

Abstract
The lumen-dwelling protozoan, Giardia, is an important parasitic cause of diarrheal disease worldwide. Infection can persist over extended periods with minimal intestinal inflammation, suggesting that Giardia may attenuate host responses to ensure its survival, although clearance eventually occurs in most cases. IL-10 is an anti-inflammatory regulator critical for intestinal homeostasis and controlling host responses to bacterial exposure, yet its potential role in coordinating antiprotozoal host defense in the intestine is not known. Here, we found that murine infection with the natural enteric pathogen, Giardia muris, induced a transient IL-10 response after 2–4 weeks at the primary site of infection in the upper small intestine, but parasite colonization and eradication were not affected by the absence of the cytokine in gene-targeted mice. However, IL-10 was critical for controlling infection-associated immunological sequelae in the colon, because severe and persistent diarrhea and colitis were observed in IL-10 deficient mice within 1–2 weeks after infection but not in uninfected littermate controls. Inflammation was characterized by epithelial hyperplasia, neutrophil and macrophage expansion, and Th1 induction, and could be prevented by blockade of IL-12/IL-23 p40 but not depletion of CD11c dendritic cells. Furthermore, the intestinal microbiota underwent characteristic shifts in composition and was required for disease, since antibiotics and loss of TLR signaling in MyD88-deficient mice protected against colitis. Together, our data suggest that transient infection by a luminal and seemingly non-inflammatory pathogen can trigger sustained colitis in genetically susceptible hosts, which has broader implications for understanding post-infectious syndromes and other chronic intestinal inflammatory conditions.
INTRODUCTION
IL-10 is a critical immune regulator that is secreted by hematopoietic cells, particularly macrophages and T cells, as well as non-hematopoietic cells. The cytokine acts on both myeloid and T cells to stimulate anti-inflammatory factors and suppress pro-inflammatory mediators (1). The importance of IL-10 in regulating homeostasis was first demonstrated by the observation that IL-10-deficient (Il10−/−) mice, which are phenotypically normal after birth and weaning, develop spontaneous, progressive enterocolitis upon aging (2–5). Colitis in this model is driven by commensal bacteria, as inflammation is not observed under germ-free conditions (4), and antibiotic treatment can prevent and attenuate disease (6, 7). Similarly, mice with impaired IL-10R signaling develop severe spontaneous colitis, driven by expansion of Th17 cells normally restricted by regulatory T cells and dendritic cells (1, 8, 9). Consistent with the findings in murine models, genome-wide association studies of inflammatory bowel disease (IBD) patients have identified IL-10 and IL-10R polymorphisms that are associated with severe, early-onset forms of the disease (1, 10). Despite its importance in immune regulation and association with IBD, trials of IL-10 treatment did not reveal clinical benefits, perhaps because disease had progressed beyond IL-10 dependent stages (1).
While loss of IL-10 signaling can cause excessive immune and inflammatory responses to commensals, its impact on host interactions with pathogens is more complex. Thus, on the one hand, IL-10 deficiency can promote clearance of bacterial pathogens, such as Salmonella, Klebsiella pneumoniae, Listeria monocytogenes, and Chlamydia trachomatis, as well as certain viruses, parasites, and fungi, by induction of heightened Th1 responses, thereby improving host survival (11–14). On the other hand, enhanced Th1 responses can lead to exacerbation of inflammation after infection. For instance, intestinal colonization of Il10−/− mice with the enteric pathogens Helicobacter hepaticus or Campylobacter jejuni causes chronic inflammatory lesions in the cecum and colon (15, 16). Inflammation under these conditions is not only caused by excessive Th1 responses, but is also related to blooms of distinct families of intestinal commensals that correlate with increased susceptibility to colitis (17, 18).
Intestinal infection with the common protozoan parasite Giardia leads to diarrhea and abdominal pain, and engages adaptive immunity, but causes only minimal mucosal inflammation (19). The parasite colonizes the small intestine, but does not invade the mucosa, indicating that it can impact host responses from an “off-shore” location in the lumen (20). Infection elicits both Th1 and Th17 responses at the site of infection, yet only IL-17 producing cells are required for parasite elimination (21, 22). Importantly, this effector response does not appear to be associated with significant inflammation, raising the possibility that any potential pro-inflammatory responses that are typically associated with microbial infections may be attenuated by effective suppression during Giardia infection.
Given the importance of IL-10 in regulating intestinal inflammation and immunity, we set out to test the hypothesis that IL-10 is essential for dampening effector T cell responses induced by Giardia infection. Unexpectedly, we found that IL-10 is not critical for limiting T cell responses at the site of infection in the small intestine, but is required later for controlling excessive immune responses to the microbiota in the colon. The results suggest that a seemingly innocuous pathogen has the potential to trigger severe colitis in genetically susceptible hosts, which has broader conceptual implications for understanding the onset of IBD in humans.
MATERIALS AND METHODS
Mice and infection protocol.
Wild-type, Il10−/− (5), Ccr7−/− (23), and Tcra−/− (24) mice on a C57BL/6 background were obtained from The Jackson Laboratory, and bred and maintained under specific pathogen-free conditions. To generate double mutant mice, Il10−/− mice were crossed with either Myd88−/− (25) or Cd11c-diphtheria toxin receptor (DTR) transgenic mice (26). Littermates served as controls. At 7–9 weeks of age, mice were infected by oral gavage with G. muris cysts (104 cysts/mouse) prepared from infected wild-type mice as previously described (27). Parasite burden in infected mice was determined by enumerating trophozoites in the small intestine (27). All mice were monitored daily for weight loss, stool consistency, and rectal bleeding. No significant differences were observed in the course of infection or disease severity between males and females, so results were combined and are reported together. All animal studies were approved by the Institutional Animal Care and Use Committees of the University of California, San Diego and the University of Texas Medical Branch.
Histological analysis.
Jejunum, ileum and colon were removed, open longitudinally and processed as Swiss rolls before overnight fixation in either Bouin’s solution or 10% formalin. Fixed tissues were embedded in paraffin, and 5 μm-sections were prepared and stained with hematoxylin and eosin. Histological scores (range 0–20) were obtained in a blinded fashion by assessing the following parameters: a. Mucosal architecture (0, normal; 1, focally abnormal; 2, diffusely abnormal; 3, severely abnormal); b. Inflammatory cell infiltration of mucosa (0, normal; 1, mild infiltration; 2, moderate infiltration; 3, severe infiltration); c. Epithelial hyperplasia (0, normal; 1, mild; 2, moderate; 3, extensive); d. Epithelial erosions and ulcerations (0, absent; 1, present); e. Crypt abscesses (0, absent; 1, present); f. Goblet cells (0, present; 1, absent); g. Muscle thickening (0, normal; 1, mild; 2, moderate; 3, severe); and h. Extent of damage and inflammation (0, normal; 1, damage present in <25% of the tissue; 2, damage in 25–75%; 3, damage in >75%). Cell numbers in the lamina propria were quantified per 40x visual field in an area equivalent to 0.1 mm2. Crypt depth was measured using NIS-Elements software (Nikon). For each colon section, quantification was performed on the two most affected areas at least 10 crypts apart, and the scores were averaged.
Analysis of mRNA expression.
Full-thickness pieces of jejunum, ileum, and colon were treated with RNAlater (Ambion) at 4°C for 24 h, snap-frozen, and stored at −80°C until use. Total RNA was isolated with TRIzol reagent (Thermo Fisher), and analyzed individually for each tissue by RT-PCR (21). Briefly, RNA was reverse-transcribed into cDNA using the High Capacity cDNA Reverse Transciption kit (Applied Biosystems) and quantitative RT-PCR analysis (qRT-PCR) was done with Mesa Green 2 x SYBR mix (Eurogentec) in a real-time PCR machine. Primers and expected PCR product size were as follows: IL-10, 5’-GCT CTT ACT GAC TGG CAT GAG-3’ (sense), 5’-CGC AGC TCT AGG AGC ATG TG-3’ (antisense), 108 bp; IL-17A, 5’-ACC AGC TGA TCA GGA CGC GC-3’ (sense), 5’-CCA GGC TCA GCA GCA ACA-3’ (antisense), 84 bp; IFN-γ, 5’-TCA AGT GGC ATA GAT GTG GAA GAA-3’ (sense), 5’-TGG CTC TGC AGG ATT TTC ATG-3’ (antisense), 84 bp; IL-4, 5’-AGA AGG GAC GCC ATG CAC GG-3’ (sense), 5’-ATG CGA AGC ACC TTG GAA GCC C-3’ (antisense), 105 bp; IL-9, 5’-CAG CTG ACC AAT GCC ACA CAG AAA-3’ (sense), 5’-GAA ATG ACA GTG TGT TGC CTG CCA-3’ (antisense), 151 bp; IL-12 p40, 5’-CGG ACG GTT CAC GTG CTC ATG G-3’ (sense), 5’-TCC ACA TGT CAC TGC CCG AGA GT-3’ (antisense), 103 bp; CCL2, 5’-GCT CAG CCA GAT GCA GTT AAC GC-3’ (sense), 5’-TGG GGT CAG CAC AGA CCT CTC T-3’ (antisense), 169 bp; CCL3, 5’-GGT CTC CAC CAC TGC CCT TGC-3’ (sense), 5’-AGG CAG TCG GGG TGT CAG CT-3’ (antisense), 95 bp; CXCL5, 5’-GCA TTT CTG TTG CTG TTC ACG CTG-3’ (sense), 5’-TGC AGG GAT CAC CTC CAA ATT AGC-3’ (antisense), 150 bp; MPO, 5’-CAG CGA GGA CCC CCT AGC CA-3’ (sense), 5’-GGC ATC TCG CTG GAG CGC AT-3’ (antisense), 198 bp; GAPDH, 5’-TGT GAT GGG TGT GAA CCA CGA CAA-3’ (sense), 5’-AGT GAT GGC ATG GAC TGT GGT CAT-3’ (antisense), 209 bp. Relative changes in target mRNA levels were calculated by the 2ΔΔCt method, with GAPDH mRNA as the reference standard.
Isolation and analysis of immune cells.
Single-cell suspensions were prepared from mesenteric lymph nodes and intestinal tissues by collagenase digestion as previously described (21). After removing the epithelial layer with EDTA/DTT, tissues were digested in RMPI 1640 medium containing 1 mg/ml collagenase D (Roche Applied Science) and 100 μg/ml DNase I (Worthington Biochemical). Liberated cells were collected and pooled. To assess changes in dendritic cells and macrophages, cells were stained with Alexa Fluor 700-conjugated rat anti-mouse MHC II (clone M5/114.15.2, eBioscience), Horizon V450-conjugated Armenian hamster anti-mouse CD11c (clone HL3, BD Bioscience), Texas Red-conjugated rat anti-mouse CD11b (clone M1/70, Thermo Fisher), PE-Cy7-conjugated rat anti-mouse B220 (clone RA3–6B2, eBioscience), and FITC-conjugated Armenian hamster anti-mouse CD103 (clone 2E7, eBioscience). Viability was assessed by staining with Fixable Aqua Dead Cell Stain (Thermo Fisher). For functional evaluation of T cell subsets, cells were stimulated for 6 h with 50 ng/ml phorbol myristate acetate and 750 ng/ml ionomycin in complete RPMI 1640 medium at 37°C. After an additional 2 h of incubation with GolgiStop (BD Biosciences), stimulated cells were washed and stained with PE-Cy5-conjugated rat anti-mouse CD4 (clone RM4–5, eBioscience). Following fixation, cells were permeabilized and stained with FITC-conjugated rat anti-mouse IFN-γ (clone XMG1.2, eBioscience) and PE-conjugated rat anti-mouse IL-17A (clone eBio17B7, eBioscience). Cells were analyzed on a FACSCalibur flow cytometer (BD Biosciences) followed by data analysis with FlowJo software (TreeStar Inc.).
Luminex immunoassays.
For quantitation of tissue CCL2 and CCL3 levels, colon tissue was harvested, snap-frozen, and stored at −80°C until use. The tissue was weighed and homogenized in PBS containing 1% Triton X-100 and Complete Protease Inhibitor (Roche). Following centrifugation, supernatants were collected and assayed by Luminex™ xMAP technology (Thermo Fisher) according to the manufacturer’s instructions. Results were normalized to total protein as determined by BCA assay (Biorad).
Generation of bone marrow chimeras and DC depletion.
To generate bone marrow chimeric mice, Il10−/− recipients were exposed to a single lethal dose of 9 Gy total body irradiation from a caesium-137 source. Within 6–8 hours after irradiation, mice were given 4–6 × 106 bone marrow cells from sex-matched Il10−/− x Cd11c-DTR mice via tail vein injection, and were rested for 8–10 weeks to allow reconstitution. To deplete dendritic cells, Cd11c-DTR mice were treated with 4 ng/g diphtheria toxin i.p. (Sigma-Aldrich) 24 h prior to infection and every 48 h thereafter. Cell depletion was confirmed by FACS.
Neutralization of IL-12 p40.
Anti-IL-12 p40 (clone C17.8, rat IgG2a) and its isotype control were purchased from Bio x Cell (West Lebanon, NH). Mice were given 1 mg of antibody subcutaneously 2 days prior to infection and every 7 days for up to 3 weeks.
16S rRNA sequencing and analysis.
For metagenomic studies, total DNA was extracted from unwashed colon tissue using MO BIO PowerSoil DNA Isolation Kit (MO BIO Laboratories). The V4 region of the bacterial 16S rRNA gene was amplified and sequenced in the MiSeq platform (Illumina) using a 2×250 bp paired-end protocol. The primers used for amplification contained adapters for MiSeq sequencing and single-index barcodes, allowing the PCR products to be pooled and sequenced directly (28). 16Sv4 rDNA sequences were clustered into Operational Taxonomic Units (OTUs) at a similarity cutoff value of 97% using the UPARSE algorithm (29), and the OTUs subsequently mapped using the SILVA Database (30). Abundances were recovered by mapping the demultiplexed reads to the UPARSE OTUs. Analysis and visualization of the microbial composition data were conducted in R. Significance was determined by non-parametric tests that included the Mann-Whitney test and Kruskal-Wallis test. Correlations were determined with R’s base “lm” function for linear regression models, where p-values indicate the probability that the slope of the regression line is zero. The Monte Carlo permutation test was used to estimate p-values for PCoA plots. All p-values were adjusted for multiple comparisons with the FDR algorithm (31).
Antibiotic treatment.
For modulation of the intestinal microbiota, an antibiotic cocktail of 500 mg/l vancomycin hydrochloride, 500 mg/l imipenem, and 1,000 mg/l neomycin was added to the drinking water every 3 days for 2–3 weeks.
Statistical analysis.
Statistical analysis was performed using SigmaPlot12 (Systat Software Inc.). To compare parasite loads, trophozoite numbers were log10 transformed, and mean and SEM were calculated. Samples without detectable trophozoites were assigned a log value of 3, which represents half of the detection limit of the assay. Differences in trophozoite numbers were evaluated by Wilcoxon rank-sum test, survival data were examined by Kaplan-Meier analysis, and all other comparisons between groups were evaluated by Student’s t test. Differences with a p-value of <0.05 were considered significant.
RESULTS
Giardia infection triggers colonic inflammation in IL-10 deficient mice
Giardia infection induces IL-17A and IFN-γ expressing CD4+ T cells (21, 32), although only the former are important for parasite control (21, 22). Despite the pro-inflammatory potential of these effectors, wild-type mice and humans generally exhibit little mucosal inflammation in the small intestine after acute infection (19). To determine whether IL-10 may be involved in this outcome, we first examined expression of the cytokine during the course of G. muris infection in adult C57BL/6 mice. At different times after infection, total RNA was extracted from the jejunum (the primary site of infection), and assayed for IL-10 mRNA by qRT-PCR. IL-10 expression was significantly increased between 2–4 weeks, with a maximum >60-fold induction after three weeks (Fig. 1A), a time that coincides with a steep decrease in trophozoite burden in wild-type mice (Fig. 1B) and a reduction of elevated IL-17A expression to pre-infection levels (21).
FIG. 1.
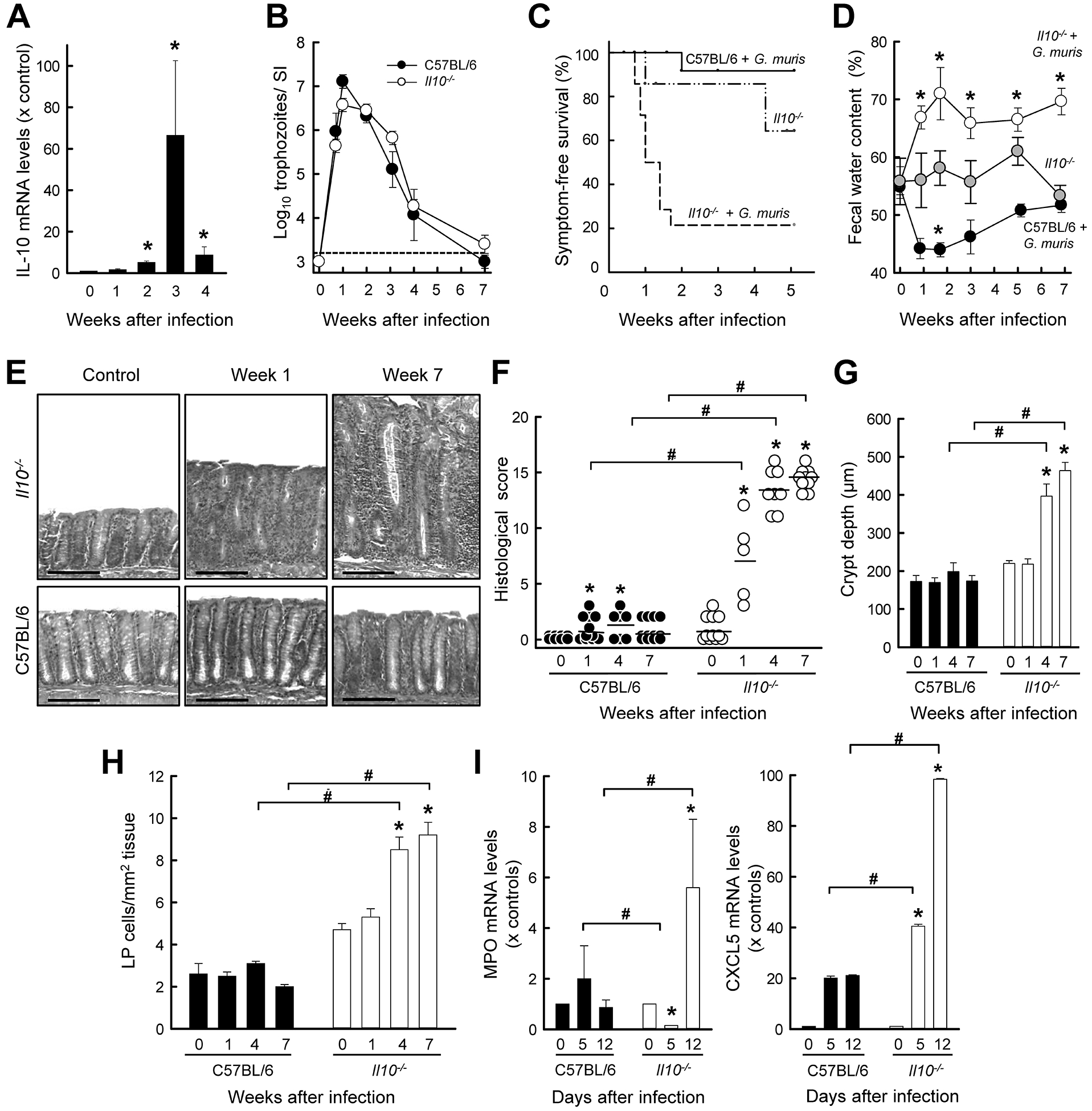
G. muris infection triggers colitis in IL-10−/− mice. (A) Wild-type (C57BL/6) mice were orally infected with 104 G. muris cysts, or left uninfected as controls (Week 0). At the indicated times, RNA was extracted from the small intestine, pooled from 3–5 mice, and analyzed for IL-10 mRNA expression by qRT-PCR. Expression levels are shown relative to uninfected controls (mean ± SE, n=3 experiments, *p<0.05 vs. week 0). (B-I) Il10−/− mice (open circles and bars, dashed line) and wild-type mice (solid circles, bars and lines) were infected with G. muris. Uninfected Il10−/− littermate mice (gray circles, dash-dotted line) served as controls. Trophozoite numbers in the small intestine were assessed at the indicated times (B; mean ± SE; n≥5 mice/time point). The horizontal dashed line depicts the detection limit of the assay. Time after infection until the development of significant clinical symptoms (>20% body weight loss or frank diarrhea) was recorded as symptom-free survival (C; n≥9 mice/group, *p<0.05 vs. uninfected Il10−/− littermate mice). Fecal water content of freshly collected stool was determined as a measure of diarrhea (D; mean ± SEM; n≥5 mice/time point; *p<0.05 vs. uninfected mice of the same genotype). (E-H) Histological analysis of the colon from infected mice and uninfected (Week 0) littermate controls. Representative H&E-stained sections are shown in panel E (size bar, 100 μm), histological damage scores were determined (F; each data point represents one animal, means are shown as horizontal lines, *p<0.05), and crypt depths (G) and density of lamina propria cells (H) were quantified morphometrically (means ± SEM, n≥10 mice/group; *p<0.05 vs. Week 0 of the same genotype; #p<0.05 vs. wild-type mice at the same time). (I) Myeloperoxidase (MPO) and CXCL5 mRNA expression was determined by qRT-PCR analysis in total RNA from whole colon (mean ± SE, n≥5 mice/time; *p<0.05 vs. Week 0 of the same genotype, #p<0.05 vs. wild-type mice at the same time).
To determine the importance of IL-10 in the host response to Giardia infection, we compared the time course of G. muris infection in IL-10-deficient (Il10−/−) mice with wild-type controls. In both groups, peak trophozoite numbers were observed in the small intestine by one week, followed by gradual decline over the ensuing six weeks, and clearance by seven weeks (Fig. 1B). No significant difference was observed between the groups at any time after infection, indicating that IL-10 was not required for antigiardial host defense. However, 80% of the infected Il10−/− mice, but very few (<10%) of the infected wild-type controls, developed significant clinical symptoms, including diarrhea, lethargy, and weight loss within two weeks (Fig. 1C). Induction of symptoms was largely dependent on infection, because only a minority (28%) of uninfected Il10−/− littermate controls spontaneously developed any symptoms during the same follow-up period. Furthermore, marked diarrhea, as measured by a significant increase in fecal water content, was evident as early as one week after infection of Il10−/− animals and persisted until the end of the 7 week experiment, but was not observed at any time in parallel cohorts of uninfected Il10−/− or infected wild-type mice (Fig. 1D).
For clues about the underlying pathophysiology of the clinical symptoms, we examined the histology of the intestinal tract after infection. Il10−/− mice, but not the wild-type controls, displayed modest mucosal inflammation in the small intestine one week after G. muris infection (Supplemental Fig. 1), but the changes were transient, and thus not consistent with the observed symptom persistence. Surprisingly, the colon, which is not a major site of Giardia infection, showed marked and progressive inflammation in Il10−/− mice after infection, while infected wild-type mice had no signs of inflammation (Fig. 1E,F). Inflammatory changes occurred in the knock-out mice within one week after infection and showed steady progression for 7 weeks. Inflammation was characterized by epithelial hyperplasia, lamina propria infiltration with leukocytes, crypt abscesses, and submucosal edema (Fig. 1E–G). Quantitative histological analysis confirmed a significant increase in lamina propria leukocytes by 4–7 weeks after infection (Fig. 1H), while increased neutrophil recruitment, as determined by tissue myeloperoxidase levels, was already apparent by 12 days (Fig. 1I, left). Preceding this recruitment was a strong increase in the major neutrophil chemokine, CXCL5, in the colon (Fig. 1I, right). In contrast, wild-type controls showed no colonic neutrophil recruitment and only modest CXCL5 induction after infection. These pathological features were similar to the spontaneous colitis known to develop in Il10−/− mice with increasing age (5) and confirmed in our animal facility in aged (>6 months) Il10−/− mice with spontaneous colitis (Supplemental Fig. 2).
To rule of the possibility that any bystander microbes in the G. muris cyst preparations may be responsible for disease, we treated the preparations with broad-spectrum antibiotics in vitro prior to infection, but did not observe significant differences in the course of infection or disease outcome. We also isolated and prepared cysts from in vivo antibiotic-treated animals, and again saw no impact on G. muris infection or colitis induction. Together, these results suggest that in young adult Il10−/− mice, Giardia infection triggers and exacerbates colitis with 100% penetrance, which would have developed spontaneously in only a small subset of these mice in a delayed fashion with age.
TCRα deficiency renders mice susceptible to Giardia-induced colitis
Intrigued that Giardia, an enteric pathogen which typically causes only minor and transient enteritis, triggered severe persistent colitis in Il10−/− mice, we next sought to determine whether this observation was unique to that particular colitis-prone model. Mice lacking the T-cell receptor α (Tcrα−/−) develop aging-related spontaneous inflammation that resembles human ulcerative colitis (24), so we investigated the impact of Giardia infection in these mice at a young, pre-disease age. Parasite levels were similar in these mice and wild-type controls one week after infection, but Tcrα−/− mice failed to clear infection thereafter, with ~10,000-fold higher trophozoite numbers at week 7 (Fig. 2A), which confirms that intact T cell immunity is required for Giardia eradication (32). Similar to what was observed in Il10−/− animals, fecal water content was significantly increased after one week (62.1 ± 3.5% on week 1 vs. 50.0 ± 2.3% pre-infection; p=0.01) and steadily rose thereafter (78.4 ± 3.5% by week 7). No increase in fecal water was found in uninfected littermate controls. Histological examination of infected Tcrα−/− mice revealed significant colon inflammation within one week and steady progression to severe colitis, characterized by extensive thickening of the mucosa, striking crypt hyperplasia, marked leukocyte infiltration in the lamia propria and numerous crypt abscesses, over the 7-week course of infection (Fig. 2B–D). As for Il10−/− mice, histological disease penetrance was 100% in Tcrα−/− mice at week 7 (Fig. 2C). These data show that Giardia-triggered colitis can occur with different underlying susceptibility mechanisms of inflammation.
FIG. 2.
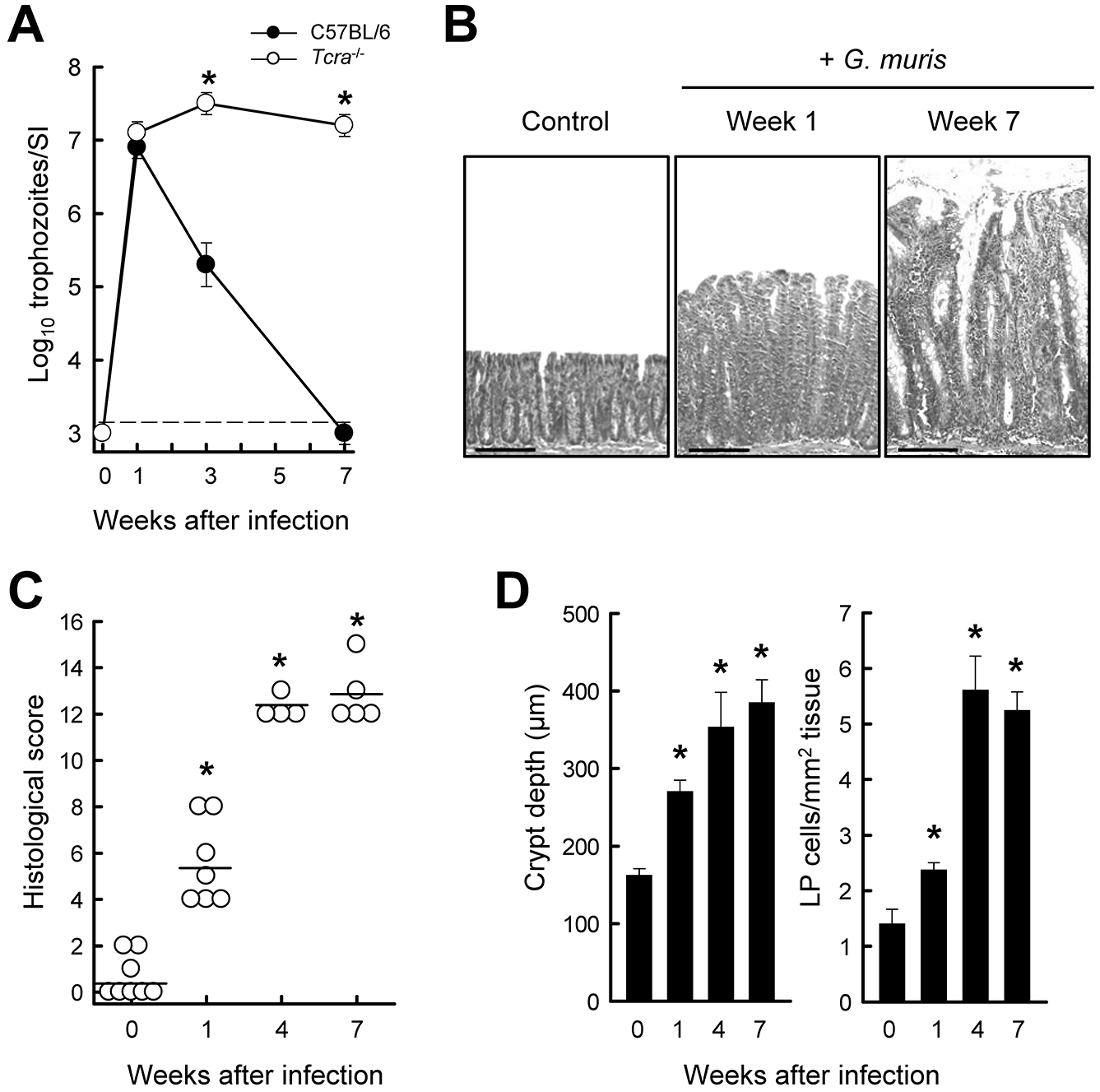
Susceptibility of Tcrα−/− mice to G. muris-induced colitis. Healthy age-matched (7- to 8-week-old) littermates of Tcrα−/− (open circles) and wild-type (solid circles) mice were infected with G. muris or left uninfected as controls, and followed for 7 weeks. (A) Trophozoite numbers in the small intestine were determined at the indicated times (mean ± SE; n≥3 mice/time point; *p<0.05 vs. wild-type at the same time). (B-D) Histological analysis of the colon in Tcrα−/− mice. Representative H&E-stained sections are shown in panel B (scale bars, 100 μm). Histological scores of colon damage and inflammation were determined (C; each data point represents one animal, horizontal lines represent mean values for each group; *p<0.05 vs. uninfected mice). Crypt depths and cell density in the lamina propria (LP) were quantified morphometrically (D; means ± SE, n≥4 mice/group; *p< 0.05 vs. uninfected mice).
Role of adaptive immune cells and IL-12/IL-23 p40 in Giardia-triggered colitis
Both Il10−/− and Tcrα−/− mice developed colitis after Giardia infection, yet the latter lack normal T cells, raising the question whether T cells play any role in the colitis observed in the absence of IL-10. To address this, we generated double knock-out mice for IL-10 and Rag2 (Il10−/− x Rag2−/−), and challenged them with G. muris. As expected, the double knock-out mice and Rag2−/− single knock-out controls were unable to clear infection (Fig. 3A), confirming that adaptive immune cells are required for effective host defense against Giardia. The double knock-out mice had a significant reduction in the severity of Giardia-triggered colitis relative to Il10−/− mice, although they still exhibited modest inflammation (Fig. 3B). In contrast, Rag2−/− mice, like wild-type mice, had no signs of colon inflammation after infection (Fig. 3B). These findings show that adaptive immune cells, most likely T cells, are major mediators of colitis after infection, but also suggest that innate immune cells contribute to inflammation.
FIG. 3.
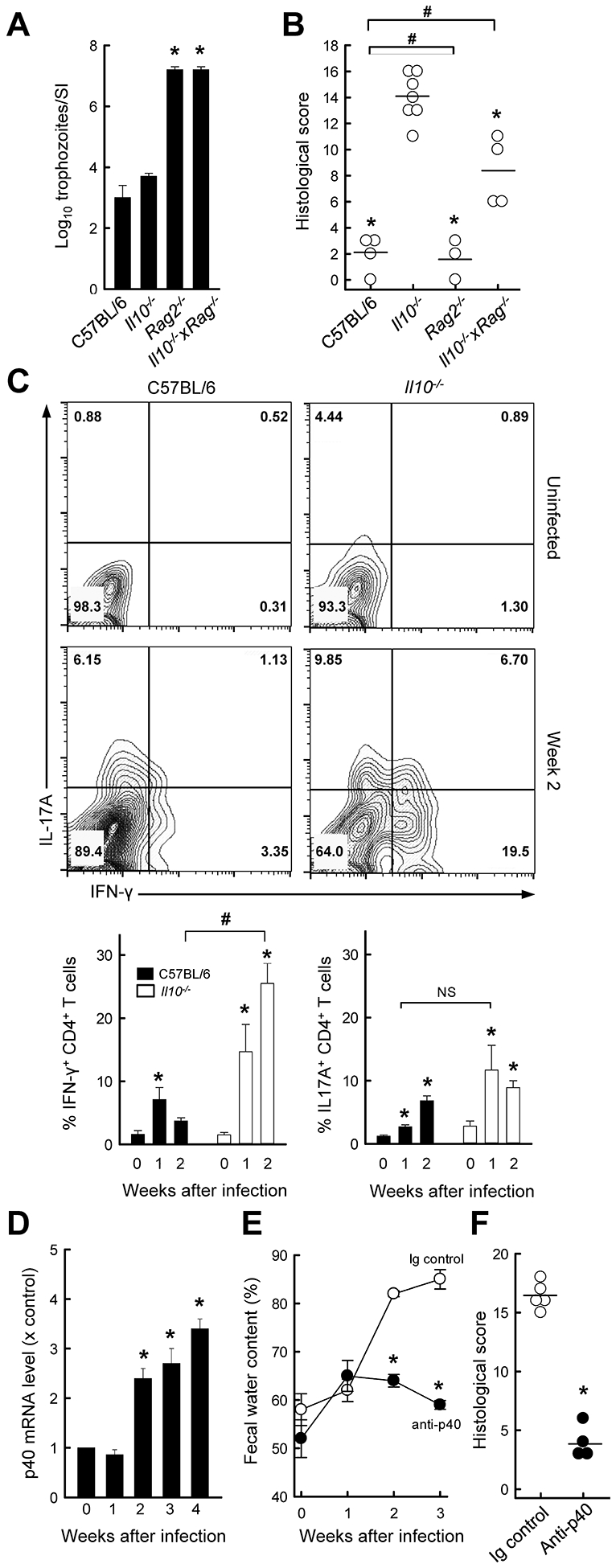
IL-12 p40 promotes colitis development after Giardia infection in IL-10−/− mice. (A, B) Il10−/−, Rag−/−, Il10−/− x Rag−/−, and C57BL/6 control mice were infected with G. muris. Three weeks after infection, parasites were enumerated in the small intestine (A), and histological analysis of the colon was done (B; each data point represent one animal, horizontal lines show mean values for each group; *p<0.05 vs IL-10−/− mice, #p<0.05 vs wild-type mice). (C) IL-10−/− mice (open bars) and wild-type C57BL/6 mice (solid bars) were infected with G. muris or left uninfected as controls (Week 0), and single cells were isolated from the colon lamina propria and analyzed by flow cytometry for IFN-γ and IL-17A expressing CD4+ T cells. Representative FACS contour plots are shown (top panel), and percentages of Th1 and Th17 cells are given (bottom panel; means ± SE, n≥7 mice/time; *p<0.05 vs. age-matched uninfected mice; #p<0.05 vs. wild-type mice at the same time; NS, not significant). (D) Quantitative RT-PCR analysis of IL-12 p40 mRNA in the colon of Il10−/− mice infected with G. muris (means ± SE, n≥7 mice/time; *p<0.05 vs. uninfected controls). (E, F) IL-10−/− mice were treated with anti-p40 or an isotype control IgG (Ig), and infected with G. muris. Fecal water content was assessed at the indicated times (E). Histological damage scores were determined three weeks after infection (F; each data point represents one animal, horizontal lines shown means for each group; *p<0.05 vs. Ig control).
To characterize the involvement of T cells in Giardia-triggered colitis in more detail, we examined T effector subsets by flow cytometry. The frequency of both Th1 and Th17 cells increased in the colon of Il10−/− mice after 1–2 weeks (Fig. 3C). Similarly, infected wild-type mice showed an increase of both Th subsets in the colon, although the increase in Th1 cells was significantly lower compared to Il10−/− mice. A differential Th1 response was also observed in the small intestine, with the increase in Th1 cells peaking at one week after infection in Il10−/− mice (12.3 ± 0.9 in Il10−/− mice vs. 1.2 ± 0.4% in wild-type mice; p<0.001).
Induction of a transient Th1 response in the small intestine and a more persistent response in the colon suggested that Th1 cells may contribute to Giardia-induced colitis. IL-12/IL-23 p40 is a key cytokine involved in inducing and sustaining effector Th1 cells (33). Consistent with Th1 involvement, we found that IL-23 p40 mRNA expression was significantly increased in Il10−/− mice after 2–4 weeks (Fig. 3D). To determine if IL-12/IL-23 p40 was important in mediating the host response to infection, we treated G. muris-infected Il10−/− mice with anti-p40 neutralizing or control antibodies. Anti-p40 significantly attenuated the development of diarrhea (Fig. 3E) and colitis (Fig. 3F) three weeks after infection compared with control antibodies, whereas it had no significant impact on host defense against the parasite (log10 trophozoites = 5.8 ± 0.3 in Il10−/− mice treated with anti-p40 vs. 6.1 ± 0.3 in untreated controls; p=0.7). These results indicate that colonic Th1 responses are unchecked upon Giardia infection in the absence of IL-10, and that IL-12/IL-23 p40 is major mediator of the ensuing colitis.
Dendritic cells are not required for Giardia-triggered colitis
Development of T effector cells is controlled to a large extent by dendritic cells, so we questioned whether these cells played a role in colitis after Giardia infection of susceptible mice. Characterization of dendritic cell numbers by flow cytometry revealed only minimal changes in the numbers of CD11c+ MHCII+ B220− dendritic cells in the colon, small intestine, and mesenteric lymph nodes (Fig. 4A), suggesting that dendritic cells were not particularly impacted by Giardia infection or the infection-induced inflammation. To directly determine dendritic cell functions in the model, we crossed Il10−/− mice to mice expressing the diphtheria toxin receptor under the control of the dendritic cell-specific Cd11c promoter. Administration of diphtheria toxin resulted in effective CD11c+ cell deletion (>99% in the spleen and MLN). Depletion of CD11c+ dendritic cells impaired parasite clearance, as shown by 100-fold higher trophozoite numbers at week 3 (Fig. 4B), indicating that the cells played a role in mounting an effective host defense against the parasite. However, depletion had no significant impact on the development or severity of colitis induced by G. muris infection (Fig. 4B), indicating that colitis was not controlled by CD11c+ dendritic cells.
FIG. 4.
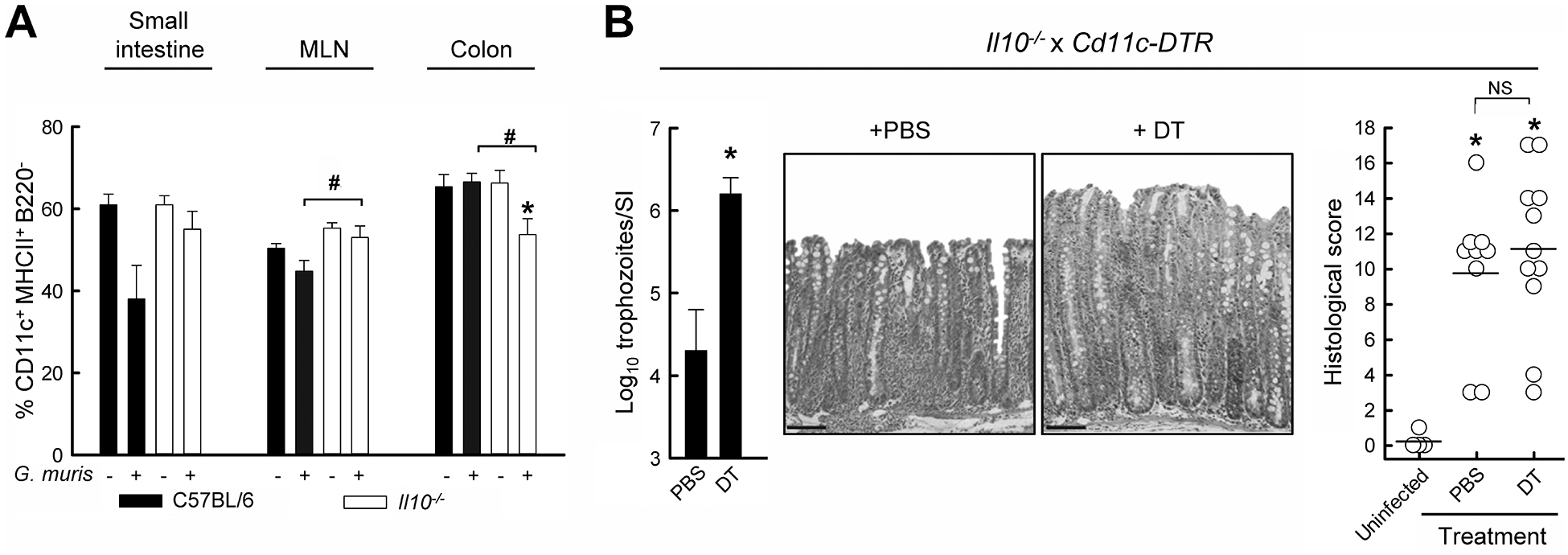
Colitis development after Giardia infection is independent of dendritic cells. (A) IL-10−/− mice (open bars) and wild-type C57BL/6 mice (solid bars) were infected with G. muris or left uninfected as controls, and dendritic cell numbers in the indicated organs were assessed by flow cytometry (means ± SE, n≥6 mice/group; *p<0.05 vs. uninfected littermate controls, #p<0.05 vs. wild-type mice at the same time). (B) Il10−/− x Cd11c-Dtr double-mutant mice were infected with G. muris and subsequently treated every 48 h with PBS or diphtheria toxin (DT) for two weeks, after which parasite numbers were determined in the small intestine (SI, left; means ± SE, n≥9 mice/group; *p<0.05 vs. PBS-treated mice). Histological analysis was performed on H&E stained colon sections (representative sections in the middle panels; scale bar, 100 μm), with each data point representing an individual animal (right graph; horizontal lines represent mean values for each group; *p<0.05 vs. uninfected DT-treated controls; NS, not significant).
Mucosal macrophage expansion and role of CCR7 signaling in Giardia-induced colitis
The unexpected lack of dendritic cell involvement in Giardia-triggered colitis prompted us to explore other innate immune cells in the model. Macrophages, in particular, not only serve as a link to adaptive immunity, but also promote inflammation independent of T cells. Flow cytometry of mucosal leukocyte populations revealed that G. muris infection induced a marked 4-fold increase in CD11b+ CD11c− macrophages in the colon of Il10−/− mice after two weeks (Fig. 5A), which was paralleled by induction of macrophage chemokines CCL2 and CCL3 (Fig. 5B). By comparison, no change in macrophage numbers was observed in the small intestine or mesenteric lymph nodes of G. muris infected Il10−/− mice.
FIG. 5.
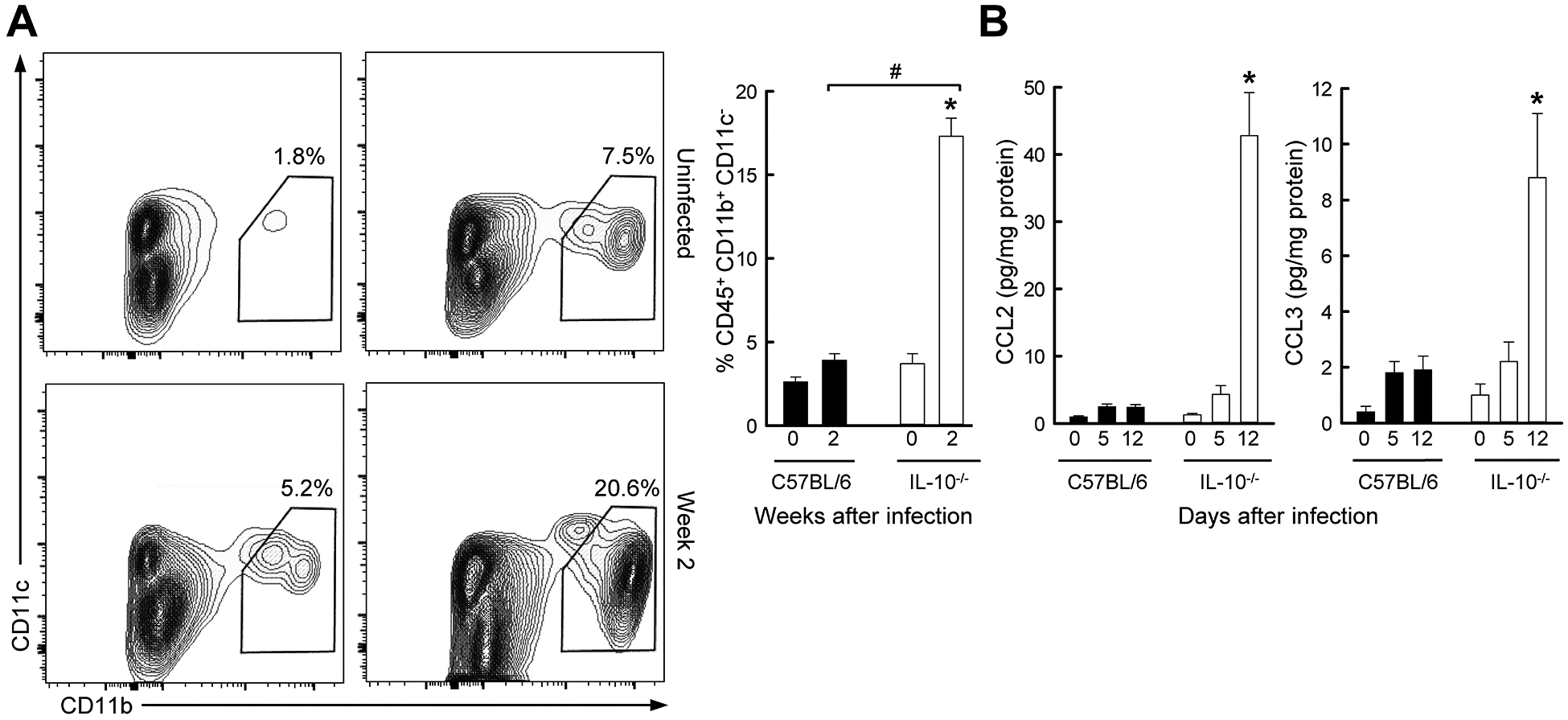
Colon infiltration with macrophages in Giardia-infected IL-10 deficient mice. Il10−/− mice (open bars) and wild-type C57BL/6 mice (solid bars) were infected with G. muris, or left uninfected (Week 0). (A) Single cells were isolated from the colon lamina propria and analyzed by flow cytometry for macrophages. Representative FACS contour plots are shown on the left, and percentages of macrophages are given on the right (mean ± SE, n≥6 mice/group; *p<0.05 vs. age-matched uninfected mice; #p<0.05 vs. wild-type mice at the same time). (B) Levels of CCL2 and CCL3 were determined in colon extracts by multiplex immunoassay (mean ± SE, n=5 mice/time; *p<0.05 vs. age-matched uninfected mice of the same genotype, #p<0.05 vs. wild-type mice at the same time).
The marked macrophage expansion in the colon raised the possibility that their local activation might induce or sustain colon inflammation. Although multiple pathways have been proposed for the inflammation-promoting functions of certain macrophage subsets, two mechanisms are particularly relevant for intestinal homeostasis. One involves down-regulation of inflammation-inducing genes by IL-10 dependent macrophage deactivation (34). In this context, loss of IL-10 leads to intestinal accumulation of activated macrophages and subsequent inflammation, findings consistent with our observations in Il10−/− mice. Another mechanism relates to CCR7-dependent migration of activated macrophages out of the mucosa (35). In the absence of CCR7 signaling, activated cells accumulate in the intestine and may cause inflammation. To explore whether this process might be relevant to Giardia-triggered colitis, we infected CCR7 knock-out mice (Ccr7−/−) with G. muris. Surprisingly, Ccr7−/− mice were relatively protected against acute infection, with a 100-fold lower peak trophozoite burden after 7 days compared to wild-type controls (Fig. 6A). Despite the reduced infectious load, mice developed marked colitis that commenced within one week and reached maximal severity by 3–7 weeks (Fig. 6B). These results are consistent with the notion that CCR7-dependent cell migration of macrophages and perhaps other potentially colitogenic immune cells out of the mucosa normally attenuate inflammation, but if this homeostatic process is disturbed, inflammation can ensue upon mucosal microbial challenge.
FIG. 6.
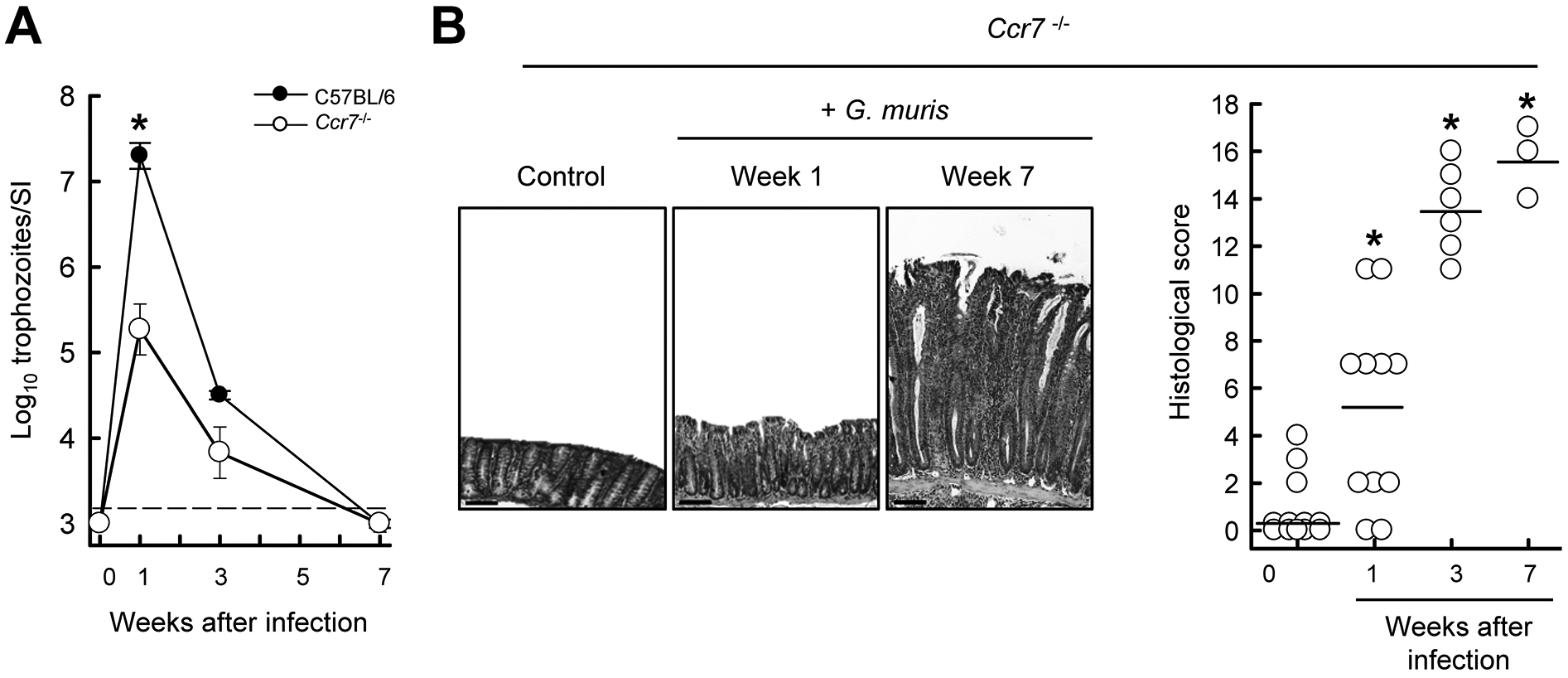
Giardia-induced colitis in CCR7-deficient mice. Ccr7−/− mice were infected with G. muris or left uninfected as controls (Week 0). (A) Trophozoite numbers were determined in the small intestine at the indicated times. (B) H&E stained colon sections were prepared (representative sections shown in left panel; scale bars, 100 μm) and histological damage scores were determined (right panel; each data point represents one animal, horizontal lines depict means for each group; *p<0.05 vs. uninfected Ccr7−/− controls).
Giardia-triggered colitis is dependent on the microbiota
Alterations in the intestinal microbiota are associated with and contribute to intestinal inflammation (36). To determine if Giardia infection impacts the microbiome in Il10−/− and Tcra−/− mice, DNA from unwashed colon tissues were analyzed using the Illumina MiSeq platform. Principal coordinate analysis to assess overall compositional differences between individual mice revealed that prior to Giardia infection, Il10−/− and Tcra−/− mice had distinct microbiome profiles compared to wild-type mice (Fig. 7A, left). Two weeks after infection, clear shifts in the composition were observed, with the microbiota of Il10−/− and Tcra−/− mice converging in the coordinate space and becoming more similar to that in wild-type mice (Fig. 7A, right). Giardia infection of Il10−/− mice also led to a significant reduction in microbial diversity, a trend also observed in Tcra−/− but not wild-type mice (Fig. 7B). Analysis of the dominant phyla revealed that Giardia infection caused a reduction in Bacteroidetes and Firmicutes, but increases in Proteobacteria in both Il10−/− and Tcra−/− mice (Fig. 7C). Expansion of Proteobacteria had previously been associated with intestinal inflammation (37). At the family level, major changes in abundance were observed for Provotellaceae, Rickenellaceae, Deferribacteraceae, Lachnospiraceae and Helicobacteraceae, the most dominantly detected family in wild-type controls. Further analysis of the OTUs classified in the Helicobacteraceae family demonstrated >99% identity to two particular species, Helicobacter bilis and Helicobacter mastomyrinus, that were increased in abundance after Giardia infection, although they were also identified at lower levels in uninfected controls.
FIG. 7.
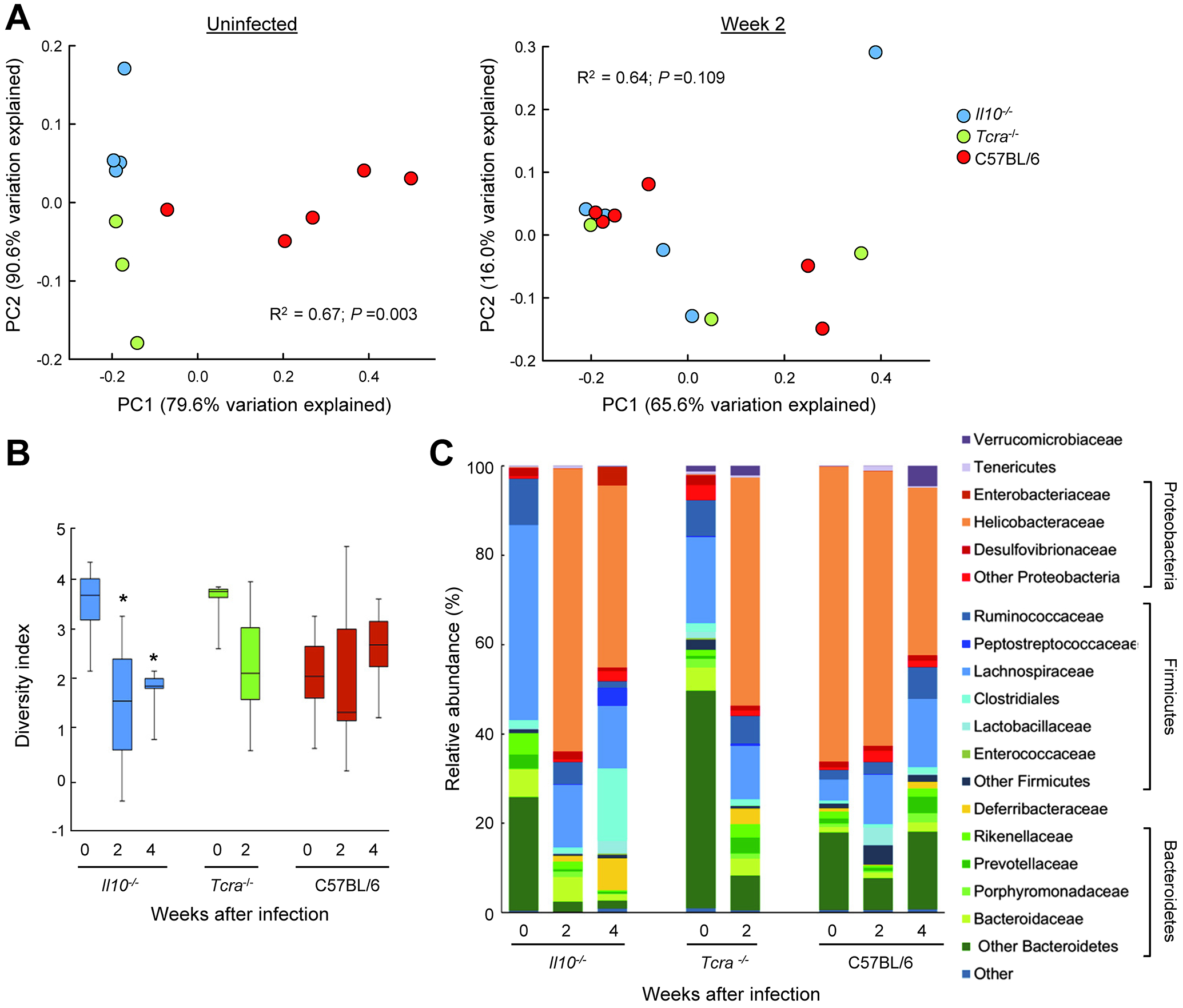
Effect of Giardia infection on intestinal microbiota. Age-matched Il10−/− mice (blue circles and bars in A and B), Tcra−/− (green circles and bars), and wild-type C57BL/6 littermate controls (red circles and bars) were infected with G. muris, or left uninfected (Week 0). DNA from colon tissues was analyzed using Illumina MiSeq System. (A) Principal coordinate analysis of the weighted UniFrac matrix of microbial composition in the colon. Each dot corresponds to an individual mouse. (B) Shannon Diversity Index analysis of microbial complexity in the colon of the indicated groups of mice. Data are shown as box plots (*p<0.05 vs. Week 0 of the same genotype). (C) Average relative microbial abundance at the phylum and family levels at different times after Giardia infection.
To determine whether the microbiota contributes to the development of Giardia-triggered colitis, we treated Il10−/− mice with a combination of broad-spectrum antibiotics before and during G. muris infection. The antibiotics were chosen to have no direct antigiardial activity, and sustained administration to wild-type mice had no significant impact on the course of G. muris infection (Supplemental Fig. 3). Il10−/− mice given antibiotics two days prior to Giardia infection showed significantly less colon inflammation, as evidenced by reductions in mucosal thickening, crypt hyperplasia, and leukocytic infiltration compared to infected but untreated Il10−/− mice (Fig. 8A). Administration of antibiotics three weeks after G. muris infection also reduced the severity of colitis, although not as effectively as the pre-Giardia infection regimen (Fig. 8B). These findings strongly suggest that Giardia-triggered colitis can be prevented or at least attenuated by reducing or modifying the intestinal microbiota.
FIG. 8.
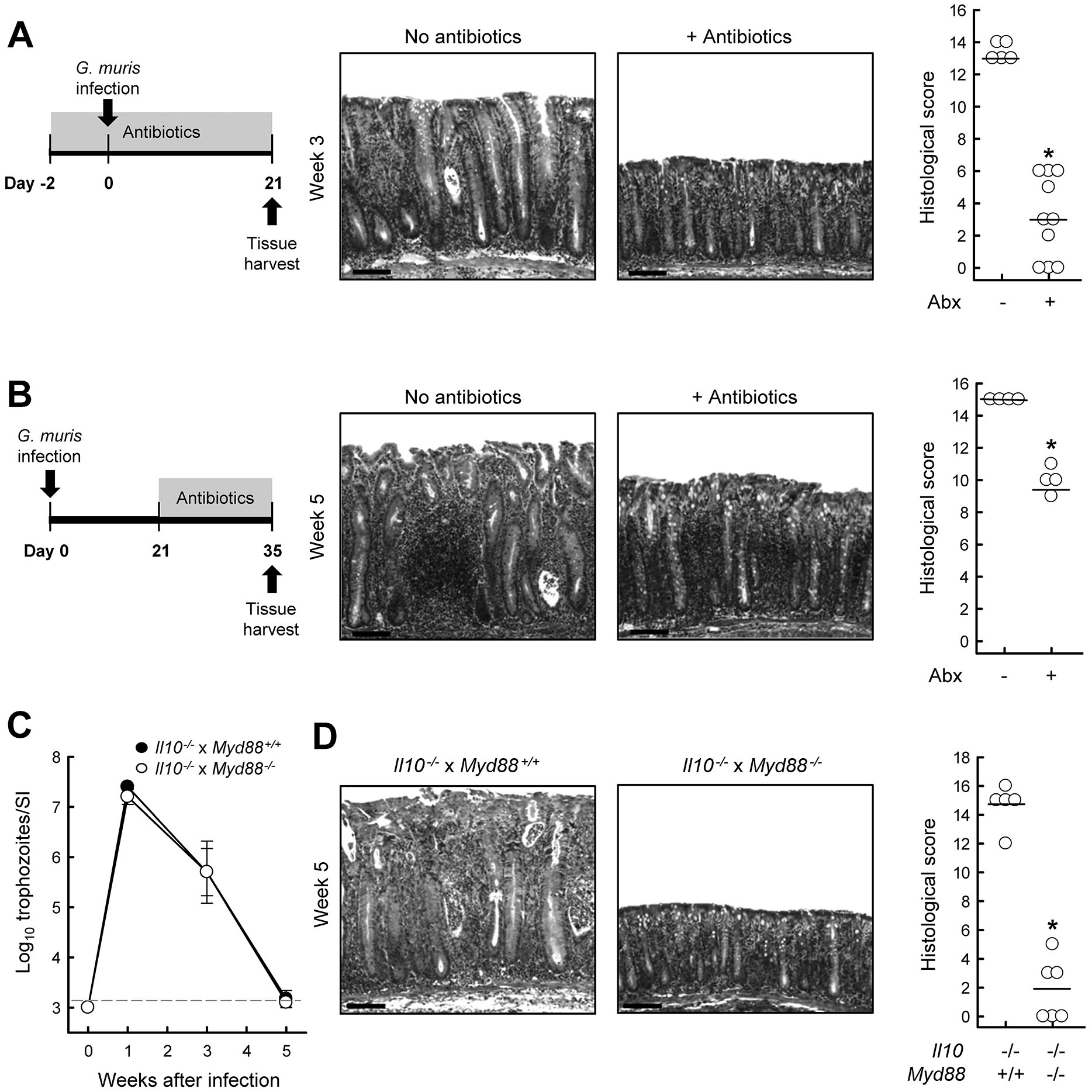
Requirement for microbiota in Giardia-triggered colitis. Il10−/− and wild-type C57BL/6 mice were infected with G. muris, or left uninfected (Week 0). (A) Administration of broad-spectrum antibiotics prior to and during infection prevents onset of colitis. Representative H&E stained colon sections (middle; scale bars, 100 μm) and histological scores are shown (right; each data point represents one animal, horizontal lines are means of each group; *p<0.05 relative to Giardia-infected littermates not treated with antibiotics). (B) Delayed administration of broad-spectrum antibiotics also attenuates colitis. Representative H&E sections are (middle; scale bars, 100 μm) and histological scores are shown (right; *p<0.05 vs. Giardia-infected but untreated littermates). (C, D) Il10−/− x Myd88−/− double-mutant mice (open circles) and Il10−/− single mutant mice (solid circles) mice were orally infected with G. muris, and trophozoite numbers in the small intestine were determined at the indicated times (C; mean ± SE, n=5–6 mice/time). Histological analysis of the colon five weeks after infection. Representative H&E stained sections (D, left; scale bars, 100 μm) and histological scores are shown (D, right; each data point represents an individual animal, horizontal lines depict means; *p<0.05 vs. Il10−/− single mutants).
MyD88 is required for driving Giardia-triggered colitis but not antigiardial defense
To further define the role of the bacterial microbiota in mediating Giardia-triggered colitis, we explored the functions of the common TLR signal transducers, MyD88. This transducer is central to the recognition of bacterial products and in antibacterial host defense (25). We constructed double-mutant mice lacking both IL-10 and MyD88 (Il10−/− x Myd88−/−), and infected them with G. muris. The double-mutant mice were equally infected after one week and cleared the parasites as effectively thereafter as Il10−/− controls (Fig. 8C), indicating that MyD88 signaling is not required for normal host defense against Giardia. However, infected double-mutants had strongly attenuated colitis after 5 weeks compared to the infected Il10−/− single-mutants (Fig. 8D). These results provide independent support for the conclusion of the antibiotics studies that the intestinal bacterial microbiota, as recognized by signaling through MyD88, mediates Giardia-triggered colitis in the absence of IL-10.
DISCUSSION
Our studies demonstrate that colonization of the small intestine by a lumen-dwelling protozoan parasite, which by itself causes minimal mucosal inflammation in a normal host (19), can trigger chronic colitis in genetically susceptible mice. Giardia-triggered colitis in at least one of the models, Il10−/− mice, was associated with mucosal expansion of neutrophils and macrophages, and induction of a persistent Th1 response that contributed to elevated IL-12/IL-23 p40 expression critical for the disease manifestation. Our findings parallel prior observations that subclinical norovirus infection can initiate chronic intestinal inflammation in Il10−/− mice (38). In both models, the acute pathogen infection is transient and minimally symptomatic, while the intestinal microbiota was required for disease induction and persistence beyond the initial infection. Important differences exist between these microbial triggers, as norovirus infects multiple cell types throughout the intestinal tract, while Giardia is non-invasive and largely restricted to the proximal small intestine. Nonetheless, the data suggest that the development of chronic inflammation can result in a susceptible host from complex temporal host-pathogen-microbiota interactions that are not unique to particular enteric pathogens.
Synergistic effects of genetic susceptibility and pathogen exposure have also been implicated in the development of IBD in humans. Several viral pathogens have been associated with IBD (38–41). Similarly, a number of bacterial pathogens, such as Salmonella and Campylobacter, have been linked to IBD (42–44). For example, adherent invasive Escherichia coli is commonly found in Crohn’s disease patients with ileal involvement (45), whereas Yersinia enterocolitica has been associated with ulcerative colitis in adults (46). Yet, the available evidence does not suggest that a single viral or bacterial pathogen is the definitive cause of IBD (4, 42, 47). Instead and consistent with our findings, a range of microbial pathogens can initiate inflammatory processes in the intestinal tract that ultimately become self-perpetuating independent of the original trigger.
Colitis induction after Giardia infection required the resident microbiota and MyD88 signaling, indicating that host interactions with the endogenous microbiota are essential for disease induction and persistence. Similarly, development of spontaneous colitis in aged Il10−/− mice relies on the intestinal microbiota and microbial sensing through MyD88-dependent pathways (48, 49). Besides the overall importance of the microbiota, we also found that the composition of the intestinal microbiota changed in parallel with disease development in both Il10−/− and Tcra−/− mice. Although additional studies are required to determine why certain commensal species expand following Giardia infection, the parasite promoted bloom of two colitogenic pathobionts, H. bilis and H. mastomyrinus. These two Helicobacter species have been associated with colitis in other models (50, 51), although it is not clear that either is sufficient to cause colitis. Inflammation induced in Il10−/− mice with another colitogenic Helicobacter species, Helicobacter hepaticus, is accompanied and modulated by the composition of the intestinal microbiota (4), yet the altered microbiota alone cannot induce inflammation after colonization of germ-free Il10−/− mice (52). These findings underline that the causal relationship between microbiota changes and inflammation observed in our and prior studies remains to be firmly established. Mono-association studies in gnotobiotic mice have shown that several different bacterial members of the normal microbiota can induce colitis in genetically susceptible hosts (52, 53), so it is likely that a constellation of commensal bacteria with relatively greater potential to act as pathobionts rather than a single dominant bacterial pathobiont are responsible for colitis induction upon microbial triggering in a susceptible host.
Our data on persistent mucosal inflammation upon transient Giardia infection in mice are consistent with clinical reports that G. lamblia infection can cause protracted intestinal symptoms and even mucosal inflammation (54–56). Following a large waterborne outbreak of giardiasis in Norway, over a third of patients presented with irritable bowel syndrome and chronic fatigue six years after successful treatment of the infection (56). Post-infectious syndromes can occur after infection with other enteric pathogens, particularly Salmonella spp., Escherichia coli O157 and Campylobacter jejuni (57). The underlying mechanisms are not well understood in most cases, but may involve localized residual inflammation impacting the composition of the microbiota, or functions of the enteric nervous systems and serotonin transporters (58, 59). In giardiasis, it has been postulated that disruptions in the microbiota predispose individuals to post-infectious gastrointestinal disorders (60). Our data suggest that post-infectious complications after giardiasis can result from interactions between the microbiota and dysregulated mucosal immunity, suggesting that functional and genetic testing of patients with protracted post-giardiasis intestinal disorders may reveal subtle immunological abnormalities.
Giardia infection caused a marked induction of IL-10 in the jejunum, the primary site of infection, yet the cytokine was not required for the development of protective immunity against giardiasis. Similar observations have been made in other infections with enteric pathogens. For example, infection with Salmonella enterica serovar Typhimurium led to increased IL-10 expression in the spleen, but neutralization of IL-10 did not modify the course of infection (61). For another enteric pathogen, Citrobacter rodentium, IL-10-deficient mice also had no clearance defect, and could, in fact, eradicate intestinal infection moderately faster than wild-type controls, suggesting that the cytokine was not only not essential but modestly inhibitory for adaptive immunity against this pathogen (62). For Giardia infection, we can conclude that IL-10 had no positive or negative role in acute intestinal pathogen defense, but was critical for controlling the long-term, indirect immunological sequelae of the infection. This intriguing concept - a key role of an immune regulator in maintaining long-term homeostasis upon transient microbial exposure without contributing to acute host defense - may have relevance to understanding other protracted post-infectious syndromes.
Supplementary Material
ACKNOWLEDGMENTS
We thank Kim Nguyen, Lauren Gima, MaChesha Banks, Raymond Dann, and the staff of the VA San Diego Flow Cytometer Core Laboratory and the Alkek Center for Metagenomics and Microbiome Research at Baylor College of Medicine in Houston for technical assistance, and Lucia Hall and the staff of the UCSD and UTMB Animal Care Programs for animal husbandry.
This work was supported by NIH grants AI075527 and AI125860. SMD was supported by the Institute for Translational Sciences at the University of Texas Medical Branch, an NIH Clinical and Translational Science Award (UL1TR000071, KL2TR000072), and a Career Development Award from the Crohn’s and Colitis Foundation of America.
Footnotes
DISCLOSURES
The authors have no financial conflicts of interest.
REFERENCES
- 1.Shouval DS, Ouahed J, Biswas A, Goettel JA, Horwitz BH, Klein C, Muise AM, and Snapper SB. 2014. Interleukin 10 receptor signaling: master regulator of intestinal mucosal homeostasis in mice and humans. Adv Immunol 122: 177–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sellon RK, Tonkonogy S, Schultz M, Dieleman LA, Grenther W, Balish E, Rennick DM, and Sartor RB. 1998. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect Immun 66: 5224–5231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sydora BC, Tavernini MM, Wessler A, Jewell LD, and Fedorak RN. 2003. Lack of interleukin-10 leads to intestinal inflammation, independent of the time at which luminal microbial colonization occurs. Inflamm Bowel Dis 9: 87–97. [DOI] [PubMed] [Google Scholar]
- 4.Keubler LM, Buettner M, Hager C, and Bleich A. 2015. A Multihit Model: Colitis Lessons from the Interleukin-10-deficient Mouse. Inflamm Bowel Dis 21: 1967–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuhn R, Lohler J, Rennick D, Rajewsky K, and Muller W. 1993. Interleukin-10-deficient mice develop chronic enterocolitis. Cell 75: 263–274. [DOI] [PubMed] [Google Scholar]
- 6.Madsen KL, Doyle JS, Tavernini MM, Jewell LD, Rennie RP, and Fedorak RN. 2000. Antibiotic therapy attenuates colitis in interleukin 10 gene-deficient mice. Gastroenterology 118: 1094–1105. [DOI] [PubMed] [Google Scholar]
- 7.Hoentjen F, Harmsen HJ, Braat H, Torrice CD, Mann BA, Sartor RB, and Dieleman LA. 2003. Antibiotics with a selective aerobic or anaerobic spectrum have different therapeutic activities in various regions of the colon in interleukin 10 gene deficient mice. Gut 52: 1721–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huber S, Gagliani N, Esplugues E, O’Connor W Jr., Huber FJ, Chaudhry A, Kamanaka M, Kobayashi Y, Booth CJ, Rudensky AY, Roncarolo MG, Battaglia M, and Flavell RA. 2011. Th17 cells express interleukin-10 receptor and are controlled by Foxp3(−) and Foxp3+ regulatory CD4+ T cells in an interleukin-10-dependent manner. Immunity 34: 554–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chaudhry A, Samstein RM, Treuting P, Liang Y, Pils MC, Heinrich JM, Jack RS, Wunderlich FT, Bruning JC, Muller W, and Rudensky AY. 2011. Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity 34: 566–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Engelhardt KR, and Grimbacher B. 2014. IL-10 in humans: lessons from the gut, IL-10/IL-10 receptor deficiencies, and IL-10 polymorphisms. Curr Top Microbiol Immunol 380: 1–18. [DOI] [PubMed] [Google Scholar]
- 11.Arai T, Hiromatsu K, Nishimura H, Kimura Y, Kobayashi N, Ishida H, Nimura Y, and Yoshikai Y. 1995. Endogenous interleukin 10 prevents apoptosis in macrophages during Salmonella infection. Biochem Biophys Res Commun 213: 600–607. [DOI] [PubMed] [Google Scholar]
- 12.Greenberger MJ, Strieter RM, Kunkel SL, Danforth JM, Goodman RE, and Standiford TJ. 1995. Neutralization of IL-10 increases survival in a murine model of Klebsiella pneumonia. J Immunol 155: 722–729. [PubMed] [Google Scholar]
- 13.Dai WJ, Kohler G, and Brombacher F. 1997. Both innate and acquired immunity to Listeria monocytogenes infection are increased in IL-10-deficient mice. J Immunol 158: 2259–2267. [PubMed] [Google Scholar]
- 14.Igietseme JU, Ananaba GA, Bolier J, Bowers S, Moore T, Belay T, Eko FO, Lyn D, and Black CM. 2000. Suppression of endogenous IL-10 gene expression in dendritic cells enhances antigen presentation for specific Th1 induction: potential for cellular vaccine development. J Immunol 164: 4212–4219. [DOI] [PubMed] [Google Scholar]
- 15.Kullberg MC, Andersen JF, Gorelick PL, Caspar P, Suerbaum S, Fox JG, Cheever AW, Jankovic D, and Sher A. 2003. Induction of colitis by a CD4+ T cell clone specific for a bacterial epitope. Proc Natl Acad Sci U S A 100: 15830–15835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mansfield LS, Bell JA, Wilson DL, Murphy AJ, Elsheikha HM, Rathinam VA, Fierro BR, Linz JE, and Young VB. 2007. C57BL/6 and congenic interleukin-10-deficient mice can serve as models of Campylobacter jejuni colonization and enteritis. Infect Immun 75: 1099–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagalingam NA, Robinson CJ, Bergin IL, Eaton KA, Huffnagle GB, and Young VB. 2013. The effects of intestinal microbial community structure on disease manifestation in IL-10−/− mice infected with Helicobacter hepaticus. Microbiome 1: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang I, Eibach D, Kops F, Brenneke B, Woltemate S, Schulze J, Bleich A, Gruber AD, Muthupalani S, Fox JG, Josenhans C, and Suerbaum S. 2013. Intestinal microbiota composition of interleukin-10 deficient C57BL/6J mice and susceptibility to Helicobacter hepaticus-induced colitis. PLoS One 8: e70783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oberhuber G, Kastner N, and Stolte M. 1997. Giardiasis: a histologic analysis of 567 cases. Scand J Gastroenterol 32: 48–51. [DOI] [PubMed] [Google Scholar]
- 20.Lopez-Romero G, Quintero J, Astiazaran-Garcia H, and Velazquez C. 2015. Host defences against Giardia lamblia. Parasite Immunol 37: 394–406. [DOI] [PubMed] [Google Scholar]
- 21.Dann SM, Manthey CF, Le C, Miyamoto Y, Gima L, Abrahim A, Cao AT, Hanson EM, Kolls JK, Raz E, Cong Y, and Eckmann L. 2015. IL-17A promotes protective IgA responses and expression of other potential effectors against the lumen-dwelling enteric parasite Giardia. Exp Parasitol 156: 68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dreesen L, De Bosscher K, Grit G, Staels B, Lubberts E, Bauge E, and Geldhof P. 2014. Giardia muris infection in mice is associated with a protective interleukin 17A response and induction of peroxisome proliferator-activated receptor alpha. Infect Immun 82: 3333–3340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Forster R, Schubel A, Breitfeld D, Kremmer E, Renner-Muller I, Wolf E, and Lipp M. 1999. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell 99: 23–33. [DOI] [PubMed] [Google Scholar]
- 24.Mombaerts P, Clarke AR, Rudnicki MA, Iacomini J, Itohara S, Lafaille JJ, Wang L, Ichikawa Y, Jaenisch R, Hooper ML, and et al. 1992. Mutations in T-cell antigen receptor genes alpha and beta block thymocyte development at different stages. Nature 360: 225–231. [DOI] [PubMed] [Google Scholar]
- 25.Hou B, Reizis B, and DeFranco AL. 2008. Toll-like receptors activate innate and adaptive immunity by using dendritic cell-intrinsic and -extrinsic mechanisms. Immunity 29: 272–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jung S, Unutmaz D, Wong P, Sano G, De los Santos K, Sparwasser T, Wu S, Vuthoori S, Ko K, Zavala F, Pamer EG, Littman DR, and Lang RA. 2002. In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell-associated antigens. Immunity 17: 211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davids BJ, Palm JE, Housley MP, Smith JR, Andersen YS, Martin MG, Hendrickson BA, Johansen FE, Svard SG, Gillin FD, and Eckmann L. 2006. Polymeric immunoglobulin receptor in intestinal immune defense against the lumen-dwelling protozoan parasite Giardia. J Immunol 177: 6281–6290. [DOI] [PubMed] [Google Scholar]
- 28.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, Gormley N, Gilbert JA, Smith G, and Knight R. 2012. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6: 1621–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Edgar RC 2013. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10: 996–998. [DOI] [PubMed] [Google Scholar]
- 30.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, and Glockner FO. 2013. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41: D590–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Benjamini Y, and Hochberg Y. 1995. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J Royal Stat Soc 57: 289–300. [Google Scholar]
- 32.Singer SM, and Nash TE. 2000. T-cell-dependent control of acute Giardia lamblia infections in mice. Infect Immun 68: 170–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hsieh CS, Macatonia SE, Tripp CS, Wolf SF, O’Garra A, and Murphy KM. 1993. Development of TH1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science 260: 547–549. [DOI] [PubMed] [Google Scholar]
- 34.Zigmond E, Bernshtein B, Friedlander G, Walker CR, Yona S, Kim KW, Brenner O, Krauthgamer R, Varol C, Muller W, and Jung S. 2014. Macrophage-restricted interleukin-10 receptor deficiency, but not IL-10 deficiency, causes severe spontaneous colitis. Immunity 40: 720–733. [DOI] [PubMed] [Google Scholar]
- 35.Diehl GE, Longman RS, Zhang JX, Breart B, Galan C, Cuesta A, Schwab SR, and Littman DR. 2013. Microbiota restricts trafficking of bacteria to mesenteric lymph nodes by CX(3)CR1(hi) cells. Nature 494: 116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sheehan D, and Shanahan F. 2017. The Gut Microbiota in Inflammatory Bowel Disease. Gastroenterol Clin North Am 46: 143–154. [DOI] [PubMed] [Google Scholar]
- 37.Litvak Y, Byndloss MX, Tsolis RM, and Baumler AJ. 2017. Dysbiotic Proteobacteria expansion: a microbial signature of epithelial dysfunction. Curr Opin Microbiol 39: 1–6. [DOI] [PubMed] [Google Scholar]
- 38.Basic M, Keubler LM, Buettner M, Achard M, Breves G, Schroder B, Smoczek A, Jorns A, Wedekind D, Zschemisch NH, Gunther C, Neumann D, Lienenklaus S, Weiss S, Hornef MW, Mahler M, and Bleich A. 2014. Norovirus triggered microbiota-driven mucosal inflammation in interleukin 10-deficient mice. Inflamm Bowel Dis 20: 431–443. [DOI] [PubMed] [Google Scholar]
- 39.Khan RR, Lawson AD, Minnich LL, Martin K, Nasir A, Emmett MK, Welch CA, and Udall JN Jr. 2009. Gastrointestinal norovirus infection associated with exacerbation of inflammatory bowel disease. J Pediatr Gastroenterol Nutr 48: 328–333. [DOI] [PubMed] [Google Scholar]
- 40.Rahbar A, Bostrom L, Lagerstedt U, Magnusson I, Soderberg-Naucler C, and Sundqvist VA. 2003. Evidence of active cytomegalovirus infection and increased production of IL-6 in tissue specimens obtained from patients with inflammatory bowel diseases. Inflamm Bowel Dis 9: 154–161. [DOI] [PubMed] [Google Scholar]
- 41.Yanai H, Shimizu N, Nagasaki S, Mitani N, and Okita K. 1999. Epstein-Barr virus infection of the colon with inflammatory bowel disease. Am J Gastroenterol 94: 1582–1586. [DOI] [PubMed] [Google Scholar]
- 42.Gradel KO, Nielsen HL, Schonheyder HC, Ejlertsen T, Kristensen B, and Nielsen H. 2009. Increased short- and long-term risk of inflammatory bowel disease after salmonella or campylobacter gastroenteritis. Gastroenterology 137: 495–501. [DOI] [PubMed] [Google Scholar]
- 43.Schultz BM, Paduro CA, Salazar GA, Salazar-Echegarai FJ, Sebastian VP, Riedel CA, Kalergis AM, Alvarez-Lobos M, and Bueno SM. 2017. A Potential Role of Salmonella Infection in the Onset of Inflammatory Bowel Diseases. Front Immunol 8: 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bosca-Watts MM, Tosca J, Anton R, Mora M, Minguez M, and Mora F. 2015. Pathogenesis of Crohn’s disease: Bug or no bug. World J Gastrointest Pathophysiol 6: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hansen R, Thomson JM, El-Omar EM, and Hold GL. 2010. The role of infection in the aetiology of inflammatory bowel disease. J Gastroenterol 45: 266–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saebo A, Vik E, Lange OJ, and Matuszkiewicz L. 2005. Inflammatory bowel disease associated with Yersinia enterocolitica O:3 infection. Eur J Intern Med 16: 176–182. [DOI] [PubMed] [Google Scholar]
- 47.Garcia Rodriguez LA, Ruigomez A, and Panes J. 2006. Acute gastroenteritis is followed by an increased risk of inflammatory bowel disease. Gastroenterology 130: 1588–1594. [DOI] [PubMed] [Google Scholar]
- 48.Rakoff-Nahoum S, Hao L, and Medzhitov R. 2006. Role of toll-like receptors in spontaneous commensal-dependent colitis. Immunity 25: 319–329. [DOI] [PubMed] [Google Scholar]
- 49.Karrasch T, Kim JS, Muhlbauer M, Magness ST, and Jobin C. 2007. Gnotobiotic IL-10−/−;NF-kappa B(EGFP) mice reveal the critical role of TLR/NF-kappa B signaling in commensal bacteria-induced colitis. J Immunol 178: 6522–6532. [DOI] [PubMed] [Google Scholar]
- 50.Liu Z, Ramer-Tait AE, Henderson AL, Demirkale CY, Nettleton D, Wang C, Hostetter JM, Jergens AE, and Wannemuehler MJ. 2011. Helicobacter bilis colonization enhances susceptibility to Typhlocolitis following an inflammatory trigger. Dig Dis Sci 56: 2838–2848. [DOI] [PubMed] [Google Scholar]
- 51.Eaton KA, Opp JS, Gray BM, Bergin IL, and Young VB. 2011. Ulcerative typhlocolitis associated with Helicobacter mastomyrinus in telomerase-deficient mice. Vet Pathol 48: 713–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dieleman LA, Arends A, Tonkonogy SL, Goerres MS, Craft DW, Grenther W, Sellon RK, Balish E, and Sartor RB. 2000. Helicobacter hepaticus does not induce or potentiate colitis in interleukin-10-deficient mice. Infect Immun 68: 5107–5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hansen AK, Hansen CH, Krych L, and Nielsen DS. 2014. Impact of the gut microbiota on rodent models of human disease. World J Gastroenterol 20: 17727–17736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Halliez MC, and Buret AG. 2013. Extra-intestinal and long term consequences of Giardia duodenalis infections. World J Gastroenterol 19: 8974–8985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hanevik K, Hausken T, Morken MH, Strand EA, Morch K, Coll P, Helgeland L, and Langeland N. 2007. Persisting symptoms and duodenal inflammation related to Giardia duodenalis infection. J Infect 55: 524–530. [DOI] [PubMed] [Google Scholar]
- 56.Hanevik K, Wensaas KA, Rortveit G, Eide GE, Morch K, and Langeland N. 2014. Irritable bowel syndrome and chronic fatigue 6 years after giardia infection: a controlled prospective cohort study. Clin Infect Dis 59: 1394–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Klem F, Wadhwa A, Prokop LJ, Sundt WJ, Farrugia G, Camilleri M, Singh S, and Grover M. 2017. Prevalence, Risk Factors, and Outcomes of Irritable Bowel Syndrome After Infectious Enteritis: A Systematic Review and Meta-analysis. Gastroenterology 152: 1042–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dupont HL 2014. Review article: evidence for the role of gut microbiota in irritable bowel syndrome and its potential influence on therapeutic targets. Aliment Pharmacol Ther 39: 1033–1042. [DOI] [PubMed] [Google Scholar]
- 59.Halliez MC, and Buret AG. 2015. Gastrointestinal Parasites and the Neural Control of Gut Functions. Front Cell Neurosci 9: 452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Beatty JK, Akierman SV, Motta JP, Muise S, Workentine ML, Harrison JJ, Bhargava A, Beck PL, Rioux KP, McKnight GW, Wallace JL, and Buret AG. 2017. Giardia duodenalis induces pathogenic dysbiosis of human intestinal microbiota biofilms. Int J Parasitol 47: 311–326. [DOI] [PubMed] [Google Scholar]
- 61.Pie S, Matsiota-Bernard P, Truffa-Bachi P, and Nauciel C. 1996. Gamma interferon and interleukin-10 gene expression in innately susceptible and resistant mice during the early phase of Salmonella typhimurium infection. Infect Immun 64: 849–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dann SM, Le C, Choudhury BK, Liu H, Saldarriaga O, Hanson EM, Cong Y, and Eckmann L. 2014. Attenuation of intestinal inflammation in interleukin-10-deficient mice infected with Citrobacter rodentium. Infect Immun 82: 1949–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.