

SUMMARY
Inborn errors of metabolism (IEMs) link metabolic defects to human phenotypes. Modern genomics has accelerated IEM discovery, but assessing the impact of genomic variants is still challenging. Here, we integrate genomics and metabolomics to identify a cause of lactic acidosis and epilepsy. The proband is a compound heterozygote for variants in LIPT1, which encodes the lipoyltransferase required for 2-ketoacid dehydrogenase (2KDH) function. Metabolomics reveals abnormalities in lipids, amino acids, and 2-hydroxyglutarate consistent with loss of multiple 2KDHs. Homozygous knock-in of a LIPT1 mutation reduces 2KDH lipoylation in utero and results in embryonic demise. In patient fibroblasts, defective 2KDH lipoylation and function are corrected by wild-type, but not mutant, LIPT1 alleles. Isotope tracing reveals that LIPT1 supports lipogenesis and balances oxidative and reductive glutamine metabolism. Altogether, the data extend the role of LIPT1 in metabolic regulation and demonstrate how integrating genomics and metabolomics can uncover broader aspects of IEM pathophysiology.
In Brief
Ni et al. investigate human LIPT1 deficiency, which results in developmental delay, epilepsy, and broad metabolic abnormalities, including lactic acidosis, L- and D-2-hydroxyglutaric aciduria, defective lipogenesis, and an altered balance between oxidative and reductive glutamine metabolism.
Graphical Abstract
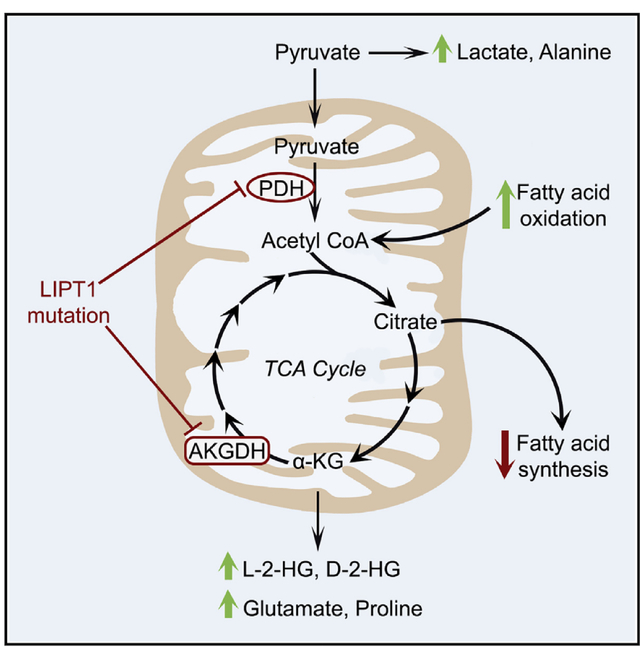
INTRODUCTION
Inborn errors of metabolism (IEMs) are genetic disorders characterized by abnormal metabolism (DeBerardinis and Thompson, 2012). Most IEMs are caused by single-enzyme, cofactor, or transporter deficiencies, resulting in metabolite depletion or toxin accumulation. IEMs are diagnosed by assessing the phenotype and pedigree, along with clinical biochemical and molecular testing (Vernon, 2015). However, establishing a specific diagnosis is challenging, because many lEMs have non-specific symptoms (Cleary and Green, 2005; EI-Hattab, 2015; Pascual, 2017; Raghuveer et al., 2006). Many lEMs are treatable if the underlying metabolic anomaly is known. Accurate counseling and treatment require identifying the mode of inheritance, ideally by pinpointing the mutant gene.
Whole exome sequencing (WES) identifies causative mutations in about 25% of patients with undiagnosed Mendelian disorders, even for non-specific phenotypes (Farwell et al., 2015; Lee et al., 2014). However, ascribing causality to rare genetic variants relies on imperfect computational predictions about the impact of a mutation on protein function and on connecting the projected consequences of altered protein function to incompletely characterized human phenotypes (Farwell Hagman et al., 2017). Metabolomics can provide another dimension of human phenotyping to enhance interpretation of rare variants. For example, detecting altered intermediates from a metabolic pathway in a patient with sequence variants in the same pathway would prioritize those variants for functional evaluation. Focused analysis of a few metabolites (e.g., amino acids, organic acids, and acylcarnitines) has long been used to evaluate IEMs, and increasing the scope of metabolic phenotyping through metabolomics has great diagnostic potential (Miller et al., 2015).
Here, we describe a familial form of lactic acidosis, epilepsy, developmental delay, and early death. These symptoms occur in many IEMs and thus do not point to a specific defect (Davison and Rahman, 2017). Metabolomics suggested a defect in multiple 2-ketoacid dehydrogenase (2KDH) enzymes, and WES identified variants in LIPT1, the gene encoding lipoyltransferase-1 (LIPT1), which catalyzes transfer of lipoic acid from the H-protein of the glycine cleavage system to E2 subunits of 2KDHs. Functional analysis in cells and mice confirmed LIPT1 deficiency and provided insights into the metabolic anomalies caused by this disease.
RESULTS
Compound Heterozygous LIPT1 Variants in a Child with Lactic Acidosis, Epilepsy, and Developmental Delay
We consulted on an 8-year-old female (P3–002,II.5 in Figure 1A) with abnormal development, seizures, and lactic acidemia. She was born to non-consanguineous Caucasian parents and had no complications during pregnancy or delivery. Two older sisters died of a similar condition at 7 months and 3 years, before the birth of the proband (II.1 and II.2 in Figure 1A). The proband came to attention at 3 months with hypotonia and elevated lactate, followed by seizures and minimal acquisition of developmental milestones. A brain MRI at 3.5 months revealed an abnormal signal on diffusion-weighted images in the supratentorial white matter. Subsequent imaging revealed progressive white matter loss, bilateral thalamic cysts, and gliosis. Magnetic resonance (MR) spectroscopy revealed lactate accumulation, including in the basal ganglia.
Figure 1. Metabolic Defects in a Patient with Compound Heterozygous Mutations of LIPT1.

(A) Patient (P3–002) and affected siblings are indicated in black. The LIPT1 genotype is indicated under the individuals subjected to molecular analysis.
(B) Chromatogram of LIPT1 sequences from the patient (II.5, P3–002) and healthy sibling (II.3).
(C) Schematic of LIPT1 proteins encoded from paternal and maternal alleles. The N44S and S292X mutations in the lipoyltransferase domain are indicated in red.
(D) Conservation of N44 (red frame) and three interacting residues (blue frames) in 16 species.
(E) Three-dimensional structure of bovine Liptl (left panel) and an enlargement (right panel) showing hydrogen bonding among Asn44, Ser36, Ser38, and Asn68. The locations of cofactors, AMP-conjugated lipoic acid (lipoyl-AMP) and magnesium ion, are also indicated.
(F) Plasma lactate concentrations from P3–002 over 12 years. The upper limit of the reference range (0.7–2.5 mmol/L) is indicated in red.
(G) Plasma alanine, proline, and glutamate in P3–002. The dashed lines indicate the upper limit of the normal range for each amino acid.
(H) Principal-component analysis (PCA) of plasma metabolomics from patient P3–002, the healthy sibling (II.3), and healthy controls (n = 60}.
(I) Quantitative plasma abundance of total 2-hydroxyglutarate (2-HG), L-2-hydroxyglutarate (L-2-HG), and D-2-hydroxyglutarate (D-2-HG) in P3–002, the healthy sibling (II.3), and controls (n = 6). Data are the mean and SD from two (sick) or six (control) replicates.
(J) Heatmap of plasma lipidomics showing species with variable importance in the projection (VIP) scores ≥1.0 between patient P3–002 and healthy controls (n = 16).
The clinical workup revealed increased lactate and α-ketoglutarate (α-KG) in urine, typical of disordered oxidative metabolism. A provisional diagnosis of pyruvate dehydrogenase (PDH) deficiency was made based on a 70% reduction in PDH activity in skin fibroblasts {0.81 nmol/min/mg protein compared to 1.264.42 nmol/min/mg protein in controls). No mutations were identified in the mitochondrial DNA orin the X-linked PDHA1 gene. The fibroblasts also had low-normal α-ketoglutarate dehydrogenase (AKGDH)activity (1.23nmol/min/mg compared toacontrol range of 1.08–4.42 nmol/min/mg). Ketogenic diets are used to reduce lactate in PDH deficiency. However, initiating a ketogenic diet in the proband at age 4 precipitated severe ketoacidosis. The cause of this paradoxical response was unknown but seemed inconsistent with isolated PDH deficiency.
WES revealed two LIPT1 variants in the proband, a maternally inherited C.875C > G (p.S292X) nonsense mutation and a paternally inherited variant of unknown significance (VUS), C.131A > G (p.N44S) (Figures 1A–1C). Heterozygous variants in six other genes were also reported (Table S1). None of these seemed likely explanations for the phenotype, because the genes were unrelated to the perturbed pathways and because heterozygosity of the variants was inconsistent with the expected autosomal recessive inheritance of the phenotype.
Both N44 and S292 are within the lipoyltransferase domain of human LIPT1 (Figure 1C). S292X is a rare allele (allele frequency 0.0003 in the Exome Aggregation Consortium [ExAC]) and was reported in a previous case of LIPT1 deficiency (Soreze et al., 2013). N44 is highly conserved (Figure 1D), and Polymorphism Phenotyping v.2 (PolyPhen-2), Sorting Intolerant From Tolerant (SIFT), and Combined Annotation-Dependent Depletion (CADD) predict that the N44S mutation would compromise LIPT1 function. Structural modeling of bovine Liptl (PDB: 2E5A) (Fujiwara et al., 2007), with 88% similarity to human LIPT1. showed that the AMP-conjugated lipoic acid (lipoyl-AMP) substrate is buried in a hydrophobic pocket formed by the N-terminal domain (Figure 1E). N44 is located in this pocket and is predicted to form a hydrogen bond with three other conserved residues: S36, S38, and N68 (Figure 1D) (Venselaar et al., 2010). These interactions might contribute to substrate binding and/or catalysis. The N44S substitution abrogates formation of hydrogen bonds due to serine’s smaller size and reduced hydrophobicity. Thus, P3–002 has two potentially deleterious LIPT1 alleles, although the precise impact of N44S was unknown.
PDH lipoylation enables transfer of pyruvate’s acyl group from thiamine pyrophosphate (TPP) in E1 to coenzyme A at the E2 active site, producing acetyl-coenzyme A (CoA) for the tricarboxylic acid (TCA) cycle (Reed, 1974). LIPT1 deficiency impairs pyruvate oxidation and promotes its conversion to lactate and alanine. P3–002’s plasma had periodic elevations of both lactate and alanine (Figures 1F and 1G). Modest elevations of glutamate and proline, both derived from α-KG, were consistent with impaired AKGDH, another lipoylation-dependent 2KDFI (Figure 1G). The branched-chain amino acids leucine, isoleucine, and valine were normal despite their oxidation by a third 2KDH, branched-chain ketoacid dehydrogenase (BCKDH) (Figures S1A–S1C). In contrast to glycine accumulation in patients with lipoic acid synthesis defects (Tort et al., 2016), P3–002’s plasma glycine was normal (Figure S1D).
Plasma Metabolomic Profiling Broadens the Phenotype of LIPT1 Deficiency
To further characterize the phenotype, we used metabolomic and lipidomic profiling in P3–002’s plasma during periods of clinical stability and metabolic acidosis. A targeted analysis reported relative quantities of 93 aqueous metabolites (Table S2). Unsupervised clustering differentiated P3–002 from controls, including her healthy sister, regardless of whetherthe samples were obtained during periods of health or illness (Figure 1H). Because lactate and alanine were normal during periods of relative health, the data indicate that metabolomics detects the disease more sensitively than these two conventional diagnostic metabolites. A supervised analysis revealed 36 metabolites that discriminated P3–002 from controls (variable importance in the projection [VIP] score ≥ 1) (Figure S1E). Both L-2-hydroxyglutarate (L-2-HG) and D-2-hydroxyglutarate (D-2-HG) were elevated in the patent, with marked accumulation of L-2-HG during illness (Figure 1I).
Because PDH provides acetyl-CoA for lipogenesis, we also assessed more than 680 plasma lipids in P3–002 and 16 controls (Table S3). P3–002’s plasma contained reduced levels of membrane lipids, including phosphatidylcholines (PCs), ceramides (CERs), and sphingomyelins (SMs) (Figures 1J and S1F). Conversely, triacylglycerols (TAGs) were higher in P3–002, possibly reflecting dietary intake (Quehenberger et al., 2010).
LIPT1 Mutations Impair Mitochondrial Protein Lipoylation and the TCA Cycle
To functionally assess P3–002's LIPT1 variants, we used fibroblasts derived from a punch biopsy of the patient’s skin. An antibody directed against lipoylated proteins detected lipoyl-DLAT and lipoyl-DLST, the lipoylated E2 subunits of the PDH and AKGDH complexes, respectively (Figure 2A). Compared to five normal controls, P3–002’s fibroblasts had lower levels of lipoylation on both proteins, whereas total DLAT, DLST, and LIPT1 were similar. Lipoylation of DBT, the BCKDH E2 subunit, was not detected.
Figure 2. Functional Analysis of LIPT1 Variants in Patient-Derived Fibroblasts.

(A) Immunobiot analysis of primary human fibroblasts from five healthy subjects and patient P3–002. DLAT, DLST, and DBT are the E2 subunit of PDH, AKGDH, and BCKDH complexes, respectively.
(B) Metabolite set enrichment analysis of metabolites decreased in the P3–002 fibroblasts compared to the control primary fibroblasts. The metabolomic profiling was performed on primary human fibroblasts from five healthy subjects and patient P3–002.
(C) Comparison of selected metabolites determined by liquid chromatography-tandem mass spectrometry (LC-MS/MS) in the fibroblasts from five controls and patient P3–002. Data are the mean and SD from 3 replicates for P3–002 and 15 replicates for controls. An unpaired, two-tailed t test was used to assess statistical significance. ****p < 0.0001, ***p < 0.001.
(D) PDH activity determined by the amount of CO2 released from 14C-pyruvate in control primary fibroblasts and P3–002 fibroblasts. Data are the mean and SD from four replicates. ****p < 0.0001, ***p < 0.001, calculated by unpaired two-tailed t test.
(E) PDH activity measured by the amount of CO2 released from 14C-pyruvate in P3–002 fibroblasts with or without ectopic expression of WT LIPT1. Data are the mean and SD from four replicates. ****p < 0.0001, calculated by unpaired two-tailed t test.
(F) Seahorse assay to measure the maximal palmitate-stimulated respiration in P3–002 fibroblasts with or without ectopic expression of LIPT1. Data are the mean and SEM from six replicates. **p < 0.01, *p < 0.05, calculated by unpaired two-tailed t test.
(G) Lipogenesis rate measured with 3H2O in H460 cells containing or lacking LIPT1. Data are the mean and SD from nine replicates. ****p < 0.0001, calculated by unpaired two-tailed t test. The immunobiot analysis of LIPT1 is shown in the insert panel.
(H) Immunobiot analysis of human neonatal fibroblast cell (HNFC), P3–002 fibroblasts, and P3–002 fibroblasts with ectopic expression of WT LIPT1 or either of the two LIPT1 variants (N44S and S292X) observed in the patient.
(I) Relative PDH activity in P-3002 fibroblasts and in P3–002 fibroblasts with ectopic expression of the WT LIPT1 or the LIPT1 mutants (N44S and S292X). Data are the mean and SD from five replicates. ****p < 0.0001, ***p < 0.001, **p < 0.01, calculated by unpaired two-tailed t test.
Metabolomics identified 44 metabolites that discriminated between P3–002 cells and fibroblasts from five healthy subjects (VIP score ≥ 1) (Table S4). Metabolic pathway enrichment analysis (Xia et al., 2015) revealed that TCA cycle metabolites, including malate and fumarate, were depleted in P3–002 fibroblasts, whereas 2-hydroxyglutarate (2-HG) was elevated (Figures 2B and 2C). As expected from clinical testing, P3–002 fibroblasts had reduced PDH activity measured by 14CO2 release from 14C-pyruvate, and expressing wild-type (WT) LIPT1 enhanced PDH activity (Figures 2D and 2E). BCKDH activity was also reduced (Figure S2A). These results, together with the clinical analysis of AKGDH, indicate decreased LIPT1 function in P3–002 fibroblasts, with the most striking defect in PDH.
Fatty acid oxidation supplies mitochondrial respiration and acetyl-CoA formation, particularly when oxidation of other fuels like glucose is limited. Addition of BSA-conjugated palmitate stimulated respiration in P3–002 fibroblasts, but this was blunted in cells re-expressing WT LIPT1 (Figure 2F). We also examined the effect of LIPT1 mutation on lipid synthesis. Acetyl-CoA, the precursor for de novo fatty acid synthesis, was depleted in PS002 cells (Figure S2B). Because de novo fatty acid synthesis was barely detectable in these fibroblasts, we used CRISPR/ Cas9 gene editing to create an isogenic pair of H460 lung cancer cells containing or lacking LIPT1 expression (Figure 2G). These cells were cultured with 3H2O, which results in the incorporation of 3H in newly synthesized fatty acids. LIPT1 deficiency resulted in suppressed 3H labeling in lipids (Figure 2G). These data indicate that LIPT1 deficiency enhances fatty acid oxidation and suppresses fatty acid synthesis, both of which are predicted to reduce total lipid levels as observed in the patient’s plasma.
We then transduced P3–002 cells with vectors expressing WT, N44S, or S292X LIPT1. WT LIPT1 increased lipoylation of both DLAT and DLST (Figure 2H). N44S modestly increased lipoylation, but not nearly to the level of the WT allele. S292X had essentially no effect on lipoylation, partly because the nonsense mutation prevented stable LIPT1 expression in fibroblasts and H460 cells (Figures 2H and S2C). Ectopic expression of WT LIPT1 more than doubled PDH activity, but neither of the mutants had this effect (Figure 2I).
PDH is regulated by reversible phosphorylation states through pyruvate dehydrogenase kinases (PDKs) and pyruvate dehydrogenase phosphatase (PDP). PDKs inhibit PDH by phosphorylating the E1 subunit (PDHA1) at S293 (Yeaman et al., 1973; Dahl et al., 1987). Despite their low PDH activity, P3–002 cells had very low levels of PDHA1 phosphorylation (Figure 2A), likely because lipoylation of E2 is necessary for PDK binding to E1 (Liu et al., 1995). Ectopic WT LIPT1 enhanced PDHA1 phosphorylation, together with DLAT lipoylation (Figure 2H), and enhanced PDH activity (Figure 2I). The N44S mutant restored PDHA1 phosphorylation to about 50% of that induced by the WT LiPT1, consistent with this mutation’s hypomorphic effect. Given the opposite effect of E1 phosphorylation and E2 lipoylation on PDH activity, the results indicate that defective lipoylation caused by LIPT1 mutations is responsible for impaired PDH activity.
Homozygous Lipt1 N44S Mutations in Mice Cause Embryonic Demise
To evaluate the N44S mutation in vivo, we used CRISPR/Cas9 (Yang et al., 2014c) to edit this mutation into the mouse genome. Progeny from founder germline chimeras carrying heterozygous Lipt1N44S/+ alleles were normal in growth and fecundity. Initially, breeding between heterozygous Lipt1N44S/+ mice failed to yield any viable homozygous Lipt1N44S/N44S mice (260 pups from >25 litters) (Figure 3A). About 70% of the progeny were heterozygous for N44S and the rest were homozygous WT, consistent with prenatal loss of Lipt1N44S/N44S embryos.
Figure 3. Homozygosity of the Lipt1 N44S Variant Causes Mid-embryonic Demise in Mice.

(A) Genotype frequencies of embryos derived from Lipt1N44S/+ intercrosses.
(B) Lateral view of mouse embryos at embryonic day (E) 10.5 with the denoted Liptl genotypes. Scale bars: 1 mm.
(C) Immunoblot analysis of embryos with WT Liptl (Liptl+/+) or with heterozygosity {LiptTN44S/+) or homozygosity (Lipt1N44S/N44S) for the N44S variant. All embryos on the blot are from the same litter; these results are representative of two independent litters.
(D) PDH activity in mouse embryos with the denoted genotypes. The data are the mean and SD of Lipt1+/+ (n = 3) or Lipt1N44S/+ (n = 8) embryos. The data from two individual Lipt1N44S/N44S embryos are also shown. Unpaired two-tailed t tests were used to assess statistical significance. *p < 0.05; n.s., not significant.
To establish the timing of embryonic demise, timed matings were established between Lipt1N44S/+ littermates. Genotype analysis detected Lipt1N44S,N44S embryos at 10.5 dpc (17.1% of 188 embryos) but not later (0 of 51 genotyped embryos at 11.5 dpc) (Figure 3A). Lipt1N44S/N44S embryos collected at 10.5 dpc were pale and small compared to Lipt1+/+ or Lipt1N44S/+ littermates (Figure 3B). Immunoblotting revealed defective lipoylation of both PDH and AKGDFI in the Lipt1N44S/N44S embryos, although LIPT1 protein was expressed in all genotypes (Figure 3C). Lipt1N44S/N44S embryos also had decreased PDH activity compared to Lipt1+/+ and Lipt1N44S/+ embryos (Figure 3D). Altogether, these data indicate that homozygous N44S mutations severely impair 2KDH lipoylation and enzyme function in vivo and impair mouse embryonic development.
LIPT1 Regulates Glutamine Metabolism in Support of Respiration, the TCA Cycle, and Fatty Acid Synthesis
We next challenged P3–002 and control fibroblasts to grow in nutrient-replete and nutrient-deprived conditions. In medium with abundant glucose and glutamine and 5% fetal bovine serum, P3–002 fibroblasts had normal morphology and proliferated at a rate only modestly lower than that of controls (Figures S3A and S3B). Growth was enhanced by expressing WT LIPT1 in these cells (Figure S3B). However, in conditional medium with 2 mM glucose, no glutamine, 8 mM galactose, and 2.5% dialyzed fetal bovine serum, P3–002 cells lost their elongated shape, detached from the dishes (Figure S3A), and displayed greater growth suppression than control fibroblasts or P3–002 cells with ectopic WT LIPT1 expression (Figure S3B).
We then supplemented conditional medium with various metabolites to test how they affected energetics and growth. Glutamine supplementation improved cell morphology and enhanced ATP content, as assessed by CellTiter Glo, by 6-fold, similar to the effects of WT LIPT1 (Figures 4A and 4B). Adding TCA cycle metabolites downstream of glutamine also improved morphology and ATP content (Figures 4A and 4B). Aspartate and nucleotides can stimulate growth of cells with impaired electron transport chain (ETC) activity (Birsoy et al., 2015; Sullivan et al., 2015). Aspartate modestly rescued LIPT1-deficient fibroblasts in conditional medium, but adding the nucleotides adenosine, guanosine, and uridine together had no effect (Figure S3C). Unlike WT LIPT1, none of these supplements increased lipoylation (Figures 4C and S3D). Expressing WT LIPT1 or supplementing with glutamine or glutamine-derived metabolites normalized the metabolome in P3–002 cells grown in conditional medium, including by replenishing the depleted TCA cycle intermediates (Figure 4D; Table S5).
Figure 4. Oxidative and Reductive Glutamine Metabolism Support TCA Cycle Replenishment, Fatty Acid Synthesis, and Growth of LIPT1Deficient Fibroblasts.

(A) Representative phase contrast images showing the morphology of P3–002 fibroblasts in culture when LIPT1 was ectopically expressed or when the indicated metabolites were supplemented in conditional medium (2 mM glucose, 0 mM glutamine, 8mM galactose, and 2.5% dialyzed fetal bovine serum [FBS]). Scale bar: 100
(B) P3–002 fibroblast viability and bioenergetics were evaluated in conditional medium supplemented with the indicated metabolites for 48 h. The results are shown as mean and SD from three replicates, and p values are calculated by unpaired two-tail t test. ***p < 0.001, **p < 0.01, *p < 0.05.
(C) Immunoblot analysis of P3–002 fibroblasts cultured with the indicated metabolites in the conditional medium for 48 h.
(D) Heatmap of metabolic pathways enriched after LIPT1 expression or metabolite supplementation in P3–002 fibroblasts. The color scale represents the impact score on pathways using metabolomic pathway analysis (MetPA). Gray indicates no enrichment of the indicated pathway.
(E) Cellular respiration rate measured in P3–002 fibroblasts in the presence or absence of glutamine (2 mM). Data are the mean and SEM from eight replicates. ****p < 0.0001, calculated by unpaired two-tail t test.
{F) Fractional enrichment of the citrate isotopologues in P3–002 fibroblasts with or without ectopic LIPT1 expression (P3–002+LIPT1). The cells were cultured in [U-13C]glutamine for 24 h. Data are the mean and SD from three replicates. **p < 0.01, calculated by unpaired two-tail t test.
(G) Fractional enrichment of the malate isotopologues in P3–002 or P3–002+LIPT1 fibroblasts after culture with [U-13C]glutarmine for 24 h. Data are the mean and SD from three replicates. **p < 0.01, calculated by unpaired two-tail t test.
(H) Fractional enrichment of the citrate isotopologues in WT or L1PT1-knockout (KO) H460 cell clones after culture with [U-13C]glutamine for 6 h.
(I) Fractional enrichment of the malate isotopologues in WT or LIPT1-KO H460 cell clones after cultured with [U-13C]glutamine for 6 h.
(J) Mass isotopologue distribution in palmitate from WT or LIPT1-KO H460 cells after culture with [U-13C]glutamine for 24 h. Data are the mean and SD from three replicates.
(K) Mass isotopologue distribution in palmitate from WT or LIPT1-KO H460 cells after culture with [U-13C]glucose for 24 h. Data are the mean and SD from three replicates.
(L) Major metabolic alterations associated with LIPT1 deficiency in humans. Green arrows indicate elevation, and red arrows indicate reduction.
We assessed how P3–002 cells metabolize glutamine. Glutamine stimulated respiration (Figure 4E), indicating persistent glutamine oxidation. We next cultured the fibroblasts in [U-13C] glutamine for 24 h and extracted metabolites for 13C analysis. Various defects in oxidative metabolism result in reductive labeling of TCA cycle intermediates through reversible isocitrate dehydrogenase-1 (IDH1) and/or isocitrate dehydrogenase-2 (IDH2) function (Mullen et al., 2011, 2014; Rajagopalan et al., 2015). During culture with [U-13C]glutamine, oxidative TCA cycle function results in m+4 labeling in malate and citrate, whereas reductive labeling results in m+5 citrate and m+3 malate (Mullen et al., 2011). In P3–002 fibroblasts, culture with [U-13C]glutamine produced a combination of oxidative and reductive labeling, with oxidative labeling predominating (Figures 4F and 4G; isotopologue distributions in Table S6). Expressing WT LIPT1 diminished the fraction of reductive labeling, particularly in citrate, indicating that impaired LIPT1 function contributes to reductive TCA cycle function (Figures 4F and 4G). Persistent glutamine oxidation likely reflects residual LIPT1 function from one or both LIPT1 variants; this is consistent with the residual IDLST lipoylation and AKGDH enzyme activity in these cells (Figure 2A). Eliminating LIPT1 function in H460 cells resulted in a larger skew toward reductive labeling (Figures 4H and 4I).
LIPT1 deficiency also enhanced glutamine’s contribution to fatty acid synthesis, as demonstrated by the shift toward higher-order 13C labeling of palmitate (Figure 4J). Conversely, incorporation of 13C from glucose into palmitate was suppressed in LIPT1 -deficient cells (Figure 4K). Both changes are consistent with enhanced reductive glutamine metabolism in LIPT1-deficient cells (Mullen et al., 2011). Altogether, these data indicate the importance of LIPT1 in regulating the fate of carbon from both glucose and glutamine and demonstrate that glutamine’s contributions to the TCA cycle and other pathways support LIPT1 mutant cell growth and viability.
DISCUSSION
Many IEMs are detected through clinical measurement of a handful of metabolites from the plasma and urine. These relatively straightforward tests are important in detecting treatable diseases, including those in population-based newborn screening programs. However, two key factors determining the efficiency with which IEMs are diagnosed are the specificity of the phenotype and the underlying genetic heterogeneity of the disorder. For example, medium-chain acyl-CoA dehydrogenase deficiency (MCADD; OMIM: 607008) is a disorder of fatty acid p-oxidation resulting in an easily recognizable pattern of medium-chain acylcarnitines and dicarboxylic acids. MCADD patients have bi-allelic mutations in ACADM on human chromosome 1p31.1. By contrast, OMIM lists more than 200 monogenic disorders associated with lactic acidosis, many with overlapping phenotypes. Pinpointing the molecular cause of these diseases is challenging and increasingly involves advanced tools like WES. Our analysis of LIPT1 deficiency suggests that broad metabolomic profiling can help prioritize genetic variants for functional analysis.
LIPT1 mutations have been observed in a few previous reports of children with lactic acidosis (Soreze et al., 2013; Stowe et al., 2018;Tacheetal., 2016; Tort et al., 2014). All patients had prominent metabolic acidosis and neurologic involvement, with three dying in the neonatal period. Elevated glutamate and proline fevels were reported in two children (Soreze et al., 2013; Stowe et al., 2018). In agreement with our patient’s phenotype, glycine levels were normal when reported. This distinguishes LIPT1 deficiency from defects in lipoic acid synthesis, which prevent lipoyiation of the H-protein of the glycine cleavage system and cause combined hyperglycinemia and 2KDH dysfunction (Solmonson and DeBerardinis, 2018). Unlike bacteria, eukaryotic cells appear to lack an efficient system to scavenge free lipoic acid to modify 2KDHs, making LIPT1-mediated transfer of the lipoyl moiety from H-protein essential to activate these enzymes (Hermes and Cronan, 2013; Schonauer et al., 2009; Yi and Maeda, 2005). Supplementing LIPT1-defective cells with free lipoic acid was reported to result in a modest suppression of lactate production, although lipoylation of 2KDH complexes was unchanged (Soreze et al., 2013). This may indicate an unknown role for lipoate beyond its function as a covalently bound 2KDH cofactor. As is often the case in children suspected to have mitochondrial diseases, our patient was treated with lipoic acid during her workup. Her parents noted a reduced frequency of myoclonic seizures, so the treatment was continued, although the subsequent plasma analysis reported here revealed evidence of persistent 2KDH dysfunction.
Metabolomics broadened the phenotype of LIPT1 deficiency (Figure 4L). First, an unsupervised analysis of aqueous metabolites differentiated our patient from controls and a healthy sibling. Second, plasma lipidomics revealed depletion of PCs, SMs, and CERs, and cultured LIPT1-deficient cells displayed defective lipid synthesis and enhanced fatty acid oxidation, both of which are predicted to cause lipid depletion.
Third, we observed accumulation of both isomers of 2-HG. This metabolite is best known in cancers with mutations in IDH1 or IDH2 (Ward and Thompson, 2012). These mutations impart a neomorphic activity, allowing the enzyme to catalyze the NADPH-dependent reduction of α-KG to D-2-HG (Dang et al., 2009). IEMs associated with elevated 2-HG include mutations in L2HGDH or D2HGDH, the dehydrogenases that convert naturally occurring L- or D-2-HG to α-KG (Kranendijk et al., 2012). The only IEMs reported to cause elevations of both isomers are defects in the mitochondrial citrate carrier encoded by SLC25A1 (Notaetal., 2013). In LIPT1 deficiency, combined L- and D-2-HG accumulation may result from elevated α-KG caused by reduced AKGDH lipoylation in some tissues. The striking rise in L-2-HG during acidosis may be related to the propensity of malate dehydrogenase and lactate dehydrogenase to convert α-KG to L-2-HG at low pH (Intlekoferetal., 2017; Nadtochiy et al., 2016). At high levels, both L- and D-2-HG are thought to elicit epigenetic effects through their influence on histone demethylases and TET family 5-methylcytosine hydroxylases, which require α-KG as a substrate (Ansó et al., 2017; Figueroa et al., 2010; Shim et al., 2014). Tissue levels of 2-HG were not measured in our patient, but her plasma levels were lower than the millimolar concentrations observed in IDH1/2 mutant tumors. It is unknown whether 2-HG elevations in LIPT1 deficiency lead to clinically relevant epigenetic alterations.
The effect of LIPT1 deficiency on substrate utilization has not been studied in detail. P3–002 cells were sensitive to glutamine deprivation, and glutamine replenishment was similar to expression of WT LIPT1 in its ability to restore growth. Although P3–002 fibroblasts oxidized glutamine, this was accompanied by reductively labeled citrate (citrate m+5), and complementing with WT LIPT1 reduced this fraction. These findings, coupled with the depletion of 4-carbon TCA cycle intermediates, emphasize altered anaplerosis in LIPT1 deficiency. Accordingly, feeding glutamine-deprived fibroblasts with single anaplerotic intermediates enhanced ATP content. Glutamine’s contribution to fatty acid synthesis was also increased in LIPT1-deficient cells, as commonly observed in settings of reductive metabolism (Metallo etal., 2011; Mullen etal.,2011; Rajagopalanetal., 2015). Thus, in LIPT1 deficiency, glutamine continues to supply pathways that contribute to energy formation and biosynthesis.
Poor 2KDH lipoylation and mid-gestation demise of LIPT7N44S/N44S homozygotes verify LIPT1's importance in embryonic development. Defects in lipoate biosynthesis also cause embryonic lethality (Yi and Maeda, 2005). It is unclear why N44S homozygosity caused embryonic lethality in these mice (mixed C57BL/6J and C3H/HeJ background) while the proband survived development. The proband is a compound heterozygote, perhaps resulting in a different level of enzyme activity than N44S homozygosity. Furthermore, although N44S and S292X had similar effects on PDH in fibroblasts, the cells and embryos differed markedly in 2KDH lipoylation. P3–002 cells displayed near-complete loss of PDH lipoylation but retained some AKGDH lipoylation and activity, whereas the embryos displayed more prominent effects on AKGDH. This may explain the seemingly more severe phenotype in the mice. In humans, isolated PDH deficiency causes postnatal lactic acidosis, but most patients lack major anomalies in organ development. In contrast, isolated AKGDH deficiency is associated with defects in embryonic development, in addition to metabolic dysfunction after birth (Surendran et al., 2002).
Although there is no definitive treatment for LIPT1 deficiency, this case provides an example of the benefits of establishing the diagnosis and clarifying the functional significance of rare genetic variants. First, establishing the molecular diagnosis enables specific counseling regarding recurrence risk. Second, because a few other LIPT1-deficient patients have been reported, it becomes possible to assess the clinical spectrum of this disease and to begin to establish genotype-phenotype correlations that might predict outcomes. We believe the disorder is underdiagnosed based on the high frequency of unexplained syndromes involving lactic acidosis. Incorporating metabolomics into the diagnostic workflow should help identify patients from this group with defects in LIPT1 and lipoic acid synthesis (Tort et al., 2016). Finally, confirming LIPT1 deficiency helps direct some aspects of therapy. Because these children have reduced PDH activity, it is reasonable to consider therapies used in isolated PDFI deficiency. These include ketogenic diets and dichloroacetate (DCA), which reduces inhibitory phosphorylation of PDH’s E1 complex by inh ibiting PDKs. But neither of these therapies are expected to be effective in LIPT1 deficiency. Ketogenesis from branched-chain amino acids is compromised by poor lipoylation of BCKDH. Our patient experienced severe metabolic acidosis on a ketogenic diet, perhaps because branched-chain ketoacids accumulated and/or because of excessive ketogenesis from fatty acids in the setting of compromised utilization of other fuels. DCA lowers lactic acidosis in isolated PDH deficiency (Stacpoole et al., 2003), but here we demonstrate that PDH phosphorylation is barely detectable in the absence of lipoylation. Other modalities, such as the nutritional provision of anaplerotic precursors and pharmacologic inhibition of delipoylating enzymes, may benefit patients with LIPT1 deficiency.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed and will be fulfilled by the Lead Contact, Ralph J. DeBerardinis (ralph.deberardinis@utsouthwestern.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Clinical samples
Plasma samples were obtained from fresh blood collected in heparinized tubes from the patient, her family and 60 children admitted to the newborn screening follow-up clinic at the Children’s Medical Center at Dallas. Punch biopsies of the skin for fibroblast culture were obtained from the patient, following the standard of procedure for clinical diagnostics. All subjects were enrolled in the study (NCT02650622) approved by the Institutional Review Board (IRB) at University of Texas Southwestern Medical Center (UTSW). informed consent was obtained from all patients and their families. The gender and age information of human subjects were included in the data tables for plasma metabolomics (Table S2) and lipidomics (Table S3).
Cell lines
Primary patient fibroblasts were grown from skin biopsy specimen and the immortalized line was generated by stable expression of hTERT. The human normal fibroblast lines were obtained from ATCC. The fibroblasts were maintained in low-glucose DMEM (Sigma, D6046) supplemented with 5% heat-inactivated fetal bovine serum (FBS), and cultured at 37°C with 5% CO2.
Mice
All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at UTSW, and conducted in accordance with institutionally approved protocols and guidelines for animal care and use. Liptl N44S knock-in mice were generated by the Mouse Genome Engineering Core of the Children’s Research Institute at UTSW using CRISPR/Cas9 genome editing technology. Guide RNA was designed to target the sequence that is 24 bp downstream of the N44S point mutation site. Guide RNA, S. pyogenes Cas9 mRNA, and a single-strand oligo donor containing the N44S point mutation were injected into single celled zygotes. B6C3F1/J mice were used to generate founder mice. To generate Lipt1N44S/+ study mice, C57BL/6J WT females were crossed to Lipt1N44S/+ founder males to confirm germline transmission. Interbreeding of Lipt1N44S/+ male and female littermates was performed to obtain Lipt1+/+, Lipt1N44S/+ and Lipt1N44S/ N44S mice or embryos.
METHOD DETAILS
Genotyping
The mutation status of the LIPT1 gene was identified by clinical whole exome sequencing and verified by Sanger sequencing at Baylor Genetics Laboratory using genomic DNA from the proband and her parents. In this study, we obtained genomic DNA from whole blood from the proband and her healthy sibling using a QIAGEN kit, following the manufacturer’s instructions. 420 bp regions covering the N44 or S292 sites were PCR amplified using the primer pairs listed above. The genotypes were then determined by Sanger sequencing (Genewiz).
Targeted metabolomics
Fifty μL of patient plasma was added to 950 μL of ice-cold methanol/water 80% (vol/vol) for metabolite extraction. After vigorous mixing and centrifuging, the metabolite-containing supernatant was dried in a SpeedVac concentrator (Thermo Savant) for 5–7 hours. For analysis of patient fibroblasts, 500,000 cells were collected and lysed with ice-cold 80% methanol. After three freeze-thaw cycles in liquid nitrogen, the lysate was centrifuged to remove debris and the supernatant was collected and dried in a SpeedVac. The metabolite pellets obtained from either plasma samples or fibroblasts were analyzed by targeted metabolomics using a liquid chromatography-tandem mass spectrometry (LC-MS/MS) approach as described previously (Mullen et al., 2014). Briefly, metabolites were reconstituted in 100 μL of 0.03% formic acid in LCMS-grade water, vortex-mixed and centrifuged to remove debris. LCMS/MS and data acquisition were performed using an AB QTRAP 5500 liquid chromatography/triple quadrupole mass spectrometer (Applied Biosystems SCIEX) with an injection volume of 20 μL Chromatogram review and peak area integration were performed using MultiQuant software version 2.1 (Applied Biosystems SCIEX). The peak area for each detected metabolite was normalized against the total ion count of each sample and the mean for each metabolite within the sample batch of each run to correct for variations introduced by sample handling through instrument analysis. The normalized areas were used as variables for the multivariate analyses and modeling using SIMCA-P (version 13.0.1; Umetrics). The processed datasets were mean-centered, unit-variance scaled and then applied to principal component analysis to evaluate the clustering and to detect outliers. Univariate statistical differences of the metabolites between two groups were analyzed using Student’s t test. The pathway enrichment analysis of differential metabolites between two groups was performed using Metaboanalyst 3.0 (http://www.metaboanalyst.ca).
Quantitation of L-2-HG and D-2-HG
Measurement of L-2-HG and D-2-HG was performed as described previously (Rakheja et al., 2011). Briefly, the metabolites were extracted from plasma samples and derivatized with (+)-Di-O-acetyl-L-tartaric anhydride and injected for chromatographic separation on an Agilent Hypersil ODS 4.0 3 250 mm, 5 mm column followed by detection and measurement using an API 3000 triple-quadrupole mass spectrometer equipped with an ESI source (Applied Biosystems). MRM transitions were monitored at 363.2 > 147.2 for both L-2-HG and D-2-HG.
Lipidomics
The lipid extracts were infused into a Sciex quadrupole time of flight (QTOF) TripleTOF 6600 mass spectrometer (Framingham, MA, USA) via a custom configured LEAP InfusePAL HTS-xt autosampler (Morrisville, NC, USA). The sample was infused over 3 minutes to the QTOF at a flow rate of 10μL/min. Chronos XT® software from Axel Semrau (Srockhovel, Germany) was used to control the infusePAL system. The electrospray ionization (ESI) source parameters were, GS1 at 25, GS2 at 55, curtain gas (Cur) at 20, temperature at 300°C and ion spray voltage at 5500V and −4500V in positive and negative ionization mode, respectively. The optimal declustering potential and collision energy settings were 120V and 40eVfor positive ionization mode and −90V and −50eV for negative ionization mode. For the MS/MSALL analysis, a production spectrum was collected at each mass unit from 200–1200 Da with an accumulation time of 0.3 s per mass. Analyst® TF 1.7.1 software (Sciex) was used for the TOF MS and MS/MSALL data acquisition. The obtained data were processed by in-house software, LipPY (Dallas, Tx, USA). The intensity of each peak was normalized to the total lipid signal and the z scores were determined over the whole sample set. The heatmap of lipid species was generated by Pleatmap 3 (https://cran.r-project.org/web/packages/heatmap3/index.html) using the R package.
2KDH activity assay
Enzyme activities of PDH and BCKDH complexes were assessed as described previously (Yang et al., 2014a). Briefly. Micro-bridges (Hampton Research) were placed into wells of a 24-well plate with one piece of 0.6 × 1 cm2 chromatography paper per well. PBS with 0.2% of BSA was used as basic assay medium. One million cells per well were then suspended in 350 μLof the assay medium on ice, supplemented with 4 mM [1-14C]pyruvate(0.1 μCi/well), or 100 μM [U-14C]valine (0.25 μCi/well), respectively. Each micro-bridge was moistened with 20 μL of phenethylamine, and the plate was sealed with adhesive film. Reactions were initiated by transferring the plate to a 37°C water bath. After 60 min of incubation, metabolism was terminated by adding 50 μL of 20% trichloroacetic acid. The plate was re-sealed with adhesive film and incubated at room temperature for another 60 min to release 14CO2. Then the 14CO2-containing chromatography papers were collected for scintillation counts. The medium aliquot from each sample was used to quantify radioactivity on a scintillation counter. This value was used to determine the specific activity of each substrate, respectively. Samples containing 350 pg BSA without cells in 350 μl assay medium were used to establish background levels of 14CO2. This level was used as a background value to be subtracted from assayed samples. Enzyme activities were presented as 14CO2 production rates as nmol/hr of 10e6 cells.
Lipogenesis assay
Lipogenesis was assayed using tritium-labeled water described previously (Mullen et al., 2011) with slight modifications. 4–8 million cells were plated in to 225 cm2 flasks with regular DMEM culture medium on day 0. On day 1, cells were switched to 35 mL fresh culture medium supplemented with 0.5 mCi tritium water (PerkinElmer, 1 mCi / ml). After 48 hours of incubation, cells were collected by trypsinization and total lipids were extracted by Bligh/Dyer method (Bligh and Dyer, 1959). Newly synthesized lipids were determined by measurement of radio activities from 3H radioisotope using scintillation counter.
Fatty acid oxidation assay
Fatty acid oxidation was measured using Seahorse FAO assay, following the manufacturer’s protocol. Briefly, 1×104 cells per well were seeded into a 96-well Seahorse assay plate with regular growth medium. After 24 hours, the medium was replaced with substrate-limited medium (0.5 mM glucose, 0.5 mM carnitine and 1% dialyzed FBS) supplemented with or without 1mM glutamine. After cultured for another 24 hours, the cells were washed twice with FAO assay medium (111 mM NaCl, 4.7 mM KCl, 1.25 mM CaCl2, 2 mM MgSO4, 1.2 mM NaH2PO4) supplemented with 2.5 mM glucose, 0.5mM carnitine and 5 mM HEPES, and then incubated in a non-CO2 incubator for 45 minutes at 37°C. At the same time, the assay cartridge with cell mito stress compounds (final concentrations: 2 uM oligormycin, 1 uM CCCP, 2 uM antimycin A) were prepared and loaded onto the machine. 15 minutes prior to starting the assay, Etomoxir was added to the designated wells at the final concentration of 17 uM. After incubate for 15 minutes, BSA or palmitate-BAS (1 mM) was added to the designated wells and the plate was immediately loaded onto the machine to run the assay. The maximal respiration rate was analyzed using Seahorse Wave software.
PDH activity colorimetric assay
Mitochondria were isolated from ten million fibroblasts using the Mitochondria Isolation Kit (Biovision, K288–50) and the mitochondrial PDH activity was assessed using the Pyruvate Dehydrogenase Activity Colorimetric Assay kit (Biovision, K679–100), following the manufacturer’s instructions. Protein concentration was determined using the BCA protein assay kit (Pierce, 23227).
Acetyl-CoA assay
Two million primary fibroblasts were lysed and deproteinized using a perchloric acid (PCA)/KOH protocol (Deproteinizing Sample Preparation Kit, BioVision). 10 μL of the samples were added into assay wells in triplicate to measure the acetyl-CoA level using the PicoProbeAcetyl-CoA Fluorometric Assay kit (BioVision) according to the manufacturer’s manual. Standard curves were generated to calculate the amount of acetyl-CoA in each sample with background correction.
Lentiviral production and transduction
Lentivirus was produced by transfection of HEK293T cells using the LENTI-Smart reagent (Invivogen, ltslnt-10). Viral supernatants were harvested at 48 hr and 72 hr, filtered through a 0.45 um filter and concentrated using PEG-it Virus Precipitation Solution (System Biosciences, LV810A-1). For transduction, the lentiviral pellets were suspended in culture medium and added to fibroblasts at 70%80% confluency per well in 6-well plates. After 48 hr of transduction, the fibroblasts were selected under puromycin for one week for stable expression of wild-type or mutant LIPT1.
Cell viability assay
Fibroblasts were seeded into 96-well plates at a density of 3×103 cells per well. After 48 hr, the cells were cultured in the conditional DMEM containing no L-glutamine, 2 mM glucose, 8 mM galactose and 5% dialyzed FBS (Gemini Bio-Products, 100–108) with or without supplementation of the indicated metabolites. After incubation for 48 hr, the cells were subjected to the CellTiter-Glo Luminescent Cell Viability Assay (Promega, G7573) according to the manufacturer’s instruction.
Immunoblotting
Whole cell lysates were extracted from cells or embryos in RIPA buffer followed by three freeze/thaw cycles. The protein supernatants were quantified using the BCA protein assay (Pierce, 23227). Proteins were separated on 4%-20% SDS-PAGE gels, transferred to PVDF membranes, and probed with primary antibodies against the indicted proteins as listed above and secondary antibodies. Immunoreactive proteins were visualized by enhanced chemiluminescence (Pierce, 32106).
Stable isotope tracing
Isotope tracing was modified from a previously described procedure (Cheng et al., 2011; Faubert et al., 2017; Yang et al., 2014b). Briefly, 5×105 H460 lung cancer cells or fibroblasts were traced with 4 mM [U-13C]glutamine in phenol-red free DMEM with 5% dialyzed FBS forthe indicated time. Then cells were rinsed in ice-cold saline and collected in 80% ice-cold methanol. After three freeze-thaw cycles, the lysates were centrifuged to remove proteins and cell debris. The purified lysates, with 1 ml of d27-myristic acid added as an internal control, were evaporated and derivatized using TBDMS. Two ul_ of the derivatized samples were injected and analyzed using an Agilent 7890 gas chromatograph coupled to an Agilent 5975C Mass Selective Detector, respectively. The observed distributions of mass isotopologues were corrected for natural abundance of 13C. To assay 13C enrichment in palmitate, cells were cultured with 10 mM [U-13C]glucose or 4 mM [U-13C]glutaminefor 24 hours, and then lipids were extracted in methanol:chloroform: water (1:1:1, v/v/v). The organic phase was evaporated with air dryer. Samples were processed with 100 μL of 1N HCl, followed by adding of 200 μL toluene to form two layers after vortexing and centrifuging. The toluene layers were transferred to glass tubes, evaporated to dry and derivatized using TBDMS. Two μL of the derivatized samples were injected and analyzed using an Agilent 7890 gas chromatograph coupled to an Agilent 5975C Mass Selective Detector, respectively. Fragment ion m/z 313–329 was used to monitor enrichment in palmitate.
QUANTIFICATION AND STATISTICAL ANALYSIS
No method was used to predetermine sample sizes. Metabolomics and lipidomics of patient plasma samples or human fibroblasts were performed once with multiple replicates. Biochemical assays of 2KDH activities, fatty acid oxidation, lipogenesis, CTG activity and respiration were performed two or three times with multiple replicates. Biochemical assays of PDH activity in mouse embryos were performed once with multiple replicates. Isotope tracing assays were performed 2–3 times with multiple replicates. Samples for metabolomics, lipidomics and isotope tracing were randomized before analysis. All other experiments were nonrandomized and no blinding of the investigators was involved. To assess statistical significance between two groups, a two-tailed unpaired t test was used, and data were presented as mean ± SEM or SD (indicated in the figure legend). In all figures, the p values were shown as: *, p < 0.05; **, p < 0.01; ***, p < 0.001, ****, p < 0.0001.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Lipoic acid | Millipore | 437695-100UL; RRID:AB_212120 |
| Anti-LIPT1 | Sigma-Aldrich | AV48784; RRID:AB_1852908 |
| Anti-DLAT | Cell Signaling | #12362; RRID:AB_2797893 |
| Anti-DLAT | Santa Cruz | sc-271534; RRID:AB_10649809 |
| Anti-DLST | Cell Signaling | #5556; RRID:AB_10695157 |
| Anti-DBT | GeneTex | GTX113412; RRID:AB_2036741 |
| Anti-p-PDHA1(Ser293) | Millipore | ABS204; RRID:AB_11213668 |
| Anti-PDHA1 | Cell Signaling | #3205; RRID:AB_2162926 |
| Anti-ACTB | Sigma-Aldrich | A3854; RRID:AB_262011 |
| Anti-GAPDH | Cell Signaling | #8884; RRID:AB_11129865 |
| Biological Samples | ||
| Patients’ blood samples | This paper | N/A |
| Patient’s skin biopsies | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Puromycin | ThermoFisher | A1113803 |
| Etomoxir sodium salt hydrate | Sigma-Aldrich | E1905 |
| α-Ketoglutarate | Sigma-Aldrich | K1128 |
| Citrate | Sigma-Aldrich | 251275 |
| Fumarate | Sigma-Aldrich | F19353 |
| Malate | Sigma-Aldrich | 240176 |
| Succinate | Sigma-Aldrich | S3674 |
| L-Aspartate | Sigma-Aldrich | A7219 |
| Adenosine | Sigma-Aldrich | A9251 |
| Guanosine | Sigma-Aldrich | G6264 |
| Uridine | Sigma-Aldrich | U3003 |
| [U-13C5]Glutamine | Cambridge Isotope Laboratories | CLM-1822 |
| [U-13C6]Glucose | Cambridge Isotope Laboratories | CLM-1396 |
| [1-14C]Pyruvate | PerkinElmer | NEC255050UC |
| L-[U-14C]Valine | PerkinElmer | NEC291EU050UC |
| 3H2O | PerkinElmer | 6004052 |
| Critical Commercial Assays | ||
| Kit Lenti-Smart INT | Invivogen | ItIns-10 |
| Kit mammalian mitochondria isolation | BioVision | K288 |
| Kit PDH activity colorimetric assay | BioVision | K679 |
| Deproteinizing Sample Preparation Kit | BioVision | K808 |
| Kit PicoProbeAcetyl-CoA Fluorometric Assay | BioVision | K317 |
| Kit Q5 Site-Directed Mutagenesis Kit | NEB | E0554S |
| Kit CellTiter-Glo luminescent cell viability assay | Promega | G7572 |
| Deposited Data | ||
| Human LIPT1 mutation N44S | ClinVar | SCV000588895 |
| Human LIPT1 mutation S292X | ClinVar | SCV000588895 |
| Experimental Models: Cell Lines | ||
| Patient-derived fibroblast and ctrl-4917 control fibroblast | This paper | N/A |
| Primary normal human neonatal fibroblasts | ATCC | PCS-201-010 |
| Primary normal human adult fibroblasts | ATCC | PCS-201-012 |
| Detroit 551 | ATCC | CCL-110 |
| BJ | ATCC | CRL-2522 |
| H460 | ATCC | HTB-177 |
| Experimental Models: Organisms/Strains | ||
| B6C3F1/J mice | The Jackson Laboratory | 100010 |
| Lipt1N44S/+ mice | This paper | N/A |
| Oligonucleotides | ||
| Genotyping primer LIPT1-N44S forward: 5′-AAAGCCAAAAGCAACTGCAC-3′ | This paper | N/A |
| Genotyping primer LIPT1-N44S reverse: 5′-GCCAAGGATTTTGATGCCTA-3′ | This paper | N/A |
| Genotyping primer LIPT1-S292X forward: 5′-AAACCCAACGGATGAGACAC-3′ | This paper | N/A |
| Genotyping primer LIPT1-S292X reverse: 5′-TGTTTAGTTTGTGGTCTTGTGGA-3′ | This paper | N/A |
| Cloning primer LIPT1 forward: 5′-CCGGCGATCGCATGCTGATCCCATTTTC –3′ | This paper | N/A |
| Cloning primer LIPT1 reverse: 5′-CGGACGCGTCATTATTCCCTTAATTTTTTC-3′ | This paper | N/A |
| Cloning primer LIPT1-S292X reverse: 5′-CGGACGCGTCTGTTCATATAACACATGAAAGG-3′ | This paper | N/A |
| Mutagenesis primer LIPT1-N44S forward: 5′-GTCTATCAAAGTCTGGCTGTG-3′ | This paper | N/A |
| Mutagenesis primer LIPT1-N44S reverse: 5′-ATCATTGGAAATTGACTGTAAAATG-3′ | This paper | N/A |
| Genotyping primer Lipt1-N44S forward: 5′-AAGAAGGTCTGGACCCTTTGA-3′ | This paper | N/A |
| Genotyping primer Lipt1-N44S reverse: 5′-GCCAGTTTGATTCCTTCCTG-3′ | This paper | N/A |
| sgRNA target sequence of Lipt1 for CRISPR knock-in of N44S: 5′-CTTCTAGATGTATGTGGTCGTGG-3′ | This paper | N/A |
| HR template single-strand oligo for CRISPR knock-in of N44S: 5′-CTTTGTCAGCACAAGGTCCCAGCAGCTGGCTTTAAAAGCCCACCCACACATGGGCTCATTTTACAGTCGATCTCCAATGATGTATATGAAAGTCTGGCTTTTGAAGACTGGATTCATGACCACATACATCTCGAAGGCAAGCCGATTCTTTTCCTTTGGAGAAATTCTCCTTCTGTCGTCATC-3′ | This paper | N/A |
| Recombinant DNA | ||
| pLOX-TERT-iresTK | Addgene | 12245 |
| pLenti-EF1a-C-Myc-DDK-IRES-Puro | OriGene | PS10085 |
| pLenti-EF1a-C-Myc-DDK-IRES-Puro-LIPT1-WT | This paper | N/A |
| pLenti-EF1a-C-Myc-DDK-IRES-Puro-LIPT1-N44S | This paper | N/A |
| pLenti-EF1a-C-Myc-DDK-IRES-Puro-LIPT1-S292X | This paper | N/A |
| Software and Algorithms | ||
| MultiQuant software v2.1 | Applied Biosystems | N/A |
| SIMCA-P v13.0.1 | Umetrics | N/A |
| Heatmap3 | https://cran.r-project.org/web/packages/heatmap3/index.html | N/A |
| MetaboAnalyst 3.0 | http://www.metaboanalyst.ca/ | N/A |
| HOPE-CMBI | http://www.cmbi.ru.nl/hope/ | N/A |
| PyMol | https://pymol.org/2/ | N/A |
Highlights.
Human LIPT1 mutations impair 2-ketoacid dehydrogenase lipoylation and activity
LIPT1 deficiency increases 2-HG and depletes structural lipids in plasma
LIPT1 deficiency impedes lipogenesis but increases fatty acid oxidation
LIPT1 regulates the balance between oxidative and reductive glutamine metabolism
ACKNOWLEDGMENTS
The Genetic and Metabolic Disease Program is supported by the Children’s Medical Center Research Institute and the UT Southwestern Department of Pediatrics. R.J.D. is supported by the Howard Hughes Medical Institute, Welch Foundation (grant I-1733), and Once Upon a Time Foundation.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.Org/10.1016/j.celrep.2019.04.005.
REFERENCES
- Ansó E, Weinberg SE, Diebofd LP, Thompson BJ, Malinge S, Schumacker PT, Liu X, Zhang Y, Shao Z, Steadman M, et al. (2017). The mitochondrial respiratory chain is essential for haematopoietic stem cell function. Nat. Cell Biol. 19, 614–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birsoy K, Wang T, Chen WW, Freinkman E, Abu-Remaileh M, and Sabatini DM (2015). An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 162, 540–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bligh EG, and Dyer WJ (1959). A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917. [DOI] [PubMed] [Google Scholar]
- Cheng T, Sudderth J, Yang C, Mullen AR, Jin ES, Mates JM, and DeBerardinis RJ (2011). Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc. Natl. Acad. Sci. USA 108, 8674–8679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary MA, and Green A (2005). Developmental delay: when to suspect and how to investigate for an inborn error of metabolism. Arch. Dis. Child. 90, 1128–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl HH, Hunt SM, Hutchison WM, and Brown GK (1987). The human pyruvate dehydrogenase complex. Isolation of cDNA clones for the E1 alpha subunit, sequence analysis, and characterization of the mRNA. J. Biol. Chem. 262, 7398–7403. [PubMed] [Google Scholar]
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et al. (2009). Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davison JE, and Rahman S (2017). Recognition, investigation and management of mitochondrial disease. Arch. Dis. Child. 102,1082–1090. [DOI] [PubMed] [Google Scholar]
- DeBerardinis RJ, and Thompson CB (2012). Cellular metabolism and disease: what do metabolic outliers teach us? Cell 148,1132–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EI-Hattab AW (2015). Inborn errors of metabolism. Clin. Perinatol. 42, 413–439. [DOI] [PubMed] [Google Scholar]
- Farwell KD, Shahmirzadi L, El-Khechen D, Powis Z, Chao EG, Tippin Davis B, Baxter RM, Zeng W, Mroske C, Parra MC, et al. (2015). Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet. Med. 17, 578–586. [DOI] [PubMed] [Google Scholar]
- Farwell Hagman KD, Shinde DN, Mroske C, Smith E, Radtke K, Shahmirzadi L, EI-Khechen D, Powis Z, Chao EC, Alcaraz WA, et al. (2017). Candidate-gene criteria for clinical reporting: diagnostic exome sequencing identifies altered candidate genes among 8% of patients with undiagnosed diseases. Genet. Med. 19, 224–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faubert B, Li KY, Cai L, Hensley CT, Kim J, Zacharias LG, Yang C, Do QN, Doucette S, Burguete D, et al. (2017). Lactate Metabolism in Human Lung Tumors. Cell 171, 358–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, et al. (2010). Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18, 553–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara K, Hosaka H, Matsuda M, Okamura-Ikeda K, Motokawa Y, Suzuki M, Nakagawa A, and Taniguchi H (2007). Crystal structure of bovine lipoyltransferase in complex with lipoyl-AMP. J. Mol. Biol. 371, 222–234. [DOI] [PubMed] [Google Scholar]
- Hermes FA, and Cronan JE (2013). The role of the Saccharomyces cerevisiae lipoate protein ligase homologue, Lip3, in lipoic acid synthesis. Yeast 30, 415–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intlekofer AM, Wang B, Liu H, Shah H, Carmona-Fontaine C, Rustenburg AS, Salah S, Gunner MR, Chodera JD, Cross JR, and Thompson CB (2017). L-2-Hydroxyglutarate production arises from noncanonical enzyme function at acidic pH. Nat. Chem. Biol. 13, 494–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranendijk M, Struys EA, Salomons GS, Van der Knaap MS, and Jakobs C (2012). Progress in understanding 2-hydroxyglutaric acidurias. J. Inherit. Metab. Dis. 35, 571–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Deignan JL, Dorrani N, Strom SP, Kantarci S, Quintero-Rivera F, Das K, Toy T, Harry B, Yourshaw M, et al. (2014). Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA 312, 1880–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Baker JC, and Roche TE (1995). Binding of the pyruvate dehydrogenase kinase to recombinant constructs containing the inner lipoyl domain of the dihydrolipoyl acetyltransferase component. J. Biol. Chem. 270, 793–800. [DOI] [PubMed] [Google Scholar]
- Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L, et al. (2011). Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481, 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MJ, Kennedy AD, Eckhart AD, Burrage LG, Wulff JE, Miller LA, Milbum MV, Ryals JA, Beaudet AL, Sun Q, et al. (2015). Untargeted metabolomic analysis for the clinical screening of inborn errors of metabolism. J. Inherit. Metab. Dis. 38,1029–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS, and DeBerardinis RJ (2011). Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 481, 385–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen AR, Hu Z, Shi X, Jiang L, Boroughs LK, Kovacs Z, Boriack R, Rakheja D, Sullivan LB, Linehan WM, et al. (2014). Oxidation of alpha-ketoglutarate is required for reductive carboxylation in cancer cells with mitochondrial defects. Cell Rep. 7,1679–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadtochiy SM, Schafer X, Fu D, Nehrke K, Munger J, and Brookes PS (2016). Acidic pH Is a Metabolic Switch for 2-Hydroxyglutarate Generation and Signaling. J. Biol. Chem. 291, 20188–20197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nota B, Struys EA, Pop A, Jansen EE, Fernandez Ojeda MR, Kanhai WA, Kranendijk M, van Dooren SJ, Bevova MR, Sistermans EA, et al. (2013). Deficiency in SLC25A1, encoding the mitochondrial citrate carrier, causes combined D-2- and L-2-hydroxyglutaric aciduria. Am. J. Hum. Genet. 92, 627–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual JM (2017). Pyruvate Dehydrogenase Deficiency In Progressive brain disorders in childhood (Cambridge University Press; ), pp. 48–51. [Google Scholar]
- Quehenberger O, Armando AM, Brown AH, Milne SB, Myers DS, Merrill AH, Bandyopadhyay S, Jones KN, Kelly S, Shaner RL, et al. (2010). Lipidomics reveals a remarkable diversity of lipids in human plasma. J. Lipid Res. 51, 3299–3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghuveer TS, Garg U, and Graf WD (2006). Inborn errors of metabolism in infancy and early childhood: an update. Am. Fam. Physician 73,1981–1990. [PubMed] [Google Scholar]
- Rajagopalan KN, Egnatchik RA, Calvaruso MA, Wasti AT, Padanad MS, Boroughs LK, Ko B, Hensley CT, Acar M, Hu Z, et al. (2015). Metabolic plasticity maintains proliferation in pyruvate dehydrogenase deficient cells. Cancer Metab. 3, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakheja D, Mitui M, Boriack RL, and DeBerardinis RJ (2011). Isocitrate dehydrogenase 1/2 mutational analyses and 2-hydroxyglutarate measurements in Wilms tumors. Pediatr. Blood Cancer 56, 379–383. [DOI] [PubMed] [Google Scholar]
- Reed LJ (1974). Multienzyme complexes. Acc. Chem. Res. 7, 40–46. [Google Scholar]
- Schonauer MS, Kastaniotis AJ, Kursu VA, Hiltunen JK, and Dieckmann CL (2009). Lipoic acid synthesis and attachment in yeast mitochondria. J. Biol. Chem. 284, 23234–23242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim EH, Livi CB, Rakheja D, Tan J, Benson D, Parekh V, Kho EY, Ghosh AP, Kirkman R, Velu S, et al. (2014). L-2-Hydroxyglutarate: an epigenetic modifier and putative oncometabolite in renal cancer. Cancer Discov. 4, 1290–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solmonson A, and DeBerardinis RJ (2018). Lipoic acid metabolism and mitochondrial redox regulation. J. Biol. Chem. 293, 7522–7530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soreze Y, Boutron A, Habarou F, Barnerias C, Nonnenmacher L, Delpech H, Mamoune A, Chretien D, Hubert L, Bole-Feysot C, et al. (2013). Mutations in human lipoyltransferase gene LIPT1 cause a Leigh disease with secondary deficiency for pyruvate and alpha-ketoglutarate dehydrogenase. Orphanet J. Rare Dis. 8, 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacpoole PW, Nagaraja NV, and Hutson AD (2003). Efficacy of dichloroacetate as a lactate-lowering drug. J. Clin. Pharmacol. 43, 683–691. [PubMed] [Google Scholar]
- Stowe RG, Sun Q, Elsea SH, and Scaglia F (2018). LIPT1 deficiency presenting as early infantile epileptic encephalopathy, Leigh disease, and secondary pyruvate dehydrogenase complex deficiency. Am. J. Med. Genet. A. 176, 1184–1189. [DOI] [PubMed] [Google Scholar]
- Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, and Vander Heiden MG (2015). Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 162, 552–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surendran S, Michals-Matalon K, Krywawych S, Qazi QH, Tuchman R, Rady PL, Tyring SK, and Matalon R (2002). DOOR syndrome: deficiency of E1 component of the 2-oxoglutarate dehydrogenase complex. Am. J. Med. Genet. 113, 371–374. [DOI] [PubMed] [Google Scholar]
- Tache V, Bivina L, White S, Gregg J, Deignan J, Boyadjievd SA, and Poulain FR (2016). Lipoyltransferase 1 Gene Defect Resulting in Fatal Lactic Acidosis in Two Siblings. Case Rep. Obstet. Gynecol. 2016, 6520148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tort F, Ferrer-Gortès X, Thió M, Navarro-Sastre A, Matalonga L, Quintana E, Bujan N, Arias A, García-Villoria J, Acquaviva C, et al. (2014). Mutations in the lipoyltransferase LIPT1 gene cause a fatal disease associated with a specific lipoylafion defect of the 2-ketoacid dehydrogenase complexes. Hum. Mol. Genet. 23,1907–1915. [DOI] [PubMed] [Google Scholar]
- Tort F, Ferrer-Cortes X, and Ribes A (2016). Differential diagnosis of lipoic acid synthesis defects. J. Inherit. Metab. Dis. 39, 781–793. [DOI] [PubMed] [Google Scholar]
- Venselaar H, Te Beek TA, Kuipers RK, Hekkelman ML, and Vriend G (2010). Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinformatics 11, 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernon HJ (2015). Inborn Errors of Metabolism: Advances in Diagnosis and Therapy. JAMA Pediatr. 169, 778–782. [DOI] [PubMed] [Google Scholar]
- Ward PS, and Thompson CB (2012). Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell 21, 297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J, Sinelnikov IV, Han B, and Wishart DS (2015). MetaboAnalyst 3.0—making metabolomics more meaningful. Nucleic Acids Res. 43, W251–W257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Harrison C, Jin ES, Chuang DT, Sherry AD, Malloy CR, Merritt ME, and DeBerardinis RJ (2014a). Simultaneous steady-state and dynamic 13C NMR can differentiate alternative routes of pyruvate metabolism in living cancer cells. J. Biol. Chem. 289, 6212–6224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Ko B, Hensley CT, Jiang L, Wasti AT, Kim J, Sudderth J, Calvaruso MA, Lumata L, Mitsche M, et al. (2014b). Glutamine oxidation maintains the TCA cycle and cell survival during impaired mitochondrial pyruvate transport. Mol. Cell 56, 414–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wang H, and Jaenisch R (2014c). Generating genetically modified mice using CRISPR/Cas-mediated genome engineering. Nat. Protoc. 9,1956–1968.. [DOI] [PubMed] [Google Scholar]
- Yeaman SJ, Hutcheson ET, Roche TE, Pettit FH, Brown JR, Reed LJ, Watson DC, and Dixon GH (1978). Sites of phosphorylation on pyruvate dehydrogenase from bovine kidney and heart. Biochemistry 17, 2364–2370.. [DOI] [PubMed] [Google Scholar]
- Yi X, and Maeda N (2005). Endogenous production of lipoic acid is essential for mouse development. Mol. Cell. Biol. 25, 8387–8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.