genes with mouse-specific new exons (pink) in CD4+ T cells using PRO-seq data. Distributions were smoothed with kernel density estimation. E, Distribution of the number of polyadenylation sites used per gene located 2 kb upstream/downstream of a control set of mouse genes with skipped exons (grey, sePCPA) and genes with mouse-specific new exons (pink, nePCPA). In the inset, distribution of the number of polyadenylation sites used 2 kb upstream/downstream of new exons per gene in tissues where new exon is excluded (PSI < 0.05, grey) or included (PSI > 0.05, pink), for genes with new exons and at least one nePCPA. Distributions are not significantly different by Kolmogorov-Smirnov test.
See also Figure S3.
We next sought to confirm the directionality of this effect and to ask how splicing of new exons impacts the usage of different TSSs. We chose for analysis the mouse Stoml1 (Stomatin Like 1) gene, which has three active alternative TSSs as well as a new exon. Using CRISPR/Cas9 mutagenesis to generate cell lines with mutations abolishing the inclusion of the new exon (Figure S3B) we observed that the three alternative TSSs of the gene responded differently to inhibition of splicing of the new exon. The upstream TSS −1 was down-regulated by 4-fold, while downstream +1 and +2 TSSs were up-regulated to a similar extent in the mutant cell lines (Figure 3C). Effects on antisense transcription in these mutant cell lines mirrored those observed for sense transcription (Figure 3C), suggesting that inclusion of the new exon enhances transcription from the upstream promoter in both directions. This pattern is distinct from a report of intron-mediated enhancement in which sense-oriented introns specifically inhibited antisense transcription (Agarwal and Ansari, 2016), but is consistent with reported impacts on transcription initiation resulting from changes in the position of an intron in a reporter gene (Gallegos and Rose, 2017). Levels of H3K4me3 and RNAPII decreased in the upstream TSS and increased in the downstream TSSs in the mutant cell lines, consistent with the observed effects on nascent transcript production (Figure S3C).
To assess the relationship between splicing and nearby transcription initiation on a genome-wide scale, we analyzed precision run-on sequencing (PRO-seq) data from mouse and rat CD4+ T cells (Danko et al., 2018). Genes with evolutionarily new internal exons had increased nascent RNA expression compared to all expressed genes (Figure 3D). Furthermore, the relative increase in nascent RNA is driven by transcripts initiating upstream of the position of the new exon, specifically from TSSs within two kb upstream of new exons (Figure S3D and S3E).
PCPA can produce truncated, unstable transcripts, but can be inhibited by binding of U1 snRNP near of a PCPA site (Gunderson et al., 1998; Kaida et al., 2010). If the observations above reflected effects of splicing machinery on PCPA rather than on transcription, this would require the presence of new exon proximal PCPA (“nePCPA”) sites in affected genes. Using available polyA-seq data from five mouse tissues, we observed that only 8.6% of genes with new exons had evidence of a nePCPA site, no higher than in control genes (Figure 3E). For the subset of genes that contain nePCPA site(s), we did not observe differences in usage of the site between tissues where the new exon was spliced in and those where it was spliced out (Figure 3E inset and S3F). We also saw no relationship between the number of nePCPA sites and gene expression changes between mouse and rat (Figure S3G). Thus, our results suggest that effects on PCPA do not contribute significantly to EMATS and that EMATS impacts transcription initiation rather than later steps.
Creation of a new splice site activates the use of a cryptic promoter nearby
We next sought to explore how splicing might affect the use of different upstream TSSs. In the Tsku gene, the mouse-specific TSS in position −1 is located within 1 kb upstream of the mouse-specific exon, while the conserved TSS −2 is located further upstream. Analysis by 5′ RACE showed that both TSSs are used at similar levels in mouse fibroblasts. However, inhibiting splicing of the new exon by MO preferentially suppressed TSS −1 (Figure 4A, S4A,B). This shift was accompanied by a 3-fold decrease in H3K4me3 levels near TSS −1 in MO-treated cells (Figure 4B). However, levels of H3K4me3 near TSS −2 were unchanged, confirming that transcription from TSS −2 is not affected (Figure 4B). In cells treated with MOs, levels of TFIIF and RNAPII decreased by almost 3-fold near TSS −1 but were unchanged near TSS −2 (Figure 4C and S4C). These observations suggest that splicing of the new exon may contribute to recruitment of core transcription machinery to the proximal TSS −1. Moreover, the loss of signal for TFIIF and RNAPII near the new exon following MO treatment suggests that inclusion of the new exon is associated with recruitment of transcription factors, consistent with functional interactions between GTFs and splicing machinery (Damgaard et al., 2008; Das et al., 2007). These observations confirm that splicing of new exons can regulate the usage of alternative TSSs, with predominant effects on proximal upstream promoters.
Figure 4. Creation of a new splice site activates the use of a cryptic promoter nearby.
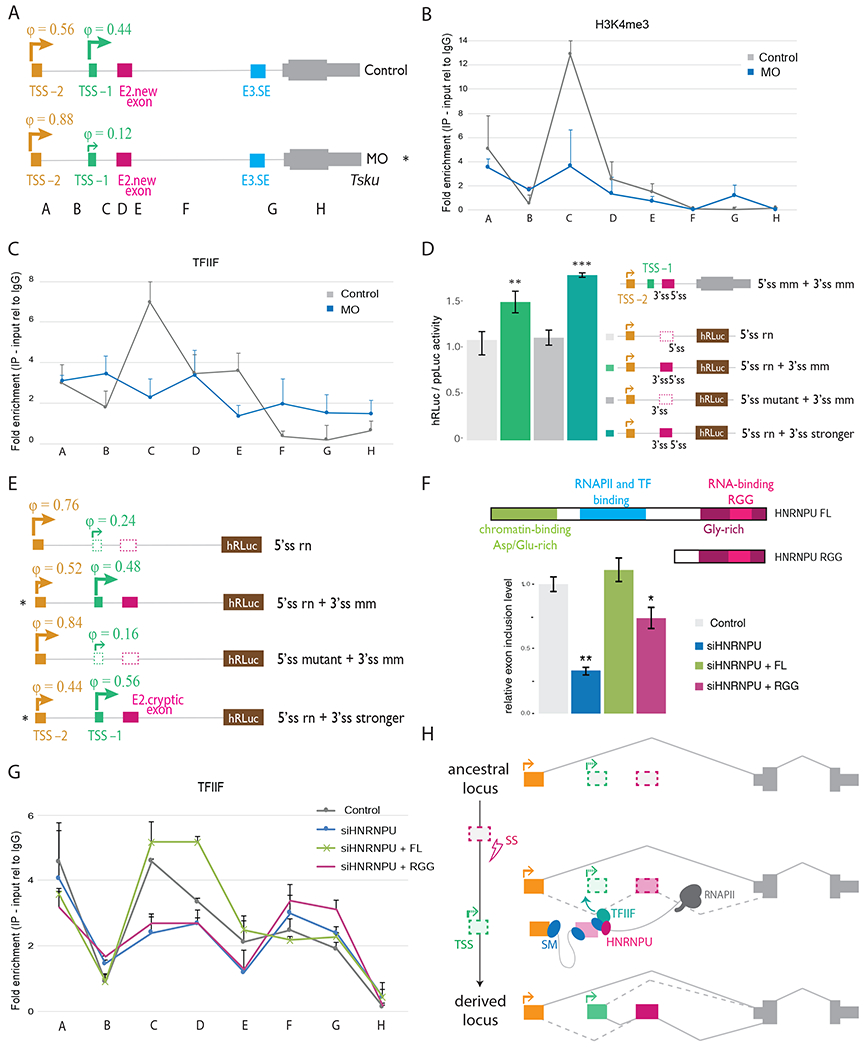
A, Schematic of 5′ RACE products showing TSS usage defined by the fraction of clones (φ, “phi”) corresponding to each TSS in control NIH3T3 cells and cells transfected with MO targeting the 3′ and 5′ splice sites of the new exon in Tsku, with a minimum of 25 clones for each sample from 2 biological replicates, * p < 0.05, one-tailed Fisher exact test. B,C, ChIP-PCR analysis of H3K4me3 (B) and TFIIF (subunit TFIIF-alpha) (C) in Tsku gene in NIH3T3 cells for regions indicated in (A). Mean ± SD of two independent immunoprecipitations normalized to input and mean value for control IgG antibody are shown. Data shown for control cells (grey) and cells treated with MOs targeting the 3′ and 5′ splice sites of the new exon (blue). D, Luciferase activity in HeLa cells transfected with the Tsku minigene reporters shown (right). Promoter activities of the corresponding constructs (corrected for transfection efficiency) are presented as fold increase of Renilla luciferase activity relative to firefly luciferase activity (both encoded on the same plasmid). Mean ± SD for n=3 independent experiments. E, 5′ RACE analysis as in (A). NIH3T3 mouse cells were transfected with plasmids expressing the corresponding rat Tsku mutants. F, (top) Schematic of HNRNPU constructs are shown, (bottom) qRT-PCR analysis of fold change in new exon PSI value following treatment with a control siRNA or an siRNA targeting HNRNPU (siHNRNPU) and rescues in NIH3T3 cells. Mean ± SEM of displayed distributions, n=3 biological replicates. G, ChIP-PCR analysis of TFIIF (subunit TFIIF-alpha) in Tsku gene in NIH3T3 cells for regions indicated in (A). Mean ± SD of two independent immunoprecipitations normalized to input and mean value for control IgG antibody are shown. Data shown for control cells (grey) and cells treated with siHNRNPU (blue) rescued with HNRNPU RNA binding domain (RGG, pink) or HNRNPU full-length (FL, green). H, Model in which creation of a splice site during evolution triggers inclusion of a new internal exon which activates use of an upstream cryptic TSS. In the model, exon recognition by HNRNPU in transcripts from the distal promoter recruits TFIIF that activates a TSS located proximal and upstream of the exon. Transcripts initiating from the proximal promoter also include the exon, further boosting activity of this promoter.
See also Figure S4.
To dissect the impacts of individual splice sites and splicing levels, we created an exon corresponding to the mouse-specific new exon in the rat Tsku gene and assessed effects on transcription. In the rat Tsku locus, transcripts are predominantly transcribed from the distal TSS −2. However, the regions homologous to TSS −1 and the mouse-specific new exon have high sequence identity with the mouse genome: both 5′ splice sites are present in rat, but no YAG is present in rat near the location of the mouse 3′ splice site, likely preventing splicing (Figure S4D). To introduce the desired mutations, we cloned the 5′ end of the rat Tsku gene upstream of the coding sequence of Renilla luciferase and recreated the 3′ splice site that is present in the mouse genome (5′ss rn + 3′ss mm), as well as a stronger 3′ splice site (5′ss rn + 3′ss stronger), while either maintaining or mutating the native rat 5′ splice site sequence (5′ss mutant + 3′ss mm). Strikingly, the creation of a 3′ splice site promoted the inclusion of an exon analogous to that observed in mouse in constructs with an intact 5′ splice site (Figure S4E), indicating that this mutation is sufficient to create a new exon in the rat gene. In the presence of both 3′ and 5′ splice sites, but not when either splice site was absent, total gene expression levels increased, as measured by luciferase activity (Figure 4D). By 5′ RACE analysis, TSS −1 is used at basal levels in the minigene. However, the mouse-specific exon in the rat context activates the usage of TSS −1 by 3-fold in the presence of a 5′ splice site, demonstrating that the effect on TSS usage depends on splicing of the mouse-specific exon rather than merely the presence of a 3′ splice site sequence (Figure 4E and S4F).
Our findings above imply the existence of mechanisms that coordinate splicing with TSS usage. To explore factors that might be involved in this coordination, we analyzed the enrichment of binding motifs for splicing factors in mouse novel exons. We observed that the binding motifs of splicing factors RBM22, HNRNPU and ELAVL1 were at least two-fold enriched in new exons whose inclusion was correlated with usage of nearby TSS (ρ > 0.3 compared to ρ < 0.3) (Figure S4G). This observation raised the possibility that some splicing factors may contribute to splicing-dependent regulation of TSSs, perhaps by recruitment of GTFs near sites of RNA splicing as seen above (Figure 4C). To explore this possibility, we analyzed the recruitment of TFIIF to the Tsku locus following depletion of HNRNPU, which is known to interact with TFIIF via its N-terminal domain (Kim and Nikodem, 1999). HNRNPU motifs were enriched downstream of the mouse-specific exon in Tsku (Figure S4H), and splicing of this exon was reduced by about 3-fold following depletion of HNRNPU (Figure 4F). Levels of TFIIF near TSS −1 and near the new exon decreased following HNRNPU depletion, while levels near TSS −2 and the constitutive exon were not affected (Figure 4G). Consistently, by qRT-PCR we observed down-regulation of transcripts from TSS −1 and no change in transcripts from TSS −2 following HNRNPU depletion (Figure S4I). The effects of HNRNPU depletion on exon splicing, transcription from TSS −1 and on TFIIF levels at the proximal promoter were rescued by overexpression of full length HNRNPU. However, a truncated version of HNRNPU that contained the C-terminal RNA binding and splicing regulatory domain but lacked the N-terminal domain that interacts with TFIIF partially rescued splicing but failed to rescue TFIIF levels and TSS −1 expression (Figure 4G and S4I). Together, these observations and those above (Figure 4C) suggest that HNRNPU acts both to activate splicing of the new exon and to mediate splicing-dependent activation of transcription, perhaps by recruitment of TFIIF to the proximal promoter.
In some examples studied previously, species-specific alternative splicing alters protein function (Gracheva et al., 2011; Gueroussov et al., 2015). Our observations support the existence of a distinct evolutionary pathway in which, following a mutation that generates a new internal exon, splicing of the new exon in transcripts from a distal upstream promoter activates transcription from a cryptic proximal upstream promoter. A possible model consistent with our data proposes that the new exon recruits the spliceosome and perhaps also splicing factors such as HNRNPU that act to increase the concentration of GTFs nearby. Transcripts from the new promoter will also include the exon, further activating the new promoter in a sort of positive feedback loop. The newly activated TSS will also produce novel transcript isoforms and generate higher gene expression in tissues where the upstream promoter is active and the exon is included (Figure 4H).
Efficiently spliced exons activate use of weak proximal TSSs
To investigate the genomic scope of the relationship between splicing and alternative TSS usage observed above, we asked whether the inclusion of alternative skipped exons (SE) in general – not just those that evolved recently – can influence TSS selection. We identified 49,488 SEs in mouse RNA-seq data, distributed across 13,491 genes using conservative criteria (Table S4). Analyzing unique SEs with TSS-exon distances matching those of new exons, we observed no significant association between SE inclusion and use of proximal upstream TSSs overall (Figure 5A). In addition, we observed a symmetrical distribution of TSSs around the locations of SEs, distinct from the upstream-biased distribution seen relative to new exons (Figure 5B). These differences suggest that genes with new exons have distinct properties that favor the linkage of splicing and transcription.
Figure 5. Efficiently spliced exons activate weak proximal TSSs.
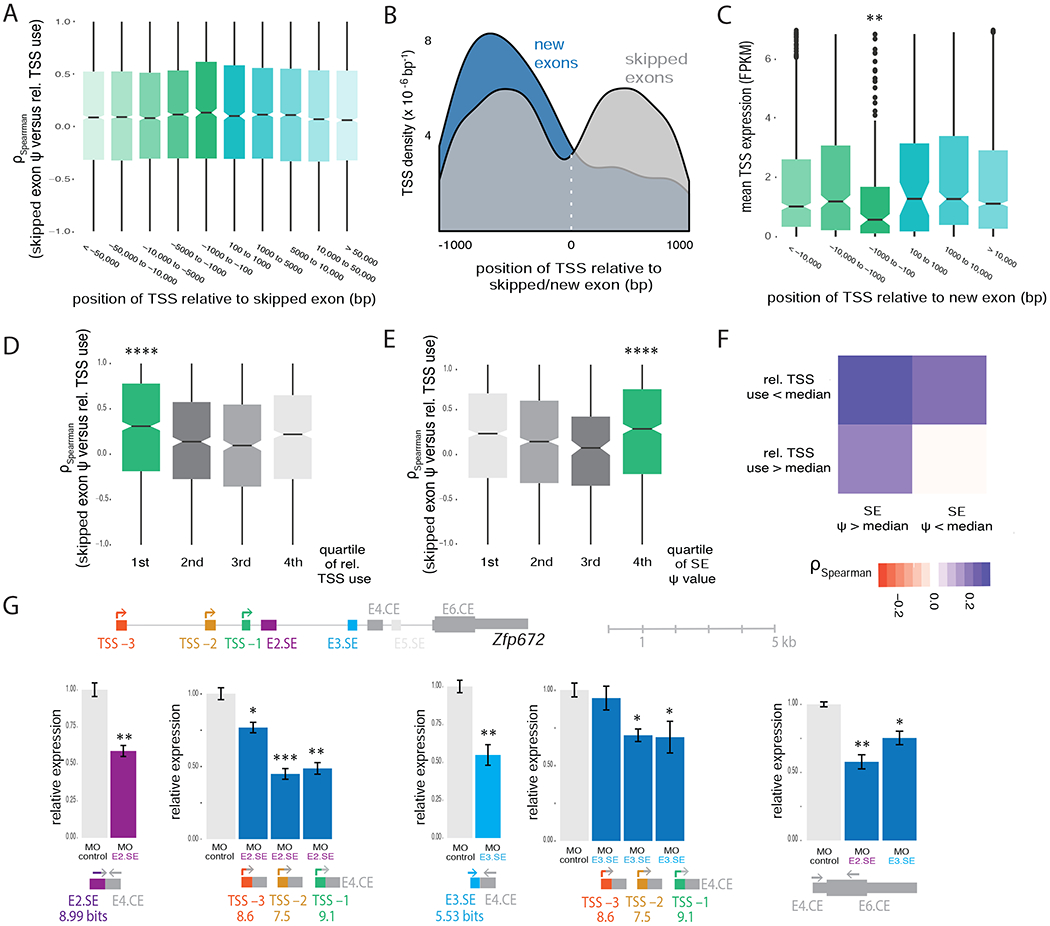
A, Spearman correlations between relative TSS usage (n = 49,911) and skipped exon PSE (SE, n = 13,491) in the same gene across mouse tissues for all expressed TSSs in genes with SEs, binned by genomic position relative to the SE. B, Comparison of distributions of TSS positions in 9 tissues for genes with mouse-specific new exons (blue) and genes with SEs in mouse (grey). Position 0 is set to the start coordinate of the new exon/skipped exon. Distributions were smoothed with Kernel density estimation. C, Expression of alternative first exons (AFE) for all TSSs in genes with mouse-specific new exons in tissues where the new exon is included (PSI > 0.05), binned by position relative to the new exon. D, Spearman correlation between relative TSS use and SE PSI in the same gene across mouse tissues for TSSs within 1kb upstream of the SE, binned by quartiles of mean relative TSS use. E, Same as (D) but binned by quartiles of mean SE PSI. F, Heat map showing the median Spearman correlation between relative TSS use and SE PSI in the same gene across mouse tissues for SEs with at least one TSS located upstream, in four groups, according to whether the mean relative TSS use (across tissues) and the mean SE PSI were greater than or less than the corresponding median values (across all TSSs and SEs analyzed). G, Exon-intron organization of mouse Zfp672 gene. qRT-PCR analysis of expression of Zfp672 in NIH3T3 cells normalized to expression of housekeeping genes Hprt and Hspcb. Data for control cells and cells treated with MO targeting the indicated splice sites (E4.CE and E6.CE). E5.SE is not included in NIH3T3 cells. Inclusion levels of the skipped exons, as well as levels of exon-excluding transcripts from the alternative TSSs (TSS −3, TSS −2, TSS −1) and total gene expression are shown. Scores of 5′ splice sites of skipped exons and first exons are listed in bits. Mean ± SEM of displayed distributions for n=3 independent experiments.
See also Figure S5.
Examining other features of gene loci with new exons, we observed that, although new exons tend to have lower PSI values than SEs overall (Figure S5A), those new exons with proximal upstream TSSs tended to have higher PSI values and stronger 5′ splice sites (Figure S5B). Furthermore, although the distribution of relative TSS usage (PSI) values was similar in genes with new exons and genes with SEs generally (Figure S5C), those TSSs located proximal and upstream of new exons had lower average PSI across tissues than TSSs in other locations (Figure 5C). Thus the link between splicing and TSS usage is most pronounced when the promoter is intrinsically weak and splicing activity is high. Consistently, previous studies have observed stronger intron-mediated enhancement in the presence of weaker promoters (Callis et al., 1987). To test this idea, we grouped SEs and their most proximal and upstream TSS into four bins from weak to strong on the basis of the relative TSS usage value, and separately for the SE PSI value, and analyzed the correlation between relative TSS usage and SE PSI separately within each bin. Notably, we observed that TSS usage was most highly correlated with exon inclusion for the lowest quartile of relative TSS usage values (Figure 5D, S5D and S5E) and for the highest quartile of SE PSI (Figure 5E and S5F). This observation provides evidence that the EMATS observed for new exons may occur for a subset of SEs generally. Robust effects were observed when a weak promoter is located upstream of a highly included SE – an arrangement we call “EMATS organization” – which occurred in 3,833 mouse genes. The strongest effects were observed when the weak promoter was within 2 kb of the SE – a pattern we call “EMATS structure” – which occurred in 1777 mouse genes (Figure 5F and Table S4). In humans, we identified 3548 genes with EMATS organization and 1413 genes with EMATS structure. Considering also constitutive exons, the number of genes increases 3-fold.
To further investigate the distance-dependence of splicing effects on TSS use, we analyzed changes in TSS usage when inhibiting the inclusion of a SE in the mouse Tsku locus that is located more than 6 kb downstream of the TSSs. Perturbations of the splicing of this exon yielded no detectable changes in TSS usage (Figure S5G), consistent with a requirement for proximity of the spliced exon and TSS for EMATS activity. Considering another mouse gene, Zfp672 (Zinc Finger Protein 672) – chosen because it contained multiple TSSs and SEs expressed in mouse fibroblasts – we observed that inhibition of the stronger upstream SE in the locus affected the usage of TSSs more dramatically than inhibition of the weaker downstream SE (Figure 5G). A weaker distal TSS (TSS −2) was impacted to a similar degree as a stronger proximal TSS (TSS −1) by perturbations of the splicing of these SEs (Figure 5G). Together, these observations provide further evidence that splicing of SEs can impact TSS usage, particularly when the TSS is intrinsically weak, the SE is highly included and the TSS is located proximal and upstream of the SE. The generalization of EMATS from new exons to the much larger class of highly included SEs implies that gene expression may commonly be regulated through effects on the splicing of promoter-proximal exons.
Splicing factors impact TSS use and EMATS connection to neurogenesis
A mechanistic link between splicing and nearby transcription initiation could potentially be mediated by core splicing machinery, splicing factors, or exon junction complex (EJC) components deposited during splicing, particularly those factors that interact with transcription machinery, transcription factors or chromatin. To explore functional links between RNA-binding proteins (RBPs) and TSS use, we analyzed transcriptome-wide changes in alternative TSS usage following knockdown of RBPs using data from a recent ENCODE project (Van Nostrand et al., 2018). Consistent with previous observations in Drosophila cells showing that up to 30% of alternative splicing events that were affected by knockdown of 56 RBPs involved changes in promoter use (Brooks et al., 2015), our analysis detected large numbers of TSS changes (Figure S6A). Depletion of EJC components and factors involved in RNA splicing impacted larger numbers of TSSs than did depletion of other RBPs (Figure 6A). Previous studies have linked the EJC to gene expression regulation at the level of RNAPII pausing, enhancement of 3′ end processing, increasing mRNA steady-state levels and translational utilization (Akhtar et al., 2019; Wiegand et al., 2003).
Figure 6. A subset of splicing factors have wide impacts on TSS usage and interact with transcription machinery.
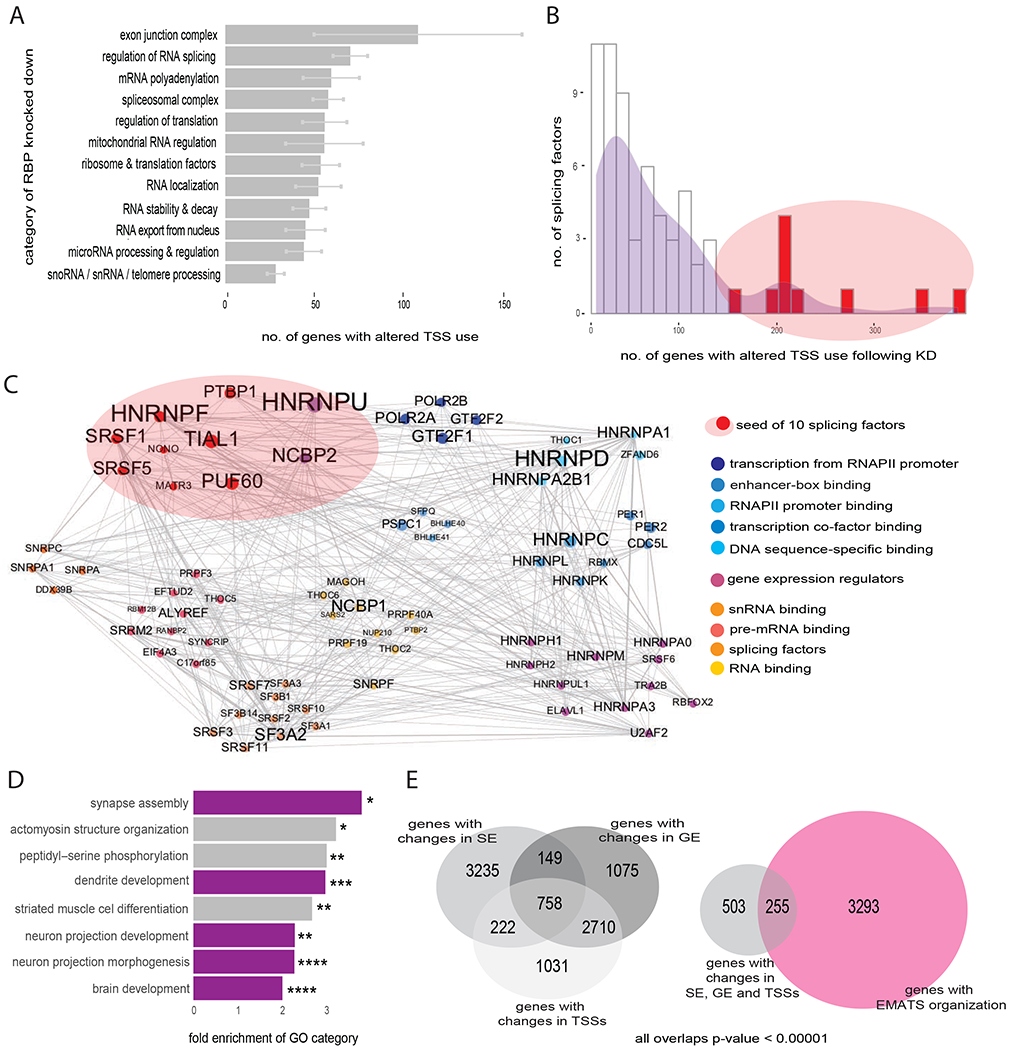
A, Distribution of the number of genes with significant changes in promoter usage associated with depletion of 250 RBPs, binned by Gene Ontology Biological Process categories of RBPs. Mean ± SEM between all RBPs in each GO category for two cell lines (HepG2 and K562) is plotted. B, Histogram of number of genes with significant changes in TSS usage following depletion of 67 splicing factors, top ten splicing factors with greatest number of changes shown in red. C, PPI network for the top 10 splicing factors from (B), colored by Gene Ontology category. Nodes represent proteins and edges represent PPIs. Node and label size are proportional to protein connectivity. The 10 selected splicing factors in red primarily interact with other 65 proteins, generating a network with 75 nodes and 424 edges. PPI data are from STRING database (Szklarczyk et al., 2015). Networks were built using Gephi (http://gephi.org). D, Gene Ontology analysis of 1777 mouse genes with the strongest EMATS potential. Fold enrichments shown for the most significant categories with asterisk indicating adjusted p-values and color indicating relation to neuron development. E, (left) Venn diagram showing the overlap between genes with significant changes in gene expression (GE), alternative splicing of SEs and relative usage of TSSs following knockdown of PTBP1 in human HepG2 cells. (right) Venn diagram showing the overlap between genes with changes in GE, SE and TSSs following knockdown of PTBP1, for human genes with EMATS organization. The overlap is 1.7-fold above background expectation (p < 1.6e-20, hypergeometric test).
See also Figure S6.
Based on our results indicating that splicing factors can regulate the recruitment of transcription machinery near alternative exons, we focused on the ten splicing factors associated with the largest numbers of changes in TSS usage (Figure 6B and S6B), which included the HNRNPU factor studied above. Using protein-protein interaction (PPI) data from the STRING database (Szklarczyk et al., 2015), we observed that these ten splicing factors interact with 65 other proteins, including subunits of RNAPII and GTFs such as TFIIF (Figure 6C). Compared with the PPI partners of the ten splicing factors whose depletion affected the fewest TSSs, these 65 proteins were enriched for functions in enhancer binding, transcription factor activity and promoter proximal binding (Figure S6C). Together, these observations indicate that some splicing factors may broadly impact promoter choice and identify extensive interactions between these factors and core transcription machinery, consistent with a recent study (Xiao et al., 2019).
To investigate potential biological roles of gene expression regulation via EMATS, we analyzed the functions of genes with EMATS structure. In both human and mouse, these genes were enriched for functions in brain development, neuron projection, synapse organization and related functions (Figure 6D and S6D). This observation raised the possibility that regulation via EMATS might contribute to neuronal differentiation. For example, in the Ehmt2 (Euchromatic histone-lysine N-methyltransferase 2) gene, inclusion of a SE contributes to neuronal differentiation (Fiszbein et al., 2016). Consistent with EMATS regulation of this locus, we observed that up-regulation of the SE during differentiation of mouse neuro2A (N2a) cells was accompanied by increased usage of upstream TSSs, and that usage of these TSSs decreased following inhibition of exon splicing by MO (Figure S6E). To investigate whether neuro-related splicing factors regulate expression via EMATS, we analyzed transcriptome-wide changes following depletion of PTBP1, which plays a central role in neurogenesis (Linares et al., 2015), using available ENCODE data (Van Nostrand et al., 2019). Following PTBP1 knockdown, 758 genes had significant changes in SE splicing, TSS usage and gene expression, including 255 genes with EMATS organization, a 1.7-fold enrichment over the background frequency of EMATS genes (Figure 6E). For example, in the human BMF (Bcl2 Modifying Factor) gene we observed reduced exon inclusion accompanied by decreased use of upstream proximal TSSs and decreased gene expression following PTBP1 knockdown (Figure S6F).
EMATs impacts transcription initiation and translation efficiency globally
To investigate whether splicing of SEs affects gene expression by regulation of transcription on a genome-wide scale, we analyzed transient transcriptome sequencing (TT-seq) data that sensitively monitors rapid changes in transcription following stimulation of human T-cells (Michel et al., 2017; Schwalb et al., 2016). We observed that genes with decreased inclusion of SEs after 15 minutes of activation tend to have decreased transcription (lower TT-seq read density) (Figure 7A). This trend was stronger in genes with EMATS structure (Figure 7A), suggesting that EMATS contributes to gene regulation by modulation of transcription globally.
Figure 7. EMATS impacts transcription and translation initiation globally.
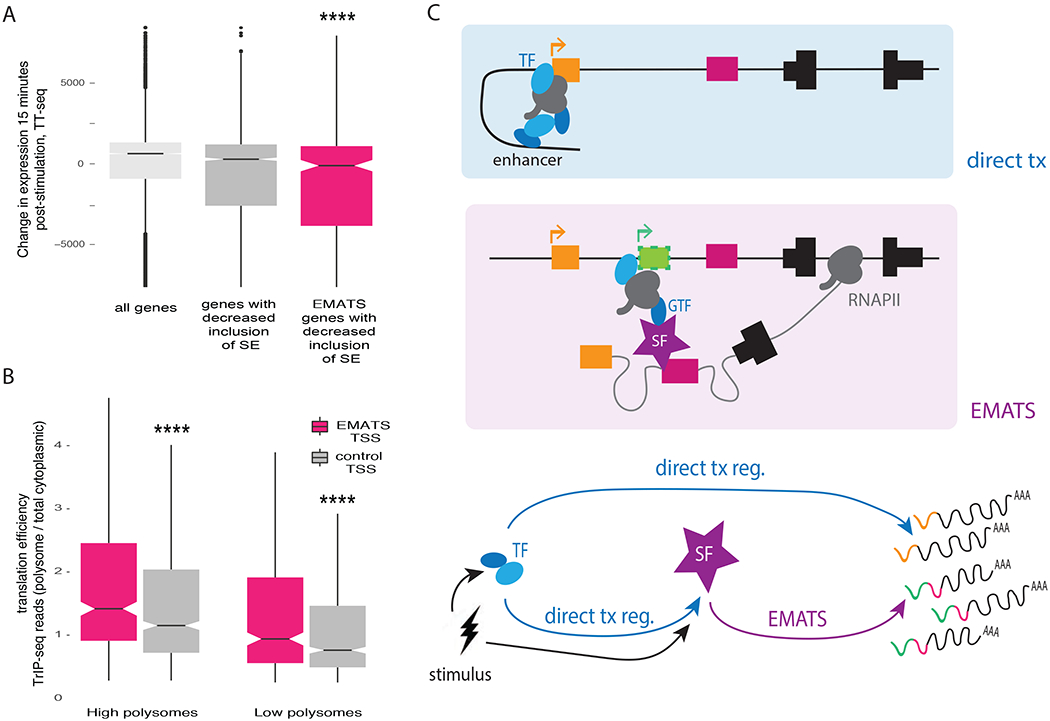
A, Change in nascent RNA levels (TT-seq read counts) after 15 minutes of T-cell stimulation for all genes expressed in humans, genes with increased inclusion of a skipped exon (SE) and genes with EMATS structure and increased inclusion of SE. B, EMATS TSS isoforms (pink) have increased translation efficiency (TE) relative to matched control isoforms from the same gene (grey). For each AFE, the TE was calculated across high (left, polysomes 6-8) or low (right, monosomes and polysomes 2-4) polysomes. Box plots show the distributions of TE values. C, Model for the role of EMATS in dynamic gene expression programs. Growth factor or other stimuli activate transcription factors (TF) and splicing factors (SF). TFs influence gene expression by direct effects on transcription (tx) and indirectly by regulating levels of SFs. Effects of SFs on splicing contribute to gene expression programs by EMATS. In genes with EMATS structure, SFs recruit general TFs (GTF) or RNAPII to activate weak TSS(s) proximal and upstream of the exon.
Switching between alternative 5′ UTR isoforms by altered TSS choice has recently been identified as an important regulator of translation efficiency in yeast (Cheng et al., 2018). To ask whether the splicing-dependent regulation of TSS selection by EMATS impacts translation, we analyzed transcript isoforms in polysomes sequencing (TrIP-seq) data from human HEK293T cells (Floor and Doudna, 2016) to assess the ribosome occupancy of EMATS isoforms. We found that first exons with EMATS-associated TSSs are significantly more ribosome-associated by a median fold-change of about 1.3-fold than gene-matched controls that lack EMATS structure (Figure 7B and S7A). These observations indicate that isoforms activated transcriptionally by EMATS tend to have enhanced translational activity, amplifying the impact of EMATS on protein production.
Discussion
Here, we have shown that inclusion of a new internal exon in a gene can activate transcription from an upstream TSS and thereby increase gene expression levels, a phenomenon which we refer to as EMATS. Our study highlights several features of this relationship: (i) it requires exon splicing, not merely presence of a 5′ or 3′ splice site; (ii) it is more potent when the exon is highly included and (iii) when the promoter is intrinsically weak; (iv) it is sensitive to genomic distance, occurring most robustly when exon and promoter are within 1-2 kb; and (v) the above features occur in thousands of mammalian genes (Table S4).
The most straightforward model to explain the above properties would involve direct positive effects of cotranscriptionally recruited splicing components on recruitment of transcription machinery to nearby upstream promoters (Figure 7D). Splicing machinery can recruit GTFs or modulate transcription activity (Damgaard et al., 2008; Fong and Zhou, 2001; Kwek et al., 2002), and depletion of RBPs can impact promoter selection on a large scale ((Brooks et al., 2015) and Figure 6B). The involvement of splicing machinery or proteins deposited on the transcript in connection with splicing would explain feature (i) above, while the more efficient recruitment of splicing machinery to more efficiently spliced exons would explain feature (ii). Recruitment of RNAPII or GTFs might be expected to activate transcription more effectively at weaker promoters where RNAPII recruitment is limiting than at strong promoters with higher intrinsic RNAPII occupancy, explaining feature (iii). A requirement for direct physical interaction between splicing machinery and RNAPII or GTFs might constrain the genomic distances involved, feature (iv). However, the varied chromatin conformations of different gene loci – which in some cases may involve chromatin loops between promoters and alternative exons (Mercer et al., 2013; Ruiz-Velasco et al., 2017) – might alter distance requirements for different genes. Frequent occurrence of the evolutionary path outlined above (Figure 4I) and of alternative 5′ UTRs (Singer et al., 2008) may explain widespread EMATS organization in mammalian genomes, feature (v).
Recent studies have broadened the definition of enhancers, showing that some gene promoters also function as enhancers (Engreitz et al., 2016; Scruggs et al., 2015); our findings support further broadening of this definition to include some exons as well. It is possible that a 5′ UTR intron may also be able to activate use of proximal upstream promoters.
We propose that emergence of new internal exons and of new TSSs are linked (Figure 4I). Once so activated, the new TSS produces new transcript isoforms and higher overall expression of the gene in specific tissues, providing a substrate for the regulatory evolution of the gene. The most obvious regulatory role for EMATS would be as a means for splicing factors to contribute to gene expression programs involved in differentiation or cellular responses to stimuli (Figure 7C). Specifically, we propose that external stimuli such as growth factors trigger gene expression changes not only via direct effects on TF activity (Malladi et al., 2016; Rajbhandari et al., 2018) but also by changes in splicing factor levels downstream of affected TFs or effects on splicing factor activity (Reinhardt et al., 2011; van der Houven van Oordt et al., 2000), triggering additional gene expression changes via EMATS. Another implication of our findings is that targeted activation of the expression of a gene for research or therapeutic purposes may be achievable by use of compounds such as antisense oligonucleotides or small molecules (Havens and Hastings, 2016) that enhance the splicing of appropriately located alternative promoter-proximal exon.
STAR*METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Please direct any requests for further information and resources to the Lead Contact, Christopher B. Burge (cburge@mit.edu), Department of Biology, Massachusetts Institute of Technology, Cambridge MA 02138.
Materials Availability Statement
All reagents generated in this study are available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines, cell culture and treatments
NIH3T3 and HeLa cells were grown in DMEM, with high glucose and pyruvate (Gibco), supplemented with 10% fetal bovine serum (FBS). Mouse CAD (Cath.-a-differentiated) cells were grown in DMEM/F12 (Gibco) supplemented with 10% FBS. N2a cells were grown in DMEM, with high glucose and pyruvate (Invitrogen), supplemented with 10% fetal bovine serum (FBS). N2a cells were differentiated with retinoic acid as in Fiszbein et al., 2016. For morpholino oligonucleotide (MO) treatment (Gene Tools), 20 μM of morpholino targeting 5′ or 3′ splice site or MO control was added with Endo-Porter (Gene Tools) following manufacturer’s instructions to cells plated at low confluence and left for 24 h.
METHOD DETAILS
RNA-seq analysis and genome builds
We used the strand-specific paired-end RNA-seq data from 9 tissues from mouse and rat (3 individuals each) associated with Merkin et al. (Merkin et al., 2012), available at NCBI Gene Expession Omnibus (GEO) (accession no. GSE41637). Reads were mapped to the mm9 and rn4 genome builds, respectively, and processed using TopHat (Trapnell et al., 2009) and Cufflinks (Trapnell et al., 2012). Cufflinks was used to estimate transcript abundance in each library (in standard FPKM units), and these values were used for splicing estimates or summed to obtain gene expression values. Alternative splicing patterns and PSI values were validated using MISO (Katz et al., 2010). Exons were defined as in Merkin et al. (Merkin et al., 2012), requiring FPKM ≥ 2 and meeting splice site junction read requirements implicit in the TopHat mapping. Exons with 0.05 < PSI < 0.97 in at least one tissue and two individuals were categorized as skipped exons (SE). Exons with PSI > 0.97 in all expressed tissues were defined as constitutive exons (CE), if the gene was expressed in at least three tissues and two individuals. Genomic and splicing ages were defined as previously described (Merkin et al., 2015) by the pattern of species with genomic regions aligned to the exon or with an expressed exon in the orthologous gene overlapping the aligned region, respectively, using the principle of evolutionary parsimony. As in Merkin et al. (Merkin et al., 2015), orthologous exons were identified by finding annotated exons that overlapped with the query exonic region in Ensembl Pecan 19 amniota genome alignments (Paten et al., 2008). Exon groups with multiple overlapping exons in any species were excluded. Exons were considered “lost” in a species if there was no syntenic region in that species or if no exon overlapping the syntenic region was identified and spliced into transcripts identified herein with a PSI ≥ 0. Open reading frames (ORFs) were annotated as described previously (Merkin et al., 2012) and used to classify exons as located in the 5′ UTR, 3′ UTR or coding region.
CRISPR sgRNA design, genetic deletions and genotyping
CRISPR-Cas cell lines with the 5′ splice site of Stoml1 deleted were generated using the protocol described by Ran and coworkers (Ran et al., 2013). The single-guide RNA was designed in silico to target the 5′ splice site using the CRISPR Design Tool (http://tools.genome-engineering.org) and cloned into a Cas9 expression plasmid (pSpCas9). After transfecting CAD cells with the plasmid expressing Cas9 and the appropriate sgRNA, clonal cell lines were isolated and insertion/deletion mutations were detected by the Surveyor nuclease assay (IDT). Positive clones detected were amplified by PCR, subcloned into TOPO-TA plasmids, and individual colonies were sequenced to reveal the clonal genotype.
RNA Extraction, RT-PCR and qPCR
Total RNA was extracted using the RNA-easy kit (Qiagen) according to the manufacturer’s protocol. Reverse transcription using M-MLV reverse transcriptase (Invitrogen) and random primers was performed according to the manufacturer’s instructions. For nascent RNA extraction, RNA was metabolically labeled with 5-Ethynil Uridine for 10 minutes using Click-iT (Invitrogen) and labeled RNA was extracted and amplified according to the manufacturer’s instructions. Quantitative PCR analyses were performed with SYBR green labeling using a LightCycler 480 II (Roche).
ChIP and antibodies
Chromatin immunoprecipitation was performed using the MAGnify™ Chromatin Immunoprecipitation System (Invitrogen) according to the manufacturer’s recommendations. For each immunoprecipitation, we used 10 μg of H3K4me3 antibody (PA5-17420 from Invitrogen), 10 μg of RNA polymerase II (CTD repeat YSPTSPS) antibody (Ab817 from Abcam), 10 μg of Transcription Factor IIF1 (TFIIF-alpha) antibody (PA5-30050 from Invitrogen) and 10 μg of Rabbit IgG antibody (Invitrogen) as a negative control. DNA was purified and quantitative PCR analysis was performed with SYBR green labeling using a LightCycler 480 II (Roche). Immunoprecipitated chromatin was normalized to input chromatin and control IgG antibody.
5′ RACE
5′ RACE experiments were performed with 5′ RACE System for Rapid Amplification of cDNA Ends (Invitrogen) using three gene-specific primers (GSP) that anneal to the known region and an adapter primer that targets the 5′ end. Products generated by 5′ RACE were subcloned into TOPO-TA vectors and individual colonies were sequenced.
Plasmids, RNAi and luciferase activity assay
Rat Tsku genomic region and mutants were cloned into the psiCHECK backbone. HNRNPU full-length and mutant were cloned intro the RG6 plasmid (pcDNA3.1 backbone). For transfection assays, 1 μg plasmid was transfected into each well of a 6-well culture plate using Lipofectamine 2000 (Life Technologies) according to the manufacturer’s recommendations and cells were harvested after 24 h. For knock-down experiments, a siRNA targeting the 3’UTR of HNRNPU or a scrambled siRNA was transfected together with either the control (empty) RG6 plasmid or with one of the HNRNPU-expressing constructs. To measure luciferase activity, we used the Dual-Luciferase® Reporter Assay System (Promega).
PRO-seq data analysis
PRO-seq reads in mouse and rat CD4+ T-cells were mapped as in Danko et al. (Danko et al., 2018) counting reads in the interval between 500 bp downstream of the annotated TSS and whichever was shorter: either the end of the gene or 60,000 bp into the gene body. Following the analysis in Danko et al., reads were transferred to the hg19 coordinates to be compared between mouse and rat using liftOver. For each gene PRO-seq reads were defined by the sum of read counts within the gene in the interval described above. The number of reads mapping to a gene (r) were then divided by the number of reads in the library (L). RPM values were calculated for each gene as r/L × 1,000,000 and divided by gene length in bp (and multiply by 1000) to get RPKM values for both species.
Motif enrichment analysis
The number of binding motifs for each splicing factor was calculated using RBPmap (Paz et al., 2014) by mapping each binding motif to the query sequence. We used the 94 RNA binding proteins present in the RBPmap database and added 30 additional RNA binding proteins whose binding motifs were identified by RNA Bind-n-Seq (Dominguez et al., 2018). The whole sequence of the novel exons and 20bp into the upstream and downstream introns was taken for the analysis. The enrichment of splicing factors binding motifs in mouse novel exons for each protein was calculated by dividing the mean number of binding motifs in new exons with a correlation above 0.3 with the nearby TSS by the mean number of binding moths in new exons with a correlation below 0.3 with the analogue TSS.
TT-seq data analysis
TT-seq reads in human T-cells were taken from Michel et al. (Michel et al., 2017) and fold change of nascent gene expression was calculated between 0 and 15 minutes of T-cell activation. Genes expressed in both samples were then assigned to EMATS or control genes depending of their genomic structure. For splicing analyses, total RNA-seq samples from Michel et al. (Michel et al., 2017) were mapped to the hg19 genome build using STAR (Dobin et al., 2013) and splicing fold changes were processed using MATS (Shen et al., 2014). After filtering for an FDR < 0.1, we obtained 9,379 significantly changing internal exons between 0 and 15 minutes after T-cell stimulation. Distribution of fold change in TT-seq reads after 15 minutes of T-cell stimulation were assessed for all genes, genes with significant decreased SE inclusion and EMATS genes with significant decreased SE inclusion.
Polysome profile analysis
Cytoplasmic, monosomal, and polysomal samples from Floor et al. (Floor and Doudna, 2016) were mapped using STAR aligner (Dobin et al., 2013) with standard ENCODE specifications, and with the requirement that each read map uniquely to the genome. Perfectly mapping reads were then assigned to EMATS or control alternative first exons (AFEs) if they overlapped at least 25 bases with the AFE. Each instance of an overlapping read was then tallied, and AFEs with at least 5 reads assigned were considered for further analysis. AFEs were then filtered for gene representation in both EMATS and control sets to preserve a gene-matched analysis, ultimately including 177 EMATS AFEs and 313 control AFEs in the analysis. Normalized read counts were used to create a “translational efficiency” (TE) score for each AFE by dividing the normalized read counts in a given sample by the reads counts in the cytoplasm for that AFE. Results were assessed by Wilcoxon Rank-Sum.
QUANTIFICATION AND STATISTICAL ANALYSIS
Definition of species-specific exons
Evolutionarily new exons were identified as in Merkin et al. (Merkin et al., 2015). Genomic mappings of mouse and rat RNA-seq data were combined with whole-genome alignments to classify the species distribution of exons. Only internal exons were considered in this analysis, excluding first and last exons, and only unique exons were considered, excluding exons that arose from intra-genic duplications to avoid issues related to possibly inaccurate genome assemblies, annotations or read mappings. In all, 1,089 mouse exons were classified as mouse-specific exons and 1,571 rat exons were classified as rat-specific exons, as they were detected in RNA-seq data from mouse or rat, respectively, but not from any other species analyzed (Supplementary Tables 1, 2). Most genes that contained a new exon had only one, with 159 mouse genes and 276 rat genes containing more than one new exon.
Transcription start site annotation
TSSs in the same RNA-seq data used to classify new exons, were identified using data from Merkin et al. (Merkin et al., 2012) (GEO accession no. GSE41637) mapped with TopHat combined with Ensembl annotations. As in Merkin et al. (Merkin et al., 2012), Cufflinks version 1.0.2 was used to identify novel transcripts. The set of TSSs from each library identified from transcripts as the start site of the first exon were combined with the existing Ensembl annotations and merged into a single set of annotations using Cuffcompare (Roberts et al., 2011). Cufflinks was then applied to each library to quantitate the same set of transcripts. TSS FPKM was calculated by summing the FPKM of transcripts that used the TSS. The TSS FPKM was then divided by the sum of FPKM of transcripts that used any other TSS to calculate the relative TSS usage. Thus, relative TSS usage was calculated dividing the FPKM of transcripts that used the TSS by that of transcripts that used a different TSS. Extensive data has accumulated that relative TSS usage derived from RNA-seq data correlate with methods that assess 5′ ends of nascent RNA. Expression in FPKM from different TSSs estimated by Cufflinks from RNA-seq data strongly correlates with those derived from TT-seq analysis of nascent RNA (r = 0.76). In this manuscript, TSSs in mouse were also identified using Start-seq data from Scruggs and coworkers (Scruggs et al., 2015) downloaded from GEO (accession no. GSE62151); Start-seq uses high-throughput sequencing of nascent capped RNA species from the 5′-end, allowing for definition of TSSs at nucleotide resolution. TSSs were defined in 2,000 bp search windows centered on RefSeq-annotated TSSs, using the location to which the largest number of Start-RNA reads aligned. Very closely spaced TSSs separated by less than 50 bp were considered as a single TSS in Figure 1D. The number of TSSs was also estimated by the number of H3K4me3 peaks assigned to each gene with ChIP data from Yu et al. (Yu et al., 2015) (GEO accession nos. GSE59896 and GSE59998).
Software for data analysis, graphical plots and statistical analyses
For data analysis we used R Bioconductor, BEDTools, SamTools, GenomicRanges, the Integrative Genomics Viewer, MISO, Cufflinks, STAR and MATS. All statistical analyses were performed in R (v.3.4.2) and graphical plots were made using the R package ggplot2. Lower and upper hinges of box plots correspond to the 25th and 75th percentiles, respectively. The upper and lower whiskers extend from the hinge to the largest and lowest value no further than 1.5 χ IQR (interquartile range), respectively. Notches give approximate 95% confidence interval for comparing the medians. Statistical significance of one-way ANOVA, Tukey post hoc test, is indicated by asterisks (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, *****p < 0.00001), unless otherwise indicated.
DATA AND CODE AVAILABILITY
Data availability
Data of evolutionarily new exons is available in Merkin et al. (Merkin et al., 2015) as well as here in Supplementary Tables 1 and 2. The RNA-seq data from 9 tissues from mouse and rat associated with Merkin et al. (Merkin et al., 2012) is available at GEO (accession no. GSE41637). The Start-seq data from Scruggs et al. (Scruggs et al., 2015) is available at GEO (accession no. GSE62151), as well as the H3K4me3 data from Yu et al. (Yu et al., 2015) (accession no. GSE59896 and GSE59998). PolyA-seq data from five mouse tissues is available in Derti et al (Derti et al., 2012) (accession no. GSE30198). PRO-seq data from mouse and rat CD4+ T cells from Danko et al. (Danko et al., 2018) is available at GEO (accession no. GSE93229). TT-seq data in human T-cells from Michel et al. (Michel et al., 2017) is available at GEO (accession no. GSE85201). Polysomes sequencing (TrIP-seq) data in human cells from Floor et al. (Floor and Doudna, 2016) is available at GEO (accession no. GSE69352).
ADDITIONAL RESOURCES
New exon inclusion, TSS usage, and species-specific expression
We considered genes with new exons as all genes with a new exon with PSI > 0.05 in any of the 9 tissues sequenced. We grouped genes as control genes with no new exons and genes with new exons divided by whether the exon was included or excluded in a given tissue. We calculated the number of TSSs used in each gene in each tissue and considered genes that gained TSSs in mouse, genes that gained TSSs in rat, and genes with same number of TSSs in both species based on the numbers of TSSs for each species in each gene in each tissue, or when considering all tissues together. Gene expression was calculated by estimating transcript abundance with Cufflink and summing standard FPKM units per gene. The new exons were included in the length normalization for species with the exons. The FPKM normalization was done by Cufflinks and was species-specific and isoform-specific. Each tissue was run individually and transcript expression was length normalized before combining. Gene expression in mouse was compared to that in rat by taking the ratio of expression in mouse to expression of the orthologous gene in the analogous tissue in rat.
Definition of new exon-proximal cleavage and polyadenylation sites
Polyadenylation sites were identified using available polyA-seq data from five mouse tissues (brain, liver, kidney, muscle, testis) (Derti et al., 2012). Only reads aligning to unique loci were retained and ends of reads within 25 nt of each other on the same strand were clustered. Polyadenylation sites were considered to be new exon-proximal cleavage and polyadenylation (nePCPA) sites if they were located within 2 kb upstream or downstream of a new exon, and as skipped exon-proximal cleavage and polyadenylation (sePCPA) sites if they were located within 2 kb upstream or downstream of skipped exons.
Effects on nascent and steady state RNA levels
Effects on transcription initiation should be reflected in nascent RNA, while effects on RNA stability would only be visible in steady state mRNA. In the Tsku gene, nascent RNA levels were reduced to a similar extent as steady state mRNA (Figure 2d, Extended data Figure 3b, Extended data Figure 5a-d), in both sense and antisense orientations. For other genes studied here, Stoml1 and Gper1, we also observed similar effects on nascent RNA in sense and antisense directions (Figure 2c, Extended data Figure 3b, Extended data Figure 4a-c). Furthermore, the model invoking inhibition of PCPA involves U1 snRNP binding at a 5′ splice site, but we observed increased gene expression from creation of a 3′ splice site. Thus, our observations are consistent with splicing-dependent regulation of transcription initiation but not with models involving PCPA.
Supplementary Material
Figure S3. Related to Figure 3
A, A diagram representing the technique used to label nascent RNA with 5-ethynyl uridine and pull down the nascent RNA with the click-it method. Fold change in RNA levels of Gpr30 (left) and Tsku (right) in NIH3T3 cells measured by qPCR. NIH3T3 cells were transfected with 20 μM morpholino (MO) targeting the 5′ splice site of the new exon in Gpr30 or 20 μM MO targeting the 3′ and/or the 5′ splice sites of the new exon in Tsku for 24 h. Mean ± SEM of displayed distributions, n=3 biological replicates. Statistical significance indicated by asterisks corresponds to one-way ANOVA, Tukey post hoc test. B, Relative exon inclusion / exon exclusion of the mouse-specific new exon in Stoml1 gene is shown, measured by qPCR of nascent RNA in wild type CAD cells and cells with CRISPR/cas-mediated mutations in the 5′ splice site of the new exon. Mean ± SEM is shown for n=3 biological replicates. Statistical significance indicated by asterisks corresponds to one-way ANOVA, Tukey post hoc test. C, H3K4me3 and RNAPII (CTD repeat YSPTSPS) profiles in Stoml1 gene in CAD cells determined by ChIP assay followed by qPCR with the regions indicated in the top panel. Values of two independent immunoprecipitations normalized to input and the mean value for control IgG antibody are shown for each region. Wild type cells (grey) and cells with CRISPR/cas-mediated mutations in the 5′ splice site of the new exon (blue) are shown. D, Fold change in nascent RNA expression (PRM units) between mouse and rat relative to the start coordinate of the new exon for genes with mouse-specific new exons in CD4+ T cells using PRO-seq data. E, Distribution of nascent RNA expression levels in mouse genes with new exons binned by the position relative to the start coordinate of the new exon in CD4+ T cells using PRO-seq data. F, Distribution of the number of polyadenylation sites used 2 kb upstream/downstream of new exons per gene in tissues new exon is excluded (PSI < 0.05, grey) and tissues with inclusion of new exons (PSI > 0.05, pink) for all genes with new exons. Distributions are not significantly different by Kolmogorov-Smirnov test. G, Scatter plot showing the relationship between the number of nePCPA sites and the fold change in gene expression levels between mouse and rat. These variables are not significantly associated by Spearman correlation test. Polyadenylation sites for 5 tissues in mouse were analyzed using polyA-seq data (Derti et al., 2012).
Figure S4. Related to Figure 4
A, Isoform expression for Tsku gene in NIH3T3 cells, measured by RT-PCR with primers targeting TSS −2 (top), TSS −1 (bottom) and exon E4.CE. Cells were transfected for 24 h with 20 μM MO targeting the 3′ and/or 5′ splice sites of the new exon as indicated. B, Fold change in inclusion and exclusion levels of the mouse-specific new exon in Tsku gene and antisense transcription levels from both TSSs measured by isoform-specific qPCR of nascent RNA with primers illustrated by arrows. Exon exclusion levels are measured from both alternative first exons to the following skipped exon downstream the mouse-specific new exon. NIH3T3 cells were transfected with control MO or MO targeting the 3′ and/or 5′ splice sites of the new exon. A decrease in the inclusion level of transcripts starting at TSS −2 is compensated by an increase in the exclusion levels, while total level of transcripts starting at TSS −1 is reduced by MO treatment. Mean ± SEM of displayed distributions, n=3 biological replicates. Statistical significance indicated by asterisks corresponds to one-way ANOVA, Tukey post hoc test. C, RNAPII profile in Tsku gene in NIH3T3 cells determined by ChIP assay followed by qPCR with the regions indicated in panel A. Values of two independent immunoprecipitations normalized to input and the mean value for control IgG antibody are shown for each region. NIH3T3 cells transfected for 24 hours with 20 μM control MO or MO targeting both 3′ and 5′ splice sites of the new exon. D, Alignments and identity between mouse (mm) and rat (rn) of the DNA sequence of TSS −1 and mouse-specific new exon in the Tsku gene. e, Splicing patterns of the Tsku gene in HeLa cells transfected with the hybrid constructs shown in Figure 4d. The creation of the mouse 3′ splice site (rn + mm 3′ss) or of a stronger 3′ splice site (rn + strong 3′ss) of the mouse-specific new exon in the rat sequence promotes the inclusion of the mouse-specific new exon in the rat context only when the wild type 5′ splice site is maintained (but not in the mm 3′ss + mut 5′ss construct). F, Sequence of the 5′ end of Tsku transcripts generated by 5′ RACE in HeLa cells transfected with rat Tsku constructs with the 3′ splice site of the mouse-specific new exon (5′ RACE clone A, clone B) aligned to the mouse sequence (mm) and the rat sequence (rn). For 80% of the sequenced transcripts, the 5′ end mapped 1 bp upstream of the position of mouse TSS −1 (clone A), while in the remainder the 5′ end mapped 19 bp upstream of (clone B). G, Ratio of mean density of RBPmap binding motifs (Paz et al., 2014) in mouse-specific new exons with mild/high correlation with usage of proximal upstream TSS (Spearman correlation > 0.3) to density of motifs in mouse-specific new exons with low correlation (spearman correlation < 0.3) for selected splicing proteins (top ten splicing proteins by ratio are shown). H, Z-scores of splicing factors binding sites relative to the position of the new exon in mouse Tsku. I, Isoform-specific qRT-PCR analysis of fold change in exclusion levels of the mouse-specific new exon in Tsku gene from both TSSs following treatment with a control siRNA or an siRNA targeting HNRNPU (siHNRNPU) and rescues in NIH3T3 cells. Mean ± SEM of displayed distributions, n=3 biological replicates. Statistical significance indicated by asterisks corresponds to one-way ANOVA, Tukey post hoc test.
Figure S5. Related to Figure 5
A, Distribution of the PSI values of mouse-specific new exons (blue) and SE (grey) in mouse across 9 tissues. B, Distribution of 5′ splice site scores of mouse-specific new exons, binned by the relative position to the next upstream TSS used in the same gene. 5′ splice site scores were calculated using MaxEntScan (Yeo and Burge, 2004). C, Distribution of the PSI values of first exons associated with TSSs in genes with mouse-specific new exons (blue) and in genes with SEs in mouse (grey). D, E. Difference in TSS usage based on relative TSS use value (d) and FPKM (e) in tissues with high versus low inclusion of skipped exons (SE), in the same gene across multiple tissues for proximal and upstream TSSs (within 1kb upstream the SE) used in genes with SEs in mouse, binned by quartiles of PSI values of the TSSs. F, Difference between relative TSS usage values in tissues with high versus low inclusion of skipped exons (SE), for all weak TSSs (bottom quartile) used in genes with skipped exons in mouse, binned by their position relative to the SE. G, Fold change in inclusion (far left), exclusion levels (left center, right center) and total levels (far right) of the skipped exon in Tsku gene (E3.SE) from both TSSs measured by isoform-specific qPCRs with primers shown by arrows. Exclusion levels were measured from both alternative first exons relative to the next constitutive exon downstream of the skipped exon. NIH3T3 cells were transfected with control MO or MO targeting the 5′ splice site of E3.SE. Mean ± SEM of displayed distributions, n=3 biological replicates. Statistical significance indicated by asterisks corresponds to one-way ANOVA, Tukey post hoc test.
Figure S6. Related to Figure 6
A, Histogram and smoothed density of number of TSSs with significant expression change following depletion of each of 250 RNA binding protein genes. Mean two cell lines (HepG2 and K562) is plotted for each RBP. B, Number of genes with significant difference in promoter usage associated with depletion of 67 splicing factors (SF). The red line indicates the cutoff for the top ten splicing factors driving the largest changes in promoter usage, while the green line indicates the cutoff for bottom ten control splicing factors driving the fewest changes in promoter usage. C, Protein interaction network for 10 control splicing factors driving the fewest changes in promoter usage. The control 10 splicing factors in red primarily interact with 88 other proteins, generating a network with 98 nodes and 410 edges, a diameter of 3, an average weighted degree of 4.29, an average clustering coefficient of 0.43 and an average path length of 1.32. Nodes represent proteins and links represent the interactions among them. Node size and label size is proportional to the protein connectivity (number of interactions a protein establishes with others). Protein interaction data were collected from STRING (Szklarczyk et al., 2015) and networks were built using Gephi (http://gephi.org). Colors represent the Gene Ontology analysis of 88 proteins included in the interaction network, excluding the 10 seed control splicing factors. D, Gene Ontology analysis of 1414 human genes with the strongest EMATS potential. Fold enrichments shown for the most significant categories with asterisk indicating adjusted p-values and color indicating relation to neuron development. E, Exon-intron organization of EHMT2 gene. qRT-PCR analysis of expression of TSS-2 and TSS-1 in (left) N2a differentiated cells and (right) cells treated with MO targeting the SE (E9.SE) normalized to expression of housekeeping genes Hprt and GAPDH. Mean ± SEM of displayed distributions for n=3 independent experiments. In Fiszbein et al., 2016 the authors only worked with Ehmt2 isoforms expressed from the gene-distal promoter that contain only the one constitutive NLS near the internal alternative exon. Here, we also analyze the expression of isoforms starting from the gene-proximal promoter that contains another NLS in the alternative first exon. F, Exon-intron organization of human BMF gene. RNA-seq analysis of expression of BMF in HepG2 cells following PTBP1 knockdown normalized to expression of control cells. Inclusion levels of the skipped exon, as well as levels of relative usage of the alternative TSSs (TSS −2, TSS −1) and total gene expression are shown. Mean ± SEM of displayed distributions for n=2 replicates.
Figure S7. Related to Figure 7
EMATS TSSs (pink) have increased translation efficiency (TE) relative to gene-matched control TSSs (grey). For each TSS, the TE was calculated for each polysomal fraction. Boxplots show the distributions of TEs.
Table S1. Related to Figure 1
Mouse genes with new internal exons and their rat homologs. Column A shows the locus of the new exon in mouse while column D shows the position of the new exon in the gene. Columns B and H show the IDs for homologous mouse and rat genes with mouse-specific new exons. Column C shows the splice site of the new exon and column E the new exon length. Columns F and I show the average gene expression levels in brain for 3 individuals in mouse and rat, respectively. Column G shows the average PSI values in the mouse brain for 3 individuals. Column J shows the homologues locus of the mouse new exons in the rat genome.
Table S3. Related to Figure 2
Coordinates of first exons associated with TSSs used in mouse. Column A shows the ID of mouse genes and column B shows coordinated of first exons associated with TSSs used in mouse corresponding to each gene.
Figure S1. Related to Figure 1
A, Fold change in gene expression between mouse and rat for mouse control genes with no evolutionarily new exons (black, dotted line), genes with mouse-specific new exons in tissues where inclusion of the new exon is not detected, PSI < 0.05 (grey), and genes with new mouse-specific exons in tissues were the exon is included, PSI > 0.05 (pink). Statistical significance by Mann-Whitney U test is indicated between genes with mouse-specific new exons in tissues with PSI < 0.05 and tissues with PSI > 0.05. B, Distribution of the number of TSSs per gene using RNA-seq data across multiple species and multiple tissues, for all genes expressed in mouse and genes with mouse-specific new exons. Distributions are significantly different by Kolmogorov-Smirnov test. C, Distribution of the number of TSSs per gene in the mouse brain using RNA-seq data, for all genes expressed in mouse and genes with mouse-specific new exons, after matching the distribution of gene expression levels between the two groups using the MatchIt package in R. Distributions remain significantly different by Kolmogorov-Smirnov test after matching the gene expression levels between the groups, demonstrating that, independent of gene expression, genes with mouse-specific new exons are enriched in multiple TSSs. D, Distribution of the number of -see3 peaks per gene using H3K4me3 ChIP-seq data for all genes expressed in mouse (grey) and genes with mouse-specific new exons (dark red). Distributions are significantly different by Kolmogorov-Smirnov test. Genes with mouse-specific new exons are enriched in H3K4me3 peaks. E, Distribution of the number of TSSs per gene for all genes expressed in rat (grey) and genes with rat-specific new exons (green). Genes with rat-specific new exons are enriched in multiple TSSs (by Kolmogorov-Smirnov test). F, The proportion of genes with fewer TSSs in mouse (genes w/ mouse TSS < rat TSS), genes with the same number of TSSs in both species (genes we mouse TSSs = rat TSS), and genes that have more TSSs in mouse, for all genes expressed in both species (gray) and for genes with rat-specific new exons (green). Statistical significance indicated by asterisks corresponds to one-way ANOVA, Tukey post hoc test (NS = not significant). G, Fold change in the number of TSSs used per gene between tissues where mouse-specific exons are included (PSI > 0.05) and excluded (PSI < 0.05), for mouse genes and for the same tissues in rat. Evolutionary gain of internal exons and of TSSs are associated, but only in those tissues where new exons are included. H, Distribution of PSI values of new exons binned by the number of TSSs used in the same gene, for 9 tissues pooled together in mouse. I, Fold change in the number of TSSs used per gene between mouse and rat for 9 tissues, for genes with mouse-specific new exons, binned by ψ value of the new exon in each tissue. J, Fold change in gene expression between rat and mouse, for all genes expressed in rat (grey) and genes with rat-specific lost exons (blue).
Figure S2. Related to Figure 2
A, Distribution of the fold change in gene expression levels between mouse and rat for genes with fewer or same number of TSSs used in mouse than rat (white) and genes with more TSSs used in mouse than rat (brown), before (left panel) and after (right panel) balancing the distribution of gene expression levels in mouse between the groups by MatchIt. Evolutionarily change in gene expression remain significantly different when balancing gene expression levels in mouse between groups, demonstrating that, independently of gene expression levels in one species, genes gaining TSSs in mouse have increased gene expression levels compared to rat. B, Venn diagram showing the overlap between genes that gained mouse-specific new exons and genes that gained new TSSs in mouse. C, TSS position relative to the start coordinate of the new exon in genes with mouse-specific new exons, for all TSSs used in 9 tissues in mouse. D, Spearman correlations between the usage of a particular TSS and the PSI value of the new exon across multiple tissues for all TSSs used in genes with mouse-specific new exons, binned by their relative position to the new exon with negative numbers for TSSs located upstream of the new exon and positive numbers for TSSs located downstream of the new exon.
Table S2. Related to Figure 1
Numbers of TSSs used in mouse and rat genes. Columns A and B show the IDs for homologous mouse and rat genes. Columns C and D show the numbers of TSSs used in mouse and rat, respectively, in a specific gene, pooling all nine sequenced tissues together.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| H3K4me3 antibody | Invitrogen | PA5-17420 |
| RNA polymerase II (CTD repeat YSPTSPS) antibody | Abcam | Ab817 |
| Transcription Factor IIF1 (TFIIF-alpha) antibody | Invitrogen | PA5-30050 |
| Rabbit IgG antibody | Cell Signaling Technology | 2729 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| M-MLV reverse transcriptase | Invitrogen | 28025013 |
| Lipofectamine 2000 | Life Technologies | 11668027 |
| Dual-Luciferase® Reporter Assay System | Promega | E1910 |
| DMEM | Gibco | 11965118 |
| Fetal bovine serum (FBS) | Gibco | A31406-02 |
| Endo-Porter (in PEG) | Gene Tools | OT-EP-PEG-1 |
| Lipofectamine RNAiMAX | Thermo | 13778150 |
| DMEM/F12 | Gibco | 11320033 |
| Critical Commercial Assays | ||
| RNA-easy kit | Qiagen | 74104 |
| Click-iT | Invitrogen | C10365 |
| 5′ RACE System for Rapid Amplification of cDNA Ends | Invitrogen | 18374058 |
| Experimental Models: Cell Lines | ||
| NIH3T3 | ATCC | CRL-1658 |
| HeLa | ATCC | CCL-2 |
| CAD | ECACC | 08100805 |
| N2a | ATCC | CCL-131 |
| Oligonucleotides | ||
| Primers for qPCR experiments | ||
| Tsku_INC_F | This study | GTGTCCTGCCAAAGCAAGTG |
| Tsku_INC_R | This study | CAGGAACAGAGAGCACAGCA |
| Tsku_INC_F_juntCE | This study | CCTGGCTGAGCAGGTGT |
| Tsku_INC_R_juntCE | This study | ATCCAAAGGGATGGGCACAG |
| Tsku_INC_TSS1_F | This study | GACCTGCCAGGACGCTG |
| Tsku_INC_TSS1_R | This study | TCAGCCAGGTCTGCTCCTAT |
| Tsku_antisense_TSS1_F | This study | GCTCAGGGAGCGTCGTTAAA |
| Tsku_antisense_TSS1_R | This study | GGGAACCGCGCACTTTTTAG |
| Tsku_INC_TSS2_F | This study | TGGCCAGGCTCAGAGGAC |
| Tsku_INC_TSS2_R | This study | TCAGCCAGGTCTGCTCCTAT |
| Tsku_antisense_TSS2_F | This study | CCCCTTTTCATCACAGCCCA |
| Tsku_antisense_TSS2_R | This study | GGGACGAACCTTCCAATCCA |
| Stoml1_TSS1_F | This study | GGAGTAAAGCCGGAAGCAGT |
| Stoml1_TSS1_R | This study | TCATGCTTGGAAGGTCTGGC |
| Stoml1_TSS2_F | This study | ATATGGGACCTCCGTGTCCA |
| Stoml1_TSS2_R | This study | AGCATGCCACACTCCTTACC |
| Stoml1_TSS3_F | This study | CATGCAGAGCACTGACCTAGT |
| Stoml1_TSS3_R | This study | CTGGAGGCTGTACTCAAGGC |
| Stoml1_TSS1_antisense_F | This study | TCCTGACCACCTCCTACCTG |
| Stoml1_TSS1_antisense_R | This study | TGGCCTCAAACCATTCCTCC |
| Stoml1_TSS2_antisense_F | This study | CTGGGGAGAACTGAGGGTTC |
| Stoml1_TSS2_antisense_R | This study | GAACCCCAGAGGGGAGTCTAT |
| Stoml1_TSS3_antisense_F | This study | CTGCCTCTTGATTCCCAGCA |
| Stoml1_TSS3_antisense_R | This study | CCCTTCCAAGACTGTGGCTT |
| Primers for 5′ RACE experiments | ||
| Tsku_GSP1 | This study | TGAATGGTAGGTGCAGGCAG |
| Tsku_GSP2 | This study | GGGAAGCAGGCGATGGATAA |
| Tsku_nestedGSP | This study | GATGTCACTCAAGGGGGAGC |
| hRLUC_GST1 | This study | GAACCAAGCGGTGAGGTACT |
| hRLUC_GST2 | This study | CGATATGAGCCATTCCCGCT |
| hRLUC_nested | This study | ATGATGCATCTAGCCACGGG |
| Primers for ChIP experiments | ||
| Tsku_Promoter_F | This study | ACTTTAACGACGCTCCCTGA |
| Tsku_Promoter_R | This study | ATGGGCCGGCGCTTTT |
| Tsku_TSS1_Intron1_F | This study | GAGGCGACAACTGCAGACC |
| Tsku_TSS1_Intron1_R | This study | CGACTCTATGGCTCGGTGTC |
| Tsku_Intron1_TSS2_F | This study | TTCCCAAGGGATGGCCAATG |
| Tsku_Intron1_TSS2_R | This study | AGTGACCGAATCTCAACGGG |
| Tsku_TSS2_Intron2_F | This study | GTGGCGAGCTTAGCTGAAAG |
| Tsku_TSS2_Intron2_R | This study | ACCCAGGATCAAAAGCTCGG |
| Tsku_Intron2_NEx_F | This study | ACAGACTCGGCAAGAGATGGA |
| Tsku_Intron2_NEx_R | This study | CTTCAGGAAACTCCCAGGCTCA |
| Tsku_NEx_F | This study | ACGCTGAGCCTGGGAGTTTC |
| Tsku_NEx_R | This study | TAGCACTTGCTTTGGCAGGA |
| Tsku_NEx_Intron3_F | This study | GTCCCATAGGAGCAGACCTGG |
| Tsku_NEx_Intron3_R | This study | TCCCAGCCTTTGGGTAACTC |
| Tsku_Intron3_AltEx_F | This study | GCTCAGTTCTCCCTTAGTGGG |
| Tsku_Intron3_AltEx_R | This study | TGGGGGCTTCATTCACCTTT |
| Tsku_AltEx_Intron4_F | This study | ACCGTCCGGTCTAACAGATTT |
| Tsku_AltEx_Intron4_R | This study | ACGGTTAAGGGTTGGACCAG |
| Tsku_LastEx_F | This study | AGGGCATCCTCCATCTACCA |
| Tsku_LastEx_R | This study | GCAAACCCAGGCCTGAAAAC |
| Morpholino sequences | ||
| G9a_mouse_E10_5ss | This study | GTCCCGGCAGTTGGCAATTAATTAC |
| G9a_mouse_E10_3ss | This study | CCATTCACTCCTGACACAGAGACAG |
| zfp672_m_ex2_5′ss | This study | CTGCATACATCTCACATTACCTTTG |
| zfp672_m_ex3_5′ss | This study | GGGTGTTTGTTCTGCCATACCAATA |
| zfp672_m_ex5_3′ss | This study | GATCCTATGGAAGGACAGTATGTAT |
| mTsku_2ndSE_3′ss | This study | CTTTGCTGAAATGAAACCACAGGTC |
| Tsku_3′ss | This study | TCTGAGAAAGGATAGGGAACCCAAT |
| Tsku_5′ss | This study | ACCCCTGAGTAGAGAGAGTCACCTG |
| Gpr30_5′ss | This study | ACCTGAAAATTTAAAAGTACTCACG |
| Deposited Data | ||
| RNA-seq data from 9 tissues from mouse and rat | Merkin et al., 2012 | GEO: GSE41637 |
| Start-seq data from murine macrophages | Scruggs et al., 2015 | GEO: GSE62151 |
| H3K4me3 ChIP-seq data from mouse | Yu et al., 2015 | GEO: GSE59896 |
| PolyA-seq data from five mouse tissues | Derti et al., 2012 | GEO: GSE30198 |
| PRO-seq data from mouse and rat CD4+ T cells | Danko et al., 2018 | GEO: GSE93229 |
| TT-seq data from human T-cells | Michel et al., 2017 | GEO: GSE85201 |
| TrIP-seq data from human cells | Floor, Doudna, 2016 | GEO: GSE69352 |
| Software and Algorithms | ||
| TopHat | Trapnell et al., 2009 | http://ccb.jhu.edu/software/tophat/index.shtml |
| STAR | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| MATS | Shen et al., 2014 | http://rnaseq-mats.sourceforge.net/mats3.0.8/ |
| Cufflinks | Trapnell et al., 2012 |
http://cole-trapnell-lab.github.io/cufflinks/ |
| MISO | Katz et al., 2010 | http://genes.mit.edu/burgelab/miso/ |
| Cuffcompare | Roberts et al., 2011 | http://cole-trapnell-lab.github.io/cufflinks/cuffcompare/ |
| Bioconductor | N/A | https://www.bioconductor.org/ |
| BEDTools | Quinlan and Hall, 2010 | http://bedtools.readthedocs.io/en/latest/ |
| SamTools | Li et al., 2009 | http://samtools.sourceforge.net/ |
| GenomicRanges | Lawrence et al., 2013 | https://www.bioconductor.org/packages/release/bioc/html/GenomicRanges.html |
| Integrative Genomics Viewer | Robinson et. al, 2011 | https://igv.org/ |
| R (v.3.4.2) | N/A | https://www.r-project.org/ |
Highlights.
New promoters arise near evolutionarily new internal exons
Splicing of internal exons activates proximal upstream weak promoters
Splicing recruits transcription machinery locally to influence promoter selection
These impacts of splicing on transcription are widespread
Acknowledgments
We thank Karla Neugebauer, Alberto R. Kornblihtt, Phillip A. Sharp, and members of the Burge lab for helpful discussions and comments, as well as Maria Alexis, Jason Merkin, Peter Freese, Charles Danko and Brenton R. Graveley for assistance with access to genomic data and analysis pipelines. This work was supported by grants from the NIH to C.B.B. (grant numbers HG002439, GM085319), and by the Pew Latin American Fellows Program in the Biomedical Sciences to A.F.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
CBB and AF have filed a patent application related to this work. The authors declare no other competing interests. Correspondence and requests for materials should be addressed to cburge@mit.edu
References
- Agarwal N, and Ansari A (2016). Enhancement of Transcription by a Splicing-Competent Intron Is Dependent on Promoter Directionality. PLoS Genet. 12, e1006047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhtar J, Kreim N, Marini F, Mohana G, Brüne D, Binder H, and Roignant J-Y (2019). Promoter-proximal pausing mediated by the exon junction complex regulates splicing. Nat Commun 10, 521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almada AE, Wu X, Kriz AJ, Burge CB, and Sharp PA (2013). Promoter directionality is controlled by U1 snRNP and polyadenylation signals. Nature 499, 360–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anvar SY, Allard G, Tseng E, Sheynkman GM, de Klerk E, Vermaat M, Yin RH, Johansson HE, Ariyurek Y, Dunnen, den JT, et al. (2018). Full-length mRNA sequencing uncovers a widespread coupling between transcription initiation and mRNA processing. 19, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley DL (2014). Coupling mRNA processing with transcription in time and space. Nat. Rev. Genet 15, 163–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieberstein NI, Carrillo Oesterreich F, Straube K, and Neugebauer KM (2012). First exon length controls active chromatin signatures and transcription. Cell Rep 2, 62–68. [DOI] [PubMed] [Google Scholar]
- Braunschweig U, Gueroussov S, Plocik AM, Graveley BR, and Blencowe BJ (2013). Dynamic integration of splicing within gene regulatory pathways. Cell 152, 1252–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks AN, Duff MO, May G, Yang L, Bolisetty M, Landolin J, Wan K, Sandler J, Booth BW, Celniker SE, et al. (2015). Regulation of alternative splicing in Drosophila by 56 RNA binding proteins. Genome Res. 25, 1771–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callis J, Fromm M, and Walbot V (1987). Introns increase gene expression in cultured maize cells. Genes Dev. 1, 1183–1200. [DOI] [PubMed] [Google Scholar]
- Chathoth KT, Barrass JD, Webb S, and Beggs JD (2014). A splicing-dependent transcriptional checkpoint associated with prespliceosome formation. Mol. Cell 53, 779–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Z, Otto GM, Powers EN, Keskin A, Mertins P, Carr SA, Jovanovic M, and Brar GA (2018). Pervasive, Coordinated Protein-Level Changes Driven by Transcript Isoform Switching during Meiosis. Cell 172, 910–923. e916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Custódio N, and Carmo-Fonseca M (2016). Co-transcriptional splicing and the CTD code. Crit. Rev. Biochem. Mol. Biol 51, 395–411. [DOI] [PubMed] [Google Scholar]
- Damgaard CK, Kahns S, Lykke-Andersen S, Nielsen AL, Jensen TH, and Kjems J (2008). A 5′ splice site enhances the recruitment of basal transcription initiation factors in vivo. Mol. Cell 29, 271–278. [DOI] [PubMed] [Google Scholar]
- Danko CG, Choate LA, Marks BA, Rice EJ, Wang Z, Chu T, Martins AL, Dukler N, Coonrod SA, Tait Wojno ED, et al. (2018). Dynamic evolution of regulatory element ensembles in primate CD4+ T cells. Nat Ecol Evol 2, 537–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R, Yu J, Zhang Z, Gygi MP, Krainer AR, Gygi SP, and Reed R (2007). SR proteins function in coupling RNAP II transcription to pre-mRNA splicing. Mol. Cell 26, 867–881. [DOI] [PubMed] [Google Scholar]
- Derti A, Garrett-Engele P, Macisaac KD, Stevens RC, Sriram S, Chen R, Rohl CA, Johnson JM, and Babak T (2012). A quantitative atlas of polyadenylation in five mammals. Genome Res. 22, 1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez D, Freese P, Alexis MS, Su A, Hochman M, Palden T, Bazile C, Lambert NJ, Van Nostrand EL, Pratt GA, et al. (2018). Sequence, Structure, and Context Preferences of Human RNA Binding Proteins. Mol. Cell 70, 854–867. e859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emili A, Shales M, McCracken S, Xie W, Tucker PW, Kobayashi R, Blencowe BJ, and Ingles CJ (2002). Splicing and transcription-associated proteins PSF and p54nrb/nonO bind to the RNA polymerase II CTD. 8, 1102–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engreitz JM, Haines JE, Perez EM, Munson G, Chen J, Kane M, McDonel PE, Guttman M, and Lander ES (2016). Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature 539, 452–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiszbein A, Giono LE, Quaglino A, Berardino BG, Sigaut L, Bilderling, von C, Schor IE, Steinberg JHE, Rossi M, Pietrasanta LI, et al. (2016). Alternative Splicing of G9a Regulates Neuronal Differentiation. Cell Rep 14, 2797–2808. [DOI] [PubMed] [Google Scholar]
- Floor SN, and Doudna JA (2016). Tunable protein synthesis by transcript isoforms in human cells. Elife 5, 1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong YW, and Zhou Q (2001). Stimulatory effect of splicing factors on transcriptional elongation. Nature 414, 929–933. [DOI] [PubMed] [Google Scholar]
- Furger A, O’Sullivan JM, Binnie A, Lee BA, and Proudfoot NJ (2002). Promoter proximal splice sites enhance transcription. Genes Dev. 16, 2792–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallegos JE, and Rose AB (2017). Intron DNA Sequences Can Be More Important Than the Proximal Promoter in Determining the Site of Transcript Initiation. Plant Cell 29, 843–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gracheva EO, Cordero-Morales JF, González-Carcacía JA, Ingolia NT, Manno C, Aranguren CI, Weissman JS, and Julius D (2011). Ganglion-specific splicing of TRPV1 underlies infrared sensation in vampire bats. Nature 476, 88–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gueroussov S, Gonatopoulos-Pournatzis T, Irimia M, Raj B, Lin ZY, Gingras AC, and Blencowe BJ (2015). An alternative splicing event amplifies evolutionary differences between vertebrates. Science 349, 868–873. [DOI] [PubMed] [Google Scholar]
- Gunderson SI, Polycarpou-Schwarz M, and Mattaj IW (1998). U1 snRNP Inhibits Pre-mRNA Polyadenylation through a Direct Interaction between U1 70K and Poly(A) Polymerase. Mol. Cell 1, 255–264. [DOI] [PubMed] [Google Scholar]
- Havens MA, and Hastings ML (2016). Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 44, 6549–6563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaida D, Berg MG, Younis I, Kasim M, Singh LN, Wan L, and Dreyfuss G (2010). U1 snRNP protects pre-mRNAs from premature cleavage and polyadenylation. Nature 468, 664–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kameoka S, Duque P, and Konarska MM (2004). p54(nrb) associates with the 5′ splice site within large transcription/splicing complexes. Embo J. 23, 1782–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz Y, Wang ET, Airoldi EM, and Burge CB (2010). Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat. Methods 7, 1009–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MK, and Nikodem VM (1999). hnRNP U inhibits carboxy-terminal domain phosphorylation by TFIIH and represses RNA polymerase II elongation. Mol. Cell. Biol 19, 6833–6844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornblihtt AR, Schor IE, Alló M, Dujardin G, Petrillo E, and Muñoz MJ (2013). Alternative splicing: a pivotal step between eukaryotic transcription and translation. Nat. Rev. Mol. Cell Biol 14, 153–165. [DOI] [PubMed] [Google Scholar]
- Kwek KY, Murphy S, Furger A, Thomas B, O’Gorman W, Kimura H, Proudfoot NJ, and Akoulitchev A (2002). U1 snRNA associates with TFIIH and regulates transcriptional initiation. Nat. Struct. Biol 9, 800–805. [DOI] [PubMed] [Google Scholar]
- Linares AJ, Lin C-H, Damianov A, Adams KL, Novitch BG, and Black DL (2015). The splicing regulator PTBP1 controls the activity of the transcription factor Pbx1 during neuronal differentiation. Elife 4, e09268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malladi S, Macalinao DG, Jin X, He L, Basnet H, Zou Y, de Stanchina E, and Massagué J (2016). Metastatic Latency and Immune Evasion through Autocrine Inhibition of WNT. Cell 165, 45–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer TR, Edwards SL, Clark MB, Neph SJ, Wang H, Stergachis AB, John S, Sandstrom R, Li G, Sandhu KS, et al. (2013). DNase I-hypersensitive exons colocalize with promoters and distal regulatory elements. Nat. Genet 45, 852–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkin JJ, Chen P, Alexis MS, Hautaniemi SK, and Burge CB (2015). Origins and impacts of new mammalian exons. Cell Rep 10, 1992–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkin J, Russell C, Chen P, and Burge CB (2012). Evolutionary dynamics of gene and isoform regulation in Mammalian tissues. Science 338, 1593–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel M, Demel C, Zacher B, Schwalb B, Krebs S, Blum H, Gagneur J, and Cramer P (2017). TT-seq captures enhancer landscapes immediately after T-cell stimulation. Mol. Syst. Biol 13, 920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris DP, and Greenleaf AL (2000). The splicing factor, Prp40, binds the phosphorylated carboxyl-terminal domain of RNA polymerase II. J. Biol. Chem 275, 39935–39943. [DOI] [PubMed] [Google Scholar]
- Mortillaro MJ, Blencowe BJ, Wei X, Nakayasu H, Du L, Warren SL, Sharp PA, and Berezney R (1996). A hyperphosphorylated form of the large subunit of RNA polymerase II is associated with splicing complexes and the nuclear matrix. Proc. Natl. Acad. Sci. U.S.a 93, 8253–8257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neugebauer KM, and Roth MB (1997). Transcription units as RNA processing units. Genes Dev. 11, 3279–3285. [DOI] [PubMed] [Google Scholar]
- Paten B, Herrero J, Beal K, Fitzgerald S, and Birney E (2008). Enredo and Pecan: genome-wide mammalian consistency-based multiple alignment with paralogs. Genome Res. 18, 1814–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz I, Kosti I, Ares M, Cline M, and Mandel-Gutfreund Y (2014). RBPmap: a web server for mapping binding sites of RNA-binding proteins. Nucleic Acids Res. 42, W361–W367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajbhandari P, Thomas BJ, Feng A-C, Hong C, Wang J, Vergnes L, Sallam T, Wang B, Sandhu J, Seldin MM, et al. (2018). IL-10 Signaling Remodels Adipose Chromatin Architecture to Limit Thermogenesis and Energy Expenditure. Cell 172, 218–233. e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt HC, Cannell IG, Morandell S, and Yaffe MB (2011). Is post-transcriptional stabilization, splicing and translation of selective mRNAs a key to the DNA damage response? Cell Cycle 10, 23–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A, and Huber W (2018). Alternative start and termination sites of transcription drive most transcript isoform differences across human tissues. Nucleic Acids Res. 46, 582–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts A, Pimentel H, Trapnell C, and Pachter L (2011). Identification of novel transcripts in annotated genomes using RNA-Seq. 27, 2325–2329. [DOI] [PubMed] [Google Scholar]
- Ruiz-Velasco M, Kumar M, Lai MC, Bhat P, Solis-Pinson AB, Reyes A, Kleinsorg S, Noh K-M, Gibson TJ, and Zaugg JB (2017). CTCF-Mediated Chromatin Loops between Promoter and Gene Body Regulate Alternative Splicing across Individuals. Cell Syst 5, 628–637. e6. [DOI] [PubMed] [Google Scholar]
- Schor IE, Fiszbein A, Petrillo E, and Kornblihtt AR (2013). Intragenic epigenetic changes modulate NCAM alternative splicing in neuronal differentiation. Embo J. 32, 2264–2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwalb B, Michel M, Zacher B, Fruhauf K, Demel C, Tresch A, Gagneur J, and Cramer P (2016). TT-seq maps the human transient transcriptome. Science 352, 1225–1228. [DOI] [PubMed] [Google Scholar]
- Scruggs BS, Gilchrist DA, Nechaev S, Muse GW, Burkholder A, Fargo DC, and Adelman K (2015). Bidirectional Transcription Arises from Two Distinct Hubs of Transcription Factor Binding and Active Chromatin. Mol. Cell 58, 1101–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaul O (2017). How introns enhance gene expression. Int. J. Biochem. Cell Biol 91, 145–155. [DOI] [PubMed] [Google Scholar]
- Shen S, Park JW, Lu Z-X, Lin L, Henry MD, Wu YN, Zhou Q, and Xing Y (2014). rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. U.S.a 111, E5593–E5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer GAC, Wu J, Yan P, Plass C, Huang TH-M, and Davuluri RV (2008). Genome-wide analysis of alternative promoters of human genes using a custom promoter tiling array. BMC Genomics 9, 349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, et al. (2015). STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–D452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, and Salzberg SL (2009). TopHat: discovering splice junctions with RNA-Seq. 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Houven van Oordt W, Diaz-Meco MT, Lozano J, Krainer AR, Moscat J, and Cáceres JF (2000). The MKK(3/6)-p38-signaling cascade alters the subcellular distribution of hnRNP A1 and modulates alternative splicing regulation. J. Cell Biol 149, 307–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nostrand EL, Freese P, Pratt GA, Wang X, Wei X, Xiao R, Blue SM, Chen J-Y, Cody NA, Dominguez D, et al. (2018). Biorxiv 1–111. [Google Scholar]
- Vincent M, Lauriault P, Dubois MF, Lavoie S, Bensaude O, and Chabot B (1996). The nuclear matrix protein p255 is a highly phosphorylated form of RNA polymerase II largest subunit which associates with spliceosomes. Nucleic Acids Res. 24, 4649–4652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegand HL, Lu S, and Cullen BR (2003). Exon junction complexes mediate the enhancing effect of splicing on mRNA expression. Proc. Natl. Acad. Sci. U.S.a 100, 11327–11332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao R, Chen J-Y, Liang Z, Luo D, Chen G, Lu ZJ, Chen Y, Zhou B, Li H, Du X, et al. (2019). Pervasive Chromatin-RNA Binding Protein Interactions Enable RNA-Based Regulation of Transcription. Cell 178, 107–121. e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo G, and Burge CB (2004). Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol 11, 377–394. [DOI] [PubMed] [Google Scholar]
- Yu H-B, Yurieva M, Balachander A, Foo I, Leong X, Zelante T, Zolezzi F, Poidinger M, and Ricciardi-Castagnoli P (2015). NFATc2 mediates epigenetic modification of dendritic cell cytokine and chemokine responses to dectin-1 stimulation. Nucleic Acids Res. 43, 836–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S3. Related to Figure 3
A, A diagram representing the technique used to label nascent RNA with 5-ethynyl uridine and pull down the nascent RNA with the click-it method. Fold change in RNA levels of Gpr30 (left) and Tsku (right) in NIH3T3 cells measured by qPCR. NIH3T3 cells were transfected with 20 μM morpholino (MO) targeting the 5′ splice site of the new exon in Gpr30 or 20 μM MO targeting the 3′ and/or the 5′ splice sites of the new exon in Tsku for 24 h. Mean ± SEM of displayed distributions, n=3 biological replicates. Statistical significance indicated by asterisks corresponds to one-way ANOVA, Tukey post hoc test. B, Relative exon inclusion / exon exclusion of the mouse-specific new exon in Stoml1 gene is shown, measured by qPCR of nascent RNA in wild type CAD cells and cells with CRISPR/cas-mediated mutations in the 5′ splice site of the new exon. Mean ± SEM is shown for n=3 biological replicates. Statistical significance indicated by asterisks corresponds to one-way ANOVA, Tukey post hoc test. C, H3K4me3 and RNAPII (CTD repeat YSPTSPS) profiles in Stoml1 gene in CAD cells determined by ChIP assay followed by qPCR with the regions indicated in the top panel. Values of two independent immunoprecipitations normalized to input and the mean value for control IgG antibody are shown for each region. Wild type cells (grey) and cells with CRISPR/cas-mediated mutations in the 5′ splice site of the new exon (blue) are shown. D, Fold change in nascent RNA expression (PRM units) between mouse and rat relative to the start coordinate of the new exon for genes with mouse-specific new exons in CD4+ T cells using PRO-seq data. E, Distribution of nascent RNA expression levels in mouse genes with new exons binned by the position relative to the start coordinate of the new exon in CD4+ T cells using PRO-seq data. F, Distribution of the number of polyadenylation sites used 2 kb upstream/downstream of new exons per gene in tissues new exon is excluded (PSI < 0.05, grey) and tissues with inclusion of new exons (PSI > 0.05, pink) for all genes with new exons. Distributions are not significantly different by Kolmogorov-Smirnov test. G, Scatter plot showing the relationship between the number of nePCPA sites and the fold change in gene expression levels between mouse and rat. These variables are not significantly associated by Spearman correlation test. Polyadenylation sites for 5 tissues in mouse were analyzed using polyA-seq data (Derti et al., 2012).
Figure S4. Related to Figure 4
A, Isoform expression for Tsku gene in NIH3T3 cells, measured by RT-PCR with primers targeting TSS −2 (top), TSS −1 (bottom) and exon E4.CE. Cells were transfected for 24 h with 20 μM MO targeting the 3′ and/or 5′ splice sites of the new exon as indicated. B, Fold change in inclusion and exclusion levels of the mouse-specific new exon in Tsku gene and antisense transcription levels from both TSSs measured by isoform-specific qPCR of nascent RNA with primers illustrated by arrows. Exon exclusion levels are measured from both alternative first exons to the following skipped exon downstream the mouse-specific new exon. NIH3T3 cells were transfected with control MO or MO targeting the 3′ and/or 5′ splice sites of the new exon. A decrease in the inclusion level of transcripts starting at TSS −2 is compensated by an increase in the exclusion levels, while total level of transcripts starting at TSS −1 is reduced by MO treatment. Mean ± SEM of displayed distributions, n=3 biological replicates. Statistical significance indicated by asterisks corresponds to one-way ANOVA, Tukey post hoc test. C, RNAPII profile in Tsku gene in NIH3T3 cells determined by ChIP assay followed by qPCR with the regions indicated in panel A. Values of two independent immunoprecipitations normalized to input and the mean value for control IgG antibody are shown for each region. NIH3T3 cells transfected for 24 hours with 20 μM control MO or MO targeting both 3′ and 5′ splice sites of the new exon. D, Alignments and identity between mouse (mm) and rat (rn) of the DNA sequence of TSS −1 and mouse-specific new exon in the Tsku gene. e, Splicing patterns of the Tsku gene in HeLa cells transfected with the hybrid constructs shown in Figure 4d. The creation of the mouse 3′ splice site (rn + mm 3′ss) or of a stronger 3′ splice site (rn + strong 3′ss) of the mouse-specific new exon in the rat sequence promotes the inclusion of the mouse-specific new exon in the rat context only when the wild type 5′ splice site is maintained (but not in the mm 3′ss + mut 5′ss construct). F, Sequence of the 5′ end of Tsku transcripts generated by 5′ RACE in HeLa cells transfected with rat Tsku constructs with the 3′ splice site of the mouse-specific new exon (5′ RACE clone A, clone B) aligned to the mouse sequence (mm) and the rat sequence (rn). For 80% of the sequenced transcripts, the 5′ end mapped 1 bp upstream of the position of mouse TSS −1 (clone A), while in the remainder the 5′ end mapped 19 bp upstream of (clone B). G, Ratio of mean density of RBPmap binding motifs (Paz et al., 2014) in mouse-specific new exons with mild/high correlation with usage of proximal upstream TSS (Spearman correlation > 0.3) to density of motifs in mouse-specific new exons with low correlation (spearman correlation < 0.3) for selected splicing proteins (top ten splicing proteins by ratio are shown). H, Z-scores of splicing factors binding sites relative to the position of the new exon in mouse Tsku. I, Isoform-specific qRT-PCR analysis of fold change in exclusion levels of the mouse-specific new exon in Tsku gene from both TSSs following treatment with a control siRNA or an siRNA targeting HNRNPU (siHNRNPU) and rescues in NIH3T3 cells. Mean ± SEM of displayed distributions, n=3 biological replicates. Statistical significance indicated by asterisks corresponds to one-way ANOVA, Tukey post hoc test.
Figure S5. Related to Figure 5
A, Distribution of the PSI values of mouse-specific new exons (blue) and SE (grey) in mouse across 9 tissues. B, Distribution of 5′ splice site scores of mouse-specific new exons, binned by the relative position to the next upstream TSS used in the same gene. 5′ splice site scores were calculated using MaxEntScan (Yeo and Burge, 2004). C, Distribution of the PSI values of first exons associated with TSSs in genes with mouse-specific new exons (blue) and in genes with SEs in mouse (grey). D, E. Difference in TSS usage based on relative TSS use value (d) and FPKM (e) in tissues with high versus low inclusion of skipped exons (SE), in the same gene across multiple tissues for proximal and upstream TSSs (within 1kb upstream the SE) used in genes with SEs in mouse, binned by quartiles of PSI values of the TSSs. F, Difference between relative TSS usage values in tissues with high versus low inclusion of skipped exons (SE), for all weak TSSs (bottom quartile) used in genes with skipped exons in mouse, binned by their position relative to the SE. G, Fold change in inclusion (far left), exclusion levels (left center, right center) and total levels (far right) of the skipped exon in Tsku gene (E3.SE) from both TSSs measured by isoform-specific qPCRs with primers shown by arrows. Exclusion levels were measured from both alternative first exons relative to the next constitutive exon downstream of the skipped exon. NIH3T3 cells were transfected with control MO or MO targeting the 5′ splice site of E3.SE. Mean ± SEM of displayed distributions, n=3 biological replicates. Statistical significance indicated by asterisks corresponds to one-way ANOVA, Tukey post hoc test.
Figure S6. Related to Figure 6
A, Histogram and smoothed density of number of TSSs with significant expression change following depletion of each of 250 RNA binding protein genes. Mean two cell lines (HepG2 and K562) is plotted for each RBP. B, Number of genes with significant difference in promoter usage associated with depletion of 67 splicing factors (SF). The red line indicates the cutoff for the top ten splicing factors driving the largest changes in promoter usage, while the green line indicates the cutoff for bottom ten control splicing factors driving the fewest changes in promoter usage. C, Protein interaction network for 10 control splicing factors driving the fewest changes in promoter usage. The control 10 splicing factors in red primarily interact with 88 other proteins, generating a network with 98 nodes and 410 edges, a diameter of 3, an average weighted degree of 4.29, an average clustering coefficient of 0.43 and an average path length of 1.32. Nodes represent proteins and links represent the interactions among them. Node size and label size is proportional to the protein connectivity (number of interactions a protein establishes with others). Protein interaction data were collected from STRING (Szklarczyk et al., 2015) and networks were built using Gephi (http://gephi.org). Colors represent the Gene Ontology analysis of 88 proteins included in the interaction network, excluding the 10 seed control splicing factors. D, Gene Ontology analysis of 1414 human genes with the strongest EMATS potential. Fold enrichments shown for the most significant categories with asterisk indicating adjusted p-values and color indicating relation to neuron development. E, Exon-intron organization of EHMT2 gene. qRT-PCR analysis of expression of TSS-2 and TSS-1 in (left) N2a differentiated cells and (right) cells treated with MO targeting the SE (E9.SE) normalized to expression of housekeeping genes Hprt and GAPDH. Mean ± SEM of displayed distributions for n=3 independent experiments. In Fiszbein et al., 2016 the authors only worked with Ehmt2 isoforms expressed from the gene-distal promoter that contain only the one constitutive NLS near the internal alternative exon. Here, we also analyze the expression of isoforms starting from the gene-proximal promoter that contains another NLS in the alternative first exon. F, Exon-intron organization of human BMF gene. RNA-seq analysis of expression of BMF in HepG2 cells following PTBP1 knockdown normalized to expression of control cells. Inclusion levels of the skipped exon, as well as levels of relative usage of the alternative TSSs (TSS −2, TSS −1) and total gene expression are shown. Mean ± SEM of displayed distributions for n=2 replicates.
Figure S7. Related to Figure 7
EMATS TSSs (pink) have increased translation efficiency (TE) relative to gene-matched control TSSs (grey). For each TSS, the TE was calculated for each polysomal fraction. Boxplots show the distributions of TEs.
Table S1. Related to Figure 1
Mouse genes with new internal exons and their rat homologs. Column A shows the locus of the new exon in mouse while column D shows the position of the new exon in the gene. Columns B and H show the IDs for homologous mouse and rat genes with mouse-specific new exons. Column C shows the splice site of the new exon and column E the new exon length. Columns F and I show the average gene expression levels in brain for 3 individuals in mouse and rat, respectively. Column G shows the average PSI values in the mouse brain for 3 individuals. Column J shows the homologues locus of the mouse new exons in the rat genome.
Table S3. Related to Figure 2
Coordinates of first exons associated with TSSs used in mouse. Column A shows the ID of mouse genes and column B shows coordinated of first exons associated with TSSs used in mouse corresponding to each gene.
Figure S1. Related to Figure 1
A, Fold change in gene expression between mouse and rat for mouse control genes with no evolutionarily new exons (black, dotted line), genes with mouse-specific new exons in tissues where inclusion of the new exon is not detected, PSI < 0.05 (grey), and genes with new mouse-specific exons in tissues were the exon is included, PSI > 0.05 (pink). Statistical significance by Mann-Whitney U test is indicated between genes with mouse-specific new exons in tissues with PSI < 0.05 and tissues with PSI > 0.05. B, Distribution of the number of TSSs per gene using RNA-seq data across multiple species and multiple tissues, for all genes expressed in mouse and genes with mouse-specific new exons. Distributions are significantly different by Kolmogorov-Smirnov test. C, Distribution of the number of TSSs per gene in the mouse brain using RNA-seq data, for all genes expressed in mouse and genes with mouse-specific new exons, after matching the distribution of gene expression levels between the two groups using the MatchIt package in R. Distributions remain significantly different by Kolmogorov-Smirnov test after matching the gene expression levels between the groups, demonstrating that, independent of gene expression, genes with mouse-specific new exons are enriched in multiple TSSs. D, Distribution of the number of -see3 peaks per gene using H3K4me3 ChIP-seq data for all genes expressed in mouse (grey) and genes with mouse-specific new exons (dark red). Distributions are significantly different by Kolmogorov-Smirnov test. Genes with mouse-specific new exons are enriched in H3K4me3 peaks. E, Distribution of the number of TSSs per gene for all genes expressed in rat (grey) and genes with rat-specific new exons (green). Genes with rat-specific new exons are enriched in multiple TSSs (by Kolmogorov-Smirnov test). F, The proportion of genes with fewer TSSs in mouse (genes w/ mouse TSS < rat TSS), genes with the same number of TSSs in both species (genes we mouse TSSs = rat TSS), and genes that have more TSSs in mouse, for all genes expressed in both species (gray) and for genes with rat-specific new exons (green). Statistical significance indicated by asterisks corresponds to one-way ANOVA, Tukey post hoc test (NS = not significant). G, Fold change in the number of TSSs used per gene between tissues where mouse-specific exons are included (PSI > 0.05) and excluded (PSI < 0.05), for mouse genes and for the same tissues in rat. Evolutionary gain of internal exons and of TSSs are associated, but only in those tissues where new exons are included. H, Distribution of PSI values of new exons binned by the number of TSSs used in the same gene, for 9 tissues pooled together in mouse. I, Fold change in the number of TSSs used per gene between mouse and rat for 9 tissues, for genes with mouse-specific new exons, binned by ψ value of the new exon in each tissue. J, Fold change in gene expression between rat and mouse, for all genes expressed in rat (grey) and genes with rat-specific lost exons (blue).
Figure S2. Related to Figure 2
A, Distribution of the fold change in gene expression levels between mouse and rat for genes with fewer or same number of TSSs used in mouse than rat (white) and genes with more TSSs used in mouse than rat (brown), before (left panel) and after (right panel) balancing the distribution of gene expression levels in mouse between the groups by MatchIt. Evolutionarily change in gene expression remain significantly different when balancing gene expression levels in mouse between groups, demonstrating that, independently of gene expression levels in one species, genes gaining TSSs in mouse have increased gene expression levels compared to rat. B, Venn diagram showing the overlap between genes that gained mouse-specific new exons and genes that gained new TSSs in mouse. C, TSS position relative to the start coordinate of the new exon in genes with mouse-specific new exons, for all TSSs used in 9 tissues in mouse. D, Spearman correlations between the usage of a particular TSS and the PSI value of the new exon across multiple tissues for all TSSs used in genes with mouse-specific new exons, binned by their relative position to the new exon with negative numbers for TSSs located upstream of the new exon and positive numbers for TSSs located downstream of the new exon.
Table S2. Related to Figure 1
Numbers of TSSs used in mouse and rat genes. Columns A and B show the IDs for homologous mouse and rat genes. Columns C and D show the numbers of TSSs used in mouse and rat, respectively, in a specific gene, pooling all nine sequenced tissues together.
Data Availability Statement
Data availability
Data of evolutionarily new exons is available in Merkin et al. (Merkin et al., 2015) as well as here in Supplementary Tables 1 and 2. The RNA-seq data from 9 tissues from mouse and rat associated with Merkin et al. (Merkin et al., 2012) is available at GEO (accession no. GSE41637). The Start-seq data from Scruggs et al. (Scruggs et al., 2015) is available at GEO (accession no. GSE62151), as well as the H3K4me3 data from Yu et al. (Yu et al., 2015) (accession no. GSE59896 and GSE59998). PolyA-seq data from five mouse tissues is available in Derti et al (Derti et al., 2012) (accession no. GSE30198). PRO-seq data from mouse and rat CD4+ T cells from Danko et al. (Danko et al., 2018) is available at GEO (accession no. GSE93229). TT-seq data in human T-cells from Michel et al. (Michel et al., 2017) is available at GEO (accession no. GSE85201). Polysomes sequencing (TrIP-seq) data in human cells from Floor et al. (Floor and Doudna, 2016) is available at GEO (accession no. GSE69352).
Data of evolutionarily new exons is available in Merkin et al. (Merkin et al., 2015) as well as here in Supplementary Tables 1 and 2. The RNA-seq data from 9 tissues from mouse and rat associated with Merkin et al. (Merkin et al., 2012) is available at GEO (accession no. GSE41637). The Start-seq data from Scruggs et al. (Scruggs et al., 2015) is available at GEO (accession no. GSE62151), as well as the H3K4me3 data from Yu et al. (Yu et al., 2015) (accession no. GSE59896 and GSE59998). PolyA-seq data from five mouse tissues is available in Derti et al (Derti et al., 2012) (accession no. GSE30198). PRO-seq data from mouse and rat CD4+ T cells from Danko et al. (Danko et al., 2018) is available at GEO (accession no. GSE93229). TT-seq data in human T-cells from Michel et al. (Michel et al., 2017) is available at GEO (accession no. GSE85201). Polysomes sequencing (TrIP-seq) data in human cells from Floor et al. (Floor and Doudna, 2016) is available at GEO (accession no. GSE69352).