

Abstract
Certain cognitive deficits in schizophrenia, such as impaired working memory, are thought to reflect alterations in the neural circuitry of the dorsolateral prefrontal cortex (DLPFC). Gamma oscillations in the DLPFC appear to be a neural corollary of working memory function, and the power of these oscillations during working memory tasks is lower in individuals with schizophrenia. Thus, gamma oscillations represent a potentially useful biomarker to index dysfunction in the DLPFC circuitry responsible for working memory in schizophrenia. Postmortem studies, by identifying the cellular basis of DLPFC dysfunction, can help inform the utility of biomarker measures obtained in vivo. Given that gamma oscillations reflect network activity of excitatory pyramidal neurons and inhibitory GABA neurons, we review postmortem findings of alterations to both cell types in the DLPFC and discuss how these findings might inform future biomarker development and use.
Keywords: GABA, gamma oscillations, Glutamate, Cognition, Working memory, Schizophrenia
1. Introduction
Although positive symptoms are usually the presenting clinical feature of schizophrenia, cognitive deficits have long been regarded as core features of the illness [1]. Cognitive deficits occur with high frequency, are present prior to the initial onset of psychosis, are relatively stable over time, and are independent of psychotic symptoms [1,2]. Additionally, the degree of cognitive dysfunction is the best predictor of long-term functional outcome [1].
The cognitive abnormalities in schizophrenia include marked impairments in working memory [3,4], which is the ability to transiently maintain and manipulate a limited amount of information to guide thought or behavior [3,4]. Substantial empirical findings [5], confirmed by meta-analyses [3], indicate that schizophrenia is associated with large working memory deficits (~1.5–2.0 standard deviations below healthy subjects). These deficits cannot be explained by effects of IQ, duration of illness, or antipsychotic medications [3]. In fact, some studies suggest that working memory impairments contribute to disturbances in other cognitive domains in schizophrenia [6]. Thus, the development of novel interventions targeting the affected neural circuitry responsible for working memory deficits in schizophrenia remains a major unmet need.
Working memory is dependent on the integrity of neural circuitry in the dorsolateral prefrontal cortex (DLPFC) [7], and subjects with schizophrenia show altered activation of the DLPFC when performing working memory tasks [8]. Within the DLPFC, gamma oscillatory activity (30–80 Hz) appears to be a neural corollary of mental representation during working memory. For example, gamma oscillatory power scales with working memory load [9], and individuals with schizophrenia fail to increase gamma oscillatory power in the DLPFC in response to working memory demands [10].
Gamma oscillations reflect synchronous activity of large groups of glutamatergic pyramidal neurons located in cortical layers 2–3 [11]. In particular, pyramidal neurons located in layer 3 furnish horizontal connections that provide recurrent excitatory connections among pyramidal neurons and to local inhibitory neurons [12]. These inhibitory GABA neurons spatially tune this excitatory network [13] and play a key role in the generation of gamma oscillations [14] (Fig. 1A). Indeed, disturbances to either excitatory or inhibitory strength in the microcircuit disrupts gamma oscillations [15,16] and working memory function [13,17]. Thus, alterations to the neural components of this layer 3 microcircuit in the DLPFC of individuals with schizophrenia might represent the neural substrate for impaired gamma oscillations and working memory. Consequently, novel therapeutics designed to normalize the function of this circuit would be expected to improve working memory.
Fig. 1.
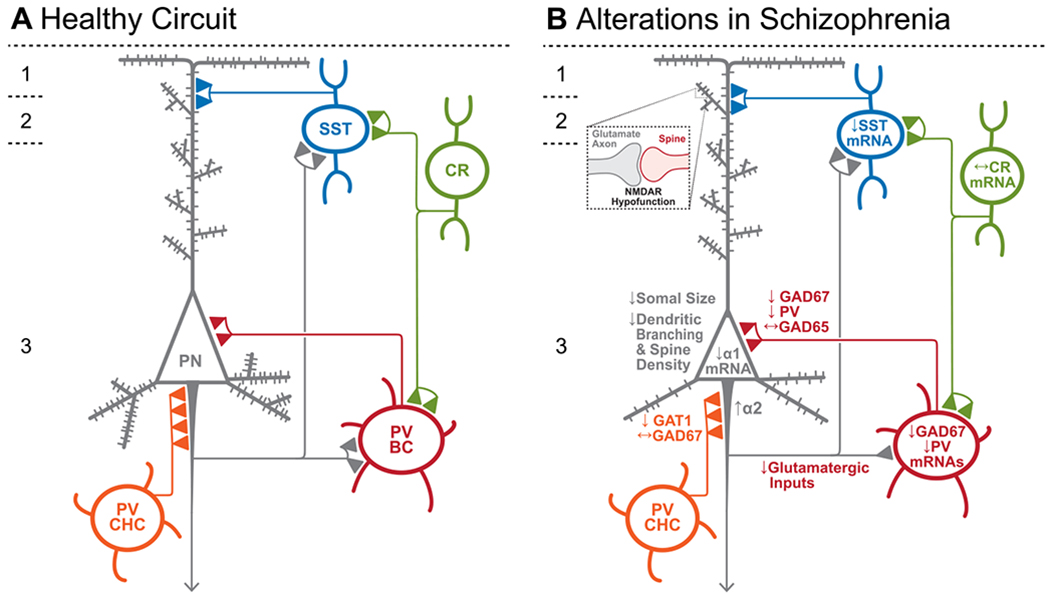
Schematic of components of the DLPFC layer 3 microcircuitry thought to be responsible for gamma oscillations and working memory. (A) In the unaffected circuit, current data support the notion that excitatory inputs recruit the activity of parvalbumin basket cells (PV BC) which inhibit large groups of pyramidal neurons (PN) simultaneously, thus synchronizing their subsequent activity. This synchronous activity occurs as gamma frequency oscillations which is thought to reflect the neural representation of working memory. Excitatory axon terminals from layer 3 pyramidal neurons also synapse onto somatostatin (SST)-expressing interneurons in layers 2–3, which target the distal dendrites of other groups of pyramidal neurons and inhibit pyramidal neurons that encode stimuli not related to the working memory memoranda. Calretinin (CR)-expressing interneurons target both PV and SST neurons, serving to disinhibit pyramidal neurons. PV chandelier cells (PV CHC) form connections directly onto the axon initial segment, and thus are poised to rapidly suppress the output of pyramidal neurons. (B) In schizophrenia, the morphological alterations to pyramidal neurons, which include a lower complement of dendritic spines, lower dendritic spine density and smaller somal sizes, suggest that these cells are hypoactive, leading to weaker recruitment of PVBCs and thus lower gamma oscillatory power. The hypoactivity of pyramidal neurons in layer 3 might arise from NMDAR hypofunction on dendrites receiving other excitatory inputs. Lower GABAA α1 receptor subunit mRNA expression in pyramidal neurons, together with lower PV and GAD67 in the PV BC terminals, suggests that both pre- and postsynaptic inhibition of PV BCs onto pyramidal neurons is lower. While the cell type-specific deficits in SST neurons are less well-defined, lower SST mRNA expression suggests these neurons may have an impaired ability to laterally inhibit other populations of pyramidal cells. CR cells do not exhibit lower levels of CR mRNA or protein in the illness, suggesting that these cells may not be affected in schizophrenia. PV CHCs express normal levels of GAD67, but lower GAT1 in their cartridges and higher levels of the GABAA receptor α2 subunit on the axon initial segment of the pyramidal neuron. Together, these alterations suggest that GABA signaling from PV CHCs might be higher in schizophrenia.
Assessing the potential efficacy of these novel therapeutics requires biomarkers, such as gamma oscillations, which reflect the functional integrity of the DLPFC circuitry responsible for working memory. Understanding the basis for altered gamma oscillatory activity and other biomarker measurements in schizophrenia can be informed by postmortem studies, which are able to interrogate the cellular constituents responsible for in vivo alterations in the disease state. Consequently, in this review, we summarize findings of altered glutamate and GABA signaling, and the affected cell types that likely give rise to this altered signaling, in the DLPFC of subjects with schizophrenia. We focus on postmortem findings which index the synaptic function, activity, and neurotransmitter synthesis in the glutamate and GABA neurotransmitter systems responsible for gamma oscillations in the DLPFC, and then consider how such findings might inform the development and use of biomarkers in schizophrenia.
2. Glutamatergic Abnormalities in the DLPFC in Schizophrenia
The primary site of glutamatergic neurotransmission, the tripartite synapse, is composed of an excitatory presynaptic bouton that releases glutamate, a postsynaptic cellular membrane loaded with glutamatergic receptors, and an astrocytic process that takes up and deactivates glutamate [18] (Fig. 2A). Glutamate is cycled through the tripartite synapse, beginning in the presynaptic bouton by the glutaminase-mediated conversion of glutamine to glutamate, which is then packaged into vesicles by vesicular glutamate transporters (vGLUTs) localized at the vesicular membrane. Following calcium influx triggered by presynaptic depolarization, these vesicles fuse with the presynaptic membrane, releasing glutamate into the synaptic cleft where it binds to postsynaptic glutamatergic receptors or is cleared by astrocytic excitatory amino acid transporters (EAATs). Within the astrocyte, glutamate is converted by glutamine synthetase to glutamine, which is then released and taken up by the presynaptic bouton via glutamine transporters, beginning the cycle once again.
Fig. 2.
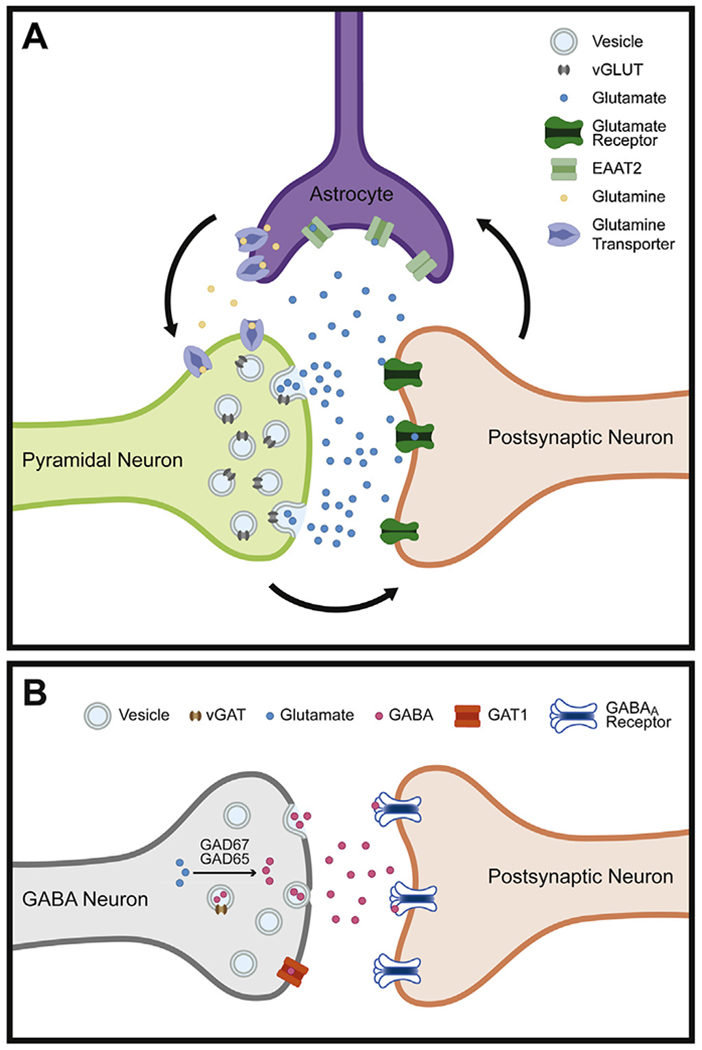
Schematic of excitatory and inhibitory synapses. (A) The tripartite glutamate synapse. In the presynaptic pyramidal neuron, glutamine is converted to glutamate by glutaminase and packaged into synaptic vesicles by the vesicular glutamate transporter (vGLUT). Following release into the synaptic cleft, glutamate binds to postsynaptic glutamate receptors located on a postsynaptic neuron. Glutamate is then cleared from the synaptic space through excitatory amino acid transporters (EAATs) on nearby astrocytes. Within the astrocyte, glutamate is converted to glutamine by glutamine synthetase before being transported to presynaptic neurons and beginning the process again. (B) The inhibitory GABA synapse. In the presynaptic terminal of a GABA neuron, GABA is synthesized from glutamate by the enzymes glutamic acid decarboxylase 67 and 65 (GAD67 and GAD65, respectively). GABA is packaged into vesicles via the vesicular GABA transporter (vGAT). After release into the synaptic cleft, GABA acts on GABAA receptors located on the postsynaptic neuron, opening chloride channels and hyperpolarizing the membrane. The action of GABA in the synapse is terminated, in part, by the reuptake of GABA into the presynaptic axon by GABA membrane transporter 1 (GAT1).
2.1. Markers of glutamate in presynaptic boutons
In the DLPFC of subjects with schizophrenia, levels of glutaminase activity were found to be four-fold higher [19], suggesting greater conversion of glutamine to glutamate within the presynaptic bouton. However, DLPFC levels of glutaminase mRNA were reported to be unaltered in schizophrenia [20,21].
The two vGLUT proteins are located in different types of axonal boutons: vGLUT1 is predominantly localized within the terminals of cortical and hippocampal projections, whereas efferents from the thalamus and hypothalamus contain vGLUT2 [22,23]. vGLUT levels are a key determinant of the amount of glutamate in vesicles primed for exocytosis within the presynaptic bouton and thus also directly indicate the quantal size of glutamate release [24]. Therefore, analysis of vGLUT1 levels can provide insight into the strength of glutamatergic neurotransmission in the DLPFC. However, findings in the DLPFC of schizophrenia subjects differ across studies. Eastwood and Harrison [25] observed significantly lower vGLUT1 mRNA in layers 1–3 and a nonsignificant decrease in layers 4–6 of the DLPFC. Similarly, a study restricted to DLPFC layer 3 reported significantly lower vGLUT1 mRNA expression in schizophrenia subjects [21]. In contrast, two other investigations of total DLPFC gray matter found no change in either vGLUT1 mRNA [26,27] or protein [27]. Therefore, deficits in vGLUT1 mRNA expression may be pronounced selectively in layer 3 and thus masked when assessing all cortical layers.
2.2. Glutamatergic receptors
Glutamate can bind to any of several post-synaptic receptors upon its release: ionotropic AMPA, NMDA, and kainite receptors, as well as metabotropic receptors [28]. Ionotropic glutamate receptors are voltage-gated ion channels that quickly open and close upon the binding of glutamate.
Ionotropic AMPA receptors are tetramers composed of two GluA subunit dimers [28]. Findings of altered AMPA receptor subunit mRNA levels in schizophrenia have varied across studies, differences which might reflect the impact of age. For example, lower levels of GluA2 and GluA4 [29], or unaltered levels of GluA2 [21], transcripts in the DLPFC were found in a middle-aged cohort of subjects with schizophrenia, whereas in an elderly cohort, higher levels of GluA2 and GluA4 mRNA were found [30]. Together, these results suggest that AMPA receptor expression level may depend on an interaction between age and disease state.
NMDA receptors are composed of two obligatory GluN1 subunits and two GluN2 and/or GluN3 subunits [28]. In adult PFC, NMDAR-containing synapses predominantly comprise di-heteromeric GluN1/GluN2A and tri-heteromeric GluN1/GluN2A/GluN2B receptors, as GluN3A expression peaks at birth and decreases progressively into adulthood [31,32]. Therefore, the glycine binding subunit GluN3A has been infrequently studied in DLPFC, with reports of elevated [33] or unaltered [34] mRNA levels. However, multiple studies have assessed the required GluN1 subunit and the glutamate binding GluN2 subunit, although these results have also been varied. Multiple studies have found lower levels of NMDA receptor mRNA and protein in the DLPFC of schizophrenia subjects. Specifically, downregulated GluN1, GluN2A, GluN2C mRNA [34–37], and GluN1 protein [34] have been observed in schizophrenia, although such findings have not always achieved statistical significance. Downregulated NMDAR subunits might contribute to working memory impairments in schizophrenia, as lower GluN1 could mean fewer functional receptor units, and lower synaptic GluN2A has been associated with poorer working memory performance [38]. Thus, lower levels of GluN1 and/or GluN2A in schizophrenia could contribute to glutamatergic signaling abnormalities and working memory deficits in the disease.
However, other studies have found elevated levels of GluN1 mRNA with no differences in other subunits [39,40], and one study found no difference in GluN1 mRNA [21]. These varied findings might stem from cohort-specific factors such as age and exposure to antipsychotic medications. For example, the studies reporting higher levels of GluN1 primarily used tissue from antipsychotic-treated, elderly subjects [39,40], whereas the studies that found levels that were lower or not different in schizophrenia [21,39,40] used tissue from middle-aged subjects. Furthermore, Sokolov and colleagues [37] found a negative relationship between duration of time off antipsychotic treatment prior to death and GluN1, such that the longer the duration of no treatment, the lower the levels of GluN1. In contrast, schizophrenia subjects treated within 72 h prior to death exhibited levels of GluN1 that were similar to unaffected comparison subjects [37]. Therefore, it appears that GluN1 may be elevated with both age and treatment duration.
Activation of the NMDA receptor is dependent upon the binding of its ligand and a co-agonist such as D-serine or glycine. Unfortunately, D-serine and its precursor L-serine, have yet to be assessed in postmortem tissue. Serine racemase, which synthesizes D-serine, has been assessed in multiple studies, but with varied results. Protein levels of the D-serine synthesis enzyme have been observed to be lower [41], unaltered [42], or elevated (but mRNA expression did not differ) [43]. In contrast, the degradation of serine racemase has consistently been reported to be unaltered in schizophrenia [41,43].
Kynurenic acid is a noncompetitive antagonist at the glycine site on NMDA receptors [44], such that the presence of kynurenic acid at the glycine site results in the receptor failing to activate when glutamate binds. Thus, if available in excess, kynurenic acid can downregulate NMDA receptor activity. Levels of kynurenic acid protein have been reported to be elevated in the DLPFC in schizophrenia [45,46], potentially indicating higher antagonism of the NMDA receptor glycine site.
Although levels of NMDA receptor signaling in postmortem tissue are often inferred by assessing related activity-dependent markers, other approaches have been developed to assess NMDA receptor activation in postmortem tissue, including postmortem tissue-stimulation [47] and microtransplantation of postmortem cellular membranes into Xenopus oocytes [48]. Although glutamate receptors desensitized without additional pharmacostimulation [48], a more pronounced neuregulin-1 induced suppression of NMDA receptor activity in the postmortem prefrontal cortex of schizophrenia subjects was observed using tissue-stimulation [47]. Neuregulin-1 and its receptor erbB have been implicated in the pathology of schizophrenia [49–51] (for review please see [52]) due to their modulation of glutamatergic signaling [53,54]. Therefore, these results suggest a potential mechanism for lower NMDA receptor activation (i.e., NMDA receptor hypofunction) in the prefrontal cortex of schizophrenia subjects.
Unlike other ionotropic receptors, kainate receptors exhibit slow response recovery and are pre-synaptically localized [28]. Studies of kainate receptors in schizophrenia have produced varied findings. Assessment of the mRNA of subunits 1 −5 in a middle-aged cohort found that only GRIK1 was lower [55], but assessment in an elderly cohort revealed unaltered GRIK1, elevated GRIK3, and downregulated GRIK5 mRNA levels [56]. In a second study of elderly subjects that also assessed the role of antipsychotic treatment, schizophrenia subjects who were untreated for 6 months prior to death exhibited the lowest levels of GRIK3 and GRIK4 mRNA; subjects who had stopped treatment in the 11 weeks prior to death had intermediate mRNA levels; and schizophrenia subjects who had been treated within 72 h prior to death exhibited levels of GRIK3 and GRIK4 transcripts that did not differ from comparison subjects [37]. Therefore, like the other ionotropic receptors, the expression of kainate receptors may depend upon age and treatment status.
Metabotropic glutamate receptors are transmembrane G-protein-coupled receptors (GPCRs) that respond more slowly to glutamate binding, but can have a longer duration of action by initiating signaling cascades [28]. Metabotropic glutamate receptors are categorized into subfamilies: I, II, and III [28]. Group I mGluRs (mGluR1 and mGluR5) are primarily postsynaptic, where they modulate the activity of NMDA receptors, whereas group II (mGluR2 and mGluR3) and III (mGluR4, mGluR6–8) receptors are presynaptic and modulate presynaptic glutamatergic release [28]. Transcript and protein levels of group I receptors are higher in prefrontal cortex of schizophrenia subjects [57–59], although the tissue used in these studies were not limited to DLPFC. In contrast, one group observed downregulated mGluR5 protein in DLPFC, although these results did not reach significance [60]. Group II mGluR3 mRNA and mGluR2/3 protein were upregulated (although not always significantly so) in DLPFC [58,60,61], indicating higher presynaptic glutamatergic modulation.
Glutamatergic signaling also depends on the proper glutamate receptor localization within the synapse and the integrity of the proteins responsible for receptor clustering within postsynaptic microdomains. Intracellular molecules enriched in the postsynaptic density (PSD) target glutamate receptors to the synaptic membrane, modulate receptor activity, and regulate synaptic plasticity. The predominant PSD protein, PSD-95 (for review please see [62]) interacts with NMDA receptor subunits, AMPA receptors, tyrosine kinase receptors, ion channels, and other cytoplasmic proteins, and therefore can modulate signaling cascades that congregate within the PSD [63]. Lower levels of PSD-95 transcripts and protein have been observed in the DLPFC of schizophrenia subjects [40,62,64–71], potentially impacting PSD-related neuregulinerbB signaling [72], dendritic spine enlargement, synaptic strength, and neuronal connectivity [73].
2.3. Cortical astrocytic clearance of glutamate in schizophrenia
Synaptic glutamate is tightly regulated through several mechanisms in the tripartite synapse to maintain homeostatic balance [74]. Different subtypes of excitatory amino acid transporters (EAATs) are located on all parts of the tripartite synapse (for review please see [75]); however, astrocytic EAAT2 transports ~90% of extracellular glutamate to the intracellular space of the astrocyte, preventing hyperexcitation of nearby neurons and maintaining homeostatic balance. The density of EAAT2 at the synaptic membrane also appears to shift with changes to the excitatory/inhibitory balance as it can be increased or decreased, respectively, via pharmacological stimulation or blockade of neuronal activity in vitro [76,77]. Therefore, altered glutamate transport can result in glutamate spillover, potential “runaway” excitatory transmission, and the promotion of neuronal cell death (for review please see [78,79]).
Analysis of total DLPFC gray matter has not detected alterations in EAAT2 protein or mRNA levels in schizophrenia [80,81]; however, when the analysis was limited to cortical layers 3 and 5, EAAT2 transcripts were found to be lower, although non-significantly [58]. Additionally, age may influence EAAT2 expression in schizophrenia. For example, Ohnuma et al. [58] found lower EAAT2 in layer 3 of the DLPFC in elderly schizophrenia subjects, whereas higher EAAT2 mRNA levels were observed in DLPFC layer 3 in a younger (~45 years of age) cohort of schizophrenia subjects [21]. However, the effect of age may also impact EAAT2 localization, as elevated protein levels were observed in fractionated extrasynaptic membranes in a second elderly cohort [82].
Lower glutamine synthetase protein has been found in the DLPFC [83], suggesting downregulated glutamate-to-glutamine conversion within astrocytes following EAAT2 intake [84]. However, this finding was not replicated by other groups in studies of various prefrontal cortical regions [19,84,85].
2.4. Cell type-specific alterations in morphology of cortical glutamatergic neurons
Despite differences across studies for some findings, in aggregate the data reviewed above support the idea that glutamatergic signaling is altered in the DLPFC in schizophrenia subjects, especially in layer 3 (Fig. 1B). This apparent laminar-specificity is supported by evidence of pyramidal cell type-specific morphological alterations in schizophrenia. For example, dysregulated glutamatergic activity within the DLPFC would be expected to alter cellular and dendritic spine morphology on glutamatergic pyramidal neurons. Dendritic spines, which are almost exclusively located on pyramidal neurons, are the primary site of glutamatergic receptors, and their morphology and number are activity-dependent [86]. Consistent with molecular evidence of altered glutamate signaling in DLPFC layer 3, all studies published to date observed findings of lower spine density and shorter dendritic length in DLPFC layer 3 pyramidal neurons from schizophrenia subjects [87–89] (Fig. 1B). In contrast, such alterations were not observed in layer 5 or 6 pyramidal neurons [90].
Somal size has often been considered a proxy measure for other structural and cytoskeletal abnormalities, including dendritic length and spine density [88,91,92]. Like the deficits in dendritic spine density, pyramidal neuron cell body size in schizophrenia is smaller in layer 3 [88,93,94] (Fig. 1B) but unaltered in layers 5 and 6 [90,94].
In concert, the findings from molecular studies support the provisional conclusion from molecular studies that deficits in glutamatergic signaling in DLPFC layer 3 could contribute to impaired gamma oscillations and working memory in schizophrenia.
3. Prefrontal Cortical GABA Abnormalities in Schizophrenia
Although gamma oscillations are thought to reflect the synchronized activity of large groups of pyramidal neurons, the rhythmic firing of inhibitory GABA neurons at gamma frequency is essential for this synchronized activity [95], as activation of GABA neurons is sufficient to drive oscillatory activity in vitro [15,96]. Altered GABA signaling in schizophrenia may therefore also contribute to impaired frontal gamma oscillations and working memory disturbances in schizophrenia.
GABA levels are technically challenging to measure directly in human postmortem tissue, but the strength of GABA neurotransmission can be inferred by measuring levels of mRNA transcripts and their cognate proteins that regulate GABA in presynaptic terminals (Fig. 2B). The availability of synaptic GABA is controlled by the 67 and 65 kDa isoforms (GAD67 and GAD65, respectively) of the synthesizing enzyme glutamic acid decarboxylase, which converts glutamic acid into GABA. Many, but not all, GABA neurons appear to contain both isoforms [97]. GAD67 is responsible for most cortical GABA synthesis [98]; GAD67 largely exists in the cofactor-saturated active form and is therefore regulated primarily by transcription. In contrast, GAD65 appears to be active only during periods of high neuronal activity [99]. In addition to the availability of GABA via synthesis, the strength of GABAergic neurotransmission is also dependent on the accumulation of GABA into synaptic vesicles, mediated by the vesicular GABA transporter (vGAT), which can serve as a key regulator of inhibitory strength in response to homeostatic signals of network activity [100,101]. Finally, the strength of GABA neurotransmission is regulated by clearance of GABA from the synaptic cleft, largely carried out by GABA transporter 1 (GAT1) on the presynaptic terminal. In the primate DLPFC, GAT1 is known to regulate the inhibitory post synaptic potentials of perisomatic GABA inhibition and to prevent GABA spillover at both perisomatic and dendritic-targeting GABA inputs [102] (Fig. 2B). Together, levels of these three presynaptic markers of GABA neurotransmission in the postmortem brain can provide insight into the integrity of GABAergic signaling in schizophrenia.
3.1. GABA in presynaptic boutons in schizophrenia
In the DLPFC of schizophrenia subjects, lower levels of GAD67 mRNA in total gray matter have been consistently reported across cohorts of subjects and between research groups [103]. Two in situ hybridization studies revealed a GAD67 deficit across cortical layers 2–5, with pronounced deficits in layers 3–4 [104,105]. Protein levels of GAD67, measured by Western blot and immunofluorescence, are also lower in the disease [106], consistent with the idea that the regulation of the functional enzyme is principally through transcription. In contrast, levels of GAD65 have not been reported to be lower in the illness, although GAD65 may be lower in subjects with schizoaffective disorder [107]. Findings of lower levels of GAD67 mRNA and protein support the notion that presynaptic measures of inhibitory strength are diminished in the DLPFC of schizophrenia subjects.
Despite marked reductions in GAD67 and a likely impairment of GABA synthesis in schizophrenia, levels of vGAT mRNA appear to be normal or only modestly lower in most studies of total tissue homogenates [26,108] or in samples restricted to layer 3 [21]. Consistent with mRNA findings, levels of vGAT protein per bouton are unaltered in schizophrenia [109]. Thus, in contrast to layer-specific deficits in vGLUT1 mRNA, schizophrenia does not appear to be associated with deficits in vesicular packaging of GABA. Similar to vGAT, mRNA levels of GAT1 are not altered in total gray matter samples of DLPFC from individuals with schizophrenia [108], although a subset of GABAergic neurons may express lower levels of GAT1, suggested by a ~25% deficit in the density of GAT1 mRNA positive neurons in schizophrenia [110]. These findings highlight the importance of cell type-specific studies in schizophrenia [111], as measures of GAT1 mRNA in total tissue homogenates might mask deficits in a particular subset of neurons.
Indeed, these in situ hybridization findings of undetectable GAT1 [110] and GAD67 [105] mRNA in a subset of neurons suggest that specific subtypes of GABA neurons may be differentially affected in schizophrenia. GABA neurons are typically classified based on the expression of certain molecular markers that are largely distinct: the calcium binding proteins parvalbumin (PV) or calretinin (CR), and the neuropeptide somatostatin (SST). Understanding the functional consequences of alterations in transcripts and proteins related to GABA neurotransmission depends on knowing in which subtypes of GABA neurons these alterations are present. Thus, we review cell type-specific findings of altered levels of markers that index GABA neurotransmission in schizophrenia (Fig. 1B).
3.2. PV interneurons in the DLPFC are altered in schizophrenia
In the DLPFC of schizophrenia subjects, deficits in PV mRNA have been consistently replicated [112–117]. The deficit is apparent in total gray matter tissue homogenates, but laminar analysis reveals a pronounced deficit in layers 3–4, where PV cell bodies are enriched [115]. The deficits in PV mRNA are accompanied by comparable decrements in levels of PV protein in cell bodies [118] and axon terminals [119]. These changes to do not appear to reflect reduced numbers of PV neurons in the illness, as the density of PV mRNA-positive neurons [115] and PV-immunoreactive neurons [118,120–122] are not altered in the DLPFC of schizophrenia subjects. In the few studies which report a deficit in PV neuron density in the DLPFC of schizophrenia subjects [123–125], lower levels of PV immunoreactivity per neuron may have rendered these neurons undetectable at lower levels of magnification [120]. Together, these findings of lower PV in schizophrenia suggest that PV neurons may be one of the subtypes of GABA neurons that exhibit deficits in GABA signaling in schizophrenia.
Indeed, PV neurons exhibit other alterations, including pronounced deficits in GAD67 expression such that ~45% of PV neurons lack detectable GAD67 mRNA [115]. Although PV neurons can be further subclassified based on their characteristic morphology and the targets of their axon terminals, studies of PV and GAD67 mRNA have not revealed whether both subtypes of PV neurons exhibit similar alterations, but studies at the protein level have been informative. The first subtype, the PV basket cell, primarily targets the perisomatic region of pyramidal neurons where the postsynaptic membrane is enriched in GABAA receptors containing the α1 subunit. The second subtype, PV chandelier cells, form distinctive cartridges of axon terminals that innervate the axon initial segment (AIS) of pyramidal neurons (thus termed ‘axo-axonic’ neurons) which are enriched with GABAA receptors containing the α2 subunit [126,127]. Importantly, chandelier cells exclusively use GAD67 for GABA synthesis, whereas basket cells use both GAD65 and GAD67 [97], a difference supported by single cell-transcriptomic findings [128].
Further studies of these two subtypes of PV neurons have revealed that PV basket cells, and not chandelier cells, exhibit deficits in GAD67 levels. In PV terminals that are also GAD65 positive, and therefore must represent the axon terminals of PV basket cells, protein levels of PV [119] and GAD67 [106] are lower. However, GAD67 protein levels are not altered in the distinctive axon cartridges of chandelier cells [109]. PV basket cells therefore represent a likely source of the GAD67 mRNA deficit observed in the middle layers of the DLPFC in schizophrenia subjects. The basis for the PV basket cell-selective deficits in GAD67 and PV in schizophrenia might be fewer excitatory inputs to PV basket cells in the middle layers of the DLPFC [118] or alterations in the perineuronal nets [120] that surround basket cells and not chandelier cells [129].
In addition to deficits in presynaptic GABA function in PV basket cells, most studies also report a deficit in the GABAA receptor subunits that are postsynaptic to these inputs. Both the α1 subunit and the β2 subunit, which colocalizes with the a1 subunit at basket cell inputs [130], are lower in schizophrenia especially in layers 3–4 [131], where PV basket cell terminals are highest and PV chandelier cell terminals are lowest [132]. Indeed, α1 mRNA levels are lower specifically within pyramidal neurons in deep layer 3 [133], strongly supporting the notion that in addition to deficits in presynaptic GABA function in PV basket cells, the postsynaptic efficacy of basket cell inputs to pyramidal cells is weaker in schizophrenia.
Although PV chandelier cells may not exhibit deficits in GABA synthesis, the density of chandelier cell cartridges, which are identified by the immunoreactivity of GAT1 in distinctive vertical arrays, is lower across layers 2–4 in the DLPFC of schizophrenia subjects [134,135]. The lower density of cartridges is thought to reflect their lower detectability due to reduced GAT1 immunoreactivity, rather than an inherent structural deficit. Indeed, the density of cartridges identified by vGAT is not lower in the illness [136]. Thus, chandelier cells may be among the subtypes of GABA neurons that exhibit undetectable GAT1 mRNA levels, although this deficit is also likely present within PV basket cells: lower GAT1 is observed in samples of layer 3 PV basket cells, identified by a characteristic perineuronal net [113].
The postsynaptic side of chandelier cell inputs also appears to be altered in schizophrenia, but in ways that are distinct from alterations to the postsynaptic side of PV basket cell inputs. As previously mentioned, chandelier cell inputs tend to be populated by GABAA receptors enriched in the α2 subunit [126,127], and both the mRNA expression of this subunit and the density of cartridges positive for α2 [137] are upregulated in schizophrenia. Importantly, this upregulation is observed prominently in layer 2 [131,137], where the majority of chandelier cell inputs are located in human neocortex [132].
In sum, studies of PV neurons in schizophrenia have consistently revealed disease-associated alterations in markers that index their function in GABA neurotransmission (Fig. 1B). These studies highlight the critical importance of cell type-specific studies of the postmortem human brain, as measures at total tissue levels do not reveal in which subtypes of neurons these differences may be occurring. Together, these data strongly suggest that PV basket cells are a major contributor to the GAD67 deficit in the middle layers of the DLPFC in schizophrenia, and that impairments in GABA signaling from these neurons underlie altered gamma oscillatory function in the supragranular layers of the DLPFC in the disease.
3.3. The CR interneuron subtype does not appear to be altered in the DLPFC of schizophrenia
While PV neurons likely represent the source of the GAD67 deficit in the middle layers, GAD67 is lower in superficial layer 2 as well [105], raising the possibility that other GABA neuron subtypes contribute to this deficit. Interneurons expressing calretinin (CR) are also enriched in the superficial layers and primarily target other interneurons, disinhibiting pyramidal cells [138]. However, convergent lines of evidence suggest that this subtype may not be altered in schizophrenia. First, tissue levels of CR mRNA [112,114–116] and protein [114] are unaltered in schizophrenia (Fig. 1B). Second, neither the density of CR mRNA-expressing [115] nor of CR-immunoreactive neurons [124,139] are altered in schizophrenia. Third, alterations that are present within the PV neuron population, such as a lower complement of excitatory inputs onto the soma, are not present within the CR neuron population [118]. The lack of alteration in CR suggest that this subtype might not contribute to the alterations in markers of GABA neurotransmission, including GAD67, observed in total tissue. However, GAD67 levels have yet to be directly assessed within CR neurons.
3.4. Deficits in SST neurons in the DLPFC of schizophrenia are robust and highly replicated
Thus, another subtype of GABA neuron expressing SST, enriched in the superficial layers 2–3 as well as layer 5 and the superficial white matter, might be the source of the GAD67 deficit in these layers. SST cells primarily target the distal dendrites of pyramidal neurons and gate the excitatory inputs that pyramidal neurons receive [140,141]. However, these neurons also provide dendritic inhibition onto PV neurons, and the functional consequences of alterations to SST neurons might depend on whether they target excitatory or inhibitory cells [142,143]. SST cells in layers 2 and superficial layer 3 appear to receive a large proportion of the excitatory inputs from layer 3 pyramidal neurons [12], suggesting that SST cells participate in the layer 3 microcircuit that generates working memory and gamma oscillatory activity.
In schizophrenia, lower levels of SST mRNA have been consistently reported in the DLPFC across multiple cohorts and multiple research groups. Indeed, these deficits are robust relative to other GABA deficits and are present in a large proportion of subjects with schizophrenia [114,117,144,145]. Thus, these disease-related deficits in SST mRNA support the notion that these cells contribute to the GAD67 deficit in schizophrenia (Fig. 1B). However, identifying these and other alterations of markers related to GABA neurotransmission within SST neurons is methodologically challenging, as the density of DLPFC SST mRNA-positive neurons is 25% lower in schizophrenia [145]. Given the lack of a total neuron deficit in the illness [146] and the presence of lower mean SST mRNA levels in the remaining neurons [145], it is likely that the deficit in SST mRNA-positive neuron density in the illness reflects an inability to detect SST in some neurons, rather than a lower number of SST neurons in schizophrenia. Although this assumption has yet to be directly tested, if true, future studies should utilize proxy markers of SST neurons that are unaltered in schizophrenia in order to directly assess disease-related alterations in this population.
Although SST neurons are present across cortical gray matter layers and the subcortical white matter, both SST and GAD67 levels are lower in the superficial gray matter, but not in deep layer 6 or the superficial white matter [105,145]. This layer-specific deficit suggests that DLPFC SST neuron alterations in schizophrenia reflect alterations to the microcircuit in the superficial layers of the neocortex that involves these SST neurons as well as PV neurons and deep layer 3 pyramidal neurons. Thus, alterations in SST neurons could also contribute to impaired gamma oscillatory power in the DLPFC and working memory deficits in schizophrenia.
4. Conclusions
As reviewed above, alterations in glutamate and GABA neurotransmission in schizophrenia are most marked in the supragranular layers, and especially in a microcircuit composed of excitatory layer 3 pyramidal neurons, deep layer 3-layer 4 PV basket cells and layer 2-superficial layer 3 SST neurons (Fig. 1B). Given the apparent key role of this circuit in gamma oscillations and working memory, working memory task-related gamma oscillatory activity might represent a useful biomarker for (1) identifying which individuals with schizophrenia might benefit from pro-cognitive therapeutics targeting this circuit and (2) for assessing the efficacy of such therapeutic agents.
One example of this approach has been the investigation of a positive allosteric modulator with relative selectivity for GABAA receptors containing α2/3 subunits. In an initial study, a drug with these features was associated with both greater gamma power during a task requiring working memory and improved performance on two different working memory tasks in subjects with schizophrenia [147]. A subsequent study, which did not include any biomarker measures, failed to show improvement in cognition [148], although a number of limitations in the potency of the medication and the study design may have contributed to the negative findings [149]. Non-pharmacological interventions are also using this strategy for biomarkers. For example, an ongoing clinical trial utilizing direct current stimulation to enhance frontal cognitive function in individuals with schizophrenia includes both behavioral assessments and changes in gamma oscillatory function as outcome measures (e.g., ClinicalTrials.gov identifier NCT02739347). Gamma oscillations might therefore not only be a useful physiologic measure of frontal cortical networks during working memory tasks as secondary measures in clinical trials, but also provide a translatable measure from preclinical models for the cognitive dysfunction of individuals with schizophrenia [16,150].
Other in vivo measures of DLPFC glutamate and GABA signaling may also serve as biomarkers in schizophrenia, but the currently available tools appear to lack the resolution needed to specifically reflect the integrity of the microcircuit. For example, magnetic resonance spectroscopy (MRS) can measure the levels of glutamate and GABA in frontal regions of the cortex. However, in contrast to consistent and replicated findings of alterations to these neurotransmitter systems reported in postmortem studies, the MRS literature on glutamate and GABA levels in schizophrenia is mixed [151,152]. This variability may be due to the fact that glutamate and GABA peaks measured in MRS are difficult to distinguish at commonly used magnetic field strengths (1.5 or 3 Tesla). Indeed, results of recent 7T MRS studies have been more consistent in reporting lower glutamate and GABA levels in the frontal cortex of subjects with schizophrenia [153,154]. However, MRS measures total tissue levels of glutamate and GABA which includes levels associated with metabolism and not just neurotransmission. Approaches utilizing novel PET radioligands may overcome these limitations by directly indexing the availability of GABA in the synapse. For example, studies utilizing this approach support the idea that GABA synthesis in the frontal cortex is impaired in schizophrenia subjects [155]. Future studies utilizing other novel PET radioligands may provide more specificity to index the neural circuitry responsible for working memory in schizophrenia subjects in vivo.
Another approach to indexing frontal circuit function in vivo is through task-related alterations in cerebral blood flow in the DLPFC, indexed by fMRI, in schizophrenia subjects. Indeed, these studies were the first to provide insight into how DLPFC dysfunction could underlie deficits in working memory [156]. However, this imaging measure is similarly less specific than task-evoked gamma, as differences in fMRI measures can be driven by numerous factors other than alterations in glutamatergic and GABAergic signaling in the superficial layers. Recent advances in laminar-specific fMRI in humans have supported the importance of supragranular layers in the maintenance of working memory function [157], raising the possibility that this technique might be applied to subjects with schizophrenia to reveal layer-specific, task-evoked alterations in fMRI measures during working memory.
As these and other approaches in in vivo measurements advance, they may provide greater resolution into the function of specific neural circuits and thus offer new biomarkers that are informed by the neurobiology of schizophrenia. Such biomarkers offer the promise of important linkages between circuit-based molecular targets for new medications, as revealed by postmortem studies, and the means to better index the function of the circuit targeted by pro-cognitive therapeutics.
Acknowledgment
This work was supported by NIH grant MH043784 (D.A.L) and F99/K00 MH122943 (K.E.S).
Declaration of competing interest
David A. Lewis currently receives investigator-initiated research support from Pfizer and Merck. Kirsten E. Schoonover and Samuel J. Dienel declare no conflicts of interest.
References
- [1].Keefe RS, Harvey PD, Cognitive impairment in schizophrenia, Handb. Exp. Pharmacol (2012) 11–37. [DOI] [PubMed] [Google Scholar]
- [2].Meier MH, Caspi A, Reichenberg A, Keefe RS, Fisher HL, Harrington H, Houts R, Poulton R, Moffitt TE, Neuropsychological decline in schizophrenia from the premorbid to the postonset period: evidence from a population-representative longitudinal study, Am. J. Psychiatry 171 (2014) 91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Forbes NF, Carrick LA, Mcintosh AM, Lawrie SM, Working memory in schizophrenia: a meta-analysis, Psychol. Med 39 (2009) 889–905. [DOI] [PubMed] [Google Scholar]
- [4].Keefe RS, Roitman SE, Harvey PD, Blum CS, Dupre RL, Prieto DM, Davidson M, Davis KL, A pen-and-paper human analogue of a monkey prefrontal cortex activation task: spatial working memory in patients with schizophrenia, Schizophr. Res 17 (1995) 25–33. [DOI] [PubMed] [Google Scholar]
- [5].Barch DM, Ceaser A, Cognition in schizophrenia: core psychological and neural mechanisms, Trends Cognit. Sci 16 (2012) 27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Silver H, Feldman P, Bilker W, Gur RC, Working memory deficit as a core neuropsychological dysfunction in schizophrenia, Am. J. Psychiatry 160 (2003) 1809–1816. [DOI] [PubMed] [Google Scholar]
- [7].Miller EK, Cohen JD, An integrative theory of prefrontal cortex function, Annu. Rev. Neurosci 24 (2001) 167–202. [DOI] [PubMed] [Google Scholar]
- [8].Kraguljac NV, Srivastava A, Lahti AC, Memory deficits in schizophrenia: a selective review of functional magnetic resonance imaging (FMRI) studies, Behav. Sci. Basel 3 (2013) 330–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Howard MW, Rizzuto DS, Caplan JB, Madsen JR, Lisman J, Aschenbrenner-Scheibe R, Schulze-Bonhage A, Kahana MJ, Gamma oscillations correlate with working memory load in humans, Cereb. Cortex 13 (2003) 1369–1374. [DOI] [PubMed] [Google Scholar]
- [10].Cho RY, Konecky RO, Carter CS, Impairments in frontal cortical gamma synchrony and cognitive control in schizophrenia, Proc. Natl. Acad. Sci. U.S.A 103 (2006) 19878–19883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Bastos AM, Loonis R, Kornblith S, Lundqvist M, Miller EK, Laminar recordings in frontal cortex suggest distinct layers for maintenance and control of working memory, Proc. Natl. Acad. Sci. U.S.A (2018) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Melchitzky DS, Lewis DA, Pyramidal neuron local axon terminals in monkey prefrontal cortex: differential targeting of subclasses of GABA neurons, Cereb. Cortex 13 (2003) 452–460. [DOI] [PubMed] [Google Scholar]
- [13].Constantinidis C, Williams GV, Goldman-Rakic PS,A role for inhibition in shaping the temporal flow of information in prefrontal cortex, Nat. Neurosci 5 (2002) 175–180. [DOI] [PubMed] [Google Scholar]
- [14].Gonzalez-Burgos G, Cho RY, Lewis DA, Alterations in cortical network oscillations and parvalbumin neurons in schizophrenia, Biol. Psychiatry 77 (2015) 1031–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sohal VS, Zhang F, Yizhar O, Deisseroth K, Parvalbumin neurons and gamma rhythms enhance cortical circuit performance, Nature 459 (2009) 698–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Tatard-Leitman VM, Jutzeler CR, Suh J, Saunders JA, Billingslea EN, Morita S, White R, Featherstone RE, Ray R, Ortinski PI, Banerjee A, Gandal MJ, Lin R, Alexandrescu A, Liang Y, Gur RE, Borgmann-Winter KE, Carlson GC, Hahn CG, Siegel SJ, Pyramidal cell selective ablationofN-methyl-d-aspartate receptor 1 causes increase in cellular and network excitability, Biol. Psychiatry 77 (2015) 556–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wang M, Yang Y, Wang CJ, Gamo NJ, Jin LE, Mazer JA, Morrison JH, Wang XJ, Arnsten AF, NMDA receptors subserve persistent neuronal firing during working memory in dorsolateral prefrontal cortex, Neuron 77 (2013) 736–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Rudy CC, Hunsberger HC, Weitzner DS, Reed MN, The role of the tripartite glutamatergic synapse in the pathophysiology of Alzheimer’s disease, Aging Dis. 6 (2015) 131–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Gluck MR, Thomas RG, Davis KL, Haroutunian V, Implications for altered glutamate and GABA metabolism in the dorsolateral prefrontal cortex of aged schizophrenic patients, Am. J. Psychiatry 159 (2002) 1165–1173. [DOI] [PubMed] [Google Scholar]
- [20].Gandal MJ, Zhang P, Hadjimichael E, Walker RL, Chen C, Liu S, Won H, Van bakel H, Varghese M, Wang Y, Shieh AW, Haney J, Parhami S, Belmont J, Kim M, Moran Losada P, Khan Z, Mleczko J, Xia Y, Dai R, Wang D, Yang YT, Xu M, Fish K, Hof PR, Warrell J, Fitzgerald D, White K, Jaffe AE, Peters MA, Gerstein M, Liu C, Iakoucheva LM, Pinto D, Geschwind DH, Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder, Science (2018) 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hoftman GD, Dienel SJ, Bazmi HH, Zhang Y, Chen K, Lewis DA, Altered gradients of glutamate and gamma-aminobutyric acid transcripts in the cortical visuospatial working memory network in schizophrenia, Biol. Psychiatry 83 (2018) 670–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Fremeau RT JR, Troyer MD, Pahner I, Nygaard GO, Tran CH, Reimer RJ, Bellocchio EE, Fortin D, Storm-Mathisen J, Edwards RH, The expression of vesicular glutamate transporters defines two classes of excitatory synapse, Neuron 31 (2001) 247–260. [DOI] [PubMed] [Google Scholar]
- [23].Herzog E, Bellenchi GC, Gras C, Bernard V, Ravassard P, Bedet C, Gasnier B, Giros B, El Mestikawy S, The existence of a second vesicular glutamate transporter specifies subpopulations of glutamatergic neurons, J. Neurosci 21 (2001) Rc181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wojcik SM, Rhee JS, Herzog E, Sigler A, Jahn R, Takamori S, Brose N, Rosenmund C, An essential role for vesicular glutamate transporter 1 (VGLUT1) in postnatal development and control of quantal size, Proc. Natl. Acad. Sci. U.S.A 101 (2004) 7158–7163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Eastwood SL, Harrison PJ, Decreased expression of vesicular glutamate transporter 1 and complexin II mRNAs in schizophrenia: further evidence for a synaptic pathology affecting glutamate neurons, Schizophr. Res 73 (2005) 159–172. [DOI] [PubMed] [Google Scholar]
- [26].Fung SJ, Sivagnanasundaram S, Weickert CS, Lack of change in markers of presynaptic terminal abundance alongside subtle reductions in markers of presynaptic terminal plasticity in prefrontal cortex of schizophrenia patients, Biol. Psychiatry 69 (2011) 71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Oni-Orisan A, Kristiansen LV, Haroutunian V, Meador-Woodruff JH, Mccullumsmith RE, Altered vesicular glutamate transporter expression in the anterior cingulate cortex in schizophrenia, Biol. Psychiatry 63 (2008) 766–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Reiner A, Levitz J, Glutamatergic signaling in the central nervous system: ionotropic and metabotropic receptors in concert, Neuron 98 (2018) 1080–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Beneyto M, Meador-Woodruff JH, Lamina-specific abnormalities of AMPA receptor trafficking and signaling molecule transcripts in the prefrontal cortex in schizophrenia, Synapse 60 (2006) 585–598. [DOI] [PubMed] [Google Scholar]
- [30].Dracheva S, Mcgurk SR, Haroutunian V, mRNA expression of AMPA receptors and AMPA receptor binding proteins in the cerebral cortex of elderly schizophrenics, J. Neurosci. Res 79 (2005) 868–878. [DOI] [PubMed] [Google Scholar]
- [31].Catts VS, Fung SJ, Long LE, Joshi D, Vercammen A, Allen KM, Fillman SG, Rothmond DA, Sinclair D, Tiwari Y, Tsai SY, Weickert TW, Shannon Weickert C, Rethinking schizophrenia in the context of normal neurodevelopment, Front. Cell. Neurosci 7 (2013) 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH, Developmental and regional expression in the rat brain and functional properties of four NMDA receptors, Neuron 12 (1994) 529–540. [DOI] [PubMed] [Google Scholar]
- [33].Mueller HT, Meador-Woodruff JH, NR3A NMDA receptor subunit mRNA expression in schizophrenia, depression and bipolar disorder, Schizophr. Res 71 (2004) 361–370. [DOI] [PubMed] [Google Scholar]
- [34].Weickert CS, Fung SJ, Catts VS, Schofield PR, Allen KM, Moore LT, Newell KA, Pellen D, Huang XF, Catts SV, Weickert TW, Molecular evidence of N-methyl-D-aspartate receptor hypofunction in schizophrenia, Mol. Psychiatry 18 (2013) 1185–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Akbarian S, Kim JJ, Potkin SG, Hetrick WP, Bunney WE JR., Jones EG, Maldistribution of interstitial neurons in prefrontal white matter of the brains of schizophrenic patients, Arch. Gen. Psychiatry 53 (1996) 425–436. [DOI] [PubMed] [Google Scholar]
- [36].Beneyto M, Meador-Woodruff JH, Lamina-specific abnormalities of NMDA receptor-associated postsynaptic protein transcripts in the prefrontal cortex in schizophrenia and bipolar disorder, Neuropsychopharmacology 33 (2008) 2175–2186. [DOI] [PubMed] [Google Scholar]
- [37].Sokolov BP, Expression of NMDAR1, GluR1, GluR7, and KA1 glutamate receptor mRNAs is decreased in frontal cortex of “neuroleptic-free” schizophrenics: evidence on reversible up-regulation by typical neuroleptics, J. Neurochem 71 (1998) 2454–2464. [DOI] [PubMed] [Google Scholar]
- [38].Mcquail JA, Beas BS, Kelly KB, Simpson KL, Frazier CJ, Setlow B, Bizon JL, NR2A-containing NMDARs in the prefrontal cortex are required for working memory and associated with age-related cognitive decline, J. Neurosci 36 (2016) 12537–12548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Dracheva S, Marras SA, Elhakem SL, Kramer FR, Davis KL, Haroutunian V, N-methyl-D-aspartic acid receptor expression in the dorsolateral prefrontal cortex of elderly patients with schizophrenia, Am. J. Psychiatry 158 (2001) 1400–1410. [DOI] [PubMed] [Google Scholar]
- [40].Kristiansen LV, Beneyto M, Haroutunian V, Meador-Woodruff JH, Changes in NMDA receptor subunits and interacting PSD proteins in dorsolateral prefrontal and anterior cingulate cortex indicate abnormal regional expression in schizophrenia, Mol. Psychiatry 11 (737-47) (2006) 705. [DOI] [PubMed] [Google Scholar]
- [41].Bendikov I, Nadri C, Amar S, Panizzutti R, De Miranda J, Wolosker H, Agam G, A CSF and postmortem brain study of D-serine metabolic parameters in schizophrenia, Schizophr. Res 90 (2007) 41–51. [DOI] [PubMed] [Google Scholar]
- [42].Steffek AE, Haroutunian V, Meador-Woodruff JH, Serine racemase protein expression in cortex and hippocampus in schizophrenia, Neuroreport 17 (2006) 1181–1185. [DOI] [PubMed] [Google Scholar]
- [43].Verrall L, Walker M, Rawlings N, Benzel I, Kew JN, Harrison PJ, Burnet PW, d-Amino acid oxidase and serine racemase in human brain: normal distribution and altered expression in schizophrenia, Eur. J. Neurosci 26 (2007) 1657–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Stone TW, Neuropharmacology of quinolinic and kynurenic acids, Pharmacol. Rev 45 (1993) 309–379. [PubMed] [Google Scholar]
- [45].Sathyasaikumar KV, Stachowski EK, Wonodi I, Roberts RC, Rassoulpour A, Mcmahon RP, Schwarcz R, Impaired kynurenine pathway metabolism in the prefrontal cortex of individuals with schizophrenia, Schizophr. Bull 37 (2011) 1147–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Schwarcz R, Rassoulpour A, Wu HQ, Medoff D, Tamminga CA, Roberts RC, Increased cortical kynurenate content in schizophrenia, Biol. Psychiatry 50 (2001) 521–530. [DOI] [PubMed] [Google Scholar]
- [47].Hahn CG, Wang HY, Cho DS, Talbot K, Gur RE, Berrettini WH, Bakshi K, Kamins J, Borgmann-Winter KE, Siegel SJ, Gallop RJ, Arnold SE, Altered neuregulin 1-erbB4 signaling contributes to NMDA & receptor hypofunction in schizophrenia, Nat. Med 12 (2006) 824–828. [DOI] [PubMed] [Google Scholar]
- [48].Limon A, Reyes-Ruiz JM, Miledi R, Microtransplantation of neurotransmitter receptorsfrom postmortem autistic brains toXenopus oocytes, Proc Natl Acad Sci U S A 105 (2008) 10973–10977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Hashimoto R, Straub RE, Weickert CS, Hyde TM, Kleinman JE, Weinberger DR, Expression analysis of neuregulin-1 in the dorsolateral prefrontal cortex in schizophrenia, Mol. Psychiatry 9 (2004) 299–307. [DOI] [PubMed] [Google Scholar]
- [50].Law AJ, Lipska BK, Weickert CS, Hyde TM, Straub RE, Hashimoto R, Harrison PJ, Kleinman JE, Weinberger DR, Neuregulin 1 transcripts are differentially expressed in schizophrenia and regulated by 5’ SNPs associated with the disease, Proc. Natl. Acad. Sci. U.S.A 103 (2006) 6747–6752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Stefansson H, Sigurdsson E, Steinthorsdottir V, Bjornsdottir S, Sigmundsson T, Ghosh S, Brynjolfsson J, Gunnarsdottir S, Ivarsson O, Chou TT, Hjaltason O, Birgisdottir B, Jonsson H, Gudnadottir VG, Gudmundsdottir E, Bjornsson A, Ingvarsson B, Ingason A, Sigfusson S, Hardardottir H, Harvey RP, Lai D, Zhou M, Brunner D, Mutel V, Gonzalo A, Lemke G, Sainz J, Johannesson G, Andresson T, Gudbjartsson D, Manolescu A, Frigge ML, Gurney ME, Kong A, Gulcher JR, Petursson H, Stefansson K, Neuregulin 1 and susceptibility to schizophrenia, Am. J. Hum. Genet 71 (2002) 877–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Buonanno A, The neuregulin signaling pathway and schizophrenia: from genes to synapses and neural circuits, Brain Res. Bull 83 (2010) 122–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Fisahn A, Neddens J, Yan L, Buonanno A, Neuregulin-1 modulates hippocampal gamma oscillations: implications for schizophrenia, Cereb. Cortex 19 (2009) 612–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Li B, Woo RS, Mei L, Malinow R, The neuregulin-1 receptor erbB4 controls glutamatergic synapse maturation and plasticity, Neuron 54 (2007) 583–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Scarr E, Beneyto M, Meador-Woodruff JH, Dean B, Cortical glutamatergic markers in schizophrenia, Neuropsychopharmacology 30 (2005) 1521–1531. [DOI] [PubMed] [Google Scholar]
- [56].Meador-Woodruff JH, Davis KL, Haroutunian V, Abnormal kainate receptor expression in prefrontal cortex in schizophrenia, Neuropsychopharmacology 24 (2001) 545–552. [DOI] [PubMed] [Google Scholar]
- [57].Devon RS, Anderson S, Teague PW, Burgess P, Kipari TM, Semple CA, Millar JK, Muir WJ, Murray V, Pelosi AJ, Blackwood DH, Porteous DJ, Identification of polymorphisms within disrupted in Schizophrenia 1 and disrupted in Schizophrenia 2, and an investigation of their association with schizophrenia and bipolar affective disorder, Psychiatr. Genet 11 (2001) 71–78. [DOI] [PubMed] [Google Scholar]
- [58].Ohnuma T, Augood SJ, Arai H, Mckenna PJ, Emson PC, Expression of the human excitatory amino acid transporter 2 and metabotropic glutamate receptors 3 and 5 in the prefrontal cortex from normal individuals and patients with schizophrenia, Brain Res. Mol. Brain Res 56 (1998) 207–217. [DOI] [PubMed] [Google Scholar]
- [59].Volk DW, Eggan SM, Lewis DA, Alterations in metabotropic glutamate receptor 1alpha and regulator of G protein signaling 4 in the prefrontal cortex in schizophrenia, Am. J. Psychiatry 167 (2010) 1489–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Gupta DS, Mccullumsmith RE, Beneyto M, Haroutunian V, Davis KL, Meador-Woodruff JH, Metabotropic glutamate receptor protein expression in the prefrontal cortex and striatum in schizophrenia, Synapse 57 (2005) 123–131. [DOI] [PubMed] [Google Scholar]
- [61].Crook JM, Akil M, Law BC, Hyde TM, Kleinman JE, Comparative analysis of group II metabotropic glutamate receptor immunoreactivity in Brodmann’s area 46 of the dorsolateral prefrontal cortex from patients with schizophrenia and normal subjects, Mol. Psychiatry 7 (2002) 157–164. [DOI] [PubMed] [Google Scholar]
- [62].De Bartolomeis A, Latte G, Tomasetti C, Iasevoli F, Glutamatergic postsynaptic density protein dysfunctions in synaptic plasticity and dendritic spines morphology: relevance to schizophrenia and other behavioral disorders pathophysiology, and implications for novel therapeutic approaches, Mol. Neurobiol 49 (2014) 484–511. [DOI] [PubMed] [Google Scholar]
- [63].Kim E, Sheng M, PDZ domain proteins of synapses, Nat. Rev. Neurosci 5 (2004) 771–781. [DOI] [PubMed] [Google Scholar]
- [64].Boeckers TM, The postsynaptic density, Cell Tissue Res. 326 (2006) 409–422. [DOI] [PubMed] [Google Scholar]
- [65].Föcking M, Lopez LM, English JA, Dicker P, Wolff A, Brindley E, Wynne K, Cagney G, Cotter DR, Proteomic and genomic evidence implicates the postsynaptic density in schizophrenia, Mol. Psychiatry 20 (2015) 424–432. [DOI] [PubMed] [Google Scholar]
- [66].Funk A, Greis K, Meller J, Mccullumsmith R, 32.2 Abnormalities of Synaptic Proteomes in Schizophrenia, (2018) .
- [67].Funk AJ, Mielnik CA, Koene R, Newburn E, Ramsey AJ, Lipska BK, Mccullumsmith RE, Postsynaptic Density-95 isoform abnormalities in schizophrenia, Schizophr. Bull 43 (2017) 891–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Funk AJ, Rumbaugh G, Harotunian V, Mccullumsmith RE, Meador-Woodruff JH, Decreased expression of NMDA receptor-associated proteins in frontal cortex of elderly patients with schizophrenia, Neuroreport 20 (2009) 1019–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Kristiansen LV, Patel SA, Haroutunian V, Meador-Woodruff JH, Expression of the NR2B-NMDA receptor subunit and its Tbr-1/CINAP regulatory proteins in postmortem brain suggest altered receptor processing in schizophrenia, Synapse 64 (2010) 495–502. [DOI] [PubMed] [Google Scholar]
- [70].Meador-Woodruff JH, Clinton SM, Beneyto M, Mccullumsmith RE, Molecular abnormalities of the glutamate synapse in the thalamus in schizophrenia, Ann. N. Y. Acad. Sci 1003 (2003) 75–93. [DOI] [PubMed] [Google Scholar]
- [71].Murakoshi H, Yasuda R, Postsynaptic signaling during plasticity of dendritic spines, Trends Neurosci. 35 (2012) 135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Abel T, Nickl-jockschat T, in: Abel T, Nickl-jockschat T (Eds.), The Neurobiology of Schizophrenia, Academic Press, 2016. [Google Scholar]
- [73].Brennand KJ, Simone A, Jou J, Gelboin-Burkhart C, Tran N, Sangar S, Li Y, Mu Y, Chen G, Yu D, Mccarthy S, Sebat J, Gage FH, Modelling schizophrenia using human induced pluripotent stem cells, Nature 473 (2011) 221–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Robinson MB, The family of sodium-dependent glutamate transporters: a focus on the GLT-1/EAAT2 subtype, Neurochem. Int 33 (1998) 479–491. [DOI] [PubMed] [Google Scholar]
- [75].O’donovan SM, Sullivan CR, Mccullumsmith RE, The role of glutamate transporters in the pathophysiology of neuropsychiatric disorders, NPJ Schizophr. 3 (2017) 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Benediktsson AM, Marrs GS, TU JC, Worley PF, Rothstein JD, Bergles DE, Dailey ME, Neuronal activity regulates glutamate transporter dynamics in developing astrocytes, Glia 60 (2012) 175–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Wong M, Ess KC, Uhlmann EJ, Jansen LA, Li W, Crino PB, Mennerick S, Yamada KA, Gutmann DH, Impaired glial glutamate transport in a mouse tuberous sclerosis epilepsy model, Ann. Neurol 54 (2003) 251–256. [DOI] [PubMed] [Google Scholar]
- [78].Hardingham GE, Bading H, Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders, Nat. Rev. Neurosci 11 (2010) 682–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Tzingounis AV, Wadiche JI, Glutamate transporters: confining runaway excitation by shaping synaptic transmission, Nat. Rev. Neurosci 8 (2007) 935–947. [DOI] [PubMed] [Google Scholar]
- [80].Bauer D, Gupta D, Harotunian V, Meador-woodruff JH, Mccullumsmith RE, Abnormal expression of glutamate transporter and transporter interacting molecules in prefrontal cortex in elderly patients with schizophrenia, Schizophr. Res 104 (2008) 108–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Lauriat TL, Dracheva S, Chin B, Schmeidler J, Mcinnes LA, Haroutunian V, Quantitative analysis of glutamate transporter mRNA expression in prefrontal and primary visual cortex in normal and schizophrenic brain, Neuroscience 137 (2006) 843–851. [DOI] [PubMed] [Google Scholar]
- [82].Shan D, Mount D, Moore S, Haroutunian V, Meador-Woodruff JH, Mccullumsmith RE, Abnormal partitioning of hexokinase 1 suggests disruption of a glutamate transport protein complex in schizophrenia, Schizophr. Res 154 (2014) 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Prabakaran S, Swatton JE, Ryan MM, Huffaker SJ, Huang JT, Griffin JL, Wayland M, Freeman T, Dudbridge F, Lilley KS, Karp NA, Hester S, Tkachev D, Mimmack ML, Yolken RH, Webster MJ, Torrey EF, Bahn S, Mitochondrial dysfunction in schizophrenia: evidence for compromised brain metabolism and oxidative stress, Mol. Psychiatry 9 (684-97) (2004) 643. [DOI] [PubMed] [Google Scholar]
- [84].Steffek AE, Mccullumsmith RE, Haroutunian V, Meador-Woodruff JH, Cortical expression of glial fibrillary acidic protein and glutamine synthetase is decreased in schizophrenia, Schizophr. Res 103 (2008) 71–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Toro CT, Hallak JE, Dunham JS, Deakin JF, Glial fibrillary acidic protein and glutamine synthetase in subregions of prefrontal cortex in schizophrenia and mood disorder, Neurosci. Lett 404 (2006) 276–281. [DOI] [PubMed] [Google Scholar]
- [86].Holtmaat A, Svoboda K, Experience-dependent structural synaptic plasticity in the mammalian brain, Nat. Rev. Neurosci 10 (2009) 647–658. [DOI] [PubMed] [Google Scholar]
- [87].Garey LJ, Ong WY, Patel TS, Kanani M, Davis A, Mortimer AM, Barnes TR, Hirsch SR, Reduced dendritic spine density on cerebral cortical pyramidal neurons in schizophrenia, J Neurol Neurosurg Psychiatry 65 (1998) 446–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Glantz LA, Lewis DA, Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia, Arch. Gen. Psychiatry 57 (2000) 65–73. [DOI] [PubMed] [Google Scholar]
- [89].Konopaske GT, Lange N, Coyle JT, Benes FM, Prefrontal cortical dendritic spine pathology in schizophrenia and bipolar disorder, JAMA Psychiatry 71 (2014) 1323–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Kolluri N, Sun Z, Sampson AR, Lewis DA, Lamina-specific reductions in dendritic spine density in the prefrontal cortex of subjects with schizophrenia, Am. J. Psychiatry 162 (2005) 1200–1202. [DOI] [PubMed] [Google Scholar]
- [91].Amatrudo JM, Weaver CM, Crimins JL, Hof PR, Rosene DL, Luebke JI, Influence of highly distinctive structural properties on the excitability of pyramidal neurons in monkey visual and prefrontal cortices, J. Neurosci 32 (2012) 13644–13660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Gonzalez-Albo MC, Elston GN, Defelipe J, The human temporal cortex: characterization of neurons expressing nitric oxide synthase, neuropeptides and calcium-binding proteins, and their glutamate receptor subunit profiles, Cereb. Cortex 11 (2001) 1170–1181. [DOI] [PubMed] [Google Scholar]
- [93].Pierri JN, Volk CL, Auh S, Sampson A, Lewis DA, Decreased somal size of deep layer 3 pyramidal neurons in the prefrontal cortex of subjects with schizophrenia, Arch. Gen. Psychiatry 58 (2001) 466–473. [DOI] [PubMed] [Google Scholar]
- [94].Rajkowska G, Selemon LD, Goldman-Rakic PS, Neuronal and glial somal size in the prefrontal cortex: a postmortem morphometric study of schizophrenia and Huntington disease, Arch. Gen. Psychiatry 55 (1998) 215–224. [DOI] [PubMed] [Google Scholar]
- [95].Whittington MA, Cunningham MO, Lebeau FE, Racca C, Traub RD, Multiple origins of the cortical gamma rhythm, Dev. Neurobiol 71 (2011) 92–106. [DOI] [PubMed] [Google Scholar]
- [96].Whittington MA, Traub RD, Jefferys JG, Synchronized oscillations in interneuron networks driven by metabotropic glutamate receptor activation, Nature 373 (1995) 612–615. [DOI] [PubMed] [Google Scholar]
- [97].Fish KN, Sweet RA, Lewis DA, Differential distribution of proteins regulating GABA synthesis and reuptake in axon boutons of subpopulations of cortical interneurons, Cereb. Cortex 21 (2011) 2450–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Asada H, Kawamura Y, Maruyama K, Kume H, Ding RG, Kanbara N, Kuzume H, Sanbo M, Yagi T, Obata K, Cleft palate and decreased brain gamma-aminobutyric acid in mice lacking the 67-kDa isoform of glutamic acid decarboxylase, Proc. Natl. Acad. Sci. U.S.A 94 (1997) 6496–6499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Patel AB, De Graaf RA, Martin DL, Battaglioli G, Behar KL, Evidence that GAD65 mediates increased GABA synthesis during intense neuronal activity in vivo, J. Neurochem 97 (2006) 385–396. [DOI] [PubMed] [Google Scholar]
- [100].Erickson JD, De Gois S, Varoqui H, Schafer MK, Weihe E, Activity-dependent regulation of vesicular glutamate and GABA transporters: a means to scale quantal size, Neurochem. Int 48 (2006) 643–649. [DOI] [PubMed] [Google Scholar]
- [101].Hartmann K, Bruehl C, Golovko T, Draguhn A, Fast homeostatic plasticity of inhibition via activity-dependent vesicular filling, PLoS One 3 (2008) e2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Gonzalez-Burgos G, Rotaru DC, Zaitsev AV, Povysheva NV, Lewis DA, GABA transporter GAT1 prevents spillover at proximal and distal GABA synapses onto primate prefrontal cortex neurons, J. Neurophysiol 101 (2009) 533–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Lewis DA, Curley AA, Glausier JR, Volk DW, Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia, Trends Neurosci. 35 (2012) 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Akbarian S, Kim JJ, Potkin SG, Hagman JO, Tafazzoli A, Bunney WE JR., Jones EG, Gene expression for glutamic acid decarboxylase is reduced without loss of neurons in prefrontal cortex of schizophrenics, Arch. Gen. Psychiatry 52 (1995) 258–266. [DOI] [PubMed] [Google Scholar]
- [105].Volk DW, Austin MC, Pierri JN, Sampson AR, Lewis DA, Decreased glutamic acid decarboxylase67 messenger RNA expression in a subset of prefrontal cortical gamma-aminobutyric acid neurons in subjects with schizophrenia, Arch. Gen. Psychiatry 57 (2000) 237–245. [DOI] [PubMed] [Google Scholar]
- [106].Curley AA, Arion D, Volk DW, Asafu-Adjei JK, Sampson AR, Fish KN, Lewis DA, Cortical deficits of glutamic acid decarboxylase 67 expression in schizophrenia: clinical, protein, and cell type-specific features, Am. J. Psychiatry 168 (2011) 921–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Glausier JR, Kimoto S, Fish KN, Lewis DA, Lower glutamic acid decarboxylase 65-kDa isoform messenger RNA and protein levels in the prefrontal cortex in schizoaffective disorder but not schizophrenia, Biol. Psychiatry 77 (2015) 167–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Hoftman GD, Volk DW, Bazmi HH, Li S, Sampson AR, Lewis DA, Altered cortical expression of GABA-related genes in schizophrenia: illness progression vs developmental disturbance, Schizophr. Bull 41 (2015) 180–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Rocco BR, Lewis DA, Fish KN, Markedly lower glutamic acid decarboxylase 67 protein levels in a subset of boutons in schizophrenia, Biol. Psychiatry 79 (2016) 1006–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Volk D, Austin M, Pierri J, Sampson A, Lewis D, GABA transporter-1 mRNA in the prefrontal cortex in schizophrenia: decreased expression in a subset of neurons, Am. J. Psychiatry 158 (2001) 256–265. [DOI] [PubMed] [Google Scholar]
- [111].Dienel SJ, Enwright JF 3RD, Hoftman GD, Lewis DA, Markers of glutamate and GABA neurotransmission in the prefrontal cortex of schizophrenia subjects: disease effects differ across anatomical levels of resolution, Schizophr. Res (2019) in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Chung DW, Volk DW, Arion D, Zhang Y, Sampson AR, Lewis DA, Dysregulated ErbB4 splicing in schizophrenia: selective effects on parvalbumin expression, Am. J. Psychiatry 173 (2016) 60–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Enwright JFI, Huo Z, Arion D, Corradi JP, Tseng G, Lewis DA, Transcriptome alterations of prefrontal cortical parvalbumin neurons in schizophrenia, Mol. Psychiatry 23 (2018) 1606–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Fung SJ, Webster MJ, Sivagnanasundaram S, Duncan C, Elashoff M, Weickert CS, Expression of interneuron markers in the dorsolateral prefrontal cortex of the developing human and in schizophrenia, Am. J. Psychiatry 167 (2010) 1479–1488. [DOI] [PubMed] [Google Scholar]
- [115].Hashimoto T, Volk DW, Eggan SM, Mirnics K, Pierri JN, Sun Z, Sampson AR, Lewis DA, Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia, J. Neurosci 23 (2003) 6315–6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Tsubomoto M, Kawabata R, Zhu X, Minabe Y, Chen K, Lewis DA, Hashimoto T, Expression of transcripts selective for GABA neuron subpopulations across the cortical visuospatial working memory network in the healthy state and schizophrenia, Cereb. Cortex (2018) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Volk DW, Sampson AR, Zhang Y, Edelson JR, Lewis DA, Cortical GABA markers identify a molecular subtype of psychotic and bipolar disorders, Psychol. Med 46 (2016) 2501–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Chung DW, Fish KN, Lewis DA, Pathological basis for deficient excitatory drive to cortical parvalbumin interneurons in schizophrenia, Am. J. Psychiatry 173 (2016) 1131–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Glausier JR, Fish KN, Lewis DA, Altered parvalbumin basket cell inputs in the dorsolateral prefrontal cortex of schizophrenia subjects, Mol. Psychiatry 19 (2014) 30–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Enwright JF, Sanapala S, Foglio A, Berry R, Fish KN, Lewis DA, Reduced labeling of parvalbumin neurons and perineuronal nets in the dorsolateral prefrontal cortex of subjects with schizophrenia, Neuropsychopharmacology 41 (2016) 2206–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Tooney PA, Chahl LA, Neurons expressing calcium-binding proteins in the prefrontal cortex in schizophrenia, Prog. Neuropsychopharmacol. Biol. Psychiatry 28 (2004) 273–278. [DOI] [PubMed] [Google Scholar]
- [122].Woo TU, Miller JL, Lewis DA, Schizophrenia and the parvalbumin-containing class of cortical local circuit neurons, Am. J. Psychiatry 154 (1997) 1013–1015. [DOI] [PubMed] [Google Scholar]
- [123].Beasley CL, Reynolds GP, Parvalbumin-immunoreactive neurons are reduced in the prefrontal cortex of schizophrenics, Schizophr. Res 24 (1997) 349–355. [DOI] [PubMed] [Google Scholar]
- [124].Beasley CL, Zhang ZJ, Patten I, Reynolds GP, Selective deficits in prefrontal cortical GABAergic neurons in schizophrenia defined by the presence of calcium-binding proteins, Biol. Psychiatry 52 (2002) 708–715. [DOI] [PubMed] [Google Scholar]
- [125].Reynolds GP, Zhang ZJ, Beasley CL, Neurochemical correlates of cortical GABAergic deficits in schizophrenia: selective losses of calcium binding protein immunoreactivity, Brain Res. Bull 55 (2001) 579–584. [DOI] [PubMed] [Google Scholar]
- [126].Loup F, Weinmann O, Yonekawa Y, Aguzzi A, Wieser HG, Fritschy JM, A highly sensitive immunofluorescence procedure for analyzing the subcellular distribution of GABAA receptor subunits in the human brain, J. Histochem. Cytochem 46 (1998) 1129–1139. [DOI] [PubMed] [Google Scholar]
- [127].Nusser Z, Sieghart W, Benke D, Fritschy JM, Somogyi P, Differential synaptic localization of two major gamma-aminobutyric acid type A receptor alpha subunits on hippocampal pyramidal cells, Proc. Natl. Acad. Sci. U.S.A 93 (1996) 11939–11944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Hodge RD, Bakken TE, Miller JA, Smith KA, Barkan ER, Graybuck LT, Close JL, Long B, Johansen N, Penn O, Yao Z, Eggermont J, Hollt T, Levi BP, Shehata SI, Aevermann B, Beller A, Bertagnolli D, Brouner K, Casper T, Cobbs C, Dalley R, Dee N, Ding SL, Ellenbogen RG, Fong O, Garren E, Goldy J, Gwinn RP, Hirschstein D, Keene CD, Keshk M, Ko AL, Lathia K, Mahfouz A, Maltzer Z, Mcgraw M, Nguyen TN, Nyhus J, Ojemann JG, OLDRE A, PARRY S, Reynolds S, Rimorin C, Shapovalova NV, Somasundaram S, Szafer A, Thomsen ER, Tieu M, Quon G, Scheuermann RH, Yuste R, SUNKIN SM, Lelieveldt B, Feng D, Ng L, Bernard A, Hawrylycz M, Phillips JW, Tasic B, Zeng H, Jones AR, Koch C, Lein ES, Conserved cell types with divergent features in human versus mouse cortex, Nature 573 (2019) 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Dienel SJ, Lewis DA, Alterations in cortical interneurons and cognitive function in schizophrenia, Neurobiol. Dis 131 (2019) 104208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Mohler H, GABA(A) receptor diversity and pharmacology, Cell Tissue Res. 326 (2006) 505–516. [DOI] [PubMed] [Google Scholar]
- [131].Beneyto M, Abbott A, Hashimoto T, Lewis DA, Lamina-specific alterations in cortical GABAA receptor subunit expression in schizophrenia, Cereb. Cortex 21 (2011) 999–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Fish KN, Rocco BR, Lewis DA, Laminar distribution of subsets of GABAergic axon terminals in human prefrontal cortex, Front. Neuroanat 12 (2018) 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Glausier JR, Bazmi HH, Lewis DA, GABA A alpha 1 subunit mRNA expression in pyramidal cells and interneurons in the dorsolateral prefrontal cortex of schizophrenia subjects, Biol. Psychiatry 67 (2010) 74S. [Google Scholar]
- [134].Pierri JN, Chaudry AS, Woo TU, Lewis DA, Alterations in chandelier neuron axon terminals in the prefrontal cortex of schizophrenic subjects, Am. J. Psychiatry 156 (1999) 1709–1719. [DOI] [PubMed] [Google Scholar]
- [135].Woo TU, Whitehead RE, Melchitzky DS, Lewis DA, A subclass of prefrontal gamma-aminobutyric acid axon terminals are selectively altered in schizophrenia, Proc. Natl. Acad. Sci. U.S.A 95 (1998) 5341–5346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Rocco BR, Dedionisio AM, Lewis DA, Fish KN, Alterations in a unique class of cortical chandelier cell axon cartridges in schizophrenia, Biol. Psychiatry 82 (2017) 40–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Volk DW, Pierri JN, Fritschy JM, Auh S, Sampson AR, Lewis DA, Reciprocal alterations in pre- and postsynaptic inhibitory markers at chandelier cell inputs to pyramidal neurons in schizophrenia, Cereb. Cortex 12 (2002) 1063–1070. [DOI] [PubMed] [Google Scholar]
- [138].Pfeffer CK, Xue M, He M, Huang ZJ, Scanziani M, Inhibition of inhibition in visual cortex: the logic of connections between molecularly distinct interneurons, Nat. Neurosci 16 (2013) 1068–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Daviss SR, Lewis DA, Local circuit neurons of the prefrontal cortex in schizophrenia: selective increase in the density of calbindin-immunoreactive neurons, Psychiatry Res. 59 (1995) 81–96. [DOI] [PubMed] [Google Scholar]
- [140].Horn ME, Nicoll RA, Somatostatin and parvalbumin inhibitory synapses onto hippocampal pyramidal neurons are regulated by distinct mechanisms, Proc Natl Acad Sci U S A 115 (2018) 589–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Melchitzky DS, Lewis DA, Dendritic-targeting GABA neurons in monkey prefrontal cortex: comparison of somatostatin- and calretinin-immunoreactive axon terminals, Synapse 62 (2008) 456–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Gentet LJ, Kremer Y, Taniguchi H, Huang ZJ, Staiger JF, Petersen CC, Unique functional properties of somatostatin-expressing GABAergic neurons in mouse barrel cortex, Nat. Neurosci 15 (2012) 607–612. [DOI] [PubMed] [Google Scholar]
- [143].Xu H, Jeong HY, Tremblay R, Rudy B, Neocortical somatostatin-expressing GABAergic interneurons disinhibit the thalamorecipient layer 4, Neuron 77 (2013) 155–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Hashimoto T, Arion D, Unger T, Maldonado-Aviles JG, Morris HM, Volk DW, Mirnics K, Lewis DA, Alterations in GABA-related transcriptome in the dorsolateral prefrontal cortex of subjects with schizophrenia, Mol. Psychiatry 13 (2008) 147–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Morris HM, Hashimoto T, Lewis DA, Alterations in somatostatin mRNA expression in the dorsolateral prefrontal cortex of subjects with schizophrenia or schizoaffective disorder, Cereb. Cortex 18 (2008) 1575–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Thune JJ, Uylings HBM, Pakkenberg B, No deficit in total number of neurons in the prefrontal cortex in schizophrenics, J. Psychiatr. Res 35 (2001) 15–21. [DOI] [PubMed] [Google Scholar]
- [147].Lewis DA, Cho RY, Carter CS, Eklund K, Forster S, Kelly MA, Montrose D, Subunit-selective modulation of GABA type A receptor neurotransmission and cognition in schizophrenia, Am. J. Psychiatry 165 (2008) 1585–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Buchanan RW, Keefe RS, Lieberman JA, Barch DM, Csernansky JG, Goff DC, Gold JM, Green MF, Jarskog LF, Javitt DC, Kimhy D, Kraus MS, Mcevoy JP, Mesholam-Gately RI, Seidman LJ, Ball MP, Mcmahon RP, Kern RS, Robinson J, Marder SR, A randomized clinical trial of MK-0777 for the treatment of cognitive impairments in people with schizophrenia, Biol. Psychiatry 69 (2011) 442–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Lewis DA, Pharmacological enhancement of cognition in individuals with schizophrenia, Biol. Psychiatry 69 (2011) 397–398. [DOI] [PubMed] [Google Scholar]
- [150].Featherstone RE, Mcmullen MF, Ward KR, Bang J, Xiao J, Siegel SJ, EEG biomarkers of target engagement, therapeutic effect, and disease process, Ann. N. Y. Acad. Sci 1344 (2015) 12–26. [DOI] [PubMed] [Google Scholar]
- [151].Egerton A, Modinos G, Ferrera D, Mcguire P, Neuroimaging studies of GABA in schizophrenia: a systematic review with meta-analysis, Transl. Psychiatry 7 (2017) e1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Merritt K, Egerton A, Kempton MJ, Taylor MJ, Mcguire PK, Nature of glutamate alterations in schizophrenia: a meta-analysis of proton magnetic resonance spectroscopy studies, JAMA Psychiatry 73 (2016) 665. [DOI] [PubMed] [Google Scholar]
- [153].Marsman A, Mandl RC, Klomp DW, Bohlken MM, Boer VO, Andreychenko A, Cahn W, Kahn RS, Luijten PR, Hulshoff Pol HF, GABA and glutamate in schizophrenia: a 7T (1)H-MRS study, Neuroimage Clin. 6 (2014) 398–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Thakkar KN, Rosler L, Wijnen JP, Boer VO, Klomp DW, Cahn W, Kahn RS, Neggers SF, 7T proton magnetic resonance spectroscopy of gamma-aminobutyric acid, glutamate, and glutamine reveals altered concentrations in patients with schizophrenia and healthy siblings, Biol. Psychiatry 81 (2017) 525–535. [DOI] [PubMed] [Google Scholar]
- [155].Frankle WG, Cho RY, Prasad KM, Mason NS, Paris J, Himes ML, Walker C, Lewis DA, Narendran R, In vivo measurement of GABA transmission in healthy subjects and schizophrenia patients, Am. J. Psychiatry 172 (2015) 1148–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Weinberger DR, Berman KF, Zec RF, Physiologic dysfunction of dorsolateral prefrontal cortex in schizophrenia. I. Regional cerebral blood flow evidence, Arch. Gen. Psychiatry 43 (1986) 114–124. [DOI] [PubMed] [Google Scholar]
- [157].Finn ES, Huber L, Jangraw DC, Molfese PJ, Bandettini PA, Layer-dependent activity in human prefrontal cortex during working memory, Nat. Neurosci 22 (2019) 1687–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]