

SUMMARY
Bacterial CRISPR-Cas systems employ RNA-guided nucleases to destroy phage (viral) DNA. Phages, in turn, have evolved diverse “anti-CRISPR” proteins (Acrs) to counteract acquired immunity. In Listeria monocytogenes, prophages encode 2–3 distinct anti-Cas9 proteins, with acrIIA1 always present. However, the significance of AcrIIA1s pervasiveness and its mechanism are unknown. Here, we report that AcrIIA1 binds with high affinity to Cas9 via the catalytic HNH domain. During lysogeny in Listeria, AcrIIA1 triggers Cas9 degradation, but during lytic infection, AcrIIA1 fails to block Cas9 due to its multi-step inactivation mechanism. Thus, phages require an additional Acr that rapidly binds and inactivates Cas9. AcrIIA1 also uniquely inhibits a highly-diverged Cas9 found in Listeria (similar to SauCas9) and Type II-C Cas9s, likely due to Cas9 HNH domain conservation. In summary, Listeria phages inactivate Cas9 in lytic growth using variable, narrow-spectrum inhibitors, while the broad-spectrum AcrIIA1 stimulates Cas9 degradation for protection of the lysogenic genome.
Graphical Abstract
eTOC
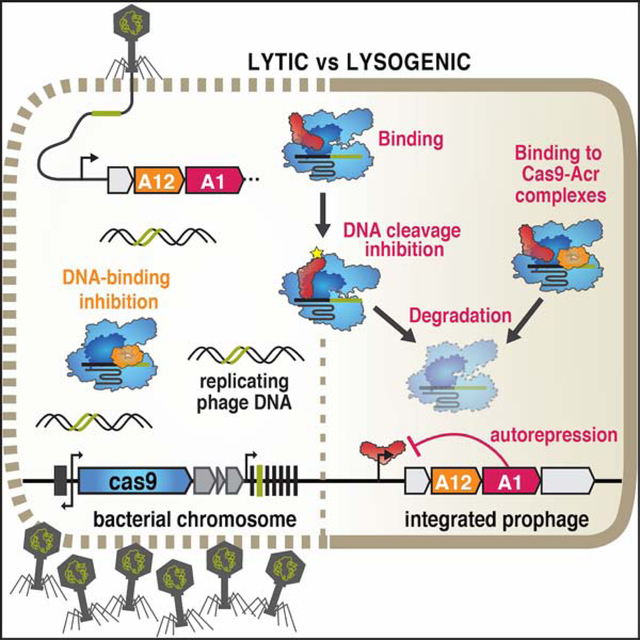
Bacteriophages inactivate CRISPR-Cas immunity by encoding “anti-CRISPR” proteins. Osuna et al. reveal that a protein commonly encoded by Listeria phages, AcrIIA1, directly binds to the Cas9 HNH domain and stimulates its degradation to stabilize the lysogenic state, while the phages use an independent Acr protein for lytic replication.
INTRODUCTION
All cells must combat viral infections to survive. Bacteria have evolved innate and adaptive defense mechanisms against bacterial viruses (phages), which constantly pose a risk of infection. One such defense mechanism is CRISPR-Cas, a common and diverse adaptive immune system in prokaryotes that encompasses two distinct classes and six types (I-VI) (Koonin et al., 2017; Makarova et al., 2015). The CRISPR array maintains a genetic record of past viral infections with phage DNA fragments (spacers) retained between clustered regularly interspaced short palindromic repeats (CRISPR) (Mojica et al., 2005). These phage-derived spacers are transcribed into CRISPR RNAs (crRNAs) that complex with Cas nucleases to guide the sequence-specific destruction of invading nucleic acids (Brouns et al., 2008; Garneau et al., 2010). The CRISPR-associated (cas) genes typically neighbor the CRISPR array and encode proteins that facilitate spacer acquisition into the CRISPR array (Nuñez et al., 2014; Yosef et al., 2012), generate mature crRNAs (Deltcheva et al., 2011; Haurwitz et al., 2010), and cleave invading genomes (Garneau et al., 2010).
To counteract bacterial immunity, phages have evolved multiple mechanisms of CRISPR-Cas evasion (Borges et al., 2017). Phage-encoded anti-CRISPR proteins have been shown to directly inhibit the type I-C, I-D, I-E, I-F, II-A, II-C, III-B, and V-A CRISPR-Cas systems (Hwang and Maxwell, 2019; Trasanidou et al., 2019), and they all have distinct protein sequences, structures, and mechanisms. Some anti-CRISPRs such as AcrIIA2 and AcrIIA4, encoded by Listeria phages, block CRISPR-Cas target DNA binding by steric occlusion and DNA mimicry (Bondy-Denomy et al., 2015; Dong et al., 2017; Jiang et al., 2019; Liu et al., 2019), while others interfere with guide-RNA loading (Thavalingam et al., 2019; Zhu et al., 2019), induce effector dimerization (Fuchsbauer et al., 2019; Harrington et al., 2017; Zhu et al., 2019), or prevent DNA cleavage by interacting with the catalytic domains of Cas nucleases (Bondy-Denomy et al., 2015; Harrington et al., 2017). Type II CRISPR-Cas systems have been widely investigated for genome editing applications. However, few studies have examined Cas9-anti-CRISPR interactions in the natural context of phage-bacteria warfare (Hynes et al., 2017, 2018).
In the lytic cycle, phage replication causes host cell lysis, whereas in lysogeny, temperate phages integrate into the bacterial chromosome and become prophages. The bacterial host and prophage replicate together during lysogeny and prophages can contribute novel genes that provide fitness benefits or even serve as regulatory switches (Argov et al., 2017; Bondy-Denomy et al., 2016; Feiner et al., 2015; Rabinovich et al., 2012). In Listeria monocytogenes, prophages inactivate CRISPR-Cas9 through the expression of anti-CRISPR proteins (Rauch et al., 2017). In lysogens with CRISPR arrays encoding spacers that target the prophage (i.e. self-targeting), anti-CRISPRs are essential for host and prophage survival. However, whether anti-CRISPRs play distinct roles during lysogeny or lytic growth when expressed by temperate phages is unknown.
Here, we show that the Listeria phage protein AcrIIA1 selectively triggers degradation of catalytically active Cas9, through a direct interaction between the AcrIIA1CTD (C-terminal domain) unstructured loop and Cas9 HNH domain. AcrIIA1 is sufficient to prevent CRISPR-targeting of prophages, but is ineffective during lytic replication due to its multi-step Cas9 inactivation mechanism. This latter property necessitates the co-existence of AcrIIA1 with an anti-CRISPR (e.g. AcrIIA2, AcrIIA4, or AcrIIA12, identified here) that rapidly binds and simultaneously blocks Cas9 during lytic infection.
RESULTS
AcrIIA1 specifically induces degradation of catalytically active Cas9
To determine the AcrIIA1 mechanism of action, we first attempted to immunoprecipitate Cas9 from L. monocytogenes (Lmo10403s) strains where AcrIIA1 was expressed from one of three prophages (ΦA006, ΦA118, and ΦJ0161a). Surprisingly, upon immunoblotting for Cas9 protein, we observed highly reduced Cas9 levels in these lysogens (Figure 1A, top). Cas9 transcriptional and translational reporters revealed that transcript levels were unaffected, while the protein reporter levels decreased by ~70% (Figure 1A, bottom). RT-qPCR experiments confirmed cas9 mRNA levels were unaffected in each lysogen (Figure S1A). AcrIIA1 alone, but not AcrIIA4, was sufficient to mediate decreased Cas9 levels in both the immunoblotting (Figure 1B, top) and reporter assays (Figure 1B, bottom left and S1B). The well-studied orthologue, SpyCas9 (53% amino acid identity to LmoCas9), displayed the same post-transcriptional AcrIIA1-dependent loss of Cas9 when introduced into L. monocytogenes (Figure 1B, bottom right and S1B). To test whether AcrIIA1 stimulates Cas9 degradation post-translationally, we measured the stability of SpyCas9 protein in L. monocytogenes. Upon induction of AcrIIA1, we observed an accelerated decay of SpyCas9 protein in comparison to treatment with a translation inhibitor, gentamicin (Figures 1C and S1C). In contrast, SpyCas9 protein increased over time when AcrIIA1 was not induced, similar to strains expressing AcrIIA4 or lacking an anti-CRISPR (Figures 1C and S1C). Thus, Cas9 degradation is specifically triggered by AcrIIA1 and is not a general consequence of Cas9 inhibition by any anti-CRISPR.
Figure 1. AcrIIA1 Induces Cas9 Degradation in Listeria.
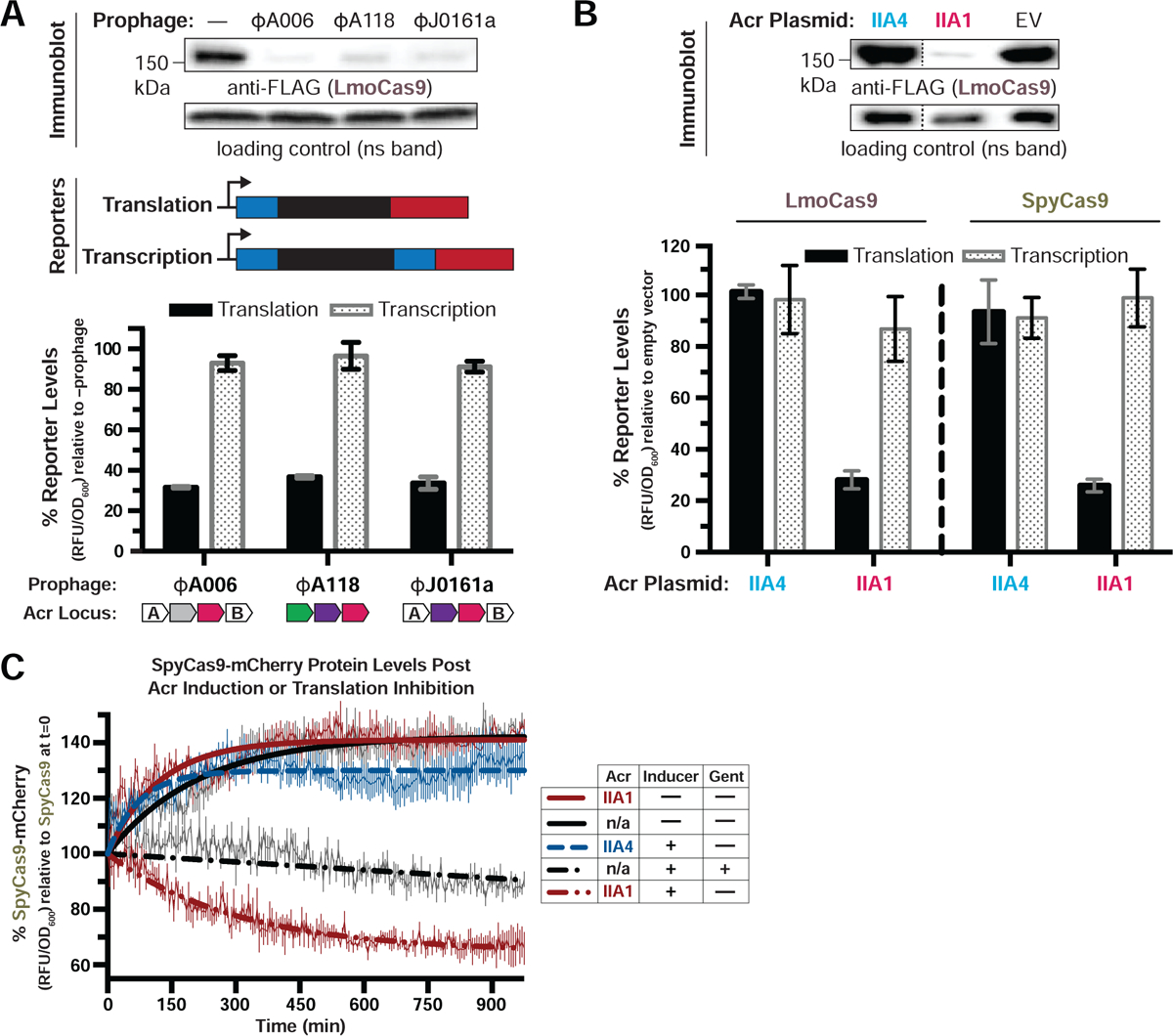
(A-B) Immunoblots detecting FLAG-tagged LmoCas9 protein and a non-specific (ns) protein loading control in Listeria monocytogenes strain 10403s (Lmo10403s) lysogenized with the indicated wild-type prophages (A, top) or Lmo10403s containing Acr-expressing plasmids (B, top). Dashed lines indicate where intervening lanes were removed for clarity (B, top). Representative blots of at least three biological replicates are shown (A and B). Schematics of translational and transcriptional reporters used to measure Lmo or Spy Cas9 protein and mRNA levels in Lmo10403s (A, middle). Cas9 translational (black bars) and transcriptional (gray shaded bars) reporter measurements reflect the mean percentage mCherry relative fluorescence units (RFU/OD600) in the indicated lysogens (A, bottom) or strains with Acr-expressing plasmids (B, bottom) relative to the control strain lacking a prophage (−prophage) (A, bottom) or containing an empty vector (B, bottom). Error bars represent the mean ± SD of at least three biological replicates. (C) SpyCas9-mCherry protein levels post Acr induction or translation inhibition. Lmo10403s expressing SpyCas9-mCherry from the constitutively active pHyper promoter and AcrIIA1 or AcrIIA4 from an inducible promoter were grown to mid-log phase and treated with 100 mM rhamnose to induce Acr expression (dashed lines) or 100 mM glycerol as a neutral carbon source control (solid lines) and 5 μg/mL gentamicin (Gent) to inhibit translation (+) or water (−) as a control. SpyCas9-mCherry protein measurements reflect the mean percentage fluorescence (RFU/OD600) relative to the SpyCas9-mCherry levels at the time translation inhibition was initiated (0 min). Error bars (vertical lines) represent the mean ± SD of at least three biological replicates. Data were fitted by nonlinear regression to generate best-fit decay curves. See Figure S1C for additional controls and S1B for data showing tight repression of the pRha promoter under non-inducing conditions. Note: Lmo doubling time is significantly slower in LB media containing glycerol and/or rhamnose carbon sources (Fieseler et al., 2012).
Given that AcrIIA1 induces Cas9 degradation, we expected that this anti-CRISPR would inhibit catalytically-dead Cas9 (dCas9) in a Listeria CRISPRi assay. However, we observed that AcrIIA1 did not inhibit Lmo- or Spy- dCas9 engineered to repress RFP expression (Figures 2A and S2A), but did inhibit active Cas9 in an isogenic self-targeting strain (Figure 2B). Consistent with these findings, lysogens expressing AcrIIA1 or AcrIIA4 alone or together also revealed no significant decrease in dCas9 levels, whereas active Cas9 protein diminished by ~70% in all AcrIIA1-expressing lysogens (Figures 2C, S2B and S2C). Therefore, AcrIIA1 has a mechanism to detect catalytically active Cas9 protein and induce its degradation.
Figure 2. AcrIIA1 Selectively Binds Catalytically Active Cas9 to Trigger its Degradation.
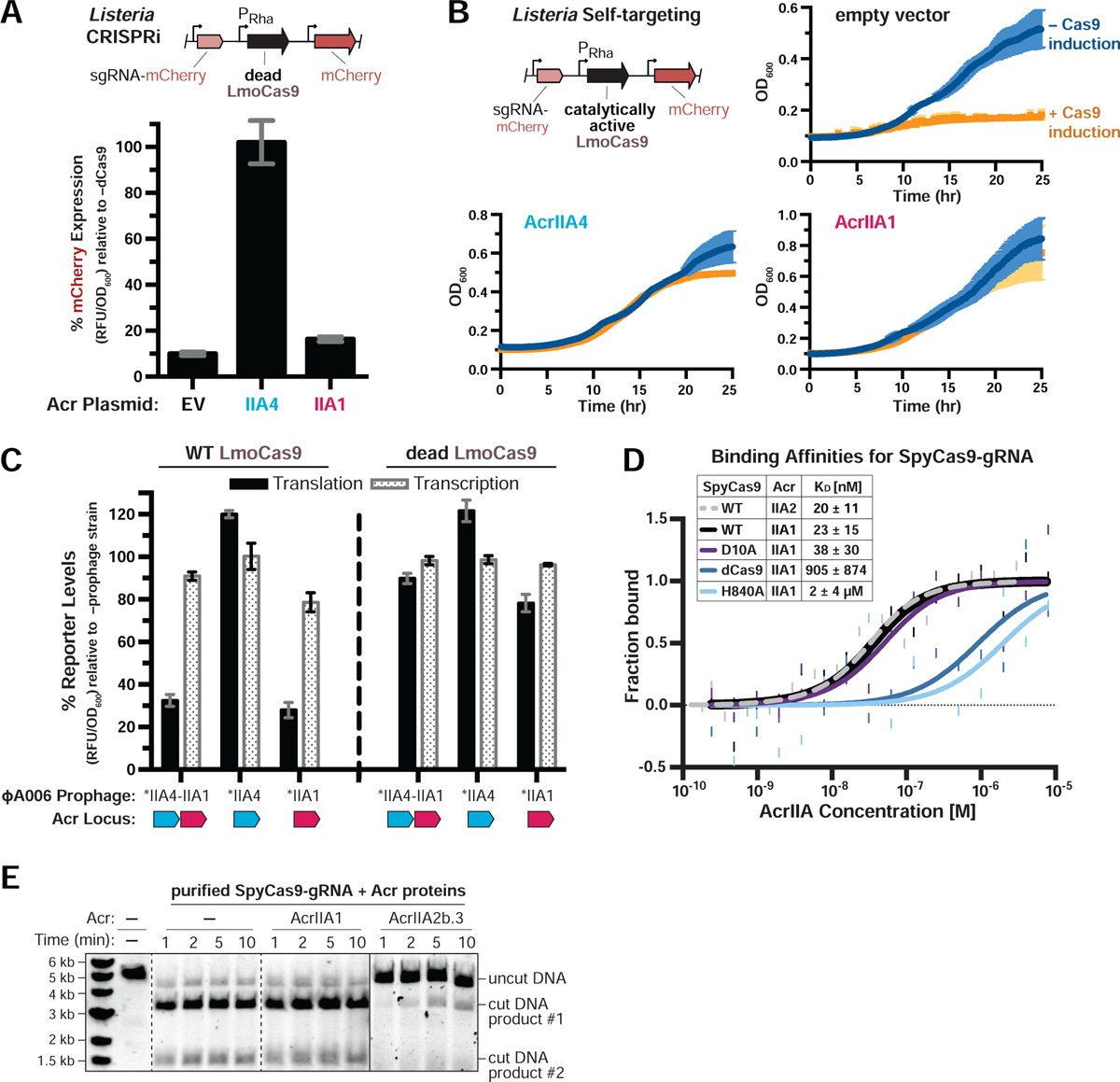
(A-B) Acr-mediated inhibition of CRISPRi (A) or self-targeting (B). Lmo10403s contains chromosomally-integrated constructs expressing dead (A) or catalytically active (B) LmoCas9 from the inducible pRha-promoter and sgRNA that targets the pHelp-promoter driving mCherry expression. For CRISPRi, mCherry expression measurements reflect the mean percentage fluorescence (RFU/OD600) in deadCas9-induced cells relative to uninduced controls (−dCas9) of three biological replicates ± SD (error bars) (A). For self-targeting, bacterial growth was monitored after LmoCas9 induction (orange lines) or no induction (blue lines) and data are displayed as the mean OD600 of three biological replicates ± SD (error bars) (B). See Figure S2A for CRISPRi data with Lmo10403s expressing deadSpyCas9. (C) Translational (black bars) and transcriptional (gray shaded bars) reporter levels of catalytically active (left) and dead LmoCas9 (right) in Lmo10403s lysogenized with engineered isogenic ΦA006 prophages. Cas9 reporter measurements reflect the mean percentage mCherry relative fluorescence units (RFU/OD600) in the indicated lysogens relative to the control strain lacking a prophage (−prophage). Error bars represent the mean ± SD of at least three biological replicates. Asterisk (*) denotes genes containing the native orfA RBS (strong) in ΦA006 and unmarked genes contain their native RBS. See Figure S2B for equivalent data with Lmo10403s expressing SpyCas9. (D) Quantification of the binding affinities (KD; boxed inset) of Acr proteins for WT, catalytically dead (dCas9), or nickase (D10A or H840A) SpyCas9-gRNA complexes using microscale thermophoresis. Data shown are representative of three independent experiments. See Figure S2D for additional controls with AcrIIA2b.3 (IIA2, dashed line). (E) Time course of SpyCas9 DNA cleavage reactions in the presence of Acr proteins that were recombinantly purified from E. coli. Dashed lines indicate where intervening lanes were removed for clarity. Solid lines indicate a separate image. Data shown are representative of three independent experiments.
AcrIIA1 binds directly to Cas9 via the catalytic HNH domain
Due to the discrepant inhibition of Cas9 and dCas9 by AcrIIA1, we assessed the ability of AcrIIA1 to bind these proteins in vitro using microscale thermophoresis (MST). AcrIIA1 and SpyCas9-gRNA (SpyCas9 was used because LmoCas9 was insoluble) interacted with high affinity (KD = 23 ± 15 nM), comparable to AcrIIA2b.3 (KD = 20 ± 11 nM, Figure 2D), a well-characterized Cas9-interactor (Jiang et al., 2019). AcrIIA1 co-purified with RNA, as seen previously (Ka et al., 2018) however, benzonase treatment to remove nucleic acids did not impact the binding affinity (data not shown). Only two residues differ between catalytically active Cas9 and dCas9 (D10A and H840A). AcrIIA1 binding to dCas9 and Cas9(H840A) was reduced ~40-fold (KD = 905 ± 874 nM) and ~80-fold (KD = 2 ± 4 μM) respectively, whereas binding to Cas9(D10A) (KD = 38 ± 30 nM) was similar to wild-type Cas9 (Figure 2D). AcrIIA2b.3, which binds the PAM-interacting domain, displayed no difference in binding affinity to the four Cas9 variants (KD = 18 – 38 nM, Figure S2D). These results were consistent with an in vitro pull-down assay using the four Cas9 variants and AcrIIA1 or AcrIIA2b.3 (Figure S2E). Additionally, AcrIIA1 interacted well with ApoCas9 (KD = 18 ± 7 nM, Figure S2F), Cas9-gRNA prebound to a DNA substrate (KD = 31 ± 22 nM, Figure S2G), and Cas9-gRNA prebound to AcrIIA2b.3 (KD = 24 ± 16 nM, Figure S2H), demonstrating a unique binding mechanism for AcrIIA1. Interestingly, AcrIIA1 binding in vitro was not sufficient to degrade Cas9 or destabilize it when subjected to limited proteolysis (Figure S2I), nor did AcrIIA1 inhibit DNA cleavage by Cas9 (Figure 2E). Thus, we conclude that AcrIIA1 directly interacts with the catalytic Cas9 HNH domain (where H840 resides), but requires an additional stimulus, presumably provided by the L. monocytogenes cellular environment, that activates Cas9 degradation (see Discussion).
AcrIIA1 protects CRISPR-targeted prophages but fails during lytic replication
We next sought to determine when AcrIIA1 anti-CRISPR activity manifests during the phage life cycle. Isogenic ΦA006 phages were engineered to encode no anti-CRISPR, acrIIA1, acrIIA4, or acrIIA1 and acrIIA4 together, and assessed along with wild-type (WT) phage, during lytic infection (Figure 3A) and lysogenic replication (Figure 3B). When infecting Lmo10403s expressing Cas9 and a native ΦA006-targeting spacer sequence, phages encoding only acrIIA1 surprisingly failed to replicate, similar to a Δacr phage (efficiency of plaquing, EOP ≤ 3×10−5, Figures 3A and S3A). Phages encoding acrIIA4 replicated well (EOP = 0.1 – 0.7, depending on acrIIA4 expression strength), similar to WT ΦA006 (EOP ≥ 0.7), with no added benefit derived from acrIIA1 (Figures 3A and S3A). In contrast, after the establishment of lysogeny, acrIIA1 was sufficient for prophage survival. ΦA006 prophages encoding only acrIIA1 completely prevented self-targeting when WT Cas9 was induced, whereas lysogens lacking an anti-CRISPR (Δacr) died (Figure 3B). These data suggest that AcrIIA1 is optimal for lysogeny, but ineffective during lytic infection (where phage DNA is rapidly cleaved), perhaps due to the additional rate-limiting stimulus required to induce degradation after AcrIIA1 binds Cas9 (see Discussion).
Figure 3. AcrIIA1 Inhibits Cas9 to Protect Prophages During Lysogeny.
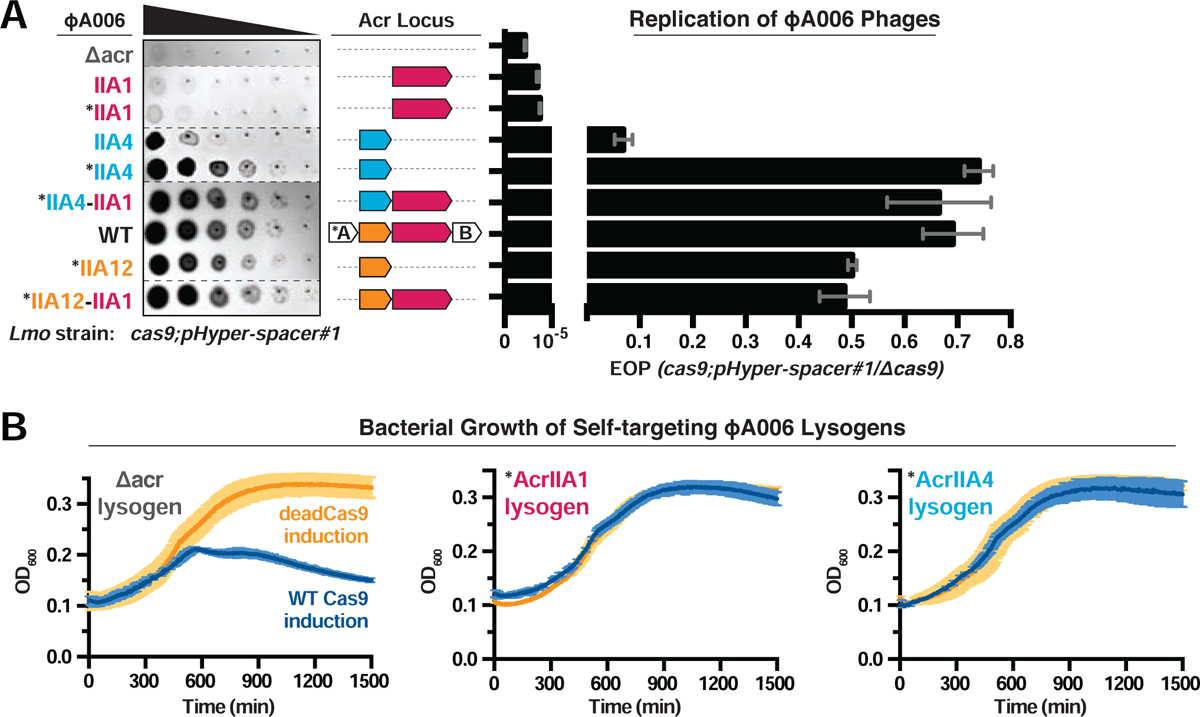
(A) Left: Representative image of plaquing assays where isogenic ΦA006 phages are titrated in ten-fold serial dilutions (black spots) on a lawn of Lmo10403s (gray background). Dashed lines indicate where intervening rows were removed for clarity. Right: Efficiency of plaquing (EOP) of isogenic ΦA006 phages expressing the indicated Acrs on Lmo10403s. Plaque forming units (PFUs) were quantified on Lmo10403s overexpressing the first spacer in the native CRISPR array that targets ΦA006 (cas9;pHyper-spacer#1) and normalized to the number of PFUs measured on a non-targeting Lmo10403s-derived strain (Δcas9). Data are displayed as the mean EOP of at least three biological replicates ± SD (error bars). See Figure S3A for EOP measurements of additional ΦA006 phages. (B) Bacterial growth curves of self-targeting Lmo10403s::ΦA006 isogenic lysogens expressing the indicated Acrs and rhamnose-inducible WT or dead LmoCas9. WT LmoCas9 (blue lines) is lethal in an Acr-deficient (Δacr) strain because the Lmo10403s CRISPR array contains a spacer targeting the ΦA006 prophage integrated in the bacterial genome. Data are displayed as the mean OD600 of at least three biological replicates ± SD (error bars) as a function of time (min). Asterisk (*) indicates the native orfA RBS (strong) in ΦA006 was used for Acr expression.
The inability of AcrIIA1 to independently inhibit Cas9 during lytic infection, suggests that phages need additional Cas9 inhibitors to establish lysogeny. In 119 Listeria prophage genomes analyzed, 77% encode acrIIA1 with at least one additional acrIIA gene (i.e. acrIIA2-A4), 13% possess acrIIA1 without a known acrIIA neighbor (including WT ΦA006), and 10% encode orfD (a distant acrIIA1 orthologue) (Rauch et al., 2017). The WT ΦA006 phage, which has acrIIA1 and no other known acr, replicated far better (EOP ≥ 0.7) than an engineered phage encoding acrIIA1 alone, suggesting an additional Cas9 inhibitor in this phage (Figures 3A and S3A). Engineered phages encoding the gene adjacent to acrIIA1 restored phage lytic replication (EOP ≥ 0.5, Figures 3A and S3A) and revealed anti-CRISPR, AcrIIA12, which also inhibited Lmo (but not Spy) dCas9-based CRISPRi (Figure S3B). Notably, we observed the presence of acrIIA12 in every acr locus previously reported to encode only acrIIA1, indicating that phages do not encode acrIIA1 alone. Therefore, Listeria prophages most commonly encode acrIIA1, which triggers Cas9 degradation to ensure stable lysogeny, in combination with a Cas9 interactor that blocks DNA binding (AcrIIA2, AcrIIA4, or AcrIIA12) for successful lytic replication.
AcrIIA1 utilizes an unstructured C-terminal loop to inactivate Cas9
The AcrIIA1 crystal structure revealed a two-domain architecture, with a helix-turn-helix (HTH)-containing AcrIIA1NTD similar to known transcriptional repressors and an extended AcrIIA1CTD of unknown function (Ka et al., 2018). Surprisingly, AcrIIA1CTD was both necessary and sufficient for anti-CRISPR function, protecting Listeria cells from self-targeting (Figure S3C) and inducing Cas9 protein degradation (Figures S3D and S3E). The AcrIIA1NTD displayed no evidence of CRISPR-Cas9 inhibition or regulation (Figures S3C and S3D), and is instead required for autoregulation of the phage anti-CRISPR locus (see companion manuscript; Osuna et al., 2020b). To further characterize the AcrIIA1 mechanism of Cas9 inactivation, AcrIIA1 homologs were tested in our P. aeruginosa anti-SpyCas9 screening platform (Jiang et al., 2019) and sequence conservation was used to guide the construction of mutants. First, we confirmed that similar to in Listeria, AcrIIA1ΦA006 displayed robust inhibition of active Cas9 in P. aeruginosa (Figure S4A, left) and did not interfere with dCas9 (Figure S4A, right). The AcrIIA1 homologs tested for Cas9 inhibition were identified in mobile genetic elements of Listeria, Enterococcus, Lactobacillus, and Leuconostoc species, with protein sequence identity ranging from 22% to 77% (Figure 4A). As expected, only homologs with conserved CTDs displayed anti-SpyCas9 activity, whereas the three proteins with highly diverged CTDs (including orfD) did not (Figures 4B and S4B). Alanine scanning mutagenesis of amino acids conserved across the AcrIIA1 homologs identified a stretch of aromatic and charged residues in an unstructured region of the AcrIIA1CTD (P112 to R117) that were required for complete anti-CRISPR activity (Figures 4C and S4B). Expression levels of each mutant protein were unperturbed relative to WT AcrIIA1 (Figure S4C). Notably, the AcrIIA1(F115A) and AcrIIA1(T114A/F115A) mutants completely lost anti-CRISPR function (Figures 4C and S4B), and very weakly interacted with Cas9 in vitro (Figures S4D). In Listeria, these same mutants failed to protect cells from genomic self-targeting (Figure S3C) and completely (T114A/F115A) or partially (F115A) lost the ability to induce Cas9 protein degradation (Figure S3E). Thus, AcrIIA1 uses highly conserved residues in its CTD to interact with the Cas9 HNH domain and trigger Cas9 protein degradation in Listeria.
Figure 4: AcrIIA1 Uses its C-terminal Domain to Lock Cas9 in an Inhibited State.
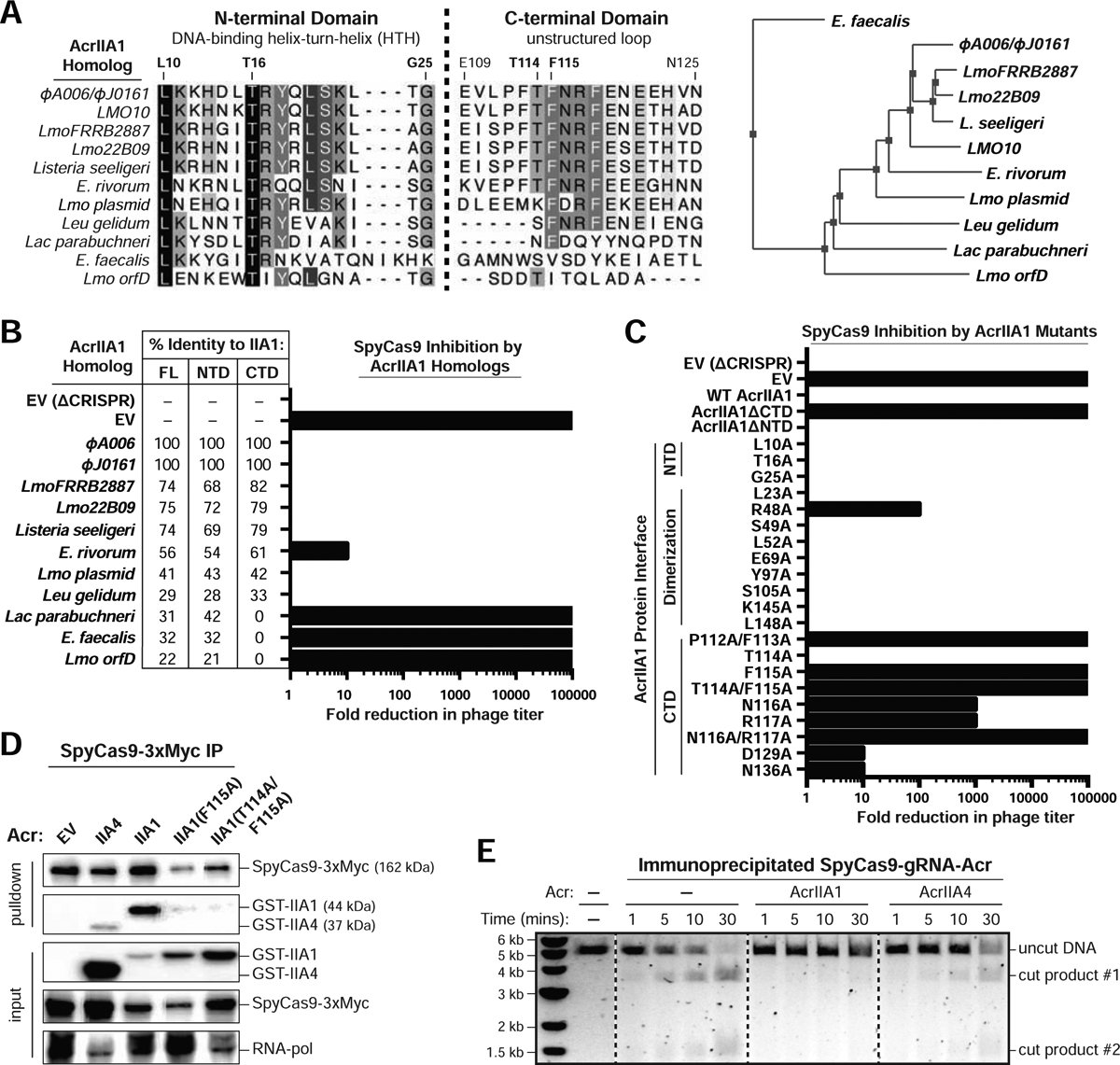
(A) Left: Alignment of AcrIIA1 homolog protein sequences denoting key residues. Right: Phylogenetic tree of the protein sequences of AcrIIA1 homologs. See companion manuscript for a complete alignment of the AcrIIA1 homolog protein sequences (Osuna et al., 2020b). (B-C) Fold reduction in phage titer in response to SpyCas9 targeting of a P. aeruginosa DMS3m-like phage in the presence of AcrIIA1 homologs (B) or mutants (C). The percent protein sequence identities of each homolog to the full-length (FL) or domains (NTD or CTD) of AcrIIA1ΦA006 are listed in (B). The displayed fold reductions in phage titer were qualitatively determined by examining three biological replicates of each phage-plaquing experiment. See Figure S4B for representative pictures of the corresponding phage-plaquing experiments. (D) Immunoblots detecting GST-tagged anti-CRISPR proteins that co-immunoprecipitated with Myc-tagged SpyCas9 in a P. aeruginosa strain heterologously expressing Type II-A SpyCas9-gRNA and the indicated Acrs. For input samples, one-hundredth lysate volume was analyzed to verify tagged protein expression and RNA-polymerase was used as a loading control. Representative blots of three biological replicates are shown. See Figure S4E for the reciprocal GST-Acr pulldown. (E) Time course of SpyCas9 DNA cleavage reactions conducted with SpyCas9-gRNA-Acr (or no Acr, −) complexes immunoprecipitated from P. aeruginosa. Dashed lines indicate where intervening lanes were removed for clarity. Data shown are representative of three independent experiments. See Figure S4F for reactions with AcrIIA1 mutants.
Cas9 inactivation by AcrIIA1 is a multi-step process
To determine if AcrIIA1 mutants differentially impact Cas9 protein levels, immunoblots were performed, surprisingly revealing that wild-type AcrIIA1 can inhibit Cas9 in P. aeruginosa without triggering its degradation (Figure S4C). When immunoprecipitated from this heterologous host, Cas9 co-purified with AcrIIA1 or the control AcrIIA4 (Figure 4D) and these complexes failed to cleave DNA (Figure 4E), whereas Cas9-gRNA alone or Cas9 bound to AcrIIA1(F115A) and AcrIIA1(T114A/F115A) was functional (Figures 4E, S4E and S4F). No other proteins stoichiometrically co-immunoprecipitated with Cas9-AcrIIA1 (Figure S4G), suggesting that inhibition in vivo does not require an additional stable protein interactor. Thus, AcrIIA1 inhibits Cas9 using a multi-step mechanism that first involves a high affinity interaction, a subsequent inhibited state locked in by the cellular environment (seen ex vivo in Figure 4E, but not recapitulated in vitro in Figure 2E), and finally Cas9 degradation mediated by an additional Listeria-specific stimulus. The non-native host P. aeruginosa apparently lacks this Cas9-degrading pathway. This multi-step inactivation mechanism likely explains the inefficiency of AcrIIA1 during phage lytic infection and the necessary co-existence with another anti-CRISPR that rapidly binds and directly inactivates Cas9 in a single-step process (a mechanism that is recapitulated in vitro in Figure 2E).
AcrIIA1 is a broad-spectrum Cas9 inhibitor
Listeria species also contain a highly diverged Type II-A Cas9 (LivCas9, 1,078 a.a., Hupfeld et al., 2018), which shares similarities with other small Cas9 proteins (e.g. SauCas9) and Type II-C orthologues (Figure 5A). The AcrIIA1 Cas9 inactivation mechanism, which involves recognition of a highly conserved catalytic residue (H840), suggested it might inhibit diverged Cas9 orthologues. Indeed, AcrIIA1 inhibited LivCas9 in Listeria strains programmed to target phage ΦP35 or ΦA511, while AcrIIA4 and AcrIIA12 failed to inhibit (Figures 5B and S5A). To ascertain the extent of AcrIIA1s spectrum of inhibition, we tested Escherichia coli strains expressing Type II-A, II-B, and II-C Cas9 proteins targeting phage Mu. AcrIIA1 intermediately or completely inhibited four Type II-C (Geo, Cje, Hpa, and Boe) and two Type II-A (Sau and Spy) Cas9s (Figures 5C and S5B). In contrast, AcrIIA2 only weakly inhibited Hpa and SpyCas9, and AcrIIA4 only inactivated SpyCas9 (Figure 5C). Thus, AcrIIA1 displays broad-spectrum activity against diverged Cas9 nucleases, which further explains the utility of AcrIIA1 to phages infecting Listeria, where at least two distinct Cas9 orthologues are encountered.
Figure 5. AcrIIA1 is a Broad-Spectrum Cas9 Inhibitor.
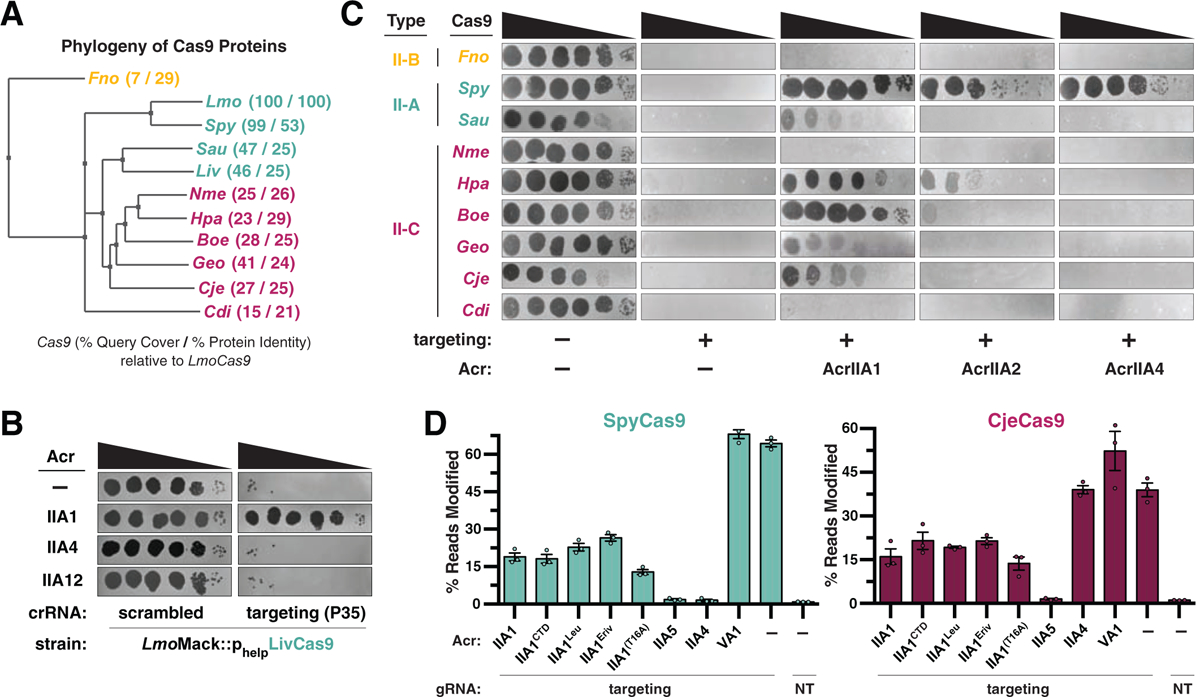
(A) Phylogenetic tree of the protein sequences of Cas9 orthologues. The percent query coverage and percent protein sequence identities relative to LmoCas9 are listed in parentheses. Cas9 orthologue names: Francisella novicida (Fno), Listeria monocytogenes (Lmo), Streptococcus pyogenes (Spy), Staphylococcus aureus (Sau), Listeria ivanovii (Liv), Neisseria meningitidis (Nme), Haemophilus parainfluenzae (Hpa), Brackiella oedipodis (Boe), Geobacillus stearothermophilus (Geo), Campylobacter jejuni (Cje), Corynebacterium diphtheriae (Cdi). (B) Plaquing assays where the Listeria phage ΦP35 is titrated in ten-fold serial dilutions (black spots) on lawns of L. monocytogenes Mack (gray background) strains that express chromosomally-integrated LivCas9/tracrRNA and contain pLEB plasmids expressing two components: an Acr or no Acr (−) and a crRNA that targets phage DNA or a scrambled crRNA (non-targeting control). (C) Plaquing assays where the E. coli phage Mu is titrated in ten-fold serial dilutions (black spots) on lawns of E. coli (gray background) expressing the indicated anti-CRISPR proteins and Type II-A, II-B and II-C Cas9-sgRNA programmed to target phage DNA. Representative pictures of at least 3 biological replicates are shown. (D) Gene editing activities of SpyCas9 and CjeCas9 in human cells in the presence of AcrIIA1 variants and orthologues. Control inhibitors (references in Methods): AcrIIA4 selective inhibitor of SpyCas9; AcrIIA5 broad-spectrum Cas9 inhibitor; AcrVA1 Cas12 inhibitor (negative control for Cas9 orthologues). Editing assessed by targeted sequencing. NT indicates a no-sgRNA control condition. Error bars indicate SEM for three independent biological replicates. See Figure S5C for editing experiments with additional Cas9 orthologues.
The robust AcrIIA1 activity observed in various heterologous hosts led us to assess inhibition of Cas9 gene editing in human cells. We employed a deep sequencing-based approach to improve the dynamic range of edit detection, in comparison to our previous GFP-disruption assay (Rauch et al., 2017). HEK 293T cells were co-transfected with plasmids encoding acrIIA1, cas9, and sgRNAs targeting endogenous human sequences and editing efficacy was evaluated after 3 days. AcrIIA1 blocked the gene editing activity of SpyCas9 by 50–70% and of CjeCas9, SauCas9, St3Cas9, and NmeCas9 by 20–40%, whereas AcrIIA4 only inhibited SpyCas9 (Figures 5D and S5C). Titration of anti-CRISPR expression plasmids over a range of concentrations confirmed that AcrIIA1 was a less potent inhibitor than cognate Acrs for SpyCas9 and CjeCas9 (Figure S5D). However, these experiments also revealed that AcrIIA1CTD alone is more effective than full-length AcrIIA1, and as expected the AcrIIA1(T114A/F115A) double mutant was inactive (Figure S5D). Thus, AcrIIA1 inactivates diverse Cas9 orthologues in many heterologous systems, including bacteria (L. monocytogenes, P. aeruginosa, E. coli), yeast (Nakamura et al., 2019), and human cells, providing a genome editing modulator that specifically inhibits active Cas9, but not deadCas9. Future work is needed to enhance its efficiency, however. Our attempts to increase anti-CRISPR function in human cells by weakening DNA interactions with the T16A mutation or AcrIIA1NTD removal (companion manuscript; Osuna et al., 2020b) were only modestly successful (Figures 5D, S5C and S5D). Future engineering of AcrIIA1 could generate a more potent inhibitor, as recently achieved with AcrIIC1 (Mathony et al., 2019).
DISCUSSION
Listeria temperate phages commonly encode the multifunctional AcrIIA1 protein for protection against CRISPR-Cas and autorepression of anti-CRISPR transcription (companion manuscript; Osuna et al., 2020b). The broad-spectrum AcrIIA1 is sufficient for maintaining Cas9 inactivation after lysogeny has been established, but is nonfunctional during lytic growth, likely due to the multi-step inactivation mechanism that first requires Cas9 binding, a second rate-limiting step that leads to inhibition, and lastly, Cas9 degradation. Cas9 has been shown to cleave phage DNA in as little as 2 min post-infection (Garneau et al., 2010). Thus, a distinct anti-Cas9 protein (e.g. AcrIIA2, AcrIIA4, AcrIIA12) that is much narrower in its inhibitory spectrum, but uses a one-step mechanism to rapidly bind and inactivate Cas9, coexists with AcrIIA1 to allow lytic replication and the initial establishment of lysogeny. Although AcrIIA4 and AcrIIA12 also protect CRISPR-targeted prophages, only AcrIIA1 triggers Cas9 degradation. Listeria lysogens were devoid of Cas9 protein even when acrIIA1 was co-encoded with other acrs, supporting that Cas9 degradation is the dominant inactivation mechanism in lysogeny. Given that Cas9 is required for the acquisition of functional new spacers (Heler et al., 2015), eliminating it could also prevent acquisition of lethal self-targeting spacers. Altogether, these data suggest Listeria temperate phages employ a “division of labor” approach, evolving multiple Acrs with distinct Cas9 binding sites and inactivation mechanisms because they synergistically grant unique advantages in the temperate phage life cycle and together ensure long-term stability in lysogeny (see Graphical Abstract).
Much remains to be elucidated about AcrIIA1’s mechanism of action. Given that AcrIIA1 binds both ApoCas9 and gRNA-bound Cas9 and co-purifies with RNA (our observations and Ka et al., 2018), AcrIIA1 may also impact gRNA biogenesis or loading. While previous work (Ka et al., 2018) ruled out direct interactions between AcrIIA1 and CRISPR-Cas nucleic acids in vitro (e.g. tracrRNA, crRNA, tracrRNA:crRNA duplex), AcrIIA1 bound to ApoCas9 may occlude gRNA processing or loading. We were unable to directly test this possibility as recombinantly-purified AcrIIA1 protein lacks inhibition or degradative activity in vitro, even when preincubated with ApoCas9. While we demonstrate that AcrIIA1 triggers degradation of pre-expressed Cas9-gRNA in vivo (Figure 1C), it is possible that AcrIIA1 may also destabilize ApoCas9 directly or by preventing loading, similar to AcrIIC2, a Type II-C inhibitor that blocks gRNA loading (Thavalingam et al., 2019; Zhu et al., 2019).
We also observed many parallels between AcrIIA1 and AcrIIC1, another Type II-C Cas9 inhibitor. AcrIIC1 binds to the Cas9 HNH domain with strong affinity (KD = 6.3 nM; Harrington et al., 2017), but is a rather weak anti-CRISPR in comparison to AcrIIC3–5 (Lee et al., 2018; Mathony et al., 2019). In contrast to the narrow-spectrum DNA binding inhibitors AcrIIC3–5, AcrIIC1 blocks a broad spectrum of Type II-C orthologues by directly binding Cas9 (Apo or gRNA-bound) via the HNH domain (Harrington et al., 2017). Similarly, AcrIIA1 also targets the highly conserved Cas9 HNH domain catalytic site, likely explaining its capacity to inhibit a broad-spectrum of Cas9 orthologues in comparison to the narrow-spectrum DNA binding inhibitors AcrIIA2, AcrIIA4, AcrIIA12. This AcrIIA1 feature provides a unique advantage to Listeria phages, allowing inhibition of a small LivCas9 variant (25% amino acid identity to large LmoCas9) that is also found in L. monocytogenes strains. Thus, Cas9 HNH-domain interactors may tend to be weaker anti-CRISPRs, but they considerably bolster the phage defense arsenal by targeting a highly conserved, and potentially immutable feature amongst bacterial Cas nucleases.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Please direct any requests for further information or reagents to the lead contact, Joseph Bondy-Denomy (joseph.bondy-denomy@ucsf.edu).
Materials Availability
Listeria strains, plasmids, and phages constructed and used in this study are disclosed in Table S2 (Excel spreadsheet). Nuclease, sgRNA, and Acr plasmids used in human cell experiments are disclosed in Table S3 (Excel spreadsheet). Oligonucleotides used in human cell experiments are disclosed in Table S4 (Excel spreadsheet).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Microbe Strains
Listeria monocytogenes strains (10403s) were cultured in brain-heart infusion (BHI) medium at 30°C. All Lmo strains containing pPL2oexL-Rhamnose-inducible constructs were cultured in Luria broth (LB) supplemented with 50–150 mM glycerol (neutral carbon source; no induction/repression) and 0–100 mM rhamnose (inducer) as indicated. To ensure plasmid maintenance in Listeria strains, BHI or LB was supplemented with tetracycline (2 μg/mL) for the pPL2oexL integrated construct or erythromycin (7.5 μg/mL) for pLEB579. Escherichia coli (DH5α, XL1Blue, NEB 10-beta, or NEB Turbo for plasmid maintenance and SM10 for conjugation into Listeria) and Pseudomonas aeruginosa (PAO1) were cultured in LB medium at 37°C. To maintain plasmids, LB was supplemented with chloramphenicol (25 μg/mL) for pPL2oexL in E. coli, erythromycin (250 μg/mL) for pLEB579 in E. coli, gentamicin (30 μg/mL) for pHERD30T in E. coli and P. aeruginosa, or carbenicillin (250 μg/mL for P. aeruginosa, 100 μg/mL for E. coli) for pMMB67HE. For maintaining pHERD30T and pMMB67HE in the same P. aeruginosa strain, media was supplemented with 30 μg/mL gentamicin and 100 μg/mL carbenicillin. Listeria strains, plasmids, and phages constructed and used in this study are listed in Table S2.
Phages
Listeria phages A006, A118, J0161a, and their derivatives were all propagated at 30°C on acrIIA1NTD-expressing L. monocytogenes 10403sϕcure (Δcas9, ΔtRNAArg::pPL2oexL-acrIIA1NTD) to allow optimal lytic growth of phages lacking their own acrIIA1NTD. A511 was propagated on L. ivanovii WSLC 3009 at 30°C and P35 on L. monocytogenes Mack at 20°C. The Pseudomonas DMS3m-like phage (JBD30) was propagated on PAO1 at 37°C. All phages were stored in SM buffer (100 mM NaCl, 8 mM MgSO4•7H2O, 50 mM Tris-HCl pH 7.5, 0.01% (w/v) gelatin), supplemented with 10 mM CaCl2 for Listeria phages, at 4°C.
Human cell lines
Human HEK 293T cells (ATCC) were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% heat-inactivated FBS (HI-FBS) and 1% penicillin/streptomycin. Media supernatant from cell cultures was analyzed monthly for the presence of mycoplasma using MycoAlert PLUS (Lonza).
METHOD DETAILS
Listeria phage tittering
A mixture of 150 μL stationary Listeria culture and 3 mL molten LC top agar (10 g/L tryptone, 5 g/L yeast extract, 10 g/L glucose, 7.5 g/L NaCl, 10 mM CaCl2, 10 mM MgSO4, 0.5% agar) was poured onto a BHI plate (1.5% agar) to generate a bacterial lawn, 3 μL of phage ten-fold serial dilutions were spotted on top, and after 24 hr incubation at 30°C, plate images were collected using the Gel Doc EZ Documentation system (BioRad) and Image Lab (BioRad) software.
Construction of Lmo10403s lysogens
Lysogens were isolated from plaques that emerged after titering phages ϕA006, ϕA118, ϕJ0161a, and their derivatives on a lawn of Lmo10403sϕcureΔcas9 (see “Listeria phage titering”). Lysogeny was confirmed by prophage induction with mitomycin C (0.5 μg/mL) treatment as previously described (Estela et al., 1992) and by PCR amplification and Sanger sequencing of the phage anti-CRISPR locus. All Lmo10403s strains containing prophages were lysogenized and verified prior to introducing additional constructs (integrated pPL2oexL).
Listeria and Pseudomonas strain construction
DNA fragments were PCR-amplified from genomic, plasmid, or synthesized DNA and cloned by Gibson Assembly into Listeria plasmids: episomal pLEB579 (Beasley et al., 2004) or the pPL2oexL single-copy integrating plasmid derived from pPL2 (Lauer et al., 2002) or P. aeruginosa plasmids: pMMB67HE or pHERD30T. To generate all Listeria strains, pPL2oexL plasmids were conjugated (Lauer et al., 2002; Simon et al., 1983) and pLEB579 plasmids were electroporated (Hupfeld et al., 2018; Park and Stewart, 1990) into Lmo10403s. For all Pseudomonas strains, plasmids were electroporated into PAO1 (Choi et al., 2006).
Listeria protein samples for immunoblotting
Saturated overnight cultures of Lmo10403s strains overexpressing FLAG-tagged Cas9 (Δcas9, ΔtRNAArg::pPL2oexL-LmoCas9–6xHis-FLAG) were diluted 1:10 in BHI with appropriate antibiotic selection (see “microbes”), grown to log phase (OD600 0.2–0.6), harvested by centrifugation at 8000 g for 5 min at 4°C, and lysed by bead-beating or lysozyme treatment. For bead-beating: 4 OD600 units of each culture were harvested, cell pellets were resuspended in 500 μL ice cold lysis buffer (50 mM Tris-HCl pH 8.0, 650 mM NaCl, 10 mM MgCl2, 10% glycerol, 1x cOmplete mini EDTA-free protease inhibitor cocktail [Roche]), combined with ~150 μL 0.1 mm glass beads, and vortexed for 1 hr at 4°C. Cell debris was cleared by centrifugation at 21000 g for 5 min at 4°C and supernatant was mixed with one-third volume 4X Laemmli Sample Buffer (Bio-Rad). For lysozyme lysis: 1.6 OD600 units were harvested, cell pellets were resuspended in 200 μL of TE buffer supplemented with 2.5 mg/mL lysozyme and 1x cOmplete mini EDTA-free protease inhibitor cocktail (Roche), samples were incubated at 37°C for 30 min, quenched with one-third volume of 4X Laemmli Sample Buffer (Bio-Rad), and boiled for 5 min at 95°C.
Immunoblotting
Protein samples were separated by SDS-PAGE using 4–20% Mini-PROTEAN TGX gels (Bio-Rad) and transferred in 1X Tris/Glycine Buffer onto 0.22 micron PVDF membrane (Bio-Rad). Blots were probed with the following antibodies diluted 1:5000 in 1X TBS-T containing 5% nonfat dry milk: rabbit anti-FLAG (Sigma-Aldrich Cat# F7425, RRID:AB_439687), mouse anti-FLAG (Sigma-Aldrich Cat# F1804, RRID:AB_262044), mouse anti-Myc (Cell Signaling Technology Cat# 2276, RRID:AB_331783), rabbit anti-GST (Cell Signaling Technology Cat# 2625, RRID:AB_490796), mouse anti-E.coli RNA polymerase β (BioLegend Cat# 663903, RRID:AB_2564524), HRP-conjugated goat anti-Rabbit IgG (Bio-Rad Cat# 170–6515, RRID:AB_11125142), and HRP-conjugated goat anti-mouse IgG (Santa Cruz Biotechnology Cat# sc-2005, RRID:AB_631736). Blots were developed using Clarity ECL Western Blotting Substrate (Bio-Rad) and chemiluminescence was detected on an Azure c600 Imager (Azure Biosystems).
Bacterial growth & fluorescence measurements
Saturated overnight cultures were diluted 1:100 in 150 μL BHI or LB media with appropriate antibiotic selection (see “microbes”) in a 96-well special optics microplate (Corning). Listeria cells were incubated at 30°C and Pseudomonas at 37°C with continuous double-orbital rotation for 16–48 hr in the Synergy H1 Hybrid Multi-Mode Reader (BioTeK) and measurements of OD600 and mCherry (excitation 570 nm, emission 610 nm) relative fluorescence units (RFU) recorded every 5 min with the Gen5 (BioTek) software. For bacterial growth curves, data are displayed as the mean OD600 of at least three biological replicates ± SD (error bars) as a function of time (min or hr, as indicated). For Cas9-mCherry or mCherry fluorescence levels, background fluorescence of growth media was subtracted and the resulting RFU values were normalized to OD600 for each time point. Data are displayed as the mean normalized fluorescence of three biological replicates ± SD.
Cas9 protein and mRNA reporter quantification
Cas9 (WT or dead; Lmo or Spy) reporters (see Figure 1A schematic) designed to measure protein levels in Listeria contain a single RBS generating a fused Cas9-mCherry protein. Reporters for mRNA levels contain two ribosomal binding sites, one for Cas9 and a second for mCherry, generating two separate proteins. All reporters were conjugated into Lmo10403s devoid of endogenous cas9 generating strains with the genotype Δcas9, ΔtRNAArg::pPL2oexL-pHyper-Cas9Reporter. Cells were grown and data collected and processed as in “bacterial growth and fluorescence measurements.” Data are shown as the percentage of Cas9 translation and transcription levels (mCherry fluorescence averaged across 6 hr of logarithmic growth) relative to control strains (no prophage (−prophage) or empty vector, as indicated) of at least three biological replicates ± SD (error bars).
RT-qPCR of cas9 mRNA levels
WT or Cas9-overexpressing Lmo10403s (Δcas9, ΔtRNAArg::pPL2oexL-LmoCas9–6xHis-FLAG) strains were grown to early (OD600 0.2–0.3) or mid-log (OD600 0.4–0.6) phase and 1.6 OD600 units of cells were harvested as in “Listeria protein samples.” Cell pellets were resuspended in 100 μL TE buffer supplemented with 0.2 U/μL SUPERase•In RNase Inhibitor (Thermo Fisher Scientific) and 5 mg/mL lysozyme, and incubated at 37°C for 10 min. Each sample was mixed with solutions pre-heated to 65°C for 15 min: 600 μL hot 1.2X lysis buffer (60 mM NaOAc, 1.2% SDS, 12 mM EDTA) and 700 μL hot acid-phenol:chloroform pH 4.5 (with IAA, 125:24:1) (Ambion). After incubating at 65°C for 30 min with shaking at 1500 rpm, followed by centrifugation at 12000 g for 15 min at 4°C, 500 μL aqueous phase was recovered for each sample. RNA was extracted with neutral phenol:chloroform:isoamyl alcohol (25:24:1) (Sigma) three times, precipitated with ethanol, and resuspended in nuclease-free water. Residual DNA was removed using the TURBO DNA-free Kit (Invitrogen). RT-qPCR was conducted in technical triplicate using the Luna Universal One-Step RT-qPCR Kit (New England Biolabs) according to the manufacturer’s instructions in 10 μL reaction volumes and reactions were run on a CFX Real-Time PCR Detection System (BioRad). cas9 mRNA and 16srRNA were analyzed with the following primers: cas9-FWD: 5′-ATGCCGCGATAGATGGTTAC-3′ and cas9-REV: 5′-CGCCTTCGATGTTCTCCAATA-3′; 16s-FWD: 5′-CCTGGTAGTCCACGCCGT-3′ and 16s-REV: 5′-TGCGTTAGCTGCAGCACTAAG-3′.
SpyCas9 protein decay measurements in Lmo
Saturated overnight cultures of Lmo10403s strains devoid of endogenous cas9 and expressing AcrIIA1 or AcrIIA4 from a tightly regulated rhamnose-inducible promoter (Fieseler et al., 2012) and SpyCas9-mCherry from the constitutively active pHyper promoter (Δcas9, ΔtRNAArg::pPL2oexL-pHyper-SpyCas9-mCherry-GyrA_terminator-pRha-AcrIIA) were diluted 1:100 in fresh LB supplemented with 50 mM glycerol and tetracycline (2 μg /mL) and grown to mid-log (OD600 ~0.5). Cultures were then diluted 1:2 in LB containing 50 mM glycerol and tetracycline (2 μg /mL) plus 200 mM rhamnose to induce Acr expression or 200 mM glycerol for uninduced controls (100 mM final concentration rhamnose or glycerol) in a 96-well microplate and treated with gentamicin (5 μg/mL) to inhibit translation or water as a control. Cells were grown and data collected and processed as in “bacterial growth and fluorescence measurements.” Data are shown as the mean percentage of SpyCas9-mCherry fluorescence relative to levels measured at “0 hr” (the beginning of translation inhibition or anti-CRISPR induction) of at least three biological replicates ± SD (error bars) as a function of time (min). Data were fitted by nonlinear regression to generate best-fit decay curves.
Listeria CRISPRi and self-targeting
Single-copy integrating CRISPRi and self-targeting constructs (see Figure 2A and 2B schematics) were designed as follows: pPL2oexL–pHyper-sgRNA [pHELP-spacer] GyrATerminator–pRhamnose-Cas9 (Lmo WT or Lmo dead or Spy dead) LambdaTerminator–pHELP-mCherry-LuxTerminator and conjugated into Lmo10403sϕcureΔcas9 containing pLEB579 plasmids expressing the indicated anti-CRISPRs. Overnight cultures were grown in LB supplemented with 50 mM glycerol (no induction/repression), 2 μg/mL tetracycline, and 7.5 μg/mL erythromycin. Cultures were then diluted 1:100 in LB containing 50 mM glycerol and the aforementioned antibiotics plus 200 mM rhamnose to induce Cas9 expression (and thus, CRISPRi or self-targeting) or 200 mM glycerol for uninduced controls (100 mM final concentration rhamnose or glycerol) in a 96-well microplate. Cells were grown and data collected and processed as in “bacterial growth and fluorescence measurements.” For self-targeting, data are displayed as the mean OD600 of at least three biological replicates ± SD (error bars) as a function of time (hr). For CRISPRi, data are shown as the mean percentage mCherry expression (mCherry fluorescence averaged across 6 hr of logarithmic growth) relative to uninduced controls of at least three biological replicates ± SD (error bars).
Construction of isogenic ϕA006 Acr phages
Isogenic ϕA006 phages encoding distinct anti-CRISPRs from the native anti-CRISPR locus were engineered by rebooting genomic bacteriophage DNA in L. monocytogenes L-form cells (EGDe strain variant Rev2) as previously described (Kilcher et al., 2018). Denoted acr genes (*) contain the strong ribosomal binding site (RBS) naturally associated with the first gene in the natural ϕA006 anti-CRISPR locus (orfA) whereas unmarked genes contain the weaker RBS associated with acrIIA1.
Cas9 and anti-CRISPR protein purifications
N-terminally 6xHis-tagged Acr proteins were expressed from the pET28 vector whereas WT SpyCas9 and mutants were expressed from 6xHis-MBP-Cas9 constructs (gifts from Jennifer Doudna, UC Berkeley) in E. coli Rosetta (DE3) pLysS cells. Recombinant protein expression was induced with 0.25 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) at 18 °C overnight. Cells were harvested by centrifugation and lysed by sonication in buffer A (50 mM Tris-HCl pH 7.5, 500 mM NaCl, 0.5 mM DTT, 20 mM imidazole, 5% glycerol) supplemented with 1 mM PMSF and 0.25 mg/mL lysozyme (Sigma). Cell debris was removed by centrifugation at 20000 g for 40 min at 4 °C and the lysate incubated with Ni-NTA Agarose Beads (Qiagen). After washing, bound proteins were eluted with Buffer A containing 300 mM imidazole and dialyzed overnight into storage buffer (20 mM HEPES-NaOH pH 7.4, 150mM KCl, 10% glycerol, 2mM DTT). GST-tagged AcrIIA1 and AcrIIA2b.3 were expressed from pGEX-6P-1 plasmids in E. coli BL21 (DE3) cells, lysed in buffer (20 mM HEPES-NaOH pH 7.4, 300 mM KCl and 5 mM DTT) supplemented with 1 mM PMSF and 0.25 mg/mL lysozyme, and clarified lysate was incubated with Glutathione Agarose Beads (Pierce). After washing, bound proteins were eluted using 100 mM Tris-HCl pH 8.5, 150 mM KCl, 15 mM reduced glutathione. The GST tag was cleaved with PreScission Protease (Millipore) and proteins were dialyzed overnight in 50 mM HEPES-NaOH pH 7.5, 150 mM KCl, 10% glycerol and 2 mM DTT to remove free glutathione. Cleaved GST was removed from dialyzed proteins with Glutathione Agarose Beads (Pierce).
in vitro binding of anti-CRISPRs to SpyCas9
The binding affinities of Acr proteins to SpyCas9 were calculated using microscale thermophoresis (MST) on the Monolith NT.115 instrument (NanoTemper Technologies GmbH, Munich, Germany). For AcrIIA1/AcrIIA2b.3 with Cas9-gRNA complexes (WT, mutant, DNA-bound, or AcrIIA2b.3-bound) 6xHis-Cas9 proteins were incubated with two-fold molar excess gRNA (Integrated DNA Technologies) and labeled with RED-tris-NTA using the His-Tag labeling kit. To form the Cas9-gRNA-DNA or Cas9-gRNA-AcrIIA2b.3 complexes, two fold molar excess of a bubbled DNA substrate (whose target strand cannot be cleaved by the HNH domain due to mismatches in the seed) or AcrIIA2b.3 was incubated with Cas9-gRNA complex for 10 min at room temperature (RT). The substrate proteins AcrIIA1/AcrIIA2b.3 at 0.09 nM to 3 μM concentrations were then incubated with 25 nM labeled Cas9-gRNA complexes at RT for 10 min in MST buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 15 mM MgCl2, 0.05% Tween-20). For AcrIIA1/AcrIIA2b.3 with ApoCas9, the substrate protein ApoCas9 (QB3 Macrolab) at 0.61 nM to 10 μM concentrations was incubated with 25 nM NT-647-NHS-labeled AcrIIA1/A2b.3 proteins. For AcrIIA1 mutants with WT Cas9-gRNA, the substrate protein Cas9-gRNA (QB3 Macrolab) at 15 pM to 0.5 μM concentrations was incubated with 25 nM RED-tris-NTA-labeled 6xHis-AcrIIA1 mutant proteins. Samples were loaded into Monolith NT.115 Capillaries and measurements were performed at 25 °C using 40% LED power and medium microscale thermophoresis power. All experiments were repeated three times for each measurement. Data analysis was conducted using NanoTemper analysis software. For the Cas9-gRNA-DNA complex, the target dsDNA sequence for the top and bottom strands are as follows:
5’-CTCAGCCTGGAAGAGATCGTAACGCGAACTACGCGGGTTGGTATACCAACATCATGACCT-3’
5’-AGGTCATGATGTTGGTATACCATGGGGCGTAGTTCGCGTTACGATCTCTTCCAGGCTGAG-3’
in vitro pull-downs of SpyCas9-Acr complexes
5 μg apoCas9 proteins (WT, dead, D10A, or H840A) were incubated with two-fold molar excess gRNA at 37°C for 15 min. Cas9-gRNA complexes were then incubated with 10-fold molar excess GST-tagged AcrIIA1 or AcrIIA2b.3 proteins for 15 min at RT in binding buffer (20 mM HEPES-NaOH pH 7.5, 150 mM NaCl, 10% glycerol, 10 mM MgSO4, 0.05% Tween 20 and 1 mM DTT). Samples were then incubated with 20 μL Glutathione sepharose beads (GE) for 15 min at 4°C and washed five times with binding buffer. Beads were boiled in 1X Laemmli Sample Buffer and proteins were analyzed by SDS-PAGE and Bio-Safe Coomassie staining (Biorad).
Cas9 DNA cleavage with purified proteins
To generate gRNAs, crRNA and tracrRNA were annealed with Nuclease-free Duplex Buffer (Integrated DNA Technologies) according to the manufacturer’s instructions. In vitro Cas9 DNA cleavage reactions were assembled in 1X MST Buffer (50 mM Tris-Cl pH 7.4, 150 mM NaCl, 20 mM MgCl2, 5 mM DTT, 5% glycerol, 0.05% (v/v) Tween-20) with 50 nM SpyCas9 and 625 nM AcrIIA, incubated for 5 min on ice, supplemented with 50 nM gRNA, and incubated for an additional 5 min at room temperature. Reactions were initiated by adding 2 nM target DNA substrate and at 1, 2, 5 and 10 min time points reaction aliquots were mixed with warm Quenching Buffer (50mM EDTA, 0.02% SDS) and boiled at 95°C for 10 min. DNA cleavage products were analyzed by agarose (1%) gel electrophoresis and staining with SYBR Safe (Thermo Fisher Scientific).
SpyCas9-AcrIIA1 limited proteolysis
20 μg purified SpyCas9 (QB3 Macrolab) in Apo form or in complex with gRNA (1.1-fold molar excess) was incubated with 1.5-fold and 4-fold molar excess AcrIIA1 and AcrIIA2b.3, respectively, in protease buffer (10 mM Tris-HCl pH 7.5, 300 mM NaCl) at 25°C for 15 min. Alternatively, ApoSpyCas9 was incubated first with AcrIIA protein followed by gRNA addition. Proteolysis reactions were performed with 20 ng α-chymotrypsin (sequencing grade, Promega) at 25°C and at 0, 10, 30, or 60 min time points, reactions were quenched with 2X SDS Laemmli Buffer and boiled for 10 min at 95°C. Samples were analyzed by SDS-PAGE and staining with Bio-Safe Coomassie (Bio-Rad).
Phage plaque forming unit quantification
Listeria phage infections were conducted using the soft agar overlay method: 10 μL phage dilution was mixed with 150 μL stationary Listeria culture in 3 mL molten LC top agar supplemented with 300 μg/mL Tetrazolium Violet (TCI Chemicals) to generate contrast for plaque visualization (Hurst et al., 1994) and poured onto a BHI-agar plate. After 24 hr incubation at 30°C, phage plaque-forming units (PFU) were quantified.
Efficiency of plaquing of Listeria phages
Efficiency of plaquing (EOP) calculations are a ratio of the number of plaque forming units (PFUs) that formed on a Lmo10403sϕcure targeting strain (endogenous cas9 with overexpression of the native CRISPR array spacer #1 that targets ϕA006) divided by the number of PFUs that formed on a non-targeting strain (Δcas9). Each PFU measurement was conducted in biological triplicate and all EOP data is displayed as the mean EOP ± SD (error bars).
Construction of self-targeting Lmo lysogens
Lmo10403sΔcas9::ϕA006 isogenic self-targeting lysogens encoding no anti-CRISPR or AcrIIA1, AcrIIA4, AcrIIA12 (alone or in combination as indicated) were isolated as in “construction of Lmo10403s lysogens.” To prevent self-targeting during strain construction, pPL2oexL constructs encoding a tightly regulated rhamnose-inducible LmoCas9 (WT or dead as a control) were conjugated into each lysogen. To assess the stability of each lysogen, cells were cultured, Cas9 induced, and data displayed as described for the self-targeting strain in “Listeria CRISPRi and self-targeting,” except erythromycin was omitted from LB media. Each lysogen stability measurement was performed in biological triplicate.
P. aeruginosa anti-SpyCas9 screening platform
The previously described P. aeruginosa anti-SpyCas9 screening platform (Jiang et al., 2019) and bacteriophage plaque assays (Borges et al., 2018; Jiang et al., 2019) were utilized to assay the anti-CRISPR activity of AcrIIA1 homologs and mutants. AcrIIA1 homolog genes were synthesized (Twist Bioscience) and cloned into the pMMB67HE-PLac vector. Protein accession numbers are listed in Table S1. Site directed mutagenesis by Gibson Assembly was used to introduce point mutations into pMMB67HE-PLac-GST-AcrIIA1. The PBAD promoter driving chromosomally integrated SpyCas9–3xMyc and pHERD30T-sgRNA was induced with 0.1% arabinose and the PLac promoter driving pMMB67HE-AcrIIA with 1 mM IPTG. Expression of AcrIIA1 mutants was confirmed by harvesting 1 OD600 unit of cells and resuspending in 200 μL 1X Laemmli Sample Buffer (Bio-Rad) followed by SDS-PAGE and immunoblotting as described above. The fold reductions in phage titer displayed were qualitatively derived by examining at least three replicates of each experiment. Plate images were acquired as in “Listeria phage titering” and a representative picture is shown.
P. aeruginosa self-targeting and CRISPRi
Strains were generated as previously described by Borges et al., 2018 under “construction of PAO1::SpyCas9 expression strain,” except the sgRNA was designed to target the PAO1 chromosomal phzM gene promoter and was integrated into the bacterial genome using the mini-CTX2 vector (Hoang et al., 2000). Cultures were grown overnight in LB supplemented with 50 μg/mL gentamicin and 0.1% arabinose to pre-induce anti-CRISPR expression and the next day diluted 1:100 with fresh LB containing 50 μg/mL gentamicin, 0.1% arabinose, and IPTG (0, 0.01, 0.1 or 1mM to titrate WT or dead SpyCas9-sgRNA expression) in a 96-well microplate (150 μL/well) for self-targeting analysis or glass tubes (3 mL) for CRISPRi. Self-targeting experiments were conducted in biological triplicate with cells grown and data collected and processed as in “bacterial growth and fluorescence measurements.” For CRISPRi, cells were grown for 8–10 hr with continuous shaking after which CRISPRi was qualitatively assessed by inspecting the culture pigment. Repression of the phzM gene by dCas9 generates a yellow culture whereas inhibition of dCas9 (e.g. by an Acr) allows phzM expression and pyocyanin production that generates a green culture. Representative pictures of at least three biological replicates are shown.
SpyCas9–3xMyc and GST-AcrIIA co-IPs
Saturated overnight cultures of P. aeruginosa strains were diluted 1:100 in 50 mL of LB supplemented with required antibiotics, grown to OD600 0.3–0.4, and induced with 0.3% arabinose (SpyCas9-gRNA) and 1mM IPTG (anti-CRISPR). Cells were harvested at OD600 1.8–2.0 by centrifugation at 6000 g for 10 min at 4°C, flash frozen on dry ice, resuspended in 1 mL lysis buffer (50 mM Tris-Cl pH 7.4, 150 mM NaCl, 20 mM MgCl2, 0.5% NP-40, 5% (v/v) glycerol, 5 mM DTT, 1 mM PMSF), lysed by sonication (20 sec pulse × 4 cycles with cooling between cycles), and lysate was clarified by centrifugation at 14 000g for 10 min at 4°C. For input samples, 10 μL lysate was mixed with one-third volume 4X Laemmli Sample Buffer. Remaining lysate (~1 mL) was mixed with pre-washed Myc-Tag Magnetic Bead Conjugate #5698 (Cell Signaling Technology) or Glutathione Magnetic Agarose Beads #78601 (Thermo Fisher Scientific) using a lysate to bead slurry volume ratio of 20:1 for Myc or 40:1 for GST. After overnight incubation at 4°C with end-over-end rotation, beads were washed five times with 1 mL cold wash buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 20 mM MgCl2, 5mM DTT) containing decreasing concentrations of NP-40 (0.5%, 0.05%, 0.01%, 0.005%, 0) and glycerol (5%, 0.5%, 0.05%, 0.005%, 0) on a magnetic stand. Bead-bound proteins were resuspended in 100 μL wash buffer without detergent and glycerol. 10 μL bead-bound protein slurry was mixed with one-third volume 4X Laemmli Sample Buffer, boiled for 5 min at 95°C, and samples were analyzed by SDS-PAGE using 4–20% Mini-PROTEAN TGX gels (Bio-Rad) and staining with Bio-Safe Coomassie (Bio-Rad) or immunoblotting.
Immunoprecipitated SpyCas9 DNA cleavage assay
SpyCas9–3xMyc DNA cleavage reactions were assembled with bead-bound protein slurry and 1.5 nM DNA substrate, incubated at 25°C with gentle shaking at 1000 rpm, and at 1, 5, 10, and 30 min time points reaction aliquots were mixed with warm Quenching Buffer (50 mM EDTA, 0.02% SDS) and boiled at 95°C for 10 min. DNA cleavage products were analyzed by agarose (1%) gel electrophoresis and staining with SYBR Safe (Thermo Fisher Scientific).
Inhibition of LivCas9 by anti-CRISPR proteins
Plaquing assays were conducted as previously described by Hupfeld et al., 2018. A Listeria monocytogenes Mack strain containing chromosomally-integrated pHelp-LivCas9/tracrRNA was transformed with pLEB579-derived plasmids constructed to express two components: an Acr protein from the pSpac promoter (or no Acr) and a crRNA that targets phage ΦP35 (or a scrambled non-targeting control). A Listeria ivanovii WSLC 30167 strain containing an endogenous Type II-A LivCas9 system was transformed with a pKSV7-derived plasmid expressing AcrIIA1 from the ΦA006 anti-CRISPR promoter (or empty vector) and a pLRSR-crRNA plasmid that targets phage ΦA511 (or a non-targeting control). A mixture of 200 μL stationary host culture and 4 mL LC top agar was poured onto an agar plate (LC for ΦP35; 1/2 BHI for ΦA511). Ten-fold serial dilutions of phage were spotted on top, plates were incubated at 20°C for ΦP35 and 30°C for ΦA511 for one day, and plate images were subsequently acquired.
E. coli phage Mu plaquing assays
Plasmids expressing Type II-A, II-B, and II-C Cas9-sgRNA combinations were previously described (Garcia et al., 2019). Cas9 plasmids containing a spacer targeting phage Mu and a pCDF-1b plasmid expressing the indicated anti-CRISPR proteins were co-transformed into E. coli BB101. After 2 hr of growth in LB at 37°C with continuous shaking, cells were treated with 0.01 mM IPTG to induce anti-CRISPR expression, and incubated for an additional 3 hr. A mixture of cells and LB top agar (0.7% agar) was poured onto an LB plate supplemented with 200 ng/mL aTc, 0.2% arabinose, and 10 mM MgSO4. Ten-fold serial dilutions of phage Mu were spotted on top and plates were incubated overnight. Anti-CRISPR expression after IPTG induction was analyzed by SDS-PAGE on a 15% Tris-Tricine gel followed by Coomassie Blue staining as previously described (Lee et al., 2018).
Generation of human cell expression plasmids
Descriptions of plasmids used for expression of sgRNAs (including sgRNA/crRNA target sequences), nucleases, and Acr proteins in human cells are available in Table S3. U6 promoter sgRNA and crRNA expression plasmids were generated by annealing and ligating oligonucleotide duplexes into BsmBI-digested BPK1520, BPK2660, KAC14, KAC27, KAC482, KAC32 and BPK4449 for SpyCas9, SauCas9, St1Cas9, St3Cas9, CjeCas9, and NmeCas9, respectively. New human cell expression plasmids for CjeCas9, St3Cas9, and NmeCas9 were generated by sub-cloning the nuclease open-reading frames of Addgene plasmids # 89752, 68337, and 119923, respectively (gifts from Seokjoong Kim, Feng Zhang and Erik Sontheimer) into the AgeI and NotI sites of pCAG-CFP (Addgene plasmid 11179; a gift from C. Cepko). Human codon optimized Acr constructs containing a C-terminal SV40 nuclear localization signal were generated by isothermal assembly of synthetic gene fragments (Twist Biosciences) into the NotI and AgeI sites of Addgene plasmid ID 43861. New human expression plasmids described in this study have been deposited with Addgene (see Table S3).
Transfection of human cells
Approximately 20 hours prior to transfection, HEK 293T cells were seeded at 2×104 cells/well in 96-well plates. Cells were transfected using 70 ng of nuclease, 30 ng sgRNA/crRNA, and 110 ng of acr expression plasmids with 1.25 μL of TransIT-X2 (Mirus Bio) in 20 μL Opti-MEM. For control conditions containing no acr plasmid, 110 ng of a pCMV-EGFP plasmid was utilized as filler DNA; for non-targeting sgRNA/crRNA conditions, 30 ng of an empty U6 promoter plasmid was used as filler DNA. For titration experiments cells were transfected using 70 ng of nuclease, 30 ng sgRNA/crRNA, varying amounts of acr expression and DNA stuffer plasmids totaling 197 ng (6 ng acr with 191 ng stuffer; 22 ng acr with 175 stuffer; 62 ng acr with 135 stuffer; 197 ng acr with no stuffer), and 1.77 μL of TransIT-X2 (Mirus Bio) in 20 μL Opti-MEM. DNA stuffer plasmids were an orthogonal and incompatible pCAG-nuclease expression plasmid. Genomic DNA was harvested from cells 72 hours post-transfection by suspending cells in 100 μL of lysis buffer (20 mM Hepes pH 7.5, 100 mM KCl, 5 mM MgCl2, 5% glycerol, 25 mM DTT, 0.1% Triton X-100, and 30 ng/uL Proteinase K (NEB)), followed by incubation at 65°C for 6 minutes and 98°C for 2 minutes. All experiments were performed with at least 3 independent biological replicates.
Cas and Acr protein activities in human cells
Genome editing efficiencies were determined by next-generation sequencing using a 2-step PCR-based Illumina library construction method (for primers see Table S4). Briefly, genomic regions were initially amplified using Q5 High-Fidelity DNA Polymerase (NEB), ~100 ng of genomic DNA lysate, and gene-specific round 1 primers. PCR products were purified using paramagnetic beads as previously described (Kleinstiver et al., 2019) and diluted 1:100 prior to the 2nd round of PCR to add Illumina barcodes and adapter sequences using Q5 polymerase. PCR amplicons were bead purified, quantified and normalized (Qiagen QIAxcel), and pooled. Final libraries were quantified using an Illumina Library qPCR Quantification Kit (KAPA Biosystems) and sequenced on a MiSeq sequencer using a 300-cycle v2 kit (Illumina). Genome editing activities were determined from the sequencing data using CRISPResso2 (Clement et al., 2019) with commands --min_reads_to_use_region 100. -w 10, and for certain sequencing data sets --ignore_substitutions. The known control inhibitors that were used included: AcrIIA4 SpyCas9 inhibitor (Dong et al., 2017), AcrIIA5 broad-spectrum Cas9 inhibitor (Garcia et al., 2019), AcrVA1 Cas12 inhibitor as a negative control for Cas9 orthologues (Marino et al., 2018).
QUANTIFICATION AND STATISTICAL ANALYSIS
All numerical data, with the exception of the microscale thermophoresis (MST) data, were analyzed and plotted using GraphPad Prism 6.0 software. The MST data were analyzed using the NanoTemper analysis software (NanoTemper Technologies GmbH) and plotted using GraphPad Prism 6.0 software. Statistical parameters are reported in the Figure Legends.
Data and Code Availability
The AcrIIA1 homolog protein accession numbers and associated promoter sequences are disclosed in Table S1.
Supplementary Material
Table S2. Listeria strains, plasmids, and phages constructed and used in this study (Excel spreadsheet), Related to STAR Methods
Table S3. Nuclease, sgRNA, and Acr plasmids used in human cell experiments (Excel spreadsheet), Related to Figures 5 and S5
Table S4. Oligonucleotides used in human cell experiments (Excel spreadsheet), Related to Figures 5 and S5
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rabbit anti-FLAG | Sigma-Aldrich | Cat# F7425; RRID: AB_439687 |
| mouse anti-FLAG | Sigma-Aldrich | Cat# F1804; RRID: AB_262044 |
| mouse anti-Myc | Cell Signaling Technology | Cat# 2276; RRID: AB_331783 |
| rabbit anti-GST | Cell Signaling Technology | Cat# 2625; RRID: AB_490796 |
| mouse anti-E.coli RNA polymerase β | BioLegend | Cat# 663903; RRID: AB_2564524 |
| HRP-conjugated goat anti-Rabbit IgG | Bio-Rad | Cat# 170–6515; RRID: AB_11125142 |
| HRP-conjugated goat anti-mouse IgG | Santa Cruz Biotechnology | Cat# sc-2005; RRID: AB_631736 |
| Bacterial and Virus Strains | ||
| Listeria monocytogenes 10403s | Rauch et al., 2017 | RefSeq: NC_017544.1 |
| Listeria monocytogenes 10403s derivatives | This paper | See Table S2 |
| Listeria ivanovii WSLC 30167 | Hupfeld et al., 2018 | RefSeq: NZ_CP009575.1 |
| Listeria ivanovii WSLC 3009 | Hupfeld et al., 2018 | RefSeq: NZ_CP007172.1 |
| Listeria monocytogenes Mack | Hupfeld et al., 2018 | https://doi.org/10.1093/nar/gky544 |
| Pseudomonas aeruginosa strain PAO1 | Laboratory of Alan Davidson | RefSeq: NC_002516.2 |
| Pseudomonas aeruginosa strain PAO1 derivatives | This paper | N/A |
| Escherichia coli DH5α | New England Biolabs | Cat #C2982I |
| E. coli Rosetta (DE3) pLysS | QB3 Macrolab | N/A |
| Escherichia coli SM10 | Laboratory of Daniel Portnoy | N/A |
| E. coli BB101 | Garcia et al., 2019 | https://doi.org/10.1016/j.celrep.2019.10.017 |
| Listeria phage A006 | This paper | RefSeq: NC_009815.1 |
| Listeria phage A006 derivatives | This paper | See Table S2 |
| Listeria phage A118 | This paper | RefSeq: NC_003216.1 |
| Listeria phage J0161a | Rauch et al., 2017 | RefSeq: NC_017545.1 |
| Listeria phage A511 | Hupfeld et al., 2018 | RefSeq: NC_009811.2 |
| Listeria phages P35 | This paper | RefSeq: NC_009814.1 |
| Pseudomonas phage JBD30 | Laboratory of Alan Davidson | RefSeq: NC_020198.1 |
| Phage Mu | Garcia et al., 2019 | https://doi.org/10.1016/j.celrep.2019.10.017 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| AcrIIA1 protein homologs tested for Cas9 inhibition | This paper | See Table S1 |
| Purified protein: SpyCas9 | QB3 Macrolab | N/A |
| Purified protein: 6xHis-MBP-SpyCas9 | This paper | N/A |
| Purified protein: 6xHis-MBP-deadSpyCas9 | This paper | N/A |
| Purified protein: 6xHis-MBP-SpyCas9(D10A) | This paper | N/A |
| Purified protein: 6xHis-MBP-SpyCas9(H840A) | This paper | N/A |
| Purified protein: 6xHis-AcrIIA1 | This paper | N/A |
| Purified protein: 6xHis-AcrIIA1(T16A) | This paper | N/A |
| Purified protein: 6xHis-AcrIIA1(T114A/F115A) | This paper | N/A |
| Purified protein: 6xHis-AcrIIA1(F115A) | This paper | N/A |
| Purified protein: GST-AcrIIA1 | This paper | N/A |
| Purified protein: GST-AcrIIA2b.3 | This paper | N/A |
| Monolith His-Tag Labeling Kit RED-tris-NTA | Nanotemper Technologies | Cat #MO-L018 |
| Monolith NT Protein Labeling kit RED-NHS | Nanotemper Technologies | Cat # L001 |
| Tetrazolium Violet | TCI Chemicals | Cat #T0174 |
| Critical Commercial Assays | ||
| Gibson Assembly Master Mix | New England Biolabs | Cat #E2611L |
| Phusion Hot Start Flex DNA Polymerase | New England Biolabs | Cat #M0535S |
| TURBO DNA-free Kit | Thermo Fisher Scientific | Cat #AM1907 |
| Luna Universal One-Step RT-qPCR Kit | New England Biolabs | Cat #E3005S |
| Myc-Tag Magnetic Bead Conjugate | Cell Signaling Technology | Cat #5698 |
| Glutathione Magnetic Agarose Beads | Thermo Fisher Scientific | Cat #78601 |
| 4–20% Mini-PROTEAN TGX gels | Bio-Rad | Cat #4561096 |
| Experimental Models: Cell Lines | ||
| Human: HEK 293T cells | ATCC | ATCC CRL-11268 |
| Oligonucleotides | ||
| RT qPCR primer cas9-FWD: ATGCCGCGATAGATGGTTAC | This paper | N/A |
| RT qPCR primer cas9-REV: CGCCTTCGATGTTCTCCAATA | This paper | N/A |
| RT qPCR primer 16s-FWD: CCTGGTAGTCCACGCCGT | This paper | N/A |
| RT qPCR primer 16s-REV: TGCGTTAGCTGCAGCACTAAG | This paper | N/A |
| Target DNA used in SpyCas9-Acr in vitro binding assays CTCAGCCTGGAAGAGATCGTAACGCGAACTACGCGGGTTGGTATACCAACATCATGACCT |
This paper | N/A |
| oligonucleotides used in human cell experiments | This paper | See Table S4 |
| Recombinant DNA | ||
| pKSV7 | Rauch et al., 2017 | addgene.org/26686/ |
| pKSV7-derivative plasmids | This paper | See Table S2 |
| pPL2oexL | Rauch et al., 2017 | https://doi.org/10.1016/j.cell.2016.12.009 |
| pPL2oexL-derivative plasmids | This paper | See Table S2 |
| pLEB579 | Beasley et al., 2004 | https://doi.org/10.1093/ps/83.1.45 |
| pLEB579-derivative plasmids | This paper | See Table S2 |
| pLRSR-crRNA plasmid | Hupfeld et al., 2018 | https://doi.org/10.1093/nar/gky544 |
| pHERD30T | Laboratory of Alan Davidson | GenBank: EU603326.1 |
| pHERD30T-derivative plasmids | This paper | N/A |
| pMMB67HE | ATCC | http://www.snapgene.com/resources/plasmid_files/basic_cloning_vectors/pMMB67HE/ |
| pMMB67HE-derivative plasmids | This paper | N/A |
| 6xHis-MBP-SpyCas9 protein expression plasmid | Laboratory of Jennifer Doudna | N/A |
| 6xHis-MBP-deadSpyCas9 protein expression plasmid | Laboratory of Jennifer Doudna | N/A |
| 6xHis-MBP-SpyCas9(D10A) protein expression plasmid | Laboratory of Jennifer Doudna | N/A |
| 6xHis-MBP-SpyCas9(H840A) protein expression plasmid | Laboratory of Jennifer Doudna | N/A |
| pET28 protein expression plasmid | Laboratory of David Morgan | N/A |
| pET28–6xHis-AcrIIA1 protein expression plasmid | This paper | N/A |
| pET28–6xHis-AcrIIA1(T16A) protein expression plasmid | This paper | N/A |
| pET28–6xHis-AcrIIA1(T114A/F115A) protein expression plasmid | This paper | N/A |
| pET28–6xHis-AcrIIA1(F115A) protein expression plasmid | This paper | N/A |
| pGEX-6P-1-GST-AcrIIA1 protein expression plasmid | This paper | N/A |
| pGEX-6P-1-GST-AcrIIA2b.3 protein expression plasmid | This paper | N/A |
| Type II-A, II-B, II-C Cas9-sgRNA plasmids in E. coli | Garcia et al., 2019 | https://doi.org/10.1016/j.celrep.2019.10.017 |
| Nuclease plasmids used in human cell experiments | This paper | See Table S3 |
| sgRNA plasmids used in human cell experiments | This paper | See Table S3 |
| Acr plasmids used in human cell experiments | This paper | See Table S3 |
| Software and Algorithms | ||
| Prism 6.0 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Gen 5 | BioTek | https://www.biotek.com/products/software-robotics-software/gen5-microplate-reader-and-imager-software/ |
| Image Lab 5.2.1 | BioRad | http://bio-rad.com/en-cn/product/image-lab-software |
| NanoTemper Analysis Software | NanoTemper Technologies | https://nanotempertech.com/monolith/ |
| CRISPResso2 | Clement et al., 2019 | http://crispresso.pinellolab.partners.org/ |
| Other | ||
| Synergy H1 Microplate Reader | BioTek | https://www.biotek.com/products/detection-hybrid-technology-multi-mode-microplate-readers/synergy-h1-hybrid-multi-mode-reader/ |
| Azure c600 Imager | Azure Biosystems | https://www.azurebiosystems.com/imaging-systems/azure-600/ |
| Monolith NT.115 | NanoTemper Technologies | https://nanotempertech.com/monolith/ |
| CFX Real-Time PCR Detection System | BioRad | https://www.bio-rad.com/en-us/product/cfx-connect-real-time-pcr-detection-system |
Highlights.
Listeria phages use independent Cas9 inhibitors to support lytic and lysogenic stages
AcrIIA1 induces degradation of Cas9 during lysogeny, but is ineffective in lytic growth
AcrIIA1 binds directly to Apo or sgRNA-loaded Cas9 via the HNH domain
AcrIIA1 has a broad spectrum of inactivation, inhibiting both Listeria Cas9 orthologues
ACKNOWLEDGEMENTS
We would like to thank Daniel A. Portnoy (UC Berkeley) for providing the pLMB3C-pRhamnose plasmid, Jennifer A. Doudna for Cas9 expression plasmids (UC Berkeley), and Jonathan Asfaha (David Morgan Lab, UCSF) and Ujjwal Rathore (Alex Marson Lab, UCSF) for experimental advice and reagents. The J.B.-D lab was supported by the UCSF Program for Breakthrough Biomedical Research funded in part by the Sandler Foundation, the Searle Fellowship, the Vallee Foundation, the Innovative Genomics Institute, an NIH Director’s Early Independence Award DP5-OD021344, and R01GM127489; S.Ki. lab was supported by an Ambizione Fellowship (Swiss National Science Foundation, PZ00P3_174108).; the B.P.K. lab by NIH R00-CA218870 and P01-HL142494, an A.S.G.C.T. Career Development Award, and the Margaret Q. Landenberger Research Foundation; and the A.R.D. lab by a CIHR Foundation grant FDN-15427.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
J.B.-D. is a scientific advisory board member of SNIPR Biome and Excision Biotherapeutics and a scientific advisory board member and co-founder of Acrigen Biosciences, and is an inventor on patents relating to anti-CRISPR proteins. B.P.K. is an inventor on various patents and patent applications that describe gene editing and epigenetic editing technologies, is a consultant for Avectas Inc., and an advisor to Acrigen Biosciences. A.R.D is a scientific advisory board member for Acrigen Biosciences and is an inventor on patents relating to anti-CRISPR proteins.
REFERENCES
- Argov T, Azulay G, Pasechnek A, Stadnyuk O, Ran-Sapir S, Borovok I, Sigal N, and Herskovits AA (2017). Temperate bacteriophages as regulators of host behavior. Curr. Opin. Microbiol 38, 81–87. [DOI] [PubMed] [Google Scholar]
- Beasley SS, Takala TM, Reunanen J, Apajalahti J, and Saris PEJ (2004). Characterization and Electrotransformation of Lactobacillus Crispatus Isolated from Chicken Crop and Intestine. Poult. Sci 83, 45–48. [DOI] [PubMed] [Google Scholar]
- Bondy-Denomy J, Garcia B, Strum S, Du M, Rollins MF, Hidalgo-Reyes Y, Wiedenheft B, Maxwell KL, and Davidson AR (2015). Multiple mechanisms for CRISPR-Cas inhibition by anti-CRISPR proteins. Nature 526, 136–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondy-Denomy J, Qian J, Westra ER, Buckling A, Guttman DS, Davidson AR, and Maxwell KL (2016). Prophages mediate defense against phage infection through diverse mechanisms. ISME J. 10, 2854–2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges AL, Davidson AR, and Bondy-Denomy J (2017). The Discovery, Mechanisms, and Evolutionary Impact of Anti-CRISPRs. Annu. Rev. Virol 4, null. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges AL, Zhang JY, Rollins M, Osuna BA, Wiedenheft B, and Bondy-Denomy J (2018). Bacteriophage cooperation suppresses CRISPR-Cas3 and Cas9 immunity. BioRxiv 279141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouns SJJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJH, Snijders APL, Dickman MJ, Makarova KS, Koonin EV, and van der Oost J (2008). Small CRISPR RNAs Guide Antiviral Defense in Prokaryotes. Science 321, 960–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi K-H, Kumar A, and Schweizer HP (2006). A 10-min method for preparation of highly electrocompetent Pseudomonas aeruginosa cells: Application for DNA fragment transfer between chromosomes and plasmid transformation. J. Microbiol. Methods 64, 391–397. [DOI] [PubMed] [Google Scholar]
- Clement K, Rees H, Canver MC, Gehrke JM, Farouni R, Hsu JY, Cole MA, Liu DR, Joung JK, Bauer DE, et al. (2019). CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol 37, 224–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, Eckert MR, Vogel J, and Charpentier E (2011). CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471, 602–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong D, Guo M, Wang S, Zhu Y, Wang S, Xiong Z, Yang J, Xu Z, and Huang Z (2017). Structural basis of CRISPR–SpyCas9 inhibition by an anti-CRISPR protein. Nature 546, 436–439. [DOI] [PubMed] [Google Scholar]
- Estela LA, Sofos JN, and Flores BB (1992). Bacteriophage Typing of Listeria monocytogenes Cultures Isolated From Seafoods. J. Food Prot 55, 13–17. [DOI] [PubMed] [Google Scholar]
- Feiner R, Argov T, Rabinovich L, Sigal N, Borovok I, and Herskovits AA (2015). A new perspective on lysogeny: prophages as active regulatory switches of bacteria. Nat. Rev. Microbiol 13, 641–650. [DOI] [PubMed] [Google Scholar]
- Fieseler L, Schmitter S, Teiserskas J, and Loessner MJ (2012). Rhamnose-Inducible Gene Expression in Listeria monocytogenes. PLoS ONE 7, e43444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchsbauer O, Swuec P, Zimberger C, Amigues B, Levesque S, Agudelo D, Duringer A, Chaves-Sanjuan A, Spinelli S, Rousseau GM, et al. (2019). Cas9 Allosteric Inhibition by the Anti-CRISPR Protein AcrIIA6. Mol. Cell [DOI] [PubMed] [Google Scholar]
- Garcia B, Lee J, Edraki A, Hidalgo-Reyes Y, Erwood S, Mir A, Trost CN, Seroussi U, Stanley SY, Cohn RD, et al. (2019). Anti-CRISPR AcrIIA5 Potently Inhibits All Cas9 Homologs Used for Genome Editing. Cell Rep. 29, 1739–1746.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garneau JE, Dupuis M-È, Villion M, Romero DA, Barrangou R, Boyaval P, Fremaux C, Horvath P, Magadán AH, and Moineau S (2010). The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468, 67–71. [DOI] [PubMed] [Google Scholar]
- Harrington LB, Doxzen KW, Ma E, Liu J-J, Knott GJ, Edraki A, Garcia B, Amrani N, Chen JS, Cofsky JC, et al. (2017). A Broad-Spectrum Inhibitor of CRISPR-Cas9. Cell 170, 1224–1233.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haurwitz RE, Jinek M, Wiedenheft B, Zhou K, and Doudna JA (2010). Sequence- and Structure-Specific RNA Processing by a CRISPR Endonuclease. Science 329, 1355–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heler R, Samai P, Modell JW, Weiner C, Goldberg GW, Bikard D, and Marraffini LA (2015). Cas9 specifies functional viral targets during CRISPR–Cas adaptation. Nature 519, 199–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang TT, Kutchma AJ, Becher A, and Schweizer HP (2000). Integration-Proficient Plasmids for Pseudomonas aeruginosa: Site-Specific Integration and Use for Engineering of Reporter and Expression Strains. Plasmid 43, 59–72. [DOI] [PubMed] [Google Scholar]
- Hupfeld M, Trasanidou D, Ramazzini L, Klumpp J, Loessner MJ, and Kilcher S (2018). A functional type II-A CRISPR–Cas system from Listeria enables efficient genome editing of large non-integrating bacteriophage. Nucleic Acids Res. 46, 6920–6933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst CJ, Blannon JC, Hardaway RL, and Jackson WC (1994). Differential Effect of Tetrazolium Dyes upon Bacteriophage Plaque Assay Titers. Appl Env. Microbiol 60, 3462–3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang S, and Maxwell KL (2019). Meet the Anti-CRISPRs: Widespread Protein Inhibitors of CRISPR-Cas Systems. CRISPR J. 2, 23–30. [DOI] [PubMed] [Google Scholar]
- Hynes AP, Rousseau GM, Lemay M-L, Horvath P, Romero DA, Fremaux C, and Moineau S (2017). An anti-CRISPR from a virulent streptococcal phage inhibits Streptococcus pyogenes Cas9. Nat. Microbiol 2, 1374. [DOI] [PubMed] [Google Scholar]
- Hynes AP, Rousseau GM, Agudelo D, Goulet A, Amigues B, Loehr J, Romero DA, Fremaux C, Horvath P, Doyon Y, et al. (2018). Widespread anti-CRISPR proteins in virulent bacteriophages inhibit a range of Cas9 proteins. Nat. Commun 9, 2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang F, Liu J-J, Osuna BA, Xu M, Berry JD, Rauch BJ, Nogales E, Bondy-Denomy J, and Doudna JA (2019). Temperature-Responsive Competitive Inhibition of CRISPR-Cas9. Mol. Cell 73, 601–610.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ka D, An SY, Suh J-Y, and Bae E (2018). Crystal structure of an anti-CRISPR protein, AcrIIA1. Nucleic Acids Res. 46, 485–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilcher S, Studer P, Muessner C, Klumpp J, and Loessner MJ (2018). Cross-genus rebooting of custom-made, synthetic bacteriophage genomes in L-form bacteria. Proc. Natl. Acad. Sci 115, 567–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinstiver BP, Sousa AA, Walton RT, Tak YE, Hsu JY, Clement K, Welch MM, Horng JE, Malagon-Lopez J, Scarfò I, et al. (2019). Engineered CRISPR–Cas12a variants with increased activities and improved targeting ranges for gene, epigenetic and base editing. Nat. Biotechnol 37, 276–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV, Makarova KS, and Zhang F (2017). Diversity, classification and evolution of CRISPR-Cas systems. Curr. Opin. Microbiol 37, 67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauer P, Chow MYN, Loessner MJ, Portnoy DA, and Calendar R (2002). Construction, Characterization, and Use of Two Listeria monocytogenes Site-Specific Phage Integration Vectors. J BACTERIOL 184, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Mir A, Edraki A, Garcia B, Amrani N, Lou HE, Gainetdinov I, Pawluk A, Ibraheim R, Gao XD, et al. (2018). Potent Cas9 Inhibition in Bacterial and Human Cells by AcrIIC4 and AcrIIC5 Anti-CRISPR Proteins. MBio 9, e02321–18, /mbio/9/6/mBio.02321–18.atom. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Yin M, Wang M, and Wang Y (2019). Phage AcrIIA2 DNA Mimicry: Structural Basis of the CRISPR and Anti-CRISPR Arms Race. Mol. Cell 73, 611–620.e3. [DOI] [PubMed] [Google Scholar]
- Makarova KS, Wolf YI, Alkhnbashi OS, Costa F, Shah SA, Saunders SJ, Barrangou R, Brouns SJJ, Charpentier E, Haft DH, et al. (2015). An updated evolutionary classification of CRISPR–Cas systems. Nat. Rev. Microbiol 13, 722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino ND, Zhang JY, Borges AL, Sousa AA, Leon LM, Rauch BJ, Walton RT, Berry JD, Joung JK, Kleinstiver BP, et al. (2018). Discovery of widespread type I and type V CRISPR-Cas inhibitors. Science 362, 240–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathony J, Harteveld Z, Schmelas C, Upmeier zu Belzen J, Aschenbrenner S, Hoffmann MD, Stengl C, Scheck A, Rosset S, Grimm D, et al. (2019). Computational design of anti-CRISPR proteins with improved inhibition potency and expanded specificity. BioRxiv 685032. [DOI] [PubMed] [Google Scholar]
- Mojica FJM, Díez-Villaseñor C, García-Martínez J, and Soria E (2005). Intervening Sequences of Regularly Spaced Prokaryotic Repeats Derive from Foreign Genetic Elements. J. Mol. Evol 60, 174–182. [DOI] [PubMed] [Google Scholar]
- Nakamura M, Srinivasan P, Chavez M, Carter MA, Dominguez AA, La Russa M, Lau MB, Abbott TR, Xu X, Zhao D, et al. (2019). Anti-CRISPR-mediated control of gene editing and synthetic circuits in eukaryotic cells. Nat. Commun 10, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuñez JK, Kranzusch PJ, Noeske J, Wright AV, Davies CW, and Doudna JA (2014). Cas1–Cas2 complex formation mediates spacer acquisition during CRISPR–Cas adaptive immunity. Nat. Struct. Mol. Biol 21, 528–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osuna BA, Karambelkar S, Mahendra C, Kilcher S, and Bondy-Denomy J (2020b). Critical anti-CRISPR locus repression by a bi-functional Cas9 inhibitor. co-submitted companion manuscript [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SF, and Stewart GSAB (1990). High-efficiency transformation of Listeria monocytogenes by electroporation of penicillin-treated cells. Gene 94, 129–132. [DOI] [PubMed] [Google Scholar]
- Rabinovich L, Sigal N, Borovok I, Nir-Paz R, and Herskovits AA (2012). Prophage Excision Activates Listeria Competence Genes that Promote Phagosomal Escape and Virulence. Cell 150, 792–802. [DOI] [PubMed] [Google Scholar]
- Rauch BJ, Silvis MR, Hultquist JF, Waters CS, McGregor MJ, Krogan NJ, and Bondy-Denomy J (2017). Inhibition of CRISPR-Cas9 with Bacteriophage Proteins. Cell 168, 150–158.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon R, Priefer U, and Pühler A (1983). A Broad Host Range Mobilization System for In Vivo Genetic Engineering: Transposon Mutagenesis in Gram Negative Bacteria. Bio/Technology 1, 784–791. [Google Scholar]
- Thavalingam A, Cheng Z, Garcia B, Huang X, Shah M, Sun W, Wang M, Harrington L, Hwang S, Hidalgo-Reyes Y, et al. (2019). Inhibition of CRISPR-Cas9 ribonucleoprotein complex assembly by anti-CRISPR AcrIIC2. Nat. Commun 10, 2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trasanidou D, Gerós AS, Mohanraju P, Nieuwenweg AC, Nobrega FL, and Staals RHJ (2019). Keeping crispr in check: diverse mechanisms of phage-encoded anti-crisprs. FEMS Microbiol. Lett 366, fnz098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yosef I, Goren MG, and Qimron U (2012). Proteins and DNA elements essential for the CRISPR adaptation process in Escherichia coli. Nucleic Acids Res. 40, 5569–5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Gao A, Zhan Q, Wang Y, Feng H, Liu S, Gao G, Serganov A, and Gao P (2019). Diverse Mechanisms of CRISPR-Cas9 Inhibition by Type IIC Anti-CRISPR Proteins. Mol. Cell 74, 296–309.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2. Listeria strains, plasmids, and phages constructed and used in this study (Excel spreadsheet), Related to STAR Methods
Table S3. Nuclease, sgRNA, and Acr plasmids used in human cell experiments (Excel spreadsheet), Related to Figures 5 and S5
Table S4. Oligonucleotides used in human cell experiments (Excel spreadsheet), Related to Figures 5 and S5
Data Availability Statement
The AcrIIA1 homolog protein accession numbers and associated promoter sequences are disclosed in Table S1.