

Abstract
SLC30A8 encodes the zinc transporter ZnT8. SLC30A8 haploinsufficiency protects against type 2 diabetes (T2D), suggesting that ZnT8 inhibitors may prevent T2D. We show here that, while adult chow fed Slc30a8 haploinsufficient and knockout (KO) mice have normal glucose tolerance, they are protected against diet-induced obesity (DIO), resulting in improved glucose tolerance. We hypothesize that this protection against DIO may represent one mechanism whereby SLC30A8 haploinsufficiency protects against T2D in humans and that, while SLC30A8 is predominantly expressed in pancreatic islet beta cells, this may involve a role for ZnT8 in extra-pancreatic tissues. Consistent with this latter concept we show in humans, using electronic health record-derived phenotype analyses, that the ‘C’ allele of the non-synonymous rs13266634 single nucleotide polymorphism, which confers a gain of ZnT8 function, is associated not only with increased T2D risk and blood glucose but also but also increased risk for hemolytic anemia and decreased mean corpuscular hemoglobin (MCH). In Slc30a8 KO mice MCH was unchanged but reticulocytes, platelets and lymphocytes were elevated. Both young and adult Slc30a8 KO mice exhibit delayed rise in insulin after glucose injection but only the former exhibit increased basal insulin clearance and impaired glucose tolerance. Young Slc30a8 KO mice also exhibit elevated pancreatic G6pc2 gene expression, potentially mediated by decreased islet zinc levels. These data indicate that the absence of ZnT8 results in a transient impairment in some aspects of metabolism during development. These observations in humans and mice suggest the potential for negative effects associated with T2D prevention using ZnT8 inhibitors.
Introduction
Genome-wide association studies (GWAS) have linked the ‘C’ allele of the common non-synonymous SLC30A8 single nucleotide polymorphism (SNP) rs13266634 with increased susceptibility for the development of type 2 diabetes (T2D) (Sladek, et al. 2007). The alternate ‘C’ and ‘T’ alleles of rs13266634 encode an arginine or a tryptophan at ZnT8 amino acid 325, respectively. The R325 variant is associated with reduced proinsulin to insulin conversion (Kirchhoff, et al. 2008), impaired glucose tolerance (Xu, et al. 2011), and impaired insulin secretion (Boesgaard, et al. 2008; Staiger, et al. 2007), relative to the W325 variant, suggesting that this R325 variant negatively affects beta cell function. The R325 variant was originally reported to have reduced zinc transporter activity (Nicolson, et al. 2009). However, more recent studies have suggested the opposite result, namely increased zinc transporter activity with the R325 variant (Li, et al. 2016; Merriman, et al. 2016), implying that it represents a deleterious gain of function. This variant is associated with increased zinc content in human islets (Wong, et al. 2017), which, if toxic, would provide a mechanism by which this variant increases T2D risk.
In contrast to these studies on the rs13266634 SNP, multiple groups have shown that, while homozygous deletion of Slc30a8 markedly reduces islet zinc content, it has little or no effect on glucose tolerance or glucose-stimulated insulin secretion (GSIS) in adult chow fed mice (Davidson, et al. 2014; Rutter and Chimienti 2015). The high levels of zinc in islets, relative to other tissues, had been reported to be important for multiple aspects of islet function including paracrine signaling from beta to alpha cells (Hardy, et al. 2011; Robertson, et al. 2011), proinsulin processing (Dunn 2005) and crystallization of insulin hexamers (Chausmer 1998; Dunn 2005). These Slc30a8 knockout (KO) mouse data suggest that, while the gain of ZnT8 function and a further increase in islet zinc content associated with the rs13266634 ‘C’ allele are deleterious, the absence of ZnT8 and presence of low islet zinc content in adult Slc30a8 KO mice are not. In addition, we have recently shown that SLC30A8 is a pseudogene in multiple species and that this correlates with low islet zinc content (Bosma, et al. 2019; Syring, et al. 2018). Consistent with the concept that high zinc content is not essential for adult mouse pancreatic islet beta cell function, we also showed that deletion of the Slc30a7 gene in adult mice, which encodes the ZnT7 zinc transporter, also markedly reduces islet zinc content, however, this lowered zinc content had no effect on GSIS in isolated Slc30a7 KO mouse islets (Syring, et al. 2016). The same marked reduction in islet zinc content was observed following individual or combined deletion of Slc30a7 and Slc30a8 (Pound, et al. 2009; Syring et al. 2016). However, combined deletion of Slc30a7 and Slc30a8 abolished GSIS in isolated islets (Syring et al. 2016). These data suggest that the actions of ZnT7 and ZnT8 in adult mouse islets are redundant, such that a role of ZnT8 in islets can be unmasked by removal of ZnT7 and vice versa.
In contrast to these studies showing a deleterious effect of the rs13266634 ‘C’ allele and little effect of Slc30a8 deletion in adult chow fed mice, Flannick et al. (Flannick, et al. 2014; Flannick, et al. 2019) have shown that SLC30A8 haploinsufficiency is protective against the development of T2D in humans. To investigate these seemingly paradoxical results, in the current study we have investigated the effect of Slc30a8 haploinsufficiency in mice on both a chow diet and when challenged with a high fat diet. We show that both heterozygous and homozygous deletion of Slc30a8 protect against diet-induced obesity (DIO), potentially providing one mechanism to explain the observations of Flannick et al. (Flannick et al. 2014; Flannick et al. 2019). The mechanism behind this protection is unclear but electronic health record-derived phenotype analyses and KO mouse studies suggest that ZnT8 activity in extra-pancreatic tissues affects blood-related parameters in humans and mice, respectively. We also show that deletion of Slc30a8 impairs glucose tolerance and insulin clearance in young but not adult chow fed mice, implying that developmental compensation accounts for the lack of a glucose tolerance phenotype in adult chow fed mice. Overall these observations imply that, while individuals who are haploinsufficient for SLC30A8 are protected against T2D, acute inhibition of ZnT8 might have negative consequences.
Materials and Methods
Animal care
The Vanderbilt University Medical Center Animal Care and Use Committee approved all protocols used. Mice were maintained on either a standard rodent chow diet (LabDiet 5001, PMI Nutrition International, which comprises 24% protein; 5% fat (linoleic acid, 1.16%; linolenic acid, total saturated fatty acids, 1.50%; total monounsaturated fatty acids, 1.58%) and 49% carbohydrate (starch, 31.9%; glucose, 0.22%); fructose 0.3%; sucrose, 3.7%; lactose 2.0%) with calories from protein (29%), fat (13%) and carbohydrate (58%)) or a high fat diet (Mouse Diet F3282; BioServ, which comprises 21% protein; 36% fat (linoleic acid, 3.7%; linolenic acid, 0.4%; total saturated fatty acids, 14.1%; total monounsaturated fatty acids, 16.2%) and 37% carbohydrate (monosaccharides, 0.1%; disaccharides, 14.6%; polysaccharides, 20%) with calories from protein (15%), fat (59%) and carbohydrate (26%)) as indicated. For high fat diet studies, mice were placed on the diet at 10 weeks of age for 10–13 weeks as indicated in the Figure Legends. Food and water were provided ad libitum. Food and water were provided ad libitum.
Generation of Germline Slc30a8 KO Mice
The generation of germline Slc30a8 KO mice and backcrossing onto the C57BL/6J genetic background have been previously described (Pound et al. 2009; Pound, et al. 2012).
In Vivo Phenotypic Analyses
Phenotypic analyses of 6 h fasted mice were performed as previously described. Briefly, mice were weighed after 5 h fasting and then allowed to recover for one hour prior to being anaesthetized using isoflurane and bled from the retro-orbital venous plexus (Pound, et al. 2013; Pound et al. 2012; Wang, et al. 2006). Whole blood glucose concentrations were determined using an Accu-Check Advantage monitor (Roche) and EDTA was added to blood samples prior to isolation of plasma by centrifugation. Plasma insulin, C peptide, cholesterol, triglyceride and NEFA were quantitated as previously described (Pound et al. 2013; Pound et al. 2012; Wang et al. 2006). Intraperitoneal glucose tolerance tests (IPGTTs) were performed by injection with 2.0 g/kg body weight dextrose in sterile PBS and blood glucose was measured in tail vein samples using a glucose meter (Freestyle; Abbott Diabetes Care Inc., Alameda, CA, USA) over a 100 minute period. For IPGTT experiments in which insulin secretion was assessed, the Accu-Check Advantage monitor (Roche) was used to measure glucose at t=0 and t=15 min and only injections where blood glucose rose to at least 300 mg/dl after 15 mins were considered successful. Insulin tolerance tests (ITTs) were performed by injection with a 0.75 U/kg body weight dose of insulin and blood glucose was then measured in tail vein samples using a glucose meter (Freestyle; Abbott Diabetes Care Inc., Alameda, CA, USA) over a 60 minute period. Body composition was measured in 6 hr fasted mice as previously described (Pound et al. 2013; Pound et al. 2012; Wang et al. 2006). The Vanderbilt Diabetes Research and Training Center Hormone Assay Core assayed plasma T3 and T4 using RIA and corticosterone using the MILLIPLEX Metabolic Hormone Multiplex Assay.
Electronic Health Record (EHR)-Based Analyses of Human Research Subjects
To conduct EHR-based analyses, the Vanderbilt University Medical Center (VUMC)-EHR was used in conjunction with BioVU, the VUMC-associated DNA repository. This biobank links genetic data genotyped on the Illumina Mega-ex Array to individuals’ deidentified electronic health records, including International Classification of Disease (ICD) codes and laboratory values. Detailed information regarding BioVU’s genotyping and quality control methods, program details, ethical rationale, and patient engagement have been previously published (Pulley, et al. 2010; Roden, et al. 2008). Individuals included in this study included a European Americans (EA) population (N = 50,115) and a African American (AA) population (N = 9,640), which were defined using genetic ancestry as determined from principal component analysis. The rs13266634 allele used was common in both of these populations.
Two EHR-based analyses were used in these studies. The first was a phenome-wide association study (PheWAS), which was used to find associations between the presence of the SLC30A8 SNP rs13266634 ‘T’ allele and any phenotypes present in the EHR, following previously published methods (Denny, et al. 2010; Denny, et al. 2013). Phenotypes in PheWAS are defined using Phecodes, which use hierarchical clustering of ICD codes (Denny et al. 2010; Denny et al. 2013). The PheWAS package in R was used to perform 1,247 logistic regressions between the presence of the rs13266634 ‘T’ allele and each phenotype after adjusting for sex, age (median age of ages at each ICD code diagnosed), and the top four principal components of ancestry. To be included in the PheWAS, a minimum number of 100 cases were required for each phenotype to be tested.
The second EHR-based analysis employed was the laboratory value-wide association study (LabWAS), which was used to determine any associations between the rs13266634 ‘T’ allele and any laboratory values from the EHR, which was conducted as described in the GitHub repository [https://bitbucket.org/juliasealock/labwas/src/master/]. The LabWAS package was used for 453 separate laboratory tests to determine, using linear regression, whether any laboratory values were associated with the presence of the rs13266634 ‘T’ allele.
Whole Blood Analyses
The comparison of multiple parameters in blood isolated from WT and Slc30a8 KO mice was performed by the Vanderbilt Translational Pathology Shared Resource using a FORCYTE analyzer. To quantify the levels of circulating metal ions, mouse plasma was incubated with 70% Optima nitric acid and 30% hydrogen peroxide overnight at 60°C. Samples were then analyzed as previously described (Hesse, et al. 2019).
Cell Culture
The mouse βTC-3 cell line (Efrat, et al. 1988) was cultured in DMEM supplemented with 2.5% FBS and 15% horse serum.
Analysis of Gene Expression
Tissue culture cell RNA were isolated using the RNAqueous kit whereas pancreatic and liver RNA were isolated using the ToTALLY RNA kit (Ambion, Carlsbad, CA). The Turbo DNA-free DNAse Treatment Kit (Ambion, Carlsbad, CA) was then used to remove trace genomic DNA followed by cDNA generation using the iScript DNA Synthesis Kit (Bio-Rad, Hercules, CA). Gene expression was then quantitated by PCR using the dUTP-containing FastStart SYBR Green Master Mix in conjunction with Uracil-DNA-Glycosylase (Roche, Nutley, NJ). Fold induction of gene expression was calculated using the 2(-ΔΔC(T)) method (Livak and Schmittgen 2001). PCR primer sequences are provided in Supplemental Material. The efficiency of primer amplification was tested in cDNA dilution analyses. These showed that correlation coefficients were ~0.99 and PCR efficiency was ~95%.
Fusion Gene Analyses
The construction of a wild type human G6PC2- and mouse G6pc2-luciferase fusion genes have been previously described (Boustead, et al. 2004; Martin, et al. 2002). The construction of ZnT8 expression vectors and fusion genes containing cAMP and metal response elements ligated to a minimal Xenopus albumin promoter is described in the Supplemental Material. Cells were transfected with the indicated plasmids using lipofectamine (Promega) according to the manufacturer’s instructions and incubated for 18–20 hrs in the presence or absence of the indicated concentrations of ZnCl2 or CaCl2 prior to harvesting. Luciferase assays were performed using the Promega Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer’s instructions. Firefly luciferase activity directed by the various fusion gene constructs was expressed relative to protein concentration in the same sample. Each construct was analyzed in triplicate in multiple transfections, as specified in the Figure Legends.
Statistical Analyses
Mouse data were analyzed either using a Student’s t-test, two sample assuming equal variance or using a one-way non-repeated or two-way repeated measures ANOVA, assuming normal distribution and equal variance, as indicated. Post hoc analyses were performed using the Bonferroni correction for multiple comparisons. The level of significance was as indicated. A p value less than 0.05 was considered significant. Data are presented as scatter plots or as mean −/+ S.E.M.
EHR associations were analyzed using logistic and linear regressions via the PheWAS package in R and the LabWAS package in R, respectively. Results in PheWAS and LabWAS were considered significant if the p value of the association passed a Bonferroni multiple testing correction to account for 1,247 logistic and 453 linear regressions that were performed, respectively.
Results
Slc30a8 Haploinsufficiency Does Not Affect Glucose Tolerance in 10 Week Old Chow Fed Male Mice
To begin to explore the molecular basis for the observation that SLC30A8 haploinsufficiency is protective against T2D in humans (Flannick et al. 2014; Flannick et al. 2019), we examined the effect of Slc30a8 haploinsufficiency on glucose metabolism in adult chow fed C57BL/6J mice. Our previous studies had not consistently included heterozygous Slc30a8 mice and had instead focused on the effects of homozygous Slc30a8 deletion (Pound et al. 2012). In 16 week old chow fed C57BL/6J male mice homozygous Slc30a8 deletion had no effects on glucose tolerance or insulin secretion from isolated islets (Pound et al. 2012). Figure 1A shows that in 10 week old chow fed C57BL/6J mice there was no difference in glucose tolerance comparing wild type (WT), Slc30a8 heterozygous and Slc30a8 knockout (KO) male mice. We have previously shown that glucose tolerance is similarly unaltered in both 20 week old C57BL/6J male and female Slc30a8 KO mice (Pound et al. 2012). Body weights were also unchanged between WT, Slc30a8 heterozygous and Slc30a8 KO female (Fig. 1B) or male mice (Fig. 1C). Fasting blood glucose (FBG) (Fig. 1D), plasma insulin (FPI) (Fig. 1E) and plasma C-peptide levels (Fig. 1F) were also unchanged in Slc30a8 heterozygous and Slc30a8 KO male mice. Blood glucose (Fig. 1D), plasma insulin (Fig. 1E) and plasma C-peptide (Fig. 1F) all increased 15 min following intraperitoneal (IP) glucose injections, but no differences were apparent between WT, Slc30a8 heterozygous and KO male mice. In addition, no differences in insulin clearance were apparent under fasting conditions or 15 min following IP glucose injection (Fig. 1G). These data suggest that Slc30a8 haploinsufficiency does not substantially affect glucose metabolism in adult chow fed C57BL/6J mice. Studies on mice expressing a floxed Slc30a8 allele and a knockin Ins1-CreER gene led to the same conclusion, namely that partial knockdown of Slc30a8 in adult animals does not negatively affect glucose tolerance (Supplemental Fig. 1).
Figure 1. Analysis of Body Weight, Glucose Tolerance, Blood Glucose, Insulin and C Peptide Plus Insulin Clearance in Adult C57BL/6J Wild Type, Slc30a8 Heterozygous and KO Mice in vivo.
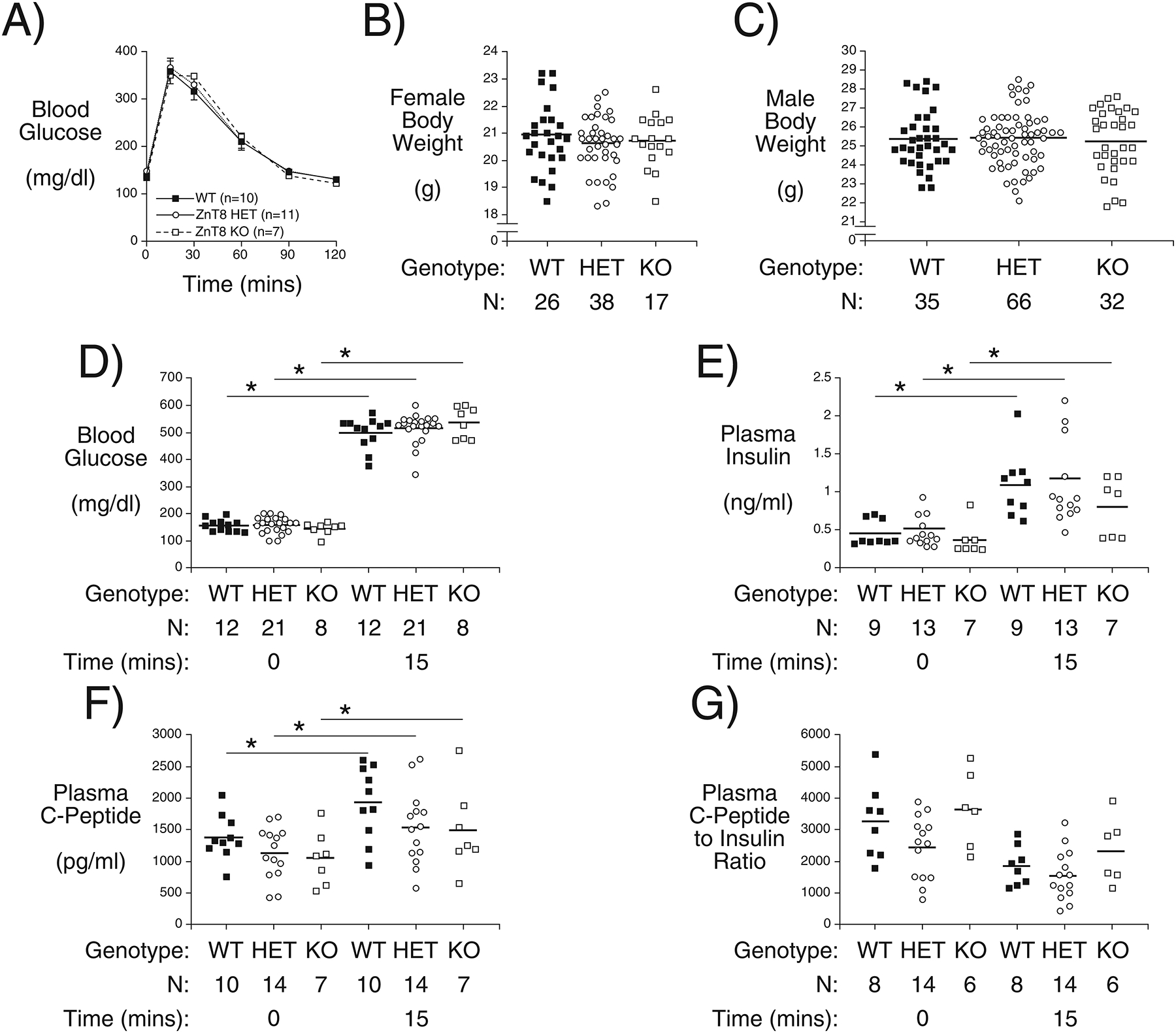
IPGTTs (Panel A) were performed on 6 hr fasted conscious 10 week old male mice. Results show the glucose concentrations in tail blood over time. Body weight was measured in non-fasted 10 week old female (Panel B) and male mice (Panel C). Panels D-G show the blood glucose (Panel D), plasma insulin (Panel E), and plasma C-peptide (Panel F) concentrations plus C-peptide/insulin ratio (Panel G) from IPGTTs performed on 6 hr fasted conscious 10 week old male mice with blood isolated from the retro-orbital plexus at t=0 and t=15 min after glucose injection. Results show the mean data ± S.E.M. with the genotype and number of animals indicated. *p < 0.05 comparing t=0 and t=15 min, two-way ANOVA. WT=wild type; HET=heterozygous; KO=knockout.
Slc30a8 Haploinsufficiency Improves Glucose Tolerance in High Fat Fed Male Mice
We next examined the effect of Slc30a8 haploinsufficiency on glucose metabolism in high fat fed C57BL/6J mice, since diet-induced obesity (DIO), which challenges the beta cell by inducing insulin resistance, is frequently required to uncover effects of gene deletion (Surwit, et al. 1988). These studies were performed on age matched mice with high fat feeding beginning at 10 weeks of age and continuing for 10–13 weeks. We observed that C57BL/6J Slc30a8 KO mice were protected against DIO and that heterozygous mice exhibited an intermediate phenotype (Fig. 2A). This protection against DIO is relative, not absolute, since the high fat fed Slc30a8 heterozygous and KO mice still gain more weight (Fig. 2A) than chow fed KO mice (Fig. 1) (Pound et al. 2012). Indeed, this protection against DIO was insufficient to result in marked changes in FBG (Fig. 2B) or fasting plasma cholesterol (Fig. 2C), triglycerides (Fig. 2D), NEFA (Fig. 2E), corticosterone (Fig. 2F), T3 (Fig. 2G) and T4 (Fig. 2H). However, FPI was reduced in Slc30a8 KO mice with a similar trend in heterozygous mice (Fig. 2I), suggesting the presence of improved insulin sensitivity. This is consistent with the results of insulin tolerance tests that suggest a trend towards improved insulin sensitivity in Slc30a8 KO mice (Fig. 2J), especially when data were expressed as a percentage of blood glucose at t=0 (Fig. 2K). Most importantly, protection against DIO in heterozygous and Slc30a8 KO mice resulted in improved glucose tolerance compared to WT littermates (Fig. 2L). In contrast, in adult chow fed mice deletion of Slc30a8 had no effect on glucose tolerance (Fig. 1A) or insulin sensitivity (Pound et al. 2012). After 15 weeks of high fat diet feeding both fat mass and muscle mass were reduced in Slc30a8 KO mice (Fig. 2M) though body composition, expressed as a percentage of body weight, was unchanged (Fig. 2N). These data suggest that protection against DIO may represent one mechanism whereby SLC30A8 haploinsufficiency confers protection against T2D.
Figure 2. Analysis of the Effects of Diet-Induced Obesity in Male Germline C57BL/6J Wild Type, Slc30a8 Heterozygous and KO Mice in vivo.
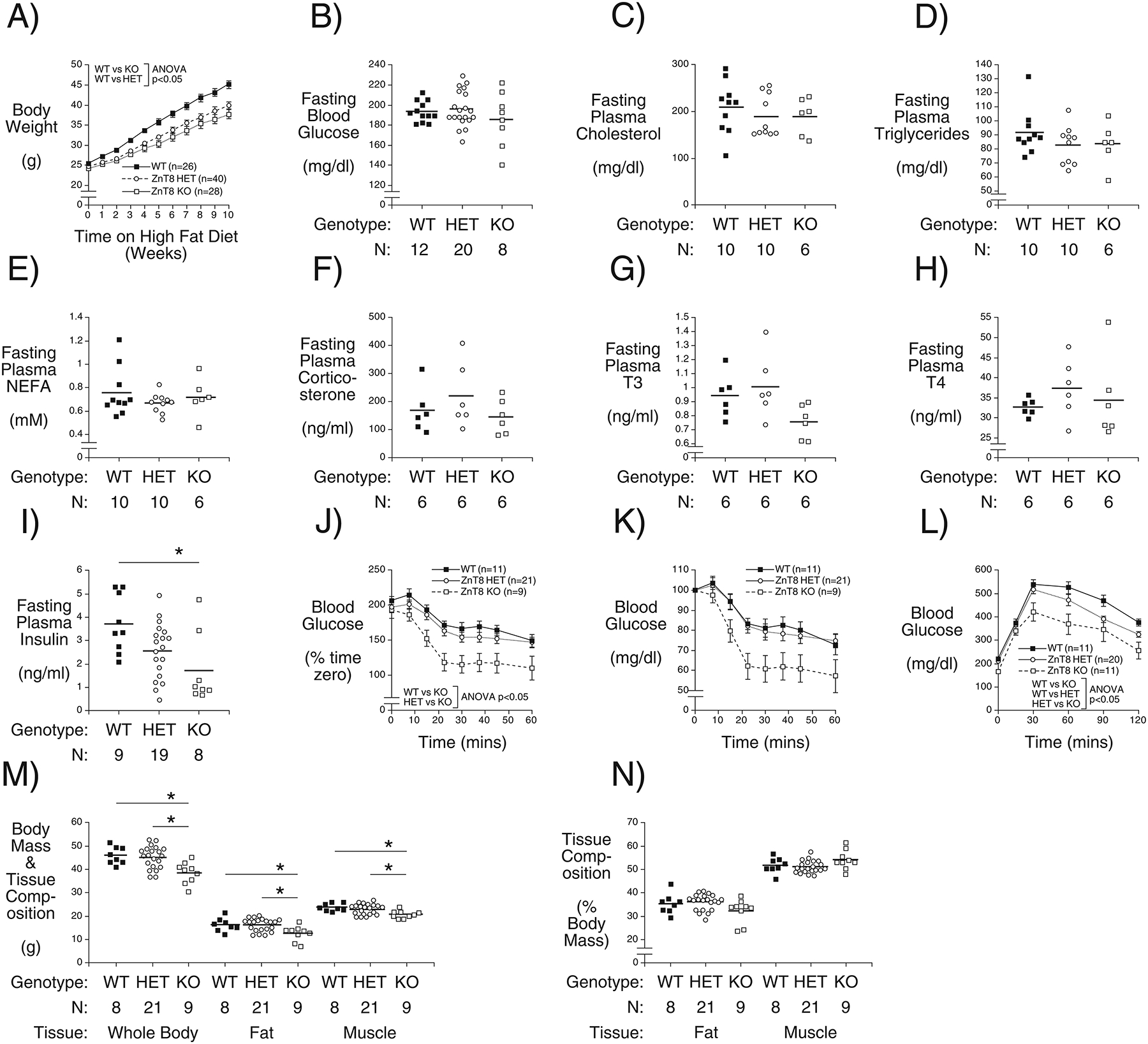
Mice were fed a high-fat diet starting at 10 weeks of age and continuing for 10–13 weeks. Non-fasting body weights were measured weekly (Panel A). Slc30a8 heterozygous and KO mice are protected against DIO: p < 0.05 versus WT; two-way ANOVA. After 11–13 weeks of high fat feeding, blood glucose (Panel B), plasma cholesterol (Panel C), plasma triglyceride (Panel D), plasma NEFA (Panel E), plasma corticosterone (Panel F), plasma T3 (Panel G), plasma T4 (Panel H), and plasma insulin (Panel I) concentrations in blood isolated from the retro-orbital plexus were measured in 6 hr fasted mice. *, p < 0.05 versus WT; one-way ANOVA. After 12 weeks of high fat feeding ITTs were performed on 5 hr fasted conscious 22 week old mice with blood glucose data expressed as mg/dl (Panel J) or as a percentage of blood glucose at t=0 (Panel K). Insulin sensitivity is higher in Slc30a8 KO mice: p < 0.05 versus WT; two-way ANOVA. After 10 weeks of high fat feeding IPGTTs were performed on 6 hr fasted conscious 20 week old mice (Panel L). Glucose tolerance is improved in Slc30a8 heterozygous and KO mice: p < 0.05 versus WT; two-way ANOVA. After 15 weeks of high fat feeding, body composition was measured in 6 hr fasted mice with data expressed as grams (Panel M) or as a percentage of body mass (Panel N). Body weight, fat mass and muscle mass were all reduced in Slc30a8 KO mice: *p < 0.05 versus WT; two-way ANOVA. Results show the mean data ± S.E.M. with the genotype and number of animals indicated. WT=wild type; HET=heterozygous; KO=knockout.
Human Biobank Studies Find Evidence for Extra-Pancreatic Actions of ZnT8
In adult chow fed mice ZnT7 appears to compensate for the absence of ZnT8 and thereby maintain normal insulin secretion and glucose tolerance (Syring et al. 2016) but if this compensation is overcome by the high fat diet the protection against DIO observed in Slc30a8 KO mice could potentially be mediated by changes in insulin secretion affecting hepatic function. However, the absence of changes in plasma lipid levels argues against this possibility (Fig. 2C–E). Instead, the data suggest that, while Slc30a8 is predominantly expressed in pancreatic islet beta cells, the mechanism whereby Slc30a8 haploinsufficiency protects against DIO may involve an action of ZnT8 in peripheral tissues. GTex database analyses and published reports (Deniro and Al-Mohanna 2012; Giacconi, et al. 2018; Liu, et al. 2011; Murgia, et al. 2009; Overbeck, et al. 2008; Smidt, et al. 2007; Zhang, et al. 2018; Zhong, et al. 2012) all show that SLC30A8 is expressed at low levels in several non-pancreatic tissues. To explore this concept and complement our mouse studies, we searched for evidence for potential extra-pancreatic functions of ZnT8 in humans using Vanderbilt University’s Medical Center (VUMC) BioVU Biobank, a DNA biobank linked to a de-identified version of the VUMC electronic health records, called the Synthetic Derivative (SD) (Pulley et al. 2010; Roden et al. 2008). Systematic and efficient approaches have been developed that involve screening the VUMC-EHR with specific SNPs to identify both novel phenotype-variant associations and plasma hormone/metabolite associations, referred to as PheWAS and LabWAS analyses, respectively (Denny et al. 2010; Denny, et al. 2011; Denny et al. 2013; Ritchie, et al. 2013; Shameer, et al. 2014). For these analyses we used the non-synonymous SLC30A8 rs13266634 SNP that has been shown by GWAS to affect T2D risk (Sladek et al. 2007), fasting plasma glucose (Dupuis, et al. 2010) and hemoglobin A1c (HbA1c) (Pare, et al. 2008). The allele frequency of SLC30A8 rs13266634 alleles in the analyzed population was as follows: homozygous ‘AA’ (0.5352), heterozygous ‘AT’ (0.3838) and homozygous ‘TT’ (0.0809).
PheWAS analyses showed that the SLC30A8 rs13266634 ‘T’ allele is associated with decreased risk for type 1 diabetes (T1D) (OR = 0.90, CI = 0.85 – 0.94, p = 3.60E-06), T2D (OR = 0.94, CI = 0.92–0.97, p = 1.04E-05) and T2D with ophthalmic manifestations (OR = 0.84, CI = 0.77–0.91, p = 4.00E-05) in the total (multi-ancestry) BioVU population after Bonferroni correction for the 1247 phenotypes tested (Table 1). SLC30A8 has not been linked with T1D risk by GWAS (Xu et al. 2011) and detailed analyses of the VUMC-EHR suggest that the apparent association observed using BioVU is an anomaly caused by a lack of specificity in coding T1D versus T2D in clinical records (i.e. both T1D and T2D are billed as possible causes of diabetes in medical charts pending further testing) (Supplemental Fig. 2). The specific link with ophthalmic manifestations is consistent with previous studies suggesting that ZnT8 is functionally active in the eye (Xu, et al. 2015).
Table 1. Analysis of the Association Between SLC30A8 SNP rs13266634 ‘T’ allele with Disease Phenotypes Using Electronic Health Record (EHR)-Derived Analyses.
Data show an association between SLC30A8 SNP rs13266634 and various diabetes and hemolytic anemia sub-groups in the total EHR population and in the European American (EA) population within the EHR. No associations were observed in the African American (AA) population or separately in the EA population or AA population when split into with- or without-T2D sub-groups (data not shown). ‘Phecodes’ refer to the numerical abbreviation for individual phenotypes in the VUMC-EHR. All associations were adjusted for the covariates median age of record, sex, and EHR-reported race, and the top 4 principal ancestry components using linear regression. SE, standard error; OR, odds ratio; FDR, false discovery rate; LCI, lower confidence interval; UCI, upper confidence interval.
| Population | Phecode | Description | Beta | SE | OR | P | N Total | N Cases | N Controls | LCI | UCI | Bonferroni | FDR |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total | 250.1 | Type 1 diabetes | −0.11 | 0.02 | 0.90 | 3.60E–06 | 31084 | 2041 | 29043 | 0.85 | 0.94 | TRUE | TRUE |
| Total | 250.2 | Type 2 diabetes | −0.06 | 0.01 | 0.94 | 1.04E–05 | 36594 | 7551 | 29043 | 0.92 | 0.97 | TRUE | TRUE |
| Total | 250.23 | Type 2 diabetes with ophthalmic manifestations | −0.18 | 0.04 | 0.84 | 4.00E–05 | 29673 | 630 | 29043 | 0.77 | 0.91 | TRUE | TRUE |
| Total | 250.22 | Type 2 diabetes with renal manifestations | −0.12 | 0.03 | 0.89 | 1.34E–04 | 30285 | 1242 | 29043 | 0.84 | 0.94 | FALSE | TRUE |
| Total | 282.9 | Other hereditary hemolytic anemias | −0.33 | 0.09 | 0.72 | 1.71E–04 | 26615 | 227 | 26388 | 0.60 | 0.85 | FALSE | TRUE |
| Total | 282 | Hereditary hemolytic anemias | −0.24 | 0.07 | 0.78 | 2.26E–04 | 26757 | 369 | 26388 | 0.69 | 0.89 | FALSE | TRUE |
| All EA | 250.1 | Type 1 diabetes | −0.11 | 0.03 | 0.89 | 1.01E–05 | 25267 | 1701 | 23566 | 0.85 | 0.94 | TRUE | TRUE |
| All EA | 250.2 | Type 2 diabetes | −0.06 | 0.01 | 0.94 | 2.95E–05 | 30041 | 6475 | 23566 | 0.91 | 0.97 | TRUE | TRUE |
| All EA | 250 | Diabetes mellitus | −0.05 | 0.01 | 0.95 | 1.13E–04 | 30390 | 6824 | 23566 | 0.92 | 0.97 | FALSE | TRUE |
Using the false discovery rate (FDR) multiple testing correction, which is less stringent than Bonferroni multiple testing correction, these PheWAS analyses showed that the SLC30A8 rs13266634 ‘T’ allele is also associated with decreased risk for T2D with renal manifestations (OR = 0.89, CI = 0.84–0.94, p = 0.00013), hereditary hemolytic anemias (OR = 0.78, CI = 0.69–0.89, p = 0.00023) and other hereditary hemolytic anemias (OR = 0.72, CI = 0.60–0.85, p = 0.00017) and (Table 1). The Phecode 282 for hereditary hemolytic anemias includes ICD9 code 282 for hereditary hemolytic anemias, while the Phecode 282.9 for other hereditary hemolytic anemias includes ICD9 codes spanning from 282.0 – 282.9, including codes for hereditary spherocytosis, elliptocytosis, and hemolytic anemias due to enzyme deficiencies of glutathione metabolism disorders.
Similar, though less statistically significant, trends were observed for T1D, T2D and diabetes mellitus in the total EA population using FDR multiple testing correction (Table 1). No associations were observed in the African American population or when EA population sub-groups with or without T2D were analyzed separately (data not shown).
LabWAS analyses showed that the SLC30A8 rs13266634 ‘T’ allele was associated with reduced blood glucose (Effect estimate = −0.03, SE = 0.005, p = 3.02E-10), elevated mean corpuscular hemoglobin (MCH) (Effect estimate = 0.03, SE = 0.005, p = 4.95E-8), elevated mean corpuscular volume (MCV) (Effect estimate = 0.02, SE = 0.005, p = 1.80E-7), reduced HbA1c (Effect estimate = −0.04, SE = 0.008, p = 1.01E-6) and reduced total blood protein (Effect estimate = −0.02, SE = 0.005, p = 4.78E-6) in the total population (Table 2).
Table 2. Analysis of the Association Between SLC30A8 SNP rs13266634 ‘T’ allele with Laboratory Analytes Using Electronic Health Record (EHR)-Derived Analyses.
Data show an association between SLC30A8 SNP rs13266634 and blood glucose, mean corpuscular hemoglobin (MCH), mean corpuscular volume (MCV), glycated hemoglobin A1c and total blood protein in the total EHR population. Some of these associations are maintained in the European American (EA) population and the EA sub-population without T2D within the EHR. No associations were observed in the African American (AA) population or in EAs with T2D (data not shown). SE, standard error; OR, odds ratio; FDR, false discovery rate; LCI, lower confidence interval; UCI, upper confidence interval. For each laboratory value, we tested for associations between the median of all lab values for an individual against the number of minor alleles for that individual. All associations were adjusted for covariates median age of record, sex, and EHR-reported race, and the top four principal ancestry components using linear regression. The units for all measurements in any given lab were consistent within the lab (e.g. all blood glucose values were reported in mg/dL).
| Population | Analyte | Effect Estimate | SE | OR | P | N | LCI | UCI | Bonferroni | FDR |
|---|---|---|---|---|---|---|---|---|---|---|
| Total | GLUCOSE BLOOD | −0.03 | 0.005 | 0.97 | 3.02E–10 | 40908 | 0.96 | 0.98 | TRUE | TRUE |
| Total | MEAN CORPUSCULAR HEMOGLOBIN (MCH) | 0.03 | 0.005 | 1.03 | 4.95E–08 | 41776 | 1.02 | 1.04 | TRUE | TRUE |
| Total | MEAN CORPUSCULAR VOLUME (MCV) | 0.02 | 0.005 | 1.03 | 1.80E–07 | 41772 | 1.02 | 1.03 | TRUE | TRUE |
| Total | HGB A1C GLYCATED | −0.04 | 0.008 | 0.96 | 1.01E–06 | 15428 | 0.95 | 0.98 | TRUE | TRUE |
| Total | PROTEIN TOTAL BLOOD | −0.02 | 0.005 | 0.98 | 4.78E–06 | 33870 | 0.97 | 0.99 | TRUE | TRUE |
| All EA | GLUCOSE BLOOD | −0.03 | 0.005 | 0.97 | 1.97E–11 | 34244 | 0.96 | 0.98 | TRUE | TRUE |
| All EA | HGB A1C GLYCATED | −0.03 | 0.008 | 0.97 | 1.11E–04 | 12996 | 0.95 | 0.98 | FALSE | TRUE |
| Non T2D EA | GLUCOSE BLOOD | −0.02 | 0.005 | 0.98 | 4.52E–05 | 22654 | 0.97 | 0.99 | TRUE | TRUE |
After FDR multiple testing correction, significant reductions in blood glucose and HbA1c were observed in the total EA population whereas only the reduction in blood glucose was significant in non-diabetic EA population (Table 2). Given the differences in sample sizes we cannot rule out the possibility that the lack of association with some of the other parameters in these sub-groups is simply related to power. No associations were observed in AAs or in EAs with T2D (data not shown). For the latter group the lack of association with blood glucose and HbA1c could again be explained by a lack of power or it may indicate that the influence of ZnT8 on blood glucose and HbA1c is lost under diabetic conditions. The specific links with MCH and MCV may relate to previous studies suggesting that ZnT8 is present in blood cells (Giacconi et al. 2018; Overbeck et al. 2008).
Slc30a8 KO Mouse Studies Find Evidence for Extra-Pancreatic Actions of ZnT8
To follow up on these observations from human population studies, we examined whether various blood-associated parameters are altered in Slc30a8 KO relative to WT mice. Circulating zinc (Fig. 3A) and calcium (Fig. 3B) levels were unchanged as were MCH (Fig. 3C) and MCV (Fig. 3D) levels, as well as those for various other parameters (Fig. 3E–I). However, platelets (Fig. 3J), reticulocytes (Fig. 3K) and lymphocytes (Fig. 3L) were all elevated in Slc30a8 KO mice. Neutrophil, monocyte, eosinophil and basophil numbers were unchanged (data not shown). LabWAS analyses showed that the SLC30A8 rs13266634 ‘T’ allele was associated with a trend towards reduced platelet numbers (p<0.006) but no change in reticulocytes or lymphocytes. These data suggest differences in the effects of altered SLC30A8 expression on blood parameters between mice and humans, with the caveat that the human studies are comparing an activated form of ZnT8, conferred by the SLC30A8 rs13266634 ‘C’ allele, with the low activity variant whereas the mouse studies are examining the effect of a complete loss of ZnT8. Overall these PheWAS and LabWAS results and Slc30a8 KO mouse blood analyses are consistent with the existence of extra-pancreatic functions of ZnT8.
Figure 3. Comparison of Multiple Blood Parameters in Male Germline C57BL/6J Wild Type and KO Mice in vivo.
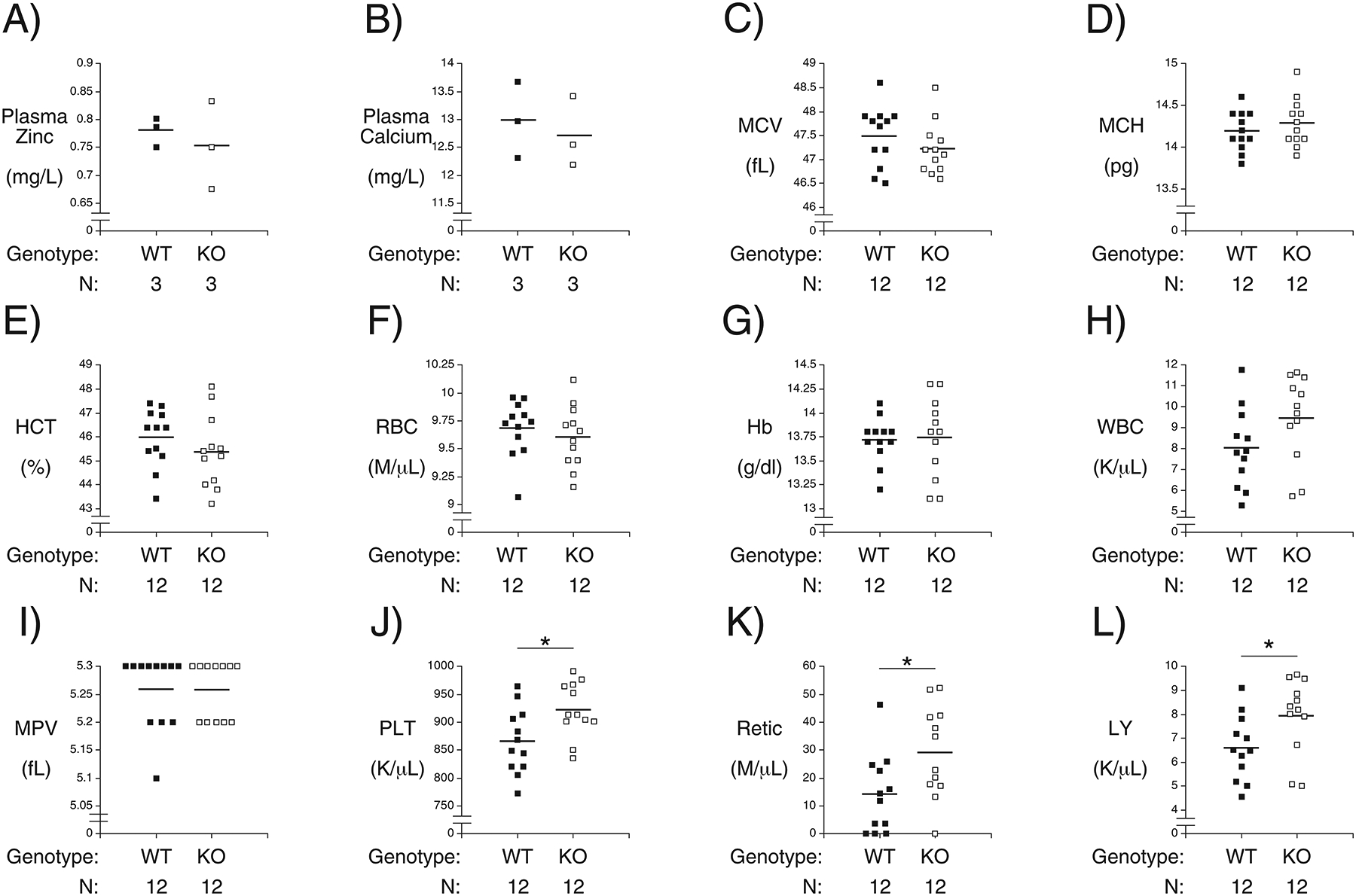
Blood from WT and Slc30a8 KO male mice was isolated from the retro-orbital plexus after a 5 hr fasted mice. The following parameters were then measured: Plasma zinc (Panel A); Plasma calcium (Panel B); mean corpuscular volume (MCV, Panel C); mean corpuscular hemoglobin (MCH, Panel D); hematocrit (HCT, Panel E); red blood cell count (RBC, Panel F); hemoglobin (Hb, Panel G); white blood cell count (WBC, Panel H); mean platelet volume (MPV, Panel I); platelet count (PLT, Panel J); reticulocyte count (Retic, Panel K) and lymphocytes count (LY, Panel L). Results show the mean data ± S.E.M. with the genotype and number of animals indicated. *p < 0.05 versus WT. WT=wild type; KO=knockout.
Slc30a8 Deletion but not Haploinsufficiency Affects Glucose Tolerance in 4 Week Old Chow Fed Mice
Another potential caveat with the use of ZnT8 inhibitors is that acute ZnT8 inhibition may have negative consequences in contrast to the effects of life-long SLC30A8 haploinsufficiency which may be associated with compensatory adaptations during development. We previously observed an impairment in glucose tolerance in 4 week old chow fed male C57BL/6J Slc30a8 KO mice (Pound et al. 2012). Similarly Nicolson et al. (Nicolson et al. 2009) and Wijesekara et al. (Wijesekara, et al. 2010) reported an impairment in glucose tolerance in 6 week old male global and beta-cell specific Slc30a8 KO mice, respectively. We therefore next examined whether Slc30a8 haploinsufficiency affects glucose metabolism at this age. Figure 4A shows that glucose tolerance was normal in male Slc30a8 heterozygous mice but an impairment in glucose tolerance was again observed in Slc30a8 KO mice. This impairment was not associated with any change in the body weight of 4 week old female (Fig. 4B) or male (Fig. 4C) Slc30a8 KO mice. These data indicate that homozygous but not heterozygous deletion of Slc30a8 affects glucose tolerance selectively in young (Fig. 4A) but not adult (Fig. 1A) chow fed mice.
Figure 4. Analysis of Body Weight, Glucose Tolerance, Blood Glucose, Insulin and C Peptide Plus Insulin Clearance in 4 Week Old C57BL/6J Wild Type, Slc30a8 Heterozygous and KO Mice in vivo.
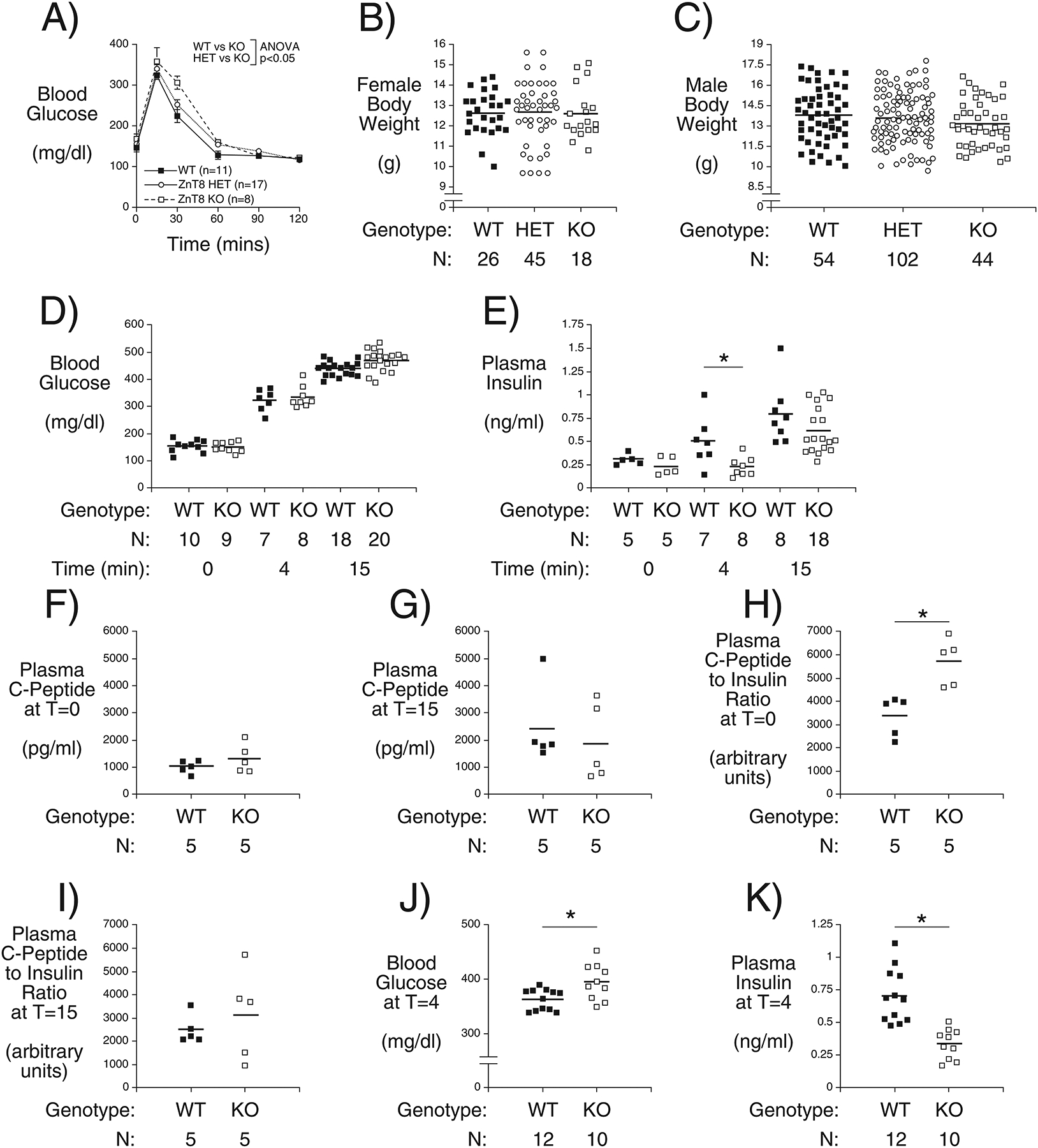
IPGTTs (Panel A) were performed on 6 hr fasted conscious 4 week old male mice. Results show the glucose concentrations in tail blood over time. Glucose tolerance is impaired in Slc30a8 KO mice at 4 weeks of age: p < 0.05, two-way ANOVA. Body weight was measured in non-fasted 4 week old female (Panel B) and male (Panel C) mice. Panels D-I show the blood glucose (Panel D), plasma insulin (Panels E), plasma C-peptide (Panels F and G) concentrations plus C-peptide/insulin ratio (Panels H and I) from IPGTTs performed on 6 hr fasted conscious 4 week old male mice with blood isolated from the retro-orbital plexus at t=0, 4 and 15 min after glucose injection as indicated. Due to the limited blood volume these experiments required using separate mice for the three time points and hence the data were analyzed using T tests. Panels J and K show the blood glucose and plasma insulin, respectively, from IPGTTs performed on 6 hr fasted conscious 10 week old male mice with blood isolated from the retro-orbital plexus at t=4 min after glucose injection. Results show the mean data ± S.E.M. with the genotype and number of animals indicated. *,p < 0.05, versus WT. WT=wild type; HET=heterozygous; KO=knockout.
Additional in vivo parameters were examined to define the molecular mechanism behind this impairment in glucose tolerance in 4 week old chow fed Slc30a8 KO mice. FBG did not differ between WT and KO mice (Fig. 4D). An increase in blood glucose from basal was observed at 4 min (Fig. 4D; p < 0.0001) and 15 min (Fig. 4D; p < 0.0001) following IP glucose injection for both WT and KO mice, but there were no differences in blood glucose between WT and KO mice at these time points. FPI also did not differ between WT and KO mice (Fig. 4E). Statistically significant increases in plasma insulin were not observed 4 min following IP glucose injection in either WT or KO mice (Fig. 4E) but a significant increase was observed after 15 min (Fig. 4E; p < 0.008 for WT, p < 0.003 for KO). While plasma insulin levels did not differ between WT and KO mice at the 15 min time point (Fig. 4E) there was a clear decrease in KO relative to WT mice at the 4 min time point (Fig. 4E). This suggests that deletion of Slc30a8 either slows the time course of insulin secretion in 4 week old mice or that the loss of beta cell zinc leads to a transient increase in hepatic insulin clearance and, hence, a delay in the rise of circulating insulin levels before liver insulin receptors become saturated (Tamaki, et al. 2013). Either mechanism could potentially explain the impaired glucose tolerance at the 30 min time point (Fig. 4A). Interestingly, Wijesekara et al. (Wijesekara et al. 2010), using in vitro perifusion assays, observed a delay in the time course of GSIS in islets isolated from beta cell-specific Slc30a8 KO mice that was associated with impaired glucose tolerance at 6 weeks of age. These authors observed little increase in in vivo insulin secretion during an oral glucose tolerance test so a difference in the timing of insulin secretion would not have been readily apparent (Wijesekara et al. 2010).
Plasma C-peptide levels were no different between fasting WT and Slc30a8 KO male mice (Fig. 4F) or 15 min after IP glucose injection (Fig. 4G), though glucose injection did elevate C-peptide levels (p < 0.0003 for WT, p < 0.0001 KO). However, an apparent difference in insulin clearance was observed under fasting conditions (Fig. 4H) but not 15 min following IP glucose injection (Fig. 4I), though it is important to note that insulin clearance was not directly measured such that other explanations for this observation, such as impaired insulin processing, are possible. Overall, these data suggest that Slc30a8 deletion clearly affects some aspects of glucose metabolism in 4 week old chow fed C57BL/6J mice.
Surprisingly, in adult 10 week old mice the increased blood glucose 4 min following IP glucose injection (Fig. 4J) was associated with lower plasma insulin levels in Slc30a8 KO relative to WT mice (Fig. 4K), just as observed in 4 week old mice (Fig. 4E). Since insulin levels do not differ between 10 week old WT and KO mice 15 min after glucose injection (Fig. 1D), this suggests that deletion of Slc30a8 either slows the time course of insulin secretion in 10 week old mice or that the loss of beta cell zinc leads to a transient increase in hepatic insulin clearance and, hence, a delay in the rise of circulating insulin levels before liver insulin receptors become saturated (Tamaki et al. 2013). In adult 10 week old mice we hypothesize that this delay in either the timing of insulin secretion or a transient increase in hepatic insulin clearance is now insufficient to result in impaired glucose tolerance 30 mins after glucose injection (Fig. 1A).
Slc30a8 Deletion Affects Pancreatic and Hepatic Gene Expression in 4 Week Old Chow Fed Mice
We have previously shown that deletion of Slc30a7 markedly reduces pancreatic insulin content and beta cell mass as well as hepatic Gys2 gene expression (Syring et al. 2016). We therefore considered the possibility that the effects of Slc30a8 deletion on the timing of insulin secretion or insulin clearance in young mice might be mediated, in part, through changes in islet and hepatic gene expression, respectively. To indirectly address the question of islet gene expression we focused on the expression of genes highly expressed in islets but not acinar cells such that their expression can be analyzed in whole pancreas RNA preparations, which then captures expression levels in the in vivo islet environment and avoids the confounding issue of gene expression changing during islet isolation. Data were normalized relative to Ppia (cyclophilin A) expression since this gene is expressed in islet, acinar and hepatic cells and is considered to be a non-hormonally regulated housekeeping gene. No decreases in pancreatic expression of Ins1 (Fig. 5B), Ins2 (Fig. 5C) or Slc2a2 (Fig. 5D), which encodes the GLUT2 glucose transporter, were observed in 4 week old Slc30a8 KO mice that might contribute to the impaired insulin secretion. In fact, Ins2 expression was slightly elevated (Fig. 5C), which might represent a compensatory response to offset the impairment in insulin secretion. However, expression of G6pc2, which encodes isoform 2 of the glucose-6-phosphatase catalytic subunit gene family (O’Brien 2013), was also elevated (Fig. 5E). Ins1 (Fig. 5G), Ins2 (Fig. 5H) and Slc2a2 (Fig. 5I) gene expression were also unaltered in 10 week old Slc30a8 KO mice and the changes in Ins2 (Fig. 5H) and G6pc2 (Fig. 5J) gene expression observed in young mice were now absent. This is consistent with the resolution of impaired glucose tolerance (Fig. 1A) and the absence of differences in gene expression comparing WT and Slc30a8 adult mouse islets using microarrays (data not shown). Resolution of gene expression changes with aging have been reported in other KO mouse models (Trefts, et al. 2019). While G6PC2 opposes the action of the glucose sensor glucokinase (O’Brien 2013), this increased expression is unlikely to contribute to the altered kinetics of insulin secretion or clearance in 4 week old Slc30a8 KO mice (Fig. 4E) since these altered kinetics still exist in 10 week old KO mice (Fig. 4K) whereas G6pc2 expression is unchanged relative to WT mice (Fig. 5J), though it will be important to examine G6PC2 protein expression to rule out this possibility.
Figure 5. Comparison of Pancreatic and Hepatic Gene Expression in 4 Week and 10 Week Old C57BL/6J Wild Type and Slc30a8 KO Male Mice.
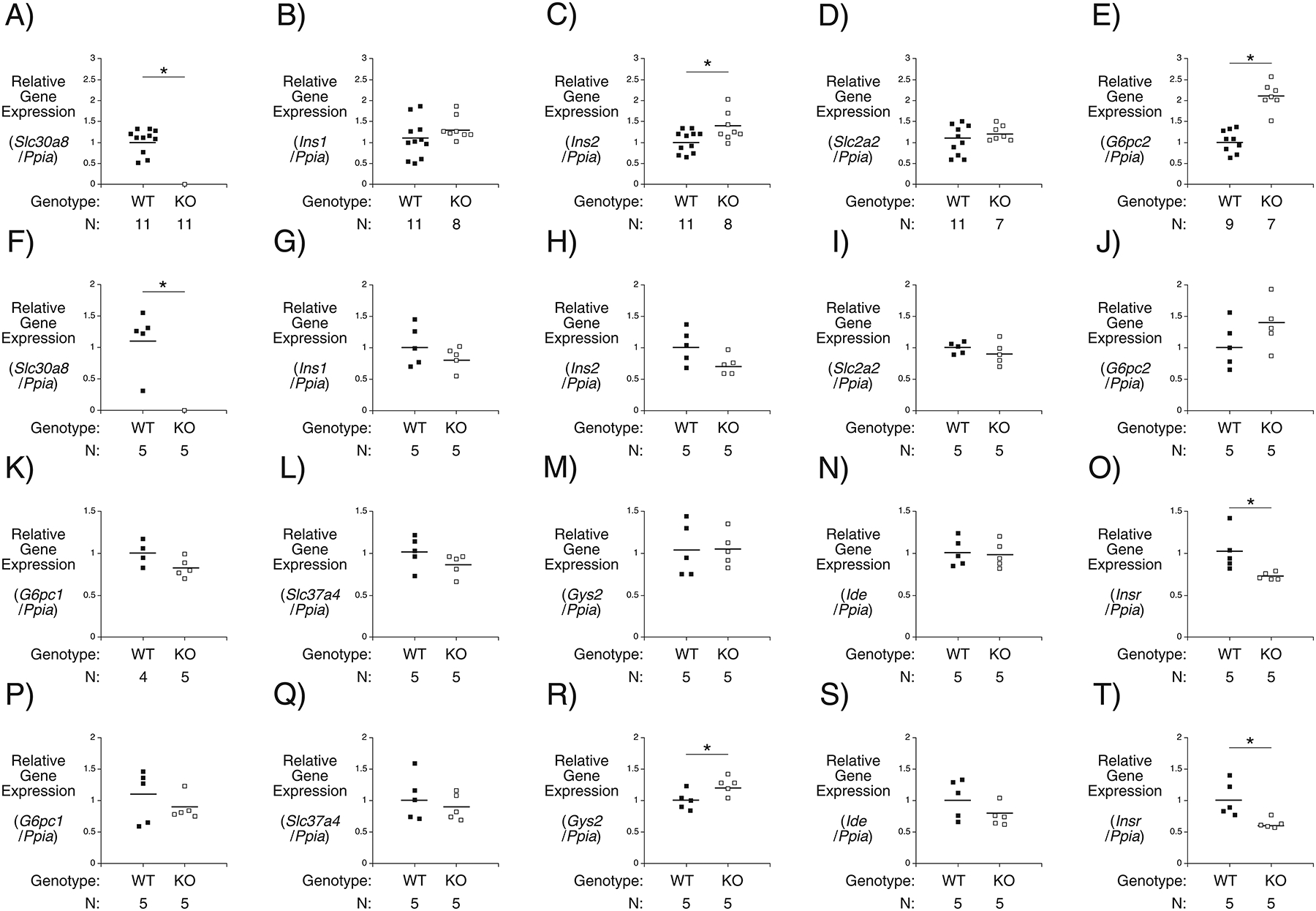
Comparison of pancreatic Slc30a8 (Panels A and F), Ins1 (Panels B and G), Ins2 (Panels C and H), Slc2a2 (Panels D and I), G6pc2 (Panels E and J) and hepatic G6pc1 (Panels K and P), Slc37a4 (Panels L and Q), Gys2 (Panels M and R), Ide (Panels N and S) and Insr (Panels O and T) gene expression in 6 hour fasted 4 week and 10 week old male mice, respectively. Gene expression was quantitated relative to Ppia (cyclophilin A) expression in the indicated tissue. Results show the mean data ± S.E.M. with the genotype and number of animals indicated. *, differences between WT and KO; p < 0.05, Student’s t-test.
Hepatic expression of G6pc1 (Fig. 5K), Slc37a4 (Fig. 5L), Gys2 (Fig. 5M), and Ide (Fig. 5N) were all unchanged in 4 week old Slc30a8 KO mice. These genes encode isoform 1 of the glucose-6-phosphatase catalytic subunit gene family, a glucose-6-phosphate transporter, glycogen synthase and the insulin degrading enzyme, respectively. At 10 weeks of age expression of G6pc1 (Fig. 5P), Slc37a4 (Fig. 5Q), and Ide (Fig. 5S) remained unchanged in Slc30a8 KO relative to WT mice though Gys2 expression was slightly elevated (Fig. 5R). Expression of the Insr gene, which encodes the insulin receptor, was decreased in both 4 week old (Fig. 5O) and 10 week old (Fig. 5T) Slc30a8 KO mice.
Zinc and Calcium Regulate G6pc2 Gene Expression
Even though loosely bound zinc is undetectable in Slc30a8 KO mouse islets (Davidson et al. 2014; Rutter and Chimienti 2015) islet zinc exists in metabolically labile compartments (Figlewicz, et al. 1984) so we hypothesized that changes in zinc distribution below detectable levels might explain the observed change in G6pc2 gene expression. To address the hypothesis that zinc might directly regulate G6pc2 gene expression we examined the effect of extracellular zinc on gene expression in the βTC-3 islet-derived cell line. Incubation in 250 μM zinc chloride induced expression of Fos (Fig. 6A) and Mt1a (Fig. 6B), which encode the c-fos transcription factor and metallothionein, respectively, and are known to be induced by zinc (Andrews, et al. 1990; Fanzo, et al. 2001). Zinc slightly induced expression of Ins2 (Fig. 6C), had little effect on Slc30a8 (Fig. 6D) but repressed expression of G6pc2 (Fig. 6E). The induction of G6pc2 in Slc30a8 KO mice (Fig. 5E) would be consistent with the fall in islet zinc content alleviating the repression of G6pc2 expression by zinc. We have previously shown that the proximal ~300 bp mouse G6pc2 promoter is sufficient to drive islet-enriched expression in transgenic mice (Frigeri, et al. 2004). However, this region mediated a minimal response to zinc chloride in fusion gene transient transfection assays (Fig. 6F). This suggests that zinc may regulate G6pc2 expression at a post-transcriptional level or through one or more of the transcriptional enhancers identified in the G6pc2 gene (Wang, et al. 2008). A fusion gene containing multimerized metal response elements (MREs) ligated to a minimal albumin promoter and the luciferase reporter gene was used as a positive control for these experiments. This fusion gene conferred a clear induction of luciferase expression by zinc (Fig. 6G), though this induction was not affected by overexpression of ZnT8, perhaps because endogenous ZnT8 is already present (Supplemental Fig. 3).
Figure 6. Comparison of Zinc- and Calcium-Regulated Endogenous and Fusion Gene Expression in the βTC-3 Cell Line.
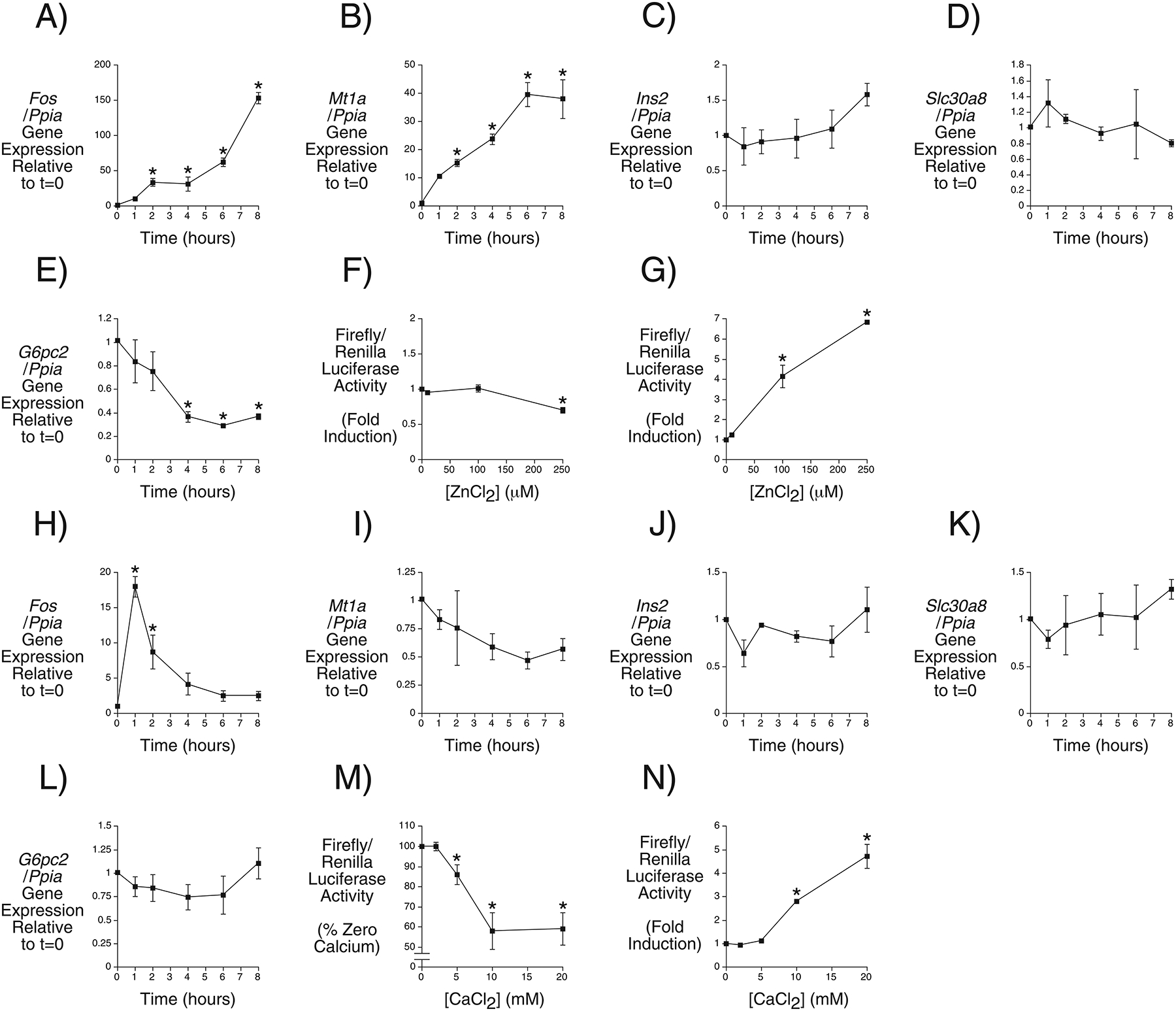
Regulation of endogenous Fos (Panels A and H), Mt1a (Panels B and I), Ins2 (Panels C and J), Slc30a8 (Panels D and K) and G6pc2 (Panels E and L) gene expression over time by 250 μM zinc chloride and 20 mM calcium chloride, respectively. Gene expression was quantitated relative to Ppia (cyclophilin A) expression. Results represent the mean data ± S.E.M. derived from 3–4 independent experiments. *, differences with t=0; p < 0.05, one-way ANOVA. Regulation of mouse G6pc2-luciferase (Panel F) and metal response element-albumin-luciferase (Panel G) fusion gene expression by the indicated concentration of zinc chloride. Regulation of mouse G6pc2-luciferase (Panel M) and cAMP response element-albumin-luciferase (Panel N) fusion gene expression by the indicated concentration of calcium chloride. Results represent the mean data ± S.E.M. derived from 3 independent experiments. *, differences with no zinc or calcium chloride; p < 0.05, one-way ANOVA.
Combined deletion of Slc30a7 and Slc30a8 abolishes GSIS at least in part through an effect on a step distal to calcium entry (Syring et al. 2016) so we also investigated whether changes in calcium distribution might potentially contribute to the observed change in G6pc2 gene expression in Slc30a8 KO mice (Fig. 5E), though it is important to note that calcium levels have not been measured in Slc30a8 KO islets, which would be necessary to support this possibility. Incubation in 20 mM calcium chloride transiently induced expression of Fos (Fig. 6H), consistent with published reports (Coulon, et al. 1999). Calcium had little effect on expression, Ins2 (Fig. 6J) or Slc30a8 (Fig. 6K) but trends towards repression of Mt1a (Fig. 6I) and G6pc2 (Fig. 6L) expression were apparent. The induction of G6pc2 in Slc30a8 KO mice (Fig. 5E) would be consistent with a loss of repression by calcium and the observation that overexpression of sorcin, which elevates cytoplasmic calcium in islets, represses G6PC2 promoter activity (Marmugi, et al. 2016). Indeed a fusion gene containing the proximal mouse G6pc2 promoter mediated an inhibitory effect of calcium chloride in fusion gene transient transfection assays (Fig. 6M) whereas a fusion gene containing multimerized cAMP response elements (CREs) ligated to a minimal albumin promoter and the luciferase reporter gene, used as a positive control, conferred a clear induction of luciferase expression (Fig. 6N). These effects were not affected by overexpression of ZnT8, again perhaps because endogenous ZnT8 is already present (Supplemental Fig. 3).
Discussion
We show here that Slc30a8 haploinsufficiency has no effect on glucose tolerance in either 4 week old (Fig. 4) or 10 week old (Fig. 1) chow fed mice but partially protects against DIO leading to an improvement in glucose tolerance (Fig. 2). Future studies will examine whether Slc30a8 haploinsufficiency protects against DIO through an effect on energy expenditure and/or food intake/storage and the site of ZnT8 action. We hypothesize that this protection against DIO is unlikely to be explained by a reduction of Slc30a8 expression in islets indirectly affecting peripheral metabolism because ZnT7 can compensate for the absence of ZnT8 in mouse islets and vice versa (Syring et al. 2016). While Slc30a8 is predominantly expressed in beta cells, it is found at low levels in multiple other cell types (Deniro and Al-Mohanna 2012; Giacconi et al. 2018; Liu et al. 2011; Murgia et al. 2009; Overbeck et al. 2008; Smidt et al. 2007; Zhang et al. 2018; Zhong et al. 2012). Our data imply that, while ZnT7 can compensate for the absence of ZnT8 in beta cells (Syring et al. 2016), such compensation does not exist, or at least is not fully effective, in relation to ZnT8 function in other tissues. Interestingly, while this compensation by ZnT7 maintains insulin secretion in static isolated islet assays in vitro (Syring et al. 2016), a defect in the timing of insulin secretion or clearance in vivo remains in adult Slc30a8 KO mice (Fig. 4K).
Our mouse studies show that Slc30a8 haploinsufficiency confers protection against DIO (Fig. 2). This potentially provides one explanation for how SLC30A8 haploinsufficiency protects against T2D in humans (Flannick et al. 2014; Flannick et al. 2019), given that obesity is a major risk factor for the development of T2D (Eckel, et al. 2011; Lu, et al. 2016). However, the key question is whether the two observations are related. In other words, does SLC30A8 haploinsufficiency confer protection against T2D in humans, in part, by conferring protection against DIO or is this protection specific to mice? Hopefully, given the increasing abundance of exome sequencing data, it will eventually be possible to identify and phenotype an adequate number of SLC30A8 haploinsufficient humans to determine whether reduced body weight might contribute to protection against T2D despite the challenges posed by the wide variation in body mass in humans and the genetic complexity of obesity (Loos and Janssens 2017). We have previously suggested an alternate possibility, namely that SLC30A8 haploinsufficiency protects against T2D by affecting cytoplasmic zinc levels in beta cells leading to a reduction in oxidative stress and hence lowering the risk for beta cell failure (Davidson et al. 2014). It has been shown that down-regulation of ZnT8 protects human insulinoma cells against inflammatory stress (Merriman and Fu 2019) and that both mice (Kleiner, et al. 2018) and humans (Dwivedi, et al. 2019) with an inactivating ZnT8 R138X mutation have increased insulin secretory capacity in vivo, although in mice the increase in insulin secretion was only apparent under hyperglycemic conditions. The mechanism behind the effect of the ZnT8 R138X mutant on insulin secretion is unclear since in both mouse (Kleiner et al. 2018) and human (Dwivedi et al. 2019) cells expression of this mutant protein was not readily detectable. Mice expressing the R138X mutant exhibit enhanced insulin secretion on a high fat diet but are not protected against DIO (Dwivedi et al. 2019), in contrast to our germline Slc30a8 heterozygous and KO mice (Fig. 2). This suggests that haploinsufficiency due to the presence of the truncated ZnT8 R138X variant may have different effects relative to haploinsufficiency that just results in a reduction in ZnT8 expression. This might also explain why expression of the ZnT8 R138X mutant promotes insulin secretion (Dwivedi et al. 2019; Kleiner et al. 2018) in contrast to Slc30a8 heterozygosity (Fig. 1). On the other hand, Dwivedi et al. (Dwivedi et al. 2019) showed that partial knockdown of ZnT8 in a human islet-derived cell line enhanced basal insulin secretion suggesting similar effects of the R138X variant and reduced ZnT8 expression. This observation supports an alternate hypothesis, namely the existence of a fundamental difference between the action of ZnT8 in humans and mice, though it will be important to repeat this acute knockdown experiment in mouse cells since germline Slc30a8 deletion is clearly associated with developmental compensation (Figs. 4–6). The observation that in the rat islet-derived INS1 cell line knockdown of ZnT8 actually has the opposite effect, impairing insulin secretion (Fu, et al. 2009; Petersen, et al. 2011), supports the presence of fundamental differences in the action of ZnT8 between species as does the surprising observation that in several species ZnT8 is not even required for insulin secretion (Bosma et al. 2019; Syring et al. 2018). In addition, SLC30A8 haploinsufficiency has no effect on insulin clearance in humans (Dwivedi et al. 2019) but we observed enhanced clearance in young (Fig. 4) though not adult mice (Fig. 1), in contrast to Tamaki et al. (Tamaki et al. 2013) who reported enhanced clearance in adult mice. Regardless of the mechanism, the observations of Flannick et al. (Flannick et al. 2014; Flannick et al. 2019) suggest that ZnT8 inhibitors should be considered as a therapy to prevent T2D.
Our data reveal two potential caveats in the use of ZnT8 inhibitors. First, we show that homozygous Slc30a8 deletion impairs glucose tolerance in 4 week old mice (Fig. 4). This impairment is associated with increased basal insulin clearance and delayed insulin secretion (Fig. 4) as well as altered Insr and G6pc2 expression (Fig. 5), the latter potentially secondary to altered islet zinc and/or calcium signaling (Fig. 6). Our data comparing 10 week old (Fig. 1) and 4 week old (Figs. 4–6) Slc30a8 KO mice suggest that some aspects of the early impairment in islet function are transient and that adaptation occurs during development in response to the complete absence of ZnT8 in mice that restores normal glucose tolerance. This suggests that full, acute ZnT8 inhibition may have negative consequences in contrast to the life-long effect of SLC30A8 haploinsufficiency which may be associated with compensatory adaptations during development. However, since heterozygous Slc30a8 deletion in 4 week old mice did not result in impaired glucose tolerance (Fig. 4) it suggests that partial, acute ZnT8 inhibition may be tolerated. Studies on mice expressing a floxed Slc30a8 allele and a knockin Ins1-CreER gene led to the same conclusion, namely that partial knockdown of Slc30a8 in adult animals does not negatively affect glucose tolerance (Supplemental Fig. 1). A second potential caveat in the use of ZnT8 inhibitors arose from our BioVU EHR analyses which showed that the SLC30A8 rs13266634 ‘T’ allele, which confers reduced ZnT8 transporter activity (Merriman et al. 2016), is associated with decreased risk for T2D and hereditary hemolytic anemias (Table 1) and reduced blood glucose but elevated MCV and MCH (Table 2). It will clearly be of interest to determine whether SLC30A8 haploinsufficient humans have altered risk for hereditary hemolytic anemias or altered MCV/MCH since the existence of negative effects of SLC30A8 haploinsufficiency might discourage the development of ZnT8 inhibitors for the prevention of T2D.
It is interesting to note that the VUMC-EHR analyses did not identify an association between the SLC30A8 rs13266634 ‘T’ allele and obesity. Furthermore, in contrast to the VUMC-EHR analyses, the Slc30a8 KO mouse studies showed no change in blood glucose (Fig. 1) and an elevation in platelets, reticulocytes and lymphocytes but unchanged MCV/MCH (Fig. 3). These observations may further support the existence of a fundamental difference between the action of ZnT8 in humans and mice. Alternatively, these discrepancies between mice and human data may arise due to quantitative differences associated with the complete loss of ZnT8 function in mice, due to Slc30a8 deletion, versus a partial reduction in ZnT8 activity in humans, comparing the SLC30A8 rs13266634 ‘T’ versus ‘C’ allele. Overall these EHR analyses, mouse DIO studies and mouse blood analyses suggest that ZnT8 has functional roles beyond beta cells.
With respect to the effect of Slc30a8 deletion on DIO, the observations reported here replicate our previous studies on a small cohort (n=5–6) of old (45–50 weeks) C57BL/6J mice where we noted that Slc30a8 deletion was protective against DIO (Pound et al. 2012). We did not appreciate the potential significance of these studies since they were published prior to the observations of Flannick et al. (Flannick et al. 2014). However, our results differ from previous studies by other groups. Lemaire et al. (Lemaire, et al. 2009) reported that Slc30a8 KO mice had impaired rather than improved glucose tolerance in response to DIO. They studied mice on a mixed genetic background, whereas the data shown in Figure 2 were derived from mice on the C57BL/6J genetic background. Another study by Hardy et al. (Hardy, et al. 2012) used mice only partially backcrossed onto the C57BL/6J genetic background. They reported that Slc30a8 KO mice were more, not less, susceptible to DIO. However, their WT C57BL/6J mice gained surprisingly little weight (Fig. 2A in Ref. (Hardy et al. 2012)) compared to our WT C57BL/6J mice (Fig. 2), even though our mice were not fed the high fat diet for as long. The fact that we used a speed congenic backcrossing strategy (Pound et al. 2012; Serreze, et al. 1996), rather than limited interbreeding with C57BL/6J mice (Hardy et al. 2012), may be significant. These differences between studies and the data shown in Figure 2 suggest that Slc30a8 haploinsufficiency protects against DIO, but only in the context of mice on a pure C57BL/6J genetic background.
Supplementary Material
Acknowledgements
We thank Susan Hajizadeh, Suzan Vaughan and Eric Allen for performing insulin, corticosterone, T3, T4 and C peptide assays. We also thank Miranda Wilkes for performing mouse blood analyses, Collen Morton for insights into the regulation of MCH/MCV and Jason Flannick for insights into the effects of SLC30A8 haploinsufficiency. This research was supported by the following grants: R.O’B., DK92589; O.P.M., DK043748 and DK078188; E.P.S 5 R01 AI101171; L.K.D., R56MH120736. The measurement of plasma hormones by the Vanderbilt Hormone Assay & Analytical Services Core was supported by NIH grants P60 DK20593 and U24-DK059637, to the Vanderbilt Diabetes Research Training Center and Mouse Metabolic Phenotyping Center, respectively. K.E.S., K.J.B. and S.B.G. were supported by the Vanderbilt Molecular Endocrinology Training Program grant 5T32 DK07563. C.A.L. was supported by the Burroughs Wellcome Fund Postdoctoral Enrichment Program and the Simons Foundation through the Jane Coffin Childs Memorial Fund for Medical Research.
R.O’B is the guarantor of this work, had full access to all the data, and takes full responsibility for the integrity of data and the accuracy of data analysis.
Abbreviations:
- T2D
type 2 diabetes
- KO
knockout
- DIO
diet-induced obesity
- MCH
mean corpuscular hemoglobin
- GWAS
genome-wide association studies
- SNP
single nucleotide polymorphism
- GSIS
glucose-stimulated insulin secretion
Footnotes
Declaration of interest
The authors have no financial interests that would result in a conflict of interest with respect to this work. D.R.P. is employed by Lexicon Pharmaceuticals and generated the germline Slc30a8 KO mice described in this study. There are no pending patents, products in development or marketed products associated with these mice or this study. This relationship between Lexicon and Vanderbilt does not alter the authors’ adherence to policies on sharing data and materials.
References
- Andrews GK, Kage K, Palmiter-Thomas P & Sarras MP Jr. 1990. Metal ions induce expression of metallothionein in pancreatic exocrine and endocrine cells. Pancreas 5 548–554. [DOI] [PubMed] [Google Scholar]
- Boesgaard TW, Zilinskaite J, Vanttinen M, Laakso M, Jansson PA, Hammarstedt A, Smith U, Stefan N, Fritsche A, Haring H, et al. 2008. The common SLC30A8 Arg325Trp variant is associated with reduced first-phase insulin release in 846 non-diabetic offspring of type 2 diabetes patients--the EUGENE2 study. Diabetologia 51 816–820. [DOI] [PubMed] [Google Scholar]
- Bosma KJ, Syring KE, Oeser JK, Lee JD, Benninger RKP, Pamenter ME & O’Brien RM 2019. Evidence that Evolution of the Diabetes Susceptibility Gene SLC30A8 that Encodes the Zinc Transporter ZnT8 Drives Variations in Pancreatic Islet Zinc Content in Multiple Species. J Mol Evol 87 147–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boustead JN, Martin CC, Oeser JK, Svitek CA, Hunter SI, Hutton JC & O’Brien RM 2004. Identification and characterization of a cDNA and the gene encoding the mouse ubiquitously expressed glucose-6-phosphatase catalytic subunit-related protein. J Mol Endocrinol 32 33–53. [DOI] [PubMed] [Google Scholar]
- Chausmer AB 1998. Zinc, insulin and diabetes. J Am Coll Nutr 17 109–115. [DOI] [PubMed] [Google Scholar]
- Coulon V, Veyrune JL, Tourkine N, Vie A, Hipskind RA & Blanchard JM 1999. A novel calcium signaling pathway targets the c-fos intragenic transcriptional pausing site. J Biol Chem 274 30439–30446. [DOI] [PubMed] [Google Scholar]
- Davidson HW, Wenzlau JM & O’Brien RM 2014. Zinc transporter 8 (ZnT8) and beta cell function. Trends Endocrinol Metab 25 415–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deniro M & Al-Mohanna FA 2012. Zinc transporter 8 (ZnT8) expression is reduced by ischemic insults: a potential therapeutic target to prevent ischemic retinopathy. PLoS One 7 e50360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denny JC, Ritchie MD, Basford MA, Pulley JM, Bastarache L, Brown-Gentry K, Wang D, Masys DR, Roden DM & Crawford DC 2010. PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinformatics 26 1205–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denny JC, Crawford DC, Ritchie MD, Bielinski SJ, Basford MA, Bradford Y, Chai HS, Bastarache L, Zuvich R, Peissig P, et al. 2011. Variants near FOXE1 are associated with hypothyroidism and other thyroid conditions: using electronic medical records for genome- and phenome-wide studies. Am J Hum Genet 89 529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denny JC, Bastarache L, Ritchie MD, Carroll RJ, Zink R, Mosley JD, Field JR, Pulley JM, Ramirez AH, Bowton E, et al. 2013. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat Biotechnol 31 1102–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn MF 2005. Zinc-ligand interactions modulate assembly and stability of the insulin hexamer -- a review. Biometals 18 295–303. [DOI] [PubMed] [Google Scholar]
- Dupuis J, Langenberg C, Prokopenko I, Saxena R, Soranzo N, Jackson AU, Wheeler E, Glazer NL, Bouatia-Naji N, Gloyn AL, et al. 2010. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet 42 105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwivedi OP, Lehtovirta M, Hastoy B, Chandra V, Krentz NAJ, Kleiner S, Jain D, Richard AM, Abaitua F, Beer NL, et al. 2019. Loss of ZnT8 function protects against diabetes by enhanced insulin secretion. Nat Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckel RH, Kahn SE, Ferrannini E, Goldfine AB, Nathan DM, Schwartz MW, Smith RJ & Smith SR 2011. Obesity and type 2 diabetes: what can be unified and what needs to be individualized? Diabetes Care 34 1424–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efrat S, Linde S, Kofod H, Spector D, Delannoy M, Grant S, Hanahan D & Baekkeskov S 1988. Beta-cell lines derived from transgenic mice expressing a hybrid insulin gene-oncogene. Proc Natl Acad Sci U S A 85 9037–9041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanzo JC, Reaves SK, Cui L, Zhu L, Wu JY, Wang YR & Lei KY 2001. Zinc status affects p53, gadd45, and c-fos expression and caspase-3 activity in human bronchial epithelial cells. Am J Physiol Cell Physiol 281 C751–757. [DOI] [PubMed] [Google Scholar]
- Figlewicz DP, Forhan SE, Hodgson AT & Grodsky GM 1984. 65Zinc and endogenous zinc content and distribution in islets in relationship to insulin content. Endocrinology 115 877–881. [DOI] [PubMed] [Google Scholar]
- Flannick J, Thorleifsson G, Beer NL, Jacobs SB, Grarup N, Burtt NP, Mahajan A, Fuchsberger C, Atzmon G, Benediktsson R, et al. 2014. Loss-of-function mutations in SLC30A8 protect against type 2 diabetes. Nat Genet 46 357–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannick J, Mercader JM, Fuchsberger C, Udler MS, Mahajan A, Wessel J, Teslovich TM, Caulkins L, Koesterer R, Barajas-Olmos F, et al. 2019. Exome sequencing of 20,791 cases of type 2 diabetes and 24,440 controls. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frigeri C, Martin CC, Svitek CA, Oeser JK, Hutton JC, Gannon M & O’Brien RM 2004. The Proximal Islet-Specific Glucose-6-Phosphatase Catalytic Subunit Related Protein (IGRP) Autoantigen Promoter is Sufficient to Initiate but not Maintain Transgene Expression in Mouse Islets In Vivo. Diabetes 53 1754–1764. [DOI] [PubMed] [Google Scholar]
- Fu Y, Tian W, Pratt EB, Dirling LB, Shyng SL, Meshul CK & Cohen DM 2009. Down-regulation of ZnT8 expression in INS-1 rat pancreatic beta cells reduces insulin content and glucose-inducible insulin secretion. PLoS One 4 e5679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacconi R, Malavolta M, Chiodi L, Boccoli G, Costarelli L, Bonfigli AR, Galeazzi R, Piacenza F, Basso A, Gasparini N, et al. 2018. ZnT8 Arg325Trp polymorphism influences zinc transporter expression and cytokine production in PBMCs from patients with diabetes. Diabetes Res Clin Pract 144 102–110. [DOI] [PubMed] [Google Scholar]
- Hardy AB, Serino AS, Wijesekara N, Chimienti F & Wheeler MB 2011. Regulation of glucagon secretion by zinc: lessons from the beta cell-specific Znt8 knockout mouse model. Diabetes Obes Metab 13 Suppl 1 112–117. [DOI] [PubMed] [Google Scholar]
- Hardy AB, Wijesekara N, Genkin I, Prentice KJ, Bhattacharjee A, Kong D, Chimienti F & Wheeler MB 2012. Effects of high-fat diet feeding on Znt8-null mice: differences between beta-cell and global knockout of Znt8. Am J Physiol Endocrinol Metab 302 E1084–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesse LE, Lonergan ZR, Beavers WN & Skaar EP 2019. The Acinetobacter baumannii Znu System Overcomes Host-Imposed Nutrient Zinc Limitation. Infect Immun 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhoff K, Machicao F, Haupt A, Schafer SA, Tschritter O, Staiger H, Stefan N, Haring HU & Fritsche A 2008. Polymorphisms in the TCF7L2, CDKAL1 and SLC30A8 genes are associated with impaired proinsulin conversion. Diabetologia 51 597–601. [DOI] [PubMed] [Google Scholar]
- Kleiner S, Gomez D, Megra B, Na E, Bhavsar R, Cavino K, Xin Y, Rojas J, Dominguez-Gutierrez G, Zambrowicz B, et al. 2018. Mice harboring the human SLC30A8 R138X loss-of-function mutation have increased insulin secretory capacity. Proc Natl Acad Sci U S A 115 E7642–E7649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaire K, Ravier MA, Schraenen A, Creemers JW, Van de Plas R, Granvik M, Van Lommel L, Waelkens E, Chimienti F, Rutter GA, et al. 2009. Insulin crystallization depends on zinc transporter ZnT8 expression, but is not required for normal glucose homeostasis in mice. Proc Natl Acad Sci U S A 106 14872–14877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Bai S & Sheline CT 2016. hZnT8 (slc30a8) Transgenic Mice Which Overexpress the R325W Polymorph Have Reduced Islet Zn2+ and Proinsulin Levels, Increased Glucose Tolerance After a High-Fat Diet, and Altered Levels of Pancreatic Zinc Binding Proteins. Diabetes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu BY, Jiang Y, Lu Z, Li S, Lu D & Chen B 2011. Down-regulation of zinc transporter 8 in the pancreas of db/db mice is rescued by Exendin-4 administration. Mol Med Rep 4 47–52. [DOI] [PubMed] [Google Scholar]
- Livak KJ & Schmittgen TD 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25 402–408. [DOI] [PubMed] [Google Scholar]
- Loos RJF & Janssens A 2017. Predicting Polygenic Obesity Using Genetic Information. Cell Metab 25 535–543. [DOI] [PubMed] [Google Scholar]
- Lu Y, Day FR, Gustafsson S, Buchkovich ML, Na J, Bataille V, Cousminer DL, Dastani Z, Drong AW, Esko T, et al. 2016. New loci for body fat percentage reveal link between adiposity and cardiometabolic disease risk. Nat Commun 7 10495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmugi A, Parnis J, Chen X, Carmichael L, Hardy J, Mannan N, Marchetti P, Piemonti L, Bosco D, Johnson P, et al. 2016. Sorcin Links Pancreatic beta-Cell Lipotoxicity to ER Ca2+ Stores. Diabetes 65 1009–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin CC, Oeser JK, Svitek CA, Hunter SI, Hutton JC & O’Brien RM 2002. Identification and Characterization of a Human cDNA and Gene Encoding a Ubiquitously Expressed Glucose-6-Phosphatase Catalytic Subunit-Related Protein. J Mol Endocrinol 29 205–222. [DOI] [PubMed] [Google Scholar]
- Merriman C & Fu D 2019. Down-regulation of the islet-specific zinc transporter-8 (ZnT8) protects human insulinoma cells against inflammatory stress. J Biol Chem. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merriman C, Huang Q, Rutter GA & Fu D 2016. Lipid-tuned Zinc Transport Activity of Human ZnT8 Protein Correlates with Risk for Type-2 Diabetes. J Biol Chem 291 26950–26957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murgia C, Devirgiliis C, Mancini E, Donadel G, Zalewski P & Perozzi G 2009. Diabetes-linked zinc transporter ZnT8 is a homodimeric protein expressed by distinct rodent endocrine cell types in the pancreas and other glands. Nutr Metab Cardiovasc Dis 19 431–439. [DOI] [PubMed] [Google Scholar]
- Nicolson TJ, Bellomo EA, Wijesekara N, Loder MK, Baldwin JM, Gyulkhandanyan AV, Koshkin V, Tarasov AI, Carzaniga R, Kronenberger K, et al. 2009. Insulin storage and glucose homeostasis in mice null for the granule zinc transporter ZnT8 and studies of the type 2 diabetes-associated variants. Diabetes 58 2070–2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien RM 2013. Moving on from GWAS: functional studies on the G6PC2 gene implicated in the regulation of fasting blood glucose. Curr Diab Rep 13 768–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overbeck S, Uciechowski P, Ackland ML, Ford D & Rink L 2008. Intracellular zinc homeostasis in leukocyte subsets is regulated by different expression of zinc exporters ZnT-1 to ZnT-9. J Leukoc Biol 83 368–380. [DOI] [PubMed] [Google Scholar]
- Pare G, Chasman DI, Parker AN, Nathan DM, Miletich JP, Zee RY & Ridker PM 2008. Novel association of HK1 with glycated hemoglobin in a non-diabetic population: a genome-wide evaluation of 14,618 participants in the Women’s Genome Health Study. PLoS Genet 4 e1000312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen AB, Smidt K, Magnusson NE, Moore F, Egefjord L & Rungby J 2011. siRNA-mediated knock-down of ZnT3 and ZnT8 affects production and secretion of insulin and apoptosis in INS-1E cells. APMIS 119 93–102. [DOI] [PubMed] [Google Scholar]
- Pound LD, Oeser JK, O’Brien TP, Wang Y, Faulman CJ, Dadi PK, Jacobson DA, Hutton JC, McGuinness OP, Shiota M, et al. 2013. G6PC2: A Negative Regulator of Basal Glucose-Stimulated Insulin Secretion. Diabetes 62 1547–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pound LD, Sarkar S, Benninger RK, Wang Y, Suwanichkul A, Shadoan MK, Printz RL, Oeser JK, Lee CE, Piston DW, et al. 2009. Deletion of the mouse Slc30a8 gene encoding zinc transporter-8 results in impaired insulin secretion. Biochem J 421 371–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pound LD, Sarkar SA, Ustione A, Dadi PK, Shadoan MK, Lee CE, Walters JA, Shiota M, McGuinness OP, Jacobson DA, et al. 2012. The physiological effects of deleting the mouse slc30a8 gene encoding zinc transporter-8 are influenced by gender and genetic background. PLoS One 7 e40972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulley J, Clayton E, Bernard GR, Roden DM & Masys DR 2010. Principles of human subjects protections applied in an opt-out, de-identified biobank. Clin Transl Sci 3 42–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie MD, Denny JC, Zuvich RL, Crawford DC, Schildcrout JS, Bastarache L, Ramirez AH, Mosley JD, Pulley JM, Basford MA, et al. 2013. Genome- and phenome-wide analyses of cardiac conduction identifies markers of arrhythmia risk. Circulation 127 1377–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson RP, Zhou H & Slucca M 2011. A role for zinc in pancreatic islet beta-cell cross-talk with the alpha-cell during hypoglycaemia. Diabetes Obes Metab 13 Suppl 1 106–111. [DOI] [PubMed] [Google Scholar]
- Roden DM, Pulley JM, Basford MA, Bernard GR, Clayton EW, Balser JR & Masys DR 2008. Development of a large-scale de-identified DNA biobank to enable personalized medicine. Clin Pharmacol Ther 84 362–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutter GA & Chimienti F 2015. SLC30A8 mutations in type 2 diabetes. Diabetologia 58 31–36. [DOI] [PubMed] [Google Scholar]
- Serreze DV, Chapman HD, Varnum DS, Hanson MS, Reifsnyder PC, Richard SD, Fleming SA, Leiter EH & Shultz LD 1996. B lymphocytes are essential for the initiation of T cell-mediated autoimmune diabetes: analysis of a new “speed congenic” stock of NOD.Ig mu null mice. J Exp Med 184 2049–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shameer K, Denny JC, Ding K, Jouni H, Crosslin DR, de Andrade M, Chute CG, Peissig P, Pacheco JA, Li R, et al. 2014. A genome- and phenome-wide association study to identify genetic variants influencing platelet count and volume and their pleiotropic effects. Hum Genet 133 95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sladek R, Rocheleau G, Rung J, Dina C, Shen L, Serre D, Boutin P, Vincent D, Belisle A, Hadjadj S, et al. 2007. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 445 881–885. [DOI] [PubMed] [Google Scholar]
- Smidt K, Pedersen SB, Brock B, Schmitz O, Fisker S, Bendix J, Wogensen L & Rungby J 2007. Zinc-transporter genes in human visceral and subcutaneous adipocytes: lean versus obese. Mol Cell Endocrinol 264 68–73. [DOI] [PubMed] [Google Scholar]
- Staiger H, Machicao F, Stefan N, Tschritter O, Thamer C, Kantartzis K, Schafer SA, Kirchhoff K, Fritsche A & Haring HU 2007. Polymorphisms within novel risk loci for type 2 diabetes determine beta-cell function. PLoS One 2 e832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surwit RS, Kuhn CM, Cochrane C, McCubbin JA & Feinglos MN 1988. Diet-induced type II diabetes in C57BL/6J mice. Diabetes 37 1163–1167. [DOI] [PubMed] [Google Scholar]
- Syring KE, Bosma KJ, Oeser JK, Shiota M & O’Brien RM 2018. The Diabetes Susceptibility Gene SLC30A8 that Encodes the Zinc Transporter ZnT8 is a Pseudogene in Guinea Pigs Potentially Contributing to Low Guinea Pig Islet Zinc Content. J Mol Evol 86 613–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syring KE, Boortz KA, Oeser JK, Ustione A, Platt KA, Shadoan MK, McGuinness OP, Piston DW, Powell DR & O’Brien RM 2016. Combined Deletion of Slc30a7 and Slc30a8 Unmasks a Critical Role for ZnT8 in Glucose-Stimulated Insulin Secretion. Endocrinology 157 4534–4541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamaki M, Fujitani Y, Hara A, Uchida T, Tamura Y, Takeno K, Kawaguchi M, Watanabe T, Ogihara T, Fukunaka A, et al. 2013. The diabetes-susceptible gene SLC30A8/ZnT8 regulates hepatic insulin clearance. J Clin Invest 123 4513–4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trefts E, Hughey CC, Lantier L, Lark DS, Boyd KL, Pozzi A, Zent R & Wasserman DH 2019. Energy metabolism couples hepatocyte integrin-linked kinase to liver glucoregulation and postabsorptive responses of mice in an age-dependent manner. Am J Physiol Endocrinol Metab 316 E1118–E1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Flemming BP, Martin CC, Allen SR, Walters J, Oeser JK, Hutton JC & O’Brien RM 2008. Long-range enhancers are required to maintain expression of the autoantigen islet-specific glucose-6-phosphatase catalytic subunit-related protein in adult mouse islets in vivo. Diabetes 57 133–141. [DOI] [PubMed] [Google Scholar]
- Wang Y, Oeser JK, Yang C, Sarkar S, Hackl SI, Hasty AH, McGuinness OP, Paradee W, Hutton JC, Powell DR, et al. 2006. Deletion of the gene encoding the ubiquitously expressed glucose-6-phosphatase catalytic subunit-related protein (UGRP)/glucose-6-phosphatase catalytic subunit-beta results in lowered plasma cholesterol and elevated glucagon. J Biol Chem 281 39982–39989. [DOI] [PubMed] [Google Scholar]
- Wijesekara N, Dai FF, Hardy AB, Giglou PR, Bhattacharjee A, Koshkin V, Chimienti F, Gaisano HY, Rutter GA & Wheeler MB 2010. Beta cell-specific Znt8 deletion in mice causes marked defects in insulin processing, crystallisation and secretion. Diabetologia 53 1656–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong WP, Allen NB, Meyers MS, Link EO, Zhang X, MacRenaris KW & El Muayed M 2017. Exploring the Association Between Demographics, SLC30A8 Genotype, and Human Islet Content of Zinc, Cadmium, Copper, Iron, Manganese and Nickel. Sci Rep 7 473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Kang D, Zhang C, Lou H, Sun C, Yang Q, Lu L, Xu GT, Zhang J & Wang F 2015. Erythropoietin Protects Retinal Cells in Diabetic Rats Through Upregulating ZnT8 via Activating ERK Pathway and Inhibiting HIF-1alpha Expression. Invest Ophthalmol Vis Sci 56 8166–8178. [DOI] [PubMed] [Google Scholar]
- Xu K, Zha M, Wu X, Yu Z, Yu R, Xu X, Chen H & Yang T 2011. Association between rs13266634 C/T polymorphisms of solute carrier family 30 member 8 (SLC30A8) and type 2 diabetes, impaired glucose tolerance, type 1 diabetes--a meta-analysis. Diabetes Res Clin Pract 91 195–202. [DOI] [PubMed] [Google Scholar]
- Zhang X, Guan T, Yang B, Chi Z, Wang ZY & Gu HF 2018. A novel role for zinc transporter 8 in the facilitation of zinc accumulation and regulation of testosterone synthesis in Leydig cells of human and mouse testicles. Metabolism 88 40–50. [DOI] [PubMed] [Google Scholar]
- Zhong ML, Chi ZH, Shan ZY, Teng WP & Wang ZY 2012. Widespread expression of zinc transporter ZnT (SLC30) family members in mouse endocrine cells. Histochem Cell Biol 138 605–616. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.