

Visual Abstract

Keywords: Genetics, APOL1, endoplasmic reticulum stress, focal and segmental glomerulosclerosis, functional genomics, genome editing, induced pluripotent stem cells, nephron progenitor cells, organoids, Basic Science
Abstract
Background
DNA variants in APOL1 associate with kidney disease, but the pathophysiologic mechanisms remain incompletely understood. Model organisms lack the APOL1 gene, limiting the degree to which disease states can be recapitulated. Here we present single-cell RNA sequencing (scRNA-seq) of genome-edited human kidney organoids as a platform for profiling effects of APOL1 risk variants in diverse nephron cell types.
Methods
We performed footprint-free CRISPR-Cas9 genome editing of human induced pluripotent stem cells (iPSCs) to knock in APOL1 high-risk G1 variants at the native genomic locus. iPSCs were differentiated into kidney organoids, treated with vehicle, IFN-γ, or the combination of IFN-γ and tunicamycin, and analyzed with scRNA-seq to profile cell-specific changes in differential gene expression patterns, compared with isogenic G0 controls.
Results
Both G0 and G1 iPSCs differentiated into kidney organoids containing nephron-like structures with glomerular epithelial cells, proximal tubules, distal tubules, and endothelial cells. Organoids expressed detectable APOL1 only after exposure to IFN-γ. scRNA-seq revealed cell type–specific differences in G1 organoid response to APOL1 induction. Additional stress of tunicamycin exposure led to increased glomerular epithelial cell dedifferentiation in G1 organoids.
Conclusions
Single-cell transcriptomic profiling of human genome-edited kidney organoids expressing APOL1 risk variants provides a novel platform for studying the pathophysiology of APOL1-mediated kidney disease.
Introduction
Apolipoprotein L1 (APOL1)-mediated kidney disease accounts for a portion of the excess risk of CKD and ESKD among black patients (1,2). The APOL1 high-risk genotype, defined as the presence of two risk alleles (G1 or G2 coding variants), increases the risk of developing CKD, but not all individuals with the high-risk genotype develop disease (3,4). Much remains unknown regarding mechanisms and modifiers that render the disease incompletely penetrant, and complex interactions underlying these mechanisms are difficult to model outside APOL1’s native genomic locus. As such, current gaps in knowledge may not be fully addressed by induction of transgenic APOL1 expression in vivo or in vitro. Additionally, because APOL1 is widely expressed across different cell types, studying APOL1 risk variants solely within a specific type of cell (e.g., podocytes) may not fully capture how these variants affect the kidney.
Human kidney organoids derived from induced pluripotent stem cells (iPSCs) can be used to model genetic disease mechanisms in the native genomic context and cell-type heterogeneity within the kidney (5–9). Using CRISPR-Cas9–mediated genome editing, we engineered iPSCs homozygous for the G1 risk allele and differentiated these cells into three-dimensional (3D) kidney organoids. To evaluate cell type–specific effects of the APOL1 high-risk genotype, we also performed single-cell RNA sequencing (scRNA-seq), which we and others have previously leveraged to uncover novel biology of how cell-specific phenotypes contribute to kidney development or disease in organoids and other models (10–14). Here we present the application of genome-edited, iPSC-derived kidney organoids and single-cell transcriptomics to profile APOL1-mediated effects on kidney organoids relevant to disease processes.
Materials and Methods
iPSC Culture
iPSC lines previously derived from fibroblasts from a non-African ancestry donor (1016SevA; Harvard Stem Cell Institute) (15–18) and PBMCs from an African ancestry donor (Penn134-61-26; WiCell) were maintained in feeder-free culture on 10-cm dishes coated with 0.5% Geltrex (Gibco) in Modified Tenneille’s Special Recipe 1 (mTeSR1; STEMCELL Technologies), supplemented with 1% penicillin/streptomycin (Gibco) and 0.02% Plasmocin (Invivogen). iPSCs were confirmed to be mycoplasma-free and below passage 48. They were passaged using 1:3 Accutase (STEMCELL Technologies).
CRISPR-Cas9 Genome Editing
APOL1 G1 risk variants (rs73885319 and rs60910145) were introduced into the 1016SevA iPSC line through a genomic footprint-free approach (Figure 1A, Supplemental Figure 1A) (19,20). Briefly, the homology-directed repair (HDR) template containing the G1 variants was engineered using the MV-PGK-Puro-TK vector (Transposagen Bio), referred to as the PMV vector, which houses a removable puromycin selection cassette flanked by two homology arms. The puromycin cassette is excisable by a piggyBac transposase, leaving only a “TTAA” sequence behind that can be seamlessly introduced into a coding sequence by carefully choosing sites where the change would be synonymous. The G1 variants were engineered by two-step PCR of G0 genomic DNA (Supplemental Figure 1A, Supplemental Table 1) to create the donor template for homology arm A, designed to flank the upstream portion of the puromycin selection cassette. Arm B, designed to flank the downstream end of the selection cassette, was amplified from G0 genomic DNA by traditional PCR. Both arm A and arm B underwent separate TOPO TA cloning reactions (Invitrogen) for insertion into a stable vector for subsequent subcloning into the PMV vector. Stepwise sequential double restriction-enzyme digests and homology-arm ligations were performed on the PMV vector with the following pairs of restriction enzymes: Not1-High Fidelity (HF) and Bbs1-HF, Nco1-HF, and Bsa1-HF (New England Biolabs). The ends of both homology arms bordering the cassette harbor the TTAA piggyBac transposase cut sequence, thus allowing for the transposase to excise the cassette from both ends and leave behind the TTAA sequence in a scarless fashion (Supplemental Figure 1B). To make this genome-editing event footprint-free, we selected a codon site that would allow the TTAA nucleotide sequence to be knocked in without altering the APOL1 amino acid sequence. We identified a leucine (an amino acid encoded by six different codons including TTA) flanked by an adenine to be the site of cassette entry and excision (Supplemental Figure 1C). A guide RNA sequence with a suitable protospacer adjacent motif was found nearby the excision site (Figure 1A, Supplemental Table 1) and cloned into gRNA_Cloning Vector (41824; Addgene) (21). The donor template incorporates a point mutation at the protospacer-adjacent-motif site to destroy it after HDR to prevent recutting. iPSCs were then electroporated with the guide vector, hCas9 (41815; Addgene) (21), and the G1-PMV donor plasmid (control lines were electroporated with the guide vector only). After 48 hours, 10 µg/ml puromycin was added to iPSC culture to select for cells expressing the donor plasmid (Supplemental Figure 1A). Seven days later, iPSC colonies were evaluated for insertion of HDR template by PCR and Sanger sequencing validation. After expansion of the successfully knocked-in colonies into separate lines, the piggyBac transposase expression vector (Transposagen Bio) was introduced by electroporation. Additional screening of genotype was performed to validate the puromycin cassette excision.
Figure 1.
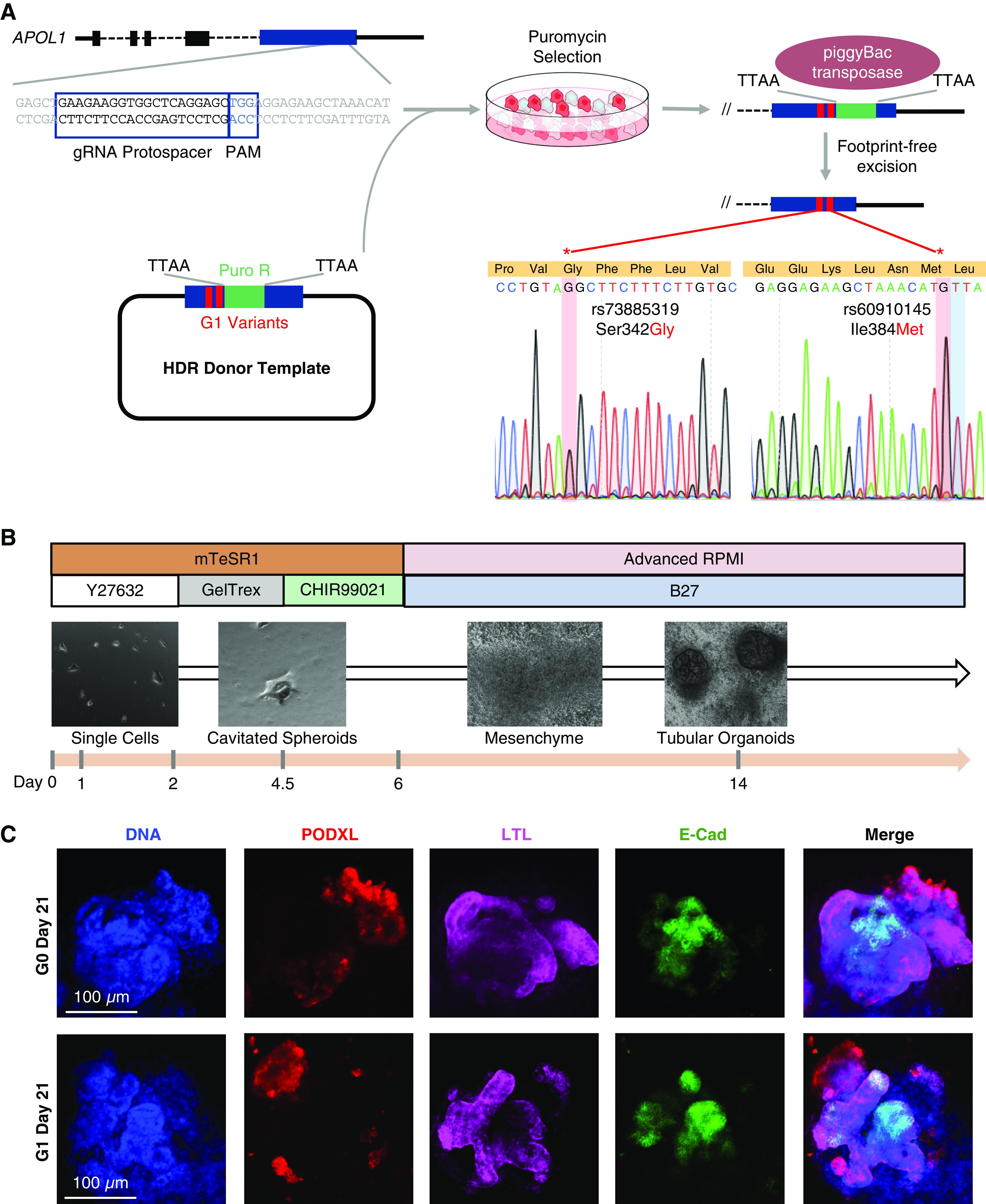
Engineering APOL1 G1 kidney organoids from induced pluripotent stem cells. (A) Schematic summarizing the CRISPR-Cas9 approach to knocking in the G1 risk variants rs73885319 and rs60910145 into the 1016SevA line, including the chosen protospacer, homology-directed repair (HDR) donor template design leveraging the piggyBac transposon system, and Sanger sequencing validation of successful variant knock-in and selection cassette excision. (B) Overview of the Freedman kidney organoid differentiation protocol, with light microscopy of the 1016SevA line forming tubular organoids. (C) Confocal immunofluorescence images of nephron markers in representative G0 and G1 organoids on day 21 of differentiation. E-Cad, E-cadherin; gDNA, genomic DNA; mTeSR1, Modified Tenneille’s Special Recipe 1.
iPSC-Derived Kidney Organoid Differentiation
iPSCs were differentiated into kidney organoids following the previously published protocol by Freedman et al. (5) (Figure 1B). Briefly, iPSCs were dissociated with 1:3 Accutase and plated onto 24-well plates precoated with 0.5% GelTrex in mTeSR1 supplemented with 10 μM Y-27632 ROCK Inhibitor (STEMCELL Technologies). After 24 hours, another layer of GelTrex at 1.5% was added in mTeSR1 media. At the end of day 4, the medium was replaced with Advanced RPMI (Gibco) supplemented with 12 µM CHIR-99021 and 10 ng/ml noggin (STEMCELL Technologies). Approximately 60 hours later, the medium was changed to Advanced RPMI with B27 (Gibco). Organoids were cultured in this medium until collection at day 25.
Induction of APOL1 Expression and Endoplasmic Reticulum Stress
Day 24 G0 and G1 kidney organoids in identically plated wells of a 24-well plate were treated with IFN-γ (25 ng/ml; PeproTech) for 24 hours to induce APOL1 expression. This dose approximates previous in vitro IFN-γ doses used for macrophage activation (22,23). Endoplasmic reticulum (ER) stress was induced by adding 5 µM tunicamycin (Tocris) for 24 hours.
scRNA-seq
We performed scRNA-seq on G0 and G1 day-25 kidney organoids on the 1016SevA background. The organoids were treated with vehicle, IFN-γ, or both IFN-γ and tunicamycin for 24 hours. Organoids were dissociated from the well with TrypLE Express (Gibco) and processed into single-cell suspension by gentle intermittent pipetting while incubating in a ThermoMixer (Eppendorf) for up to 15 minutes. Single-cell libraries were prepared using the 10× Genomics Chromium droplet-based platform and the Single Cell 5′ Library Construction Kit (10× Genomics), which was chosen to increase read coverage over the 3′ chemistry. At least three technical replicates (different wells from the same experiment) were included in each prepared library, with targeted cell recovery of 4000 cells per library. Each experimental condition was represented by at least two different libraries. These libraries were assessed for quality control, pooled, and sequenced on the NovaSeq 6000 (Illumina) through the University of Illinois Genomics Core. The libraries were processed for 150-bp paired-end reads, at an average sequencing depth of 114,000 reads per cell.
Immunofluorescence
Organoids were fixed in 4% paraformaldehyde for 15 minutes at room temperature. After fixing, samples were washed in PBS, blocked in 5% donkey serum (Millipore)/0.3% Triton X-100/PBS for 1 hour at room temperature, incubated overnight in 3% BSA (Millipore)/PBS with primary antibodies, washed, incubated with Alexa-Fluor secondary antibodies (Invitrogen) and 4′,6-diamidino-2-phenylindole, and washed into PBS for storage. Primary antibodies included PODXL (AF1658, 1:500; R&D) and ECAD (ab11512, 1:500; Abcam). Stains included fluorescein-labeled LTL (FL-1321, 1:500; Vector Labs). Fluorescence images were captured using an inverted Nikon epifluorescence Eclipse Ti or A1R confocal microscope.
Real-Time Quantitative PCR
RNA was isolated from day-25 kidney organoids using the PureLink Kit (Invitrogen). RNA was then reverse transcribed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Real-time quantitative PCR (RT-qPCR) reactions for APOL1 (Hs01066280_m1; Thermo Fisher) and ACTB (Hs01060665_g1) were run in duplicate on the QuantStudio 5 Real-Time PCR System (Applied Biosystems). Relative APOL1 expression was calculated using the 2−ΔΔCT method. Statistical significance was tested using two-way ANOVA in Graphpad (Prism).
Immunoblot
Organoids were washed with PBS and lysed in RIPA buffer (Thermo Scientific) with protease inhibitor (Thermo Scientific) and benzonase nuclease (Thermo Scientific). Cell lysates were cleared by centrifugation for 10 minutes at 14,000 × g at 4°C. Proteins (20 µg) were heated at 95°C with β-mercaptoethanol and then separated by SDS-PAGE (Invitrogen). Nitrocellulose membranes were blocked in 5% nonfat milk in PBS with Tween 20 for 1 hour, incubated in primary antibody overnight at 4°C, and then incubated with horseradish peroxidase–conjugated secondary anti-Ig (1:5000; Abcam). The primary antibodies used in this study were anti-APOL1 (HPA018885, 1:1000; Sigma) and anti–β-actin (#4970, 1:1000; Cell Signaling Technologies). Immunoblot signals were developed under chemiluminescence (Thermo Fisher) and digitally imaged (BioRad).
Bioinformatic Analyses
Approach to bioinformatic analyses of scRNA-seq data are presented in Supplemental Material.
Results
Generation of Genome-Edited, Risk-Variant Kidney Organoids
Using CRISPR-Cas9 genome editing, we engineered an iPSC line homozygous for the APOL1 G1 risk alleles, with the G0 control on an isogenic background from the 1016SevA line. Unlike previous cell lines used to study risk-variant APOL1 through transgenic expression (24–27), this engineered line houses the G1 variant at its native genomic locus under the control of APOL1’s endogenous regulatory elements. To increase HDR efficiency, the donor template contained a puromycin selection cassette flanked by 500-bp homology arms, one of which housed the G1 variants rs73885319 and rs60910145 (Figure 1A). Successful G1-variant knock-in and cassette removal were confirmed by Sanger sequencing (Figure 1A, Supplemental Figure 1, A–C). Because CRISPR-mediated genome editing can occasionally induce chromosomal changes (28), we verified that our genome-edited iPSCs maintained a normal karyotype (Supplemental Figure 1, D and E).
To model the G1 variants within a kidney context, we differentiated the G0 and G1 1016SevA lines into kidney organoids using an adherent culture protocol we have previously established (Figure 1B) (5–7,13,14). Both G0 and G1 1016SevA iPSC lines differentiated into kidney organoids without major structural differences, expressing markers of nephron structure including PODXL in glomerular epithelial cells, LTL in the proximal tubule, and E-cadherin in the distal tubule, in appropriately patterned and contiguous segments (Figure 1C).
Single-Cell Transcriptomics of G0 and G1 Kidney Organoids
To discover if risk-variant APOL1 exerts cell-specific effects in terms of APOL1 expression and function, we performed scRNA-seq on all cells collected from whole wells of G0 and G1 organoids differentiated from the 1016SevA line from three separate experiments. A total of 67,337 cells across genotype and experimental conditions passed quality-control metrics (see Supplemental Methods, Supplemental References) and were analyzed using the integrated analysis workflow of Seurat version 3 (29) (Figure 2, A and B), which was chosen to be the primary analysis pipeline to improve normalization for batch effects and evaluate differential expression across clusters.
Figure 2.
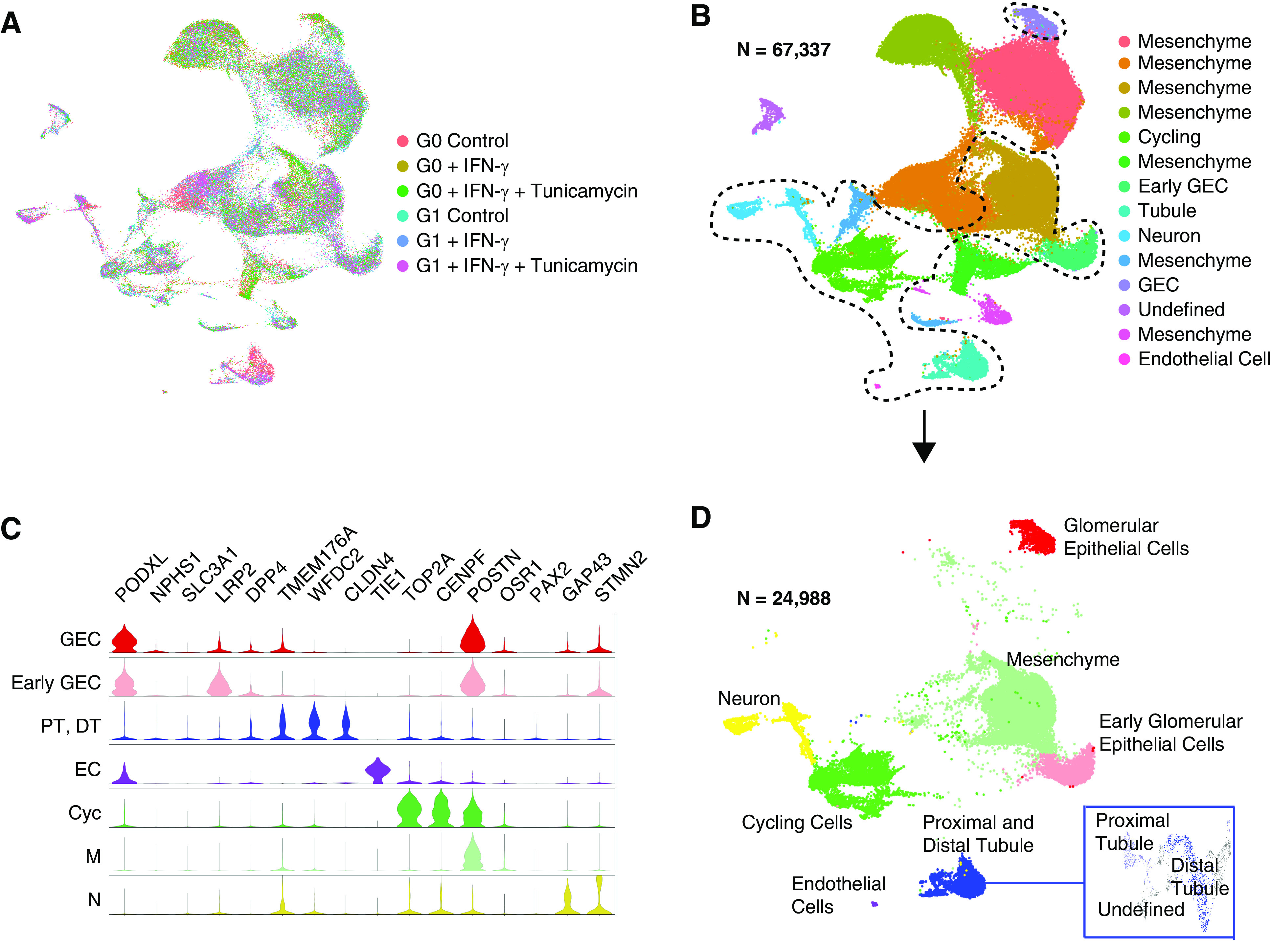
Overview of single-cell transcriptomics of G0 and G1 APOL1 organoids. (A and B) Uniform Manifold Approximation and Projection (UMAP) visualization of all whole-well G0 and G1 organoid cells profiled by single-cell RNA sequencing (scRNA-seq), integrated using Seurat version 3. (C) Violin plot of marker genes used for cluster identification of cell types, color coded according to labeling of nanodissected UMAP in (D), which separates out the main cell types seen in the organoid away from the extra mesenchyme captured in whole-well sequencing. Cyc, cycling; DT, distal tubule; EC, endothelial cells; GEC, glomerular epithelial cells; N, neuron; M, mesenchyme; PT, proximal tubule.
To further confirm our findings, we also analyzed the data separately with an unsupervised deep-embedding algorithm for single-cell clustering (DESC) (X. Li et al., unpublished data; available at https://www.biorxiv.org/content/10.1101/530378v1). Primary output of unsupervised clustering by Seurat yielded 14 clusters (Figure 2B, Supplemental Figure 3A), seven of which were determined to consist of mesenchymal cells whereas the others included glomerular epithelial cells at early and more mature stages, proximal and distal tubule, cycling cells, neurons, and endothelial cells (Figure 2, C and D, Supplemental Figure 3A). Cell type was determined by marker genes previously used in our scRNA-seq studies to identify differentiated cell types within human kidney organoids, based on direct comparison of known markers to tissue samples (Figure 2C) (13,14). The abundance of mesenchyme versus epithelial lineages in these whole-well differentiations is relatively high but within the expected range for patient-derived iPSC lines, which vary substantially in their efficiency of differentiation from one individual to the next (7,14). These clusters were present in both G0 and G1 organoids (Supplemental Figure 3B). Similar unsupervised clustering was also obtained using DESC (Supplemental Figure 4, A and B).
IFN-γ Induces APOL1 Expression in G0 and G1 Kidney Organoids
We next evaluated whether the organoids expressed APOL1. RT-qPCR on RNA from whole-well organoids differentiated from G0 and G1 1016SevA lines and the G0 Penn134-61-26 line revealed that organoids expressed little detectable APOL1 under standard culture conditions (Figure 3, A and B, Supplemental Figure 2), consistent with one study showing low levels in human kidneys in vivo (30). IFN-γ has been implicated as a factor that may induce APOL1 expression in transgenic mice, although whether this occurs in humans remains unclear (31). Robust APOL1 expression (mean >4000-fold over ACTB, P=0.004 by two-way ANOVA) was induced in both G0 and G1 organoids when exposed to 25 ng/ml IFN-γ for 24 hours (Figure 3A). APOL1 protein expression was confirmed on immunoblot of whole-well organoids, with the appearance of an approximately 40-kDa band, the expected size for APOL1, only in the IFN-γ samples (Figure 3B, Supplemental Figure 2).
Figure 3.
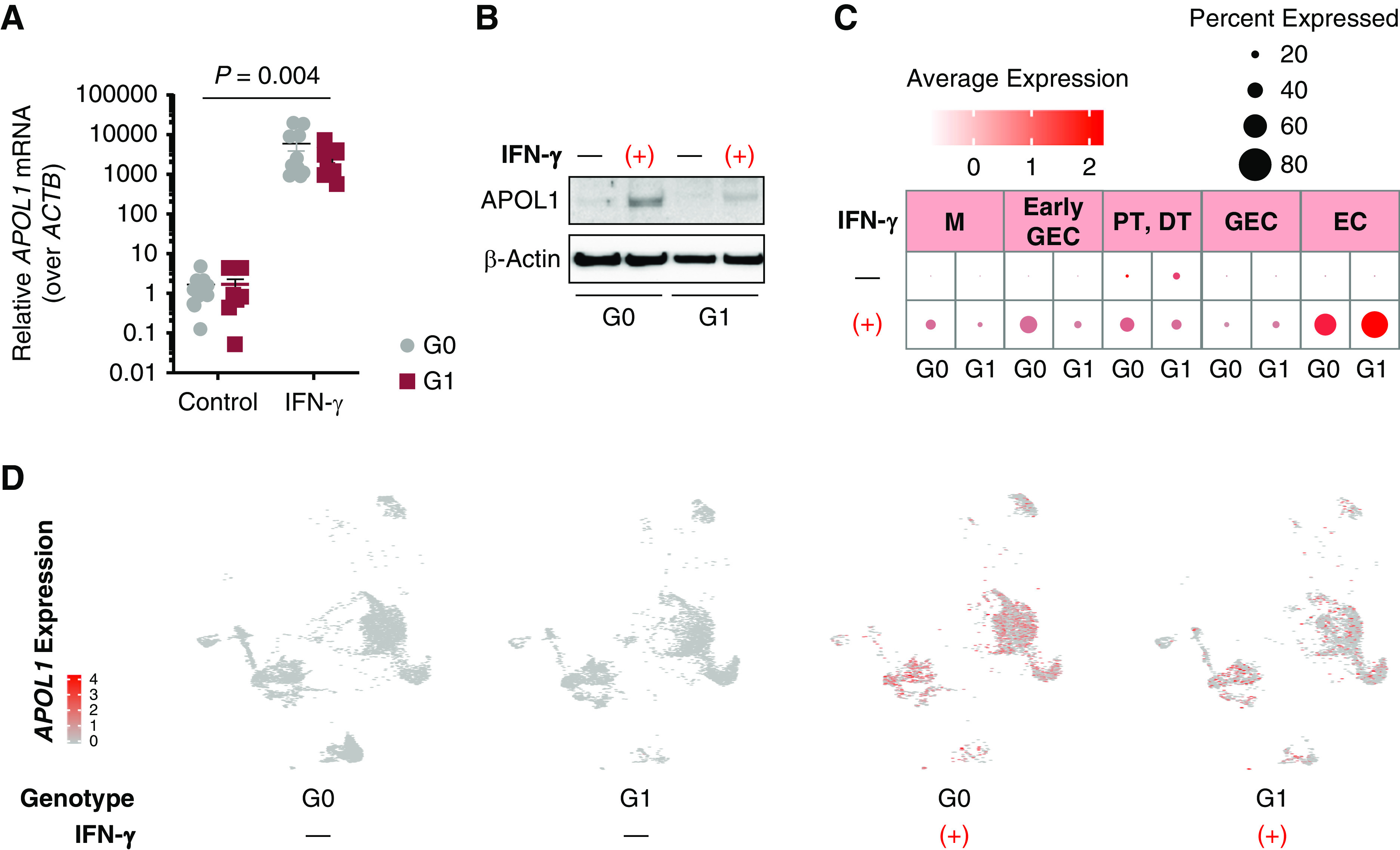
IFN-γ induces APOL1 expression in iPSC-derived kidney organoids. (A) Real-time quantitative PCR of RNA isolated from whole-well G0 and G1 organoids reveal low endogenous APOL1 expression but a >4000-fold induction of APOL1 mRNA relative to ACTB after 24 hours of 25 ng/ml IFN-γ treatment (P=0.004 by two-way ANOVA). (B) Immunoblot of APOL1 expression in G0 and G1 organoids with 25 ng/ml IFN-γ for 24 hours. (C) Dot plot of APOL1 expression in G0 and G1 kidney organoids, with and without IFN-γ treatment. Brighter red dots indicate stronger expression level across cells, and dot size reflects percentage of cells in a cluster expressing APOL1. (D) UMAP feature plots of nanodissected clusters show APOL1 expression (red dots) at baseline or when treated with 25 ng/ml IFN-γ for 24 hours.
To identify which organoid cell types express APOL1, and underlying effects of APOL1 risk variants on gene expression, scRNA-seq was performed as described above. As with the RT-qPCR and immunoblot assays, scRNA-seq revealed that untreated organoids express little detectable APOL1. However, exposure to IFN-γ for 24 hours induced APOL1 expression across multiple cell types, including glomerular epithelial cells, endothelial cells, and tubular cells (Figure 3, C and D, Supplemental Figures 5 and 6).
Gene Expression Signatures of G1 Kidney Organoids Are Specific to Cell Type
In contrast to an inducible APOL1 risk-variant overexpression cell model (25), G1 kidney organoids did not undergo appreciable cell death when APOL1 was expressed (Supplemental Figure 3B). To determine whether risk-variant APOL1 expression causes transcriptome-wide changes, we performed differential expression analysis using Seurat. When comparing G0 and G1 organoids exposed to IFN-γ, we found greater variance in gene expression patterns when examining each cell type separately than when examining the whole organoid (Figure 4A). In IFN-γ–stimulated organoids, very few genes differentially expressed between G0 and G1 glomerular epithelial cells overlapped with genes differentially expressed between G0 and G1 tubular cells (Figure 4B). Indeed, the genes differentially expressed between G0 and G1 glomerular epithelial cells stimulated with IFN-γ were enriched for biologic processes related to metabolic function, whereas tubular cells stimulated with IFN-γ exhibited differential (between G0 and G1) expression of genes related to inflammation and protein targeting and processing (Figure 4C).
Figure 4.
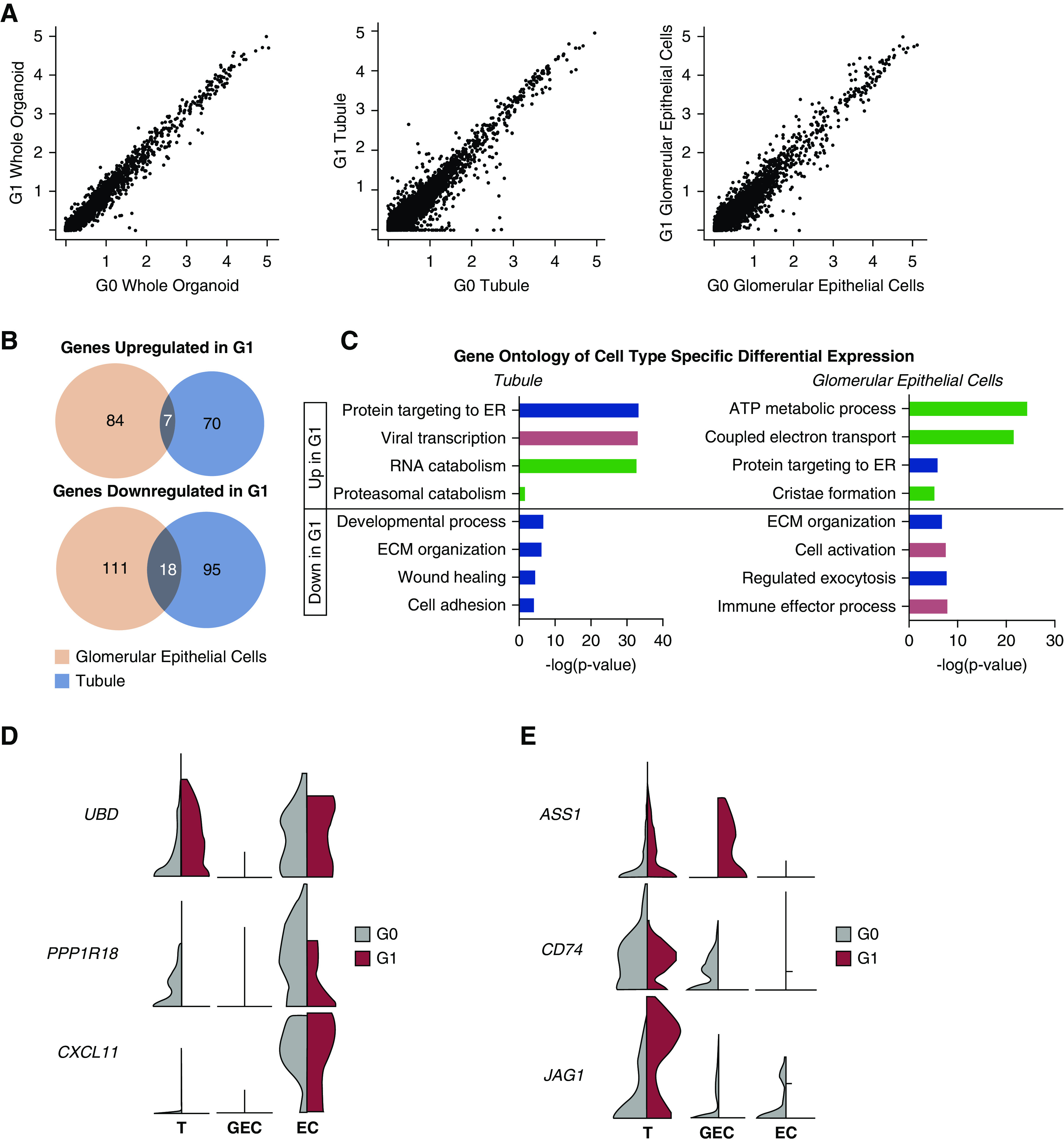
scRNA-seq reveals cell type–specific differential expression patterns between G0 and G1 organoids treated with IFN-γ. (A) Scatterplots of G0 versus G1 organoid gene expression values across all cell types (left), among tubular cells (middle), and among glomerular epithelial cells (right). Greater variance is seen in the cell type–specific plots. (B) Venn diagrams show little overlap of expression patterns across cell types when comparing which genes are upregulated in G1 IFN-γ stimulation over G0 IFN-γ stimulation. There is similarly low overlap across cell types for genes downregulated in G1 IFN-γ stimulation. (C) Gene Ontology of the differentially expressed genes within each cell type, with −log(P-value) plotted for each biologic process. Metabolic (green), stress (pink), cell and organelle function (purple), collagen/matrix (gray). (D) Violin plots visualizing cell type–specific expression patterns of putative regulators (24,33,34) of APOL1 abundance. (E) Violin plots visualizing cell type–specific expression patterns of novel genes. ECM, extracellular matrix; ER, endoplasmic reticulum; T, tubule.
To visualize these cell type–specific gene expression patterns induced by IFN-γ, we generated normalized transcript abundance on violin plots for each cluster (Figure 4, D and E). We first plotted genes previously identified by other studies to be potential modifiers of APOL1-mediated kidney disease. A large linkage disequilibrium block on chromosome 6 containing UBD and PPP1R18 may house disease-modulating genes (32,33), with visible differences between APOL1 genotypes in UBD and PPP1R18 expression in the tubule and endothelial clusters (Figure 4D). CXCL11 was previously found to be upregulated in the glomeruli of patients with APOL1-mediated kidney disease (24,33); it appears mildly more abundant in G1 endothelial cells (Figure 4D). Novel genes such as ASS1 are specifically upregulated in IFN-stimulated G1 glomerular epithelial cells, whereas CD74 and JAG1 are markedly downregulated in G1 glomerular epithelial cells compared to G0 cells (Figure 4E).
ER Stress Induces Differential Expression of Stress Response Genes in G1 Kidney Organoids
Having identified these cell type–specific differences in G1 organoid gene expression, we next evaluated whether introducing an additional stressor alters G1 cell phenotypes. Because APOL1-mediated kidney disease is incompletely penetrant, others have proposed a “second-hit” hypothesis that individuals carrying two APOL1 risk alleles develop disease after exposure to environmental or genetic modifiers (34,35). We tested whether ER stress could provide a stimulus to alter stress response and cellular phenotypes in the high-risk genotype. ER stress was chosen because ubiquitin D, encoded by UBD, is involved in the ER stress response, and risk-variant APOL1 appears to localize to the ER membrane (36). Furthermore, ER stress is becoming an increasingly recognized driver of complex diseases, including CKD (37–40). To induce ER stress, we treated organoids with 5 µM tunicamycin for 24 hours and observed increased expression of unfolded protein response genes EDEM1 and HSPA5 by RT-qPCR (Figure 5A). Likewise, select unfolded protein response and apoptosis genes exhibit a trend of differences in average cell type–cluster expression, as seen on a heatmap of G0 and G1 glomerular epithelial cell clusters (Figure 5, B and C), although these genes did not reach the statistical significance threshold established by the integrated workflow of Seurat.
Figure 5.

ER stress increases stress response in G1 kidney organoids. (A) Real-time quantitative PCR of RNA isolated from whole-well G0 and G1 organoids reveals increased unfolded protein response (UPR) gene expression (EDEM1, HSPA5) in G1 organoids exposed to both IFN-γ and tunicamycin (P=0.01 by two-way ANOVA). (B) Heatmap depicts relative average expression of UPR-relevant genes for G0 and G1 glomerular epithelial cells subjected to either IFN-γ alone or both IFN-γ and tunicamycin. (C) Heatmap depicts relative average expression of apoptosis-relevant genes for G0 and G1 glomerular epithelial cells subjected to both IFN-γ and tunicamycin.
ER Stress Induces Greater Dedifferentiation of G1 Glomerular Epithelial Cells
Heatmap and violin plots of the podocyte markers in organoids treated with IFN-γ and tunicamycin revealed relatively decreased expression of PODXL, NPHS1, and POSTN in the G1 glomerular epithelial cell cluster, suggesting the G1 organoid may acquire a less differentiated state under stress (Figure 6, A and B). We also discovered that G1 organoids treated with both IFN-γ and tunicamycin demonstrated less distinct cluster topology seen in partition-based graph abstraction (41). More specifically, G1 glomerular epithelial cells became closer to the other cell types in spatial relationship, whereas G0 glomerular epithelial cells still remained more distinct from the other cell clusters (Figure 6C). Trajectory inference of G0 and G1 organoid glomerular epithelial cells (Figure 6D) revealed that G1 organoids, compared with G0 organoids, subjected to ER stress have more cells scattered along trajectory paths between clusters, as well as a smaller proportion of mature glomerular epithelial cells compared with early glomerular epithelial cells (Figure 6, E and F). Collectively, these findings were consistent with potential dedifferentiation of G1 glomerular epithelial cells during stress.
Figure 6.
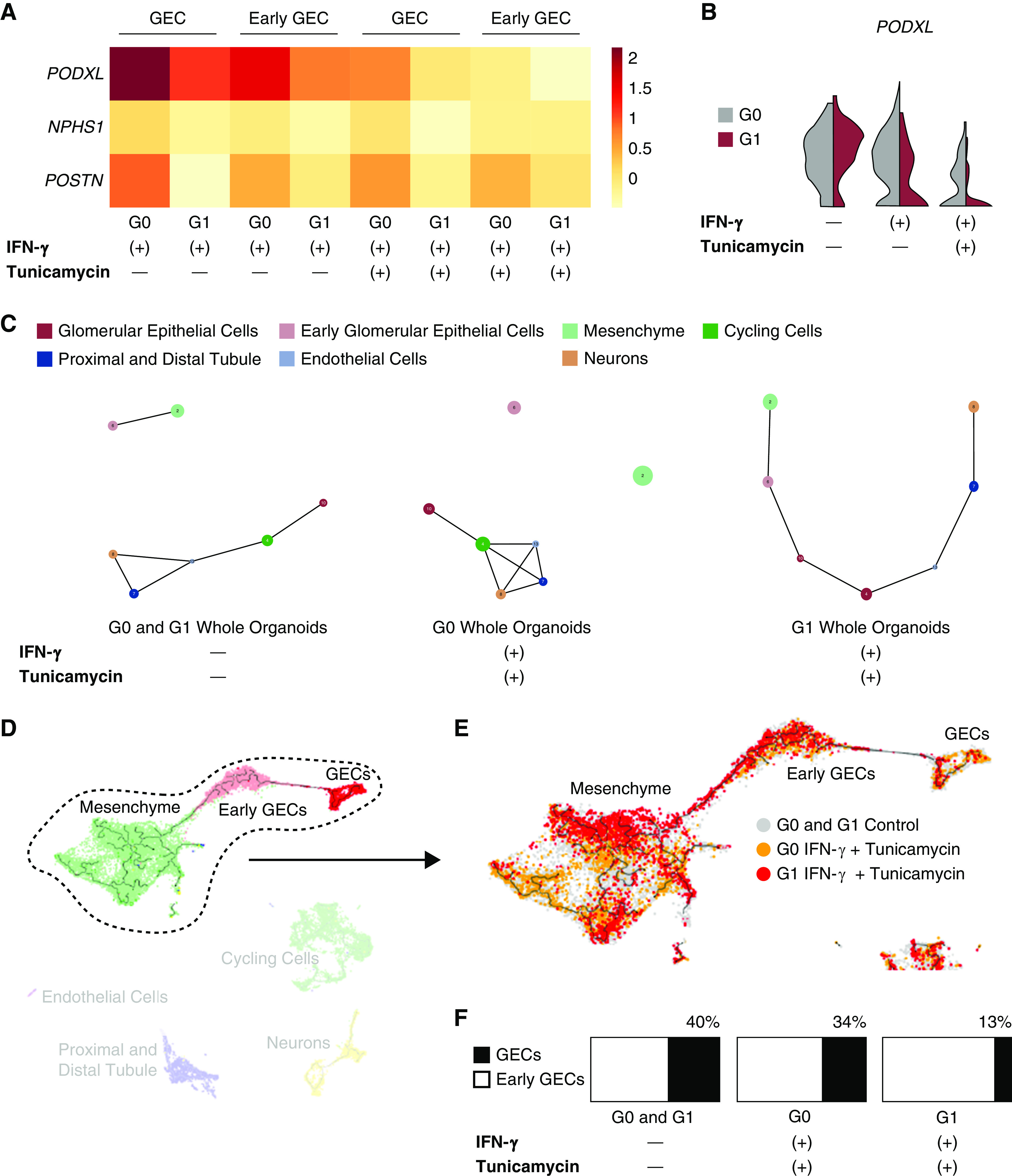
ER stress promotes dedifferentiation of GECs in G1 kidney organoids. (A) Heatmap depicts relative average expression of podocyte markers for G0 and G1 GECs and early GECs treated with either IFN-γ alone or both IFN-γ and tunicamycin. (B) Violin plots of PODXL expression in the GEC clusters of G0 and G1 organoids subjected to either IFN-γ alone or both IFN-γ and tunicamycin. (C) Partition-based graph abstraction (PAGA) (42) visualizes trajectory inference of all organoid single cells. The more mature GECs are connected to nonmesenchyme cell clusters in the topology of the control organoids. For G0 organoids subjected to both IFN-γ and tunicamycin, the more mature glomerular cells are still more distant from the mesenchyme. PAGA visualization of G1 organoids subjected to both IFN-γ and tunicamycin reveals less distinct separation of GECs from early GECs and mesenchyme. (D) Trajectory inference UMAP of all sequenced organoids was created using Monocle3. The trajectory of the relationship among mesenchyme, early GECs, and more mature GECs is circled and taken forward to (E), where G1 organoids treated with both IFN-γ and tunicamycin (red) have a relatively smaller proportion of GECs to early GECs compared with G0 organoids subjected to the same stressors (orange). (F) The relative number of GECs (black) to early GECs (white) is represented by the bar charts of G0 and G1 control organoids (left), G0 organoids treated with IFN-γ and tunicamycin (middle), and G1 organoids treated with IFN-γ and tunicamycin (right).
Discussion
We have conducted profiling of genome-edited, iPSC-derived kidney organoids to detect the potential effect of APOL1 risk variants on cellular phenotype and stress. With our novel platform of integrating genome editing, 3D kidney organoid culture, and single-cell transcriptomics, our model system recapitulates IFN-induced APOL1 expression and increased cellular stress in risk-variant organoids subjected to a second hit of ER stress. This system also demonstrated that gene expression signatures in risk-variant kidney organoids differ among cell types.
By expressing the G1 variants in their native genomic context, our approach circumvents challenges associated with studying APOL1 in model organisms and transgenic cell lines; for instance, transgenic overexpression that may introduce toxic nonphysiologic doses of APOL1 protein. Because APOL1-mediated kidney disease demonstrates incomplete penetrance (42,43), the ideal model system would allow for interrogation of native genomic regulators, including distal enhancer elements and cell-specific long noncoding RNAs (44). Use of genome editing rather than transgenesis preserves these regulatory interactions, including the response to IFN-γ, and thus would facilitate validation of molecular modifiers of risk-variant APOL1 expression and function.
Another advantage afforded by our platform is the use of a G0 control on an isogenic background, made possible through footprint-free CRISPR-Cas9 genome editing. Our footprint-free method leaves no alterations to the amino acid sequence of APOL1 protein and also does not leave behind any additional DNA (such as loxP sites) that would potentially interfere with native noncoding regulation. Isogenic backgrounds, where the only difference is the knock-in or correction of the disease-associated DNA variants, eliminates other variants as variables. Thus, this approach reduces the potential for genetic and epigenetic heterogeneity that would be expected among risk-variant, patient-derived iPSCs and age- and gender-matched, G0 patient–derived iPSCs as potential confounding modifiers of APOL1 expression and function.
Because this platform uses 3D kidney organoids, we used scRNA-seq to detect differences in molecular signatures among cell types in risk-variant organoids. Single-cell transcriptomics revealed cell type–specific gene expression differences between G0 and G1 organoids when APOL1 is induced, with little overlap between glomerular epithelial cells and tubular cells, suggesting that risk-variant APOL1 could potentially alter transcriptional programs in a cell type–specific fashion. This result is concordant with findings that human genetic variation exerts cell type–specific effects in CKD (11) and with a prior finding that glomerular and tubular compartments of APOL1-mediated FSGS biopsies exhibit distinct gene expression patterns (33).
Furthermore, we demonstrated that additional stressors can lead to more pronounced dedifferentiation of glomerular epithelial cells in G1 organoids. Unlike prior models (25,45), our model system does not lead to much cell death in the G1 kidney organoids at 24 hours, nor does it activate many proapoptotic genes, consistent with the concept that another stressor is needed to induce a phenotype. We chose ER stress as an experimental condition because previous studies have indicated that risk-variant APOL1 may be regulated by UBD (32), a gene involved in the ER stress response, and that risk-variant APOL1 localizes to the ER rather than to lipid droplets (36). Expression of risk-variant APOL1 alone does not activate a significant unfolded protein response (as seen in Figure 5B), but some differences can be seen for DDIT3, EDEM1, ERN1, GSK3A, and ERP44 upon tunicamycin stimulation, consistent with a second-hit hypothesis. With these changes in gene expression, accurate identification of cells attributed to the glomerular epithelial cell cluster among experimental conditions is essential. In this analysis, the glomerular epithelial cells were identified using the integration workflow of Seurat, which identifies conserved markers in clusters that are present in both control and stimulation conditions, decreasing the probability of cell type misclassification in the IFN-γ and/or tunicamycin-treated organoids. With the cells appropriately assigned to the main glomerular epithelial cell cluster and early glomerular epithelial cell cluster across experimental conditions, we assessed dedifferentiation by the relative ratio of “early” glomerular epithelial cells compared with both early and more mature cells in total. Because the differentiation and relative abundance of cell types did not differ between G0 and G1 organoids at baseline, the difference in this ratio between stressed G0 and G1 organoids most likely represented a dedifferentiation process induced by injury reproduced in data spanning technical replicates in experiments that were repeated. Although defining the exact mechanisms of this second hit in APOL1-mediated kidney disease is beyond the scope of this study, we demonstrate that our model system is capable of capturing the subtler phenotype of cellular dedifferentiation.
Our study also has limitations worth consideration. Our study did not include G2 variant organoids, although genome editing to create an isogenic G2 line is underway and will be important in future studies. Also, our proof-of-concept single-cell transcriptomics used iPSCs from one parent line (1016SevA) from a healthy non-African donor, so our work will require further validation from additional risk-variant APOL1 iPSC lines, preferably from patients with APOL1-mediated kidney disease, with matching isogenic genome-edited controls. In addition, the 1016SevA iPSC line yields relatively immature glomerular epithelial cells, which limits the conclusions that can be drawn in terms of APOL1-mediated podocyte biology. Although our data reveals a cell type–specific molecular signature of G1 kidney organoids, further scRNA-seq and protein-level validation in organoids derived from other iPSC lines that yield mature glomerular epithelial cells would be needed to highlight disease-relevant cellular mechanisms. Another potential limitation of our model system is the lack of detected APOL1 expression at baseline without IFN-γ stimulation, a result that is consistent with one prior study (30) but differs from a more recent study by Ma et al. (46) suggesting baseline expression on immunohistochemistry. This perceived difference in expression could be due to the altered physiologic or maturation state of the kidney organoids in vitro compared to tissue in vivo, to differential sensitivities of the various methodologies applied, or to variation of expression among different patients. It is nevertheless interesting and significant that APOL1 mRNA levels increase upon IFN-γ stimulation in organoids. Finally, the organoids generated in this study did not have vascular flow, so APOL1 expression and function in vasculature would not be fully captured despite the presence of endothelial-like cells. Likewise, although we have previously reported that organoid tubules are capable of selective solute transport, similar to proximal tubules (5), the organoids generated in this study lack a blood vessel conduit for continuous reabsorptive flux, and may not recapitulate the full range of filtrate reabsorption or specialized ion exchange, a limitation that may also affect transcriptional profiles and phenotype of the tubular cells in our experiments.
Despite these limitations, our model system, which combines the power of footprint-free genome editing with organoid culture, provides a human-relevant platform with which future studies can be executed and provides new insight into the potential mechanisms of APOL1-mediated kidney disease in diverse kidney cell types. With the power of genome editing, evolving science, and scRNA-seq, this platform could launch larger-scale mechanistic and screening studies for APOL1-mediated kidney disease, enabling the identification of pathways that could be targeted therapeutically in high-risk populations to reduce the incidence of glomerular disease.
Disclosures
B. Freedman is an inventor on patent applications related to kidney organoid differentiation and disease modeling and an advisor for Chinook Therapeutics. J. Himmelfarb reports personal fees from Gilead, personal fees from Maze Therapeutics, and personal fees from Renalytix AI, outside the submitted work. B. Chung, A. Dadi, M. Donnan, M. Li, J. Lin, E. Liu, B. Radmanesh, K. Smith, and D. Yi have nothing to disclose.
Funding
A portion of this work was supported by the National Institutes of Health (NIH) National Heart, Lung, and Blood Institute grant K08 HL135348 (to J. Lin), National Institute of Biomedical Imaging and Bioengineering grant U01 EB028892-01 (to B.S. Freedman), and NIH National Institute of Diabetes and Digestive and Kidney Diseases grant UG3TR002158 (to J. Himmelfarb).
Supplemental Material
This article contains the following supplemental material online at http://kidney360.asnjournals.org/lookup/suppl/doi:10.34067/KID.0000422019/-/DCSupplemental.
CRISPR-Cas9 engineering of G0 and G1 induced pluripotent stem cells (iPSCs) on isogenic backgrounds. Download Supplemental Figure 1, PDF file, 717 KB (716.6KB, pdf)
APOL1 protein expression induced by IFN-γ. Download Supplemental Figure 2, PDF file, 717 KB (716.6KB, pdf)
UMAP visualization by cell-type cluster and by experimental conditions. Download Supplemental Figure 3, PDF file, 717 KB (716.6KB, pdf)
Scanpy visualization of organoid scRNA-seq analyzed with the DESC pipeline. Download Supplemental Figure 4, PDF file, 717 KB (716.6KB, pdf)
APOL1 mRNA expression induced by IFN-γ. Download Supplemental Figure 5, PDF file, 717 KB (716.6KB, pdf)
Original uncropped images of Western blots for APOL1 protein expression in wholewell kidney organoid samples. Download Supplemental Figure 6, PDF file, 717 KB (716.6KB, pdf)
Oligonucleotides. Download Supplemental Table 1, PDF file, 2 MB (716.6KB, pdf) Supplemental Table 1, XLSX file, 67 KB (66.3KB, xlsx)
Details of single-cell RNA-seq (scRNA-seq) analyses. Download Supplemental Methods, PDF file, 2 MB (716.6KB, pdf)
Specific to text in this supplement. Download Supplemental References, PDF file, 2 MB (716.6KB, pdf)
Data Sharing
All scRNA-seq data have been deposited in Gene Expression Omnibus under accession number GSE135663.
Acknowledgments
We thank Dr. Kiran Musunuru for his input on footprint-free CRISPR-Cas9 genome editing. We appreciate the massively parallel sequencing services provided by the University of Illinois Genomics Core.
The views expressed in this article are those of the authors and do not necessarily reflect the position or policy of the Department of Veterans Affairs or the United States Government.
Author Contributions
B. Freedman and J. Lin conceptualized the study and provided supervision; B. Chung, A. Dadi, J. Lin, E. Liu, and K. Smith were responsible for data curation; E. Liu and B. Radmanesh were responsible for formal analysis; M. Donnan, J. Himmelfarb, J. Lin, and E. Liu were responsible for funding acquisition; B. Chung, A. Dadi, B. Freedman, J. Himmelfarb, J. Lin, E. Liu, B. Radmanesh, K. Smith, and D. Yi were responsible for the investigation; E. Liu and K. Smith were responsible for validation; B. Freedman, J. Himmelfarb, J. Lin, E. Liu, B. Radmanesh, and K. Smith wrote the original draft; M. Donnan, B. Freedman, J. Himmelfarb, B. Radmanesh, K. Smith, and D. Yi were responsible for resources; B. Chung, A. Dadi, J. Himmelfarb, and B. Radmanesh were responsible for visualization; B. Chung and M. Li were responsible for methodology and software; and all authors reviewed and edited the manuscript.
References
- 1.Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, Freedman BI, Bowden DW, Langefeld CD, Oleksyk TK, Uscinski Knob AL, Bernhardy AJ, Hicks PJ, Nelson GW, Vanhollebeke B, Winkler CA, Kopp JB, Pays E, Pollak MR: Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 329: 841–845, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tzur S, Rosset S, Shemer R, Yudkovsky G, Selig S, Tarekegn A, Bekele E, Bradman N, Wasser WG, Behar DM, Skorecki K: Missense mutations in the APOL1 gene are highly associated with end stage kidney disease risk previously attributed to the MYH9 gene. Hum Genet 128: 345–350, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parsa A, Kao WHL, Xie D, Astor BC, Li M, Hsu C-Y, Feldman HI, Parekh RS, Kusek JW, Greene TH, Fink JC, Anderson AH, Choi MJ, Wright JT Jr, Lash JP, Freedman BI, Ojo A, Winkler CA, Raj DS, Kopp JB, He J, Jensvold NG, Tao K, Lipkowitz MS, Appel LJ; AASK Study Investigators; CRIC Study Investigators: APOL1 risk variants, race, and progression of chronic kidney disease. N Engl J Med 369: 2183–2196, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Foster MC, Coresh J, Fornage M, Astor BC, Grams M, Franceschini N, Boerwinkle E, Parekh RS, Kao WHL: APOL1 variants associate with increased risk of CKD among African Americans. J Am Soc Nephrol 24: 1484–1491, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Freedman BS, Brooks CR, Lam AQ, Fu H, Morizane R, Agrawal V, Saad AF, Li MK, Hughes MR, Werff RV, Peters DT, Lu J, Baccei A, Siedlecki AM, Valerius MT, Musunuru K, McNagny KM, Steinman TI, Zhou J, Lerou PH, Bonventre JV: Modelling kidney disease with CRISPR-mutant kidney organoids derived from human pluripotent epiblast spheroids. Nat Commun 6: 8715, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim YK, Refaeli I, Brooks CR, Jing P, Gulieva RE, Hughes MR, Cruz NM, Liu Y, Churchill AJ, Wang Y, Fu H, Pippin JW, Lin LY, Shankland SJ, Vogl AW, McNagny KM, Freedman BS: Gene-edited human kidney organoids reveal mechanisms of disease in podocyte development. Stem Cells 35: 2366–2378, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cruz NM, Song X, Czerniecki SM, Gulieva RE, Churchill AJ, Kim YK, Winston K, Tran LM, Diaz MA, Fu H, Finn LS, Pei Y, Himmelfarb J, Freedman BS: Organoid cystogenesis reveals a critical role of microenvironment in human polycystic kidney disease. Nat Mater 16: 1112–1119, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cruz NM, Freedman BS: CRISPR gene editing in the kidney. Am J Kidney Dis 71: 874–883, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin J, Musunuru K: Genome engineering tools for building cellular models of disease. FEBS J 283: 3222–3231, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park J, Shrestha R, Qiu C, Kondo A, Huang S, Werth M, Li M, Barasch J, Susztak K: Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science 360: 758–763, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qiu C, Huang S, Park J, Park Y, Ko Y-A, Seasock MJ, Bryer JS, Xu X-X, Song W-C, Palmer M, Hill J, Guarnieri P, Hawkins J, Boustany-Kari CM, Pullen SS, Brown CD, Susztak K: Renal compartment-specific genetic variation analyses identify new pathways in chronic kidney disease. Nat Med 24: 1721–1731, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu H, Uchimura K, Donnelly EL, Kirita Y, Morris SA, Humphreys BD: Comparative analysis and refinement of human PSC-derived kidney organoid differentiation with single-cell transcriptomics. Cell Stem Cell 23: 869–881.e8, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Czerniecki SM, Cruz NM, Harder JL, Menon R, Annis J, Otto EA, Gulieva RE, Islas LV, Kim YK, Tran LM, Martins TJ, Pippin JW, Fu H, Kretzler M, Shankland SJ, Himmelfarb J, Moon RT, Paragas N, Freedman BS: High-Throughput screening enhances kidney organoid differentiation from human pluripotent stem cells and enables automated multidimensional phenotyping. Cell Stem Cell 22: 929–940.e4, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harder JL, Menon R, Otto EA, Zhou J, Eddy S, Wys NL, O’Connor C, Luo J, Nair V, Cebrian C, Spence JR, Bitzer M, Troyanskaya OG, Hodgin JB, Wiggins RC, Freedman BS, Kretzler M; European Renal cDNA Bank (ERCB); Nephrotic Syndrome Study Network (NEPTUNE): Organoid single cell profiling identifies a transcriptional signature of glomerular disease. JCI Insight 4: 122697, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pashos EE, Park Y, Wang X, Raghavan A, Yang W, Abbey D, Peters DT, Arbelaez J, Hernandez M, Kuperwasser N, Li W, Lian Z, Liu Y, Lv W, Lytle-Gabbin SL, Marchadier DH, Rogov P, Shi J, Slovik KJ, Stylianou IM, Wang L, Yan R, Zhang X, Kathiresan S, Duncan SA, Mikkelsen TS, Morrisey EE, Rader DJ, Brown CD, Musunuru K: Large, diverse population cohorts of hiPSCs and derived hepatocyte-like cells reveal functional genetic variation at blood lipid-associated loci. Cell Stem Cell 20: 558–570.e10, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li S, Li M, Liu X, Yang Y, Wei Y, Chen Y, Qiu Y, Zhou T, Feng Z, Ma D, Fang J, Ying H, Wang H, Musunuru K, Shao Z, Zhao Y, Ding Q: Genetic and chemical screenings identify HDAC3 as a key regulator in hepatic differentiation of human pluripotent stem cells. Stem Cell Reports 11: 22–31, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang X, Musunuru K: Confirmation of causal rs9349379- PHACTR1 expression quantitative trait locus in human-induced pluripotent stem cell endothelial cells. Circ Genom Precis Med 11: e002327, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gupta RM, Hadaya J, Trehan A, Zekavat SM, Roselli C, Klarin D, Emdin CA, Hilvering CRE, Bianchi V, Mueller C, Khera AV, Ryan RJH, Engreitz JM, Issner R, Shoresh N, Epstein CB, de Laat W, Brown JD, Schnabel RB, Bernstein BE, Kathiresan S: A genetic variant associated with five vascular diseases is a distal regulator of endothelin-1 gene expression. Cell 170: 522–533.e15, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Palmer DJ, Grove NC, Ing J, Crane AM, Venken K, Davis BR, Ng P: Homology requirements for efficient, footprintless gene editing at the CFTR locus in human iPSCs with helper-dependent adenoviral vectors. Mol Ther Nucleic Acids 5: e372, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Firth AL, Menon T, Parker GS, Qualls SJ, Lewis BM, Ke E, Dargitz CT, Wright R, Khanna A, Gage FH, Verma IM: Functional gene correction for cystic fibrosis in lung epithelial cells generated from patient iPSCs. Cell Reports 12: 1385–1390, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM: RNA-guided human genome engineering via Cas9. Science 339: 823–826, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin J, Hu Y, Nunez S, Foulkes AS, Cieply B, Xue C, Gerelus M, Li W, Zhang H, Rader DJ, Musunuru K, Li M, Reilly MP: Transcriptome-wide analysis reveals modulation of human macrophage inflammatory phenotype through alternative splicing. Arterioscler Thromb Vasc Biol 36: 1434–1447, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang H, Xue C, Shah R, Bermingham K, Hinkle CC, Li W, Rodrigues A, Tabita-Martinez J, Millar JS, Cuchel M, Pashos EE, Liu Y, Yan R, Yang W, Gosai SJ, VanDorn D, Chou ST, Gregory BD, Morrisey EE, Li M, Rader DJ, Reilly MP: Functional analysis and transcriptomic profiling of iPSC-derived macrophages and their application in modeling Mendelian disease. Circ Res 117: 17–28, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beckerman P, Bi-Karchin J, Park ASD, Qiu C, Dummer PD, Soomro I, Boustany-Kari CM, Pullen SS, Miner JH, Hu C-AA, Rohacs T, Inoue K, Ishibe S, Saleem MA, Palmer MB, Cuervo AM, Kopp JB, Susztak K: Transgenic expression of human APOL1 risk variants in podocytes induces kidney disease in mice. Nat Med 23: 429–438, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Olabisi OA, Zhang J-Y, VerPlank L, Zahler N, DiBartolo S 3rd, Heneghan JF, Schlöndorff JS, Suh JH, Yan P, Alper SL, Friedman DJ, Pollak MR: APOL1 kidney disease risk variants cause cytotoxicity by depleting cellular potassium and inducing stress-activated protein kinases. Proc Natl Acad Sci U S A 113: 830–837, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O’Toole JF, Schilling W, Kunze D, Madhavan SM, Konieczkowski M, Gu Y, Luo L, Wu Z, Bruggeman LA, Sedor JR: ApoL1 overexpression drives variant-independent cytotoxicity. J Am Soc Nephrol 29: 869–879, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Okamoto K, Rausch JW, Wakashin H, Fu Y, Chung J-Y, Dummer PD, Shin MK, Chandra P, Suzuki K, Shrivastav S, Rosenberg AZ, Hewitt SM, Ray PE, Noiri E, Le Grice SFJ, Hoek M, Han Z, Winkler CA, Kopp JB: APOL1 risk allele RNA contributes to renal toxicity by activating protein kinase R. Commun Biol 1: 188, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cullot G, Boutin J, Toutain J, Prat F, Pennamen P, Rooryck C, Teichmann M, Rousseau E, Lamrissi-Garcia I, Guyonnet-Duperat V, Bibeyran A, Lalanne M, Prouzet-Mauléon V, Turcq B, Ged C, Blouin J-M, Richard E, Dabernat S, Moreau-Gaudry F, Bedel A: CRISPR-Cas9 genome editing induces megabase-scale chromosomal truncations. Nat Commun 10: 1136, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R: Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36: 411–420, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Monajemi H, Fontijn RD, Pannekoek H, Horrevoets AJG: The apolipoprotein L gene cluster has emerged recently in evolution and is expressed in human vascular tissue. Genomics 79: 539–546, 2002 [DOI] [PubMed] [Google Scholar]
- 31.Aghajan M, Booten SL, Althage M, Hart CE, Ericsson A, Maxvall I, Ochaba J, Menschik-Lundin A, Hartleib J, Kuntz S, Gattis D, Ahlström C, Watt AT, Engelhardt JA, Monia BP, Magnone MC, Guo S: Antisense oligonucleotide treatment ameliorates IFN-γ-induced proteinuria in APOL1-transgenic mice. JCI Insight 4: 126124, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang J-Y, Wang M, Tian L, Genovese G, Yan P, Wilson JG, Thadhani R, Mottl AK, Appel GB, Bick AG, Sampson MG, Alper SL, Friedman DJ, Pollak MR: UBD modifies APOL1-induced kidney disease risk. Proc Natl Acad Sci U S A 115: 3446–3451, 2018. 10.1073/pnas.1716113115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sampson MG, Robertson CC, Martini S, Mariani LH, Lemley KV, Gillies CE, Otto EA, Kopp JB, Randolph A, Vega-Warner V, Eichinger F, Nair V, Gipson DS, Cattran DC, Johnstone DB, O’Toole JF, Bagnasco SM, Song PX, Barisoni L, Troost JP, Kretzler M, Sedor JR; Nephrotic Syndrome Study Network: Integrative genomics identifies novel associations with APOL1 risk genotypes in black NEPTUNE subjects. J Am Soc Nephrol 27: 814–823, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Freedman BI, Langefeld CD, Turner J, Núñez M, High KP, Spainhour M, Hicks PJ, Bowden DW, Reeves-Daniel AM, Murea M, Rocco MV, Divers J: Association of APOL1 variants with mild kidney disease in the first-degree relatives of African American patients with non-diabetic end-stage renal disease. Kidney Int 82: 805–811, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Langefeld CD, Comeau ME, Ng MCY, Guan M, Dimitrov L, Mudgal P, Spainhour MH, Julian BA, Edberg JC, Croker JA, Divers J, Hicks PJ, Bowden DW, Chan GC, Ma L, Palmer ND, Kimberly RP, Freedman BI: Genome-wide association studies suggest that APOL1-environment interactions more likely trigger kidney disease in African Americans with nondiabetic nephropathy than strong APOL1-second gene interactions. Kidney Int 94: 599–607, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chun J, Zhang JY, Wilkins MS, Subramanian B, Riella C, Magraner JM, Alper SL, Friedman DJ, Pollak MR: Recruitment of APOL1 kidney disease risk variants to lipid droplets attenuates cell toxicity. Proc Natl Acad Sci U S A 116: 3712–3721, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abe JI, Ko KA, Kotla S, Wang Y, Paez-Mayorga J, Shin IJ, Imanishi M, Vu HT, Tao Y, Leiva-Juarez MM, Thomas TN, Medina JL, Won JH, Fujii Y, Giancursio CJ, McBeath E, Shin JH, Guzman L, Abe RJ, Taunton J, Mochizuki N, Faubion W, Cooke JP, Fujiwara K, Evans SE, Le NT: MAGI1 as a link between endothelial activation and ER stress drives atherosclerosis. JCI Insight 4: 125570, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seimon TA, Nadolski MJ, Liao X, Magallon J, Nguyen M, Feric NT, Koschinsky ML, Harkewicz R, Witztum JL, Tsimikas S, Golenbock D, Moore KJ, Tabas I: Atherogenic lipids and lipoproteins trigger CD36-TLR2-dependent apoptosis in macrophages undergoing endoplasmic reticulum stress. Cell Metab 12: 467–482, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thorp E, Li G, Seimon TA, Kuriakose G, Ron D, Tabas I: Reduced apoptosis and plaque necrosis in advanced atherosclerotic lesions of Apoe-/- and Ldlr-/- mice lacking CHOP. Cell Metab 9: 474–481, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dickhout JG, Carlisle RE, Austin RC: Interrelationship between cardiac hypertrophy, heart failure, and chronic kidney disease: Endoplasmic reticulum stress as a mediator of pathogenesis. Circ Res 108: 629–642, 2011 [DOI] [PubMed] [Google Scholar]
- 41.Wolf FA, Hamey FK, Plass M, Solana J, Dahlin JS, Göttgens B, Rajewsky N, Simon L, Theis FJ: PAGA: Graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol 20: 59, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dummer PD, Limou S, Rosenberg AZ, Heymann J, Nelson G, Winkler CA, Kopp JB: APOL1 kidney disease risk variants: An evolving landscape. Semin Nephrol 35: 222–236, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kruzel-Davila E, Wasser WG, Skorecki K: APOL1 nephropathy: A population genetics and evolutionary medicine detective story. Semin Nephrol 37: 490–507, 2017 [DOI] [PubMed] [Google Scholar]
- 44.Mattioli K, Volders PJ, Gerhardinger C, Lee JC, Maass PG, Melé M, Rinn JL: High-throughput functional analysis of lncRNA core promoters elucidates rules governing tissue specificity. Genome Res 29: 344–355, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Granado D, Müller D, Krausel V, Kruzel-Davila E, Schuberth C, Eschborn M, Wedlich-Söldner R, Skorecki K, Pavenstädt H, Michgehl U, Weide T: Intracellular APOL1 risk variants cause cytotoxicity accompanied by energy depletion. J Am Soc Nephrol 28: 3227–3238, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ma L, Shelness GS, Snipes JA, Murea M, Antinozzi PA, Cheng D, Saleem MA, Satchell SC, Banas B, Mathieson PW, Kretzler M, Hemal AK, Rudel LL, Petrovic S, Weckerle A, Pollak MR, Ross MD, Parks JS, Freedman BI: Localization of APOL1 protein and mRNA in the human kidney: Nondiseased tissue, primary cells, and immortalized cell lines. J Am Soc Nephrol 26: 339–348, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
CRISPR-Cas9 engineering of G0 and G1 induced pluripotent stem cells (iPSCs) on isogenic backgrounds. Download Supplemental Figure 1, PDF file, 717 KB (716.6KB, pdf)
APOL1 protein expression induced by IFN-γ. Download Supplemental Figure 2, PDF file, 717 KB (716.6KB, pdf)
UMAP visualization by cell-type cluster and by experimental conditions. Download Supplemental Figure 3, PDF file, 717 KB (716.6KB, pdf)
Scanpy visualization of organoid scRNA-seq analyzed with the DESC pipeline. Download Supplemental Figure 4, PDF file, 717 KB (716.6KB, pdf)
APOL1 mRNA expression induced by IFN-γ. Download Supplemental Figure 5, PDF file, 717 KB (716.6KB, pdf)
Original uncropped images of Western blots for APOL1 protein expression in wholewell kidney organoid samples. Download Supplemental Figure 6, PDF file, 717 KB (716.6KB, pdf)
Oligonucleotides. Download Supplemental Table 1, PDF file, 2 MB (716.6KB, pdf) Supplemental Table 1, XLSX file, 67 KB (66.3KB, xlsx)
Details of single-cell RNA-seq (scRNA-seq) analyses. Download Supplemental Methods, PDF file, 2 MB (716.6KB, pdf)
Specific to text in this supplement. Download Supplemental References, PDF file, 2 MB (716.6KB, pdf)