

Abstract
Purpose
To provide evidence-based consensus recommendations on choice of end points for clinical trials in metastatic breast cancer, with a focus on biologic subtype and line of therapy.
Methods
The National Cancer Institute Breast Cancer Steering Committee convened a working group of breast medical oncologists, patient advocates, biostatisticians, and liaisons from the Food and Drug Administration to conduct a detailed curated systematic review of the literature, including original reports, reviews, and meta-analyses, to determine the current landscape of therapeutic options, recent clinical trial data, and natural history of four biologic subtypes of breast cancer. Ongoing clinical trials for metastatic breast cancer in each subtype also were reviewed from ClinicalTrials.gov for planned primary end points. External input was obtained from the pharmaceutic/biotechnology industry, real-world clinical data specialists, experts in quality of life and patient-reported outcomes, and combined metrics for assessing magnitude of clinical benefit.
Results
The literature search yielded 146 publications to inform the recommendations from the working group.
Conclusion
Recommendations for appropriate end points for metastatic breast cancer clinical trials focus on biologic subtype and line of therapy and the magnitude of absolute and relative gains that would represent meaningful clinical benefit.
INTRODUCTION
Significant heterogeneity exists in the natural history of metastatic breast cancer (MBC), particularly with regard to overall prognosis, treatment options, and benefits of therapy in biologic subtypes. Survival outcomes in clinical trials depend on many variables, including number and type of prior therapies, sites and extent of disease, and toxicity. The expected postprogression survival (PPS) after completion of protocol-specific therapy has implications for the choice of optimal end point: When overall survival (OS) is measured in years and patients receive multiple lines of therapy, progression-free survival (PFS) may be the most meaningful metric of treatment outcome. Conversely, in poor prognosis settings, such as triple-negative metastatic breast cancer (TNMBC), where expected PPS is short, OS is likely the most appropriate end point.1 The balance between incremental gain in PFS and encountered toxicity is crucial, although data are scant on this topic.2 In this context, several recent randomized trials have yielded statistically significant improvements in the primary end point with experimental therapy but did not lead to regulatory approval or practice change3 primarily because of toxicity. This highlights the need for guidance on appropriate end points for both clinical trials in the setting of MBC and incorporation of patient-reported outcomes (PROs) and toxicities into the discussion of clinical trial design, conduct, and interpretation.
Formal guidance for industry on clinical trial end points was provided by the US Food and Drug Administration (FDA) in 20074 but was not disease specific. Patient-focused drug development is mandated by the Prescription Drug User Fee Act V,5 and the integration of PROs in the assessment of benefit of new treatments is evolving. Our working group (WG) sought to create a specific consensus on end points for MBC clinical trials by focusing on biologic subtype and line of therapy with sensitivity to various stakeholders, including medical oncologists, patients, the FDA, the National Cancer Institute, biostatisticians, and industry.
METHODS
The National Cancer Institute Breast Cancer Steering Committee6 formed a WG and obtained external input from the pharmaceutic/biotechnology industry, real-world clinical data specialists, and experts in quality of life (QoL) and PROs (see Acknowledgment). We conducted a detailed curated systematic review of the literature in quarter 3 of 2016 through quarter 1 of 2017 to determine the current landscape of clinical trial data and natural history of four biologic subtypes of breast cancer: hormone receptor positive (HR+)/human epidermal growth factor receptor 2 negative (HER2–), HR–/HER2– (TNMBC), HR+/HER2+, and HR–/HER2+. The literature review was performed through PubMed using the search terms metastatic or advanced breast cancer, and clinical trial end points and included original reports, reviews, meta-analyses, and editorials (Fig 1), yielding 146 publications. Ongoing trials in each subtype also were reviewed from ClinicalTrials.gov, with particular attention to the planned primary end point of each study. External expertise was provided through Web-based teleconference. The WG achieved consensus by discussion and anonymous electronic polling/voting. Because other groups are examining the topics of brain metastases, bone metastases, immunotherapy, and cost of care,7 we did not focus on these issues. For each biologic subtype, the WG reviewed biology and prognosis, current standard treatments, recent drug approvals, and ongoing phase III trials and end points. We sought to achieve consensus on the appropriate end point and magnitude of absolute and relative gains that would represent meaningful clinical benefit. In this context, the WG critically assessed current knowledge and perception of the balance between incremental PFS gain and encountered toxicity/effect on QoL, with consideration of both categorical and combinatorial approaches. The WG examined optimal strategies to best capture toxicity and integration of PROs into such clinical benefit analysis.
Fig 1.

Preferred Reporting Items for Systematic Reviews and Meta-Analyses diagram for literature analysis. Flow diagram illustrates the selection of articles for analysis. (*) Not relevant on end point issue for randomized controlled trials.
RESULTS
Consensus on Definitions
The most objective and validated end point for clinical trials is OS, which is defined as the time from random assignment to the time of patient death as a result of any cause. The development of surrogate end points, which would allow for smaller and shorter trials, has been influenced by the pragmatic desire to reduce both the cost of trials and the timeline for an effective drug to reach the marketplace. Minor differences in definitions can have a significant effect on the reported treatment outcomes and can lead to difficulty in making cross-trial comparisons. The WG recognized the value in providing robust definitions for clinical trial end points, as was done for early-stage breast cancer in the STEEP guidelines.8 The Definition for the Assessment of Time to Event Endpoints in Cancer Trials (DATECAN) project aimed to standardize definitions of time-to-event end points to facilitate comparisons of trial results and improve the quality of trial design and reporting.9 The DATECAN group used a formal consensus (Delphi) methodology to reach agreement on appropriate end points and definitions for both metastatic and nonmetastatic disease in breast, sarcoma/GI stromal tumor, and pancreatic cancer. This group identified PFS and time to progression as the most commonly used primary end points in randomized clinical trials of MBC. For PFS, the recommended clinical events for inclusion in the definition were death as a result of breast cancer, death as a result of nonbreast cancer cause, death related to protocol treatment, death as a result of any cause, death as a result of unknown cause, regional invasive recurrence/progression, and appearance/occurrence of metastases/distant recurrence. For time to progression, the recommendation was to include death as a result of breast cancer, regional invasive recurrence/progression, and appearance/occurrence of metastases/distant recurrence.
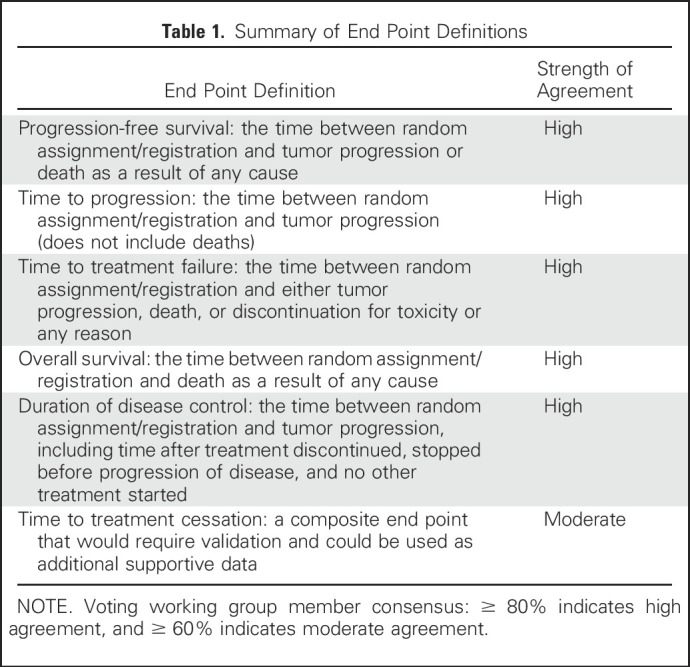
The WG believed that the DATECAN guideline definitions were appropriate and that the appropriate starting point for time-to-event determinations is time of random assignment or registration to a study rather than time of treatment initiation. We achieved consensus (defined as ≥ 80% of WG members agreeing or strongly agreeing with the statement) on definitions for several end points believed to be pertinent to MBC clinical trials (Table 1).
Table 1.
Summary of End Point Definitions
One consideration in trial design is the question of evaluating the sequencing of agents versus their combination. When new agents are being assessed, clinical trials commonly compare standard-of-care therapy with standard of care plus an investigational agent. In the absence of a compelling preclinical/biologic rationale, which often exists in investigating combinations, it may be valuable to compare the strategy that combines the standard of care and the investigational therapy versus the sequential receipt of them. The end point of time to treatment failure (TTF) spurred additional discussion. TTF usually is defined as the time since random assignment/registration to treatment discontinuation for any reason, including disease progression, treatment toxicity, patient preference, or death. The FDA has noted that TTF is a composite end point that is seldom useful for regulatory purposes because discontinuation of a drug for toxicity is not a direct reflection of its efficacy.10 The FDA posits that separate analyses of safety and efficacy are required for regulatory approval. However, given that in clinical practice patients may stop a given therapy for a multitude of reasons, TTF may have some relevance to patients. The WG believed that a composite end point that considers reasons for treatment discontinuation, such as patient preference and toxicity, although perhaps challenging to operationalize, could be clinically meaningful and useful in treatment decision making for patients and clinicians. The WG also considered several scenarios in which objective progression defined by Response Evaluation Criteria in Solid Tumors (RECIST) might not indicate treatment failure or the need to change therapy. Some examples include focal progression amenable to local therapy, indolent or asymptomatic progression, and progression while receiving immunotherapy. This issue was addressed by Oxnard et al,11 who recommended a more-detailed collection of progression characteristics, additional prospective study of treatment beyond progression, and exploration of alternate progression end points in clinical trials as potential ways to facilitate development of more-meaningful criteria for objective progression. The validated Prostate Cancer Working Group 2 guidelines for assessing progression in bone metastases, as they relate to the correlation between PFS and OS, is one recent example that breaks with traditional RECIST criteria.12
Critical Review of the Literature: Consideration of End Points by Biologic Subtype
The WG’s recommendations for preferred primary, coprimary, and secondary end points are detailed in this section and summarized in Figure 2A, with an illustration of how these recommendations were reached shown in Figure 2B. The WG strove to link recommendations for preferred end points primarily to expected PPS, which we recognize as a moving target with serial introduction of new therapeutic agents that have and will continue to increase PFS progressively and in certain circumstances, OS.
Fig 2.

(A) Working group consensus on preferred end points by biologic subtype and line of therapy. (B) Hypothetical scenarios for expected postprogression survival (PPS) and choice of preferred end point. In settings such as first-line treatment of triple-negative metastatic breast cancer (TNMBC) where expected PPS is < 12 months, overall survival (OS) is the preferred primary end point. In settings such as hormone receptor–negative (HR–)/human epidermal growth factor receptor 2–positive (HER2+) or HR+/HER2+ MBC where PPS is > 12 months, in both the first- and later-line settings, progression-free survival (PFS) is the end point of choice, and OS could be considered as a coprimary end point. In settings such as HR+/HER2– MBC, given the expected long PPS, PFS is the most appropriate end point. When such patients have disease that is refractory to endocrine therapy and have been exposed to several lines of chemotherapy, where PPS is expected to be much shorter, OS may be the most meaningful and appropriate end point. (*) Line of therapy may be endocrine therapy, chemotherapy, HER2-targeted therapy, combinations, and so forth. (†) Months shown are for illustrative purposes only. 1°, primary end point; 2°, secondary end point; PRO, patient-reported outcome; RR, response rate.
HR+/HER2–.
More than one half of all patients with MBC have HR expression, yet both intrinsic and acquired resistance limit the efficacy of anti-estrogen therapy. Loss of estrogen receptor expression, estrogen receptor mutations, altered expression of coregulators, and upregulation of alternative signal transduction pathways are all mechanisms of resistance that currently are being targeted. The development of rationally designed therapeutics has led to randomized trials that demonstrate that when added to anti-estrogen therapy, these agents can significantly extend PFS. For this population, given the expected long PPS, the WG regards PFS as the most robust and appropriate end point; the detection of an OS benefit is regarded as nice to have but not as need to have for such an approach to be clinically meaningful (Fig 2A). The FDA agreed that PFS was an acceptable primary end point and that it would be important to demonstrate no detriment in OS. When such patients have disease that is refractory to endocrine therapy and have been exposed to several lines of chemotherapy, where PPS is expected to be much shorter, OS may emerge as the preferred end point (Fig 2A).
The addition of CDK4/6 inhibitors palbociclib, ribociclib, or abemaciclib to an aromatase inhibitor (AI) as first-line therapy for postmenopausal woman with MBC yields a clinically and statistically improved PFS (Table 2). Although additional toxicities were more common with CDK4/6 inhibitors, they were uncommonly grade > 2. The addition of everolimus to exemestane significantly improved PFS without improvement in OS (Table 2). Everolimus use was associated with more grade ≥ 3 toxicity.
Table 2.
Pivotal Phase III Clinical Trials in MBC

Some trials that have not led to regulatory approval nevertheless were informative for the WG in the consideration of optimal end points in this population. Two randomized trials examined the addition of bevacizumab to endocrine therapy for MBC.3,19 In CALGB 40503 (Alliance), the experimental arm had a modest, but statistically significantly improved PFS, but grade ≥ 3 adverse events (AEs) were approximately three-fold higher with bevacizumab (Table 2). Similarly, in the Letrozole/Fulvestrant and Avastin trial, more-serious toxicities and several toxicity-related deaths occurred in the bevacizumab arm (Table 2).
The WG agreed that proportional and absolute gains in PFS need to be balanced carefully against toxicity. In the future, metrics that capture toxicity over time may be more meaningful and complement conventional worst-grade reporting.25,26 The multisymptom spider plot used by Woo et al27 is a useful tool for visualizing the levels of multiple AEs at specific time points in a composite manner. Thanarajasingam et al25 developed a toxicity over time approach to assess AEs longitudinally. Toxicity over time provides clinically meaningful data that can be visualized in several ways, such as by butterfly plot that displays side by side the mean grade for multiple AEs over all treatment cycles for two agents or by area under the curve to overlay the mean grades over treatment cycles for a specific AE for two agents. The WG regards the integration of PROs28-30 as especially relevant for patients with HR+/HER2– breast cancer as new agents with significant potential toxicities, such as phosphoinositide 3-kinase inhibitors and histone deacetylase inhibitors, are evaluated.
HR–/HER2– (TNMBC).
Approximately 15% to 20% of breast cancers are triple negative; however, this subgroup of breast cancers is quite heterogeneous by gene expression analysis.31,32 The prognosis of TNMBC is poor, with an estimated median OS of 10 to 18 months and PPS after first-line therapy of 6 months.1,32 Current standard treatment is primarily cytotoxic chemotherapy, but the development of novel biomarkers (eg, homologous recombination deficiency, androgen receptors) and novel therapeutic approaches (eg, antibody-drug conjugates, immunotherapy, targeted agents) likely will change the treatment landscape. Given limited prognosis, OS has been suggested as a valid primary end point for this patient population, irrespective of line of therapy1 (Fig 2A). The WG examined the magnitude of benefit considered to be clinically significant in simulations; an OS improvement of 4.5 to 6 months with a hazard ratio of 0.75 to 0.8 with acceptable toxicity was believed to be a reasonable target in the first-line TNMBC setting. This recommendation may change with the advent of effective novel therapies that prolong expected PPS.
HR–/HER2+.
HER2 is amplified in approximately 25% of breast cancers, and HRs are co-expressed in approximately 50% of these.33,34 Although treated similarly, some key differences exist between HR+ and HR–/HER2+ disease. HR–/HER2+ breast cancer is more likely to metastasize to viscera than HR+/HER2+ disease. In the registHER study, distant disease-free interval (defined as the time between the end of nonhormonal adjuvant treatment and metastatic diagnosis) was 26.1 months for HR+ disease and 13.1 months for HR– disease.35 PFS and OS are somewhat better for patients with HR+ disease.35,36
The prognosis for HER2+ breast cancer has dramatically improved with the standard use of HER2-targeted therapies in both the adjuvant and the metastatic setting. In the CLEOPATRA (Clinical Evaluation of Pertuzumab and Trastuzumab) trial, the addition of pertuzumab to docetaxel and trastuzumab in the first-line setting significantly improved both PFS and OS37 (Table 2). These data led to FDA approval of pertuzumab in the first-line treatment of HER2+ MBC and sets the benchmark by which future treatments in the first-line setting should be measured. The EMILIA (Trastuzumab Emtansine Versus Capecitabine + Lapatinib in Participants With HER2-Positive Locally Advanced or Metastatic Breast Cancer) trial compared ado-trastuzumab emtansine (T-DM1) with capecitabine and lapatinib in patients with prior taxane and trastuzumab treatment.20 Treatment with T-DM1 resulted in improved OS and PFS (Table 2).
In CLEOPATRA, the primary end point was PFS; OS was a secondary end point. In EMILIA, PFS and OS were coprimary end points. As of April 2017, there were eight phase III clinical trials of HER2-targeted therapies in the metastatic setting; three of these studies have OS as a coprimary end point (Table 3). In both first- and later-line settings, we conclude that PFS is the end point of choice for this patient population and that OS could be considered as a coprimary end point (Fig 2A). Studies of new agents or those that seek to move an agent from later to earlier lines of therapy should target at least a 6-month improvement in PFS. In a later-line setting, a lower benefit in PFS could be considered acceptable if the adverse event profile of the experimental therapy is significantly better than the currently approved agents. Although cost is not currently a consideration used for evaluation and drug approval by the FDA, this factor may be considered by the patient, payer, and prescriber for drugs with modest clinical benefit.5
Table 3.
Ongoing Trials in HER2+ Breast Cancer

HR+/HER2+.
For patients with HR+/HER2+ MBC, one FDA-approved regimen and at least two other approaches may be considered for postmenopausal patients for whom chemotherapy is deemed not indicated. The addition of lapatinib to letrozole significantly improves PFS at the expense of more dermatologic and GI toxicity (Table 2). The addition of trastuzumab to anastrozole also has been shown to prolong PFS (Table 2). In CLEOPATRA, the benefit of adding pertuzumab to taxane and trastuzumab did not seem to vary by HR status. The role of dual inhibition of HER2 with endocrine therapy and no chemotherapy for first-line treatment in selected patients is examined in two studies: the ALTERNATIVE (Alternate Approaches for Clinical Stage II or III Estrogen Receptor Positive Breast Cancer Neoadjuvant Treatment) trial, which met its primary objective of an improved PFS with the addition of lapatinib to AI and trastuzumab compared with AI and trastuzumab alone,23 and the randomized phase II PERTAIN (Pertuzumab in Combination With Trastuzumab Plus an AI in Participants With Metastatic HER2+ and HR+ Advanced Breast Cancer) trial in which a 3-month improvement in median PFS for the addition of pertuzumab to AI plus trastuzumab was observed for the intention-to-treat population24 (Table 2). In PERTAIN, 148 of the 258 enrolled patients received discretionary induction chemotherapy, a decision driven by patient attributes such as disease burden, symptoms, and patient preference. In terms of primary end point selection, consideration of the expected PPS still remains paramount in trial design, regardless of the choice of whether to lead with chemotherapy for such patients who have broad options. For this population, the WG consensus is that in both first- and later-line settings, PFS is the preferred end point and OS could be considered a coprimary end point (Fig 2A).
End Point Selection by Line of Therapy and/or Expected PPS
In consideration of novel primary end points other than OS (particularly PFS but also even earlier end points such as response rate), it is useful to consider both their intrinsic importance with respect to clinical benefit for the patient, which may allow approval without proven surrogacy, and their possible value as validated surrogate end points for OS. Sargent et al38 discussed the validation of such end points. Early-phase evaluation involves the establishment of the correlation of end points with established outcome variables, particularly OS, on the patient level, which can be done in the context of individual randomized or nonrandomized trials or even outside the context of formal clinical trials. After such correlation has been established, the end points can be used in individual patient management. However, to establish such end points as appropriate for the primary outcome assessment in clinical trials, it must be established that the new end point captures a substantial portion of the treatment benefit (on the trial level) associated with an established end point, such as OS.39 In other words, a meta-analysis across randomized trials that involve related treatments must be conducted to establish that the treatment benefit (eg, OS) seen across the trials is highly correlated with that of the new end point.
When PFS Is the Preferred End Point, What Are Meaningful Relative and Absolute Gains?
The WG considered a series of hypothetical case simulations to assess our internal level of consensus on the magnitude of incremental PFS gain that would be meaningful/acceptable in the context of various degrees of toxicity. Table 4 depicts one such experiment. As expected, with increasing expected toxicity, the threshold incremental PFS gain desired for meaningful benefit increases. Ultimately, it is the patients’ perspective, rather than that of this WG, that matters most in this trade-off analysis. Hurvitz et al2 used the Metastatic Breast Cancer Progression Questionnaire to assess how patients value PFS in the context of MBC, with hypothetical scenarios presented and patient feedback provided. Patients associated longer PFS with improved QoL, physical functioning, and emotional well-being and preferred a treatment that prolonged PFS from 12 to 16 months, even when adverse effects and OS were proposed to be identical. Unfortunately, a paucity of meaningful information remains on this important subject, one that begs for rigorous future investigation.
Table 4.
Balance Between Incremental PFS Gain and Degree of Encountered Toxicity

PFS/Toxicity-QoL Burden: The Balance and Meaningful Benefit
Meaningful benefit is a term used in both drug regulatory approval and patient treatment decisions. All treatment decisions in metastatic cancer depend on an informed benefit/risk (PFS or OS/toxicity) ratio. Substantial toxicities, whether short or long term, reversible, financial, or temporal, can counterbalance improvements in PFS and even OS for patients. Patients who are adjusting to an incurable illness show great variability in tolerance to treatment toxicities and QoL issues. Unique patient life experiences and different philosophical/spiritual beliefs result in a wide spectrum of value decisions about the benefit/risk balance. Patients and their clinicians must weigh the trade-off between effectiveness of treatment and QoL; a few extra months of symptomatic life prolongation may be insignificant to some and highly prized by others. Explaining and understanding the concept of average PFS improvement with unclear OS benefit and contrasting with likely toxicities pose a challenge to physicians and patients and frequently fall short of clarity. The WG considered and evaluated external attempts to quantify levels of toxicity for comparison with antitumor activity to define meaningful benefit but found this challenging. Measures that could improve toxicity reporting in clinical trials include commitment of both financial resources and statistical rigor to the study of QoL and PROs, attention to reporting of both the time course and the severity of toxicities, and visual methods for educating patients and clinicians about QoL results from clinical trials. Such measures are crucial for the evaluation and communication of meaningful benefit in the context of drug development.
Regulatory Perspective
Approval of drugs in the United States requires substantial evidence of clinical benefit, including safety and effectiveness, on the basis of adequate and well-controlled trials.40 The accelerated approval regulations41 subsequently have allowed for additional end points to support the approval of drugs or biologic products that are reasonably likely to predict clinical benefit, with postapproval trials verifying that benefit. There is no comparative efficacy requirement for regular approval. However, to meet accelerated approval requirements, a drug should demonstrate a benefit over available therapy.
The appropriate end point to support approval, therefore, depends on the pathway being sought. For regular approval, the end point should reflect direct clinical benefit, and for accelerated approval, it should be reasonably likely to predict clinical benefit. The FDA Guidance for Industry (2007) provides details of clinical trial end points for the approval of cancer drugs and biologics.4 This guidance does not include specific considerations related to breast cancer but does delineate certain statistical concepts related to preferred FDA definitions of end points, including OS, PFS, and objective response rate (ORR). These end points have factored into both regular and accelerated breast cancer approvals.42-45
OS can be challenging to assess in MBC, given that patients may receive multiple subsequent therapies after progression that can affect OS, thereby confounding its relevance as the most robust end point. However, OS should be considered as a primary or coprimary end point when expected survival is short (< 6 to 12 months).46 In many cases, the FDA has accepted PFS and ORR supported by long duration of response, especially in rare biomarker-defined subsets, as the primary end point, and depending on the context, these end points could support either regular or accelerated approval. For any end point, the magnitude of benefit that is clinically meaningful needs to be considered in the context of the safety/tolerability profile of the agent and the available therapy. The FDA does not specify a requirement for absolute or relative gains in PFS or ORR because each application is viewed independently.
From a regulatory perspective, use of alternative end points, such as TTF, could be challenging because the end point is confounded by factors unrelated to efficacy, including toxicity, patient preference, and physician reluctance to continue therapy. Nevertheless, alternative end points could be examined in concert with PFS and should track in the same direction to be viewed as supportive information.
In real-world breast cancer clinical practice, progression events by RECIST may not result in therapy discontinuation, and thus, with a prespecified plan and rationale, the continuation of treatment beyond progression in clinical trials may be acceptable from a regulatory standpoint and should be discussed with the FDA during trial design. This approach could allow patients to continue therapy if their treating physicians believe that they are deriving clinical benefit while still allowing for end point evaluation. In addition, the FDA routinely provides advice about collection of PROs to aid in the assessment of clinical benefit for new treatments.25,26,28,47
In conclusion, recognition of the heterogeneity in biology, and thus, the expected variable outcomes of patients who enroll in clinical trials of systemic therapy for MBC, mandates careful consideration of situation-sensitive and appropriate choice of primary outcome measures. The WG considered a series of key position statements related to this to assess our internal level of consensus (Table 5). In scenarios where PFS is the more appropriate end point, careful consideration of the balance between incremental PFS gain and encountered toxicity and QoL and PROs will enable stakeholders to avoid the expenditure of resources in trials that yield statistically significant P values but not clinically meaningful results. However, when outcomes are poor and PPS is short, OS is the most appropriate end point. The future development and validation of composite metrics that capture both efficacy and toxicity/effect on QoL and PROs collectively may afford a valuable means to prospectively define clinical benefit upfront in the trial design phase rather than out back after its conclusion.48,49 In addition, a more patient-centered approach to the conduct of clinical trials would provide more meaningful data to patients to inform their decision making on an individual level.
Table 5.
Summary of Key Position Statements

ACKNOWLEDGMENT
The contributions of S.B.W., L.A.K., and J.A.B. represent their opinions and not those of the US FDA. We thank Allen Melemed, MD, MBA (Eli Lilly), and Maria Koehler, MD, PhD (Pfizer), for providing a pharmaceutic/biotechnology industry perspective. We also thank Lori Minasian, MD (National Cancer Institute), and Paul Kluetz, MD (US FDA), for valuable input about PROs. Finally, we thank Amy Abernethy, MD, PhD (Flatiron Health), for sharing a unique perspective on how real-world evidence and big data analyses may inform clinical trial design in the future.
Footnotes
Supported by the Coordinating Center for Clinical Trials, Office of the Director, National Cancer Institute.
Presented at the American Society of Clinical Oncology Annual Meeting, Chicago, IL, June 2-6, 2017.
AUTHOR CONTRIBUTIONS
Conception and design: Andrew D. Seidman, Louis Fehrenbacher, William E. Barlow, Jane Perlmutter, Lawrence Rubinstein, Suparna B. Wedam, Dawn L. Hershman, Jennifer Fallas Hayes, Meredith M. Regan, Julia A. Beaver, Laleh Amiri-Kordestani, Priya Rastogi, Jo Anne Zujewski, Larissa A. Korde
Administrative support: Lynn Pearson Butler
Collection and assembly of data: Andrew D. Seidman, Louis Fehrenbacher, Jennifer Fallas Hayes, Lynn Pearson Butler, Meredith M. Regan, Julia A. Beaver, Laleh Amiri-Kordestani, Priya Rastogi, Jo Anne Zujewski, Larissa A. Korde
Data analysis and interpretation: Andrew D. Seidman, Louise Bordeleau, Louis Fehrenbacher, Lawrence Rubinstein, Dawn L. Hershman, Jennifer Fallas Hayes, Mary Lou Smith, Meredith M. Regan, Julia A. Beaver, Laleh Amiri-Kordestani, Jo Anne Zujewski, Larissa A. Korde
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
National Cancer Institute Breast Cancer Steering Committee Working Group Report on Meaningful and Appropriate End Points for Clinical Trials in Metastatic Breast Cancer
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/site/ifc.
Andrew D. Seidman
Honoraria: Genomic Health, Eisai, Genentech, Roche, Celgene, Eli Lilly, Novartis, Pfizer
Consulting or Advisory Role: Genentech, Roche, Pfizer, Celgene, Eisai, Novartis
Speakers’ Bureau: Genomic Health, Eisai, Genentech, Roche, Celgene, Eli Lilly, Novartis, Pfizer
Research Funding: Bayer AG, Odonate Therapeutics (Inst), Nektar (Inst)
Travel, Accommodations, Expenses: Genomic Health, Genentech, Roche, Eisai, Celgene
Louise Bordeleau
Consulting or Advisory Role: AstraZeneca, Novartis
Louis Fehrenbacher
No relationship to disclose
William E. Barlow
Research Funding: AbbVie (Inst), Merck (Inst), AstraZeneca (Inst)
Jane Perlmutter
No relationship to disclose
Lawrence Rubinstein
No relationship to disclose
Suparna B. Wedam
No relationship to disclose
Dawn L. Hershman
No relationship to disclose
Jennifer Fallas Hayes
No relationship to disclose
Lynn Pearson Butler
No relationship to disclose
Mary Lou Smith
Consulting or Advisory Role: Takeda
Research Funding: Genentech (Inst), Celgene (Inst), Genomic Health (Inst), Novartis (Inst)
Travel, Accommodations, Expenses: Novartis, GHI Pharma
Meredith M. Regan
Consulting or Advisory Role: Merck, Ipsen (Inst)
Research Funding: Veridex (Inst), OncoGenex Pharmaceuticals (Inst), Pfizer (Inst), Ipsen (Inst), Novartis (Inst), Merck (Inst), Ferring Pharmaceuticals (Inst), Celgene (Inst), AstraZeneca (Inst), Pierre Fabre (Inst), Ipsen (Inst), Bayer AG (Inst), Bristol-Myers Squibb (Inst), Roche (Inst)
Travel, Accommodations, Expenses: Bristol-Myers Squibb
Julia A. Beaver
No relationship to disclose
Laleh Amiri-Kordestani
No relationship to disclose
Priya Rastogi
No relationship to disclose
Jo Anne Zujewski
Consulting or Advisory Role: Leidos Biomedical Research, PMK Bioresearch
Travel, Accommodations, Expenses: Leidos Biomedical Research
Larissa A. Korde
No relationship to disclose
REFERENCES
- 1.Ellis LM, Bernstein DS, Voest EE, et al. : American Society of Clinical Oncology perspective: Raising the bar for clinical trials by defining clinically meaningful outcomes. J Clin Oncol 32:1277-1280, 2014 [DOI] [PubMed] [Google Scholar]
- 2.Hurvitz SA, Lalla D, Crosby RD, et al. : Use of the Metastatic Breast Cancer Progression (MBC-P) questionnaire to assess the value of progression-free survival for women with metastatic breast cancer. Breast Cancer Res Treat 142:603-609, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dickler MN, Barry WT, Cirrincione CT, et al. : Phase III trial evaluating letrozole as first-line endocrine therapy with or without bevacizumab for the treatment of postmenopausal women with hormone receptor-positive advanced-stage breast cancer: CALGB 40503 (Alliance). J Clin Oncol 34:2602-2609, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. US Food and Drug Administration: Guidance for Industry: Clinical trial endpoints for the approval of cancer drugs and biologics, 2007. https://www.fda.gov/downloads/drugsGuidanceComplianceRegulatoyInformation/Guida nce/UCM071590.pdf.
- 5. US Food and Drug Administration: Prescription Drug User Fee Act (PDUFA). https://www.fda.gov/forindustry/userfees/prescriptiondruguserfee.
- 6. National Cancer Institute: Breast Cancer Steering Committee. http://www.cancer.gov/about-nci/organization/ccct/steering-committees/nctn/breast-cancer.
- 7.Schnipper LE, Davidson NE, Wollins DS, et al. : American Society of Clinical Oncology Statement: A conceptual framework to assess the value of cancer treatment options. J Clin Oncol 33:2563-2577, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hudis CA, Barlow WE, Costantino JP, et al. : Proposal for standardized definitions for efficacy end points in adjuvant breast cancer trials: The STEEP system. J Clin Oncol 25:2127-2132, 2007 [DOI] [PubMed] [Google Scholar]
- 9.Gourgou-Bourgade S, Cameron D, Poortmans P, et al. : Guidelines for time-to-event end point definitions in breast cancer trials: Results of the DATECAN initiative (Definition for the Assessment of Time-to-event Endpoints in CANcer trials). Ann Oncol 26:873-879, 2015 [DOI] [PubMed] [Google Scholar]
- 10.Johnson JR, Williams G, Pazdur R: End points and United States Food and Drug Administration approval of oncology drugs. J Clin Oncol 21:1404-1411, 2003 [DOI] [PubMed] [Google Scholar]
- 11.Oxnard GR, Morris MJ, Hodi FS, et al. : When progressive disease does not mean treatment failure: Reconsidering the criteria for progression. J Natl Cancer Inst 104:1534-1541, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rathkopf DE, Beer TM, Loriot Y, et al: Radiographic progression-free survival as a clinically meaningful end point in metastatic castrate-resistant prostate cancer: The PREVAIL randomized clinical trial. JAMA Oncol 4:694-701, 2018. [DOI] [PMC free article] [PubMed]
- 13.Baselga J, Campone M, Piccart M, et al. : Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med 366:520-529, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Finn RS, Martin M, Rugo HS, et al. : Palbociclib and letrozole in advanced breast cancer. N Engl J Med 375:1925-1936, 2016 [DOI] [PubMed] [Google Scholar]
- 15.Turner NC, Ro J, André F, et al. : Palbociclib in hormone-receptor positive advanced breast cancer. N Engl J Med 373:209-219, 2015 [DOI] [PubMed] [Google Scholar]
- 16.Hortobagyi GN, Stemmer SM, Burris HA, et al. : Ribociclib as first-line treatment for HR-positive advanced breast cancer. N Engl J Med 375:1738-1748, 2016 [DOI] [PubMed] [Google Scholar]
- 17.Sledge GW, Jr, Toi M, Neven P, et al. : MONARCH 2: Abemaciclib in combination with fulvestrant in women with HR+/HER2- advanced breast cancer who had progressed while receiving endocrine therapy. J Clin Oncol 35:2875-2884, 2017 [DOI] [PubMed] [Google Scholar]
- 18.Goetz MP, Toi M, Campone M, et al. : MONARCH 3: Abemaciclib as initial therapy for advanced breast cancer. J Clin Oncol 35:3638-3646, 2017 [DOI] [PubMed] [Google Scholar]
- 19.Martín M, Loibl S, von Minckwitz G, et al. : Phase III trial evaluating the addition of bevacizumab to endocrine therapy as first-line treatment for advanced breast cancer: The Letrozole/Fulvestrant and Avastin (LEA) study. J Clin Oncol 33:1045-1052, 2015 [DOI] [PubMed] [Google Scholar]
- 20.Verma S, Miles D, Gianni L, et al. : Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med 367:1783-1791, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnston S, Pippen J, Jr, Pivot X, et al. : Lapatinib combined with letrozole versus letrozole and placebo as first-line therapy for postmenopausal hormone receptor-positive metastatic breast cancer. J Clin Oncol 27:5538-5546, 2009 [DOI] [PubMed] [Google Scholar]
- 22.Kaufman B, Mackey JR, Clemens MR, et al. : Trastuzumab plus anastrozole versus anastrozole alone for the treatment of postmenopausal women with human epidermal growth factor receptor 2-positive, hormone receptor-positive metastatic breast cancer: Results from the randomized phase III TAnDEM study. J Clin Oncol 27:5529-5537, 2009 [DOI] [PubMed] [Google Scholar]
- 23.Johnston SRD, Hegg R, Im SA, et al. : Phase III randomized study of dual human epidermal growth factor receptor 2 (HER2) blockade with lapatinib plus trastuzumab in combination with an aromatase inhibitor in postmenopausal women with HER2 positive, hormone receptor positive metastatic breast cancer: ALTERNATIVE. J Clin Oncol 36:741-748, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 24. Arpino G, Ferrero JM, de la Haba-Rodriguez J, et al: Primary analysis of PERTAIN: A randomized, two-arm, open label, multicenter phase II trial assessing the efficacy and safety of pertuzumab given in combination with trastuzumab plus an aromatase inhibitor in first-line patients with HER2-positive and hormone receptor-positive metastatic or locally advanced breast cancer. Cancer Res 77, 2017 (suppl; abstr S3-04) [Google Scholar]
- 25.Thanarajasingam G, Atherton PJ, Novotny PJ, et al. : Longitudinal adverse event assessment in oncology clinical trials: The toxicity over time (ToxT) analysis of Alliance trials NCCTG N9741 and 979254. Lancet Oncol 17:663-670, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee SM, Hershman DL, Martin P, et al. : Toxicity burden score: A novel approach to summarize multiple toxic effects. Ann Oncol 23:537-541, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Woo JA, Chen LN, Bhagat A, et al. : Clinical characteristics and management of late urinary symptom flare following stereotactic body radiation therapy for prostate cancer. Front Oncol 4:122, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Basch E, Geoghegan C, Coons SJ, et al. : Patient-reported outcomes in cancer drug development and US regulatory review: Perspectives from industry, the Food and Drug Administration, and the patient. JAMA Oncol 1:375-379, 2015 [DOI] [PubMed] [Google Scholar]
- 29.Bottomley A, Pe M, Sloan J, et al. : Analysing data from patient-reported outcome and quality of life endpoints for cancer clinical trials: A start in setting international standards. Lancet Oncol 17:e510-e514, 2016 [DOI] [PubMed] [Google Scholar]
- 30.Turner-Bowker DM, Hao Y, Foley C, et al. : The use of patient-reported outcomes in advanced breast cancer clinical trials: A review of the published literature. Curr Med Res Opin 32:1709-1717, 2016 [DOI] [PubMed] [Google Scholar]
- 31.Foulkes WD, Smith IE, Reis-Filho JS: Triple-negative breast cancer. N Engl J Med 363:1938-1948, 2010 [DOI] [PubMed] [Google Scholar]
- 32.Lehmann BD, Bauer JA, Chen X, et al. : Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest 121:2750-2767, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Slamon DJ, Godolphin W, Jones LA, et al. : Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 244:707-712, 1989 [DOI] [PubMed] [Google Scholar]
- 34.Ross JS, Slodkowska EA, Symmans WF, et al. : The HER-2 receptor and breast cancer: Ten years of targeted anti-HER-2 therapy and personalized medicine. Oncologist 14:320-368, 2009 [DOI] [PubMed] [Google Scholar]
- 35.Tripathy D, Kaufman PA, Brufsky AM, et al. : First-line treatment patterns and clinical outcomes in patients with HER2-positive and hormone receptor-positive metastatic breast cancer from registHER. Oncologist 18:501-510, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Reference deleted. [Google Scholar]
- 37.Swain SM, Baselga J, Kim SB, et al. : Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N Engl J Med 372:724-734, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sargent DJ, Rubinstein L, Schwartz L, et al. : Validation of novel imaging methodologies for use as cancer clinical trial end-points. Eur J Cancer 45:290-299, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Freedman LS, Graubard BI, Schatzkin A: Statistical validation of intermediate endpoints for chronic diseases. Stat Med 11:167-178, 1992 [DOI] [PubMed] [Google Scholar]
- 40. Government Publishing Office: Electronic Code of Federal Regulations: Title 21: §314.126: Adequate and well-controlled studies. https://www.ecfr.gov/cgi-bin/text-idx?SID=8eea61dec0ef9b17f1fa0d0d3a7736c6&mc=true&node=se21.5.314_1126&rgn =div8.
- 41. US Food and Drug Administration: Guidance for Industry: Expedited Programs for Serious Conditions—Drugs and Biologics, 2014. https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidan ces/ucm358301.pdf.
- 42.Cortazar P, Justice R, Johnson J, et al. : US Food and Drug Administration approval overview in metastatic breast cancer. J Clin Oncol 30:1705-1711, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beaver JA, Amiri-Kordestani L, Charlab R, et al. : FDA approval: Palbociclib for the treatment of postmenopausal patients wither estrogen receptor-positive, HER2-negative metastatic breast cancer. Clin Cancer Res 21:4760-4766, 2015 [DOI] [PubMed] [Google Scholar]
- 44.Amiri-Kordestani L, Blumenthal GM, Xu QC, et al. : FDA approval: Ado-trastuzumab emtansine for the treatment of patients with HER2-positive metastatic breast cancer. Clin Cancer Res 20:4436-4441, 2014 [DOI] [PubMed] [Google Scholar]
- 45.Beaver JA, Howie LJ, Pelosof L, et al. : A 25-year experience of US Food and Drug Administration accelerated approval of malignant hematology and oncology drugs and biologics: A review. JAMA Oncol 4:849-856, 2018 [DOI] [PubMed] [Google Scholar]
- 46.Broglio KR, Berry DA: Detecting an overall survival benefit that is derived from progression-free survival. J Natl Cancer Inst 101:1642-1649, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kluetz PG, Chingos DT, Basch EM, et al. : Patient-reported outcomes in cancer clinical trials: Measuring symptomatic adverse events with the National Cancer Institute’s Patient-Reported Outcomes Version of the Common Terminology Criteria for Adverse Events (PRO-CTCAE). Am Soc Clin Oncol Educ Book 35:67-73, 2016 [DOI] [PubMed] [Google Scholar]
- 48.Cherny NI, Sullivan R, Dafni U, et al. : A standardised, generic, validated approach to stratify the magnitude of clinical benefit that can be anticipated from anti-cancer therapies: The European Society for Medical Oncology Magnitude of Clinical Benefit Scale (ESMO-MCBS). Ann Oncol 26:1547-1573, 2015 [DOI] [PubMed] [Google Scholar]
- 49.Wilson MK, Karakasis K, Oza AM: Outcomes and endpoints in trials of cancer treatment: The past, present, and future. Lancet Oncol 16:e32-e42, 2015 [DOI] [PubMed] [Google Scholar]