

Abstract
The N-methyl-D-aspartate receptors (NMDARs) are ionotropic glutamate receptors that mediate the flux of calcium (Ca2+) into the postsynaptic compartment. Calcium influx subsequently triggers the activation of various intracellular signaling cascades that underpin multiple forms of synaptic plasticity. Functional NMDARs are assembled as heterotetramers composed of two obligatory GluN1 subunits and two GluN2 or GluN3 subunits. Four different GluN2 subunits (GluN2A-D) are present throughout the central nervous system; however, they are differentially expressed, both developmentally and spatially, in a cell- and synapse-specific manner. Each GluN2 subunit confers NMDARs with distinct ion channel properties and intracellular trafficking pathways. Regulated membrane trafficking of NMDARs is a dynamic process that ultimately determines the number of NMDARs at synapses, and is controlled by subunit-specific interactions with various intracellular regulatory proteins. Here we review recent progress made towards understanding the molecular mechanisms that regulate the trafficking of GluN2-containing NMDARs, focusing on the roles of several key synaptic proteins that interact with NMDARs via their carboxyl termini.
Keywords: NMDA receptors, synaptic plasticity, endosomal recycling, endocytosis, exocytosis, receptor trafficking, protein-protein interactions
Graphical Abstract
The N-methyl-D-aspartate glutamate receptors (NMDARs) mediate calcium-dependent signaling that underpins multiple forms of synaptic plasticity. Different GluN2 (GluN2A-D) subunits confer NMDARs with distinct ion channel properties and intracellular trafficking pathways. This review article summarizes the current knowledge of the molecular mechanisms that regulate the trafficking of GluN2-containing NMDARs, focusing on the roles of several key binding partners.
Glutamate is the major excitatory neurotransmitter in the brain. At most excitatory synapses, the postsynaptic specialisation that receives input from a presynaptic nerve terminal exists as a small membrane protrusion from the dendrite, known as the dendritic spine. Each spine contains glutamate receptors, including α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and N-methyl-D-aspartate (NMDA) receptors. In response to glutamate binding, the AMPA receptors (AMPARs) open and allow sodium (Na+) ions to enter the postsynaptic compartment, thereby mediating fast excitatory postsynaptic currents (EPSCs) that result in the depolarisation of the postsynaptic membrane. Under resting membrane potential, most NMDARs are inactive due to the voltage-dependent block by extracellular magnesium (Mg2+) ions. Upon heightened neuronal activity, such as during long-term potentiation (LTP), the Mg2+ blockade is relieved, leading to the opening of NMDARs that permit Ca2+ influx into the postsynaptic compartment (Nicoll & Roche 2013). The rise in intracellular Ca2+ subsequently triggers various signaling cascades, including the activation of the protein kinase Ca2+/calmodulin-dependent kinase II (CaMKII), leading to profound reorganisation of the molecular composition of the postsynaptic density (Lisman et al. 2012). This causes subsequent enlargement of dendritic spines and an increase in the number of AMPARs on the plasma membrane, producing a long-lasting increase in synaptic efficacy. Given the major function of NMDARs as “coincidence detectors” of postsynaptic depolarisation (which relieves the Mg2+ block) and the presynaptic release of glutamate (which binds to GluN2 subunits) during synaptic plasticity, pharmacological and genetic manipulations that interfere with NMDAR functions usually lead to impairments in synaptic plasticity and deficits in cognition, learning and memory in mice. Elucidating the molecular mechanisms that control NMDAR trafficking and function is therefore critical for understanding the cellular basis of neuronal plasticity and experience-dependent changes in adaptive behaviour.
NMDA Receptor Structure and Subunit Composition
Like other ionotropic glutamate receptors, NMDARs are assembled as tetrameric complexes of subunits that are permeable to Na+, potassium (K+) and Ca2+ (Traynelis et al. 2010). NMDAR subunits can be categorized into three different classes according to their sequence homology, including the glycine/D-serine binding GluN1 and GluN3 (GluN3A and GluN3B) subunits, as well as the glutamate binding GluN2 subunits (GluN2A, GluN2B, GluN2C and GluN2D) (Fig. 1a) (Traynelis et al. 2010; Paoletti et al. 2013). All NMDAR subunits consist of a highly homologous extracellular amino-terminal domain (ATD), a bi-lobed ligand-binding domain (LBD), and three transmembrane regions and a re-entrant ion pore-lining loop; as well as a more divergent intracellular carboxyl-terminal domain (CTD) (Fig. 1b). Each of these subunits are encoded by a distinct gene in the mammalian genome. Functional NMDARs are assembled as di-heteromers composed of two obligatory GluN1 subunits and two GluN2 or GluN3 subunits (Fig. 1c). However, a variety of studies have also reported the presence and prevalence of tri-heteromeric NMDARs containing GluN1 and two different GluN2 subunits in many brain regions and neuronal types, such as GluN1/2A/2B, GluN1/2A/2C and GluN1/2B/2D complexes (Fig. 1c) (Rauner & Kohr 2011; Tovar et al. 2013; Luo et al. 1997; Brickley et al. 2003; Pina-Crespo & Gibb 2002; Jones & Gibb 2005; Chazot et al. 1994; Swanger et al. 2015; Sheng et al. 1994).
Figure 1. Diversity and biophysical properties of GluN2-containing NMDARs.

A. Schematic diagram of the domain structure of the four GluN2 subunits of NMDARs, GluN2A – GluN2D. B. The modular architecture of a single GluN2 subunit comprising four distinct domains: an amino terminal domain (ATD), the ligand binding domain (LBD) that binds to glutamate, three transmembrane regions and a re-entrant loop that form the ion channel pore, as well as a carboxy terminal domain, which is the most divergent among all GluN2 subunits. C. Representative assemblies of GluN2-containing di- or tri-heteromeric NMDARs known to exist in the mammalian central nervous system. D. Schematic diagram depicting relative differences in the properties of di-heteromeric GluN2-containing NMDAR subtypes.
Activation of NMDARs requires the simultaneous binding of glutamate and glycine/D-serine to the GluN2 and GluN1 subunits respectively. Glutamate binds to GluN2 subunits in a pocket located in the extracellular ligand-binding domain. Interestingly, all four GluN2 subunits display striking differences in their spatiotemporal expression patterns in the brain, as well as unique pharmacological and functional properties (Monyer et al. 1994; Sheng et al. 1994; Hansen et al. 2017; Paoletti et al. 2013). These distinctions essentially create a repertoire of functional NMDARs with stark biophysical and signaling properties that underpin synaptic plasticity, dendritic integration and information processing in various regions of the brain.
Biophysical properties of GluN2-containing NMDARs
NMDARs possess several unique features that distinguish them from the AMPA- or kainate-type of ionotropic receptors, including: strong voltage-dependent block by extracellular Mg2+, the requirement for simultaneous binding of a co-agonist (glycine or D-serine) for activation, high Ca2+ permeability and slow deactivation kinetics (Hansen et al. 2018; Glasgow et al. 2015; Wyllie et al. 2013). These gating and ion channel properties differ among NMDAR subtypes and are determined by their subunit compositions (Fig. 1d). For instance, di-heteromeric GluN1/2A and GluN1/2B receptors have higher single-channel conductance (γ ~ 40–50 pS), Ca2+ permeability (PCa/PCs ~ 7.5), and Mg2+ sensitivity (IC50 ~ 15 μM at −100 mV) compared to GluN1/2C and GluN1/2D receptors (γ ~ 18–35 pS; PCa/PCs ~ 4.5; IC50 ~ 80 μM at −100 mV). On the other hand, GluN1/2D receptors have the highest affinity for glutamate (EC50 ~ 0.4 μM) compared to other GluN2-containing di-heteromeric receptors (GluN2C, EC50 ~ 1 μM > GluN2B, EC50 ~ 2 μM > GluN2A, EC50 ~ 4 μM). In addition, the channel open probability when agonists are fully bound to the receptors is the highest for the GluN1/2A receptors (Po ~ 0.5), followed by the GluN1/2B receptors (Po ~ 0.1) and the GluN1/2C and GluN1/2D receptors (Po ~ 0.05). Importantly, each of the GluN1/2 di-heteromeric receptor subtypes display a wide-range of deactivation time constants. GluN2A-containing receptors have the fastest deactivation kinetic (τdecay ~ 50 ms), which is more than 20-fold higher than GluN2D-containing ones (τdecay > 1 s), whereas GluN1/2B (τdecay ~ 400 ms) and GluN1/2C (τdecay ~ 290 ms) show intermediate decay times. These GluN2 subunits also impart NMDAR subtypes with differences in sensitivity to extracellular Zn2+, protons, spermine and other pharmacological agonists, antagonists and allosteric modulators, which are described in detail in several excellent reviews (Hansen et al. 2018; Traynelis et al. 2010; Zhu & Paoletti 2015; Hackos & Hanson 2017). It is interesting to note that the tri-heteromeric GluN1/GluN2A/GluN2B receptors, possibly the most abundant NMDARs in the adult forebrain, display intermediate levels of agonist sensitivity, channel open probability, and deactivation kinetics, as well as in their sensitivity to ifenprodil (GluN2B specific blocker), Zn2+ and protons (Stroebel et al. 2018; Yi et al. 2017; Hansen et al. 2014). These differences in the biophysical and pharmacological properties of various GluN2 receptor subtypes determine the time-course of NMDAR-mediated EPSCs that affect synaptic integration and plasticity in various brain regions.
Spatiotemporal expression and localisation of GluN2-containing NMDARs
The expression of GluN2 subunits is differentially regulated during development across multiple brain regions. In rodents, both GluN2B and GluN2D subunits are widely expressed in the embryonic brain (Monyer et al. 1994; Ishii et al. 1993; Akazawa et al. 1994). The expression of GluN2B remains high, but becomes restricted to the forebrain, in the adult. By contrast, the expression of GluN2D is significantly decreased in the adult and can be found mostly in the mid-brain structures, including the diencephalon and mesencephalon. GluN2A expression starts at birth, increases progressively and becomes abundant throughout the central nervous system. Thus, in higher brain structures such as the cortex and hippocampus, NMDARs change from predominantly GluN2B-containing to GluN2A-containing during development (Sheng et al. 1994). Finally, GluN2C expression starts in the second postnatal week, but is confined to the cerebellum and the olfactory bulb. A similar developmental switch from GluN2B- to GluN2C-containing NMDARs also occurs in the cerebellar granule cells where GluN2B expression drops dramatically in the adult (Monyer et al. 1994).
In addition to their regional expression patterns in the brain, many different GluN2-containing NMDAR subtypes can also be found in distinct neuronal populations. For example, while the overall expression level of GluN2C and GluN2D is low in the cortex and hippocampus, they are specifically expressed in interneurons and glial cells (Monyer et al. 1994; Perszyk et al. 2016). Similarly, GluN2B and GluN2D are present in cerebellar Golgi cells despite their low expression in the cerebellum (Brickley et al. 2003). Remarkably, heterogeneity of synaptic NMDARs within individual neurons that are segregated in an input-specific manner also occurs. In the adult hippocampal CA3 region, GluN2B subunits are present in the dendrites of pyramidal neurons that receive inputs from the entorhinal cortex, but not from the dentate gyrus (Fritschy et al. 1998). In the layer 5 pyramidal neurons, GluN2A and GluN2B are differentially enriched at synapses of the callosal and the intracortical pathways, respectively (Kumar & Huguenard 2003).
Furthermore, the localization of different GluN2-containing NMDARs also varies at the subcellular level. In the adult forebrain, GluN2A-containing receptors are predominantly localized at synaptic sites, whereas GluN2B receptors are also found at the peri- or extra-synaptic sites (Bellone & Nicoll 2007; Hardingham & Bading 2010; Gladding & Raymond 2011). However, GluN2 subunits other than GluN2A can also mediate synaptic NMDAR currents in specific brain regions, such as in the amygdala, striatum and substantia nigra pars compacta (Lopez de Armentia & Sah 2003; Brothwell et al. 2008; Logan et al. 2007). Importantly, the subunit composition of synaptic NMDARs can be dynamically altered by activity and experience, allowing for fine regulation of receptor subtypes during plasticity. Interestingly, GluN2A and GluN2B-containing receptors were recently shown to organize in distinct nanodomains in the synapse and that this organization is also developmentally regulated (Kellermayer et al. 2018).
Activity-dependent switches of NMDAR subunit composition
During development, synapses possess GluN2B-containing NMDARs (Aizenman & Cline 2007; Barria & Malinow 2002) but no functional AMPARs, rendering them silent (Isaac et al. 1995; Isaac et al. 1997; Liao et al. 1995). For synapse maturation to occur, concomitant insertion of AMPARs (Leinekugel et al. 1997) and a switch in NMDAR subunit composition from GluN2B to GluN2A are necessary (Barria & Malinow 2002). This developmental switch is evolutionarily conserved and occurs in many brain areas, including cortex, hippocampus, amygdala and cerebellum. Due to the profound difference in their ion channel and gating properties, increased contribution of GluN2A subunits markedly changes the magnitude and timing of NMDAR-mediated Ca2+ influx, resulting in subsequent changes in dendritic integration and signaling underlying synaptic plasticity (Yashiro & Philpot 2008). The switch in NMDAR subunit composition is driven in part by developmental changes in the mRNA and protein expression of GluN2A and GluN2B, but also by sensory experience, indicating a direct contribution of neuronal activity in the process. In the developing visual cortex, the change from synaptic GluN2B- to GluN2A-containing NMDARs occurs during the critical period and is not observed in dark-reared animals, until they are exposed to light, and is reversible (Quinlan et al. 1999; Philpot et al. 2001). This process coincides with synaptic maturation, which is critical for circuit refinement, synaptic- and meta-plasticity (Yashiro & Philpot 2008; Sanz-Clemente et al. 2013b; Kirkwood et al. 1996).
A rapid switching (in minutes) of GluN2B- to GluN2A-containing NMDARs can be induced by an LTP inducing protocol in acute hippocampal slices from young animals (Bellone & Nicoll 2007). This type of plasticity is reversible and is dependent on group 1 metabotropic glutamate receptor (mGluR) signaling, intracellular Ca2+ release, protein kinase C (PKC) and phospholipase C (PLC) activation (Mameli et al. 2011; Matta et al. 2011). This process is also facilitated by the phosphorylation of the GluN2B subunit by casein kinase 2 (CK2), which promotes the internalization and removal of GluN2B receptors from the neuronal plasma membrane (Sanz-Clemente et al. 2010). Selective rearrangement of the relative ratio of GluN2A- and GluN2B-containing NMDARs within synaptic nanodomains bidirectionally tunes neuronal plasticity (Kellermayer et al. 2018). These data indicate the requirement of regulated receptor trafficking as a mechanism that determines the number and subunit composition of NMDARs at synapses.
Routes of NMDA Receptor Trafficking
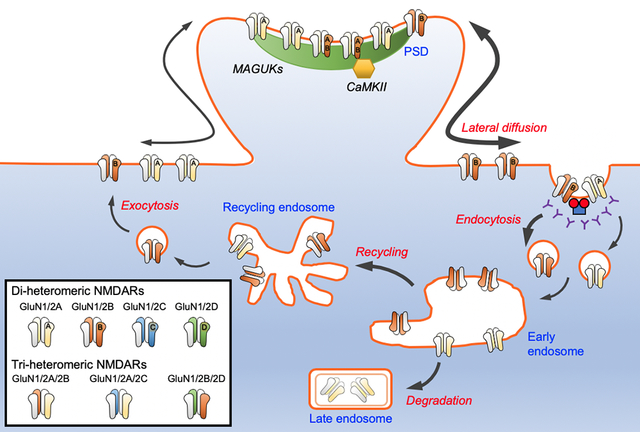
The number and subunit composition of NMDARs at synapses are tightly controlled by the delicate balance between the biosynthesis, dendritic transport, exocytosis, lateral diffusion, endocytosis, recycling and degradation of the receptors (Fig. 2). The cytoplasmic carboxyl termini of AMPA- and kainate receptors have been shown to play critical roles in orchestrating receptor trafficking to and stabilization at synapses (Anggono & Huganir 2012; Evans et al. 2019). Similarly, the mechanisms associated with NMDAR trafficking also involve the CTD, through which the receptors establish protein-protein interactions with intracellular trafficking, scaffolding and signaling molecules (Sanz-Clemente et al. 2013b). In addition, the CTD of GluN2 subunits undergoes a variety of post-translational modifications that function to regulate receptor trafficking and stabilization of NMDARs at synaptic sites (Lussier et al. 2015). The diversity of CTDs among different GluN2 subunits is therefore crucial in determining the subunit-specific trafficking of GluN2-containing NMDARs in mammalian central neurons.
Figure 2. Routes of NMDAR trafficking.

NMDARs are assembled in the ER and Golgi apparatus in the soma or in dendritic Golgi outposts. They are subsequently transported along the dendrite via kinesin-dependent vesicular trafficking on microtubule networks prior to their insertion onto the plasma membrane. Via lateral diffusion, surface NMDARs are incorporated into synapses and stabilized by PSD scaffolding proteins. NMDARs are internalized from the plasma membrane by clathrin-mediated endocytosis and trafficked to early endosomes. From early endosomes, receptors can be recycled back to the plasma membrane or can enter the degradation pathway to late endosomes. The trafficking of distinct GluN2 subunits are differentially regulated at multiple levels, including the rate of lateral diffusion, internalization and post-endocytic sorting.
Receptor biogenesis
Tetrameric NMDARs are assembled in the endoplasmic reticulum (ER) and mature through a series of glycosylation steps in the Golgi prior to their trafficking in vesicles to the plasma membrane (Horak et al. 2014). Cells have evolved strict mechanisms to ensure that no unassembled or misfolded NMDARs are transported out to the cell surface. Transfection of individual GluN2 and some GluN1 splice variants results in retention in the ER unless assembled as GluN1/GluN2 tetramers (McIlhinney et al. 1998). Importantly, genetic deletion of GluN1 leads to an accumulation of GluN2 subunits in the ER in the hippocampus (Fukaya et al. 2003). One of the underlying mechanisms is the presence of ER retention signals in different regions of GluN subunits, which are masked by complementary subunits when assembled into functional tetramers (Horak et al. 2008). For instance, the obligatory GluN1 subunit contains well characterized ER retention motifs (KKK and RRR) in the C-terminal C1 cassette (Standley et al. 2000; Scott et al. 2001; Horak & Wenthold 2009). Overexpression of GluN2A and GluN2B in the cerebellar granule neurons significantly promote the number and synaptic targeting of NMDARs (Prybylowski et al. 2002). The proximal C-terminal region of the GluN2B subunit has been proposed to also contain an ER retention motif (HLFY) (Hawkins et al. 2004). However, subsequent mutagenesis and deletion studies suggest that the HLFY motif is playing a structural role in orienting the C-terminal domain of GluN2B during ER processing, but it is not sufficient to act as a retention signal (Yang et al. 2007). The ER retention signals in GluN2 subunits are generally not well-defined to date.
An important step in the NMDAR secretory pathway lies in the interaction between GluN2 subunits with members of the membrane-associated guanylate kinase (MAGUK) family of proteins, particularly synapse-associated protein-102 kDa (SAP102) and −97 kDa (SAP97) (Sans et al. 2003; Sans et al. 2005). SAP102 is highly expressed in the hippocampus as early as postnatal day 2 and binds to both GluN2A and GluN2B subunits of NMDARs through their C-terminal postsynaptic density-95/discs-large/zona-occludens-1 (PDZ) binding motifs and a PDZ domain of SAP102 (Sans et al. 2000; Muller et al. 1996). Interestingly, SAP102 is localized not only at synapses, but also in the cytoplasm and ER (Sans et al. 2000). As a multi-PDZ domain containing protein, SAP102 acts as a scaffold for a complex containing the exocyst protein Sec8 and GluN2B (Sans et al. 2003). Furthermore, through its Src-homology 3 (SH3)/guanylate kinase (GK) domains, SAP102 can interact with mPins to stabilize the SAP102-exocyst-NMDAR complex in the ER, a process that is crucial for promoting the forward trafficking and membrane targeting of NMDARs in neurons (Sans et al. 2003; Sans et al. 2005).
NMDARs have also been proposed to bypass the conventional somatic Golgi network that is commonly associated with AMPAR trafficking. These receptors are instead sorted via an ER sub-compartment that directly merges with dendritic Golgi outposts (Jeyifous et al. 2009). Such an alternative strategy could signify a more efficient path to promote insertion of NMDARs at the postsynaptic density (PSD). Vesicles generated from this alternative pathway are highly mobile (0.76 μm/sec) as they contain large protein complexes containing the scaffolding molecules SAP97, CASK (mLin2), X11/Mint1 (mLin10) and mLin7 (MALS/Velis) that couple GluN2B to the motor protein KIF17 and facilitate the long-range transport of NMDAR-containing vesicles on microtubules along dendrites (Jeyifous et al. 2009; Setou et al. 2000; Guillaud et al. 2003). KIF17-mediated transport of NMDARs is essential for LTP, LTD, learning and memory (Yin et al. 2011). Genetic deletion of kif17 causes a significant, but not complete, loss of synaptic GluN2A and GluN2B receptors due to enhanced ubiquitin-mediated degradation of NMDARs. This suggests that other kinesin motor proteins could be involved in mediating the microtubule-based anterograde transport of NMDARs along dendrites. Indeed, KIF1B has been reported to interact directly with SAP97 and SAP102; however, its role in regulating the transport of NMDARs in the secretory pathway has not been determined (Mok et al. 2002). It is worth noting that the binding of CASK induces SAP97 into an extended confirmation that preferentially binds NMDARs relative to AMPARs (Lin et al. 2013). The function of SAP97 is tightly regulated by the phosphorylation of CaMKII on two key sites, Ser-39 (within the L27 domain) and Ser-232 (within the PDZ1 domain) (Gardoni et al. 2003; Mauceri et al. 2007). SAP97 phosphorylation at Ser-39 drives the translocation of SAP97 from the ER to the postsynaptic compartment, whereas its phosphorylation at Ser-232 disrupts SAP97 interaction with GluN2A. In addition, CaMKII-dependent phosphorylation of KIF17 has also been shown to promote the unloading of cargoes from microtubules (Yin et al. 2012). Thus, sequential phosphorylation of SAP97 and KIF17 may conceivably provide the driving force to release NMDAR complexes from the ER and facilitate the insertion of receptors at synapses.
Palmitoylation of NMDARs has recently emerged as an important regulator of post-Golgi trafficking of NMDARs (Thomas & Huganir 2013; Lussier et al. 2015). Both GluN2A and GluN2B subunits undergo activity-dependent palmitoylation at two distinct clusters in their CTDs (Hayashi et al. 2009). Palmitoylation of GluN2A and GluN2B in the second cluster of cysteines in the middle of their CTDs retains and accumulates NMDARs in the Golgi apparatus, and therefore reduces receptor surface expression (Hayashi et al. 2009). Consequently, mutations of cysteines in the second cluster leads to an increase in surface expression of NMDARs. However, these mutations do not increase the levels of synaptic NMDARs (Mattison et al. 2012). Importantly, in the YAC128 striatal neurons, aggregated mutant huntingtin proteins disrupt the interaction between two palmitoyl acetyltransferases, zDHHC13 and zDHHC17, and GluN2B, leading to a reduction in the level of GluN2B palmitoylation on cluster II (Kang et al. 2019). This subsequently causes an increase in the levels of extrasynaptic GluN2B-containing NMDARs, which may contribute to the cell death-signaling pathways underlying Huntington’s disease.
Exocytosis
Dendritic exocytosis of many neurotransmitter receptors, including NMDARs, is a tightly regulated process (Kennedy & Ehlers 2011). Exocytosis of NMDARs takes place mainly at extrasynaptic sites along the somatodendritic compartment (Gu & Huganir 2016), with the receptors then relocating to the synaptic membrane via lateral diffusion (Ladepeche et al. 2014). In general, vesicular fusion to the plasma membrane is mediated by the assembly soluble N-ethylmaleimide (NEM)-sensitive factor attachment protein receptors (SNAREs) protein family, including the target SNARE (t-SNARE) syntaxin, synaptosome-associated protein (SNAP) and the vesicular SNARE (v-SNARE) vesicle-associated membrane protein (VAMP) (Sudhof & Rothman 2009). Although the requirement of the SNARE complex in NMDAR exocytosis is well established, the precise contribution of specific members within this protein family remains controversial.
Earlier studies identified a critical role for SNAP25 in mediating the exocytosis of NMDARs, first in Xenopus oocytes and later in CA3 pyramidal neurons (Lan et al. 2001a; Lan et al. 2001b). In these studies, the authors demonstrated that mGluR1- and PKC-dependent exocytosis of NMDARs can be blocked by botulinum toxin A, a neurotoxin that specifically cleaves and inactivates SNAP25. Subsequently, SNAP25 was found to be a substrate of PKC and mutation of a single SNAP25 phosphorylation site (Ser-187) could abolish PKC-induced insertion of NMDARs (Lau et al. 2010). Consistent with this, shRNA-mediated knockdown of SNAP25 reduces NMDAR-mediated transmission in the hippocampus (Jurado et al. 2013).
In contrast, other studies have provided compelling evidence for another SNAP family member that is enriched at the excitatory synapses on dendritic spines, SNAP23, in regulating the forward trafficking of endogenous NMDARs (Washbourne et al. 2004; Suh et al. 2010). Genetic deletion of SNAP23 (SNAP23+/−) significantly reduces surface expression of endogenous GluN1, GluN2A and GluN2B subunits of NMDARs without affecting AMPAR levels (Suh et al. 2010). Furthermore, shRNA-mediated knockdown of SNAP23, but not of SNAP25, causes a similar decrease in the levels of surface NMDARs. The apparent discrepancies among these studies may arise from differences in the age and neuronal subtypes used, as well as in the choice of reagents and methodologies employed (knockout vs knockdown, endogenous vs overexpression). It is plausible that SNAP23 and SNAP25 may differentially regulate the exocytosis of NMDARs originating from separate intracellular pools (de novo insertion vs recycling) or during basal vs activity-dependent conditions.
In a more recent study, exocytosis of overexpressed super-ecliptic pH-sensitive green fluorescent protein (SEP)-tagged NMDARs was visualized directly in neurons using a total internal reflection florescence microscopy (Gu & Huganir 2016). This approach identified the SNAP25–VAMP1–syntaxin4 as the core SNARE complex essential for constitutive exocytosis of NMDARs. Consistent with this study, SNAP25 and syntaxin4 have previously been shown to mediate the group II mGluR-dependent increase in surface NMDARs in cortical pyramidal neurons (Cheng et al. 2013). Furthermore, genetic deletion of syntaxin4 leads to reduced surface expression of NMDARs and a deficit in LTP (Bin et al. 2018). Given the role of syntaxin4 as the t-SNARE for AMPARs (Kennedy et al. 2010; Bin et al. 2018), it raises the question whether both AMPARs and NMDARs are co-delivered to the plasma membrane on the same vesicles.
Lateral diffusion
NMDARs diffuse laterally on the plasma membrane between synaptic and extrasynaptic sites (Tovar & Westbrook 2002), an observation that upended the early dogma that NMDARs are static at synapses. This study employed electrophysiological recordings on cultured hippocampal neurons in the presence of NMDAR open-channel blockers, MK-801 or ketamine, and reported a rapid recovery of synaptic NMDAR currents, indicating the lateral movement and replacement of “unblocked” receptors from the extrasynaptic pools. In fact, advances in super-resolution microscopy and single-molecule tracking techniques have since provided direct evidence for the dynamic nature of NMDAR mobility on neuronal plasma membrane (Dupuis et al. 2014; Groc et al. 2004; Ladepeche et al. 2014). Lateral mobilities of synaptic AMPA- and NMDA-Rs are comparable, while extrasynaptic AMPARs are more mobile than NMDARs (Groc et al. 2004).
Remarkably, surface trafficking of NMDARs is regulated in a subunit-dependent manner that eventually determines the number and size of NMDAR nanoclusters within the synapse. There are several lines of evidence supporting the notion that extrasynaptically located GluN2B-containing NMDARs are more mobile than GluN2A, which remains more synaptically localized (Groc et al. 2006; Dupuis et al. 2014; Ferreira et al. 2017; Kellermayer et al. 2018). These two subtypes of NMDARs display substantial differences in their nanoscale organization, which changes during development in hippocampal neurons (Kellermayer et al. 2018). In mature synapses, there are considerably more GluN2A-NMDAR nanoclusters, which are also relatively larger, compared to those containing the GluN2B subunit. Synaptic retention of GluN2A requires its CTD, likely through its stable interaction with PDZ domain-containing scaffolding molecules such as PSD-95 (Kellermayer et al. 2018; Bard et al. 2010). Acute application of GluN2A-derived C-terminal peptides results in an increase in GluN2A diffusion rates and a reduction in the number of synaptic GluN2A-NMDARs. Remarkably, a similar cell-permeable peptide designed based on the GluN2B C-terminal PDZ binding motif has little effect on the synaptic retention of GluN2B-NMDARs, despite an increase in their diffusion rates that can be readily observed. These data suggest that subunit-specific nanoscale organization of NMDARs is differentially regulated by distinct sets of PDZ scaffolds at the synapse.
Furthermore, surface mobility of NMDARs can also be regulated by neuronal activity. During chemically-induced LTP (cLTP) in immature neurons, the diffusion rate of GluN2B-NMDARs increases, which results in the redistribution of GluN2B away from glutamatergic synapses, a process that requires the activities of CaMKII and CKII (Dupuis et al. 2014). In contrast, cLTP does not alter the surface diffusion of GluN2A-NMDARs and does not exert any effects on GluN2B-NMDARs in immature neurons. Interestingly, the binding of co-agonists, glycine and D-serine, also differentially affects the surface mobility of NMDARs in a subunit-specific manner (Ferreira et al. 2017; Papouin et al. 2012).
In addition to the interaction with PDZ scaffolds, surface trafficking of NMDARs is also regulated by protein phosphorylation on their CTDs. One such mechanism involves the phosphorylation of GluN2B at Ser-1480, which controls its interaction with PDZ-domain containing proteins (Bard et al. 2010; Chiu et al. 2019). Moreover, modulation of GluN2A phosphorylation at Ser-900 during development impacts NMDAR currents in retinocollicular synapses, in a calcineurin-dependent mechanism (Shi et al. 2000; Townsend et al. 2004), suggesting a role of this phosphatase in the regulation of the subunit switch. Other factors that are known to modulate NMDAR diffusion include the extracellular matrix (Michaluk et al. 2009), secreted proteases (Lesept et al. 2016), growth factors (De Rossi et al. 2016), cell surface receptors (Ladepeche et al. 2013; Mikasova et al. 2012) and hormones (Potier et al. 2016; Mikasova et al. 2017).
Endocytosis and post-endocytic sorting
Receptor internalization is a major mechanism by which neurons regulate the abundance and function of receptors at the plasma membrane. NMDAR endocytosis is developmentally and activity regulated, with younger neurons displaying a high rate of internalization, which decreases as neurons mature (Roche et al. 2001). The regulation of trafficking events like receptor endocytosis and recycling has been mostly studied in GluN2A- and GluN2B-containing NMDARs and several modulators and regulators of such events have been identified. This mechanism generally involves clathrin-coated pits, which occur laterally relative to the PSD (Petralia et al. 2003; Blanpied et al. 2002), and require the adaptor protein-2 (AP-2) complex (Racz et al. 2004; Lavezzari et al. 2004; Roche et al. 2001). Notably, glycine conditioning promotes receptor endocytosis by increasing the interaction with the endocytic AP-2 complex, thereby priming NMDARs for internalization upon receptor activation (Nong et al. 2003).
Both GluN2A and GluN2B subunits contain endocytic motifs in their CTD, but GluN2B subunits display a higher rate of endocytosis than GluN2A in mature neurons (Lavezzari et al. 2004). Clathrin-dependent internalization of GluN2B-containing NMDARs is mediated by the YEKL1472–1475 motif in the CTD, a few residues upstream of the PDZ-binding domain, which regulates MAGUK binding to the subunit (Lavezzari et al. 2003; Roche et al. 2001). This endocytic motif mediates the binding to the AP-2 complex, and this interaction is modulated by the phosphorylation state of Tyr-1472 by Fyn kinase. This phosphorylation leads to increased synaptic delivery of GluN2B-containing receptors and thereby enhanced NMDAR-mediated currents (Prybylowski et al. 2005). Notably, the analogous tyrosine in GluN2A (YKKM1454–1457) does not mediate endocytosis of the subunit. Instead, GluN2A contains a di-leucine motif (LL1319–1320) for regulating receptor internalization (Lavezzari et al. 2004). Such distinction in subunit endocytic regulation likely underlies the differential post-endocytic sorting of these two subunits.
GluN1 subunits also contribute to receptor internalization, as demonstrated by the endocytosis of heteromeric receptors containing GluN1/GluN2B-ΔCTD (Scott et al. 2004). In fact, two independent internalization motifs in the C0 cassette in GluN1 subunits are sufficient to mediate dynamin-dependent endocytosis of NMDARs: YKRH838–841 and VWRK858–861. Notably, both GluN2A (YWKL841–844) and GluN2B (YWQF842–845) contain similar motifs at the membrane proximal region of their CTDs, which have been shown to mediate dynamin-dependent endocytosis of the subunits. These membrane-proximal motifs were shown to target the receptors to the late endosome and lead to lysosomal degradation in all three subunits. Interestingly, while endocytosis can be driven by either GluN1 or GluN2 subunits, recycling of the receptors requires the functional GluN2 CTD.
A well-established mechanism of regulation of receptor surface expression is the phosphorylation/dephosphorylation of specific residues in the CTD of GluN2A and GluN2B subunits. Indeed, tyrosine phosphorylation of GluN2B at the endocytic motif YEKL was shown to contribute to receptor stabilization at synaptic sites by abrogating the interaction with the AP-2 complex subunits. Interestingly, palmitoylation of GluN2B within the first cluster of cysteines on the membrane proximal region exerts a synergistic effect in stabilizing surface NMDARs at synapses by increasing the phosphorylation of GluN2B at Tyr-1472 (Hayashi et al. 2009; Mattison et al. 2012). On the contrary, phosphorylation of Ser-1480, which lies on the PDZ-binding domain of GluN2B has the opposite effect, contributing to increased internalization of the receptor (Lavezzari et al. 2003; Prybylowski et al. 2005), due to impaired binding to MAGUK proteins (Chung et al. 2004). Phosphorylation of NMDARs on the YEKL motif can be decreased by the striatal-enriched protein tyrosine phosphatase (STEP), which directly interacts with NMDAR subunits and PSD-95 and whose downregulation leads to enhanced levels of GluN1, GluN2A and GluN2B subunits (Braithwaite et al. 2006; Won et al. 2016).
In addition to the modulatory role of post-translational modifications in surface expression (Chen & Roche 2007; Lussier et al. 2015), several scaffolding proteins have been identified as NMDAR regulators. The classical example of this is PSD-95, a member of the MAGUK family of scaffold proteins. Interaction with PSD-95 contributes to the stabilization of both GluN2 subunits at the cell surface, thereby reducing their rate of endocytosis (Lavezzari et al. 2004; Lavezzari et al. 2003; Roche et al. 2001). Another example is the protein Scribble1 (Scrib1), which was identified as an interactor of GluN2A and GluN2B subunits, via their PDZ-binding domains (Piguel et al. 2014). Scrib1 regulates surface expression of GluN2A/B subunits in an activity-dependent manner, by playing a modulatory role in endocytosis via interaction with the AP-2 complex, and recycling of these subunits.
Notably, GluN2A and GluN2B seem to display differential post-endocytic sorting. Although both subunits are initially present in early endosomes, the majority of GluN2A is sorted into late endosomes, as demonstrated by increased co-localization with the late endosomal marker Rab-9, whereas GluN2B is sorted into recycling endosomes, where it co-localizes with Rab-11 (Lavezzari et al. 2004; Scott et al. 2004). Interestingly, GluN2B seems to exert a dominant effect, as seen by preferential sorting into recycling endosomes of GluN2A/GluN2B heterodimers (Tang et al. 2010). This sorting into recycling endosomes could constitute a readily available pool of NMDARs to be inserted at the neuronal surface upon changes in synaptic activity and reflect the dynamic nature of NMDAR cycling before synaptic development when GluN2B is expressed, but not GluN2A.
As with endocytosis, neurons can respond to changes in synaptic activity by modulating the rate of vesicle recycling, which may involve specialized protein complexes, distinct from the de novo trafficking of receptors to the membrane. These mechanisms have been thoroughly studied in AMPARs but new light is now being shed on the specific regulation of NMDAR recycling. A few proteins have been proposed to participate in this process. Among these is the protein RIM1, via its interaction with Rab11, a marker of recycling endosomes. Knockdown of this protein specifically affected NMDAR transmission but had no impact on AMPAR currents (Wang et al. 2018). Furthermore, a recent report identified neurobeachin and KIF21B as regulators of GluN2B subunit recycling (Gromova et al. 2018). It should be noted that although mechanisms involving the retromer complex have not been extensively addressed in NMDAR regulation, recent evidence points to a role of this protein complex in modulation of surface expression of these receptors (Wang et al. 2013; Choy et al. 2014; Farias et al. 2014), by promoting receptor recycling to the membrane. Binding to this complex is proposed to be regulated by phosphorylation of specific residues in the PDZ-binding motif of GluN2 subunits (Clairfeuille et al. 2016).
GluN2 C-terminal Interacting Proteins
Genetic deletion of the C-termini (ΔC) of GluN2A, GluN2B and GluN2C in mice reveals an indispensable role of GluN2 CTDs for NMDAR-mediated intracellular signaling and synaptic physiology (Sprengel et al. 1998). Among the three GluN2ΔC/ΔC lines, GluN2BΔC/ΔC mice (lacking the entire or two thirds of the CTD) exhibit the most severe phenotype and die shortly after birth (Sprengel et al. 1998; Mori et al. 1998). Deletion of GluN2B CTD also reduces surface expression and synaptic retention of GluN2B receptors (Mori et al. 1998; Sprengel et al. 1998; Barria & Malinow 2002; Mohrmann et al. 2002). GluN2AΔC/ΔC mice show severe reductions in synaptic GluN2A expression and NMDAR-mediated EPSCs, as well as impairments in LTP and contextual memory (Sprengel et al. 1998; Steigerwald et al. 2000). GluN2CΔC/ΔC mice display impaired motor coordination and the GluN2AΔC/ΔC/GluN2CΔC/ΔC double mutant mice show defective cerebellar synaptic plasticity (Rossi et al. 2002; Sprengel et al. 1998). Importantly, studies using both organotypic hippocampal slices (Foster et al. 2010) or chimeric knock-in mice (GluN2A with GluN2B CTD and GluN2B with GluN2A CTD) (Ryan et al. 2013) have revealed the contribution of subunit-specific CTD of NMDARs to the regulation of synaptic signalling, plasticity and complex behaviour. Altogether, these studies provide strong evidence for the crucial roles that the CTD of GluN2 subunits and their C-terminal interacting proteins play in controlling the precise subcellular localisation, synaptic targeting and intracellular Ca2+ signaling of GluN2-containing NMDARs. In this review, we will focus only on a subset of key NMDAR-interacting proteins. Other proteins that have been reported to interact with NMDAR CTD are summarized in Table 1.
Table 1.
Other known NMDAR CTD interacting proteins
| NMDAR CTD Interacting Proteins | Subunit Specificity |
Molecular Functions | References | |||
|---|---|---|---|---|---|---|
| 2A | 2B | 2C | 2D | |||
| 14–3–3 | - | - | + y, h, b | - | Various isoforms of 14–3–3 adaptor proteins interact with GluN2C when phosphorylated at Ser-1096 by Akt to promote the surface expression of GluN2C in neurons. | (Chen & Roche 2009; Chung et al. 2015) |
| α-actinin | - | + y, h, b | n.d. | n.d. | The binding of α-actinin antagonizes the interaction of calmodulin to NMDARs (via GluN1) and is required for Ca2+-dependent inactivation of the receptors. The exact role of α-actinin binding to GluN2B is unknown. | (Wyszynski et al. 1997; Zhang et al. 1998; Krupp et al. 1999) |
| AP2 | + y, b | + y, b | n.d. | n.d. | AP2 binds to distinct endocytic motifs in the C-terminal tails of GluN2A and GluN2B to regulate the internalization of NMDARs | (Prybylowski et al. 2005; Nong et al. 2003; Lavezzari et al. 2003; Lavezzari et al. 2004) |
| BRAG1/2 | + h | + h | n.d. | n.d. | The brefeldin A-resistant ADP-ribosylation factor (Arf) guanine nucleotide exchange factors (GEF), BRAG1 and 2, interact with GluN2A and GluN2B in heterologous system. GluN2B-BRAG1 signaling is required for the tonic activation of Arf6 in young neurons, whereas GluN2A-BRAG2 drives NMDA-induced Arf6 activation in mature neurons. | (Elagabani et al. 2016) |
| C-terminal binding protein-1 (CtBP1) | + y, h | n.d. | n.d. | + y | CtBP1 is a known transcriptional repressor. Co-expression of CtBP1 results in a reduction in surface GluN1/2A expression in heterologous cells. | (Cousins & Stephenson 2019) |
| Flotillin | + y, b | + y, b | n.d. | n.d. | Flotillin binds to the distal C-terminal tails of GluN2A and GluN2B, consistent with the presence of NMDARs within the lipid rafts. The functional roles of their interaction remain elusive. | (Swanwick et al. 2009; Delint-Ramirez et al. 2010) |
| FRMPD2 | + h, b, i | + h, b. i | n.d. | n.d. | FRMDP2 is a multi-PDZ domain scaffold protein that interacts with GluN2A and GluN2B through its second PDZ domain. The function of FRMPD2 in regulating NMDAR functions is currently unknown. | (Lu et al. 2019) |
| GiPC | n.d. | + y, b | n.d. | n.d. | GiPC is a single PDZ domain protein that is associated with extrasynaptic NMDARs. GiPC regulates the surface expression of both GluN2A and GluN2B in cultured hippocampal neurons. | (Yi et al. 2007) |
| Nedd4–1 | + y | - | - | + yh, b, i | Nedd4–1 promotes poly-ubiquitination of GluN2D and reduces the number of functional cell surface GluN1/2D receptors in heterologous systems. | (Gautam et al. 2013) |
| Nck2 | n.d. | + i, h | n.d. | n.d. | Nck2 is an actin regulatory protein that interacts with phosphorylated GluN2B at Tyr-1252 to regulate NMDAR-mediated currents. | (Levy et al. 2018) |
| Rabphilin3A | + h, b | - | n.d. | n.d. | Raphilin3A directly interacts with GluN2A and PSD-95, forming a ternary complex to stabilize GluN2A-containing NMDARs at synapses. | (Stanic et al. 2015) |
| RasGRF1 | - | + h, b, i | n.d. | n.d. | RasGRF1 is a Ca2+/calmodulin-dependent Ras-guanine-nucleotide releasing factor that couples NMDARs to the downstream MAPK signaling pathway. | (Li et al. 2006; Krapivinsky et al. 2003) |
| Scribble1 | + y, h, b, i | + y, h, b, i | n.d. | n.d. | Scribble1 interacts with AP2 and together, coordinate the internalization and recycling of NMDARs. | (Piguel et al. 2014) |
| X11 and X11-L | n.d. | + h. b | n.d. | n.d. | X11 (Mint1) and X11-like (Mint2) are neuronal adaptor proteins that regulate kinesin-dependent vesicular trafficking and surface expression of extrasynaptic NMDARs. | (Motodate et al. 2019; Setou et al. 2000) |
Interactions between GluN2 subunits and their CTDs were validated by yyeast two-hybrid system, iin vitro binding assays, or co-immunoprecipitation/pull-down assays in hheterologous systems, nprimary neuronal cultures or bbrain lysates. n.d. = not determined.
MAGUKs
PSD-95 was the first member of the MAGUK family identified as the main scaffold protein that anchors and stabilizes surface NMDARs at glutamatergic synapses (Kornau et al. 1995; Niethammer et al. 1996). Other MAGUK family members, including SAP102, SAP97 and PSD-93 were subsequently shown to interact with the C-terminal type I PDZ binding motif (-ES[D/E]V) in the GluN2 subunits of NMDARs (Muller et al. 1996; Niethammer et al. 1996; Kim et al. 1996). These MAGUKs are defined by the presence of three conserved PDZ domains, one SH3 domain followed by a catalytically inactive GK modular domain (Long et al. 2003; McGee et al. 2001; Doyle et al. 1996). Although they share similar structural features, these proteins are vastly different in terms of their protein interaction and distribution profiles during development, and in different regions of the brain or subcellular compartments in neurons. For instance, both PSD-93 and PSD-95 are highly expressed at later stages of postnatal development, whereas SAP102 is expressed earlier in development (Sans et al. 2000). PSD-93 and PSD-95, as their names suggest, are found on the PSD; however, SAP97 and SAP102 are distributed along axons and dendrites as well as in cytoplasm and at synapses (Valtschanoff et al. 2000; Rumbaugh et al. 2003). With respect to protein-protein interactions, these MAGUK proteins demonstrate similar protein specificity in vitro; however, they exhibit a different specificity profile in vivo, such that PSD-95 has a preference for GluN2A, whereas SAP102 has a stronger affinity for GluN2B (Sans et al. 2000). In immature synapses, SAP102-GluN2B complexes are highly expressed, but as the synapses reach maturity, PSD-95/PSD-93-GluN2A are the predominant complexes (Sans et al. 2000; Elias et al. 2008).
MAGUKs play a critical role in the formation and maintenance of the PSD (Won et al. 2017). At the ultrastructural level, PSD-95 appears as vertically oriented filaments that anchor both AMPA- and NMDARs at the PSD (Chen et al. 2008). Genetic deletion or shRNA-mediated knockdown of PSD-95 results in the fragmentation of the PSD and a patchy loss of synaptic AMPARs, but not NMDARs (Chen et al. 2011b; Beique et al. 2006; Elias et al. 2006). Moreover, downregulation of the expression of individual MAGUK family members does not significantly affect NMDAR-mediated transmission (Elias et al. 2006; Howard et al. 2010). These data suggest that, at least for the maintenance of the number of NMDARs at synapses, these MAGUKs display some levels of functional redundancy and are able to compensate for the loss of function of an individual family member. However, simultaneous downregulation of two (PSD-95 and PSD-93) or three (PSD-95, PSD-93 and SAP102) MAGUKs causes a profound loss of functional synaptic glutamate receptors due to disintegration of the PSD structure (Elias et al. 2006; Chen et al. 2015; Levy et al. 2015). Indeed, overexpression of either SAP97 or SAP102 fully restores glutamatergic transmission in neurons derived from PSD-93/PSD-95 double-knockout mice (Elias et al. 2006; Howard et al. 2010).
SAP102 and PSD-95 both function to traffic and localise NMDARs in a subunit-dependent manner (Sans et al. 2000; Elias et al. 2008). SAP102 is expressed early in development and binds to GluN2B to mediate synaptogenesis. Following synaptogenesis, PSD-95 replaces the function of SAP102 to regulate the maturational switch of GluN2B to GluN2A. Knockdown of SAP102 during early development reduces both AMPAR- and NMDAR-mediated postsynaptic currents, whereas mice lacking PSD-95/PSD-93 fail to undergo the subunit switch from GluN2B to GluN2A (Elias et al. 2008), highlighting the role of PSD-95 and SAP102 at distinct developmental stages of excitatory synapses. Apart from the canonical PDZ binding of SAP102 to GluN2B, a secondary PDZ independent interaction to GluN2B was identified on the N-terminus of SAP102 (Chen et al. 2011a). The PDZ independent interaction is thought to mediate the removal of synaptic GluN2B containing NMDARs to the extra synaptic membrane (Chen et al. 2012a).
The clustering of NMDARs at synapses is mediated largely by an interaction between GluN2B with the PDZ domains of PSD-95. In accordance, the deletion of the C-terminal PDZ binding motif of GluN2B heightens its internalization rate (Roche et al. 2001) and alters the expression of synaptic GluN2A and GluN2B receptors (Sanz-Clemente et al. 2010). Moreover, overexpression of a C-terminally truncated form of the GluN2A and GluN2B subunits leads to impaired localization of synaptic NMDARs in vivo (Steigerwald et al. 2000). Phosphorylation of GluN2B at Ser-1480 by CK2 disrupts PSD-95 binding, leading to a significant reduction of surface and synaptic NMDARs, an important mechanism that plays a role in the GluN2B to GluN2A switch during development (Sanz-Clemente et al. 2010). More recently, PSD-95 has been shown to interact with the tyrosine phosphatase STEP61 and promotes its degradation via the ubiquitin-proteasome system (Won et al. 2016). STEP61 is known to dephosphorylate GluN2B at Tyr-1472, which is part of the YEKL endocytic motif, and induces the internalization of GluN2B (Snyder et al. 2005; Kurup et al. 2010). These data highlight multiple mechanisms exerted by PSD-95 in controlling the trafficking and stabilization of surface NMDARs.
Although binary interactions between individual GluN2 subunits with MAGUKs in vitro relies on these C-terminal PDZ binding motifs, a recent study has revealed that they are dispensable for the formation of the 1.5 mega-Dalton NMDAR-MAGUK supercomplexes in vivo (Frank et al. 2016; Frank & Grant 2017). The assembly of NMDAR-MAGUK supercomplexes follows the “tripartite rule” that describes the genetic requirement of PSD-95, PSD-93 and GluN2B. Interestingly, although the entire CTD of GluN2A is dispensable for the assembly of these supercomplexes, the C-terminal tail of GluN2B (~600 amino acids without the PDZ binding motif) is absolutely essential. Sequences upstream of the PDZ motifs are most variable among different GluN2 subunits, and they may possess different regulatory signals that confer distinct binding affinities towards different MAGUK or other PDZ domain-containing proteins (Bard et al. 2010; Clairfeuille et al. 2016; Frank et al. 2016; Chen et al. 2012b).
CaMKII
CaMKII is a multi-subunit protein kinase comprising 12 catalytically active subunits that is crucial for synaptic plasticity, learning and memory (Herring & Nicoll 2016; Lisman et al. 2012; Hell 2014). It is composed of mainly the α and β isoforms, is enriched in the PSD, and is one of the most abundant proteins in the brain (Chen et al. 2005). Each subunit present on the holoenzyme has a central organizing hub formed from its carboxy-terminal, a linker region, two rings of 6 catalytic kinase domains and a regulatory segment (S-site) that regulates the activity of the kinase domain (Hoelz et al. 2003). Under the autoinhibited state, the CaMKII dodecamer is inactive as the conformation of the enzyme is folded in a pseudosubstrate manner, in which the catalytic domain remains inaccessible for Ca2+/calmodulin binding. Upon NMDAR activation, Ca2+ binds to calmodulin, which in turns binds to the catalytic domain of CaMKII, alleviating autoinhibition and inducing autophosphorylation on Thr-286. The same stimulation also drives the translocation of CaMKII from the cytosol into the PSD (Shen & Meyer 1999; Bayer et al. 2001). This autophosphorylation allows the activity of CaMKII to persist long after the removal of Ca2+ and calmodulin in the PSD (Colbran & Brown 2004; Bayer & Schulman 2019).
A large body of evidence has demonstrated that CaMKIIα is both necessary and sufficient for NMDAR-dependent LTP (Silva et al. 1992; Giese et al. 1998; Pettit et al. 1994; Incontro et al. 2018). In one model, CaMKII-induced synaptic potentiation is associated with the phosphorylation and insertion of AMPARs into the synapses (Hayashi et al. 2000; Lee et al. 2003), as well as through an enhancement in AMPAR single channel conductance (Derkach et al. 1999; Kristensen et al. 2011). On the other hand, CaMKII also plays a critical structural role through its interaction with F-actin to maintain activity-dependent enlargement of dendritic spines during LTP (Bosch et al. 2014; Kim et al. 2016; Incontro et al. 2018). Given the localization, abundance, and catalytic activity, CaMKII is a versatile central organizer of the PSD during LTP.
One of the major interacting partners for CaMKII is the GluN2B subunit of NMDARs (Bayer et al. 2001; Leonard et al. 1999; Leonard et al. 2002; Strack & Colbran 1998). Their interaction is enhanced by NMDAR activity and requires either Ca2+/calmodulin or Thr-286 autophosphorylation. Genetic manipulations that disrupt the binding between CaMKII to NMDARs by either overexpression or the generation of knock-in mice containing point mutations on the CTD of GluN2B (R1300Q/S1303D or L1298A/R1300Q) or on the kinase domain of CaMKII (I205K) severely impair LTP (Barria & Malinow 2005; Halt et al. 2012; Incontro et al. 2018; Zhou et al. 2007). More recently, a role for CaMKIIα, but not CaMKIIβ, in controlling basal AMPAR and NMDAR neurotransmission has been reported (Incontro et al. 2018). Unlike AMPARs, NMDAR transmission does not require CaMKIIα kinase activity or its binding to GluN2B, suggesting that CaMKIIα exerts an important scaffolding role on NMDAR function.
CaMKIIα directly phosphorylates GluN2B on Ser-1303 in vitro (Omkumar et al. 1996), which is located within the CamKIIα binding site on the GluN2B CTD. Phosphomimetic GluN2B mutations on Ser-1303 reduce CaMKII/GluN2B binding (O’Leary et al. 2011; Strack et al. 2000) and subsequently result in an increase in surface GluN2B expression in neurons. Interestingly, activated CaMKII recruits CKII and GluN2B to form a tripartite complex, which is required to maintain the phosphorylation of GluN2B at Ser-1480 by CKII (Sanz-Clemente et al. 2013a). Phosphorylation of GluN2B at Ser-1480 disrupts its interaction with MAGUKs, which results in lateral diffusion of GluN2B to extrasynaptic sites and the internalization of NMDARs through a coordinated dephosphorylation of Tyr-1472 within the endocytic motif (Sanz-Clemente et al. 2010; Sanz-Clemente et al. 2013a; Chung et al. 2004). This mechanism has become a widely accepted model that underlies the developmental- and activity-dependent switches of NMDAR subunit composition at synapses. However, this model has recently been challenged by a study utilizing two lines of knock-in mice in which mutations on the CaMKII binding site on the GluN2B CTD were introduced (L1298A/R1300N/S1303D) or the entire GluN2A CTD was replaced by that of GluN2B (McKay et al. 2018). The developmental switch of GluN2B- to GluN2A-containing NMDARs proceeds normally in these mice, arguing against the need of distinct GluN2 subunit CTDs in this process. However, the effects on activity-dependent switches have not been formally tested.
Sorting nexin 27
Sorting nexin 27, which contains an N-terminal PDZ domain, a lipid-binding PX domain and three C-terminal FERM (4.1N/ezrin/radixin/moesin) domains, is a highly conserved regulator of cargo recycling from endosomes to the plasma membrane (Teasdale & Collins 2012). It is the only member of the SNX family that contains a PDZ domain through which it directly interacts with the C-terminal PDZ binding motifs of cargo proteins, including NMDARs (Clairfeuille et al. 2016; Cai et al. 2011). Loss of SNX27 leads to a profound decrease in surface expression of AMPA- and NMDA-Rs; and, as a consequence, SNX27 knockout mice display deficits in LTP and behavioural abnormalities (Wang et al. 2013; Hussain et al. 2014; Loo et al. 2014). Despite the clear involvement of SNX27 in regulating the surface expression of NMDARs, the critical question of how the interaction between SNX27 and NMDAR subunits is regulated remains elusive. Given the strong interaction between SNX27 and the retromer complex (Burd & Cullen 2014), it is likely that SNX27 is an important regulator of NMDAR recycling from endosomes to the plasma membrane.
A unique feature of the SNX27 PDZ domain binding to its cargo molecules involves the formation of an ‘electrostatic clamps’ by acidic residues at the (−3) and (−5) positions upstream of the PDZ binding motif (Clairfeuille et al. 2016). Interestingly, the corresponding amino acids at these critical positions within the CTD of all GluN2 subunits consist of mainly serine residues, suggesting that the binding affinity of SNX27 and NMDAR subunits can be modulated by protein phosphorylation of these serine residues. Indeed, the affinity of GluN2B binding to SNX27 PDZ increases 10-fold when these sites are phosphorylated or mutated to acidic residues (Clairfeuille et al. 2016), raising the possibility of a phosphorylation-dependent regulation of NMDAR recycling by SNX27.
Death-associated protein kinase 1 (DAPK1)
The role of NMDAR-mediated excitotoxicity, primarily via excessive Ca2+ influx through the extrasynaptic GluN2B-containing receptors, in the context of both acute and chronic neurological disorders including ischemic stroke and Alzheimer’s disease is well-established (Hardingham et al. 2002; Martel et al. 2012; Parsons & Raymond 2014). In a cerebral ischemia model, the death-associated protein kinase 1 (DAPK1) was discovered as a specific cell death signal when recruited to the extrasynaptic GluN2B-containing NMDARs (Tu et al. 2010). DAPK1 belongs to the CaMK family of protein kinases, which consists of an N-terminal kinase domain, a Ca2+/calmodulin-binding regulatory domain, eight ankyrin repeats, a cytoskeletal binding domain, a death domain and a Ser-rich C-terminal tail (Shiloh et al. 2014). Both the kinase and death domain are required for its proapoptotic activity (Cohen et al. 1997; Cohen et al. 1999).
DAPK1 directly interacts with the GluN2B CTD in a region that overlaps with the CaMKII binding site (Tu et al. 2010). Activated DAPK1 phosphorylates GluN2B at Ser-1303, which enhances GluN1/2B channel conductance and allows detrimental amount of Ca2+ influx, consequentially inducing infarction in the cortex of ischemic stroke mice. Genetic deletion of DAPK1 provides neuroprotective effects, including reduced infarct size and improved neurological behaviour in a stroke model in mice. However, a more recent study using an independently generated DAPK1 knockout mouse line fails to produce a similar protection of neuronal death following excitotoxic and ischemic insults (McQueen et al. 2017). Furthermore, neither DAPK1 nor ischemic insults enhance GluN2B phosphorylation at Ser-1303. Notwithstanding these differences, the CTD of GluN2B remains an important factor that mediates excitotoxic signaling in neurons (Martel et al. 2012).
The role of DAPK1 is not only confined as a mediator of cell death in neuropathology, but has recently emerged as a critical regulator of synaptic plasticity (Goodell et al. 2017). In contrast to the CaMKII/GluN2B interaction, DAPK1 binding to GluN2B is negatively regulated by Ca2+/calmodulin. As a consequence, CaMKII translocates into and accumulates in dendritic spines while DAPK1 is being removed by LTP stimuli. However, during LTD, DAPK1 is activated via a calcineurin-dependent dephosphorylation mechanism, accompanied by DAPK1-dependent GluN2B phosphorylation at Ser-1303. The phosphorylation of GluN2B strengthen its interaction with DAPK1 and prevents CaMKII binding and accumulation of in dendritic spines despite being auto-phosphorylated at Thr-286. Moreover, overexpression of DAPK1 prevents the accumulation CaMKII resulting in an inhibition of CaMKII-induced LTP. Therefore, the differential translocation of DAPK1 and CaMKII into dendritic spines provides a mechanism for bi-directional synaptic plasticity in the hippocampus.
NMDAR subunit-specific role in neurological disorders
NMDAR dysfunction has long been thought to have a paramount role in a variety of neurological disorders, ranging from acute (e.g. stroke and global cerebral ischemia) to chronic disorders such as Alzheimer’s or Huntington’s disease (Zhou & Sheng 2013; Ittner & Gotz 2011). These effects are mostly due to excitotoxicity, which results from excessive glutamate release from the presynaptic terminal. The term excitotoxicity was first used to describe neurodegeneration associated with the overactivation of excitatory amino acid receptors (Olney 1969). Indeed, a combination of excessive glutamate release from the presynaptic terminal and the failure of the mechanisms responsible for removing this neurotransmitter from the synapse leads to the accumulation of glutamate in the synaptic cleft. This subsequently leads to overactivation of iGluRs, particularly postsynaptic NMDARs, which causes excessive Ca2+ influx (Choi 1987; Choi et al. 1987). This overload of Ca2+ contributes to the activation of damaging Ca2+-dependent processes that trigger the initiation of cell death mechanisms and ensuing neurodegeneration in various brain regions (Arundine & Tymianski 2003; Choi 1988; Mehta et al. 2013; Tymianski & Tator 1996; Ong et al. 2013).
A long-standing debate in the field concerns the differential contribution of GluN2A- and GluN2B-containing NMDARs to excitotoxic neuronal death (Wyllie et al. 2013). GluN2B is generally thought to be a major mediator of neuronal degeneration (Aarts et al. 2002; Martel et al. 2012; Soriano et al. 2008). On the other hand, GluN2A is considered to have a pro-survival role (Liu et al. 2007; Terasaki et al. 2010). Notably, the CTD of GluN2 subunits is a major determinant for NMDAR-mediated toxicity (Martel et al. 2012; Vieira et al. 2016), as demonstrated by the enhanced neuronal demise in an excitotoxic context, elicited by the molecular swapping of the CTD of GluN2A for that of GluN2B. A complementary experiment involving the GluN2B chimera containing the CTD of GluN2A produces the opposite effect, promoting neuronal survival (Cepeda & Levine 2012; Martel et al. 2012; Vieira et al. 2016). Additional evidence supporting the concept of a detrimental role of the GluN2B subunit comes from several reports demonstrating a neuroprotective effect of interfering with activation of downstream signaling pathways, specifically the neuronal nitric oxide synthase (nNOS) pathway (Aarts et al. 2002; Cook et al. 2012; Cui et al. 2007). In fact, interfering peptides that disrupt the coupling of GluN2B to PSD-95 and nNOS (Aarts et al. 2002; Ittner et al. 2010), reducing nNOS activation and consequently nitrosative and oxidative stresses, have entered clinical trials and are efficacious in improving stroke patient outcome (Ballarin & Tymianski 2018; Hill et al. 2012).
Despite the data supporting a differential role of these two subunits in excitotoxic neuronal death, other reports also show evidence for a role of GluN2A in neuronal demise (Stanika et al. 2009; Zhou et al. 2013). This incongruence may be due to the contribution of triheteromeric receptors, as noted above. The most likely explanation, however, is that an interplay of factors beyond subunit composition, such as receptor localization, contributes to the decision between activation of pro-death versus pro-survival signaling pathways (Lai et al. 2011; Stanika et al. 2009; Zhou et al. 2013). In accordance with this notion, it has been observed that stimulation of extrasynaptic NMDARs contributes to activation of detrimental pathways. On the contrary, activation of synaptic NMDARs is considered to be neuroprotective (reviewed in (Hardingham & Bading 2010) and (Parsons & Raymond 2014)).
Mechanistically, synaptic NMDARs exert a neuroprotective effect via the activation of pro-survival transcription factors, such as CREB, and CREB-dependent expression of pro-survival genes. This effect is counteracted by activation of extrasynaptic NMDARs, which shuts-off CREB-dependent signaling (Dieterich et al. 2008; Hardingham & Bading 2002; Hardingham et al. 2002; Karpova et al. 2013). Extrasynaptic NMDARs also induce the activation of deleterious proteins, such as calpains (Xu et al. 2009). Overall, extrasynaptic NMDARs contribute to neuronal death by concomitantly suppressing pro-survival pathways and activating deleterious signaling cascades. Despite the plethora of evidence showing a pro-survival role of synaptic NMDARs, these have also been shown to be capable of inducing excitotoxic neuronal death (Papouin et al. 2012), thereby highlighting the intricate regulation of NMDARs and the consequences of their activation to neuronal fate.
In addition to the well-established link between NMDAR activity and excitotoxicity in neurodegeneration, recent discoveries from genetic studies have identified strong associations between mutations in genes encoding NMDAR subunits and neurological disorders, including autism-spectrum disorders (ASDs), intellectual disability and epilepsy. One of the greatest challenges in the search for the causative genes of neurodevelopmental disorders is the fact that there is not a single gene that causes autism or epilepsy. Instead, these are highly complex multigenic disorders for which hundreds of putative causative genes have been identified (Krumm et al. 2014) and share a high degree of co-morbidity (Ravizza et al. 2017; Sundelin et al. 2016). In accordance, there is a significant overlap in genes associated with ASDs, intellectual disability, epilepsy and schizophrenia (Hardingham & Do 2016; Helbig et al. 2009; International Schizophrenia 2008; Miller et al. 2009; Sharp et al. 2008; Stefansson et al. 2008). Among these, a cluster of synapse-associated genes have been identified (Gilman et al. 2011; Glessner et al. 2009; Krumm et al. 2014), which includes genes that encode various NMDAR subunits (De Rubeis et al. 2014; Sanders et al. 2015). Indeed, a significant number of rare variants of NMDAR subunits have been identified in probands of different neurological disorders (Hamdan et al. 2011; Endele et al. 2010; Hu et al. 2016; Yuan et al. 2015), which include both inherited and de novo copy number variations and single nucleotide variants.
NMDAR subunits are remarkably intolerant to variation, in particular GluN1, GluN2A and GluN2B (Ogden et al. 2017). One interesting aspect of these two genes – GRIN2A and GRIN2B – is that they seem to be preferentially associated with distinct disorders. Indeed, GRIN2A variants are closely linked with epilepsy (Carvill et al. 2013; Lesca et al. 2019; Lesca et al. 2013), whereas those found in GRIN2B are more commonly associated with ASDs and intellectual disability (Kenny et al. 2014; Sanders et al. 2015; De Rubeis et al. 2014; Iossifov et al. 2014). Functional characterization of most of GRIN2 variants is lacking, impeding our understanding on their impact on receptor function and activity. Despite this fact, it is evident that both hyper- and hypo-function of the subunits can be associated with disease (Swanger et al. 2016), including gene haploinsufficiency, which results from only one of the alleles being expressed in cells. A greater understanding of a wide range of functional outcomes resulting in mutations or variations in genes encoding NMDAR subunits may help explain the varying degrees of phenotypic severity observed in patients harboring mutations in these genes.
Such disparate observations in the functional impact of different variants can arise due to either evolutionary constraints to variation (Petrovski et al. 2013) or to the role exerted by the specific substitution in the amino acid residues. It is conceivable that any mutations in critical regions of NMDAR subunits that regulate ion channel properties or agonist binding are likely to be less tolerant to variation. Indeed, variants identified in the agonist binding domain (Swanger et al. 2016; Sceniak et al. 2019) or the channel forming regions (Ogden et al. 2017; Yuan et al. 2014; Fedele et al. 2018; Vyklicky et al. 2018) in both GRIN2A and GRIN2B are known to cause a wide spectrum of alterations in receptor function, including agonist potency, response time, channel gating, receptor biogenesis and trafficking.
Despite being generally considered less deleterious, mutations that reside in the CTD of GluN2 subunits have also been found to be damaging and potentially with disease-causing consequences. These variants are more likely to affect the molecular complex associated with the receptor, trafficking regulation and stability at synaptic sites. Therefore, mutations that affect these aspects of receptor function are likely to alter the surface expression of NMDARs, and consequently affects the downstream signaling pathways and neuronal plasticity. Accordingly, a recent study characterizing variants identified in human patients have demonstrated deficits in surface expression of NMDARs and in spine density (Liu et al. 2017). Many of these mutations have been reviewed in detail elsewhere (Burnashev & Szepetowski 2015; Hu et al. 2016), but new variants are still being identified. Accumulating evidence that supports the importance of NMDAR dysfunction in the etiology of neurodevelopmental and neuropsychiatric disorders makes them highly attractive targets for research into disease prevention and potential clinical intervention.
Closing remarks
The fact that NMDARs are critical for multiple forms of synaptic plasticity, learning and memory has continued to fascinate neuroscientists. Despite decades of research and progress in our understanding of the molecular mechanisms that underlie subunit-specific receptor assembly and trafficking into and out of synapses, many important questions remain unanswered. Research to date has focused heavily on the differential roles of GluN2A- and GluN2B-containing NMDARs, while subunit-specific regulation of GluN2C or GluN2D are not well studied. This is further complicated by the difficulty in studying the trafficking of tri-heteromeric NMDARs, which are thought to constitute a large proportion of native NMDARs in the brain. In addition, molecular mechanisms that regulate the trafficking of NMDARs in other neuronal subtypes, such as in interneurons or dopaminergic neurons are still not well understood. Future studies concerning subunit-specific interacting proteins and their roles in regulating the endosomal trafficking, supercomplex formation and neuronal plasticity-related signaling of NMDARs both in vitro and in vivo are crucial to better understand the regulation of NMDAR roles in higher brain functions, as well as the contribution of NMDAR dysfunction in neurological disorders.
Acknowledgements
This work was supported by the NINDS Intramural Research Program (KWR and MV), the Australian Research Council (DP190101390, VA), the Australian National Health and Medical Research Council (GNT1099114 and GNT1138452, VA) and the Clem Jones Centre for Ageing Dementia Research (VA). XLHY is supported by an Australian Government Research Training Scholarship. Figures were re-drawn by Marco Bazelmans in BioRender (https://biorender.com/) on the basis of a draft provided by the author. The authors have no conflict of interest to declare.
Abbreviations
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- AMPARs
AMPA receptors
- ASDs
autism-spectrum disorders
- ATD
amino terminal domain
- CaMKII
Ca2+/calmodulin-dependent kinase II
- CK2
casein kinase 2
- cLTP
chemically-induced LTP
- CTD
carboxyl terminal domain
- CREB
cAMP responsive element binding protein
- DAPK1
death-associated protein kinase 1
- EPSCs
excitatory postsynaptic currents
- ER
endoplasmic reticulum
- LBD
ligand binding domain
- LTD
long-term depression
- LTP
long-term potentiation
- MAGUKs
membrane-associated guanylate kinases
- mGluRs
metabotropic glutamate receptors
- NEM
N-ethylmaleimide
- NMDA
N-methyl-D-aspartate
- NMDARs
: NMDA receptors
- nNOS
neuronal nitric oxide synthase
- PDZ
postsynaptic density-95/discs-large/zona-occludens-1
- PKA
protein kinase A
- PKC
protein kinase C
- PSD
postsynaptic density
- SAP
synapse-associated protein
- SNARE
soluble NEM-sensitive factor attachment protein receptor
Footnotes
Conflicts of interest: none
=> if ‘none’, insert “The authors have no conflict of interest to declare.”
=> otherwise insert info unless it is already included
--Human subjects --
Involves human subjects:
If yes: Informed consent & ethics approval achieved:
=> if yes, please ensure that the info “Informed consent was achieved for all subjects, and the experiments were approved by the local ethics committee.” is included in the Methods.
ARRIVE guidelines have been followed:
Yes
=> if it is a Review or Editorial, skip complete sentence => if No, include a statement in the “Conflict of interest disclosure” section: “ARRIVE guidelines were not followed for the following reason:”
(edit phrasing to form a complete sentence as necessary).
=> if Yes, insert in the “Conflict of interest disclosure” section:
“All experiments were conducted in compliance with the ARRIVE guidelines.” unless it is a Review or Editorial
References
- Aarts M, Liu Y, Liu L, Besshoh S, Arundine M, Gurd JW, Wang YT, Salter MW and Tymianski M (2002) Treatment of ischemic brain damage by perturbing NMDA receptor- PSD-95 protein interactions. Science, 298, 846–850. [DOI] [PubMed] [Google Scholar]
- Aizenman CD and Cline HT (2007) Enhanced visual activity in vivo forms nascent synapses in the developing retinotectal projection. J Neurophysiol, 97, 2949–2957. [DOI] [PubMed] [Google Scholar]
- Akazawa C, Shigemoto R, Bessho Y, Nakanishi S and Mizuno N (1994) Differential expression of five N-methyl-D-aspartate receptor subunit mRNAs in the cerebellum of developing and adult rats. J Comp Neurol, 347, 150–160. [DOI] [PubMed] [Google Scholar]
- Anggono V and Huganir RL (2012) Regulation of AMPA receptor trafficking and synaptic plasticity. Curr Opin Neurobiol, 22, 461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arundine M and Tymianski M (2003) Molecular mechanisms of calcium-dependent neurodegeneration in excitotoxicity. Cell Calcium, 34, 325–337. [DOI] [PubMed] [Google Scholar]
- Ballarin B and Tymianski M (2018) Discovery and development of NA-1 for the treatment of acute ischemic stroke. Acta Pharmacol Sin, 39, 661–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard L, Sainlos M, Bouchet D et al. (2010) Dynamic and specific interaction between synaptic NR2-NMDA receptor and PDZ proteins. Proc Natl Acad Sci U S A, 107, 19561–19566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barria A and Malinow R (2002) Subunit-specific NMDA receptor trafficking to synapses. Neuron, 35, 345–353. [DOI] [PubMed] [Google Scholar]
- Barria A and Malinow R (2005) NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron, 48, 289–301. [DOI] [PubMed] [Google Scholar]
- Bayer KU, De Koninck P, Leonard AS, Hell JW and Schulman H (2001) Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature, 411, 801–805. [DOI] [PubMed] [Google Scholar]
- Bayer KU and Schulman H (2019) CaM Kinase: Still Inspiring at 40. Neuron, 103, 380–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beique JC, Lin DT, Kang MG, Aizawa H, Takamiya K and Huganir RL (2006) Synapse-specific regulation of AMPA receptor function by PSD-95. Proc Natl Acad Sci U S A, 103, 19535–19540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellone C and Nicoll RA (2007) Rapid bidirectional switching of synaptic NMDA receptors. Neuron, 55, 779–785. [DOI] [PubMed] [Google Scholar]
- Bin NR, Ma K, Harada H et al. (2018) Crucial Role of Postsynaptic Syntaxin 4 in Mediating Basal Neurotransmission and Synaptic Plasticity in Hippocampal CA1 Neurons. Cell Rep, 23, 2955–2966. [DOI] [PubMed] [Google Scholar]
- Blanpied TA, Scott DB and Ehlers MD (2002) Dynamics and regulation of clathrin coats at specialized endocytic zones of dendrites and spines. Neuron, 36, 435–449. [DOI] [PubMed] [Google Scholar]
- Bosch M, Castro J, Saneyoshi T, Matsuno H, Sur M and Hayashi Y (2014) Structural and molecular remodeling of dendritic spine substructures during long-term potentiation. Neuron, 82, 444–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braithwaite SP, Adkisson M, Leung J, Nava A, Masterson B, Urfer R, Oksenberg D and Nikolich K (2006) Regulation of NMDA receptor trafficking and function by striatal-enriched tyrosine phosphatase (STEP). Eur J Neurosci, 23, 2847–2856. [DOI] [PubMed] [Google Scholar]
- Brickley SG, Misra C, Mok MH, Mishina M and Cull-Candy SG (2003) NR2B and NR2D subunits coassemble in cerebellar Golgi cells to form a distinct NMDA receptor subtype restricted to extrasynaptic sites. J Neurosci, 23, 4958–4966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brothwell SL, Barber JL, Monaghan DT, Jane DE, Gibb AJ and Jones S (2008) NR2B- and NR2D-containing synaptic NMDA receptors in developing rat substantia nigra pars compacta dopaminergic neurones. J Physiol, 586, 739–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burd C and Cullen PJ (2014) Retromer: a master conductor of endosome sorting. Cold Spring Harb Perspect Biol, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnashev N and Szepetowski P (2015) NMDA receptor subunit mutations in neurodevelopmental disorders. Curr Opin Pharmacol, 20, 73–82. [DOI] [PubMed] [Google Scholar]
- Cai L, Loo LS, Atlashkin V, Hanson BJ and Hong W (2011) Deficiency of sorting nexin 27 (SNX27) leads to growth retardation and elevated levels of N-methyl-D-aspartate receptor 2C (NR2C). Mol. Cell. Biol, 31, 1734–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvill GL, Regan BM, Yendle SC et al. (2013) GRIN2A mutations cause epilepsy-aphasia spectrum disorders. Nat Genet, 45, 1073–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C and Levine MS (2012) 2B or not 2B: a tail of two NMDA receptor subunits. Neuron, 74, 426–428. [DOI] [PubMed] [Google Scholar]
- Chazot PL, Coleman SK, Cik M and Stephenson FA (1994) Molecular characterization of N-methyl-D-aspartate receptors expressed in mammalian cells yields evidence for the coexistence of three subunit types within a discrete receptor molecule. J Biol Chem, 269, 24403–24409. [PubMed] [Google Scholar]
- Chen BS, Gray JA, Sanz-Clemente A, Wei Z, Thomas EV, Nicoll RA and Roche KW (2012a) SAP102 mediates synaptic clearance of NMDA receptors. Cell Rep, 2, 1120–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BS, Gray JA, Sanz-Clemente A, Wei Z, Thomas EV, Nicoll RA and Roche KW (2012b) SAP102 Mediates Synaptic Clearance of NMDA Receptors. Cell Rep. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BS and Roche KW (2007) Regulation of NMDA receptors by phosphorylation. Neuropharmacology, 53, 362–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BS and Roche KW (2009) Growth factor-dependent trafficking of cerebellar NMDA receptors via protein kinase B/Akt phosphorylation of NR2C. Neuron, 62, 471–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BS, Thomas EV, Sanz-Clemente A and Roche KW (2011a) NMDA receptor-dependent regulation of dendritic spine morphology by SAP102 splice variants. J Neurosci, 31, 89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Levy JM, Hou A, Winters C, Azzam R, Sousa AA, Leapman RD, Nicoll RA and Reese TS (2015) PSD-95 family MAGUKs are essential for anchoring AMPA and NMDA receptor complexes at the postsynaptic density. Proc Natl Acad Sci U S A, 112, E6983–6992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Nelson CD, Li X et al. (2011b) PSD-95 is required to sustain the molecular organization of the postsynaptic density. J Neurosci, 31, 6329–6338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Vinade L, Leapman RD, Petersen JD, Nakagawa T, Phillips TM, Sheng M and Reese TS (2005) Mass of the postsynaptic density and enumeration of three key molecules. Proc Natl Acad Sci U S A, 102, 11551–11556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Winters C, Azzam R, Li X, Galbraith JA, Leapman RD and Reese TS (2008) Organization of the core structure of the postsynaptic density. Proc Natl Acad Sci U S A, 105, 4453–4458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, Liu W, Duffney LJ and Yan Z (2013) SNARE proteins are essential in the potentiation of NMDA receptors by group II metabotropic glutamate receptors. J Physiol, 591, 3935–3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu AM, Wang J, Fiske MP, Hubalkova P, Barse L, Gray JA and Sanz-Clemente A (2019) NMDAR-Activated PP1 Dephosphorylates GluN2B to Modulate NMDAR Synaptic Content. Cell Rep, 28, 332–341 e335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW (1987) Ionic dependence of glutamate neurotoxicity. J Neurosci, 7, 369–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW (1988) Glutamate neurotoxicity and diseases of the nervous system. Neuron, 1, 623–634. [DOI] [PubMed] [Google Scholar]
- Choi DW, Maulucci-Gedde M and Kriegstein AR (1987) Glutamate neurotoxicity in cortical cell culture. J Neurosci, 7, 357–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy RW, Park M, Temkin P, Herring BE, Marley A, Nicoll RA and von Zastrow M (2014) Retromer mediates a discrete route of local membrane delivery to dendrites. Neuron, 82, 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung C, Wu WH and Chen BS (2015) Identification of Novel 14–3-3 Residues That Are Critical for Isoform-specific Interaction with GluN2C to Regulate N-Methyl-D-aspartate (NMDA) Receptor Trafficking. J Biol Chem, 290, 23188–23200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung HJ, Huang YH, Lau LF and Huganir RL (2004) Regulation of the NMDA receptor complex and trafficking by activity-dependent phosphorylation of the NR2B subunit PDZ ligand. J Neurosci, 24, 10248–10259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clairfeuille T, Mas C, Chan AS et al. (2016) A molecular code for endosomal recycling of phosphorylated cargos by the SNX27-retromer complex. Nat Struct Mol Biol, 23, 921–932. [DOI] [PubMed] [Google Scholar]
- Cohen O, Feinstein E and Kimchi A (1997) DAP-kinase is a Ca2+/calmodulin-dependent, cytoskeletal-associated protein kinase, with cell death-inducing functions that depend on its catalytic activity. EMBO J, 16, 998–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen O, Inbal B, Kissil JL, Raveh T, Berissi H, Spivak-Kroizaman T, Feinstein E and Kimchi A (1999) DAP-kinase participates in TNF-alpha- and Fas-induced apoptosis and its function requires the death domain. J Cell Biol, 146, 141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colbran RJ and Brown AM (2004) Calcium/calmodulin-dependent protein kinase II and synaptic plasticity. Curr Opin Neurobiol, 14, 318–327. [DOI] [PubMed] [Google Scholar]
- Cook DJ, Teves L and Tymianski M (2012) Treatment of stroke with a PSD-95 inhibitor in the gyrencephalic primate brain. Nature, 483, 213–217. [DOI] [PubMed] [Google Scholar]
- Cousins SL and Stephenson FA (2019) Identification of C-Terminal Binding Protein 1 as a Novel NMDA Receptor Interactor. Neurochem Res, 44, 1437–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui H, Hayashi A, Sun HS et al. (2007) PDZ protein interactions underlying NMDA receptor-mediated excitotoxicity and neuroprotection by PSD-95 inhibitors. J Neurosci, 27, 9901–9915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rossi P, Harde E, Dupuis JP et al. (2016) A critical role for VEGF and VEGFR2 in NMDA receptor synaptic function and fear-related behavior. Mol Psychiatry, 21, 1768–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rubeis S, He X, Goldberg AP et al. (2014) Synaptic, transcriptional and chromatin genes disrupted in autism. Nature, 515, 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delint-Ramirez I, Fernandez E, Bayes A, Kicsi E, Komiyama NH and Grant SG (2010) In vivo composition of NMDA receptor signaling complexes differs between membrane subdomains and is modulated by PSD-95 and PSD-93. J Neurosci, 30, 8162–8170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derkach V, Barria A and Soderling TR (1999) Ca2+/calmodulin-kinase II enhances channel conductance of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate type glutamate receptors. Proc Natl Acad Sci U S A, 96, 3269–3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieterich DC, Karpova A, Mikhaylova M et al. (2008) Caldendrin-Jacob: a protein liaison that couples NMDA receptor signalling to the nucleus. PLoS Biol, 6, e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle DA, Lee A, Lewis J, Kim E, Sheng M and MacKinnon R (1996) Crystal structures of a complexed and peptide-free membrane protein-binding domain: molecular basis of peptide recognition by PDZ. Cell, 85, 1067–1076. [DOI] [PubMed] [Google Scholar]
- Dupuis JP, Ladepeche L, Seth H et al. (2014) Surface dynamics of GluN2B-NMDA receptors controls plasticity of maturing glutamate synapses. EMBO J, 33, 842–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elagabani MN, Brisevac D, Kintscher M, Pohle J, Kohr G, Schmitz D and Kornau HC (2016) Subunit-selective N-Methyl-d-aspartate (NMDA) Receptor Signaling through Brefeldin A-resistant Arf Guanine Nucleotide Exchange Factors BRAG1 and BRAG2 during Synapse Maturation. J Biol Chem, 291, 9105–9118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias GM, Elias LA, Apostolides PF, Kriegstein AR and Nicoll RA (2008) Differential trafficking of AMPA and NMDA receptors by SAP102 and PSD-95 underlies synapse development. Proc Natl Acad Sci U S A, 105, 20953–20958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias GM, Funke L, Stein V, Grant SG, Bredt DS and Nicoll RA (2006) Synapse-specific and developmentally regulated targeting of AMPA receptors by a family of MAGUK scaffolding proteins. Neuron, 52, 307–320. [DOI] [PubMed] [Google Scholar]
- Endele S, Rosenberger G, Geider K et al. (2010) Mutations in GRIN2A and GRIN2B encoding regulatory subunits of NMDA receptors cause variable neurodevelopmental phenotypes. Nat Genet, 42, 1021–1026. [DOI] [PubMed] [Google Scholar]
- Evans AJ, Gurung S, Henley JM, Nakamura Y and Wilkinson KA (2019) Exciting Times: New Advances Towards Understanding the Regulation and Roles of Kainate Receptors. Neurochem Res, 44, 572–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farias GG, Gershlick DC and Bonifacino JS (2014) Going forward with retromer. Dev Cell, 29, 3–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedele L, Newcombe J, Topf M, Gibb A, Harvey RJ and Smart TG (2018) Disease-associated missense mutations in GluN2B subunit alter NMDA receptor ligand binding and ion channel properties. Nat Commun, 9, 957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira JS, Papouin T, Ladepeche L et al. (2017) Co-agonists differentially tune GluN2B-NMDA receptor trafficking at hippocampal synapses. Elife, 6 e25492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster KA, McLaughlin N, Edbauer D, Phillips M, Bolton A, Constantine-Paton M and Sheng M (2010) Distinct roles of NR2A and NR2B cytoplasmic tails in long-term potentiation. J Neurosci, 30, 2676–2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank RA and Grant SG (2017) Supramolecular organization of NMDA receptors and the postsynaptic density. Curr Opin Neurobiol, 45, 139–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank RA, Komiyama NH, Ryan TJ, Zhu F, O’Dell TJ and Grant SG (2016) NMDA receptors are selectively partitioned into complexes and supercomplexes during synapse maturation. Nat Commun, 7, 11264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritschy JM, Weinmann O, Wenzel A and Benke D (1998) Synapse-specific localization of NMDA and GABA(A) receptor subunits revealed by antigen-retrieval immunohistochemistry. J Comp Neurol, 390, 194–210. [PubMed] [Google Scholar]
- Fukaya M, Kato A, Lovett C, Tonegawa S and Watanabe M (2003) Retention of NMDA receptor NR2 subunits in the lumen of endoplasmic reticulum in targeted NR1 knockout mice. Proc Natl Acad Sci U S A, 100, 4855–4860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardoni F, Mauceri D, Fiorentini C, Bellone C, Missale C, Cattabeni F and Di Luca M (2003) CaMKII-dependent phosphorylation regulates SAP97/NR2A interaction. J Biol Chem, 278, 44745–44752. [DOI] [PubMed] [Google Scholar]
- Gautam V, Trinidad JC, Rimerman RA, Costa BM, Burlingame AL and Monaghan DT (2013) Nedd4 is a specific E3 ubiquitin ligase for the NMDA receptor subunit GluN2D. Neuropharmacology, 74, 96–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese KP, Fedorov NB, Filipkowski RK and Silva AJ (1998) Autophosphorylation at Thr286 of the alpha calcium-calmodulin kinase II in LTP and learning. Science, 279, 870–873. [DOI] [PubMed] [Google Scholar]
- Gilman SR, Iossifov I, Levy D, Ronemus M, Wigler M and Vitkup D (2011) Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron, 70, 898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladding CM and Raymond LA (2011) Mechanisms underlying NMDA receptor synaptic/extrasynaptic distribution and function. Mol. Cell. Neurosci, 48, 308–320. [DOI] [PubMed] [Google Scholar]
- Glasgow NG, Siegler Retchless B and Johnson JW (2015) Molecular bases of NMDA receptor subtype-dependent properties. J Physiol, 593, 83–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glessner JT, Wang K, Cai G et al. (2009) Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature, 459, 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodell DJ, Zaegel V, Coultrap SJ, Hell JW and Bayer KU (2017) DAPK1 Mediates LTD by Making CaMKII/GluN2B Binding LTP Specific. Cell Rep, 19, 2231–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groc L, Heine M, Cognet L, Brickley K, Stephenson FA, Lounis B and Choquet D (2004) Differential activity-dependent regulation of the lateral mobilities of AMPA and NMDA receptors. Nat Neurosci, 7, 695–696. [DOI] [PubMed] [Google Scholar]
- Groc L, Heine M, Cousins SL, Stephenson FA, Lounis B, Cognet L and Choquet D (2006) NMDA receptor surface mobility depends on NR2A-2B subunits. Proc Natl Acad Sci U S A, 103, 18769–18774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromova KV, Muhia M, Rothammer N et al. (2018) Neurobeachin and the Kinesin KIF21B Are Critical for Endocytic Recycling of NMDA Receptors and Regulate Social Behavior. Cell Rep, 23, 2705–2717. [DOI] [PubMed] [Google Scholar]
- Gu Y and Huganir RL (2016) Identification of the SNARE complex mediating the exocytosis of NMDA receptors. Proc Natl Acad Sci U S A, 113, 12280–12285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillaud L, Setou M and Hirokawa N (2003) KIF17 dynamics and regulation of NR2B trafficking in hippocampal neurons. J Neurosci, 23, 131–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackos DH and Hanson JE (2017) Diverse modes of NMDA receptor positive allosteric modulation: Mechanisms and consequences. Neuropharmacology, 112, 34–45. [DOI] [PubMed] [Google Scholar]
- Halt AR, Dallapiazza RF, Zhou Y et al. (2012) CaMKII binding to GluN2B is critical during memory consolidation. EMBO J, 31, 1203–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdan FF, Gauthier J, Araki Y et al. (2011) Excess of de novo deleterious mutations in genes associated with glutamatergic systems in nonsyndromic intellectual disability. Am J Hum Genet, 88, 306–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KB, Ogden KK, Yuan H and Traynelis SF (2014) Distinct functional and pharmacological properties of Triheteromeric GluN1/GluN2A/GluN2B NMDA receptors. Neuron, 81, 1084–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KB, Yi F, Perszyk RE, Furukawa H, Wollmuth LP, Gibb AJ and Traynelis SF (2018) Structure, function, and allosteric modulation of NMDA receptors. J Gen Physiol, 150, 1081–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KB, Yi F, Perszyk RE, Menniti FS and Traynelis SF (2017) NMDA Receptors in the Central Nervous System. Methods Mol Biol, 1677, 1–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE and Bading H (2002) Coupling of extrasynaptic NMDA receptors to a CREB shut-off pathway is developmentally regulated. Biochim Biophys Acta, 1600, 148–153. [DOI] [PubMed] [Google Scholar]
- Hardingham GE and Bading H (2010) Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci, 11, 682–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE and Do KQ (2016) Linking early-life NMDAR hypofunction and oxidative stress in schizophrenia pathogenesis. Nat Rev Neurosci, 17, 125–134. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y and Bading H (2002) Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci, 5, 405–414. [DOI] [PubMed] [Google Scholar]
- Hawkins LM, Prybylowski K, Chang K, Moussan C, Stephenson FA and Wenthold RJ (2004) Export from the endoplasmic reticulum of assembled N-methyl-d-aspartic acid receptors is controlled by a motif in the c terminus of the NR2 subunit. J Biol Chem, 279, 28903–28910. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Thomas GM and Huganir RL (2009) Dual palmitoylation of NR2 subunits regulates NMDA receptor trafficking. Neuron, 64, 213–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC and Malinow R (2000) Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science, 287, 2262–2267. [DOI] [PubMed] [Google Scholar]
- Helbig I, Mefford HC, Sharp AJ et al. (2009) 15q13.3 microdeletions increase risk of idiopathic generalized epilepsy. Nat Genet, 41, 160–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hell JW (2014) CaMKII: claiming center stage in postsynaptic function and organization. Neuron, 81, 249–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herring BE and Nicoll RA (2016) Long-Term Potentiation: From CaMKII to AMPA Receptor Trafficking. Annu Rev Physiol, 78, 351–365. [DOI] [PubMed] [Google Scholar]
- Hill MD, Martin RH, Mikulis D et al. (2012) Safety and efficacy of NA-1 in patients with iatrogenic stroke after endovascular aneurysm repair (ENACT): a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol, 11, 942–950. [DOI] [PubMed] [Google Scholar]
- Hoelz A, Nairn AC and Kuriyan J (2003) Crystal structure of a tetradecameric assembly of the association domain of Ca2+/calmodulin-dependent kinase II. Mol Cell, 11, 1241–1251. [DOI] [PubMed] [Google Scholar]
- Horak M, Chang K and Wenthold RJ (2008) Masking of the endoplasmic reticulum retention signals during assembly of the NMDA receptor. J Neurosci, 28, 3500–3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horak M, Petralia RS, Kaniakova M and Sans N (2014) ER to synapse trafficking of NMDA receptors. Front Cell Neurosci, 8, 394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horak M and Wenthold RJ (2009) Different roles of C-terminal cassettes in the trafficking of full-length NR1 subunits to the cell surface. J Biol Chem, 284, 9683–9691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard MA, Elias GM, Elias LA, Swat W and Nicoll RA (2010) The role of SAP97 in synaptic glutamate receptor dynamics. Proc Natl Acad Sci U S A, 107, 3805–3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C, Chen W, Myers SJ, Yuan H and Traynelis SF (2016) Human GRIN2B variants in neurodevelopmental disorders. J Pharmacol Sci, 132, 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain NK, Diering GH, Sole J, Anggono V and Huganir RL (2014) Sorting nexin 27 regulates basal and activity-dependent trafficking of AMPARs. Proc Natl Acad Sci U S A, 111, 11840–11845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Incontro S, Diaz-Alonso J, Iafrati J, Vieira M, Asensio CS, Sohal VS, Roche KW, Bender KJ and Nicoll RA (2018) The CaMKII/NMDA receptor complex controls hippocampal synaptic transmission by kinase-dependent and independent mechanisms. Nat Commun, 9, 2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Schizophrenia C (2008) Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature, 455, 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, O’Roak BJ, Sanders SJ et al. (2014) The contribution of de novo coding mutations to autism spectrum disorder. Nature, 515, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaac JT, Crair MC, Nicoll RA and Malenka RC (1997) Silent synapses during development of thalamocortical inputs. Neuron, 18, 269–280. [DOI] [PubMed] [Google Scholar]
- Isaac JT, Nicoll RA and Malenka RC (1995) Evidence for silent synapses: implications for the expression of LTP. Neuron, 15, 427–434. [DOI] [PubMed] [Google Scholar]
- Ishii T, Moriyoshi K, Sugihara H et al. (1993) Molecular characterization of the family of the N-methyl-D-aspartate receptor subunits. J Biol Chem, 268, 2836–2843. [PubMed] [Google Scholar]
- Ittner LM and Gotz J (2011) Amyloid-beta and tau--a toxic pas de deux in Alzheimer’s disease. Nat Rev Neurosci, 12, 65–72. [DOI] [PubMed] [Google Scholar]
- Ittner LM, Ke YD, Delerue F et al. (2010) Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell, 142, 387–397. [DOI] [PubMed] [Google Scholar]
- Jeyifous O, Waites CL, Specht CG et al. (2009) SAP97 and CASK mediate sorting of NMDA receptors through a previously unknown secretory pathway. Nat Neurosci, 12, 1011–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S and Gibb AJ (2005) Functional NR2B- and NR2D-containing NMDA receptor channels in rat substantia nigra dopaminergic neurones. J Physiol, 569, 209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurado S, Goswami D, Zhang Y, Molina AJ, Sudhof TC and Malenka RC (2013) LTP requires a unique postsynaptic SNARE fusion machinery. Neuron, 77, 542–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang R, Wang L, Sanders SS, Zuo K, Hayden MR and Raymond LA (2019) Altered Regulation of Striatal Neuronal N-Methyl-D-Aspartate Receptor Trafficking by Palmitoylation in Huntington Disease Mouse Model. Front Synaptic Neurosci, 11, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpova A, Mikhaylova M, Bera S et al. (2013) Encoding and transducing the synaptic or extrasynaptic origin of NMDA receptor signals to the nucleus. Cell, 152, 1119–1133. [DOI] [PubMed] [Google Scholar]
- Kellermayer B, Ferreira JS, Dupuis J et al. (2018) Differential Nanoscale Topography and Functional Role of GluN2-NMDA Receptor Subtypes at Glutamatergic Synapses. Neuron, 100, 106–119 [DOI] [PubMed] [Google Scholar]
- Kennedy MJ, Davison IG, Robinson CG and Ehlers MD (2010) Syntaxin-4 defines a domain for activity-dependent exocytosis in dendritic spines. Cell, 141, 524–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy MJ and Ehlers MD (2011) Mechanisms and function of dendritic exocytosis. Neuron, 69, 856–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny EM, Cormican P, Furlong S et al. (2014) Excess of rare novel loss-of-function variants in synaptic genes in schizophrenia and autism spectrum disorders. Mol Psychiatry, 19, 872–879. [DOI] [PubMed] [Google Scholar]
- Kim E, Cho KO, Rothschild A and Sheng M (1996) Heteromultimerization and NMDA receptor-clustering activity of Chapsyn-110, a member of the PSD-95 family of proteins. Neuron, 17, 103–113. [DOI] [PubMed] [Google Scholar]
- Kim K, Saneyoshi T, Hosokawa T, Okamoto K and Hayashi Y (2016) Interplay of enzymatic and structural functions of CaMKII in long-term potentiation. J Neurochem, 139, 959–972. [DOI] [PubMed] [Google Scholar]
- Kirkwood A, Rioult MC and Bear MF (1996) Experience-dependent modification of synaptic plasticity in visual cortex. Nature, 381, 526–528. [DOI] [PubMed] [Google Scholar]
- Kornau HC, Schenker LT, Kennedy MB and Seeburg PH (1995) Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science, 269, 1737–1740. [DOI] [PubMed] [Google Scholar]
- Krapivinsky G, Krapivinsky L, Manasian Y, Ivanov A, Tyzio R, Pellegrino C, Ben-Ari Y, Clapham DE and Medina I (2003) The NMDA receptor is coupled to the ERK pathway by a direct interaction between NR2B and RasGRF1. Neuron, 40, 775–784. [DOI] [PubMed] [Google Scholar]
- Kristensen AS, Jenkins MA, Banke TG, Schousboe A, Makino Y, Johnson RC, Huganir R and Traynelis SF (2011) Mechanism of Ca2+/calmodulin-dependent kinase II regulation of AMPA receptor gating. Nat Neurosci, 14, 727–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumm N, O’Roak BJ, Shendure J and Eichler EE (2014) A de novo convergence of autism genetics and molecular neuroscience. Trends Neurosci, 37, 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupp JJ, Vissel B, Thomas CG, Heinemann SF and Westbrook GL (1999) Interactions of calmodulin and alpha-actinin with the NR1 subunit modulate Ca2+-dependent inactivation of NMDA receptors. J Neurosci, 19, 1165–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar SS and Huguenard JR (2003) Pathway-specific differences in subunit composition of synaptic NMDA receptors on pyramidal neurons in neocortex. J Neurosci, 23, 10074–10083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurup P, Zhang Y, Xu J, Venkitaramani DV, Haroutunian V, Greengard P, Nairn AC and Lombroso PJ (2010) Abeta-mediated NMDA receptor endocytosis in Alzheimer’s disease involves ubiquitination of the tyrosine phosphatase STEP61. J Neurosci, 30, 5948–5957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladepeche L, Dupuis JP, Bouchet D, Doudnikoff E, Yang L, Campagne Y, Bezard E, Hosy E and Groc L (2013) Single-molecule imaging of the functional crosstalk between surface NMDA and dopamine D1 receptors. Proc Natl Acad Sci U S A, 110, 18005–18010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladepeche L, Dupuis JP and Groc L (2014) Surface trafficking of NMDA receptors: gathering from a partner to another. Semin Cell Dev Biol, 27, 3–13. [DOI] [PubMed] [Google Scholar]
- Lai TW, Shyu WC and Wang YT (2011) Stroke intervention pathways: NMDA receptors and beyond. Trends Mol Med, 17, 266–275. [DOI] [PubMed] [Google Scholar]
- Lan JY, Skeberdis VA, Jover T, Grooms SY, Lin Y, Araneda RC, Zheng X, Bennett MV and Zukin RS (2001a) Protein kinase C modulates NMDA receptor trafficking and gating. Nat Neurosci, 4, 382–390. [DOI] [PubMed] [Google Scholar]
- Lan JY, Skeberdis VA, Jover T, Zheng X, Bennett MV and Zukin RS (2001b) Activation of metabotropic glutamate receptor 1 accelerates NMDA receptor trafficking. J Neurosci, 21, 6058–6068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau CG, Takayasu Y, Rodenas-Ruano A, Paternain AV, Lerma J, Bennett MV and Zukin RS (2010) SNAP-25 is a target of protein kinase C phosphorylation critical to NMDA receptor trafficking. J Neurosci, 30, 242–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavezzari G, McCallum J, Dewey CM and Roche KW (2004) Subunit-specific regulation of NMDA receptor endocytosis. J Neurosci, 24, 6383–6391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavezzari G, McCallum J, Lee R and Roche KW (2003) Differential binding of the AP-2 adaptor complex and PSD-95 to the C-terminus of the NMDA receptor subunit NR2B regulates surface expression. Neuropharmacology, 45, 729–737. [DOI] [PubMed] [Google Scholar]
- Lee HK, Takamiya K, Han JS et al. (2003) Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell, 112, 631–643. [DOI] [PubMed] [Google Scholar]
- Leinekugel X, Medina I, Khalilov I, Ben-Ari Y and Khazipov R (1997) Ca2+ oscillations mediated by the synergistic excitatory actions of GABA(A) and NMDA receptors in the neonatal hippocampus. Neuron, 18, 243–255. [DOI] [PubMed] [Google Scholar]
- Leonard AS, Bayer KU, Merrill MA, Lim IA, Shea MA, Schulman H and Hell JW (2002) Regulation of calcium/calmodulin-dependent protein kinase II docking to N-methyl-D-aspartate receptors by calcium/calmodulin and alpha-actinin. J Biol Chem, 277, 48441–48448. [DOI] [PubMed] [Google Scholar]
- Leonard AS, Lim IA, Hemsworth DE, Horne MC and Hell JW (1999) Calcium/calmodulin-dependent protein kinase II is associated with the N-methyl-D-aspartate receptor. Proc Natl Acad Sci U S A, 96, 3239–3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesca G, Moller RS, Rudolf G, Hirsch E, Hjalgrim H and Szepetowski P (2019) Update on the genetics of the epilepsy-aphasia spectrum and role of GRIN2A mutations. Epileptic Disord, 21, 41–47. [DOI] [PubMed] [Google Scholar]
- Lesca G, Rudolf G, Bruneau N et al. (2013) GRIN2A mutations in acquired epileptic aphasia and related childhood focal epilepsies and encephalopathies with speech and language dysfunction. Nat Genet, 45, 1061–1066. [DOI] [PubMed] [Google Scholar]
- Lesept F, Chevilley A, Jezequel J et al. (2016) Tissue-type plasminogen activator controls neuronal death by raising surface dynamics of extrasynaptic NMDA receptors. Cell Death Dis, 7, e2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy AD, Xiao X, Shaw JE et al. (2018) Noonan Syndrome-Associated SHP2 Dephosphorylates GluN2B to Regulate NMDA Receptor Function. Cell Rep, 24, 1523–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy JM, Chen X, Reese TS and Nicoll RA (2015) Synaptic Consolidation Normalizes AMPAR Quantal Size following MAGUK Loss. Neuron, 87, 534–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Tian X, Hartley DM and Feig LA (2006) Distinct roles for Ras-guanine nucleotide-releasing factor 1 (Ras-GRF1) and Ras-GRF2 in the induction of long-term potentiation and long-term depression. J Neurosci, 26, 1721–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao D, Hessler NA and Malinow R (1995) Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature, 375, 400–404. [DOI] [PubMed] [Google Scholar]
- Lin EI, Jeyifous O and Green WN (2013) CASK regulates SAP97 conformation and its interactions with AMPA and NMDA receptors. J Neurosci, 33, 12067–12076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J, Yasuda R and Raghavachari S (2012) Mechanisms of CaMKII action in long-term potentiation. Nat Rev Neurosci, 13, 169–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Zhou L, Yuan H, Vieira M, Sanz-Clemente A, Badger JD 2nd, Lu W, Traynelis SF and Roche KW (2017) A Rare Variant Identified Within the GluN2B C-Terminus in a Patient with Autism Affects NMDA Receptor Surface Expression and Spine Density. J Neurosci, 37, 4093–4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wong TP, Aarts M et al. (2007) NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J Neurosci, 27, 2846–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan SM, Partridge JG, Matta JA, Buonanno A and Vicini S (2007) Long-lasting NMDA receptor-mediated EPSCs in mouse striatal medium spiny neurons. J Neurophysiol, 98, 2693–2704. [DOI] [PubMed] [Google Scholar]
- Long JF, Tochio H, Wang P, Fan JS, Sala C, Niethammer M, Sheng M and Zhang M (2003) Supramodular structure and synergistic target binding of the N-terminal tandem PDZ domains of PSD-95. J Mol Biol, 327, 203–214. [DOI] [PubMed] [Google Scholar]
- Loo LS, Tang N, Al-Haddawi M, Stewart Dawe G and Hong W (2014) A role for sorting nexin 27 in AMPA receptor trafficking. Nat Commun, 5, 3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez de Armentia M and Sah P (2003) Development and subunit composition of synaptic NMDA receptors in the amygdala: NR2B synapses in the adult central amygdala. J Neurosci, 23, 6876–6883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Zhang Q and Wang T (2019) The second PDZ domain of scaffold protein Frmpd2 binds to GluN2A of NMDA receptors. Biochem. Biophys. Res. Commun, 516, 63–67. [DOI] [PubMed] [Google Scholar]
- Luo J, Wang Y, Yasuda RP, Dunah AW and Wolfe BB (1997) The majority of N-methyl-D-aspartate receptor complexes in adult rat cerebral cortex contain at least three different subunits (NR1/NR2A/NR2B). Mol Pharmacol, 51, 79–86. [DOI] [PubMed] [Google Scholar]
- Lussier MP, Sanz-Clemente A and Roche KW (2015) Dynamic Regulation of N-Methyl-d-aspartate (NMDA) and alpha-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic Acid (AMPA) Receptors by Posttranslational Modifications. J Biol Chem, 290, 28596–28603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mameli M, Bellone C, Brown MT and Luscher C (2011) Cocaine inverts rules for synaptic plasticity of glutamate transmission in the ventral tegmental area. Nat Neurosci, 14, 414–416. [DOI] [PubMed] [Google Scholar]
- Martel MA, Ryan TJ, Bell KF et al. (2012) The subtype of GluN2 C-terminal domain determines the response to excitotoxic insults. Neuron, 74, 543–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matta JA, Ashby MC, Sanz-Clemente A, Roche KW and Isaac JT (2011) mGluR5 and NMDA Receptors Drive the Experience- and Activity-Dependent NMDA Receptor NR2B to NR2A Subunit Switch. Neuron, 70, 339–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattison HA, Hayashi T and Barria A (2012) Palmitoylation at two cysteine clusters on the C-terminus of GluN2A and GluN2B differentially control synaptic targeting of NMDA receptors. PLoS One, 7, e49089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauceri D, Gardoni F, Marcello E and Di Luca M (2007) Dual role of CaMKII-dependent SAP97 phosphorylation in mediating trafficking and insertion of NMDA receptor subunit NR2A. J Neurochem, 100, 1032–1046. [DOI] [PubMed] [Google Scholar]
- McGee AW, Dakoji SR, Olsen O, Bredt DS, Lim WA and Prehoda KE (2001) Structure of the SH3-guanylate kinase module from PSD-95 suggests a mechanism for regulated assembly of MAGUK scaffolding proteins. Mol Cell, 8, 1291–1301. [DOI] [PubMed] [Google Scholar]
- McIlhinney RA, Le Bourdelles B, Molnar E, Tricaud N, Streit P and Whiting PJ (1998) Assembly intracellular targeting and cell surface expression of the human N-methyl-D-aspartate receptor subunits NR1a and NR2A in transfected cells. Neuropharmacology, 37, 1355–1367. [DOI] [PubMed] [Google Scholar]
- McKay S, Ryan TJ, McQueen J et al. (2018) The Developmental Shift of NMDA Receptor Composition Proceeds Independently of GluN2 Subunit-Specific GluN2 C-Terminal Sequences. Cell Rep, 25, 841–851 e844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQueen J, Ryan TJ, McKay S et al. (2017) Pro-death NMDA receptor signaling is promoted by the GluN2B C-terminus independently of Dapk1. Elife, 6 e17161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta A, Prabhakar M, Kumar P, Deshmukh R and Sharma PL (2013) Excitotoxicity: bridge to various triggers in neurodegenerative disorders. Eur J Pharmacol, 698, 6–18. [DOI] [PubMed] [Google Scholar]
- Michaluk P, Mikasova L, Groc L, Frischknecht R, Choquet D and Kaczmarek L (2009) Matrix metalloproteinase-9 controls NMDA receptor surface diffusion through integrin beta1 signaling. J Neurosci, 29, 6007–6012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikasova L, De Rossi P, Bouchet D, Georges F, Rogemond V, Didelot A, Meissirel C, Honnorat J and Groc L (2012) Disrupted surface cross-talk between NMDA and Ephrin-B2 receptors in anti-NMDA encephalitis. Brain, 135, 1606–1621. [DOI] [PubMed] [Google Scholar]
- Mikasova L, Xiong H, Kerkhofs A, Bouchet D, Krugers HJ and Groc L (2017) Stress hormone rapidly tunes synaptic NMDA receptor through membrane dynamics and mineralocorticoid signalling. Sci Rep, 7, 8053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DT, Shen Y, Weiss LA et al. (2009) Microdeletion/duplication at 15q13.2q13.3 among individuals with features of autism and other neuropsychiatric disorders. J Med Genet, 46, 242–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohrmann R, Kohr G, Hatt H, Sprengel R and Gottmann K (2002) Deletion of the C-terminal domain of the NR2B subunit alters channel properties and synaptic targeting of N-methyl-D-aspartate receptors in nascent neocortical synapses. J Neurosci Res, 68, 265–275. [DOI] [PubMed] [Google Scholar]
- Mok H, Shin H, Kim S, Lee JR, Yoon J and Kim E (2002) Association of the kinesin superfamily motor protein KIF1Balpha with postsynaptic density-95 (PSD-95), synapse-associated protein-97, and synaptic scaffolding molecule PSD-95/discs large/zona occludens-1 proteins. J Neurosci, 22, 5253–5258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B and Seeburg PH (1994) Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron, 12, 529–540. [DOI] [PubMed] [Google Scholar]
- Mori H, Manabe T, Watanabe M et al. (1998) Role of the carboxy-terminal region of the GluR epsilon2 subunit in synaptic localization of the NMDA receptor channel. Neuron, 21, 571–580. [DOI] [PubMed] [Google Scholar]
- Motodate R, Saito H, Sobu Y, Hata S, Saito Y, Nakaya T and Suzuki T (2019) X11 and X11-like proteins regulate the level of extrasynaptic glutamate receptors. J Neurochem, 148, 480–498. [DOI] [PubMed] [Google Scholar]
- Muller BM, Kistner U, Kindler S et al. (1996) SAP102, a novel postsynaptic protein that interacts with NMDA receptor complexes in vivo. Neuron, 17, 255–265. [DOI] [PubMed] [Google Scholar]
- Nicoll RA and Roche KW (2013) Long-term potentiation: peeling the onion. Neuropharmacology, 74, 18–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niethammer M, Kim E and Sheng M (1996) Interaction between the C terminus of NMDA receptor subunits and multiple members of the PSD-95 family of membrane-associated guanylate kinases. J Neurosci, 16, 2157–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nong Y, Huang YQ, Ju W, Kalia LV, Ahmadian G, Wang YT and Salter MW (2003) Glycine binding primes NMDA receptor internalization. Nature, 422, 302–307. [DOI] [PubMed] [Google Scholar]
- O’Leary H, Liu WH, Rorabaugh JM, Coultrap SJ and Bayer KU (2011) Nucleotides and phosphorylation bi-directionally modulate Ca2+/calmodulin-dependent protein kinase II (CaMKII) binding to the N-methyl-D-aspartate (NMDA) receptor subunit GluN2B. J Biol Chem, 286, 31272–31281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogden KK, Chen W, Swanger SA et al. (2017) Molecular Mechanism of Disease-Associated Mutations in the Pre-M1 Helix of NMDA Receptors and Potential Rescue Pharmacology. PLoS Genet, 13, e1006536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olney JW (1969) Brain lesions, obesity, and other disturbances in mice treated with monosodium glutamate. Science, 164, 719–721. [DOI] [PubMed] [Google Scholar]
- Omkumar RV, Kiely MJ, Rosenstein AJ, Min KT and Kennedy MB (1996) Identification of a phosphorylation site for calcium/calmodulindependent protein kinase II in the NR2B subunit of the N-methyl-D-aspartate receptor. J Biol Chem, 271, 31670–31678. [DOI] [PubMed] [Google Scholar]
- Ong WY, Tanaka K, Dawe GS, Ittner LM and Farooqui AA (2013) Slow excitotoxicity in Alzheimer’s disease. J Alzheimers Dis, 35, 643–668. [DOI] [PubMed] [Google Scholar]
- Paoletti P, Bellone C and Zhou Q (2013) NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci, 14, 383–400. [DOI] [PubMed] [Google Scholar]
- Papouin T, Ladepeche L, Ruel J et al. (2012) Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists. Cell, 150, 633–646. [DOI] [PubMed] [Google Scholar]
- Parsons MP and Raymond LA (2014) Extrasynaptic NMDA receptor involvement in central nervous system disorders. Neuron, 82, 279–293. [DOI] [PubMed] [Google Scholar]
- Perszyk RE, DiRaddo JO, Strong KL et al. (2016) GluN2D-Containing N-methyl-d-Aspartate Receptors Mediate Synaptic Transmission in Hippocampal Interneurons and Regulate Interneuron Activity. Mol Pharmacol, 90, 689–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petralia RS, Wang YX and Wenthold RJ (2003) Internalization at glutamatergic synapses during development. Eur J Neurosci, 18, 3207–3217. [DOI] [PubMed] [Google Scholar]
- Petrovski S, Wang Q, Heinzen EL, Allen AS and Goldstein DB (2013) Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet, 9, e1003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettit DL, Perlman S and Malinow R (1994) Potentiated transmission and prevention of further LTP by increased CaMKII activity in postsynaptic hippocampal slice neurons. Science, 266, 1881–1885. [DOI] [PubMed] [Google Scholar]
- Philpot BD, Sekhar AK, Shouval HZ and Bear MF (2001) Visual experience and deprivation bidirectionally modify the composition and function of NMDA receptors in visual cortex. Neuron, 29, 157–169. [DOI] [PubMed] [Google Scholar]
- Piguel NH, Fievre S, Blanc JM et al. (2014) Scribble1/AP2 complex coordinates NMDA receptor endocytic recycling. Cell Rep, 9, 712–727. [DOI] [PubMed] [Google Scholar]
- Pina-Crespo JC and Gibb AJ (2002) Subtypes of NMDA receptors in new-born rat hippocampal granule cells. J Physiol, 541, 41–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potier M, Georges F, Brayda-Bruno L, Ladepeche L, Lamothe V, Al Abed AS, Groc L and Marighetto A (2016) Temporal Memory and Its Enhancement by Estradiol Requires Surface Dynamics of Hippocampal CA1 N-Methyl-D-Aspartate Receptors. Biol Psychiatry, 79, 735–745. [DOI] [PubMed] [Google Scholar]
- Prybylowski K, Chang K, Sans N, Kan L, Vicini S and Wenthold RJ (2005) The synaptic localization of NR2B-containing NMDA receptors is controlled by interactions with PDZ proteins and AP-2. Neuron, 47, 845–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prybylowski K, Fu Z, Losi G, Hawkins LM, Luo J, Chang K, Wenthold RJ and Vicini S (2002) Relationship between availability of NMDA receptor subunits and their expression at the synapse. J Neurosci, 22, 8902–8910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan EM, Philpot BD, Huganir RL and Bear MF (1999) Rapid, experience-dependent expression of synaptic NMDA receptors in visual cortex in vivo. Nat Neurosci, 2, 352–357. [DOI] [PubMed] [Google Scholar]
- Racz B, Blanpied TA, Ehlers MD and Weinberg RJ (2004) Lateral organization of endocytic machinery in dendritic spines. Nat Neurosci, 7, 917–918. [DOI] [PubMed] [Google Scholar]
- Rauner C and Kohr G (2011) Triheteromeric NR1/NR2A/NR2B receptors constitute the major N-methyl-D-aspartate receptor population in adult hippocampal synapses. J Biol Chem, 286, 7558–7566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravizza T, Onat FY, Brooks-Kayal AR, Depaulis A, Galanopoulou AS, Mazarati A, Numis AL, Sankar R and Friedman A (2017) WONOEP appraisal: Biomarkers of epilepsy-associated comorbidities. Epilepsia, 58, 331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche KW, Standley S, McCallum J, Dune Ly C, Ehlers MD and Wenthold RJ (2001) Molecular determinants of NMDA receptor internalization. Nat Neurosci, 4, 794–802. [DOI] [PubMed] [Google Scholar]
- Rossi P, Sola E, Taglietti V, Borchardt T, Steigerwald F, Utvik JK, Ottersen OP, Kohr G and D’Angelo E (2002) NMDA receptor 2 (NR2) C-terminal control of NR open probability regulates synaptic transmission and plasticity at a cerebellar synapse. J Neurosci, 22, 9687–9697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumbaugh G, Sia GM, Garner CC and Huganir RL (2003) Synapse-associated protein-97 isoform-specific regulation of surface AMPA receptors and synaptic function in cultured neurons. J Neurosci, 23, 4567–4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan TJ, Kopanitsa MV, Indersmitten T et al. (2013) Evolution of GluN2A/B cytoplasmic domains diversified vertebrate synaptic plasticity and behavior. Nat Neurosci, 16, 25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, He X, Willsey AJ et al. (2015) Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron, 87, 1215–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sans N, Petralia RS, Wang YX, Blahos J 2nd, Hell JW and Wenthold RJ (2000) A developmental change in NMDA receptor-associated proteins at hippocampal synapses. J Neurosci, 20, 1260–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sans N, Prybylowski K, Petralia RS, Chang K, Wang YX, Racca C, Vicini S and Wenthold RJ (2003) NMDA receptor trafficking through an interaction between PDZ proteins and the exocyst complex. Nat Cell Biol, 5, 520–530. [DOI] [PubMed] [Google Scholar]
- Sans N, Wang PY, Du Q, Petralia RS, Wang YX, Nakka S, Blumer JB, Macara IG and Wenthold RJ (2005) mPins modulates PSD-95 and SAP102 trafficking and influences NMDA receptor surface expression. Nat Cell Biol, 7, 1179–1190. [DOI] [PubMed] [Google Scholar]
- Sanz-Clemente A, Gray JA, Ogilvie KA, Nicoll RA and Roche KW (2013a) Activated CaMKII couples GluN2B and casein kinase 2 to control synaptic NMDA receptors. Cell Rep, 3, 607–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz-Clemente A, Matta JA, Isaac JT and Roche KW (2010) Casein kinase 2 regulates the NR2 subunit composition of synaptic NMDA receptors. Neuron, 67, 984–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz-Clemente A, Nicoll RA and Roche KW (2013b) Diversity in NMDA receptor composition: many regulators, many consequences. Neuroscientist, 19, 62–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sceniak MP, Fedder KN, Wang Q, Droubi S, Babcock K, Patwardhan S, Wright-Zornes J, Pham L and Sabo SL (2019) An autism-associated mutation in GluN2B prevents NMDA receptor trafficking and interferes with dendrite growth. J Cell Sci, 132. [DOI] [PubMed] [Google Scholar]
- Scott DB, Blanpied TA, Swanson GT, Zhang C and Ehlers MD (2001) An NMDA receptor ER retention signal regulated by phosphorylation and alternative splicing. J Neurosci, 21, 3063–3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DB, Michailidis I, Mu Y, Logothetis D and Ehlers MD (2004) Endocytosis and degradative sorting of NMDA receptors by conserved membrane-proximal signals. J Neurosci, 24, 7096–7109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setou M, Nakagawa T, Seog DH and Hirokawa N (2000) Kinesin superfamily motor protein KIF17 and mLin-10 in NMDA receptor-containing vesicle transport. Science, 288, 1796–1802. [DOI] [PubMed] [Google Scholar]
- Sharp AJ, Mefford HC, Li K et al. (2008) A recurrent 15q13.3 microdeletion syndrome associated with mental retardation and seizures. Nat Genet, 40, 322–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K and Meyer T (1999) Dynamic control of CaMKII translocation and localization in hippocampal neurons by NMDA receptor stimulation. Science, 284, 162–166. [DOI] [PubMed] [Google Scholar]
- Sheng M, Cummings J, Roldan LA, Jan YN and Jan LY (1994) Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature, 368, 144–147. [DOI] [PubMed] [Google Scholar]
- Shi J, Townsend M and Constantine-Paton M (2000) Activity-dependent induction of tonic calcineurin activity mediates a rapid developmental downregulation of NMDA receptor currents. Neuron, 28, 103–114. [DOI] [PubMed] [Google Scholar]
- Shiloh R, Bialik S and Kimchi A (2014) The DAPK family: a structure-function analysis. Apoptosis, 19, 286–297. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Stevens CF, Tonegawa S and Wang Y (1992) Deficient hippocampal long-term potentiation in alpha-calcium-calmodulin kinase II mutant mice. Science, 257, 201–206. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG et al. (2005) Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci, 8, 1051–1058. [DOI] [PubMed] [Google Scholar]
- Soriano FX, Martel MA, Papadia S et al. (2008) Specific targeting of pro-death NMDA receptor signals with differing reliance on the NR2B PDZ ligand. J Neurosci, 28, 10696–10710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprengel R, Suchanek B, Amico C et al. (1998) Importance of the intracellular domain of NR2 subunits for NMDA receptor function in vivo. Cell, 92, 279–289. [DOI] [PubMed] [Google Scholar]
- Standley S, Roche KW, McCallum J, Sans N and Wenthold RJ (2000) PDZ domain suppression of an ER retention signal in NMDA receptor NR1 splice variants. Neuron, 28, 887–898. [DOI] [PubMed] [Google Scholar]
- Stanic J, Carta M, Eberini I et al. (2015) Rabphilin 3A retains NMDA receptors at synaptic sites through interaction with GluN2A/PSD-95 complex. Nat Commun, 6, 10181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanika RI, Pivovarova NB, Brantner CA, Watts CA, Winters CA and Andrews SB (2009) Coupling diverse routes of calcium entry to mitochondrial dysfunction and glutamate excitotoxicity. Proc Natl Acad Sci U S A, 106, 9854–9859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson H, Rujescu D, Cichon S et al. (2008) Large recurrent microdeletions associated with schizophrenia. Nature, 455, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steigerwald F, Schulz TW, Schenker LT, Kennedy MB, Seeburg PH and Kohr G (2000) C-Terminal truncation of NR2A subunits impairs synaptic but not extrasynaptic localization of NMDA receptors. J Neurosci, 20, 4573–4581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack S and Colbran RJ (1998) Autophosphorylation-dependent targeting of calcium/ calmodulin-dependent protein kinase II by the NR2B subunit of the N-methyl- D-aspartate receptor. J Biol Chem, 273, 20689–20692. [DOI] [PubMed] [Google Scholar]
- Strack S, McNeill RB and Colbran RJ (2000) Mechanism and regulation of calcium/calmodulin-dependent protein kinase II targeting to the NR2B subunit of the N-methyl-D-aspartate receptor. J Biol Chem, 275, 23798–23806. [DOI] [PubMed] [Google Scholar]
- Stroebel D, Casado M and Paoletti P (2018) Triheteromeric NMDA receptors: from structure to synaptic physiology. Curr Opin Physiol, 2, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC and Rothman JE (2009) Membrane fusion: grappling with SNARE and SM proteins. Science, 323, 474–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh YH, Terashima A, Petralia RS, Wenthold RJ, Isaac JT, Roche KW and Roche PA (2010) A neuronal role for SNAP-23 in postsynaptic glutamate receptor trafficking. Nat Neurosci, 13, 338–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundelin HE, Larsson H, Lichtenstein P, Almqvist C, Hultman CM, Tomson T and Ludvigsson JF (2016) Autism and epilepsy: A population-based nationwide cohort study. Neurology, 87, 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanger SA, Chen W, Wells G et al. (2016) Mechanistic Insight into NMDA Receptor Dysregulation by Rare Variants in the GluN2A and GluN2B Agonist Binding Domains. Am J Hum Genet, 99, 1261–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanger SA, Vance KM, Pare JF, Sotty F, Fog K, Smith Y and Traynelis SF (2015) NMDA Receptors Containing the GluN2D Subunit Control Neuronal Function in the Subthalamic Nucleus. J Neurosci, 35, 15971–15983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanwick CC, Shapiro ME, Yi Z, Chang K and Wenthold RJ (2009) NMDA receptors interact with flotillin-1 and −2, lipid raft-associated proteins. FEBS Lett, 583, 1226–1230. [DOI] [PubMed] [Google Scholar]
- Tang TT, Badger JD 2nd, Roche PA and Roche KW (2010) Novel approach to probe subunit-specific contributions to N-methyl-D-aspartate (NMDA) receptor trafficking reveals a dominant role for NR2B in receptor recycling. J Biol Chem, 285, 20975–20981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teasdale RD and Collins BM (2012) Insights into the PX (phox-homology) domain and SNX (sorting nexin) protein families: structures, functions and roles in disease. Biochem J, 441, 39–59. [DOI] [PubMed] [Google Scholar]
- Terasaki Y, Sasaki T, Yagita Y, Okazaki S, Sugiyama Y, Oyama N, Omura-Matsuoka E, Sakoda S and Kitagawa K (2010) Activation of NR2A receptors induces ischemic tolerance through CREB signaling. J Cereb Blood Flow Metab, 30, 1441–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GM and Huganir RL (2013) Palmitoylation-dependent regulation of glutamate receptors and their PDZ domain-containing partners. Biochem Soc Trans, 41, 72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar KR, McGinley MJ and Westbrook GL (2013) Triheteromeric NMDA receptors at hippocampal synapses. J Neurosci, 33, 9150–9160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar KR and Westbrook GL (2002) Mobile NMDA receptors at hippocampal synapses. Neuron, 34, 255–264. [DOI] [PubMed] [Google Scholar]
- Townsend M, Liu Y and Constantine-Paton M (2004) Retina-driven dephosphorylation of the NR2A subunit correlates with faster NMDA receptor kinetics at developing retinocollicular synapses. J Neurosci, 24, 11098–11107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ et al. (2010) Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev, 62, 405–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu W, Xu X, Peng L et al. (2010) DAPK1 interaction with NMDA receptor NR2B subunits mediates brain damage in stroke. Cell, 140, 222–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tymianski M and Tator CH (1996) Normal and abnormal calcium homeostasis in neurons: a basis for the pathophysiology of traumatic and ischemic central nervous system injury. Neurosurgery, 38, 1176–1195. [DOI] [PubMed] [Google Scholar]
- Valtschanoff JG, Burette A, Davare MA, Leonard AS, Hell JW and Weinberg RJ (2000) SAP97 concentrates at the postsynaptic density in cerebral cortex. Eur J Neurosci, 12, 3605–3614. [DOI] [PubMed] [Google Scholar]
- Vieira MM, Schmidt J, Ferreira JS et al. (2016) Multiple domains in the C-terminus of NMDA receptor GluN2B subunit contribute to neuronal death following in vitro ischemia. Neurobiol Dis, 89, 223–234. [DOI] [PubMed] [Google Scholar]
- Vyklicky V, Krausova B, Cerny J et al. (2018) Surface Expression, Function, and Pharmacology of Disease-Associated Mutations in the Membrane Domain of the Human GluN2B Subunit. Front Mol Neurosci, 11, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Lv X, Wu Y et al. (2018) Postsynaptic RIM1 modulates synaptic function by facilitating membrane delivery of recycling NMDARs in hippocampal neurons. Nat Commun, 9, 2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhao Y, Zhang X et al. (2013) Loss of sorting nexin 27 contributes to excitatory synaptic dysfunction by modulating glutamate receptor recycling in Down’s syndrome. Nat Med, 19, 473–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washbourne P, Liu XB, Jones EG and McAllister AK (2004) Cycling of NMDA receptors during trafficking in neurons before synapse formation. J Neurosci, 24, 8253–8264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won S, Incontro S, Nicoll RA and Roche KW (2016) PSD-95 stabilizes NMDA receptors by inducing the degradation of STEP61. Proc Natl Acad Sci U S A, 113, E4736–4744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won S, Levy JM, Nicoll RA and Roche KW (2017) MAGUKs: multifaceted synaptic organizers. Curr Opin Neurobiol, 43, 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyllie DJ, Livesey MR and Hardingham GE (2013) Influence of GluN2 subunit identity on NMDA receptor function. Neuropharmacology, 74, 4–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyszynski M, Lin J, Rao A, Nigh E, Beggs AH, Craig AM and Sheng M (1997) Competitive binding of alpha-actinin and calmodulin to the NMDA receptor. Nature, 385, 439–442. [DOI] [PubMed] [Google Scholar]
- Xu J, Kurup P, Zhang Y, Goebel-Goody SM, Wu PH, Hawasli AH, Baum ML, Bibb JA and Lombroso PJ (2009) Extrasynaptic NMDA receptors couple preferentially to excitotoxicity via calpain-mediated cleavage of STEP. J Neurosci, 29, 9330–9343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Zheng C, Song Q, Yang X, Qiu S, Liu C, Chen Z, Duan S and Luo J (2007) A three amino acid tail following the TM4 region of the N-methyl-D-aspartate receptor (NR) 2 subunits is sufficient to overcome endoplasmic reticulum retention of NR1–1a subunit. J Biol Chem, 282, 9269–9278. [DOI] [PubMed] [Google Scholar]
- Yashiro K and Philpot BD (2008) Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacology, 55, 1081–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi F, Traynelis SF and Hansen KB (2017) Selective Cell-Surface Expression of Triheteromeric NMDA Receptors. Methods Mol Biol, 1677, 145–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi Z, Petralia RS, Fu Z, Swanwick CC, Wang YX, Prybylowski K, Sans N, Vicini S and Wenthold RJ (2007) The role of the PDZ protein GIPC in regulating NMDA receptor trafficking. J Neurosci, 27, 11663–11675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X, Feng X, Takei Y and Hirokawa N (2012) Regulation of NMDA receptor transport: a KIF17-cargo binding/releasing underlies synaptic plasticity and memory in vivo. J Neurosci, 32, 5486–5499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X, Takei Y, Kido MA and Hirokawa N (2011) Molecular motor KIF17 is fundamental for memory and learning via differential support of synaptic NR2A/2B levels. Neuron, 70, 310–325. [DOI] [PubMed] [Google Scholar]
- Yuan H, Hansen KB, Zhang J et al. (2014) Functional analysis of a de novo GRIN2A missense mutation associated with early-onset epileptic encephalopathy. Nat Commun, 5, 3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, Low CM, Moody OA, Jenkins A and Traynelis SF (2015) Ionotropic GABA and Glutamate Receptor Mutations and Human Neurologic Diseases. Mol Pharmacol, 88, 203–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Ehlers MD, Bernhardt JP, Su CT and Huganir RL (1998) Calmodulin mediates calcium-dependent inactivation of N-methyl-D-aspartate receptors. Neuron, 21, 443–453. [DOI] [PubMed] [Google Scholar]
- Zhou Q and Sheng M (2013) NMDA receptors in nervous system diseases. Neuropharmacology, 74, 69–75. [DOI] [PubMed] [Google Scholar]
- Zhou X, Ding Q, Chen Z, Yun H and Wang H (2013) Involvement of the GluN2A and GluN2B subunits in synaptic and extrasynaptic N-methyl-D-aspartate receptor function and neuronal excitotoxicity. J Biol Chem, 288, 24151–24159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Takahashi E, Li W et al. (2007) Interactions between the NR2B receptor and CaMKII modulate synaptic plasticity and spatial learning. J Neurosci, 27, 13843–13853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S and Paoletti P (2015) Allosteric modulators of NMDA receptors: multiple sites and mechanisms. Curr Opin Pharmacol, 20, 14–23. [DOI] [PubMed] [Google Scholar]