

Abstract
Next-generation sequencing has revealed recurring somatic mutations in Waldenström macroglobulinemia (WM), including MYD88 (95%-97%), CXCR4 (30%-40%), ARID1A (17%), and CD79B (8%-15%). Deletions involving chromosome 6q are common in patients with mutated MYD88 and include genes that modulate NFKB, BCL2, Bruton tyrosine kinase (BTK), and apoptosis. Patients with wild-type MYD88 WM show an increased risk of transformation and death and exhibit many mutations found in diffuse large B-cell lymphoma. The discovery of MYD88 and CXCR4 mutations in WM has facilitated rational drug development, including the development of BTK and CXCR4 inhibitors. Responses to many agents commonly used to treat WM, including the BTK inhibitor ibrutinib, are affected by MYD88 and/or CXCR4 mutation status. The mutation status of both MYD88 and CXCR4 can be used for a precision-guided treatment approach to WM.
INTRODUCTION
Waldenström macroglobulinemia (WM) is a B-cell malignancy characterized as an immunoglobulin M (IgM)–secreting lymphoplasmacytic lymphoma using WHO criteria.1 Patients can present with morbidity related to excess malignant lymphoplasmacytic cells (LPCs) in the bone marrow (BM), lymph nodes, and spleen, as well as IgM production that can produce symptomatic hyperviscosity, cryoglobulinemia, and autoimmune-related complications.2 Despite advances in therapy, WM remains incurable. Treatment options for WM have largely represented therapeutics studied in other diseases, with more recent advances facilitated by next-generation sequencing (NGS).
Using NGS, recurring somatic mutations in MYD88, CXCR4, ARID1A, and CD79 were identified in WM LPC. Copy number alterations, including those in chromosome 6q that affect regulatory genes for NFKB, Bruton tyrosine kinase (BTK), BCL2, and apoptotic signaling, were also identified.3 Although most patients with WM (95%-97%) carry a point mutation in MYD88 that switches leucine to proline at amino acid position 265, those that are wild type for MYD88 show a more aggressive disease course and possess somatic mutations that overlap with those found in diffuse large B-cell lymphoma (DLBCL).4,5 Herein, we discuss the genomic landscape of WM and the impact of underlying genomics on disease presentation, transcriptional changes, treatment outcome, and overall survival. The use of MYD88 and CXCR4 mutation status to guide treatment in treatment-naïve and previously treated patients with WM is also discussed.
MUTATIONS IN MYD88
A recurring somatic mutation in MYD88 (L265P) was identified in 91% of patients with WM by paired tumor and normal whole-genome sequencing and subsequently confirmed by Sanger sequencing and allele-specific polymerase chain reaction (AS-PCR) assays by multiple investigators.6 Using sensitive AS-PCR testing, MYD88 L265P was found to be expressed in 93%-97% of patients with WM and was identified in both sorted B cells and plasma cells that make up the malignant clone in WM.7-11 Non-L265P MYD88 mutations have also been identified, although expression estimates for these variants are 1%-2% in WM.12,13 Mutated MYD88 was also detectable in patients with IgM but not immunoglobulin G or immunoglobulin A monoclonal gammopathy of unknown significance (MGUS), suggesting an early oncogenic role for MYD88 in WM pathogenesis.7,8,10 Patients with IgM MGUS with detectable mutated MYD88 and patients with a higher mutated allele burden are at greater risk of progression to WM.10,14
The MYD88 L265P mutation can also be detected by AS-PCR in peripheral-blood (PB) samples, particularly in treatment-naïve patients with WM.15 Prior therapy with B-cell–depleting agents can greatly decrease detection of MYD88 L265P in PB samples. MYD88 L265P can also be found in skin lesions, CSF, and pleural effusions in patients with WM, providing a means of demonstrating extramedullary disease involvement.16-18 Cell-free DNA has also been used to detect MYD88 L265P from PB samples of patients with WM and may represent a novel means for establishing MYD88 mutation status.19,20
Structural alterations on chromosome 3p can increase the allele burden of mutated MYD88.6,21 Up to 15% of untreated and 25% of previously treated patients may be homozygous for MYD88 as a result of deletions of the wild-type MYD88 allele, amplifications of the mutant MYD88 allele, and, more commonly, acquired uniparental disomy (aUPD) events.22 aUPD events resulting in homozygous MYD88 expression are associated with concurrent CXCR4 mutations, the significance of which remains to be clarified but may be related to disease length and prior ibrutinib exposure.6,23
Patients with wild-type MYD88 show similar histologic findings and transcription profile as patients with MYD88-mutated WM.4,5 However, patients with wild-type MYD88 with asymptomatic WM have a higher risk of symptomatic progression,24 and those presenting with symptomatic disease have a greater risk of disease transformation8,25 and decreased overall survival.8 Patients with wild-type MYD88 also show poor response to ibrutinib (discussed later). It is important to distinguish patients with suspected WM with wild-type MYD88 disease from patients with other IgM-secreting malignancies. In one series, 30% of patients with suspected wild-type MYD88 WM had an alternative diagnosis, including IgM multiple myeloma (MM). The presence of very high serum IgM levels, lytic lesions, and/or renal dysfunction may help identify IgM MM.8,26,27 Use of cytogenetics to evaluate for t(11;14) and cyclin D1 staining can be helpful in distinguishing IgM MM from wild-type MYD88 WM.8,27,28
MYD88 is an adaptor protein that interacts with the Toll-like and interleukin (IL)-1 receptors and dimerizes upon receptor activation. Dimerization of MYD88 provides a scaffold for recruitment of other proteins to a Myddosome complex that triggers downstream signaling, leading to nuclear factor-κB (NFKB) activation (Fig 1).29 Both IRAK1/IRAK4 and BTK are Myddosome components and trigger NFKB.30,31 Recruitment and activation of the IRAK and BTK molecules can be blocked by either knockdown or inhibition of MYD88 that leads to apoptosis of mutated MYD88 WM cells. Mutated MYD88 can also upregulate transcription of the SRC family member HCK that normally is downregulated in late stages of B-cell ontogeny and can transactivate HCK via IL-6.32 Activated HCK triggers prosurvival signaling of mutated MYD88 WM cells through BTK, PI3K/AKT, and MAPK/ERK1/2. Both BTK and HCK are targets of ibrutinib, which has shown remarkable activity in patients with mutated MYD88 WM.32,33
FIG 1.
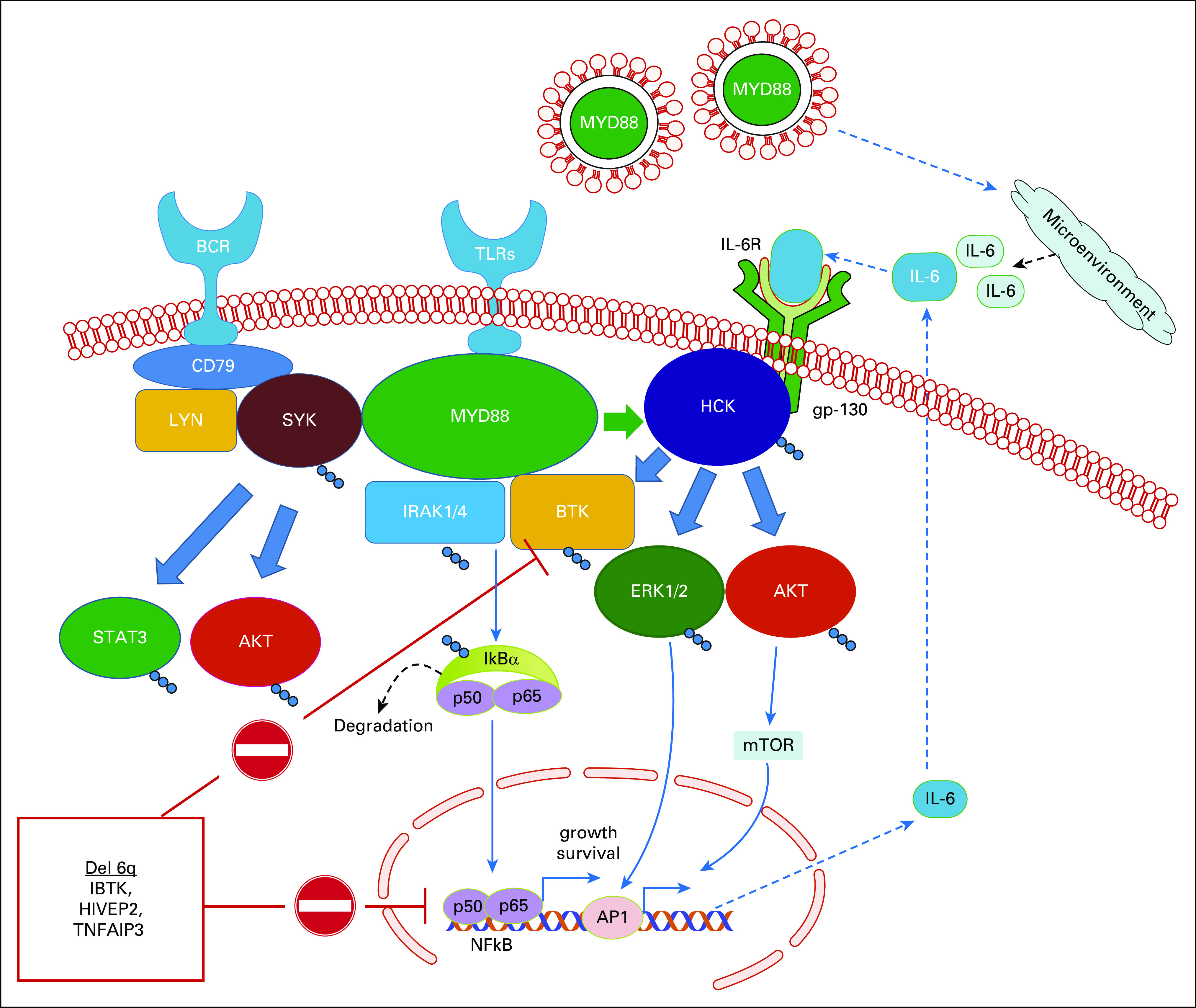
Prosurvival signaling mediated by mutated MYD88. Mutated MYD88 triggers assembly of the Myddosome, which includes activated BTK and IRAK4/IRAK1 that transactivate NFKB. Mutated MYD88 also transcriptionally upregulates and transactivates through interleukin (IL)-6 the SRC family member HCK, which triggers activation of BTK itself, as well as AKT and ERK. Mutated MYD88 also mediates cross talk through LYN-activated SYK, which activates STAT3 and AKT prosurvival signaling. Deletions in chromosome 6q result in the loss of important regulators of MYD88 signaling, including inhibitor of BTK (IBTK) and the NFKB regulators HIVEP2 and TNFAIP3. Waldenström macroglobulinemia cells also export mutated MYD88 via extracellular vesicles to induce signaling cascades in mast cells and macrophages that provide a supportive proinflammatory microenvironment. BCR, B-cell receptor; mTOR, mammalian target of rapamycin; TLRs, Toll-like receptors.
More recently, a role for mutated MYD88 as a trigger for B-cell receptor (BCR) pathway cross talk has been proposed. Phelan et al34 described the existence of a MYD88-TLR9-BCR(IgM) (MY-T-BCR) supercomplex in mutated MYD88 tumor cells. Evidence for chronic activated BCR signaling exists in WM.35,36 SYK, a component of the BCR pathway, is present in its active form in the Myddosome, and its activation is triggered by mutated MYD88.37 Importantly, mutated MYD88–directed SYK activation triggers STAT3 and AKT prosurvival signaling. In preclinical studies, the addition of an SYK inhibitor triggered synthetic lethality when combined with ibrutinib.37 Taken together, these studies show that mutated MYD88 can direct multiple prosurvival cascades that lead to NFKB, AKT, ERK, and STAT3 activation in WM cells (Fig 1). WM cells also export mutated MYD88 via extracellular vesicles, which may produce a growth-supportive proinflammatory microenvironment (Fig 1).38
MUTATIONS IN CXCR4
Somatic mutations involving the C-terminal domain of CXCR4 are present in up to 40% of patients with WM, and although they almost always occur in those with MYD88 mutations, some patients with wild-type MYD88 can also express CXCR4 mutations.21,39 CXCR4 mutations are essentially unique to WM, with only a few cases of marginal zone lymphoma and activated B-cell (ABC) subtype of DLBCL reported so far. Germline mutations in the C-terminal domain of CXCR4 are present in patients with WHIM (autosomal dominant warts, hypogammaglobulinemia, infection, and myelokathexis) syndrome.40 In patients with WHIM syndrome, activation of CXCR4 by its ligand CXCL12 induces extended chemotactic signaling and neutrophil sequestration in the BM (myelokathexis) and impairment of lymphocyte development. In WM, > 40 nonsense and frameshift mutations in the C-terminal domain of CXCR4 have been described.41,42 Mutations in the C-terminal domain of CXCR4 lead to loss of regulatory serines and promote sustained CXCL12-mediated activation of the AKT and ERK pathways and in vivo WM disease progression and dissemination in mice.43-45 Despite the autonomous prosurvival signaling associated with CXCR4 mutations, inhibition of MYD88 triggers apoptosis in both wild-type and mutated CXCR4–expressing WM cells, consistent with a primary role for mutated MYD88 survival signaling in WM.45
Unlike MYD88, mutated CXCR4 is subclonal, with highly variable clonality averaging approximately 35%. Multiple CXCR4 mutations can also exist within individual patients that can occur in separate clones or as compound heterozygous events.39 The subclonal nature of CXCR4 mutations relative to MYD88 and their paucity in IgM MGUS relative to WM suggest that CXCR4 mutations are acquired after MYD88.39 Clonal 6q deletions (del 6q21-25), which are found in 40%-50% of patients with WM, appear exclusive of CXCR4 in treatment-naïve patients.46 The increasing frequency of 6q deletions from IgM MGUS through asymptomatic and symptomatic WM suggests that loss of genes within this region (discussed later) facilitates disease progression.13,35,46 Taken together, these findings suggest that at least 2 distinct pathways mediated by either mutated CXCR4 or del 6q21-25 promote IgM MGUS progression to symptomatic WM.
Like MYD88, CXCR4 somatic mutations can affect WM disease presentation. Patients with mutated CXCR4 present with less adenopathy, and those with nonsense CXCR4 mutations have increased BM disease, serum IgM levels, and/or symptomatic hyperviscosity at presentation.41 Nonsense CXCR4 mutations are also associated with acquired von Willebrand factor deficiency.41,47 Despite differences in clinical presentation, CXCR4 mutations do not adversely affect overall survival but do seem to affect time to treatment.8,13,41
In vitro modeling of WM cells transduced with mutated CXCR4 reveals increased drug resistance in the presence of its ligand CXCL12 to multiple therapeutics including bendamustine, fludarabine, bortezomib, idelalisib, and ibrutinib.43-45 These studies also showed that resistance mediated by mutated CXCR4 could be reversed by use of CXCR4-blocking agents in both in vitro and in vivo studies.43-45
OTHER RECURRING MUTATIONS
Somatic mutations in ARID1A are present in 17% of patients with WM, including nonsense and frameshift variants.21 Patients with ARID1A and MYD88 L265P mutations have greater WM disease involvement and lower hemoglobin and platelet counts. ARID1A and its frequently deleted homolog ARID1B (see later discussion) are on chromosome 6q.46 Both are switch/sucrose nonfermentable (SWI/SNF) family members and serve as chromatin remodeling genes, thereby modulating gene regulation. Although still poorly understood, ARID1A can modulate TP53 and is thought to act as an epigenetic tumor suppressor in ovarian cancer.3
Mutations in CD79A and CD79B can be found in 8%-12% of patients with WM.13,21,42 Both are components of the BCR pathway and can form heterodimers with each other.3 The CD79A/B heterodimer associates with the immunoglobulin heavy chain required for cell surface expression of BCR and BCR-induced signaling. Activating mutations in the immunotyrosine-based activation motif of CD79A and CD79B occur in the ABC subtype of DLBCL and trigger SYK, PLCγ2, and BTK.48 Mutated MYD88 cross talk through SYK may also explain chronic BCR signaling observed in WM cells.37 Deletions of LYN, which are found in 70% of patients with WM, may contribute to hyperresponsive BCR signaling, as informed by lyn−/− transgenic mice.21,49 Although mutations in both CD79A and CD79B are mainly found in patients with mutated MYD88 including those with WM, a CD79B mutation was observed in a patient with wild-type MYD88 WM.21 In one study, mutations in CD79A and CD79B were nearly exclusive of CXCR4 mutations, suggesting that CD79A/B mutations may also have an independent role in facilitating mutated MYD88–directed progression in WM.42 In a small series of patients with WM, the coexpression of CD79B and MYD88 mutations was associated with disease transformation.50 Mutations in TP53 are more rarely observed (2%-3%) in WM13,21 and are associated with poor survival.51,52
MUTATIONS IN WILD-TYPE MYD88 WM
A small number of patients with WM (3%-7%) lack mutations in MYD88, including non-L265P mutations.5,21 Their disease course is marked by an increased risk of disease transformation5,25 and shorter overall survival.5 Moreover, patients with wild-type MYD88 show little response to ibrutinib (discussed further later). These findings point to fundamental differences in underlying genomics. NGS identified somatic mutations in patients with wild-type MYD88 WM that are predicted to trigger NFKB (TBL1XR1, PTPN13, MALT1, BCL10, NFKB1, NFKB2, NFKBIB, NFKBIZ, and UDRL1F), impart epigenomic dysregulation (KMT2D, KMT2C, and KDM6A), or impair DNA damage repair (TP53, ATM, and TRRAP).5 Predicted NFKB activating mutations were downstream of BTK, and many overlapped with somatic mutations found in DLBCL.5 Transcriptome studies revealed a distinctive transcriptional profile in patients with wild-type MYD88 WM, although the most differentially expressed genes overlapped with those found in mutated MYD88 WM.5 These findings may help explain many of the uniform characteristics shared among patients with WM, regardless of their underlying MYD88 mutation status.
COPY NUMBER ALTERATIONS
Copy number alterations are common in patients with MYD88-mutated WM and involve both chromosome 6q and non–chromosome 6q regions.13,21,35 In chromosome 6q, loss of genes that modulate NFKB activity (TNFAIP3 and HIVEP2), BCL2 (BCLAF1), apoptosis (FOXO3), BTK (IBTK), plasma cell differentiation (PRDM1), and ARID1B occur.21,46 Non–chromosome 6q genes that are commonly deleted include ETV6, a transcription repressor; BTG1, which is often deleted in DLBCL and associated with glucocorticoid resistance in acute lymphocytic leukemia; and LYN, a kinase that regulates BCR signaling.21,48,49 PRDM2 and TOP1, which participate in TP53-related signaling, are also deleted in many patients with WM. In contrast to MYD88-mutated WM, recurring copy number alterations are rare in MYD88 wild-type WM, including loss of chromosome 6q.5,13,21
IMPACT OF GENOMIC ALTERATIONS ON WM TREATMENT OUTCOME
The impact of MYD88 and CXCR4 mutations on treatment outcome has been evaluated in several clinical trials (Table 1). Patients with MYD88 mutations who are wild type for CXCR4 have better outcomes associated with ibrutinib monotherapy, including time to major response, depth of response, and progression-free survival.33,53,54 Indeed, patients with both MYD88 and CXCR4 mutations exhibit a delay by 4-5 months in attaining a major response to ibrutinib monotherapy. No major responses were observed in previously treated patients with WM with wild-type MYD88 treated with ibrutinib alone.12,55 Patients with MYD88 wild-type status also had much short progression-free survival on ibrutinib alone compared with MYD88-mutated patients, regardless of their CXCR4 mutation status.53
TABLE 1.
Summary of the Impact of MYD88 and CXCR4 Mutation Status on Treatment Outcomes in Patients With Waldenström Macroglobulinemia

Earlier disease progression has also been observed in MYD88- and CXCR4-mutated patients treated with ibrutinib alone or with ibrutinib and rituximab when compared with patients with mutated MYD88 alone.53,56 Somatic mutations at the site of ibrutinib-BTK binding (Cys481) have been observed in patients with WM who experienced progression on ibrutinib.57 Multiple BTK mutations were observed in this study within individual patients with WM, particularly those with CXCR4 mutations. Although the findings highlight the importance of BTK as a treatment target in MYD88-mutated WM, they also raise questions as to why CXCR4-mutated patients are particularly at risk for the acquisition of BTK Cys481 mutations.
The presence of CXCR4 mutations has also affected clinical outcomes with other agents. Fewer major responses were preliminarily reported with the BTK inhibitor zanubrutinib, as well as with the BCL2 inhibitor venetoclax, in prospective studies in patients with WM.58,59 Time to response was also reported to be longer in patients with WM with CXCR4 mutations receiving ixazomib-based therapy, a proteasome inhibitor (PI), in a prospective phase II study.60 Conversely, a prospective study of patients with CXCR4-mutated WM treated with carfilzomib-based therapy showed no effect on clinical outcome.61 In a retrospective study, Sklavenitis-Pistofidis et al62 showed that bortezomib-based therapy could overcome the negative impact of CXCR4 mutations on progression-free and overall survival.
Among treatment-naïve patients with WM receiving everolimus as a single agent in a prospective phase II study, nonresponders included patients who were CXCR4 mutated, as well as those who were wild type for MYD88.63 In a retrospective study by the French Innovative Leukemia Organization group, the use of bendamustine plus rituximab resulted in similar progression-free survival among MYD88-mutated patients with and without CXCR4 mutations. Conversely, those with wild-type MYD88 showed earlier progression.64 In a retrospective study, Paludo et al65 also observed a trend for shorter progression-free survival and time to next treatment among genotyped patients with MYD88 wild-type WM who received alkylator-based (bendamustine or cyclophosphamide) therapy. There were no differences in overall response rates between genotyped patients who were MYD88 mutated or wild type, and no data on major responses were reported.
Although the previously discussed studies indicate that CXCR4 represents an important target in WM, differences in response outcome could be modulated by the type of CXCR4 mutation as well as its clonal presence. In a study by Castillo et al,66 patients on ibrutinib with nonsense mutations showed fewer major responses and shorter progression-free survival compared with those with frameshift CXCR4 mutations or those wild-type for CXCR4. The clonality of CXCR4 may also be an important determinant of ibrutinib response. Gustine et al67 investigated the impact of the CXCR4 S338X nonsense variant on ibrutinib response outcome in patients with WM. Patients with a CXCR4 S338X clonality of > 25% showed greater BM disease involvement and serum IgM levels, fewer very good partial responses, and shorter progression-free survival.
Given the importance of CXCR4 mutation status in WM, a clinical trial assessing the impact of the CXCR4 inhibitor ulocuplumab with ibrutinib in patients with CXCR4-mutated WM was initiated (ClinicalTrials.gov identifier: NCT03225716). Mavorixafor, an orally administered CXCR4 antagonist, is also in clinical development for CXCR4-mutated WM. Although the use of antagonists to counter CXCR4 signaling in CXCR4-mutated patients represents a rational approach for advancing treatment in this population with WM, a targeted approach for those with MYD88 wild-type WM remains elusive and represents an urgent priority.
GENOMIC-BASED TREATMENT APPROACH TO WM
MYD88 and CXCR4 mutation status may be useful in treatment selection for symptomatic patients. Treatment algorithms recommended by the authors for the use of MYD88 and CXCR4 mutation status in the treatment approach of symptomatic untreated and previously treated patients are presented in Figures 2 and 3, respectively. In treatment-naïve patients, as well as those with previously treated WM, plasmapheresis should be considered as the first step in patients presenting with symptomatic hyperviscosity, rapidly progressing peripheral neuropathy attributed to demyelinating IgM disease, severe cryoglobulinemia, and cold agglutinemia.68,69 To minimize risks of short- and long-term treatment-related adverse effects associated with chemoimmunotherapy, including neuropathy, immunosuppression, and stem-cell damage, as well as secondary myelodysplasia and malignancies, the use of ibrutinib can be considered for treatment-naïve patients with WM who carry only the MYD88 mutation. Current nonrandomized comparisons of data from ibrutinib monotherapy and combined ibrutinib and rituximab studies show little difference in patients only have MYD88 mutation; hence, ibrutinib alone can be considered for patients without bulky disease and symptomatic amyloidosis and for whom no contraindication to ibrutinib exists.53-56
FIG 2.

Genomic-based treatment algorithm for symptomatic, treatment-naïve patients with Waldenström macroglobulinemia. Rituximab should be held for serum immunoglobulin M (IgM) ≥ 4,000 mg/dL to prevent symptomatic IgM flare. Bendamustine and rituximab (Benda-R) should be considered as primary choice in patients with bulky adenopathy or extramedullary disease. Proteasome inhibitor (PI)–based regimens should be considered in patients with symptomatic amyloidosis, with autologous stem-cell transplantation as possible consolidation. Rituximab alone or with ibrutinib if MYD88 is mutated or bendamustine may be considered in patients with IgM peripheral neuropathy (PN) depending on severity and pace of progression. Cyclophosphamide-based therapy such as dexamethasone, cyclophosphamide, and rituximab (DRC) represents an alternative to Benda-R but may be less effective. Maintenance rituximab may be considered in patients responding to rituximab-based regimens. BTK-I, Bruton tyrosine kinase inhibitor; CAGG, cold agglutinemia; CRYOS, cryoglobulinemia; HV, hyperviscosity; Mut, mutated; WT, wild type (not mutated).
FIG 3.
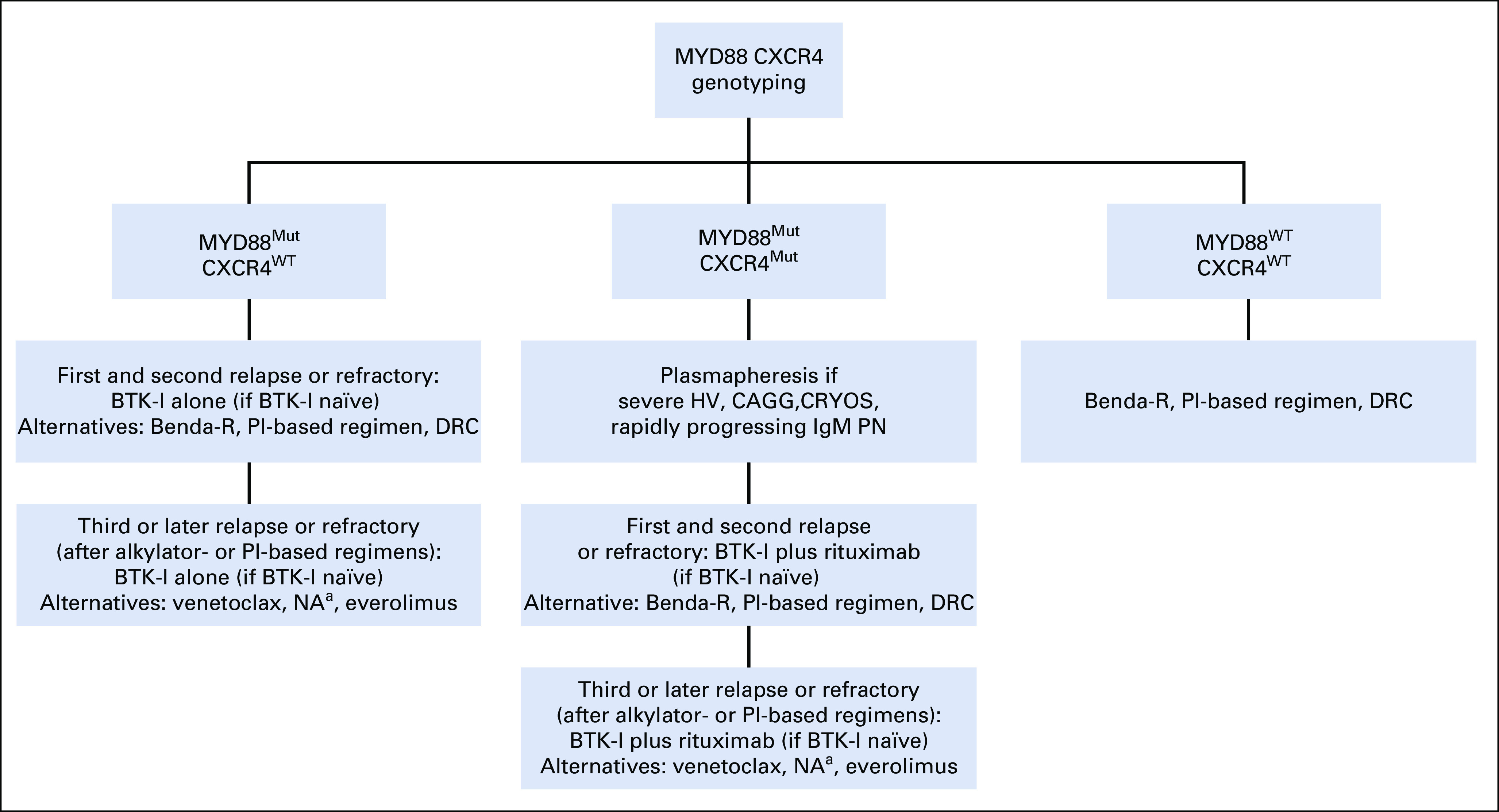
Genomic-based treatment algorithm for symptomatic, previously treated or refractory patients with Waldenström macroglobulinemia. Rituximab should be held for serum immunoglobulin M (IgM) ≥ 4,000 mg/dL to prevent symptomatic IgM flare. Bendamustine and rituximab (Benda-R) should be considered as primary choice in patients with bulky adenopathy or extramedullary disease. Proteasome inhibitor (PI)–based regimens should be considered in patients with symptomatic amyloidosis, with autologous stem-cell transplantation (ASCT) as possible consolidation. Rituximab alone or with ibrutinib if MYD88 is mutated or bendamustine may be considered in patients with IgM peripheral neuropathy (PN) depending on severity and pace of progression. Rituximab combined with cyclophosphamide and dexamethasone (DRC) represents an alternative to Benda-R, although it may be a potentially less effective option. Maintenance rituximab may be considered in patients responding to rituximab-based regimen. (a) Nucleoside analogues (NA) should be avoided in younger patients and candidates for ASCT. ASCT may be considered in patients with multiple relapses and chemotherapy-sensitive disease. BTK-I, Bruton tyrosine kinase inhibitor; CAGG, cold agglutinemia; CRYOS, cryoglobulinemia; HV, hyperviscosity; Mut, mutated; WT, wild-type (not mutated).
Alternatives to ibrutinib that can be considered include bendamustine and rituximab (Benda-R) or PI-based therapy such as bortezomib, dexamethasone, and rituximab (BDR); carfilzomib, rituximab, and dexamethasone (CaRD); or ixazomib, dexamethasone, and rituximab (IDR). Randomized and nonrandomized studies suggest that cyclophosphamide-based therapy, although active in WM, may not be as effective as Benda-R or PI-based therapy.65,70-72
Because the role of ibrutinib is not well established in patients with WM with bulky disease and has been lackluster in symptomatic patients with light chain (LC) amyloidosis, alternatives should be sought for these patients.33,73 Benda-R is a reasonable choice for those with bulky disease, ideally limited to 4 cycles to minimize prolonged cytopenias and long-term adverse events.64,65,71 Dexamethasone, rituximab, and cyclophosphamide remains an alternative if Benda-R is not available.72 A PI-based treatment such as weekly BDR to minimize treatment-related neuropathy or use of neuropathy-sparing PI regimens such as CaRD or IDR may be considered for those with symptomatic LC amyloidosis.60,61,68,69,74 In these patients, consolidation with autologous stem-cell transplantation (SCT) may be considered.75 Finally, patients with WM with IgM demyelinating neuropathy require deep and sustained responses to minimize myelin-directed monoclonal IgM deposition. For such patients, rituximab alone can be considered if the neuropathy is mild, but for more moderate to severe cases or patients with more rapid progression, the use of ibrutinib alone or with rituximab, Benda-R, or a regimen using a neuropathy-sparing PI such as CaRD or IDR may be considered.33,60,61,68,69,76
For patients with both MYD88 and CXCR4 mutations, the necessity to achieve a rapid response must be considered. Nonsense CXCR4-mutated patients in particular are at risk for presenting with symptomatic hyperviscosity requiring immediate disease control.41 Rituximab should be avoided in patients with WM with high IgM levels or symptomatic hyperviscosity until such time as the IgM has decreased to < 4,000 mg/dL to avoid a symptomatic rituximab flare.68,69 For patients requiring more emergent responses, either bendamustine or a PI-based regimen may be considered. For patients without symptomatic hyperviscosity and emergent need for disease control, ibrutinib plus rituximab may be preferred versus ibrutinib alone because the median time to achieve a major response appears to be shorter with the combination (3 v 6-7 months, respectively).53,54,56
For patients presenting with wild-type MYD88 WM determined by AS-PCR, Sanger sequencing or NGS that examines the TIR exon coding region of the MYD88 gene should be done to exclude non-L265P MYD88 mutations because patients with non-L265P MYD88 mutations show responses to ibrutinib.12 For patients with true MYD88 wild-type WM, either bendamustine or PI-based therapy with rituximab can be considered because responses have been observed with these regimens in patients with both MYD88 wild-type and mutated WM.60,64,65 In all patients with WM showing a response to a fixed-duration rituximab-containing regimen, maintenance rituximab may be considered.68,69 In patients who develop rituximab intolerance, ofatumumab may represent an option.77,78 There are no data at this time on the use of obinotuzumab in WM.
In previously treated patients with WM (Fig 3), the reuse of a front-line regimen may be considered for patients with long responses (> 2-3 years), although alternatives to alkylators or nucleoside analogs should be considered if previously used to minimize stem-cell damage. Nucleoside analogs should also be avoided in younger patients who may be candidates for autologous SCT and to minimize risk of disease transformation.69 The use of ibrutinib can be considered in symptomatic, previously treated patients with mutated MYD88 WM following the algorithm described earlier for symptomatic treatment-naïve patients with WM (Fig 2). For patients who have been exposed to ibrutinib, either Benda-R or a PI-based regimen may be considered. Alternatively, off-label use of acalabrutinib may be considered for patients not tolerating ibrutinib given findings in patients with chronic lymphocytic leukemia showing a successful switchover from ibrutinib for intolerant patients.79 The findings of a prospective randomized trial of zanubrutinib versus ibrutinib (ClinicalTrials.gov identifier NCT03053440) may also provide important guidance on the use of zanubrutinib in patients not tolerating ibrutinib.
For patients who have experienced multiple relapses and have been previously exposed to alkylators, PIs, and ibrutinib, the off-label use of venetoclax may be considered. High overall response rates and durable responses were observed in patients with WM on venetoclax, including those who were CXCR4 mutated and previously exposed to a BTK inhibitor, although response depth was affected by these factors.59,80 Everolimus, alone or in combination with bortezomib and rituximab, can also be considered in patients previously exposed to alkylators, PIs, and ibrutinib, although hematologic toxicities and autoimmune pneumonitis are noteworthy.63,81,82 Diminished activity in patients with wild-type MYD88 or mutated CXCR4 has also been reported with everolimus and should be considered in context with other available treatments.63 The use of cyclophosphamide, bortezomib, and dexamethasone, alone or with rituximab, is also an active regimen in previously treated patients with WM.82
The use of autologous SCT can be considered in patients with multiple relapses and who remain chemotherapy sensitive.69,83 As mentioned earlier, autologous SCT may also be considered as a consolidative measure in patients with symptomatic amyloidosis.75 Data on the efficacy and safety of allogenic SCT or cellular therapies are limited and should ideally be considered in the context of a clinical trial. Finally, both treatment-naïve and previously treated patients should actively be considered for clinical trials. A listing of clinical trials for WM can be found at ClinicalTrials.gov.
MOLECULAR DIAGNOSTIC APPROACHES AND LIMITATIONS
In patients with suspected WM, the use of AS-PCR on BM aspirates represents the most optimal initial assay for detection of MYD88 L265P mutations. If the patient is determined to have wild-type MYD88 by AS-PCR, Sanger sequencing or targeted NGS that examines the TIR exon coding region of MYD88 can be performed to evaluate for non-L265P MYD88 mutations.10 Targeted NGS can also be used to identify MYD88 L265P mutations, although false-negative results were observed in 30% of patients in one study when compared with AS-PCR, particularly in patients with low BM disease involvement.84 The use of CD19-selected cells can improve chances of detection of mutated MYD88, although tumor selection is not routinely performed in clinical laboratories. CD19-selected PB mononuclear cells may also be used for AS-PCR testing of mutated MYD88, although diagnostic yield is greatly reduced with prior therapy, particularly rituximab, and use of unselected PB mononuclear cells.15
Targeted NGS or Sanger sequencing represents the best approach for evaluation of C-terminal mutations in CXCR4 because > 40 nonsense and frameshift variants have been reported in WM.8,13,39,42 Because CXCR4 mutations are subclonal with a median clonal distribution of 35%, false-negative results can occur in patients with low BM disease burden and with use of unselected mononuclear cells for CXCR4 mutation testing.39 Ultra-deep NGS (> 1,000×) may improve detection of CXCR4 mutations over Sanger sequencing in patients with low tumor burden and clonality.85 However, although large insertions or deletions in CXCR4 are uncommon in WM, these may be missed with NGS but identified by Sanger sequencing.
SUMMARY
In summary, NGS has revealed recurring somatic mutations in WM that include MYD88 and CXCR4. The genomic findings have contributed to ongoing targeted drug development and the recognition of a precision-guided treatment approach to WM. MYD88 and CXCR4 mutation status can be used to guide treatment decisions in WM.
AUTHOR CONTRIBUTIONS
Conception and design: Steven P. Treon, Mark Bustoros, Antonio Sacco, Aldo Roccaro, Irene M. Ghobrial
Administrative support: Christopher J. Patterson
Collection and assembly of data: Steven P. Treon, Lian Xu, Xia Liu, Maria Demos, Joshua Gustine, Manit Munshi, Nicholas Tsakmaklis, Jiaji G. Chen, Amanda Kofides, Andrew Keezer, Kirsten Meid, Christopher J. Patterson, Guang Yang, Irene M. Ghobrial
Data analysis and interpretation: Steven P. Treon, Maria Luisa Guerrera, Cristina Jimenez, Zachary R. Hunter, Gloria Chan, Romanos Sklavenitis-Pistofidis, Andrew R. Branagan, Irene M. Ghobrial, Jorge J. Castillo
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Genomic Landscape of Waldenström Macroglobulinemia and Its Impact on Treatment Strategies
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/journal/jco/site/ifc.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Steven P. Treon
Consulting or Advisory Role: Janssen, Pharmacyclics, BeiGene
Research Funding: Janssen, Pharmacyclics, Bristol-Myers Squibb
Travel, Accommodations, Expenses: Janssen Oncology
Other Relationship: Janssen, Pharmacyclics
Zachary R. Hunter
Honoraria: Janssen
Mark Bustoros
Honoraria: Takeda, DAVAOncology
Consulting or Advisory Role: Takeda
Antonio Sacco
Consulting or Advisory Role: Celgene (I), Janssen-Cilag (I)
Research Funding: AstraZeneca (I)
Patents, Royalties, Other Intellectual Property: Pending patent: Treatment of C1013G/CXCR4-Associated Waldenström’s Macroglobulinemia With an Anti-CXCR4 Antibody; application No.: 16/271569-BMS Ref. 12242-US-PCD
Travel, Accommodations, Expenses: Janssen-Cilag (I)
Aldo Roccaro
Consulting or Advisory Role: Celgene, Janssen-Cilag
Research Funding: AstraZeneca
Patents, Royalties, Other Intellectual Property: Pending patent: Treatment of C1013G/CXCR4-Associated Waldenström’s Macroglobulinemia With an Anti-CXCR4 Antibody; application No.: 16/271569-BMS Ref. 12242-US-PCD
Travel, Accommodations, Expenses: Janssen-Cilag
Andrew R. Branagan
Consulting or Advisory Role: Surface Oncology, Pharmacyclics/Janssen
Guang Yang
Employment: AbbVie (I)
Honoraria: Janssen
Irene M. Ghobrial
Honoraria: Celgene, Bristol-Myers Squibb, Takeda, Amgen, Janssen, Karyopharm Therapeutics, Cellectar, Adaptive Biotechnologies, Sanofi, Medscape
Consulting or Advisory Role: Bristol-Myers Squibb, Novartis, Amgen, Takeda, Noxxon Pharma, Celgene, Sanofi, Genentech, GlaxoSmithKline, GNS Healthcare, Karyopharm Therapeutics, Adaptive Biotechnologies, Janssen, Medscape, AbbVie
Travel, Accommodations, Expenses: Bristol-Myers Squibb, Novartis, Onyx, Millennium, Celgene, Takeda, Janssen Oncology
Jorge J. Castillo
Consulting or Advisory Role: Pharmacyclics, Janssen, Vical, Genentech
Research Funding: Pharmacyclics (Inst), AbbVie (Inst), Janssen (Inst), BeiGene (Inst), TG Therapeutics (Inst)
No other potential conflicts of interest were reported.
REFERENCES
- 1.Swerdlow SH, Kuzu I, Dogan A, et al. The many faces of small B cell lymphomas with plasmacytic differentiation and the contribution of MYD88 testing. Virchows Arch. 2016;468:259–275. doi: 10.1007/s00428-015-1858-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Treon SP. How I treat Waldenström macroglobulinemia. Blood. 2009;114:2375–2385. doi: 10.1182/blood-2009-05-174359. [DOI] [PubMed] [Google Scholar]
- 3.Hunter ZR, Yang G, Xu L, et al. Genomics, signaling, and treatment of Waldenström macroglobulinemia. J Clin Oncol. 2017;35:994–1001. doi: 10.1200/JCO.2016.71.0814. [DOI] [PubMed] [Google Scholar]
- 4.Treon SP, Gustine J, Xu L, et al. MYD88 wild-type Waldenström macroglobulinaemia: Differential diagnosis, risk of histological transformation, and overall survival. Br J Haematol. 2018;180:374–380. doi: 10.1111/bjh.15049. [DOI] [PubMed] [Google Scholar]
- 5.Hunter ZR, Xu L, Tsakmaklis N, et al. Insights into the genomic landscape of MYD88 wild-type Waldenström macroglobulinemia. Blood Adv. 2018;2:2937–2946. doi: 10.1182/bloodadvances.2018022962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Treon SP, Xu L, Yang G, et al. MYD88 L265P somatic mutation in Waldenström’s macroglobulinemia. N Engl J Med. 2012;367:826–833. doi: 10.1056/NEJMoa1200710. [DOI] [PubMed] [Google Scholar]
- 7.Jiménez C, Sebastián E, Chillón MC, et al. MYD88 L265P is a marker highly characteristic of, but not restricted to, Waldenström’s macroglobulinemia. Leukemia. 2013;27:1722–1728. doi: 10.1038/leu.2013.62. [DOI] [PubMed] [Google Scholar]
- 8.Poulain S, Roumier C, Decambron A, et al. MYD88 L265P mutation in Waldenström macroglobulinemia. Blood. 2013;121:4504–4511. doi: 10.1182/blood-2012-06-436329. [DOI] [PubMed] [Google Scholar]
- 9.Varettoni M, Arcaini L, Zibellini S, et al. Prevalence and clinical significance of the MYD88 (L265P) somatic mutation in Waldenström’s macroglobulinemia and related lymphoid neoplasms. Blood. 2013;121:2522–2528. doi: 10.1182/blood-2012-09-457101. [DOI] [PubMed] [Google Scholar]
- 10.Xu L, Hunter ZR, Yang G, et al. MYD88 L265P in Waldenström macroglobulinemia, immunoglobulin M monoclonal gammopathy, and other B-cell lymphoproliferative disorders using conventional and quantitative allele-specific polymerase chain reaction. Blood. 2013;121:2051–2058. doi: 10.1182/blood-2012-09-454355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gustine J, Meid K, Xu L, et al. To select or not to select? The role of B-cell selection in determining the MYD88 mutation status in Waldenström macroglobulinaemia. Br J Haematol. 2017;176:822–824. doi: 10.1111/bjh.13996. [DOI] [PubMed] [Google Scholar]
- 12.Treon SP, Xu L, Hunter Z. MYD88 mutations and response to ibrutinib in Waldenström’s macroglobulinemia. N Engl J Med. 2015;373:584–586. doi: 10.1056/NEJMc1506192. [DOI] [PubMed] [Google Scholar]
- 13.Varettoni M, Zibellini S, Defrancesco I, et al. Pattern of somatic mutations in patients with Waldenström macroglobulinemia or IgM monoclonal gammopathy of undetermined significance. Haematologica. 2017;102:2077–2085. doi: 10.3324/haematol.2017.172718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Varettoni M, Zibellini S, Arcaini L, et al. MYD88 (L265P) mutation is an independent risk factor for progression in patients with IgM monoclonal gammopathy of undetermined significance. Blood. 2013;122:2284–2285. doi: 10.1182/blood-2013-07-513366. [DOI] [PubMed] [Google Scholar]
- 15.Xu L, Hunter ZR, Yang G, et al. Detection of MYD88 L265P in peripheral blood of patients with Waldenström’s macroglobulinemia and IgM monoclonal gammopathy of undetermined significance. Leukemia. 2014;28:1698–1704. doi: 10.1038/leu.2014.65. [DOI] [PubMed] [Google Scholar]
- 16. Alegría-Landa V, Prieto-Torres L, Santonja C, et al: MYD88 L265P mutation in cutaneous involvement by Waldenström macroglobulinemia. J Cutan Pathol 44:625-631, 2017. [DOI] [PubMed]
- 17. Poulain S, Boyle EM, Roumier C, et al: MYD88 L265P mutation contributes to the diagnosis of Bing Neel syndrome. Br J Haematol 167:506-513, 2014 https://doi.org/10.1158/1078-0432.CCR-15-0646PubMed. [DOI] [PubMed]
- 18. Gustine JN, Meid K, Hunter ZR, et al: MYD88 mutations can be used to identify malignant pleural effusions in Waldenström macroglobulinaemia. Br J Haematol 180:578-581, 2018 PubMed. [DOI] [PubMed]
- 19.Drandi D, Genuardi E, Dogliotti I, et al. Highly sensitive MYD88L265P mutation detection by droplet digital polymerase chain reaction in Waldenström macroglobulinemia. Haematologica. 2018;103:1029–1037. doi: 10.3324/haematol.2017.186528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bagratuni T, Ntanasis-Stathopoulos I, Gavriatopoulou M, et al. Detection of MYD88 and CXCR4 mutations in cell-free DNA of patients with IgM monoclonal gammopathies. Leukemia. 2018;32:2617–2625. doi: 10.1038/s41375-018-0197-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hunter ZR, Xu L, Yang G, et al. The genomic landscape of Waldenström macroglobulinemia is characterized by highly recurring MYD88 and WHIM-like CXCR4 mutations, and small somatic deletions associated with B-cell lymphomagenesis. Blood. 2014;123:1637–1646. doi: 10.1182/blood-2013-09-525808. [DOI] [PubMed] [Google Scholar]
- 22.Tsakmaklis N, Xu L, Manning R, et al. Mutated MYD88 homozygosity is increased in previously treated patients with Waldenström’s macroglobulinemia, and associates with CXCR4 mutation status and ibrutinib exposure. Blood. 2017;130 (abstr 1462) [Google Scholar]
- 23.Treon SP, Tsakmaklis N, Meid K, et al. Mutated MYD88 zygosity and CXCR4 mutation status are important determinants of ibrutinib response and progression free survival in Waldenström’s macroglobulinemia. Blood. 2016;128 (abstr 2984) [Google Scholar]
- 24.Bustoros M, Sklavenitis-Pistofidis R, Kapoor P, et al. Progression risk stratification of asymptomatic Waldenström macroglobulinemia. J Clin Oncol. 2019;37:1403–1411. doi: 10.1200/JCO.19.00394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abeykoon JP, Paludo J, King RL, et al. MYD88 mutation status does not impact overall survival in Waldenström macroglobulinemia. Am J Hematol. 2018;93:187–194. doi: 10.1002/ajh.24955. [DOI] [PubMed] [Google Scholar]
- 26.Castillo JJ, Jurczyszyn A, Brozova L, et al. IgM myeloma: A multicenter retrospective study of 134 patients. Am J Hematol. 2017;92:746–751. doi: 10.1002/ajh.24753. [DOI] [PubMed] [Google Scholar]
- 27.Avet-Loiseau H, Garand R, Lodé L, et al. Translocation t(11;14)(q13;q32) is the hallmark of IgM, IgE, and nonsecretory multiple myeloma variants. Blood. 2003;101:1570–1571. doi: 10.1182/blood-2002-08-2436. [DOI] [PubMed] [Google Scholar]
- 28.Willenbacher W, Willenbacher E, Brunner A, et al. Improved accuracy of discrimination between IgM multiple myeloma and Waldenström macroglobulinaemia by testing for MYD88 L265P mutations. Br J Haematol. 2013;161:902–904. doi: 10.1111/bjh.12313. [DOI] [PubMed] [Google Scholar]
- 29.Lin SC, Lo YC, Wu H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature. 2010;465:885–890. doi: 10.1038/nature09121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ngo VN, Young RM, Schmitz R, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2011;470:115–119. doi: 10.1038/nature09671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang G, Zhou Y, Liu X, et al. A mutation in MYD88 (L265P) supports the survival of lymphoplasmacytic cells by activation of Bruton tyrosine kinase in Waldenström macroglobulinemia. Blood. 2013;122:1222–1232. doi: 10.1182/blood-2012-12-475111. [DOI] [PubMed] [Google Scholar]
- 32.Yang G, Buhrlage SJ, Tan L, et al. HCK is a survival determinant transactivated by mutated MYD88, and a direct target of ibrutinib. Blood. 2016;127:3237–3252. doi: 10.1182/blood-2016-01-695098. [DOI] [PubMed] [Google Scholar]
- 33.Treon SP, Tripsas CK, Meid K, et al. Ibrutinib in previously treated Waldenström’s macroglobulinemia. N Engl J Med. 2015;372:1430–1440. doi: 10.1056/NEJMoa1501548. [DOI] [PubMed] [Google Scholar]
- 34.Phelan JD, Young RM, Webster DE, et al. A multiprotein supercomplex controlling oncogenic signalling in lymphoma. Nature. 2018;560:387–391. doi: 10.1038/s41586-018-0290-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paiva B, Corchete LA, Vidriales MB, et al. The cellular origin and malignant transformation of Waldenström macroglobulinemia. Blood. 2015;125:2370–2380. doi: 10.1182/blood-2014-09-602565. [DOI] [PubMed] [Google Scholar]
- 36.Argyropoulos KV, Vogel R, Ziegler C, et al. Clonal B cells in Waldenström’s macroglobulinemia exhibit functional features of chronic active B-cell receptor signaling. Leukemia. 2016;30:1116–1125. doi: 10.1038/leu.2016.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Munshi M, Chen JG, Liu X, et al. SYK is activated by mutated MYD88 and drives pro-survival signaling in MYD88 driven B-cell lymphomas. Blood Cancer J. 2020;10 doi: 10.1038/s41408-020-0277-6. Article 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Manček-Keber M, Lainšček D, Benčina M, et al. Extracellular vesicle-mediated transfer of constitutively active MyD88L265P engages MyD88wt and activates signaling. Blood. 2018;131:1720–1729. doi: 10.1182/blood-2017-09-805499. [DOI] [PubMed] [Google Scholar]
- 39.Xu L, Hunter ZR, Tsakmaklis N, et al. Clonal architecture of CXCR4 WHIM-like mutations in Waldenström macroglobulinaemia. Br J Haematol. 2016;172:735–744. doi: 10.1111/bjh.13897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hernandez PA, Gorlin RJ, Lukens JN, et al. Mutations in the chemokine receptor gene CXCR4 are associated with WHIM syndrome, a combined immunodeficiency disease. Nat Genet. 2003;34:70–74. doi: 10.1038/ng1149. [DOI] [PubMed] [Google Scholar]
- 41.Treon SP, Cao Y, Xu L, et al. Somatic mutations in MYD88 and CXCR4 are determinants of clinical presentation and overall survival in Waldenström macroglobulinemia. Blood. 2014;123:2791–2796. doi: 10.1182/blood-2014-01-550905. [DOI] [PubMed] [Google Scholar]
- 42. Poulain S, Roumier C, Venet-Caillault A, et al: Genomic landscape of CXCR4 mutations in Waldenström macroglobulinemia. Clin Cancer Res 22:1480-1488, 2016 https://doi.org/10.1158/1078-0432.CCR-15-0646PubMed. [DOI] [PubMed]
- 43.Roccaro AM, Sacco A, Jimenez C, et al. C1013G/CXCR4 acts as a driver mutation of tumor progression and modulator of drug resistance in lymphoplasmacytic lymphoma. Blood. 2014;123:4120–4131. doi: 10.1182/blood-2014-03-564583. [DOI] [PubMed] [Google Scholar]
- 44.Cao Y, Hunter ZR, Liu X, et al. The WHIM-like CXCR4(S338X) somatic mutation activates AKT and ERK, and promotes resistance to ibrutinib and other agents used in the treatment of Waldenström’s Macroglobulinemia. Leukemia. 2015;29:169–176. doi: 10.1038/leu.2014.187. [DOI] [PubMed] [Google Scholar]
- 45.Cao Y, Hunter ZR, Liu X, et al. CXCR4 WHIM-like frameshift and nonsense mutations promote ibrutinib resistance but do not supplant MYD88(L265P)-directed survival signalling in Waldenström macroglobulinaemia cells. Br J Haematol. 2015;168:701–707. doi: 10.1111/bjh.13200. [DOI] [PubMed] [Google Scholar]
- 46.Guerrera ML, Tsakmaklis N, Xu L, et al. MYD88 mutated and wild-type Waldenström’s Macroglobulinemia: Characterization of chromosome 6q gene losses and their mutual exclusivity with mutations in CXCR4. Haematologica. 2018;103:e408–e411. doi: 10.3324/haematol.2018.190181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Castillo JJ, Gustine JN, Meid K, et al. Low levels of von Willebrand markers associate with high serum IgM levels and improve with response to therapy, in patients with Waldenström macroglobulinaemia. Br J Haematol. 2019;184:1011–1014. doi: 10.1111/bjh.15200. [DOI] [PubMed] [Google Scholar]
- 48.Davis RE, Ngo VN, Lenz G, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010;463:88–92. doi: 10.1038/nature08638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nishizumi H, Horikawa K, Mlinaric-Rascan I, et al. A double-edged kinase Lyn: A positive and negative regulator for antigen receptor-mediated signals. J Exp Med. 1998;187:1343–1348. doi: 10.1084/jem.187.8.1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jiménez C, Alonso-Álvarez S, Alcoceba M, et al. From Waldenström’s macroglobulinemia to aggressive diffuse large B-cell lymphoma: A whole-exome analysis of abnormalities leading to transformation. Blood Cancer J. 2017;7:e591. doi: 10.1038/bcj.2017.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Poulain S, Roumier C, Bertrand E, et al. TP53 mutation and its prognostic significance in Waldenström’s macroglobulinemia. Clin Cancer Res. 2017;23:6325–6335. doi: 10.1158/1078-0432.CCR-17-0007. [DOI] [PubMed] [Google Scholar]
- 52.Gustine JN, Tsakmaklis N, Demos MG, et al. TP53 mutations are associated with mutated MYD88 and CXCR4, and confer an adverse outcome in Waldenström macroglobulinaemia. Br J Haematol. 2019;184:242–245. doi: 10.1111/bjh.15560. [DOI] [PubMed] [Google Scholar]
- 53. Treon SP, Meid K, Gustine J, et al: Ibrutinib monotherapy produces long-term disease control in previously treated Waldenström’s macroglobulinemia: Final report of the Pivotal trial ( NCT01614821). Hematol Oncol 37:184-185, 2019 (suppl; abstr 135) [Google Scholar]
- 54.Treon SP, Gustine J, Meid K, et al. Ibrutinib monotherapy in symptomatic, treatment-naïve patients with Waldenström macroglobulinemia. J Clin Oncol. 2018;36:2755–2761. doi: 10.1200/JCO.2018.78.6426. [DOI] [PubMed] [Google Scholar]
- 55.Dimopoulos MA, Trotman J, Tedeschi A, et al. Ibrutinib for patients with rituximab-refractory Waldenström’s macroglobulinaemia (iNNOVATE): An open-label substudy of an international, multicentre, phase 3 trial. Lancet Oncol. 2017;18:241–250. doi: 10.1016/S1470-2045(16)30632-5. [DOI] [PubMed] [Google Scholar]
- 56.Buske C, Tedeschi A, Trotman J, et al. Ibrutinib treatment in Waldenström’s macroglobulinemia: Follow-up efficacy and safety from the iNNOVATE study. Blood. 2018;132 (abstr 149) [Google Scholar]
- 57.Xu L, Tsakmaklis N, Yang G, et al. Acquired mutations associated with ibrutinib resistance in Waldenström macroglobulinemia. Blood. 2017;129:2519–2525. doi: 10.1182/blood-2017-01-761726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Trotman J, Tam CS, Marlton P, et al. Improved depth of response with increased follow-up for patients with Waldenström macroglobulinemia treated with Bruton’s tyrosine kinase inhibitor zanubrutinib. Proceedings of the 23rd Congress of the European Hematology Association, Stockholm, Sweden, June 14-17, 2018 (abstr PS1186) [Google Scholar]
- 59.Castillo J, Gustine J, Meid K, et al. Prospective Phase II study of venetoclax in patients with previously treated Waldenström’s Macroglobulinemia. Proceedings of the 23rd Congress of the European Hematology Association, Stockholm, Sweden, June 14-17, 2018 (abstr S854) [Google Scholar]
- 60.Castillo JJ, Meid K, Gustine JN, et al. Prospective clinical trial of ixazomib, dexamethasone, and rituximab as primary therapy in Waldenström macroglobulinemia. Clin Cancer Res. 2018;24:3247–3252. doi: 10.1158/1078-0432.CCR-18-0152. [DOI] [PubMed] [Google Scholar]
- 61.Treon SP, Tripsas CK, Meid K, et al. Carfilzomib, rituximab, and dexamethasone (CaRD) treatment offers a neuropathy-sparing approach for treating Waldenström’s macroglobulinemia. Blood. 2014;124:503–510. doi: 10.1182/blood-2014-03-566273. [DOI] [PubMed] [Google Scholar]
- 62.Sklavenitis-Pistofidis R, Capelletti M, Liu CJ, et al. Bortezomib overcomes the negative impact of CXCR4 mutations on survival of Waldenström macroglobulinemia patients. Blood. 2018;132:2608–2612. doi: 10.1182/blood-2018-07-863241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Treon SP, Meid K, Tripsas C, et al. Prospective, multicenter clinical trial of everolimus as primary therapy in Waldenström macroglobulinemia (WMCTG 09-214) Clin Cancer Res. 2017;23:2400–2404. doi: 10.1158/1078-0432.CCR-16-1918. [DOI] [PubMed] [Google Scholar]
- 64.Laribi K, Poulain S, Willems L, et al. Bendamustine plus rituximab in newly-diagnosed Waldenström macroglobulinaemia patients: A study on behalf of the French Innovative Leukaemia Organization (FILO) Br J Haematol. doi: 10.1111/bjh.15718. 186:146-149, 2019. [DOI] [PubMed] [Google Scholar]
- 65.Paludo J, Abeykoon JP, Shreders A, et al. Bendamustine and rituximab (BR) versus dexamethasone, rituximab, and cyclophosphamide (DRC) in patients with Waldenström macroglobulinemia. Ann Hematol. 2018;97:1417–1425. doi: 10.1007/s00277-018-3311-z. [DOI] [PubMed] [Google Scholar]
- 66.Castillo J, Gustine J, Xu L, et al. CXCR4 mutation subtypes impact response and survival outcomes in patients with Waldenström macroglobulinemia treated with ibrutinib. Br J Haematol. 2019;187:356–363. doi: 10.1111/bjh.16088. [DOI] [PubMed] [Google Scholar]
- 67.Gustine JN, Tsakmaklis N, Demos MG, et al. CXCR4 S338X clonality is an important determinant of ibrutinib outcomes in patients with Waldenström macroglobulinemia. Proceedings of the 10th International Workshop on Waldenström’s Macroglobulinemia, New York, NY, October 11-13, 2018 (abstr W35) [Google Scholar]
- 68.Treon SP. How I treat Waldenström macroglobulinemia. Blood. 2015;126:721–732. doi: 10.1182/blood-2015-01-553974. [DOI] [PubMed] [Google Scholar]
- 69.Leblond V, Kastritis E, Advani R, et al. Treatment recommendations from the Eighth International Workshop on Waldenström’s Macroglobulinemia. Blood. 2016;128:1321–1328. doi: 10.1182/blood-2016-04-711234. [DOI] [PubMed] [Google Scholar]
- 70.Rummel MJ, Niederle N, Maschmeyer G, et al. Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: An open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet. 2013;381:1203–1210. doi: 10.1016/S0140-6736(12)61763-2. [DOI] [PubMed] [Google Scholar]
- 71.Castillo JJ, Gustine JN, Meid K, et al. Response and survival for primary therapy combination regimens and maintenance rituximab in Waldenström macroglobulinaemia. Br J Haematol. 2018;181:77–85. doi: 10.1111/bjh.15148. [DOI] [PubMed] [Google Scholar]
- 72.Kastritis E, Gavriatopoulou M, Kyrtsonis MC, et al. Dexamethasone, rituximab, and cyclophosphamide as primary treatment of Waldenström macroglobulinemia: Final analysis of a phase 2 study. Blood. 2015;126:1392–1394. doi: 10.1182/blood-2015-05-647420. [DOI] [PubMed] [Google Scholar]
- 73.Pika T, Hegenbart U, Flodrova P, et al. First report of ibrutinib in IgM-related amyloidosis: Few responses, poor tolerability, and short survival. Blood. 2018;131:368–371. doi: 10.1182/blood-2017-09-806463. [DOI] [PubMed] [Google Scholar]
- 74.Gavriatopoulou M, García-Sanz R, Kastritis E, et al. BDR in newly diagnosed patients with WM: Final analysis of a phase 2 study after a minimum follow-up of 6 years. Blood. 2017;129:456–459. doi: 10.1182/blood-2016-09-742411. [DOI] [PubMed] [Google Scholar]
- 75.Sidiqi MH, Buadi FK, Dispenzieri A, et al. Autologous stem cell transplant for IgM-associated amyloid light-chain amyloidosis. Biol Blood Marrow Transplant. 2019;25:e108–e111. doi: 10.1016/j.bbmt.2018.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dimopoulos MA, Tedeschi A, Trotman J, et al. Phase 3 trial of ibrutinib plus rituximab in Waldenström’s macroglobulinemia. N Engl J Med. 2018;378:2399–2410. doi: 10.1056/NEJMoa1802917. [DOI] [PubMed] [Google Scholar]
- 77.Castillo JJ, Kanan S, Meid K, et al. Rituximab intolerance in patients with Waldenström macroglobulinaemia. Br J Haematol. 2016;174:645–648. doi: 10.1111/bjh.13794. [DOI] [PubMed] [Google Scholar]
- 78.Furman RR, Eradat HA, DiRienzo CG, et al. Once-weekly ofatumumab in untreated or relapsed Waldenström’s macroglobulinaemia: An open-label, single-arm, phase 2 study. Lancet Haematol. 2017;4:e24–e34. doi: 10.1016/S2352-3026(16)30166-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Awan FT, Schuh A, Brown JR, et al. Acalabrutinib monotherapy in patients with chronic lymphocytic leukemia who are intolerant to ibrutinib. Blood Adv. 2019;3:1553–1562. doi: 10.1182/bloodadvances.2018030007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Davids MS, Roberts AW, Seymour JF, et al. Phase I first-in-human study of venetoclax in patients with relapsed or refractory non-Hodgkin lymphoma. J Clin Oncol. 2017;35:826–833. doi: 10.1200/JCO.2016.70.4320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ghobrial IM, Redd R, Armand P, et al. Phase I/II trial of everolimus in combination with bortezomib and rituximab (RVR) in relapsed/refractory Waldenström macroglobulinemia. Leukemia. 2015;29:2338–2346. doi: 10.1038/leu.2015.164. [DOI] [PubMed] [Google Scholar]
- 82.Ghobrial IM, Witzig TE, Gertz M, et al. Long-term results of the phase II trial of the oral mTOR inhibitor everolimus (RAD001) in relapsed or refractory Waldenström Macroglobulinemia. Am J Hematol. 2014;89:237–242. doi: 10.1002/ajh.23620. [DOI] [PubMed] [Google Scholar]
- 83.Leblebjian H, Noonan K, Paba-Prada C, et al. Cyclophosphamide, bortezomib, and dexamethasone combination in Waldenström macroglobulinemia. Am J Hematol. 2015;90:E122–E123. doi: 10.1002/ajh.23985. [DOI] [PubMed] [Google Scholar]
- 84.Kyriakou C, Canals C, Sibon D, et al. High-dose therapy and autologous stem-cell transplantation in Waldenström macroglobulinemia: The Lymphoma Working Party of the European Group for Blood and Marrow Transplantation. J Clin Oncol. 2010;28:2227–2232. doi: 10.1200/JCO.2009.24.4905. [DOI] [PubMed] [Google Scholar]
- 85.Kofides A, Demos M, Tsakmaklis N, et al. Alternative mutations and isoform dysregulation in MYD88 in Waldenström’s macroglobulinemia. Blood. 2018;132 (abstr 1566) [Google Scholar]
- 86.Shin DW, Kim SM, Kim JA, et al. doi: 10.1016/j.clml.2019.03.009. Characteristics of Waldenström macroglobulinemia in Korean patients according to mutational status of MYD88 and CXCR4: Analysis using ultra-deep sequencing. Clin Lymphoma Myeloma Leuk 19:e496-e505, 2019. [DOI] [PubMed] [Google Scholar]
- 87.Owen RG, Kyle RA, Stone MJ, et al. Response assessment in Waldenström macroglobulinaemia: Update from the VIth International Workshop. Br J Haematol. 2013;160:171–176. doi: 10.1111/bjh.12102. [DOI] [PubMed] [Google Scholar]