

Abstract
Purpose of review:
This review summarizes recent basic science studies on homeostasis of iron, an essential dietary nutrient and potentially toxic metal, and explores the relevance of these studies to our understanding of trauma and related severe, acute events.
Recent findings:
Recent studies in experimental models of iron homeostasis have added to our understanding of how iron levels are regulated in the body and how iron levels and iron-dependent biological processes contribute to trauma and related events. Iron deficiency, a common nutritional disorder, can impair critical organ function and wound and injury repair. Iron excess, typically due to genetic defects, can cause toxicity to tissues and, like iron deficiency, impair wound and injury repair. Finally, pharmacologic inhibition of ferroptosis, a novel form of iron-dependent cell death, is beneficial in animal models of cardiac, hepatic, and intestinal injury and intracerebral hemorrhage, suggesting that ferroptosis inhibitors could serve as novel therapeutic agents for trauma and related events.
Summary:
Perturbations in iron homeostasis can contribute significantly to an individual’s predisposition to trauma and their ability to recover post-trauma, while pharmacologic targeting of ferroptosis may attenuate severity of trauma-induced organ dysfunction.
Keywords: iron, homeostasis, trauma, ferroptosis
Introduction
At first glance, there may not appear to be much overlap between iron homeostasis and trauma. However, a consideration of the roles of iron biology in predisposition to trauma and in treatment of and recovery from trauma reveals much of value in this overlap. The underlying reason is that iron is both an essential dietary nutrient yet also potentially toxic. Iron is required for erythropoiesis and other critical physiologic processes within the body. The utility of iron is due in part to its propensity to undergo oxidation-reduction reactions between Fe2+ and Fe3+. However, given that Fe2+ is toxic to cells, iron excess can have deleterious effects on health. As such, both iron deficiency and excess can render individuals more susceptible to trauma and other life-threatening acute conditions and impair their ability to recover from such events. The current review focuses on basic scientific knowledge of iron homeostasis in the context of trauma. It begins with recent discoveries regarding regulation of hepcidin, a key hormonal regulator of iron levels, and continues with recent findings linking iron homeostasis to critical organ function. The review concludes with recent reports on iron homeostasis and their relevance to trauma and other acute, life-threatening conditions.
Regulation of hepcidin expression
Iron overload has widespread impact on human health. Commonly due to increased dietary iron uptake, it can be caused by inherited defects or associated with diseases such as obesity, diabetes, alcohol use, chronic hepatitis, and β-thalassemia. Iron overload results in production of reactive oxygen species, leading to cirrhosis, hepatomas, cardiomyopathy, diabetes, hypogonadotropic hypogonadism, and arthritis. Iron deficiency anemia can also be caused by dysregulation of iron homeostasis. The anemia of chronic disease is a major complication of bacterial and viral infections, parasitic infections, and chronic inflammatory disorders. Identifying the signal transduction pathways that regulate iron homeostasis has and will continue to provide the basis for new interventions to control iron levels in the body.
The liver is the major regulator of iron homeostasis (Figure 1). It secretes hepcidin, a hormone that binds to the iron export protein ferroportin, triggering ferroportin degradation, and blocking iron export [1]. Ferroportin is essential for dietary iron uptake and release of iron from macrophages that recycle red blood cells. Iron excess and inflammation stimulate hepcidin expression to inhibit further iron uptake and release of iron from macrophages. Iron deficiency and anemia inhibit hepcidin expression to enhance dietary iron uptake and release of iron from cellular sites of storage.
Fig. 1. Regulation of hepcidin (HAMP) transcription.
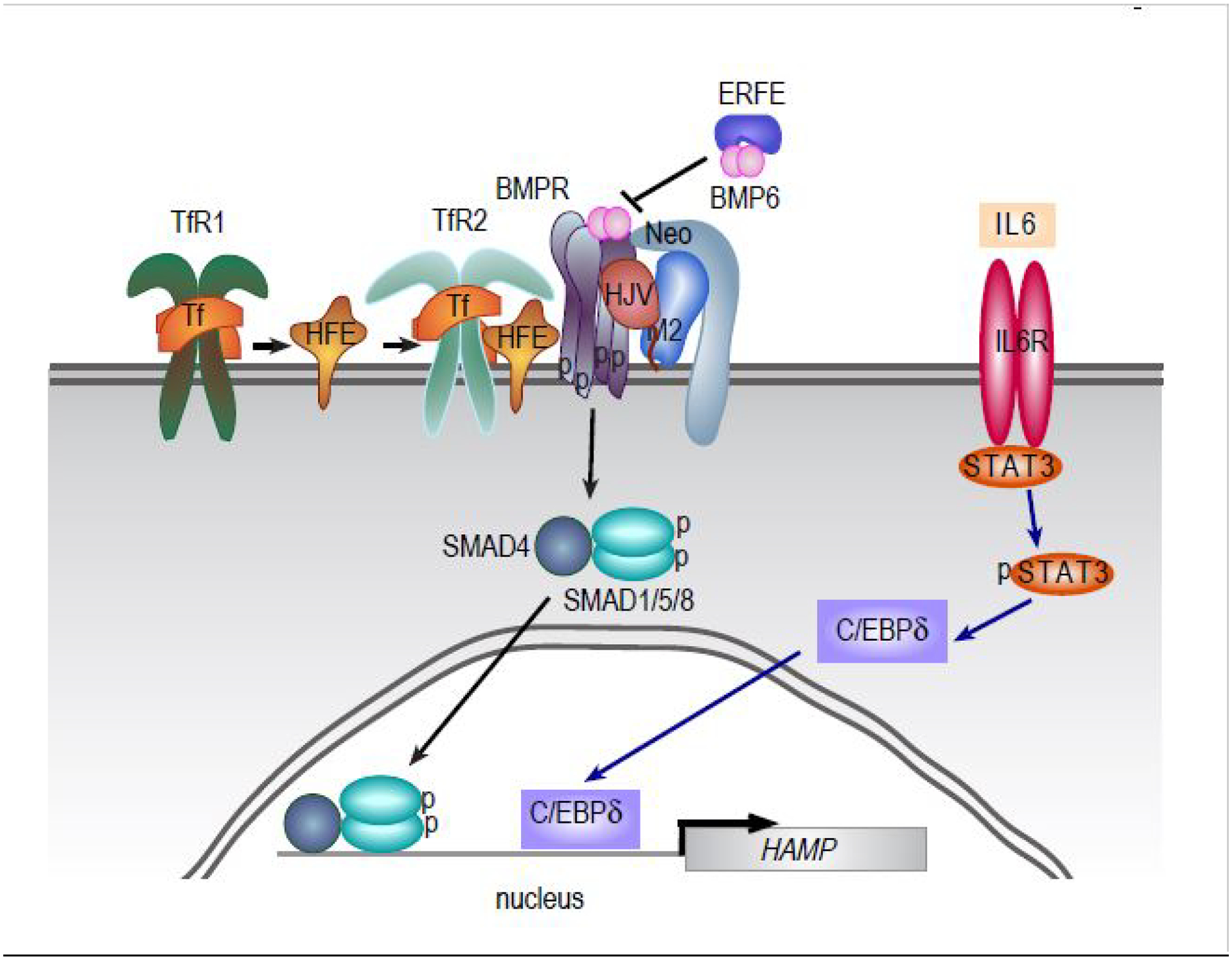
Under high iron conditions or oxidative stress, BMP6 binds to BMP receptor (BMPR) which stimulates the phosphorylation of SMAD1,5,8. SMAD4 binds to phosphorylated SMAD1,5,8 and the complex enters the nucleus to stimulate hepcidin expression. The coreceptor hemojuvelin (HJV) facilitates the binding of BMP6 to BMPR. Neogenin (Neo), transferrin receptor 2 (TfR2) and HFE increase the signaling by the BMPR. Note that TfR2 is distinct from transferrin receptor 1 (TfR1); while both TfR1 and TfR2 bind to the iron transport protein transferrin (Tf), only Tf binding to TfFR2 stimulates hepcidin expression. Matriptase 2 (M2) negatively regulates the signaling by cleaving HJV, HFE, and multiple other substrates. Hepcidin transcription can also be upregulated by inflammation through stimulation of the IL6 receptor and phosphorylation of STAT3. STAT3 increases C/EBPδ–dependent upregulation of hepcidin transcription.
Bone morphogenetic protein 6 (BMP6), a secreted protein that regulates a wide range of biological processes, is a critical stimulator of hepcidin expression in conditions of iron overload (Figure 1). BMP6 binds to BMP receptors expressed on the cell surface of hepatocytes and activates the phosphorylation of cytosolic transcription factors SMADs 1, 5, and 8 which then bind to SMAD4, translocate to the nucleus, and stimulate hepcidin expression. Earlier work found that SMAD1 and SMAD5 but not SMAD8 are critical to hepcidin expression. More recent results have indicated that SMAD8 is also required for full activation of the BMP signaling pathway [2]. However, not all SMADs stimulate hepcidin expression. BMP signaling increases expression of SMAD6 and SMAD7, two proteins that downregulate BMP signaling by interfering with phosphorylation of BMP receptors and formation of the SMAD4/SMAD1,5,8 complex. Lai and colleagues recently demonstrated that Smad7 overexpression in livers of mice results in decreased hepcidin expression and iron overload [3]. Future studies will be necessary to determine, if this finding is physiologically relevant and can be exploited therapeutically to control iron homeostasis. It should also be noted that BMP6 is not the only BMP to stimulate hepcidin expression. Wang and colleagues recently demonstrated that Bmp2 deficiency in mice partially blunts the hepcidin response to iron loading showing that Bmp2 plays a role in hepcidin expression [4].
The complexity of the relationship between BMPs and hepcidin expression is further highlighted by two other recent studies. Lim and colleagues showed that iron-induced mitochondrial stress activates NRF2, a transcription factor involved in anti-oxidative responses, which in turn activates BMP6 expression to stimulate hepcidin expression and limit iron levels [5]. This finding offers a new molecular target, NRF2, to exploit to control iron levels. The second study explores the well-known observation that hepcidin expression can be inhibited by increased erythropoiesis. Erythroid precursors secrete the hormone erythroferrone which inhibits hepcidin production in order to increase iron availability for erythropoiesis. Arezes and colleagues recently demonstrated that erythroferrone binds BMP6 and related BMP proteins to decrease BMP-stimulated hepcidin expression [6].
Important to trauma, inflammation is a potent stimulator of hepcidin expression. This stimulation involves the JAK-STAT signaling pathway which transmits inflammatory signals to the nucleus to alter gene expression. Earlier work indicated that BMP signaling could stimulate the JAK/STAT pathway, but this observation has been controversial in the field. For example, Wang and colleagues showed that endogenous BMP expression is reduced by inflammation in mice [4]. However, studies on anemia of inflammation recently showed that the decrease in iron levels observed with inflammation requires BMP signaling [7] and that deficiency in the BMP receptor Alk3 partially protects mice from the anemia of inflammation [8]. Overall, results support that basal BMP signaling is required for a full inflammatory response.
Heparan sulfate, a polysaccharide abundant in the body, is another critical regulator of hepcidin expression and iron homeostasis. Two recent papers establish a possible mechanism for this phenomenon. Poli et al. demonstrated that heparan sulfate is required for both base-line and BMP6-dependent hepcidin expression [9]. In addition, Guo et al. recently identified, using a genome-wide association study, that fibroblast growth factor 6 (FGF6) is associated with high transferrin saturation, a marker of iron overload [10]. They also demonstrated that wild-type FGF6 stimulates hepcidin expression and is stabilized by heparan sulfate. In line with these findings, cell lines expressing mutant FGF6 incapable of binding to heparan show decreased hepcidin expression. The finding that heparan and FGF6 increase hepcidin expression offers new ways to pharmacologically manipulate iron homeostasis in the body.
Cardiac and pulmonary iron homeostasis
Iron deficiency is the most common nutritional disorder in the world. Not surprisingly, it can impact the function of critical organs. Iron deficiency is present in roughly 50% of patients with heart failure, and administration of intravenous iron can benefit these patients. Iron deficiency also increases pulmonary arterial pressure in patients with pulmonary arterial hypertension. Three recent reports explore the relationship between iron deficiency and critical organ function.
The first study, by Chung et al., investigated the underlying mechanism linking iron deficiency and heart failure using mice raised on an iron-deficient diet [11]. This study demonstrated discrete molecular defects involving regulation of calcium levels and established a potential mechanism linking iron deficiency and cardiac dysfunction. As a limitation, the authors noted that the observed contractile defects were moderate and may apply more to patients with pre-existing heart failure than iron-deficient patients without heart failure. The second study, by Lakhal-Littleton et al., interrogated the link between iron deficiency and pulmonary arterial hypertension (PAH) by employing a novel mouse model of iron deficiency, in which the iron export protein ferroportin was constitutively active in pulmonary artery smooth muscle cells (PASMCs) [12]. These mice exhibited PAH and right ventricular remodeling. Intravenous iron treatment attenuated but did not reverse PAH. This study demonstrated that PASMCs are a critical cell type in iron deficiency-related PAH and right heart failure. In the third study, Yu et al. established experimental models of BOLA3 deficiency to explore the basis of pulmonary hypertension [13]. BOLA3, a regulator of iron-sulfur cluster biogenesis, is mutated in the multiple mitochondrial dysfunction syndrome, a fatal disorder characterized by pulmonary hypertension and other defects. Iron-sulfur clusters are key biological components of enzymes that mediate electron transport and oxidation-reduction processes and are particularly critical for mitochondrial functioning. The authors demonstrated that BOLA3 deficiency results in multiple defects in iron homeostasis and cell growth and metabolism. Mice with BOLA3 deficiency in pulmonary vascular endothelial cells developed pulmonary vascular proliferation and pulmonary hypertension, while mice overexpressing BOLA3 were protected against pulmonary hypertension. This study indicated that a discrete molecular factor, BOLA3, in pulmonary vascular endothelial cells is a critical determinant of pulmonary vascular biology and function.
Two recent reports of note also focused on cardiac injury, given that heart disease is a leading cause of death in the world. In the first report, Fang et al. investigated the contribution of ferroptosis, a novel form of regulated cell death caused by iron-dependent accumulation of lipid peroxides, to cardiomyocyte death, a key component of heart failure [14]. Using a mouse model of doxorubicin-induced cardiomyopathy and another of ischemia/reperfusion-induced heart failure, the authors observed that a ferroptosis inhibitor attenuated doxorubicin-induced pathology and severity of cardiac ischemia/reperfusion injury. In the second report, Zhabyeyev et al. employed mice deficient in TIMP3 (tissue inhibitor of metalloproteinase 3), a regulator of extracellular matrix remodeling and tissue inflammation, fibrosis, and repair, to demonstrate that Timp3 is a critical regulator of iron-mediated cardiac and hepatic injury [15]. Loading of liver, heart, and other tissues with iron injections into Timp3-deficient mice resulted in systolic dysfunction and worsening of diastolic dysfunction, increased markers of cardiac fibrosis, inflammation, and tissue remodeling, and exacerbated hepatic inflammation, fibrosis, and tissue remodeling, relative to wild-type mice injected with iron. Together, these studies highlighted the critical role that iron-dependent cell death and iron excess can play in organ pathophysiology.
Relevance of iron homeostasis to trauma, treatment, and recovery
Most iron in the body is dedicated to erythropoiesis. Given the toxicity of iron, the reclamation of iron from aged or damaged red cells is carefully orchestrated by macrophages in spleen and other organs. However, in events such as stroke or hemorrhage, extravasation and hemolysis of red cells can expose neighboring cells and tissues to a significant load of iron. Four recent reports explored the potential role of iron in such events. In the first study, Young et al. studied the contribution of hemojuvelin to acute ischemic stroke, where hemojuvelin is an essential component of the signaling pathway that regulates hepcidin expression [16]. The authors demonstrated that hemojuvelin-deficient mice had smaller infarct volume than wild-type mice when stroke was induced, although an underlying mechanism was not established. In the second study, Chen et al. investigated the role of neuronal ferroptosis in intracerebral hemorrhage, given that ferroptosis has been implicated in brain injury after intracerebral hemorrhage [17]. They demonstrated that the ferroptosis inhibitor ferrostatin-1 improved long-term neurobehavioral outcomes and attenuated brain atrophy in a mouse model of intracerebral hemorrhage. This finding implies inhibition of ferroptosis as a potential treatment for stroke. In the third study, García-Yébenes et al. studied the effect of iron overload on hemorrhagic transformation after tissue-type plasminogen activator administration in a mouse model of thromboembolic stroke [18]. Intriguingly, iron overload increased the rate of recanalization after thrombolytic treatment, but reduced extent of viable brain tissue after recanalization and increased the risk of thrombolytic-induced hemorrhagic transformation. This study reinforced the notion that perturbation in iron levels can exacerbate the severity of an event. In the fourth report, Chang et al. investigated the role of macrophages in hematoma clearance and brain recovery after intracerebral hemorrhage [19]. They demonstrated that macrophages, and specific receptors on the cell surface of macrophages that bind to lipids on red cells, are essential for hematoma resolution and functional recovery in a mouse model of intracerebral hemorrhage.
Ischemia/reperfusion injury is another topic of relevance explored recently. Li et al. interrogated the contribution of ferroptosis, a form of regulated cell death characterized by iron-catalyzed lipid peroxidation, to intestinal ischemia/reperfusion injury, a condition with high mortality rate observed in shock, trauma, and other conditions [20]. Using a mouse model of intestinal ischemia/reperfusion injury, they observed that the ferroptosis inhibitors alleviated intestinal ischemia/reperfusion injury and restored intestinal epithelial barrier function. This study highlighted the potential critical role of ferroptosis to ischemia/reperfusion injury and pharmacologic approaches to attenuate severity of injury.
Iron can also play critical roles in wound healing, a key phase in recovery from trauma and other severe, acute events. Using mouse models of wound healing, Wilkinson et al. observed iron accumulation during the remodeling phase of physiologic wound healing, which was delayed in diabetic wounds [21]. Using mice with myeloid cell-specific deficiency in the iron export protein ferroportin, Recalcati et al. investigated the role of iron homeostasis in skin macrophages, cell types that are critical regulators of cutaneous wound healing [22]. Mutant mice exhibited impaired wound healing after incisional skin damage which was not due to leukocyte recruitment or activation. Notably, surrounding stromal cells were iron-deficient and less proliferative with decreased markers of blood vessel formation, suggesting that macrophage ferroportin deficiency impacted stromal cells during wound healing. Using a mouse model of myocardial infarction, Zlatanova et al. demonstrated that macrophage expression of hepcidin, which is abundantly expressed with inflammation and implicated as a regulator of cardiac iron homeostasis, is essential for cardiac remodeling after myocardial infarction [23]. Overall, these studies highlighted that specific molecular determinants of iron homeostasis play pivotal roles in injury repair.
Iron homeostasis also impacts the treatment of trauma with extensive hemorrhage. Substitution of blood products including red blood cells, fresh frozen plasma, thrombocytes, and non-cellular blood products is an active area of research, with various protocols under investigation to establish optimal approaches for substitution [24–27]. In addition, researchers and clinicians aim for artificial blood products. The major challenge for the use of artificial blood components is that these products can provoke a severe inflammatory reaction [28]. (Another challenge is the fact that animal models of trauma can have limited applicability to human patients [29].) The inflammatory response leads to induction of hepcidin which restricts iron release for body stores and dietary iron absorption thereby impairing the body’s recovery from blood loss. This issue is further complicated by the observation that limited iron availability is not the only factor impairing erythropoiesis during trauma. Notably, even if iron is administered, an increase in hemoglobin is not always observed in trauma patients [30,31]. Patient Blood Management programs have been implemented that involve collection of patient`s own blood for re-transfusion to minimize inflammatory reactions [32].
Conclusion:
In this review, we have summarized how models of aberrant iron homeostasis have been employed recently to understand how defects in iron homeostasis impact trauma and other severe, acute events. For some of the studies described above, the investigators have identified specific proteins that play key roles in the body’s predisposition or response to a severe, acute event. For other studies, the investigators have also established how specific proteins contribute mechanistically to the traumatic process at hand. The ultimate goal of all these lines of questioning is to not only identify key molecular factors that contribute to trauma at the level of predisposition, treatment, and recovery but also establish how these factors act at the mechanistic level, so that new targeted treatments can be designed to diminish one’s risk of a traumatic event and increase one’s chances of recovery with minimal long-term deficits.
Key points:
The study of iron homeostasis can inform our understanding of trauma at the level of mechanisms of injury and recovery and potential treatments.
The hormone hepcidin is a central regulator of body iron levels.
Iron deficiency can contribute to trauma by impairing critical organ function and injury repair, while iron excess can contribute to trauma by causing toxicity to tissues and impairing injury repair.
Pharmacologic inhibition of ferroptosis, a novel form of iron-dependent cell death, is beneficial in animal models of cardiac, hepatic, and intestinal injury and intracerebral hemorrhage.
Iron substitution is favorable in iron deficiency anemia, anemia of chronic disease with low ferritin levels indicating iron deficiency, and chronic heart failure, but should be considered carefully in acute inflammatory conditions or hemochromatosis.
Financial support and sponsorship
T.B.B. is supported by NIH grant DK110049. A.U.S. is supported by German Research Foundation (Deutsche Forschungsgemeinschaft) grant STE 1895/4-2 and Innovative Medical Research (Innovative Medizinische Forschung) grants IMF 121617, IMF 221810, and IMF 211901 of the Medical Faculty, University of Muenster, Muenster, Germany. A.U.S. is also sponsored by research grant from Pharmacosmos, Denmark, to perform a single-center, prospective trial on preoperative anemia treatment with intravenous iron. C.A.E. is supported by NIH grants R01 DK072166 and R37 DK054488.
Footnotes
Conflicts of interest
No conflicts of interest.
References and recommended reading
Papers of particular interest, published within the last two years, have been highlighted as:
* of special interest
** of outstanding interest
- 1.Roth M-P, Meynard D, Coppin H: Regulators of hepcidin expression. Vitam Horm 2019, 110:101–129. [DOI] [PubMed] [Google Scholar]
- 2.Wang C-Y, Xiao X, Bayer A, Xu Y, Dev S, Canali S, Nair AV, Masia R, Babitt JL: Ablation of hepatocyte Smad1, Smad5 and Smad8 causes severe tissue iron loading and liver fibrosis in mice. Hepatology 2019, doi: 10.1002/hep.30780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lai D, Teng F, Hammad S, Werle J, Maas T, Teufel A, Muckenthaler MU, Dooley S, Vujić Spasić M: Hepatic Smad7 overexpression causes severe iron overload in mice. Blood 2018, 131:581–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang C-Y, Canali S, Bayer A, Dev S, Agarwal A, Babitt JL: Iron, erythropoietin, and inflammation regulate hepcidin in Bmp2-deficient mice, but serum iron fails to induce hepcidin in Bmp6-deficient mice. Am J Hematol 2019, 94:240–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lim PJ, Duarte TL, Arezes J, Garcia-Santos D, Hamdi A, Pasricha S-R, Armitage AE, Mehta H, Wideman S, Santos AG, et al. : Nrf2 controls iron homeostasis in haemochromatosis and thalassaemia via Bmp6 and hepcidin. Nat Metab 2019, 1:519–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arezes J, Foy N, McHugh K, Sawant A, Quinkert D, Terraube V, Brinth A, Tam M, LaVallie ER, Taylor S, et al. : Erythroferrone inhibits the induction of hepcidin by BMP6. Blood 2018, 132:1473–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fillebeen C, Wilkinson N, Charlebois E, Katsarou A, Wagner J, Pantopoulos K: Hepcidin-mediated hypoferremic response to acute inflammation requires a threshold of Bmp6/Hjv/Smad signaling. Blood 2018, 132:1829–1841. [DOI] [PubMed] [Google Scholar]
- 8.Gallitz I, Lofruthe N, Traeger L, Bäumer N, Hoerr V, Faber C, Kuhlmann T, Müller-Tidow C, Steinbicker AU: Deficiency of the BMP Type I receptor ALK3 partly protects mice from anemia of inflammation. BMC Physiol 2018, 18:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Poli M, Anower-E-Khuda F, Asperti M, Ruzzenenti P, Gryzik M, Denardo A, Gordts PLSM, Arosio P, Esko JD: Hepatic heparan sulfate is a master regulator of hepcidin expression and iron homeostasis in human hepatocytes and mice. J Biol Chem 2019, doi: 10.1074/jbc.RA118.007213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guo S, Jiang S, Epperla N, Ma Y, Maadooliat M, Ye Z, Olson B, Wang M, Kitchner T, Joyce J, et al. : A gene-based recessive diplotype exome scan discovers FGF6, a novel hepcidin-regulating iron-metabolism gene. Blood 2019, 133:1888–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chung YJ, Luo A, Park KC, Loonat AA, Lakhal-Littleton S, Robbins PA, Swietach P: Iron-deficiency anemia reduces cardiac contraction by downregulating RyR2 channels and suppressing SERCA pump activity. JCI Insight 2019, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lakhal-Littleton S, Crosby A, Frise MC, Mohammad G, Carr CA, Loick PAM, Robbins PA: Intracellular iron deficiency in pulmonary arterial smooth muscle cells induces pulmonary arterial hypertension in mice. Proc Natl Acad Sci USA 2019, doi: 10.1073/pnas.1822010116. [DOI] [PMC free article] [PubMed] [Google Scholar]; **This report demonstrates that iron levels in a discrete cell type can have a profound influence on organ function.
- 13.Yu Q, Tai Y-Y, Tang Y, Zhao J, Negi V, Culley MK, Pilli J, Sun W, Brugger K, Mayr J, et al. : BOLA (BolA Family Member 3) Deficiency Controls Endothelial Metabolism and Glycine Homeostasis in Pulmonary Hypertension. Circulation 2019, 139:2238–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fang X, Wang H, Han D, Xie E, Yang X, Wei J, Gu S, Gao F, Zhu N, Yin X, et al. : Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci USA 2019, 116:2672–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]; *This study demonstrates that inhibitors of ferroptosis, a form of iron-dependent cell death, should be considered in prevention and treatment of cardiomyopathies.
- 15.Zhabyeyev P, Das SK, Basu R, Shen M, Patel VB, Kassiri Z, Oudit GY: TIMP3 deficiency exacerbates iron overload-mediated cardiomyopathy and liver disease. Am J Physiol Heart Circ Physiol 2018, 314:H978–H990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Young G-H, Tang S-C, Wu V-C, Wang K-C, Nong J-Y, Huang P-Y, Hu C-J, Chiou H-Y, Jeng J-S, Hsu CY: The functional role of hemojuvelin in acute ischemic stroke. J Cereb Blood Flow Metab 2019, doi: 10.1177/0271678X19861448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen B, Chen Z, Liu M, Gao X, Cheng Y, Wei Y, Wu Z, Cui D, Shang H: Inhibition of neuronal ferroptosis in the acute phase of intracerebral hemorrhage shows long-term cerebroprotective effects. Brain Res Bull 2019, doi: 10.1016/j.brainresbull.2019.08.013. [DOI] [PubMed] [Google Scholar]
- 18.García-Yébenes I, García-Culebras A, Peña-Martínez C, Fernández-López D, Díaz-Guzmán J, Negredo P, Avendaño C, Castellanos M, Gasull T, Dávalos A, et al. : Iron Overload Exacerbates the Risk of Hemorrhagic Transformation After tPA (Tissue-Type Plasminogen Activator) Administration in Thromboembolic Stroke Mice. Stroke 2018, 49:2163–2172. [DOI] [PubMed] [Google Scholar]
- 19.Chang C-F, Goods BA, Askenase MH, Hammond MD, Renfroe SC, Steinschneider AF, Landreneau MJ, Ai Y, Beatty HE, da Costa LHA, et al. : Erythrocyte efferocytosis modulates macrophages towards recovery after intracerebral hemorrhage. J Clin Invest 2018, 128:607–624. [DOI] [PMC free article] [PubMed] [Google Scholar]; **This study dissects the molecular mechanisms by which macrophages contribute to recovery after intracerebral hemorrhage and suggests a possible novel approach for treatment based upon their findings.
- 20.Li Y, Feng D, Wang Z, Zhao Y, Sun R, Tian D, Liu D, Zhang F, Ning S, Yao J, et al. : Ischemia-induced ACSL4 activation contributes to ferroptosis-mediated tissue injury in intestinal ischemia/reperfusion. Cell Death Differ 2019, doi: 10.1038/s41418-019-0299-4. [DOI] [PMC free article] [PubMed] [Google Scholar]; **This work demonstrates that inhibition of ferroptosis should be considered for prevention and treatment of intestinal ischemia/reperfusion.
- 21.Wilkinson HN, Upson SE, Banyard KL, Knight R, Mace KA, Hardman MJ: Reduced iron in diabetic wounds: An oxidative stress-dependent role for STEAP3 in extracellular matrix deposition and remodelling. J Invest Dermatol 2019, doi: 10.1016/j.jid.2019.05.014. [DOI] [PubMed] [Google Scholar]
- 22.Recalcati S, Gammella E, Buratti P, Doni A, Anselmo A, Locati M, Cairo G: Macrophage ferroportin is essential for stromal cell proliferation in wound healing. Haematologica 2019, 104:47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]; *This work indicates that iron plays a critical and underappreciated role in wound repair.
- 23.Zlatanova I, Pinto C, Bonnin P, Mathieu JRR, Bakker W, Vilar J, Lemitre M, Voehringer D, Vaulont S, Peyssonnaux C, et al. : Iron Regulator Hepcidin Impairs Macrophage-Dependent Cardiac Repair After Injury. Circulation 2019, 139:1530–1547. [DOI] [PubMed] [Google Scholar]
- 24.Unruh M, Reyes J, Helmer SD, Haan JM: An evaluation of blood product utilization rates with massive transfusion protocol: Before and after thromboelastography (TEG) use in trauma. Am J Surg 2019, doi: 10.1016/j.amjsurg.2019.08.027. [DOI] [PubMed] [Google Scholar]
- 25.Girish A, Sekhon U, Sen Gupta A: Bioinspired artificial platelets for transfusion applications in traumatic hemorrhage. Transfusion 2019, doi: 10.1111/trf.15543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.da Luz LT, Shah PS, Strauss R, Mohammed AA, D’Empaire PP, Tien H, Nathens AB, Nascimento B: Does the evidence support the importance of high transfusion ratios of plasma and platelets to red blood cells in improving outcomes in severely injured patients: a systematic review and meta-analyses. Transfusion 2019, doi: 10.1111/trf.15540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abuzeid AM, O’Keeffe T: Review of massive transfusion protocols in the injured, bleeding patient. Curr Opin Crit Care 2019, doi: 10.1097/MCC.0000000000000668. [DOI] [PubMed] [Google Scholar]
- 28.Ferenz KB, Steinbicker AU: Artificial Oxygen Carriers-Past, Present, and Future-a Review of the Most Innovative and Clinically Relevant Concepts. J Pharmacol Exp Ther 2019, 369:300–310. [DOI] [PubMed] [Google Scholar]
- 29.Weber B, Lackner I, Haffner-Luntzer M, Palmer A, Pressmar J, Scharffetter-Kochanek K, Knöll B, Schrezenemeier H, Relja B, Kalbitz M: Modeling trauma in rats: similarities to humans and potential pitfalls to consider. J Transl Med 2019, 17:305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gómez-Ramírez S, Maldonado-Ruiz MÁ, Campos-Garrigues A, Herrera A, Muñoz M: Short-term perioperative iron in major orthopedic surgery: state of the art. Vox Sang 2019, 114:3–16. [DOI] [PubMed] [Google Scholar]
- 31.Peters F, Ellermann I, Steinbicker AU: Intravenous Iron for Treatment of Anemia in the 3 Perisurgical Phases: A Review and Analysis of the Current Literature. Anesth Analg 2018, 126:1268–1282. [DOI] [PubMed] [Google Scholar]
- 32.Mueller MM, Van Remoortel H, Meybohm P, Aranko K, Aubron C, Burger R, Carson JL, Cichutek K, De Buck E, Devine D, et al. : Patient Blood Management: Recommendations From the 2018 Frankfurt Consensus Conference. JAMA 2019, 321:983–997. [DOI] [PubMed] [Google Scholar]