

Summary
Bacteria belonging to the Lachnospiraceae family are abundant, obligate anaerobic members of the microbiota in healthy humans. Lachnospiraceae impact their hosts by producing short-chain fatty acids, converting primary to secondary bile acids, and facilitating colonization resistance against intestinal pathogens. To increase our understanding of genomic and functional diversity between members of this family, we cultured 273 Lachnospiraceae isolates representing 11 genera and 27 species from human donors and performed whole genome sequencing assembly and annotation. This analysis revealed substantial inter- and intra-species diversity in pathways that likely influence an isolate’s ability to impact host health. These differences are likely to impact colonization resistance through lantibiotic expression or intestinal acidification, influence host mucosal immune cells and enterocytes via butyrate production or contribute to synergism within a consortium by heterogenous polysaccharide metabolism. Identification of these specific functions could facilitate development of probiotic bacterial consortia that drive and/or restore in vivo microbiome functions.
Graphical Abstract

eTOC blurb
Members of the Lachnospiraceae family are abundant members of the microbiota and have health-promoting functions. Sorbara et al. describe genomic and functional diversity within a large collection of cultured Lachnospiraceae isolates. Inter-and intra-species diversity has important implications for the development of therapeutic consortia.
Introduction
The Lachnospiraceae are a family of anaerobic bacteria in the Clostridiales order within the Firmicutes phylum and include species previously identified as Clostridium cluster XIVa. This family is abundant in the unperturbed adult human microbiota(Hold et al., 2002, Lopetuso et al., 2013), however, they can be rapidly lost following common clinical antibiotic treatment protocols, such as during hematopoietic-stem cell transplant, and Lachnospiraceae abundances can be altered by changes in diet(David et al., 2014). A decrease in Lachnospiraceae abundance is likely to have negative health implications resulting from loss of the numerous beneficial functions performed by members of this family. Lachnospiraceae can contribute to the microbiota’s colonization resistance against drug-resistant pathogens through conversion of primary to secondary bile acids(Buffie et al., 2015, Studer et al., 2016), production of the short-chain fatty acids (SCFAs) acetate and butyrate(Byndloss et al., 2017, Rivera-Chavez et al., 2016), or production of lantibiotics, an important class of peptide antibiotics (Hatziioanou et al., 2017, Kim et al., 2019). In addition to functions in colonization resistance, butyrate production provides pleiotropic beneficial effects for the host in terms of metabolism and immune regulation(Arpaia et al., 2013, Furusawa et al., 2013, Velazquez et al., 1997). Furthermore, Lachnospiraceae are enriched in proximity to the mucosa (Nava et al., 2011, Riva et al., 2019, Van den Abbeele et al., 2013), thereby situating them to influence the host epithelium and mucosal immune system.
As a result of these characteristics, therapeutic interventions using defined consortia of bacterial strains to restore or promote microbiota functions will likely require the inclusion of Lachnospiraceae members. Indeed, preclinical studies in mice have demonstrated that 4-member consortia of commensal bacteria containing Lachnospiraceae can reduce enteric colonization by vancomycin-resistant Enterococci (VRE)(Caballero et al., 2017), C. difficile (Buffie et al., 2015), and L. monocytogenes (Becattini et al., 2017) while a 12 member consortium can enhance resistance against S. enterica serovar Typhimurium(Brugiroux et al., 2016). Similarly, a 17-member consortium containing Lachnospiraceae can promote regulatory T cell differentiation(Atarashi et al., 2013).
Despite their abundance and contribution to host health, bacterial species belonging to the Lachnospiraceae family remain poorly characterized in terms of inter- and intra-species diversity. Furthermore, in contrast to beneficial functions associated with Lachnospiraceae, a member of the family, [Ruminococcus] gnavus, has been implicated in Crohn’s Disease (CD) pathogenesis (Hall et al., 2017, Png et al., 2010, Willing et al., 2010), however, the intraspecies diversity and differences between this species and other related members of the family remain unknown.
To better characterize the Lachnospiraceae family, we collected 273 isolates, consisting of 11 genera and 27 species belonging to the Lachnospiraceae family from 20 human donors. Whole genome sequencing and gene annotation revealed the broad genetic repertoire and diversity of the species and strains isolated from human donors. This analysis revealed substantial inter- and intra-species diversity in pathways that are likely to influence an isolate’s ability to contribute to colonization resistance through lantibiotic expression or intestinal acidification, influence the host’s mucosal immune cells and enterocytes through its capacity to produce butyrate or to function synergistically within a consortium because of heterogeneous polysaccharide metabolism. Our results clarify the placement of [Ruminococcus] gnavus within the Lachnospiraceae family and demonstrate that this species is genetically distinct from others in the family, and that [Ruminococcus] gnavus isolates are heterogeneous in terms of loci identified as contributing to a pro-inflammatory phenotype. Overall, our results demonstrate a surprising level of diversification among bacterial species in the Lachnospiraceae family and provide support for the notion that genomic and metabolic analyses of individual strains contained within commensal bacterial strain banks will facilitate the generation of rationally designed consortia that will function effectively and persist in recipients.
Results
Identification of Lachnospiraceae isolates
To characterize the diversity within the Lachnospiraceae family, we collected stool samples from 20 human donors. Metagenomic sequence analysis of fecal samples revealed that Lachnospiraceae were abundant in all donors, with an average abundance of 23.4% (Figure 1). Fecal samples from these donors were cultured under anaerobic conditions on rich, non-selective media to enable growth of the wide range of bacterial species composing the microbiota. We obtained 956 isolates that were re-streaked for purity and whole genome sequencing (WGS). Isolates were classified using BLAST identification of the 16S rRNA gene sequence that had been assembled from shotgun sequences. This approach demonstrated that we isolated representatives from 65% of the genera present in the human donors (Figure 1) and all genera belonging to the Lachnospiraceae family that we detected by metagenomic sequencing of donor feces (Figure 1). Using the WGS-derived 16S rRNA sequence allowed us to identify commensal isolates on the basis of full length 16S rRNA sequences as opposed to using the shorter variable regions. Using longer 16S rRNA sequences has been reported to enable more accurate species identification (Bukin et al., 2019, Chakravorty et al., 2007, Fuks et al., 2018). In agreement, 21.7% of species-level BLAST identifications differed between V4-based and full-length-based sequences using in silico truncations from the same full length 16S rRNA sequence (Figure S1A). Of the commonly used regions, species identifications based on the V1-V2 region most closely matched those of the full-length sequence for these Lachnospiraceae, but even in this case 12.6% of isolates were differentially identified (Figure S1A). Based on 16S rRNA sequence analysis, we identified 273 isolates of Lachnospiraceae representing 27 species.
Figure 1:

Generation of a biobank of commensal isolates from human donors. Relative abundances of bacteria at the genus level from human donor fecal samples as determined by shotgun metagenomic sequencing in the upper plot, with genera not recovered indicated in the lower plot. Related to Table S1.
Phytogeny of the Lachnospiraceae
Next, we assembled a phylogenetic tree on the basis of 16S rRNA gene sequence alignment, which demonstrated that the Lachnospiraceae isolates are distinct from representatives of the Ruminococcaceae or Clostridiaceae families that are also in the Clostridiales order (Figure 2A-inset, Figure S1b-inset). Several members of the Blautia genus are species that have been reassigned to Blautia from the Ruminococcus genus in the Ruminococcaceae family (Lawson and Finegold, 2015, Liu et al., 2008). Consistent with its re-assignment to the Lachnospiraceae family, [Ruminococcus] gnavus isolates were positioned within the Lachnospiraceae family, however, they are the least distant of the Lachnospiraceae species from the Clostridiaceae family (Figure S1b). Unexpectedly, [Ruminococcus] gnavus was not placed with other Blautia isolates, despite its current classification as a member of this genus. Similarly, all members of the Lachnoclostridium genus formed a distinct monophyletic group, with the exception of [Clostridium] scindens (Lachnoclostridium scindens) which, based on 16S sequence, is more closely related to isolates of the genus Dorea. Based on 16S rRNA sequence alignment, isolates of each BLAST-identified species were placed within monophyletic groups with the exception of Blautia luti and Blautia glucerasea, which do not share a distinct most common ancestor from Blautia schinkii and Blautia faecis, respectively. As expected, when type strains of Lachnospiraceae species are included in the phylogenetic analysis, the majority of type strains are positioned with isolates identified as those species (Figure S1C). Unexpectedly, type strains of Blautia glucerasea and Blautia schinkii are not placed with isolates identified by BLAST as those species (Figure S1C). In addition, we identified two isolates from donor 11 that formed a distinct clade and had low percent identity to reference 16S rRNA sequences (<96%) by BLAST and were therefore labelled as unclassified Lachnospiraceae.
Figure 2:
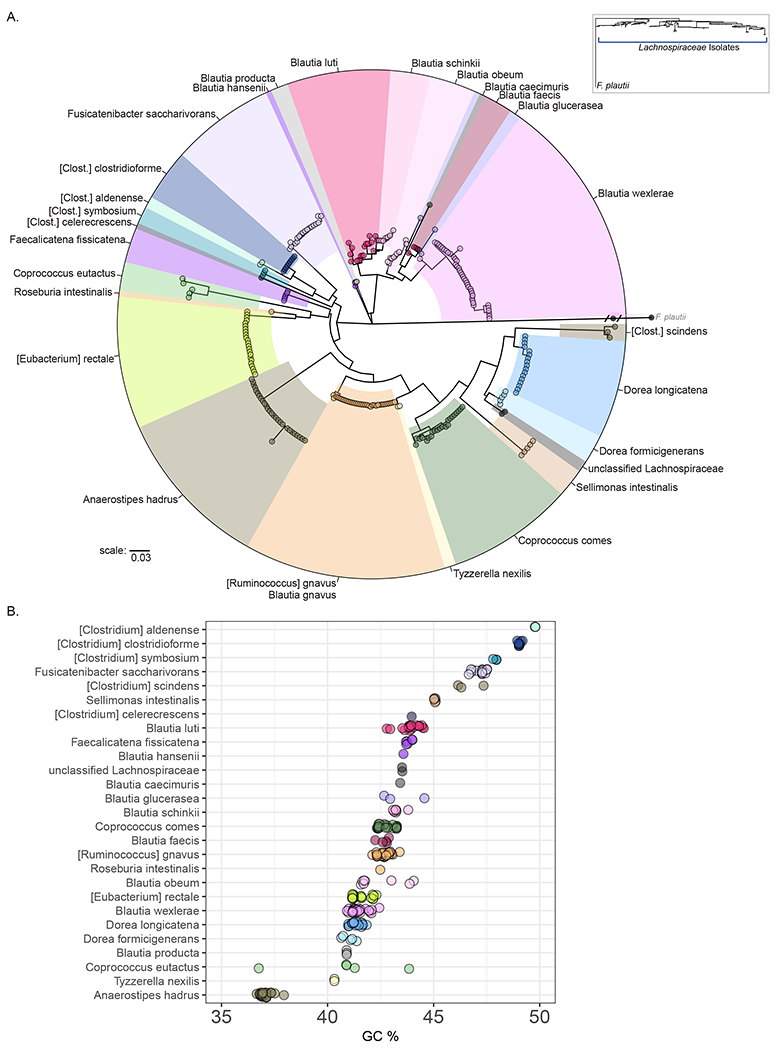
Phylogeny of Lachnospiraceae isolates from human donors. A: A 16S rRNA-derived maximum-likelihood phylogenetic dendrogram of Lachnospiraceae isolates generated using an isolate of Flavonifractor plautii, a member of the Ruminococceae, as an outgroup. Inset (upper right) shows position of Lachnospiraceae isolates relative to F. plautii. Members of the Lachnoclostridium genus are labeled [Clostridium] to reflect the names most often used to identify members of these species. B: Genomic GC content of Lachnospiraceae isolates plotted by 16S rRNA designated species.
We noted that there is variability in the distributions of intra-species phylogenetic distances between isolates (Figure S2, left panels). For some species, such as [Ruminococcus] gnavus and Anaerostipes hadrus (Figure S2, left panels), phylogenetic distances between isolates are minimal. In contrast, isolates of Blautia luti and Coprococcus eutactus have greater intraspecies phylogenetic distances indicating greater variability in 16S rRNA gene sequences (Figure S2, left panels). Furthermore, for 19/27 species, the average percent identity of the BLAST hit was > 98% (Figure S2, right panels), while 8 species were identified using a lower percentage hit from the reference database. For some groups of isolates, such as those identified as Faecalicatena fissicatena, the lower percentage match with the reference database was despite the presence of multiple isolates from multiple donors that are separated by minimal phylogenetic distance (Figure S2). 16S rRNA sequences from Blautia glucerasea or Blautia schinkii are <96% identical to the reference database, which, in addition to the separation between the type strains and these isolates (Figure S1C), suggests that these isolates may be members of the Blautia genus without a representative in the NCBI refseq database.
Next we examined the whole genome GC content across Lachnospiraceae isolates (Figure 2B, Table S1). Interestingly, this analysis revealed that there is a wide range of GC content within the Lachnospiraceae family, and that isolates of a given species have a characteristic GC content. Overall, the average species GC content ranges from a low of 37% for Anaerostipes hadrus to a high of 49.8% for [Clostridium] aldenense. Furthermore, this analysis revealed that isolates within a genus tend to have similar GC content. For example, all isolates in the Lachnoclostridium genus have relatively high GC content, despite [Clostridium] scindens not being placed with the other members of the genus on the basis of 16S rRNA derived phylogeny (Figure 2A).
Diversity in the core genomes of Lachnospiraceae isolates
We next investigated genomic diversity among Lachnospiraceae isolates. On average, using the prokka annotation pipeline(Seemann, 2014), each isolate contained 3362 protein coding sequences (CDS) (Median 3257, range 6531 – 2292) (Table S1). The range and distributions of isolate genome sizes were not significantly different from previously published genomes of Lachnospiraceae isolates (Data not shown)(Forster et al., 2019, Poyet et al., 2019). Using prokka, only 50% of all putative protein-coding sequences were annotated either with or without an associated COG identification (Figure S3A), similar to results obtained with pathogenic species (Lery et al., 2014, Lewis et al., 2017, Yang et al., 2019). Furthermore, within a single species, there was greater variability in the numbers of CDS identified as hypothetical genes compared to annotated (Figure S3B). We observed similar results using the PATRIC pipeline’s RAST annotation(data not shown)(Aziz et al., 2008, Wattam et al., 2014). Together, these results suggest that the CDS that are un-annotated during common assembly and annotation pipelines potentially make significant contributions to genomic diversity particularly at the intraspecies level. In addition, in agreement with a recent report (Sberro et al., 2019), there was a bias in the annotated protein sequences toward longer sequences, and un-annotated proteins were on average significantly shorter than annotated proteins (p<0.01, ANOVA/Tukey) (Figure S3C). In order to include potentially important diversity information contained in the un-annotated proteins, we grouped these proteins into protein clusters sharing 50% identity. Next, we attempted to identify the centroid-defining sequence of the resulting clusters using BLAST against the NCBI RefSeq database. This allowed us to identify 1093 protein clusters that are well-represented across Lachnospiraceae isolates (Figure S3D). For example, hypothetical protein clusters identified by this approach represent 18.8% of the CDS in an average Blautia wexlerae isolate (Figure S3E).
We next examined the core genome of Lachnospiraceae isolates grouped at different taxonomic ranks (Figure 3A). Surprisingly, only 397 protein-encoding genes were shared across all isolates (Family-level core genome, Figure 3A). Restricting the collection of isolates to isolates of the same genus, led to a modest increase in the size of the core genome (Genus-level core genome, Figure 3A). As has been documented for some pathogenic species such as Clostridioides difficile (Lewis et al., 2017), isolates of the same commensal Lachnospiraceae species shared on average only 62.6% of genes (Figure 3A). In addition to these taxonomic groupings of isolates, in some cases our collection of isolates contains multiple representatives of the same species from an individual donor which allowed us to ask what proportion of the genome is shared by those isolates. By assessing multiple isolates of the same species within a donor we found that some, such as [Eubacterium] rectale, share a high percentage of genome, reflecting their clonality (Figure 3A). In the case of other species, such as Blautia wexlerae, some donors were likely colonized by a single strain while others were colonized by multiple strains that shared only 80% of their protein-encoding genomes (Figure 3A, Figure S3F).
Figure 3:

Comparison of the core genome of Lachnospiraceae isolates identifies diversity between isolates. A. The number of core genes (annotated proteins and clustered hypothetical proteins) shared by isolates grouped at different taxonomic ranks is plotted. Isolates are ordered by 16S rRNA-derived phylogeny (bottom). For values of genus, species and donor level core genes, values are plotted only for isolates with more than 1 represented species in the genus (genus level), or more than one represented isolate of the species or from that donor (Species, donor level). B. Proportion of different classes of coding sequences, based on type of annotation, that become shared by subsets of isolates grouped at different taxonomic ranks. C. Proportion of genes assigned to the broad-level KEGG pathways that become shared by a subset of isolates grouped at different taxonomic ranks. D. Percentage of the genes that become shared by a subset of isolates grouped by family, genus or species that map to specific KEGG pathways. Related to Figure S3.
We next investigated what genes and pathways contribute to the core genome when isolates are grouped by different taxonomic ranks. First, we asked what proportion of CDS that become shared between isolates grouped at different taxonomic ranks are annotated (Figure 3B). The majority of the 397 genes that are shared across the Lachnospiraceae family are annotated proteins with associated COGs. This fraction decreases as more specific taxonomic ranks are used to group isolates , such that just 12.6% of the genes that only become shared when isolates are grouped by species level are annotated. This result highlights the importance of the un-annotated proteins to the functional capabilities and isolate differentiation at the species level. Genes that were initially un-annotated but were subsequently identified using our clustering/BLAST approach (identified clusters), represent 20% of the coding sequences that become core when comparing isolates from a single genus (Figure 3B).
Next we mapped the functions of annotated genes to KEGG pathways (Kanehisa and Goto, 2000). With the most general description of function, we observed that the majority of genes that are shared between isolates grouped at different taxonomic ranks were involved in metabolism (Figure 3C). However, the types of metabolism pathways that become shared by isolates grouped at different taxonomic ranks varies (Figure 3D). Fundamental metabolic processes such as peptidoglycan biosynthesis, nitrogen metabolism and gluconeogenesis/glycolysis represent large fractions of the genes that are shared by all isolates (Figure 3D). In contrast, genes contributing to pathways for butyrate (butanoate) production or starch, sucrose, fructose and mannose metabolism are not shared by all Lachnospiraceae but instead are larger contributors to the core genome when restricted to specific genera, species (Figure 3D). 29.4% of the genes shared by all Lachnospiraceae isolates were involved in the category of genetic information processing, however, genes mapping to these pathways represent smaller fractions of the genes shared between isolates when grouped by genus, species or donor (Figure 3C). As expected, when more specific pathways within this category are examined, genes that map to pathways for basic cellular functions such as ribosome formation or DNA replication and repair represent large percentages of the genes that are shared across the Lachnospiraceae family (Figure 3D). Interestingly, genes involved in environmental information processing, including pathways for sugar transport, two-component signaling, tend not to be shared at the family level but instead become substantial components of the genes that are shared only when isolates are grouped by genus or species (Figure 3C,D).
Sequence variability within the Lachnospiraceae core genome
We next asked if there was sequence variability within the genes of the Lachnospiraceae core genome and whether that variability corresponds to our species level identifications based on 16S rRNA sequence. To address this question, the protein sequence of each annotated family-level core gene was aligned, and the resulting multiple sequence alignments were concatenated into a single large protein sequence (Figure 4A). Next, a phylogenetic tree was generated following alignment of the concatenated large sequences. This analysis revealed that isolates from the same 16S-rRNA defined species are also related by the sequence variability in their core genome and that the overall relationship between isolates is consistent with 16S rRNA-derived phylogeny (Figure 4B). Notably, this analysis again revealed a difference between [Ruminococcus] gnavus and other isolates of the Blautia genus. In addition, isolates identified as C. eutactus are more related to Anaerostipes hadrus than other isolates of Coprococcus by this metric (Figure 4B). Similar results were obtained using multigene phylogeny reconstructions (ASTRAL-III) in place of this concatenation approach (Data not shown)(Zhang et al., 2018).
Figure 4:

Phylogeny of Lachnospiraceae isolates as measured by sequence variability in the core genome. A. Schematic showing the alignment of core genes and generation of core-genome dendrogram. B. A core-genome derived dendrogram of Lachnospiraceae isolates based on sequence variability in 384 prokka-annotated core genes.
Genetic repertoire of Lachnospiraceae isolates
Next, as opposed to examining genes that are shared within groups of isolates, we analyzed the pattern of the genetic repertoires of individual isolates. A uniform manifold approximation and projection analysis (UMAP) on the presence/absence of annotated genes and protein clusters within all Lachnospiraceae revealed that isolates form distinct clusters based on their whole genome (Figure 5A). In most cases, clustered isolates contain only a single species, while in rare cases, two species cannot be distinguished on the basis of their genetic repertoire, such as [Eubacterium] rectale and Roseburia intestinalis. Similar to our observations based on 16S rRNA sequence, [Ruminococcus] gnavus isolates were separated by large distances from other isolates of the Blautia genus. Interestingly, applying UMAP analysis to either the annotated coding sequences or protein clusters individually also resulted in the clustering of isolates from a given species, indicating that isolates of a species share a complement of un-annotated protein-encoding genes that distinguish them from other members of the Lachnospiraceae family (Figure S4A,B).
Figure 5:

Analysis of the genetic repertoire of individual isolates identifies clusters of related isolates. A. Plot of the a UMAP analysis of the presence/absence of unique coding sequences (annotated proteins or un-annotated protein clusters) across the Lachnospiraceae colored by 16S rRNA assigned species name as indicated. B. UMAP analysis (from A) colored to indicate acidification after 48 hours of growth for individual isolates. C. Isolates encoding a complete set of genes in a pathway to convert acetyl-CoA to butyrate. Related to Figure S4.
The Lachnospiraceae are fermentative commensals that can produce SCFAs and thereby contribute to the normal modest acidification of lumen in the proximal colon/cecum. Therefore, in designing bacterial consortia to restore normal colonic acidification and SCFA production following microbiota disruption, two factors that are important for colonization resistance against Enterobactericeae(Sorbara et al., 2019), the capacity to promote and tolerate mildly acidic conditions are likely to be critical factors in selecting Lachnospiraceae isolates for inclusion. Therefore, we asked how the capacity to acidify culture media was distributed across our isolates. When acidification was projected onto the UMAP we observed that supernatant acidification corresponded with UMAP position and varied along a combination of both axes of the UMAP (Figure 5B). With the exception of [Ruminococcus] gnavus isolates, isolates from Blautia genera are able to strongly acidify culture media. However, within Blautia there is variation in the extent of acidification with some species, such as Blautia wexlerae or Blautia luti driving greater acidification than Blautia faecis or Blautia schinkii.
The SCFA butyrate has important regulatory functions for cells of the host mucosal immune system (Arpaia et al., 2013, Atarashi et al., 2013, Furusawa et al., 2013, Lopetuso et al., 2013), and, in patients undergoing transplantation, colonization with commensal species encoding butyrate synthesis pathways is associated with resistance to viral infections(Haak et al., 2018, Lee et al., 2019). Therefore, we examined the distribution of genes enabling butyrate production from acetyl-CoA in our Lachnospiraceae isolates, having already determined that genes mapping to the more general KEGG pathway of butanoate metabolism are shared by isolates grouped by genus or species (Figure 3D). We identified complete pathways for butyrate production from acetyl-CoA in 90/273 isolates (Figure 5C). Isolates with complete pathways for butyrate production from acetyl-CoA were present in distinct clusters of the whole genome UMAP analysis corresponding to individual species (Figure 5C). Consistent with previous reports examining variation in pathways of butyrate synthesis(Vital et al., 2014), we identified groups of isolates that utilize butyrate kinase-dependent or butyrl-CoA transferase-dependent pathways to convert Butyryl-CoA to Butyrate (Figure S4B), including some, such as Coprococcus eutactus or [Clostridium] clostridioforme with genes enabling utilization of both pathways. In addition, we identified variability in transferase-dependent pathways, with genes enabling the use of either acetate or acetoacetate, or both in the case of Anaerostipes hadrus, as substrates in the reaction with butyryl-CoA(Figure S4B).
Evaluating 16S rRNA relatedness as an indication of whole-genome genetic repertoire
We noted that clusters of isolates from related taxa (on the basis of 16S rRNA) tend to be positioned close together by UMAP analysis of the coding sequences present in the whole-genome. To investigate this further, we asked whether the tip-to-tip distance in the 16S rRNA-derived phylogenetic tree was correlated with the whole genome-derived distance between isolates (Figure 6A). To ensure that conclusions drawn from these comparisons were not biased by the starting seed value for UMAP embedding, we compared the average intra-isolate UMAP distance from 100 different UMAP embeddings with the 16S rRNA phylogeny (see Materials and Methods). This analysis revealed that, in 37,128 pair-wise comparisons, there was a significant positive correlation (r=0.52) between the 16S rRNA-derived distance and whole genome-derived distance (Figure 6A). While overall there is a positive correlation, this analysis demonstrated that there are pairs of isolates with WGS distances that were proportionally higher or lower than their 16S-based distances (Figure 6B). For example, the relationship between the 16S rRNA and whole genome distances between isolates of Blautia wexlerae and other Lachnospiraceae are typical of the overall population of isolates (Figure 6B-middle). In contrast, the position of [Ruminococcus] gnavus isolates based on 16S rRNA sequence underestimates the whole-genome difference between them and most other Lachnospiraceae (Figure 6B-right). Conversely, Coprococcus eutactus are less distinct at the whole-genome level than expected based on their separation from other isolates in the 16S rRNA phylogenetic tree (Figure 6B-left).
Figure 6:

16S rRNA sequences predict whole genome differences between Lachnospiraceae isolates. A: Plot of pairwise inter-isolate tip-to-tip distances derived from the 16S rRNA phylogenetic dendrogram (Figure 2A) and the whole genome-based distance, as determined by average distance between isolates from 100 UMAP analyses of the presence of protein coding sequences (Figure 5A). The Pearson correlation coefficient is indicated. B. Colored points indicate pairwise comparisons containing a Coprococcus eutactus (left), Blautia wexlerae (middle), or [Ruminococcus] gnavus (right), all other pairwise comparisons (not containing the indicated species) are colored grey.
Intra-species Diversity
We observed that, in some instances, isolates of a single species formed multiple distinct clusters that were detectable in the context of the genomic diversity of all Lachnospiraceae (Figure 5A). To directly examine intra-species genomic diversity, we performed individual UMAP analyses on species with more than 10 representatives using altered UMAP parameters to increase sensitivity for local variations (Figure 7A). For some species including Blautia luti, Dorea longicatena, and Fusicatenibacter saccharivorans, isolates formed only a single cluster, while other species, including Blautia wexlerae, [Ruminococcus] gnavus, and Anaerostipes hadrus formed 2 or 3 distinct clusters (Figure 7A). In addition, we observed that some species form multiple clusters even if the analyses are restricted to annotated or clusters of un-annotated protein-encoding genes (Figure S5A,B). In the case of Blautia wexlerae 2 distinct clusters were observed when the analysis was restricted to either annotated or un-annotated proteins (Figure S5A,B), compared to the 3 groups of isolates identified with the inclusion of both types (Figure 7A). Similarly, isolates of Anaerostipes hadrus form a single distinct cluster on the basis of annotated proteins, but 2 clusters are formed with inclusion of the protein clusters. Interestingly, [Ruminococcus] gnavus isolates form 3 clusters based on the annotated proteins alone, protein clusters alone, or in combination, despite the fact that there is minimal distance between isolates on the basis of 16S rRNA alignment, with an average of 99.4% identity to the reference database (Figure S3). Together, these results indicate that there is significant diversity within the whole-genomes of some species of Lachnospiraceae. For some species this diversity is independent of variation in the 16S rRNA sequence, and genes that are not annotated using standard pipelines can make significant contributions to intra-species diversity.
Figure 7:

Analyses of individual Lachnospiraceae species identifies intra-species variability in pathways related to colonization resistance and disease processes. A. Plot of a UMAP analyses of individual Lachnospiraceae species with ten or greater representatives, with n-neighbors=10. B,C. For clusters of Blautia wexlerae (B) or [Ruminococcus] gnavus (C) as determined by k-means clustering, the numbers of genes that are part of the core genome of a single cluster (pink), a subset of genes involved in metabolism of carbohydrates that are part of the core genome of a single cluster (purple), and a subset of genes involved in antimicrobial production and defense that are part of the core genome of a single cluster (orange) are shown. D. The read coverage across the glucorhamnan polysaccharide cluster in [Ruminococcus] gnavus isolates. Isolates were grouped into three coverage patterns. Lines represent the average coverage for each group, and shaded areas indicate the average +/− standard deviation. Genes in the operon were identified in (Henke et al., 2019). E. The UMAP analysis of [Ruminococcus] gnavus isolates colored by glucorhamnan polysaccharide coverage group. Related to Figure S5,6.
We next examined which genes separate isolates of a species into distinct clusters in the cases of Blautia wexlerae, a prevalent member of the Blautia genus(Touyama et al., 2015), and [Ruminococcus] gnavus, a species that has recently been implicated in IBD pathogenesis (Hall et al., 2017, Png et al., 2010, Willing et al., 2010). We compared the core genomes of clusters of Blautia wexlerae or [Ruminococcus] gnavus and identified genes that were only part of the core genome in a single cluster (Figure 7B). This analysis identified 1041 and 1603 coding sequences that were differentially present in the core genomes of Blautia wexlereae and [Ruminococcus] gnavus clusters respectively. Examining these differentially represented genes revealed that all Blautia wexlerae isolates of cluster B encode several genes for the utilization of cellobiose, while all isolates of cluster A encode genes for the utilization of L-galactonate (Figure 7B). Along similar lines, isolates of cluster A of [Ruminococcus] gnavus share genes involved in lactose metabolism that are not part of the core genomes of clusters B or C (Figure 7C). These results indicate that isolates of the same species differ in their capacity for carbohydrate utilization. In support of these potential metabolic differences, isolates from different clusters of [Ruminococcus] gnavus exhibited significant differences in their ability to acidify rich culture media (Figure S6). We also identified genes involved in antimicrobial production and defense that were differentially present in the core genomes of both the Blautia wexlereae and [Ruminococcus] gnavus clusters, including bacteriocins, lantibiotics and lantibiotic immunity proteins (Figure 7B,C), consistent with an earlier report from our laboratory demonstrating variable representation of lantibiotic-encoding genes in isolates of Blautia (Kim et al., 2019).
Several potential mechanisms have been identified that could contribute to the association of [Ruminococcus] gnavus with CD and inflammation, including the production of a proinflammatory polysaccharide by genes encoded by a 30 kb operon (Henke et al., 2019). To determine if this operon is uniformly distributed across isolates and clusters of [Ruminococcus] gnavus, we mapped reads from whole-genome sequencing to the reported operon. Surprisingly, we identified three distinct coverage patterns for the biosynthetic operon within our isolates, including isolates with complete coverage (blue), intermediate coverage, where isolates lack reads aligning to a component of the cell wall remodeling machinery, a glycosyltransferase and two sugar transporters (green) and limited coverage in isolates missing reads aligning to large sections of the operon (red) (Figure 6D). Two of the [Ruminococcus] gnavus clusters contain isolates that only have low or intermediate coverage of this operon (Figure 6E), suggesting that the clusters of [Ruminococcus] gnavus may differ in their capacity to produce this proinflammatory glucorhamnan polysaccharide.
Discussion
Metagenomic sequencing of fecal samples and assembly of commensal strain genomes has revealed the remarkable diversity of bacterial taxa inhabiting healthy humans and, to a partial extent, the genomic variation between and within clonal bacterial populations (Pasolli et al., 2019, Poyet et al., 2019). Although intestinal microbiota members belonging to the Lachnospiraceae family have been shown to play important roles in promoting health and enhancing disease resistance, our understanding of the metabolic and functional diversity of strains belonging to this family remains incomplete. Because reconstitution of Lachnospiraceae family members may have eventual therapeutic potential, we generated a biobank of 273 isolates of Lachnospiraceae and sequenced their genomes. Our focused analyses of the Lachnospiraceae family extend and complement recent studies that more broadly examined isolates across the multitude of phyla constituting the intestinal microbiota (Forster et al., 2019, Poyet et al., 2019). Our fine-scale analyses revealed major genetic differences both between the represented 27 species of Lachnospiraceae, and within individual species. This study demonstrated that, in addition to reported functional variation across the entire microbiome(Forster et al., 2019, Poyet et al., 2019), there is significant diversity within a single prominent family. Isolates of Lachnospiraceae show variation in pathways relevant to colonization resistance, regulation of the host’s mucosal immune system, or proinflammatory pathways potentially relevant in Crohn’s disease.
Comparative genomic analyses of microbial isolates often rely on annotation of coding sequences using common pipelines, including prokka and PATRIC’s RAST annotation, with investigations into functional diversity across a bacterial library relying on assignment of coding sequences into COGs (Conserved Orthologous Groups). While annotating our isolates using prokka provided insight into the functional pathways encoded by different isolates (Figure 3D), and was sufficient to group isolates into distinct clusters (Figure S4A), only 50% of the coding sequences in these genomes were annotated (Figure S3). The high percentage of un-annotated proteins is in line with previous studies of model organisms(Lery et al., 2014, Lewis et al., 2017, Yang et al., 2019). However, when these un-annotated proteins are grouped into protein clusters, we found that these clusters 1.) make major contributions to the genes that become part of the core genome of different genera or species (Figure 3B); 2.) distinguish related isolates independent of the annotated proteins (Figure 5,S4); and 3.) identify diversity within a species that would otherwise be missed (Figure 7,S5).
While most of the functions assigned to species of Lachnospiraceae are beneficial for the host, [Ruminococcus] gnavus (Blautia gnavus) has been associated with inflammatory bowel disease (Hall et al., 2017), and several proinflammatory mechanisms have been described for the species (Bunker et al., 2019, Henke et al., 2019). In our strain collection, we identified 35 isolates of [Ruminococcus] gnavus. These isolates contained full length 16S rRNA sequences that have greater than 99.5% identity to the reference database (Figure 2, S2). Despite this similarity, we identified three distinct clusters of [Ruminococcus] gnavus, that differed in a large number of genes as well as in the sequence of a biosynthetic gene cluster for a pro-inflammatory glucorhamnan polysaccharide (Figure 7). Furthermore, we found the [Ruminococcus] gnavus are distinct from other members of the Blautia genus and Lachnospiraceae family in terms of their genetic repertoire (Figure 5A), and differ from other Blautia isolates in their capacity to acidify culture media (Figure 5B). Finally, while [Ruminocococcus] gnavus isolates are grouped with other Lachnospiraceae by 16S rRNA sequence (Figure 2, S1), their 16S rRNA phylogenetic distance from other isolates underestimates their whole-genome derived distance to those isolates in comparison to distances between other pairs of Lachnospiraceae species (Figure 6).
Dysbiosis or disruption of the microbiota is implicated in a wide range of host conditions, therefore targeted manipulation of the microbiota remains an attractive goal for treatments or prevention of those conditions. Clinically, fecal microbiota transplantation (FMT) has been used to transfer a diverse population of fecal microbes from a screened donor to patients with recurrent C. difficile infection(Mattila et al., 2012, Yoon and Brandt, 2010) or, for patients receiving hematopoietic stem cell transplants, in an autologous manner using samples obtained prior to microbiota perturbation (Taur et al., 2018). However, the large-scale clinical feasibility, the variability in fecal composition and the inability to completely define fecal compositions renders FMT a less than perfect medical intervention for implementation on a large scale(Pamer, 2014). Indeed, a recent report of pathogen transfer through an FMT highlights the severity of these challenges(DeFilipp et al., 2019).
An alternative approach to FMT is to assemble consortia of commensal bacteria for targeted complementation of defects in a recipient’s microbiota (Pamer, 2016). Candidate commensal bacterial strains will require extensive pre-clinical testing for effectiveness and production under conditions that adhere to Good Manufacturing Practices prior to testing for safety and effectiveness in humans. Granular analysis of related isolates, like the Lachnospiraeae, is essential for selecting appropriate isolates to test in pre-clinical studies out of the vast and expanding collections of available isolates. Effective consortia to restore colonization resistance for example, will require members that drive the desired primary mechanistic function, such as lantibiotic production, bile acid modification, SCFA production or acidification of the proximal colon. Our analysis identifies diversity at the genomic level that could alter the ability of closely related isolates to drive these primary functions. In addition, because of interdependencies of individual members of the intestinal microbiota, the consortium must also contain members that support in vivo persistence of resistance-mediating bacteria. To be optimally effective, therapeutic commensal consortia will need to optimize synergy and minimize competition. Our analysis of clusters of Blautia wexlerae for example, identified differences in genes encoding carbohydrate transport and metabolism, which may impact the competition between these isolates and others in a consortium. Overall, understanding the extent and type of variation between isolates within a single taxonomic group, such as the Lachnospiraceae, will be critical when assembling effective consortia.
STAR Methods
RESOURCE AVAILABILITY:
Lead contact
Further information and requests for resources and reagents should be directed to Eric G. Pamer, egpamer@uchicago.edu.
Materials Availability:
Assembled whole genome sequences used in this study are available under NCBI Bioproject #: PRJNA596270.
Data and Code Availability:
The code and genome annotation tables generated during this study are available at https://github.com/elittmann/lachno-cell-host-microbe
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Fecal sample donors
14 healthy donors and 5 donors prior to treatment with allogeneic hematopoietic stem cell transplantation were enrolled in a prospective fecal collection protocol. Donor age and sex were not reported. The prospective fecal collection protocol was approved by the institutional review board at MSKCC. All donors provided written and informed consent for IRB-approved biospecimen collection and analysis (protocols 06-107). The study was conducted in accordance with the Declaration of Helsinki.
Bacterial growth and pH measurements
Isolation and growth of commensal bacteria was performed under anaerobic conditions in an anaerobic chamber (Coy Labs). Fresh donor fecal samples were transferred into anaerobic conditions within 1 hour of collection. Fecal samples were resuspended in pre-reduced PBS and plated on Columbia blood agar or brain-heart infusion (BHI – Difco, BD) agar plates in three serial 10 fold dilutions and incubated at 37°C for 48 – 96 hrs. Isolated colonies were re-streaked for purity onto Columbia blood agar and frozen in pre-reduced 10% glycerol in PBS. For broth cultures, isolates were grown in pre-reduced BHI supplemented with 5g/L yeast extract (Difco, BD) and 0.1% L-cysteine. For pH measurements, after 48 hours of culture, cells were pelleted, and the supernatant pH was measured.
METHOD DETAILS
Whole Genome Sequencing, Assembly and Annotation of isolates
DNA was extracted using a phenol-chloroform extraction technique with mechanical disruption (bead-beating). Briefly, 5 mL of bacterial culture was pelleted and frozen at −80°C, and the cell pellet of each individual isolate was suspended, while frozen, in a solution containing 500μl of extraction buffer (200mM Tris, pH 8.0; 200mM NaCl; and 20mM EDTA), 210μl of 20% SDS, 500μl of phenol/chloroform/isoamyl alcohol (25:24:1), and 500μl of 0.1-mm-diameter zirconia/silica beads (BioSpec Products). Microbial cells were lysed by mechanical disruption with a bead beater (BioSpec Products) for 2 minutes, after which 2 rounds of phenol/chloroform/isoamyl alcohol extraction were performed. After extraction, DNA was precipitated in ethanol, re-suspended in 200μl of TE buffer with 100mg/mL RNase, and further purified with QIAamp mini spin columns (Qiagen).
The purified DNA was quantified using a Qubit 2.0 fluorometer. 1000ng of each sample was prepared for sequencing using the Qiagen Qiaseq FX DNA Library Kit. The protocol was carried out according to manufacturer’s instructions for a targeted fragment size of 550bp. Sequencing was performed on the HiSeq platform (Illumina) with a paired-end 100x100 bp kit in pools designed to provide 5-10 million reads per sample.
Sequences were assembled on PATRIC v3.5.0 (Wattam et al., 2014) which error corrects using BayesHammer and assembles short reads into contigs using Velvet, IDBA, and SPAdes. Assemblies are sorted by ARAST quality score to determine the highest quality assembly. GC content and other assembly statistics were tabulated from PATRIC’s genome report files for each assembly. Genomes were annotated using prokka v1.14.0 to provide protein names and clusters of orthologous gene numbers (COGs) where applicable (Seemann, 2014, Tatusov et al., 2000).
Human donor fecal samples were shotgun sequenced as described above and taxonomically profiled using Kraken2 on PATRIC v3.5.0. Results were collapsed to the Genus level and plotted using R v3.6.0 and the ggplot2 package.
Isolate Identification and 16S rRNA phylogenetic trees
The 16S rRNA gene from the WGS was blasted against NCBI RefSeq for taxonomic identification, and isolates were identified by the hit with highest bit score. In cases where the bit score was limited by the length of the sequence available in the reference database, percent identity between hits with similar bit scores was used. For most isolates, a full length 16S rRNA gene was assembled during genome assembly and annotation. In other cases, 16S rRNA sequences were present as multiple fragments of the 16S rRNA gene covering the complete 16S rRNA gene. In these cases, BLAST was used to identify each fragment, and isolates were included in subsequent analyses if fragments of a similar length gave consistent identification as a species of Lachnospiraceae. For subsequent analyses, the longest fragment (minimum of 559 bp, median of 1435 bp across all Lachnospiraceae isolates) was used. To generate phylogenetic trees, 16S rRNA sequences were aligned using MUSCLE v3.8.1551 (Edgar, 2004). Aligned sequences were then analyzed using PhyML v3.0 (Guindon et al., 2010), with automatic model selection by SMS(Lefort et al., 2017). BIONJ was used to generate the starting tree, and nearest neighbor interchange (NNI) was used for tree improvement. Branch support was determined using aLRT. Similar results were obtained using 100X bootstrap approach for branch support. The MUSCLE alignment and PhyML analysis is performed without specifying any taxonomy or an outgroup, and subsequently the resulting dendogram is rooted on either the Ruminococcaceae or Clostridiaceae isolates for presentation using ggtree.
To assess the improved accuracy of full 16S sequences over hyper variable regions, the 175 full length 16S sequences in our library were split into hypervariable regions V1-V2, V4, V4-V5 (Yang et al., 2016) and BLASTed against NCBI refseq. Hits with the highest bit score were compared to the top hit for the full length sequence.
16S rRNA sequences for a Lachnospiraceae type strains from ATCC/DSMZ were retrieved from NCBI, with the following accession numbers:____[Clostridium] aldenense: DQ279736.1, [Clostridium] celerecrescens: X71848.1, [Clostridium] clostridioforme: M59089.2, [Clostridium] scindens: AF262238.1, [Eubacterium] rectale: L34627.1, [Ruminococcus] gnavus: X94967, Anaerostipes hadrus: NR_117139, Blautia caecimuris: KR364746.1, Blautia faecis: HM626178.1, Blautia glucerasea: AB439724.1, Blautia hansenii: DSM.20583, Blautia luti: AJ133124.1, Blautia obeum: X85101.1, Blautia producta: X94966.1, Blautia schinkii: X94965.1, Blautia wexlerae: EF036467.1, Coprococcus comes: EF031542.1, Coprococcus eutactus: NR_115510., Dorea formicigenerans: L34619.2, Dorea longicatena: AJ132842.1, Faecalicatena fissicatena: NR_117142., Fusicatenibacter saccharivorans: AB698910.1, Roseburia intestinalis: AJ312385.1, Sellimonas intestinalis: KP966092.1
Core genome analysis
Protein coding sequences from the isolates were sorted by annotation type into annotated proteins with or without COGs, or proteins identified only as hypothetical proteins. To cluster the hypothetical proteins, hypothetical protein sequences from all of the isolates were sorted by length and then clustered using usearch v11.0.667 (Edgar, 2010) with a 50% identity cutoff generating 44787 clusters. Similar results and numbers of clusters were obtained using CD-HIT. Next, BLAST was used to compare the defining centroid sequence of each cluster against NCBI RefSeq. Clusters were counted as identified clusters if the top result was not a hypothetical protein or putative hypothetical protein. The cluster of hypothetical proteins corresponding to Plantarcin lantibiotic was identified by BLAST against the NCBI non-redundant protein database, after selecting for un-identified protein clusters of an appropriate length in clusters of Blautia wexlerae that also expressing nisin immunity protein. Annotated proteins or clusters of hypothetical proteins (clusters) that were present in at least one copy in every Lachnospiraceae isolate counted as part of the core genome for the family. Isolates were then grouped by genus, and annotated proteins or clusters present in every isolate of the genus, except those that were part of the core for the family, were counted as part of the genus level core genome. This process was repeated for each species, and then for individual species from a single donor. Proteins that were part of the core genome at different taxonomic ranks were functionally annotated by mapping to the KEGG database (accessed August 2019). To assess the sequence variability across the core genome, protein sequences for the 384 annotated core genes shared across all isolates were aligned to each other one gene at a time using MUSCLE. The resulting alignments for each gene were concatenated into one large protein sequence per isolate, preserving spaces in cases of different sequence lengths. These large core genome sequences were aligned using MUSCLE neighbor joining for one iteration and exported as a phylogenetic tree.
Analysis of the genetic repertoire of individual isolates
The global repertoire of all annotated proteins or protein clusters were counted as being either present or absent in each of the Lachnospiraceae isolates. A uniform manifold approximation and projection UMAP analysis was used to group isolates, with nearest neighbors set to 10 (single species) or 100 (all Lachnospiraceae) and using a manhattan distance metric (McInnes et al., 2018). Alternatively, the annotated proteins or protein clusters were used individually in the analysis. K-means clustering was used to define UMAP clusters of Blautia wexlerae and [Ruminococcus] gnavus in Figure 6.
To determine the genetic capability to produce butyrate, we determined how the genes in known pathways of butyrate production (Vital et al., 2014) were annotated in the prokka pipeline for each step from Acetyl-CoA to Butyrate (prokka product names shown in Figure S6). Isolates were counted as having the complete pathway if they had at least one copy of each gene in the pathway (Figure 4C, S6). For the conversion of butyryl-coA to butyryl-phosphate, phosphate butyryl-transferase is used in the butyrate kinase pathway (Vital et al., 2014) however this gene was annotated as either phosphate acetyltransferase or ethanolamine utilization protein EutD by prokka. In each case, where we identify a complete butyrate kinase pathway, the CDS identified as phophaste acetyltransferase or EutD were immediately upstream or downstream of butyrate kinase.
Comparison of 16S rRNA and UMAP distances
A matrix of inter-isolate tip-to-tip distances was determined using the cophenetic.phylo function from the ape R package. The Euclidean distance between isolates following UMAP analysis (Figure 4A) was determined using the dist function in R. To ensure that the settings of the UMAP analysis were preserving global structure in the embedding and were able to be used as a metric in comparison with 16S rRNA derived distances, we performed 100 independent UMAP analyses using distinct seed values. Next, we compared the average distance between isolates across these replicates with the intra-isolate distance from a single embedding, and found a direct correlation. In contrast, a direct correlation is not observed if the UMAP analyses are performed with the default settings of n_neighbors =15. We then compared the average intra-isolate distance from the 100 replicates with the 16S-rRNA derived distances. These paired distances were then plotted and analyzed using a linear regression. The Pearson correlation coefficient, calculated in R v3.6.0, is shown.
Coverage of glucorhamnan polysaccharide biosynthetic cluster in [Ruminococcus] gnavus isolates
Raw reads from the whole genome sequence were aligned to the reported biosynthetic gene cluster for the pro-inflammatory glucorhamnan plysaccharide using bowtie2 v2.3.5.1 (Henke et al., 2019). Samtools v1.9 was used to calculate sequence coverage from resulting bam files (Li et al., 2009). Average coverage per 50 basepairs was plotted in R and isolates were grouped into three coverage types.
QUNNTIFICATION AND STATISTICAL ANALYASIS
Statistical tests were performed using R v3.6.0. Details of statistical tests are provided in the results and figure legends. For analysis in Figure 6A, the Pearson correlation coefficient was calculated. In Figure 7D, the mean coverage +/− standard deviation is shown. In figure S6, a one-way ANOVA followed by a Tukey post-test was used.
Supplementary Material
Table S1: Summary statistics fo r sequencing and assembly of Lachnospiraceae isolate genomes, Related to Figures 2,3.
Key Resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological Samples and Strains | ||
| Fecal samples from healthy donors or patients prior to allogeneic hematopoietic stem cell transplant | This study | N/A |
| Lachnospiraceae isolates | This study | N/A |
| Bacterial Culturing | ||
| Gibco Bacto Brain Heart Infusion | Fisher Scientific | DF0037178 |
| BD BBL Columbia Agar with 5% sheep blood | Fisher Scientific | B21263X |
| Gibco Bacto Yeast Extract | Fisher Scientific | DF0127-17-9 |
| L-Cysteine | Sigma Aldrich | #C-7352 |
| Whole Genome Sequencing Reagents | ||
| .1mm zirconia-silica beads | Fisher Scientific | NC0362415 |
| Phenol, equal., pH 8.0 | Fisher Scientific | BP17501-40 |
| Chloroform | Fisher Scientific | C298-500 |
| Isoamyl Alcohol | Fisher Scientific | A393-500 |
| Sodium Chloride, 1M | Teknova | S0254 |
| TRIS, 1M, pH 8.0 | Fisher Scientific | E199-500ML |
| EDTA, .5M, pH 8.0 | Fisher Scientific | E177-500ML |
| 20% SDS | Fisher Scientific | BP1311-1 |
| Sodium Acetate, 3M, pH 5.2 | Teknova | S0298 |
| TRIS-EDTA, 1X, pH 8.0 | Fisher Scientific | BP2473-500 |
| Rnase A | Fisher Scientific | 50-100-3354 |
| Quit broad-range dsDNA kit (500) | Invitrogen | Q32853 |
| QIAamp DNA mini Kit (250) | Qiagen | 51306 |
| QIAseq FX DNA Library Kit (96) | Qiagen | 180475 |
| Agilent HS D1000 ScreenTape | Agilent | 5067-5584 |
| Agilent HS D1000 Reagents | Agilent | 5067-5585 |
| Quit 2.0 Fluorometer | Life Technologies | Q32866 |
| Deposited Data | ||
| Lachnospiraceae genomes | This study. | NCBI Bioproject #: PRJNA596270 |
| Software and Algorithms | ||
| PATRIC | Wattam et al., 2017 | http://www.patricbrc.org/portal/portal/patric/Home; RRID:SCR_004154 |
| Prokka | Seemann, 2014 | http://www.vicbioinformatics.com/software.prokka.shtml; RRID:SCR_014732 |
| MUSCLE | Edgar, 2014 | http://www.ebi.ac.uk/Tools/msa/muscle/; RRID:SCR_011812 |
| PhyML | Guindon et al, 2010 | http://www.atac-montpellier.fr/phyml/; RRID:SCR_014629 |
| USEARCH | Edgar, 2010 | https://www.drive5.com/usearch/ |
| CD-HIT | Limin et al., 2012 | http://weizhonali-lab.ora/cd-hit/ref.php; RRID:SCR_007105 |
| Bowtie2 | Salzberg et al 2012 | http:/bowtie-bio.sourceforge.net/index.shtml; RRID:SCR_005476 |
| samtools | Li et al 2009 | http://samtools.sourceforge.net/; RRID:SCR_002105 |
Highlights.
Characterization of 273 human-derived Lachnospiraceae isolates
There is significant inter- and intra-species genomic diversity
Strain level differences may influence the development of therapeutic consortia
Acknowledgements
This work was supported by grants RO1 AI42135, RO1 AI95706, UO1 AI124275 to E.G.P, and P30 CA008748 from the US National Institutes of Health (NIH), the MSK Center for Microbes, Inflammation and Cancer, and the University of Chicago Duchossois Family Institute. M.T.S. is supported by a Canadian Institute of Health Research Fellowship (FRN#152527). We thank members of the Pamer laboratory for discussion and comments on the manuscript.
Competing interests:
E.G.P. has received speaker honoraria from Bristol-Myer Squibb, Celgene, Seres Therapeutics, MedImmune, Novartis, and Ferring Pharmaceuticals; is an inventor on patent application no. WPO2015179437A1, entitled ‘Methods and compositions for reducing Clostridium difficile infection ’and no. WPO2017091753A1, entitled ‘Methods and compositions for reducing vancomycin-resistant Enterococci infection or colonization’; and holds patents that receive royalties from Seres Therapeutics Inc. The remaining authors declare no competing interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- ARPAIA N, CAMPBELL C, FAN X, DIKIY S, VAN DER VEEKEN J, DEROOS P, LIU H, CROSS JR, PFEFFER K, COFFER PJ & RUDENSKY AY 2013. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature, 504, 451–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ATARASHI K, TANOUE T, OSHIMA K, SUDA W, NAGANO Y, NISHIKAWA H, FUKUDA S, SAITO T, NARUSHIMA S, HASE K, KIM S, FRITZ JV, WILMES P, UEHA S, MATSUSHIMA K, OHNO H, OLLE B, SAKAGUCHI S, TANIGUCHI T, MORITA H, HATTORI M & HONDA K 2013. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature, 500, 232–6. [DOI] [PubMed] [Google Scholar]
- AZIZ RK, BARTELS D, BEST AA, DEJONGH M, DISZ T, EDWARDS RA, FORMSMA K, GERDES S, GLASS EM, KUBAL M, MEYER F, OLSEN GJ, OLSON R, OSTERMAN AL, OVERBEEK RA, MCNEIL LK, PAARMANN D, PACZIAN T, PARRELLO B, PUSCH GD, REICH C, STEVENS R, VASSIEVA O, VONSTEIN V, WILKE A & ZAGNITKO O 2008. The RAST Server: rapid annotations using subsystems technology. BMC Genomics, 9, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BECATTINI S, LITTMANN ER, CARTER RA, KIM SG, MORJARIA SM, LING L, GYALTSHEN Y, FONTANA E, TAUR Y, LEINER IM & PAMER EG 2017. Commensal microbes provide first line defense against Listeria monocytogenes infection. J Exp Med, 214, 1973–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRUGIROUX S, BEUTLER M, PFANN C, GARZETTI D, RUSCHEWEYH HJ, RING D, DIEHL M, HERP S, LOTSCHER Y, HUSSAIN S, BUNK B, PUKALL R, HUSON DH, MUNCH PC, MCHARDY AC, MCCOY KD, MACPHERS ON AJ, LOY A, CLAVEL T, BERRY D & STECHER B 2016. Genome-guided design of a defined mouse microbiota that confers colonization resistance against Salmonella enterica serovar Typhimurium. Nat Microbiol, 2, 16215. [DOI] [PubMed] [Google Scholar]
- BUFFIE CG, BUCCI V, STEIN RR, MCKENNEY PT, LING L, GOBOURNE A, NO D, LIU H, KINNEBREW M, VIALE A, LITTMANN E, VAN DEN BRINK MR, JENQ RR, TAUR Y, SANDER C, CROSS JR, TOUSSAINT NC, XAVIER JB & PAMER EG 2015. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature, 517, 205–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUKIN YS, GALACHYANTS YP, MOROZOV IV, BUKIN SV, ZAKHARENKO AS & ZEMSKAYA TI 2019. The effect of 16S rRNA region choice on bacterial community metabarcoding results. Sci Data, 6, 190007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUNKER JJ, DREES C, WATSON AR, PLUNKETT CH, NAGLER CR, SCHNEEWIND O, EREN AM & BENDELAC A 2019. B cell superantigens in the human intestinal microbiota. Sci Transl Med, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BYNDLOSS MX, OLSAN EE, RIVERA-CHAVEZ F, TIFFANY CR, CEVALLOS SA, LOKKEN KL, TORRES TP, BYNDLOSS AJ, FABER F, GAO Y, LITVAK Y, LOPEZ CA, XU G, NAPOLI E, GIULIVI C, TSOLIS RM, REVZIN A, LEBRILLA CB & BAUMLER AJ 2017. Microbiota-activated PPAR-gamma signaling inhibits dysbiotic Enterobacteriaceae expansion. Science, 357, 570–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CABALLERO S, KIM S, CARTER RA, LEINER IM, SUSAC B, MILLER L, KIM GJ, LING L & PAMER EG 2017. Cooperating Commensals Restore Colonization Resistance to Vancomycin-Resistant Enterococcus faecium. Cell Host Microbe, 21, 592–602 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAKRAVORTY S, HELB D, BURDAY M, CONNELL N & ALLAND D 2007. A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria. J Microbiol Methods, 69, 330–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAVID LA, MAURICE CF, CARMODY RN, GOOTENBERG DB, BUTTON JE, WOLFE BE, LING AV, DEVLIN AS, VARMA Y, FISCHBACH MA, BIDDINGER SB, DUTTON RJ & TURNBAUGH PJ 2014. Diet rapidly and reproducibly alters the human gut microbiome. Nature, 505, 559–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEFILIPP Z, BLOOM PP, TORRES SOTO M, MANSOUR MK, SATER MRA, HUNTLEY MH, TURBETT S, CHUNG RT, CHEN YB & HOHMANN EL 2019. Drug-Resistant E. coli Bacteremia Transmitted by Fecal Microbiota Transplant. N Engl J Med, 381, 2043–2050. [DOI] [PubMed] [Google Scholar]
- EDGAR RC 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res, 32, 1792–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EDGAR RC 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics, 26, 2460–1. [DOI] [PubMed] [Google Scholar]
- FORSTER SC, KUMAR N, ANONYE BO, ALMEIDA A, VICIANI E, STARES MD, DUNN M, MKANDAWIRE TT, ZHU A, SHAO Y, PIKE LJ, LOUIE T, BROWNE HP, MITCHELL AL, NEVILLE BA, FINN RD & LAWLEY TD 2019. A human gut bacterial genome and culture collection for improved metagenomic analyses. Nat Biotechnol, 37, 186–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FUKS G, ELGART M, AMIR A, ZEISEL A, TURNBAUGH PJ, SOEN Y & SHENTAL N 2018. Combining 16S rRNA gene variable regions enables high-resolution microbial community profiling. Microbiome, 6, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FURUSAWA Y, OBATA Y, FUKUDA S, ENDO TA, NAKATO G, TAKAHASHI D, NAKANISHI Y, UETAKE C, KATO K, KATO T, TAKAHASHI M, FUKUDA NN, MURAKAMI S, MIYAUCHI E, HINO S, ATARASHI K, ONAWA S, FUJIMURA Y, LOCKETT T, CLARKE JM, TOPPING DL, TOMITA M, HORI S, OHARA O, MORITA T, KOSEKI H, KIKUCHI J, HONDA K, HASE K & OHNO H 2013. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature, 504, 446–50. [DOI] [PubMed] [Google Scholar]
- GUINDON S, DUFAYARD JF, LEFORT V, ANISIMOVA M, HORDIJK W & GASCUEL O 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol, 59, 307–21. [DOI] [PubMed] [Google Scholar]
- HAAK BW, LITTMANN ER, CHAUBARD JL, PICKARD AJ, FONTANA E, ADHI F, GYALTSHEN Y, LING L, MORJARIA SM, PELED JU, VAN DEN BRINK MR, GEYER AI, CROSS JR, PAMER EG & TAUR Y 2018. Impact of gut colonization with butyrate-producing microbiota on respiratory viral infection following allo-HCT. Blood, 131, 2978–2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HALL AB, YASSOUR M, SAUK J, GARNER A, JIANG X, ARTHUR T, LAGOUDAS GK, VATANEN T, FORNELOS N, WILSON R, BERTHA M, COHEN M, GARBER J, KHALILI H, GEVERS D, ANANTHAKRISHNAN AN, KUGATHASAN S, LANDER ES, BLAINEY P, VLAMAKIS H, XAVIER RJ & HUTTENHOWER C 2017. A novel Ruminococcus gnavus clade enriched in inflammatory bowel disease patients. Genome Med, 9, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HATZIIOANOU D, GHERGHISAN-FILIP C, SAALBACH G, HORN N, WEGMANN U, DUNCAN SH, FLINT HJ, MAYER MJ & NARBAD A 2017. Discovery of a novel lantibiotic nisin O from Blautia obeum A2-162, isolated from the human gastrointestinal tract. Microbiology, 163, 1292–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HENKE MT, KENNY DJ, CASSILLY CD, VLAMAKIS H, XAVIER RJ & CLARDY J 2019. Ruminococcus gnavus, a member of the human gut microbiome associated with Crohn’s disease, produces an inflammatory polysaccharide. Proc Natl Acad Sci U SA, 116, 12672–12677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOLD GL, PRYDE SE, RUSSELL VJ, FURRIE E & FLINT HJ 2002. Assessment of microbial diversity in human colonic samples by 16S rDNA sequence analysis. FEMS Microbiol Ecol, 39, 33–9. [DOI] [PubMed] [Google Scholar]
- KANEHISA M & GOTO S 2000. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res, 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIM SG, BECATTINI S, MOODY TU, SHLIAHA PV, LITTMANN ER, SEOK R, GJONBALAJ M, EATON V, FONTANA E, AMORETTI L, WRIGHT R, CABALLERO S, WANG ZX, JUNG HJ, MORJARIA SM, LEINER IM, QIN W, RAMOS R, CROSS JR, NARUSHIMA S, HONDA K, PELED JU, HENDRICKSON RC, TAUR Y, VAN DEN BRINK MRM & PAMER EG 2019. Microbiota-derived lantibiotic restores resistance against vancomycin-resistant Enterococcus. Nature, 572, 665–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAWSON PA & FINEGOLD SM 2015. Reclassification of Ruminococcus obeum as Blautia obeum comb. nov. Int J Syst Evol Microbiol, 65, 789–93. [DOI] [PubMed] [Google Scholar]
- LEE JR, HUANG J, MAGRUDER M, ZHANG LT, GONG C, SHOLI AN, ALBAKRY S, EDUSEI E, MUTHUKUMAR T, LUBETZKY M, DADHANIA DM, TAUR Y, PAMER EG & SUTHANTHIRAN M 2019. Butyrate-producing gut bacteria and viral infections in kidney transplant recipients: A pilot study. Transpl Infect Dis, e13180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEFORT V, LONGUEVILLE JE & GASCUEL O 2017. SMS: Smart Model Selection in PhyML. Mol Biol Evol, 34, 2422–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LERY LM, FRANGEUL L, TOMAS A, PASSET V, ALMEIDA AS, BIALEK-DAVENET S, BARBE V, BENGOECHEA JA, SANSONETTI P, BRISSE S & TOURNEBIZE R 2014. Comparative analysis of Klebsiella pneumoniae genomes identifies a phospholipase D family protein as a novel virulence factor. BMC Biol, 12, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEWIS BB, CARTER RA, LING L, LEINER I, TAUR Y, KAMBOJ M, DUBBERKE ER, XAVIER J & PAMER EG 2017. Pathogenicity Locus, Core Genome, and Accessory Gene Contributions to Clostridium difficile Virulence. MBio, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI H, HANDSAKER B, WYSOKER A, FENNELL T, RUAN J, HOMER N, MARTH G, ABECASIS G, DURBIN R & GENOME PROJECT DATA PROCESSING, S. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25, 2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIU C, FINEGOLD SM, SONG Y & LAWSON PA 2008. Reclassification of Clostridium coccoides, Ruminococcus hansenii, Ruminococcus hydrogenotrophicus, Ruminococcus luti, Ruminococcus productus and Ruminococcus schinkii as Blautia coccoides gen. nov., comb. nov., Blautia hansenii comb. nov., Blautia hydrogenotrophica comb. nov., Blautia luti comb. nov., Blautia producta comb. nov., Blautia schinkii comb.nov. and description of Blautia wexlerae sp. nov., isolated from human faeces. Int J Syst Evol Microbiol, 58, 1896–902. [DOI] [PubMed] [Google Scholar]
- LOPETUSO LR, SCALDAFERRI F, PETITO V & GASBARRINI A 2013. Commensal Clostridia: leading players in the maintenance of gut homeostasis. Gut Pathog, 5, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MATTILA E, UUSITALO-SEPPALA R, WUORELA M, LEHTOLA L, NURMI H, RISTIKANKARE M, MOILANEN V, SALMINEN K, SEPPALA M, MATTILA PS, ANTTILA VJ & ARKKILA P 2012. Fecal transplantation, through colonoscopy, is effective therapy for recurrent Clostridium difficile infection. Gastroenterology, 142, 490–6. [DOI] [PubMed] [Google Scholar]
- NAVA GM, FRIEDRICHSEN HJ & STAPPENBECK TS 2011. Spatial organization of intestinal microbiota in the mouse ascending colon. ISME J, 5, 627–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAMER EG 2014. Fecal microbiota transplantation: effectiveness, complexities, and lingering concerns. Mucosal Immunol, 7, 210–4. [DOI] [PubMed] [Google Scholar]
- PAMER EG 2016. Resurrecting the intestinal microbiota to combat antibiotic-resistant pathogens. Science, 352, 535–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PASOLLI E, ASNICAR F, MANARA S, ZOLFO M, KARCHER N, ARMANINI F, BEGHINI F, MANGHI P, TETT A, GHENSI P, COLLADO MC, RICE BL, DULONG C, MORGAN XC, GOLDEN CD, QUINCE C, HUTTENHOWER C & SEGATA N 2019. Extensive Unexplored Human Microbiome Diversity Revealed by Over 150,000 Genomes from Metagenomes Spanning Age, Geography, and Lifestyle. Cell, 176, 649–662 e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PNG CW, LINDEN SK, GILSHENAN KS, ZOETENDAL EG, MCSWEENEY CS, SLY LI, MCGUCKIN MA & FLORIN TH 2010. Mucolytic bacteria with increased prevalence in IBD mucosa augment in vitro utilization of mucin by other bacteria. Am J Gastroenterol, 105, 2420–8. [DOI] [PubMed] [Google Scholar]
- POYET M, GROUSSIN M, GIBBONS SM, AVILA-PACHECO J, JIANG X, KEARNEY SM, PERROTTA AR, BERDY B, ZHAO S, LIEBERMAN TD, SWANSON PK, SMITH M, ROESEMANN S, ALEXANDER JE, RICH SA, LIVNY J, VLAMAKIS H, CLISH C, BULLOCK K, DEIK A, SCOTT J, PIERCE KA, XAVIER RJ & ALM EJ 2019. A library of human gut bacterial isolates paired with longitudinal multiomics data enables mechanistic microbiome research. Nat Med, 25, 1442–1452. [DOI] [PubMed] [Google Scholar]
- RIVA A, KUZYK O, FORSBERG E, SIUZDAK G, PFANN C, HERBOLD C, DAIMS H, LOY A, WARTH B & BERRY D 2019. A fiber-deprived diet disturbs the fine-scale spatial architecture of the murine colon microbiome. Nat Commun, 10, 4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RIVERA-CHAVEZ F, ZHANG LF, FABER F, LOPEZ CA, BYNDLOSS MX, OLSAN EE, XU G, VELAZQUEZ EM, LEBRILLA CB, WINTER SE & BAUMLER AJ 2016. Depletion of Butyrate-Producing Clostridia from the Gut Microbiota Drives an Aerobic Luminal Expansion of Salmonella. Cell Host Microbe, 19, 443–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SBERRO H, FREMIN BJ, ZLITNI S, EDFORS F, GREENFIELD N, SNYDER MP, PAVLOPOULOS GA, KYRPIDES NC & BHATT AS 2019. Large-Scale Analyses of Human Microbiomes Reveal Thousands of Small, Novel Genes. Cell, 178, 1245–1259 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SEEMANN T 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics, 30, 2068–9. [DOI] [PubMed] [Google Scholar]
- SORBARA MT, DUBIN K, LITTMANN ER, MOODY TU, FONTANA E, SEOK R, LEINER IM, TAUR Y, PELED JU, VAN DEN BRINK MRM, LITVAK Y, BAUMLER AJ, CHAUBARD JL, PICKARD AJ, CROSS JR & PAMER EG 2019. Inhibiting antibiotic-resistant Enterobacteriaceae by microbiota-mediated intracellular acidification. J Exp Med, 216, 84–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STUDER N, DESHARNAIS L, BEUTLER M, BRUGIROUX S, TERRAZOS MA, MENIN L, SCHURCH CM, MCCOY KD, KUEHNE SA, MINTON NP, STECHER B, BERNIER-LATMANI R & HAPFELMEIER S 2016. Functional Intestinal Bile Acid 7alpha-Dehydroxylation by Clostridium scindens Associated with Protection from Clostridium difficile Infection in a Gnotobiotic Mouse Model. Front Cell Infect Microbiol, 6, 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TATUSOV RL, GALPERIN MY, NATALE DA & KOONIN EV 2000. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res, 28, 33–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAUR Y, COYTE K, SCHLUTER J, ROBILOTTI E, FIGUEROA C, GJONBALAJ M, LITTMANN ER, LING L, MILLER L, GYALTSHEN Y, FONTANA E, MORJARIA S, GYURKOCZA B, PERALES MA, CASTRO-MALASPINA H, TAMARI R, PONCE D, KOEHNE G, BARKER J, JAKUBOWSKI A, PAPADOPOULOS E, DAHI P, SAUTER C, SHAFFER B, YOUNG JW, PELED J, MEAGHER RC, JENQ RR, VAN DEN BRINK MRM, GIRALT SA, PAMER EG & XAVIER JB 2018. Reconstitution of the gut microbiota of antibiotic-treated patients by autologous fecal microbiota transplant. Sci Transl Med, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TOUYAMA M, JIN JS, KIBE R, HAYASHI H & BENNO Y 2015. Quantification of Blautia wexlerae and Blautia luti in human faeces by real-time PCR using specific primers. Benef Microbes, 6, 583–90. [DOI] [PubMed] [Google Scholar]
- VAN DEN ABBEELE P, BELZER C, GOOSSENS M, KLEEREBEZEM M, DE VOS WM, THAS O, DE WEIRDT R, KERCKHOF FM & VAN DE WIELE T 2013. Butyrate-producing Clostridium cluster XIVa species specifically colonize mucins in an in vitro gut model. ISME J, 7, 949–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VELAZQUEZ OC, LEDERER HM & ROMBEAU JL 1997. Butyrate and the colonocyte. Production, absorption, metabolism, and therapeutic implications. Adv Exp Med Biol, 427, 123–34. [PubMed] [Google Scholar]
- VITAL M, HOWE AC & TIEDJE JM 2014. Revealing the bacterial butyrate synthesis pathways by analyzing (meta)genomic data. MBio, 5, e00889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WATTAM AR, ABRAHAM D, DALAY O, DISZ TL, DRISCOLL T, GABBARD JL, GILLESPIE JJ, GOUGH R, HIX D, KENYON R, MACHI D, MAO C, NORDBERG EK, OLSON R, OVERBEEK R, PUSCH GD, SHUKLA M, SCHULMAN J, STEVENS RL, SULLIVAN DE, VONSTEIN V, WARREN A, WILL R, WILSON MJ, YOO HS, ZHANG C, ZHANG Y & SOBRAL BW 2014. PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res, 42, D581–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WILLING BP, DICKSVED J, HALFVARSON J, ANDERSSON AF, LUCIO M, ZHENG Z, JARNEROT G, TYSK C, JANSSON JK & ENGSTRAND L 2010. A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology, 139, 1844–1854 e1. [DOI] [PubMed] [Google Scholar]
- YANG B, WANG Y & QIAN PY 2016. Sensitivity and correlation of hypervariable regions in 16S rRNA genes in phylogenetic analysis. BMC Bioinformatics, 17, 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG Z, ZENG X & TSUI SK 2019. Investigating function roles of hypothetical proteins encoded by the Mycobacterium tuberculosis H37Rv genome. BMC Genomics, 20, 394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YOON SS & BRANDT LJ 2010. Treatment of refractory/recurrent C. difficile-associated disease by donated stool transplanted via colonoscopy: a case series of 12 patients. J Clin Gastroenterol, 44, 562–6. [DOI] [PubMed] [Google Scholar]
- ZHANG C, RABIEE M, SAYYARI E & MIRARAB S 2018. ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinformatics, 19, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Summary statistics fo r sequencing and assembly of Lachnospiraceae isolate genomes, Related to Figures 2,3.
Data Availability Statement
The code and genome annotation tables generated during this study are available at https://github.com/elittmann/lachno-cell-host-microbe