

Abstract
PURPOSE
Urothelial cancers (UCs) have a substantial hereditary component, but, other than their association with Lynch syndrome, the contribution of genetic risk factors to UC pathogenesis has not been systematically defined. We sought to determine the prevalence of pathogenic/likely pathogenic (P/LP) germline variants in patients with UC and identify associated clinical factors.
PATIENTS AND METHODS
Overall, 586 patients with UC underwent prospective, matched tumor-normal DNA sequencing. Seventy-seven genes associated with cancer predisposition were analyzed; allele frequencies were compared with publicly available database.
RESULTS
P/LP germline variants were identified in 80 (14%) of 586 individuals with UC. The most common P/LP variants in high- or moderate-penetrance genes were BRCA2 (n = 9; 1.5%), MSH2 (n = 8; 1.4%), BRCA1 (n = 8; 1.4%), CHEK2 (n = 6; 1.0%), ERCC3 (n = 4; 0.7%), and NBN and RAD50 (n = 3; 0.5% each). Sixty-six patients (83%) had germline P/LP variants in DNA-damage repair (DDR) genes, of which 28 (42%) had biallelic inactivation. Patients with P/LP variants were more commonly diagnosed at an early age (22% v 6% in those without variants; P = .01). BRCA2 and MSH2 were significantly associated with an increased risk for UC (odds ratio, 3.7 [P = .004] and 4.6 [P = .001], respectively). Current clinical guidelines for referral for genetic testing failed to identify 6 (26%) patients with high-penetrance variants.
CONCLUSION
Clinically significant P/LP germline variants in DDR genes frequently are present in patients with advanced UC. The presence of DDR germline variants could guide cancer screening for patients and their families and serve as predictive biomarkers of response to targeted or immunotherapies. Family history–based criteria to identify patients with hereditary UC susceptibility are insensitive. Broader germline testing in UC, particularly in those of young ages, should be considered.
INTRODUCTION
Urothelial cancer (UC) has a substantial hereditary component, with an estimated 30% heritable fraction according to epidemiologic studies.1 Family history of UC confers a twofold increased risk, with numerous reports of multiple-case UC kindreds.2-6 The heritable mechanisms underlying familial aggregations and early-onset UC remain unknown, and there are few syndromic associations with known cancer susceptibility genes. Highly penetrant cancer susceptibility genes, such as those in the mismatch-repair (MMR) pathway, only account for a small fraction of inherited UC susceptibility; mutations in MMR-associated genes are found primarily in patients with tumors of the ureter and renal pelvis.7,8
Beyond their role in tumor pathogenesis, germline variants can be predictors of response to cancer therapies that have activities enhanced by DNA repair defects.9,10 Deficient MMR status and high microsatellite instability (MSI-H), hallmarks of tumors in patients with Lynch syndrome, are predictive of response to the PD-1 inhibitor pembrolizumab.11 In UC, inactivating somatic mutations in ERCC2, ATM, RB1, and FANCC have been associated with response to cisplatin-based neoadjuvant chemotherapy.12,13 DNA-damage repair (DDR) mutations, both somatic and germline, may independently predict response to checkpoint inhibitors in patients with UC.14 The substantial heritability of UC, the incomplete understanding of the genes and pathways responsible for this increased heritable risk, and the potential that identification of germline variants could help guide therapeutic decisions provide a strong rationale to investigate putative UC susceptibility genes.
We have shown that germline mutations are commonly identified in individuals undergoing tumor-normal next-generation sequencing (NGS) and may reveal previously unknown genetic associations.15,16 Using a matched tumor-germline NGS platform, we determined the prevalence of pathogenic/likely pathogenic (P/LP) germline variants in 77 cancer-associated genes in patients with UC, and we examined associations between germline status and somatic mutational profile.
PATIENTS AND METHODS
Patient Cohort
Beginning in May 2014, patients with UC seen at Memorial Sloan Kettering Cancer Center were offered matched tumor-germline DNA sequencing at physician discretion under an institutional protocol (ClinicalTrials.gov identifier: NCT01775072), with only somatic variants reported. Baseline clinical characteristics for all enrolled patients were collected from institutional electronic medical records. Age ≤ 45 years was prospectively defined as early onset (outside the 95% CI of the median age of UC diagnosis).17 Starting in May 2015, patients could opt in to receive results of a secondary germline analysis of genes associated with increased heritable cancer risk; 169 patients consented to disclosure of germline results. For these 169 patients (identified cohort), germline variants were associated with a broad range of clinical features. For the remaining 417 patients (anonymized cohort), analysis was performed in a permanently anonymized fashion with no clinical annotation beyond tumor subtype. The study was approved by the Memorial Sloan Kettering Cancer Center institutional review board.
Sequencing and Results Reporting
Paraffin-embedded tumor and blood from patients were obtained and sequenced using the MSK-IMPACT platform, a capture-based NGS assay capable of identifying mutations, copy number alterations, and select gene fusions involving 341 cancer-associated genes in the first iteration and 468 in the more recent iteration, as described previously (gene list; Appendix Table A1, online only).18,19 For the anonymized cases, sequence data were assigned a unique study identifier and irretrievably de-linked from personal identifiers before variant calling.
Variant Interpretation
Pathogenicity was determined according to American College of Medical Genetics criteria (updated as of January 2018).20 Germline variants in 77 cancer predisposition genes were prioritized using PathoMAN, an automated germline variant classification tool for cancer; variants from the Exome Aggregation Consortium (ExAC) database without The Cancer Genome Atlas (TCGA) alleles were similarly prioritized.21 In addition, manual curation of the dataset was performed by a research genetic counselor (Y.K.), and any differences in variant calls were resolved with review by a molecular geneticist (O.C.B.) and a cancer geneticist (M.I.C.). For these 77 genes, all coding regions were sequenced in both the germline and the tumor. P/LP germline variants (associated with disease causation) were included in this analysis; variants of unknown significance were reviewed but were not reported. According to known disease risks and prior modeling, P/LP variants were classified at the gene level as high penetrance (relative risk [RR] of disease, > 4), moderate penetrance (RR, 2-4), low-penetrance (RR, < 2), or uncertain penetrance, or they were associated with an autosomal recessive condition.22 For CHEK2, APC, and ERCC3, classification was performed at the variant level: APC p.Ile1307Lys and CHEK2 p.Ile157Thr were considered low or uncertain penetrance, and the ERCC3 p.Arg109X, moderate penetrance.23 Tumor sequencing results were available for all patients. The FACETS algorithm was used to evaluate loss of heterozygosity (LOH) at the locus of the germline variant.24
We identified 34 genes within the MSK-IMPACT panel as related to DDR, as previously described (Appendix Table A2, online only).25 Within DDR genes, MSH2, MSH6, MLH1, and PMS2 were classified as MMR pathway genes; the remaining DDR genes were classified as other DDR.
Comparison of Guidelines Based Versus Agnostic Testing for Cancer Predisposition Syndromes in the Identified Cohort
Family history, religion, and race/ethnicity were self-reported and collected from the medical record or at time of genetic counseling. Published guidelines based on personal and family history were used to determine which genetic tests would be indicated according to the age at onset of cancer, personal or family history of cancer, and self-reported Ashkenazi Jewish ancestry.26,27 A pathogenic variant was considered incremental if it was detected by sequencing in this study but would not have been identified by genetic testing through application of current clinical guidelines.
Statistical Analysis
Allele frequencies of P/LP variants in the identified and anonymized cohorts were compared with allele frequencies of P/LP variants in the ExAC without cases from TCGA, as described previously.21 Ashkenazi Jewish founder mutations were excluded (Appendix Table A3, online only). Fisher’s exact 2-sided test was performed to assess differences in frequency; odds ratios with 95% intervals were reported. The α used to determine statistical significance in the Fisher’s test and the 95% confidence limits was adjusted using the Bonferroni correction for multiple comparisons. In the identified cohort, clinical characteristics of patients with germline P/LP variants were compared with those without them using Fisher’s exact test. Statistical analysis was performed using SAS 9.4 (Cary, NC) and R version 3.3.3. Response to therapy in identified patients with MMR mutations treated with immunotherapy was assessed according to RECIST v1.1 criteria.
RESULTS
Patient Characteristics
Five hundred eighty-six patients had tumor-germline profiling using MSK-IMPACT; 417 consented to receive only tumor sequencing results (anonymized cohort), and 169 consented to receive both tumor and germline sequencing results (identified cohort). Clinical characteristics of patients are listed in Table 1. Patients were primarily men (74.1%) and had a median age of 63 years (range, 25-87 years); 42 (7.2%) were ≤ 45 years of age. Only 42 patients (7.2%) had a family history of bladder or UC, and 112 (19.1%) had a history of a second malignancy other than UC. Most patients (79.0%) had bladder as the primary tumor site, and 59.7% had or developed metastatic disease during the period of clinical follow-up.
TABLE 1.
Patient Characteristics

Frequency and Spectrum of Germline Variants
Eighty-six P/LP variants were identified in 80 individuals (13.7%), including 62 patients (13.4%) with bladder and 18 (15.8%) with upper tract (UT) tumors (Fig 1). Eleven patients had two P/LP variants each. The most frequently mutated moderate- or high-penetrance genes were BRCA2 (n = 9; 1.5%), MSH2 (n = 8; 1.4%), BRCA1 (n = 8; 1.4%), CHEK2 (n = 6; 1.0%), ERCC3 (n = 4; 0.7%), and NBN and RAD50 (n = 3; 0.5% each; Appendix Table A4, online only). The low-penetrance variant APC p.Ile1307Lys was the most prevalent overall (n = 11), and 6 (35.3%) of the 17 BRCA1/2 variants were Ashkenazi Jewish founder mutations. Of all variants, 35 (40.7%) were of high penetrance, 24 (27.9%) were of moderate penetrance, 18 (20.9%) were of low or uncertain penetrance, and 10 (11.6%) were in a gene associated with an autosomal recessive cancer-associated syndrome. Sixty-five variants (75.6%) were in genes associated with DDR; of these, 12 were in the MMR-associated genes MSH2 (n = 8), MSH6 (n = 2), and MLH1 (n = 2; Fig 2A).
FIG 1.

Frequency and penetrance of germline variants in patients with urothelial cancer. Frequency of pathogenic/likely pathogenic (P/LP) germline variants identified in 586 patients with UC. Pie chart shows that 13.7% (n = 80 patients) had a germline P/LP variant; 9.7% (n = 57) had high- or moderate-penetrance variants.
FIG 2.

Germline pathogenic/likely pathogenic variants by site of tumor origin and tumors with loss of heterozygosity (LOH) or second somatic mutation in the tumor in 586 patients. (A) Number of mutations in each gene by tumor type and (B) in germline-positive occurrences with LOH or second somatic mutation in same locus. DDR, DNA-damage repair; MMR, mismatch repair.
Correlation Between Germline Genotype and Tumor Phenotype
Of 54 germline variants in DDR genes (excluding MMR genes), LOH/somatic mutation in the tumor was present in 18 patients (33.3%), including carriers of ATM (n = 2 of 3), ERCC2 (n = 3 of 3), BRCA2 (n = 6 of 9), and BRCA1 (n = 2 of 8), among others (Fig 2B). In the identified cohort, all tumors from patients with a germline MMR variant had either MSI-H status or immunohistochemistry showing deficient MMR protein staining.
Estimate of UC Risk Associated With Observed Variants
To estimate population frequencies of germline P/LP variants in genes seen in the UC cohort, we analyzed allele frequencies of these genes in the ExAC dataset. BRCA2 and MSH2 showed statistically significant increased risk in UC patients compared with ExAC (odds ratio, 3.7; 95% CI, 1.5 to 7.8; P < .003 for BRCA2; and odds ratio, 4.6; 95% CI, 1.8 to 9.8; P < .001 for MSH2; Table 2).
TABLE 2.
Comparison of Allele Frequencies in All Patients Versus ExAC

Clinical Characteristics Associated With P/LP Variants
The only clinical characteristic available for analysis for patients in the anonymized cohort was site of tumor. In patients with bladder primaries from both the anonymized and identified cohorts, 39 (8.4%) had moderate- or high-risk P/LP variants. Conversely, in patients with UT primaries, 16 (14.0%) had moderate- or high-risk P/LP variants, including 10 (8.8%) with MMR mutations.
In the identified cohort, patients with P/LP variants were more likely to have early age of onset (age ≤ 45 years) compared with patients with no germline variant (22% v 6%; P = 0.01; Table 3). Six of 14 patients with early-onset UC had germline variants, including in MSH2 (n = 2), BRCA1 (n = 2), MSH6 (n = 1), and BRCA2 (n = 1). Presence of P/LP variants was not statistically significantly associated with a positive family history of UC, non-UC malignancy, smoking history, or metastasis at diagnosis, but presence was associated with Ashkenazi Jewish ancestry, reflective of founder mutations in BRCA1, BRCA2, and CHEK2.
TABLE 3.
Clinical Characteristics by Moderate-/High-Penetrance Variants in Identified Cohort
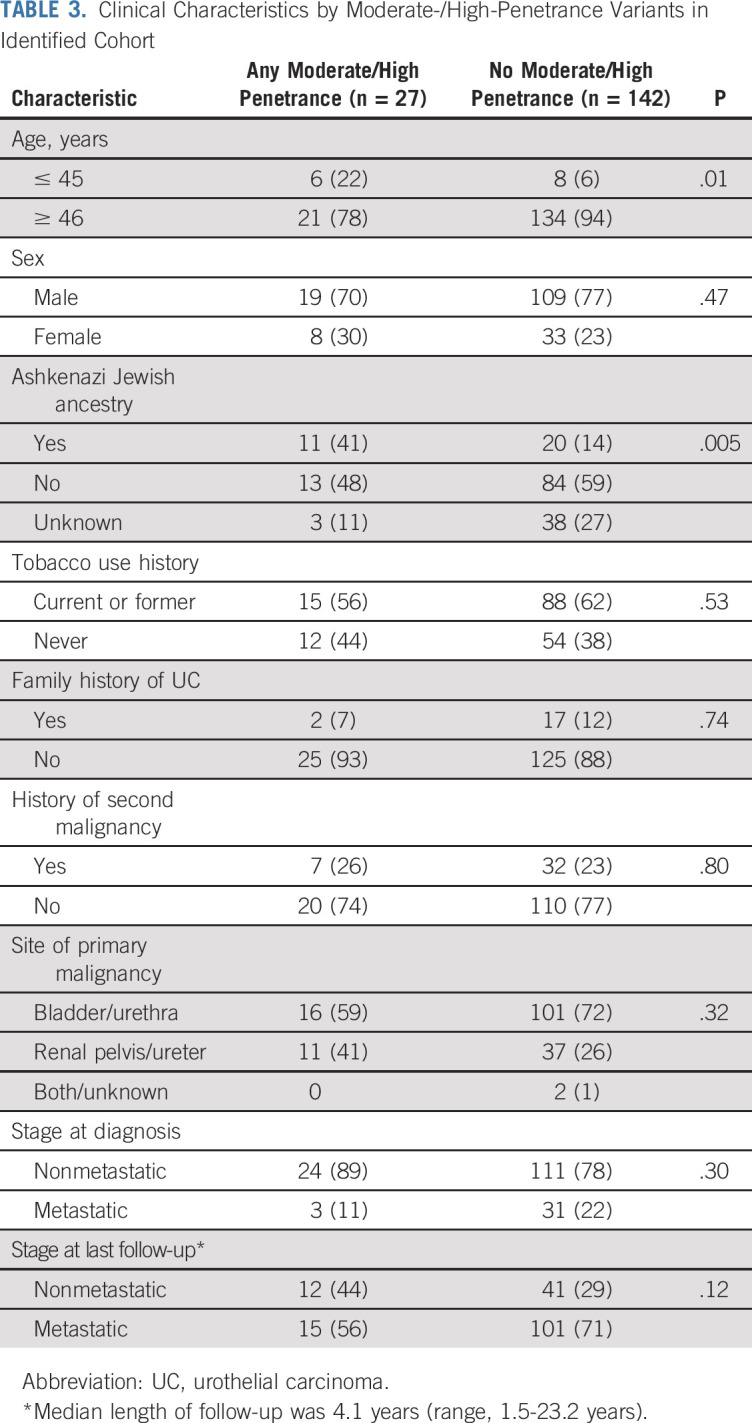
Of 9 patients with MMR variants in the identified cohort, all but one had a UT tumor as the primary site of malignancy. The median age of diagnosis of UC was 58 years (range, 31-84 years), and UC was the first malignancy in 6 patients. Although none reported a family history of bladder or UT cancer, 4 reported relatives with cancers of unknown origin or kidney cancer, not otherwise specified.
Given the association of MSI-H status or deficient MMR tumors and response to immunotherapy in other malignancies, we explored response in the four patients in the identified cohort who had germline MMR variants (all MSI-H tumors) and who had received immunotherapy. All 4 had presented initially with metastatic disease and had received platinum chemotherapy as first-line treatment. Three experienced progression of disease after platinum chemotherapy followed by a complete response after immunotherapy (Appendix Fig A1, online only). The fourth patient experienced progression of disease on both platinum chemotherapy and immunotherapy.
Comparisons to Clinical Genetics Referral Criteria
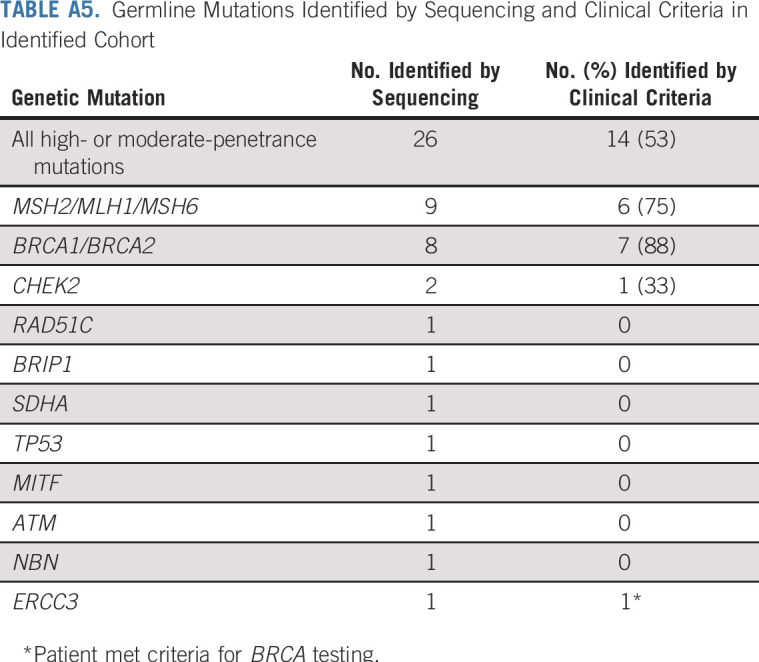
In the identified cohort, detailed family history was obtained by interview for 29 (85.3%) of the patients with P/LP variants; for the remainder of patients, data were extracted from the electronic medical record. Only 9 of 27 patients with high- or moderate-penetrance variants had undergone clinical genetic testing or attended clinical genetic counseling before receiving genetic test results through the protocol. Of patients with high-penetrance P/LP variants, 6 patients (26.3%) would not have been referred for germline testing according to published guidelines (one each with MSH2, MSH6, BRCA1, SDHA, and TP53). The patient with a TP53 variant, diagnostic of Li-Fraumeni syndrome, had a father with bladder cancer, but the patient did not meet criteria for any genetic testing. Of patients with moderate-penetrance variants, 7 (87.5%) would not have been referred (Appendix Table A5, online only).
DISCUSSION
This study shows that, in patients with UC, clinically significant P/LP germline variants, particularly in DDR genes, frequently are present. Multiple epidemiologic studies have identified an increased familial risk of UC. However, to date, the only identified hereditary cancer syndrome associated with increased UC risk is Lynch syndrome, which is caused by inactivating mutations in the MMR-associated genes MSH2, MSH6, MLH1, PMS2, and EPCAM.3,28,29 Patients with Lynch syndrome have an up to 12% cumulative risk of urinary tract cancer; although the risk is greater for UT UC, there also may be an increased risk for bladder UC.7,8,30,31 Studies estimating prevalence of Lynch syndrome in patients with UC, however, have been limited. In two studies looking at unselected patients with UT UC, 7% of tumors had deficient MMR protein expression or were MSI-H, usually a necessary but not sufficient biomarker for Lynch syndrome.32,33 In our study, 2.1% overall and 8.7% of patients with UT tumors had Lynch syndrome, and, for those in the identified cohort, 6 of 9 had UC as the first malignancy. Immunohistochemistry analysis of tumors for loss of MMR protein expression or tumor MSI analysis is standard practice for colorectal and endometrial cancers, for which the incidence of Lynch syndrome ranges from 2% to 5%.34 A recent pan-cancer study showed that, among patients with MSI-high/intermediate UC, 37.5% had a germline MMR variant diagnostic of Lynch syndrome, the highest prevalence of all cancer types analyzed.35 The increased incidence of Lynch syndrome in our cohort, and the high prevalence of Lynch syndrome in those with MSI-high/intermediate tumors, supports consideration of germline or tumor screening for those with UT UC.
In our cohort, 9% of patients had a germline DDR mutation in a gene other than those in MMR. Dysregulation of DNA repair is implicated in the carcinogenesis of UC. DDR somatic mutations are frequent in UC.25,36 For example, the nucleotide excision repair pathway gene ERCC2 was somatically mutated in 9% of UC in TCGA, and common polymorphisms in ERCC2, NBN, and XPC are associated with bladder cancer risk.13,37,38 To further investigate the pathogenic role of DDR genes, we compared allele frequencies of the most frequently mutated genes in UC with frequencies in individuals in ExAC without cancer, and we found significantly increased frequencies of mutations in BRCA2 and MSH2 in UC. We also found LOH in 6 of 9 tumors in patients with germline variants in BRCA2 and 3 of 3 tumors in patients with germline ERCC2 variants. Of note, the current number of carriers is too small to analyze enrichment for second allele loss in tumors for germline events compared with the background loss of first somatic allele in these genes. Pathogenic nucleotide excision repair germline mutations have not been described previously in UC, although heterozygous germline mutations in ERCC2 and ERCC3 have been associated with increased risk of sarcoma and breast cancer.38,39 Additional studies will be needed to determine the role of these germline mutations in the pathogenicity of UC.
Germline MMR and other DDR mutations have potential implications for treatment selection. Somatic mutations in ERCC2, ATM, RB1, FANCC, and other DDR genes are correlated with improved responses to platinum-based chemotherapy in patients with UC.12,13,40,41 Whether germline pathogenic mutations in these genes also are associated with improved outcomes must be explored. Preclinical models with ERCC2 and ERCC3 heterozygous knockouts show a hypomorphic functionality when exposed to DNA damaging agents, supporting a plausible mechanism for sensitivity to platinum therapies.23,25 Although tumor MMR deficiency predicts response to PD-1 blockade in other solid tumors, to date, no large studies correlating MMR deficiency and response to PD-1 or PD-L1 blockade have included patients with UC.11 In this study, all patients with germline MMR mutations had MSI-H or MMR-deficient tumors, and 3 of 4 patients with Lynch syndrome had complete responses to immunotherapy after they experienced progression on chemotherapy.11,42 Our findings suggest that germline DDR alterations should be included with somatic alterations when assessing for correlations between therapeutic benefit.
Finally, a quarter of patients with high-penetrance germline variants would not have been detected by guidelines-directed testing. Identification of germline mutations in these patients may allow for enhanced screening and early detection of hereditary cancers in those families for whom testing would not have been undertaken. Several individuals became the index cases (first detected cancer) in their families, which then led to cascade testing of other family members. Interestingly, among those with a family history of UC or personal history of other cancers, there was no increased incidence of P/LP germline variants. This may be explained in part by incomplete information available to patients—for example, of 8 patients with Lynch syndrome, none reported a family history of bladder or UT cancer, but 4 did report relatives with possible kidney cancer or of unclear origin, which may reflect UT cancers. Analysis in a larger cohort also may reveal whether personal history of cancer is associated with presence of germline alterations.
This study had several limitations. It was retrospective in nature and had a limited sample size; its findings will require validation in other cohorts. Given the smaller number of identified patients with clinically annotated data, associations between clinical features and prevalence of mutations may be limited by numbers. The study was conducted at a comprehensive cancer center with regional referral patterns, so the patient population may differ from that in the general community. For example, the median age of diagnosis in the cohort was 63 years, compared with approximately 70 years in the United States, and there was under-representation of nonwhite patients.43 We did observe a lower prevalence of germline mutations in the anonymized cohort, in which germline results were not returned. Although physicians were not instructed to select patients according to suspicion of an inherited syndrome, individuals consenting to return of germline results may have be more motivated to do so because of family history. We performed targeted exome sequencing of known cancer-predisposition genes; agnostic whole-exome sequencing could yield associations among novel genes or variants and risk of UC. LOH analysis was exploratory and was not corrected for possible increased background LOH in those genes, which also merits more study in larger cohorts.
This study demonstrates that germline mutations in patients with UC occur predominantly in DDR genes, with increased frequency of BRCA2 and MSH2. Traditional criteria to identify those at risk for hereditary syndromes only identified a fraction of patients. There is potential value of expanded germline analysis in UC, particularly in patients of young age at diagnosis and those with UT tumors. The genes found to be mutated were associated with increased risk for cancers other than UC; thus, their identification likely will have substantial implications for directed cancer screening in patients and their families.
APPENDIX
FIG A1.

Response to chemotherapy and immunotherapy in two patients with Lynch syndrome. (A) 18-Fluorodeoxyglucose positron emission tomography/computer tomography (PET/CT) image reveals a left para-aortic lymph node measuring 1.9 cm in largest dimension (red arrow) before initiation of paclitaxel. (B) Post-treatment CT after six infusions of paclitaxel shows enlargement of lymph node to 2.4 cm. (C) Post-treatment CT after six infusions of atezolizumab shows reduction in lymph node size to 0.9 cm, considered complete response by RECIST v 1.1 criteria. (D) CT image in another patient reveals right and left external iliac lymph nodes measuring 2.8 cm and 2.3 cm in largest dimensions, respectively (red arrows), before initiation of platinum-based chemotherapy. (E) Post-treatment CT after three cycles of platinum-based therapy shows right lymph node now to 2.7 cm and left to 3.2 cm. Treatment with chemotherapy was stopped, and the patient proceeded to treatment with a programmed death ligand 1 (PDL-1) inhibitor. (F) Post-treatment CT after 24 infusions of PDL-1 inhibitor shows disappearance of right lymph node and reduction of left lymph node to 0.6 cm. After 26 months on immune therapy, the patient proceeded to radical cystectomy with lymph node resection. Pathology review showed a complete pathologic response without residual carcinoma.
TABLE A1.
Genes on MSK-IMPACT and Syndromes Associated With Germline Mutations

TABLE A2.
DDR Gene Panel
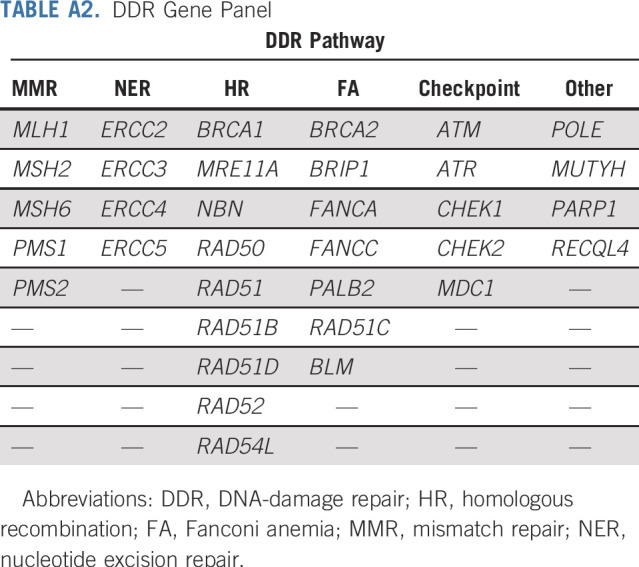
TABLE A3.
Ashkenazi Jewish and European Founder Mutations
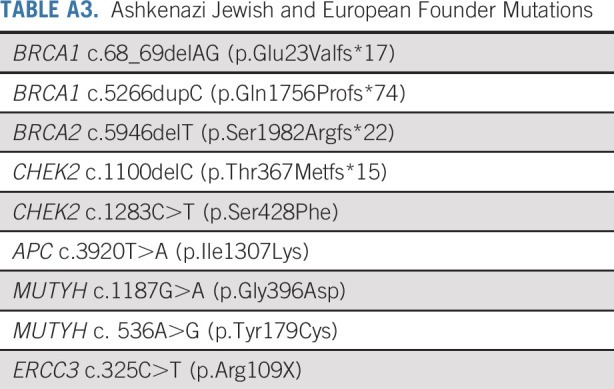
TABLE A4.
Detail on Pathogenic/Likely Pathogenic Germline Variants

TABLE A5.
Germline Mutations Identified by Sequencing and Clinical Criteria in Identified Cohort
Footnotes
Presented at the 2018 Genitourinary Cancers Symposium, San Francisco, CA, February 8-10, 2018, and American Society of Clinical Oncology 2017 Annual Meeting, Chicago, IL, June 2-6, 2019.
Supported by the Bladder Specialized Program of Research Excellence grant No. P50 CA221745-01A1 from the National Cancer Institute, The Robert and Kate Niehaus Center for Inherited Cancer Genomics at Memorial Sloan Kettering Cancer Center, the Marie-Josée and Henry R. Kravis Center for Molecular Oncology, the Andrew Sabin Cancer Research Fund, the Carmel Family Cancer Research Fund, and Cancer Center Support Grant No. P30 CA008748-50.
AUTHOR CONTRIBUTIONS
Conception and design: Maria I. Carlo, Vignesh Ravichandran, A. Ari Hakimi, Guido Dalbagni, Samuel A. Funt, David B. Solit, Jonathan E. Rosenberg, Mark E. Robson, Joseph Vijai, Dean F. Bajorin, Kenneth Offit
Collection and assembly of data: Maria I. Carlo, Vignesh Ravichandran, Chaitanya Bandlamudi, Yelena Kemel, Joshua Chaim, Zarina Fnu, Angela G. Arnold, Aliya Khurram, Kaitlyn Tkachuk, Catharine K. Cipolla, Ashley Regazzi, Hikmat Al-Ahmadie, Michael F. Walsh, Min-Yuen Teo, Samuel A. Funt, Bernard H. Bochner, David B. Solit, Liying Zhang, Jonathan E. Rosenberg, Barry S. Taylor, Mark E. Robson, Michael F. Berger, Joseph Vijai, Dean F. Bajorin, Kenneth Offit
Administrative support: Aliya Khurram, Catharine K. Cipolla, Ashley Regazzi
Data analysis and interpretation: Maria I. Carlo, Vignesh Ravichandran, Preethi Srinavasan, Chaitanya Bandlamudi, Yelena Kemel, Ozge Ceyhan-Birsoy, Semanti Mukherjee, Diana Mandelker, Joshua Chaim, Andrea Knezevic, Satshil Rana, Kelsey Breen, Hikmat Al-Ahmadie, Karen A. Cadoo, Samuel A. Funt, Jonathan A. Coleman, Bernard H. Bochner, Gopa Iyer, David B. Solit, Zsofia K. Stadler, Liying Zhang, Jonathan E. Rosenberg, Mark E. Robson, Michael F. Berger, Joseph Vijai, Dean F. Bajorin, Kenneth Offit
Provision of study material or patients: Guido Dalbagni, Min-Yuen Teo, Bernard H. Bochner, David B. Solit, Liying Zhang, Jonathan E. Rosenberg, Kenneth Offit
Administrative support: David B. Solit, Dean F. Bajorin, Kenneth Offit
Financial support: David B. Solit, Kenneth Offit
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Cancer Susceptibility Mutations in Patients With Urothelial Malignancies
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/journal/jco/site/ifc.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Maria I. Carlo
Consulting or Advisory Role: Pfizer
Other Relationship: Prostate Cancer Foundation, Robert Wood Johnson Foundation
Semanti Mukherjee
Stock and Other Ownership Interests: Regeneron
Catharine K. Cipolla
Stock and Other Ownership Interests: Roche
Travel, Accommodations, Expenses: Flatiron Health
Hikmat Al-Ahmadie
Consulting or Advisory Role: Bristol-Myers Squibb, EMD Serono, AstraZeneca/MedImmune
Karen A. Cadoo
Research Funding: AstraZeneca (Inst)
Research Funding: Syndax (Inst)
Travel, Accommodations, Expenses: AstraZeneca
Min-Yuen Teo
Research Funding: Bristol-Myers Squibb, Clovis Oncology
Samuel A. Funt
Stock and Other Ownership Interests: Kite Pharma, Urogen Pharma (I), Hubble (I), Second Science, Allogene Therapeutics, Neogene Therapeutics, Kronos Bio (I), Vida Ventures (I)
Consulting or Advisory Role: AstraZeneca (Inst), MedImmune (Inst)
Research Funding: Genentech (Inst), Roche (Inst), AstraZeneca (Inst), Decibel Therapeutics (Inst)
Travel, Accommodations, Expenses: Bristol-Myers Squibb, AstraZeneca, MedImmune
Jonathan A. Coleman
Travel, Accommodations, Expenses: Digital Angiography Reading Center (I)
Other Relationship: Steba Biotech
Bernard H. Bochner
Honoraria: Genentech, Roche
Consulting or Advisory Role: Genentech, Roche, Olympus
Gopa Iyer
Consulting or Advisory Role: Bayer, Janssen, Mirati Therapeutics
Research Funding: Mirati Therapeutics (Inst), Novartis (Inst), Debiopharm Group (Inst), Bayer (Inst)
David B. Solit
Stock and Other Ownership Interests: Loxo
Consulting or Advisory Role: Pfizer, Loxo, Illumina, Intezyne Technologies, Vivideon Therapeutics, Lilly Oncology
Travel, Accommodations, Expenses: Merck KGaA
Zsofia K. Stadler
Consulting or Advisory Role: Allergan (I), Genentech (I), Roche (I), Regeneron (I), Optos (I), Adverum (I), Biomarin (I), Alimera Sciences (I), Novartis (I), Spark Therapeutics (I), Fortress Biotech (I), Regenxbio (I)
Liying Zhang
Employment: Shanghai Genome Center (I)
Leadership: Shanghai Genome Center (I)
Stock and Other Ownership Interests: Shanghai Genome Center (I)
Honoraria: Future Technology Research, BGI, Illumina, Roche Diagnostics Asia Pacific
Travel, Accommodations, Expenses: Shanghai Genome Center (I), Roche Diagnostics Asia Pacific
Jonathan E. Rosenberg
Stock and Other Ownership Interests: Merck, Illumina
Honoraria: UpToDate, Bristol-Myers Squibb, AstraZeneca, Medscape, Vindico, Peerview, Chugai Pharma, Research to Practice, Intellisphere, Clinical Care Options, Clinical Mind
Consulting or Advisory Role: Lilly, Merck, Agensys, Roche, Genentech, Sanofi, AstraZeneca, Medimmune, Bristol-Myers Squibb, EMD Serono, Seattle Genetics, Bayer, Inovio Pharmaceuticals, BioClin Therapeutics, QED Therapeutics, Adicet Bio, Sensei Biotherapeutics, Fortress Biotech, Pharmacyclics, Western Oncolytics, GlaxoSmithKline, Janssen Oncology, Astellas Pharma
Research Funding: Oncogenex (Inst), Agensys (Inst), Mirati Therapeutics (Inst), Novartis (Inst), Viralytics (Inst), Genentech (Inst), Roche (Inst), Incyte (Inst), Seattle Genetics (Inst), Bayer (Inst), AstraZeneca (Inst), QED Therapeutics (Inst), Astellas Pharma (Inst)
Patents, Royalties, Other Intellectual Property: Predictor of platinum sensitivity (Inst)
Travel, Accommodations, Expenses: Genentech, Roche, Bristol-Myers Squibb
Barry S. Taylor
Consulting or Advisory Role: Boehringer Ingelheim
Research Funding: Genentech
Mark E. Robson
Honoraria: AstraZeneca
Consulting or Advisory Role: McKesson, AstraZeneca
Research Funding: AstraZeneca (Inst), Myriad Genetics (Inst), InVitae (Inst), AbbVie (Inst), Tesaro (Inst), Medivation (Inst)
Travel, Accommodations, Expenses: AstraZeneca, Pfizer
Other Relationship: Research to Practice, Clinical Care Options, Physician Education Resource
Uncompensated Relationships: Merck, Pfizer, Daiichi Sankyo
Open Payments Link: https://openpaymentsdata.cms.gov/physician/612669/summary
Michael F. Berger
Consulting or Advisory Role: Roche
Research Funding: Grail
Joseph Vijai
Patents, Royalties, Other Intellectual Property: Title: Diagnosis & treatment of ERCC3-mutant cancer; Inventors: Vijai Joseph, Sabine Topka, Kenneth Offit; US National Stage Patent Application No.: 16/493,214; Filing Date: September 11, 2019 (Inst)
Dean F. Bajorin
Honoraria: Merck Sharp & Dohme
Consulting or Advisory Role: Bristol-Myers Squibb, Novartis, Roche, Genentech, Merck, Lilly, Fidia Farmaceutici, Urogen Pharma, Pfizer, EMD Serono
Research Funding: Novartis (Inst), Genentech (Inst), Roche (Inst), Merck (Inst), Bristol-Myers Squibb (Inst), AstraZeneca (Inst), Astellas Pharma (Inst), Seattle Genetics (Inst), Astellas (Inst)
Travel, Accommodations, Expenses: Roche, Genentech, Merck, Bristol-Myers Squibb, Lilly, Urogen Pharma
No other potential conflicts of interest were reported.
REFERENCES
- 1.Mucci LA, Hjelmborg JB, Harris JR, et al. Familial risk and heritability of cancer among twins in Nordic countries. JAMA. 2016;315:68–76. doi: 10.1001/jama.2015.17703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kramer AA, Graham S, Burnett WS, et al. Familial aggregation of bladder cancer stratified by smoking status. Epidemiology. 1991;2:145–148. doi: 10.1097/00001648-199103000-00010. [DOI] [PubMed] [Google Scholar]
- 3.Lin J, Spitz MR, Dinney CP, et al. Bladder cancer risk as modified by family history and smoking. Cancer. 2006;107:705–711. doi: 10.1002/cncr.22071. [DOI] [PubMed] [Google Scholar]
- 4.Lynch HT, Walzak MP, Fried R, et al. Familial factors in bladder carcinoma. J Urol. 1979;122:458–461. doi: 10.1016/s0022-5347(17)56461-7. [DOI] [PubMed] [Google Scholar]
- 5.Mahboubi AO, Ahlvin RC, Mahboubi EO. Familial aggregation of urothelial carcinoma. J Urol. 1981;126:691–692. doi: 10.1016/s0022-5347(17)54693-5. [DOI] [PubMed] [Google Scholar]
- 6.McCullough DL, Lamma DL, McLaughlin AP, III, et al. Familial transitional cell carcinoma of the bladder. J Urol. 1975;113:629–635. doi: 10.1016/s0022-5347(17)59540-3. [DOI] [PubMed] [Google Scholar]
- 7.Joost P, Therkildsen C, Dominguez-Valentin M, et al. Urinary tract cancer in Lynch syndrome: Increased risk in carriers of MSH2 mutations. Urology. 2015;86:1212–1217. doi: 10.1016/j.urology.2015.08.018. [DOI] [PubMed] [Google Scholar]
- 8.van der Post RS, Kiemeney LA, Ligtenberg MJ, et al. Risk of urothelial bladder cancer in Lynch syndrome is increased, in particular among MSH2 mutation carriers. J Med Genet. 2010;47:464–470. doi: 10.1136/jmg.2010.076992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robson M, Im SA, Senkus E, et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med. 2017;377:523–533. doi: 10.1056/NEJMoa1706450. [DOI] [PubMed] [Google Scholar]
- 10.Mateo J, Carreira S, Sandhu S, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373:1697–1708. doi: 10.1056/NEJMoa1506859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409–413. doi: 10.1126/science.aan6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Plimack ER, Dunbrack RL, Brennan TA, et al. Defects in DNA repair genes predict response to neoadjuvant cisplatin-based chemotherapy in muscle-invasive bladder cancer. Eur Urol. 2015;68:959–967. doi: 10.1016/j.eururo.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Allen EM, Mouw KW, Kim P, et al. Somatic ERCC2 mutations correlate with cisplatin sensitivity in muscle-invasive urothelial carcinoma. Cancer Discov. 2014;4:1140–1153. doi: 10.1158/2159-8290.CD-14-0623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Teo MY, Seier K, Ostrovnaya I, et al. Alterations in DNA damage response and repair genes as potential marker of clinical benefit from PD-1/PD-L1 blockade in advanced urothelial cancers. J Clin Oncol. 2018;36:1685–1694. doi: 10.1200/JCO.2017.75.7740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schrader KA, Cheng DT, Joseph V, et al. Germline variants in targeted tumor sequencing using matched normal DNA. JAMA Oncol. 2016;2:104–111. doi: 10.1001/jamaoncol.2015.5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mandelker D, Zhang L, Kemel Y, et al. Mutation detection in patients with advanced cancer by universal sequencing of cancer-related genes in tumor and normal DNA vs guideline-based germline testing. JAMA. 2017;318:825–835. doi: 10.1001/jama.2017.11137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Howlader N, Noone AM, Krapcho M.(eds)SEER Cancer Statistics Review, 1975-2016, Bethesda, MD, National Cancer Institute. https://seer.cancer.gov/csr/1975_2016/ [Google Scholar]
- 18.Cheng DT, Mitchell TN, Zehir A, et al. Memorial Sloan Kettering-integrated mutation profiling of actionable cancer targets (MSK-IMPACT): A hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. 2015;17:251–264. doi: 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zehir A, Benayed R, Shah RH, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23:703–713. doi: 10.1038/nm.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ravichandran V, Shameer Z, Kemel Y, et al. Toward automation of germline variant curation in clinical cancer genetics. Genet Med. 2019;21:2116–2125. doi: 10.1038/s41436-019-0463-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tung N, Domchek SM, Stadler Z, et al. Counselling framework for moderate-penetrance cancer-susceptibility mutations. Nat Rev Clin Oncol. 2016;13:581–588. doi: 10.1038/nrclinonc.2016.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vijai J, Topka S, Villano D, et al. A Recurrent ERCC3 truncating mutation confers moderate risk for breast cancer. Cancer Discov. 2016;6:1267–1275. doi: 10.1158/2159-8290.CD-16-0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen R, Seshan VE. FACETS: Allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016;44:e131. doi: 10.1093/nar/gkw520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Teo MY, Bambury RM, Zabor EC, et al. DNA damage response and repair gene alterations are associated with improved survival in patients with platinum-treated advanced urothelial carcinoma. Clin Cancer Res. 2017;23:3610–3618. doi: 10.1158/1078-0432.CCR-16-2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. National Comprehensive Cancer Network: Guidelines: Genetic/familial high-risk assessment—breast and ovarian, 2019. https://www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf.
- 27. National Comprehensive Cancer Network: Guidelines: Genetic/familial high-risk assessment—Colorectal, 2018. https://www.nccn.org/professionals/physician_gls/pdf/genetics_colon.pdf.
- 28.Murta-Nascimento C, Silverman DT, Kogevinas M, et al. Risk of bladder cancer associated with family history of cancer: Do low-penetrance polymorphisms account for the increase in risk? Cancer Epidemiol Biomarkers Prev. 2007;16:1595–1600. doi: 10.1158/1055-9965.EPI-06-0743. [DOI] [PubMed] [Google Scholar]
- 29.Plna K, Hemminki K. Familial bladder cancer in the National Swedish Family Cancer Database. J Urol. 2001;166:2129–2133. [PubMed] [Google Scholar]
- 30.Vasen HF, Stormorken A, Menko FH, et al. MSH2 mutation carriers are at higher risk of cancer than MLH1 mutation carriers: A study of hereditary nonpolyposis colorectal cancer families. J Clin Oncol. 2001;19:4074–4080. doi: 10.1200/JCO.2001.19.20.4074. [DOI] [PubMed] [Google Scholar]
- 31.Aarnio M, Sankila R, Pukkala E, et al. Cancer risk in mutation carriers of DNA-mismatch-repair genes. Int J Cancer. 1999;81:214–218. doi: 10.1002/(sici)1097-0215(19990412)81:2<214::aid-ijc8>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 32.Harper HL, McKenney JK, Heald B, et al. Upper tract urothelial carcinomas: Frequency of association with mismatch repair protein loss and lynch syndrome. Mod Pathol. 2017;30:146–156. doi: 10.1038/modpathol.2016.171. [DOI] [PubMed] [Google Scholar]
- 33.Audenet F, Isharwal S, Cha EK, et al. Clonal relatedness and mutational differences between upper tract and bladder urothelial carcinoma. Clin Cancer Res. 2018;25:967–976. doi: 10.1158/1078-0432.CCR-18-2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kwon JS, Scott JL, Gilks CB, et al. Testing women with endometrial cancer to detect Lynch syndrome. J Clin Oncol. 2011;29:2247–2252. doi: 10.1200/JCO.2010.32.9979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Latham A, Srinivasan P, Kemel Y, et al. Microsatellite instability is associated with the presence of Lynch syndrome pan-cancer. J Clin Oncol. 2019;37:286–295. doi: 10.1200/JCO.18.00283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yap KL, Kiyotani K, Tamura K, et al. Whole-exome sequencing of muscle-invasive bladder cancer identifies recurrent mutations of UNC5C and prognostic importance of DNA repair gene mutations on survival. Clin Cancer Res. 2014;20:6605–6617. doi: 10.1158/1078-0432.CCR-14-0257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Robertson AG, Kim J, Al-Ahmadie H, et al. : Comprehensive molecular characterization of muscle-invasive bladder cancer. Cell 171:540-556.e25, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stern MC, Lin J, Figueroa JD, et al. Polymorphisms in DNA repair genes, smoking, and bladder cancer risk: Findings from the international consortium of bladder cancer. Cancer Res. 2009;69:6857–6864. doi: 10.1158/0008-5472.CAN-09-1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cancer Genome Atlas Research Network Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507:315–322. doi: 10.1038/nature12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu D, Plimack ER, Hoffman-Censits J, et al. Clinical validation of chemotherapy response biomarker ERCC2 in muscle-invasive urothelial bladder carcinoma. JAMA Oncol. 2016;2:1094–1096. doi: 10.1001/jamaoncol.2016.1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Teo MY, Seier K, Ostrovnaya I, et al. Alterations in DNA damage response and repair genes as potential marker of clinical benefit from PD-1/PD-L1 blockade in advanced urothelial cancers. J Clin Oncol. 2018;36:1685–1694. doi: 10.1200/JCO.2017.75.7740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scosyrev E, Noyes K, Feng C, et al. Sex and racial differences in bladder cancer presentation and mortality in the US. Cancer. 2009;115:68–74. doi: 10.1002/cncr.23986. [DOI] [PubMed] [Google Scholar]