

SUMMARY
Bacteriophages must rapidly deploy anti-CRISPR proteins (Acrs) to inactivate the RNA-guided nucleases that enforce CRISPR-Cas adaptive immunity in their bacterial hosts. Listeria monocytogenes temperate phages encode up to three anti-Cas9 proteins, with acrIIA1 always present. AcrIIA1 inhibits Cas9 with its C-terminal domain; however, the function of its highly conserved N-terminal domain (NTD) is unknown. Here, we report that the AcrIIA1NTD is a critical transcriptional repressor of the anti-CRISPR promoter. The strong anti-CRISPR promoter generates a rapid burst of transcription during phage infection and the subsequent negative feedback from AcrIIA1NTD is required for optimal phage replication, even in the absence of CRISPR-Cas immunity. In the presence of CRISPR-Cas immunity, the AcrIIA1 two-domain fusion acts as a “Cas9 sensor,” tuning acr expression according to Cas9 levels. Finally, we identify AcrIIA1NTD homologues in other Firmicutes, and demonstrate that they have been co-opted by hosts as “anti-anti-CRISPRs,” repressing phage anti-CRISPR deployment.
eTOC
Bacterial viruses (phages) infecting Listeria encode a suite of “anti-CRISPR” (Acr) proteins that inhibit Cas9 immunity. Osuna et al. show that AcrIIA1 is both an autorepressor, silencing the strong acr promoter is key for phage fitness, and binds to Cas9, allowing phages to tune Acr expression to match Cas9 levels.
Graphical Abstract
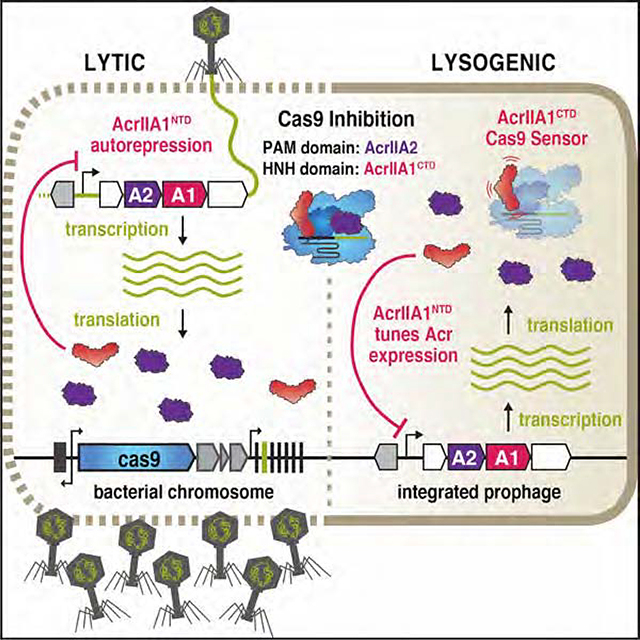
INTRODUCTION
The constant battle for survival between bacterial predators (phages) and their hosts has led to the evolution of numerous defensive and offensive strategies in both phages and bacteria (Stern and Sorek, 2011). Bacteria employ various mechanisms to combat phages, including CRISPR-Cas adaptive immune systems that keep a record of past viral infections in a CRISPR array with phage DNA fragments (spacers) stored between repetitive DNA sequences (Mojica et al., 2005). These spacers are transcribed into CRISPR RNAs (crRNAs), which bind CRISPR-associated (Cas) proteins to guide the sequence-specific detection and nucleolytic destruction of infecting phage genomes (Brouns et al., 2008; Garneau et al., 2010).
To evade this bacterial immunity, phages have evolved many tactics, including anti-CRISPR (Acr) proteins (Borges et al., 2017). Anti-CRISPRs are highly diverse and share no protein characteristics in common; they contain distinct amino acid sequences structures (Hwang and Maxwell, 2019; Trasanidou et al., 2019). However, the anti-CRISPR genomic locus displays some recurring features, containing up to three small anti-CRISPR genes and a signature anti-CRISPR-associated (aca) gene within a single operon (Borges et al., 2017). aca genes are almost invariably present in anti-CRISPR loci and they encode repressor proteins that contain a characteristic helix-turn-helix (HTH) DNA-binding motif (Birkholz et al., 2019; Stanley et al., 2019).
Listeria monocytogenes prophages contain a unique anti-CRISPR locus without an obvious standalone aca gene. These phages do, however, encode acrIIA1, a signature anti-CRISPR gene, which contains an HTH motif in its N-terminal domain (NTD) (Rauch et al., 2017). The AcrIIA1 HTH motif is highly conserved across orthologues, yet it is completely dispensable for anti-CRISPR activity, which resides in the C-terminal domain (CTD) (companion manuscript; Osuna et al., 2020a). Thus, the role and function of the AcrIIA1NTD remains unknown. Here, we show that AcrIIA1 is a bi-functional anti-CRISPR protein that performs a crucial regulatory role as an autorepressor of acr locus transcription that is required for optimal phage fitness. AcrIIA1NTD orthologues in phages and plasmids across the Firmicutes phylum also display autorepressor activity. We also show that the bacterial host can exploit the highly conserved anti-CRISPR locus repression mechanism, using the AcrIIA1NTD as an “anti-anti-CRISPR” to block phage anti-CRISPR expression during phage infection and lysogeny.
RESULTS
AcrIIA1NTD promotes general lytic growth and prophage induction
While interrogating anti-CRISPR phages in Listeria, we observed that two phage mutants displayed a lytic replication defect when their anti-CRISPR locus was deleted (ΦJ0161aΔacrIIA1–2 and ΦA006Δacr), even in a host lacking Cas9 (Figure 1A and 1B). The only gene that was removed from both phages was acrIIA1, suggesting that aside from acting as an anti-CRISPR, AcrIIA1 is also generally required for optimal phage replication. AcrIIA1 is a two-domain protein with a CTD that inhibits Cas9 (companion manuscript; Osuna et al., 2020a) and an NTD of uncharacterized function that contains a helix-turn-helix (HTH) motif similar to known transcriptional repressors (Ka et al., 2018). We hypothesized that the putative transcriptional repressor activity of AcrIIA1NTD is necessary for phage replication, even in the absence of CRISPR-Cas immunity. Indeed, complementation with acrIIA1NTD in trans rescued the lytic growth defects of both phages containing anti-CRISPR locus deletions (Figure 1A and 1B). Rare spontaneous mutants (~10−5 frequency) of the ΦJ0161aΔacrIIA1–2 phage that grew in the absence of acrIIA1NTD complementation were isolated, revealing that mutations in the −35 and −10 promoter elements suppressed the growth defect, as did a large deletion of the region, consistent with a vital cis-acting role for AcrIIA1 (Figure 1C).
Figure 1. Phages Require the AcrIIA1NTD (N-terminal Domain) for Optimal Replication.

(A-B) Left: Representative images of plaquing assays where Listeria phages were titrated in ten-fold serial dilutions (black spots) on lawns of Lmo10403s (gray background) lacking Cas9 (Δcas9) and encoding AcrIIA1NTD (Δcas9;IIA1NTD). Dashed lines indicate where intervening rows were removed for clarity. Right: Cas9-independent replication of isogenic ΦJ0161a or ΦA006 phages containing distinct anti-CRISPRs. Asterisk (*) indicates genes that contain the strong RBS associated with orfA in WT ΦA006, whereas unmarked genes contain their native RBS. Plaque forming units (PFUs) were quantified on Lmo10403s lacking cas9 (Δcas9, gray shaded bars) and expressing AcrIIA1NTD (Δcas9;IIA1NTD, black bars). Data are displayed as the mean PFU/mL of at least three biological replicates ± SD (error bars). See Figure S1A for phage titers of additional ΦA006 phages. (C) Top: Acr promoter mutations that suppress the ΦJ0161aΔIIA1–2 growth defect that manifests in the absence of AcrIIA1NTD. Bottom: Representative images of suppressor (Supp) phage plaquing assays conducted as in A-B. (D) Induction efficiency of ΦJ0161 prophages. Prophages were induced with mitomycin C from Lmo10403s::ΦJ0161 lysogens expressing cis-acrIIA1 from the prophage Acr locus (WT) or lacking acrIIA1 (ΔIIA1–2) and trans-acrIIA1 from the bacterial host genome (+) or not (−). Plaque forming units (PFUs) were quantified on Lmo10403s lacking cas9 and expressing AcrIIA1NTD (Δcas9;IIA1NTD). Data are displayed as the mean PFU/mL after prophage induction of four biological replicates ± SD (error bars).
A panel of ΦA006-derived phages engineered to study anti-CRISPR deployment during phage infection (see companion manuscript; Osuna et al., 2020a) was next examined in a host lacking Cas9. The lytic growth defect was again apparent in each phage that lacked AcrIIA1 or AcrIIA1NTD and providing acrIIA1NTD in trans or in cis (i.e. encoded in the phage acr locus) ameliorated this growth deficiency (Figure 1B and S1A). The phage engineered to express acrIIA1CTD alone (ΦA006-IIA1CTD), which is naturally always fused to acrIIA1NTD, displayed the strongest lytic defect amongst the ΦA006 phages and generated minuscule plaques (see spot titration, Figure 1B). The plaque size and phage titer deficiencies of ΦA006-IIA1CTD were fully restored with acrIIA1NTD supplemented in trans and most notably, when acrIIA1NTD was added to the phage genome as a separate gene (ΦA006-IIA1NTD+CTD, Figure 1B). Together, these data suggest that the HTH-containing AcrIIA1NTD enacts an activity that is a key determinant of phage fitness, irrespective of CRISPR-Cas immunity.
To test whether AcrIIA1NTD is also important during lysogeny, prophages were induced with mitomycin C treatment and the resulting phage titer was assessed. The ΦJ0161aΔacrIIA1–2 prophage displayed a strong induction deficiency, yielding 25-fold less phage, compared to the WT prophage or the acrIIA1-complemented mutant (Figure 1D). Attempts to efficiently induce ΦA006 prophages were unsuccessful, as previously observed (Loessner, 1991; Loessner et al., 1991). Therefore, AcrIIA1 is a bi-functional protein that not only acts as an anti-CRISPR, but also plays a critical role in the phage life cycle, promoting optimal lytic replication and lysogenic induction irrespective of CRISPR-Cas9.
AcrIIA1NTD is a repressor of the anti-CRISPR promoter and a Cas9 “sensor”
The AcrIIA1NTD domain bears close structural similarity to the phage 434 cI protein (Ka et al., 2018), an autorepressor that binds specific operator sequences in its own promoter (Johnson et al., 1981). Analysis of the anti-CRISPR promoters in ΦA006, ΦJ0161, and ΦA118 revealed a conserved palindromic operator sequence (Figures 2A and S2A), suggesting transcriptional control by a conserved regulator such as AcrIIA1. An RFP transcriptional reporter assay showed that full-length AcrIIA1 and AcrIIA1NTD, but not AcrIIA1CTD, repress the ΦA006 anti-CRISPR promoter (Figure 2B, left panel). In vitro MST binding assays also confirmed that AcrIIA1 (KD = 26 ± 10 nM) or AcrIIA1NTD (KD = 28 ± 3 nM), but not the AcrIIA1CTD, bind the anti-CRISPR promoter with high affinity (Figures 2C and S2B). Moreover, mutagenesis of the terminal nucleotides of the palindromic operator sequence prevented AcrIIA1-mediated repression of the ΦA006 anti-CRISPR promoter (Figure 2B, right panel) and abolished promoter binding in vitro (Figure 2C). Alanine scanning mutagenesis of conserved residues predicted to be important for DNA binding and dimerization (Ka et al., 2018) identified AcrIIA1NTD residues L10, T16, and R48 as critical for transcriptional repression, whereas AcrIIA1CTD mutations had little effect (Figure 2D). These data show that AcrIIA1NTD represses anti-CRISPR transcription by binding a highly conserved operator, and together with the suppressors isolated above, we conclude that this repression is important due to the need to silence a strong promoter (see Discussion).
Figure 2. AcrIIA1NTD autorepresses the anti-CRISPR locus promoter.

(A) Alignment of the phage anti-CRISPR promoter nucleotide sequences denoting the −35 and −10 elements (gray boxes) and conserved palindromic sequence (yellow boxes). See Figure S2A for a complete alignment of the promoters. (B) Expression of RFP transcriptional reporters containing the wild-type (left) or mutated (right) ΦA006-Acr.-promoter in the presence of AcrIIA1 (IIA1) or each domain (IIA1NTD or IIA1CTD). Representative images of three biological replicates are shown. (C) Quantification of the binding affinity (KD; boxed inset) of AcrIIA1 for the palindromic sequence within the acr promoter using microscale thermophoresis. ND indicates no binding detected. The nucleotide mutations (red letters) introduced into each promoter substrate are listed above the graph. Data shown are representative of three independent experiments. (D) Repression of the ΦA006Acr.–promoter RFP transcriptional reporter by AcrIIA1ΦA006 mutant proteins. Data are shown as the mean percentage RFP repression in the presence of the indicated AcrIIA1 variants relative to controls lacking AcrIIA1 of at least three biological replicates ± SD (error bars). (E) Nanoluciferase (NLuc) expression from the anti-CRISPR locus promoter in Listeria strains lysogenized with an ΦA006 reporter prophage (ΦA006acr::nluc) expressing AcrIIA1 (1) or AcrIIA1NTD (1N), in the presence of differing levels of Cas9: none (Δcas9), endogenous (PEND), overexpressed (PHYPER). Data are shown as the mean fold change in RLU (relative luminescence units) of three biological replicates, i.e., independent lysogens ± SEM (error bars). p-values: ***<0.001, ****<0.0001 (F) Immunoblots detecting FLAG-tagged LmoCas9 protein and a non-specific (ns) protein loading control in Lmo10403s::ΦJ0161a lysogens or non-lyosgenic strains containing plasmids expressing AcrIIA1 (IIA1) or AcrIIA1NTD (IIA1NTD). Dashed lines indicate where intervening lanes were removed for clarity. Representative blots of at least three biological replicates are shown.
We next hypothesized that the ability of AcrIIA1 to repress transcription with one domain and inactivate Cas9 with another would enable the tuning of acr transcripts to match the levels of Cas9 in the native host, L. monocytogenes. A reporter lysogen was engineered by inserting a nanoluciferase (nluc) gene in the acr locus. Low acr expression was seen in the absence of Cas9, or during low levels of Cas9 expression, however acr reporter levels increased by ~5-fold when Cas9 was overexpressed (Figure 2E, left). acr induction was not seen in the absence of AcrIIA1CTD (Figure 2E, right), the Cas9 binding-domain, supporting a model where Cas9 “sensing” de-represses the acr promoter. After confirming de-repression through an increase in Cas9 levels, we sought to confirm that AcrIIA1NTD is also capable of further repressing lysogenic anti-CRISPR expression. We therefore expressed the AcrIIA1NTD repressor in trans and assessed anti-CRISPR function. The Cas9 degradation normally induced by prophage-expressed AcrIIA1 activity (companion manuscript; Osuna et al., 2020a) was successfully prevented by AcrIIA1NTD (Figure 2F). These data collectively demonstrate that AcrIIA1 autoregulates acr transcript levels in L. monocytogenes and can increase acr expression in response to increased Cas9 expression.
Transcriptional autoregulation is a general feature of the AcrIIA1 superfamily
Recent studies have reported transcriptional autoregulation of anti-CRISPR loci by HTH-proteins in mobile genetic elements of Gram-negative Proteobacteria (Birkholz et al., 2019; Stanley et al., 2019). To determine whether anti-CRISPR locus regulation is similarly pervasive amongst mobile genetic elements in the Gram-positive Firmicutes phylum, we assessed AcrIIA1 homologs for transcriptional repression of their predicted cognate promoters and our model ΦA006 phage promoter. Homologs sharing 21% (i.e. Lmo orfD) to 72% amino acid sequence identity with AcrIIA1NTD were selected from mobile elements in Listeria, Enterococcus, Leuconostoc, and Lactobacillus (Figure 3A and S3A). All AcrIIA1 homologs repressed transcription of their cognate promoters by 42–99%, except AcrIIA1 from Lactobacillus parabuchneri, where promoter expression was undetectable (Figures 3A and S3B). Strong repression of the model ΦA006 promoter was only enacted by Listeria orthologues possessing ≥68% protein sequence identity (Figure 3A). Likewise, AcrIIA1ΦA006 only repressed the promoters associated with orthologues that repressed the ΦA006 promoter (Figure 3B). Interestingly, an AcrIIA1NTD palindromic binding site resides in the protein-coding sequence of the AcrIIA1LMO10 homolog, which displayed no anti-CRISPR activity despite possessing 85% AcrIIA1CTD sequence identity (Figures 3C and S3A). When this AcrIIA1NTD binding site was disrupted with silent mutations, AcrIIA1LMO10 anti-CRISPR function manifested (Figure 3C), confirming that intragenic anti-CRISPR repression can also occur. Altogether, these findings demonstrate that the anti-CRISPR promoter-AcrIIA1NTD repressor relationship is highly conserved and likely performs a vital repressive function in these diverse mobile genetic elements.
Figure 3. Autorepression is a General Feature of the AcrIIA1 Superfamily.

(A-B) Repression of RFP transcriptional reporters containing the ΦA006Acr.–promoter (gray bars) or cognate-AcrIIA1homolog.–promoters (black bars) by the indicated AcrIIA1Homolog proteins (A) or AcrIIA1ΦA006 protein (B). Data are shown as the mean percentage RFP repression in the presence of the indicated AcrIIA1 variants relative to controls lacking AcrIIA1 of at least three biological replicates ± SD (error bars). The percent protein sequence identities of each homolog to the ΦA006AcrIIA1NTD are listed in (A). (C) Top: Schematic of the wild-type (WT) and mutated AcrIIA1NTD binding site within the C-terminal protein coding sequence (CDS) of AcrIIA1LMO10. Bottom: Plaquing assays where the P. aeruginosa DMS3m-like phage JBD30 is titrated in ten-fold dilutions (black spots) on a lawn of P. aeruginosa (gray background) expressing the indicated anti-CRISPR proteins and Type II-A SpyCas9-sgRNA programmed to target phage DNA. Representative pictures of at least 3 biological replicates are shown.
Host-encoded AcrIIA1NTD blocks phage anti-CRISPR deployment
AcrIIA1NTD orthologues are encoded by many Firmicutes including Enterococcus, Bacillus, Clostridium, and Streptococcus (Rauch et al., 2017). In most cases, AcrIIA1NTD is fused to distinct AcrIIA1CTDs in mobile genetic elements, which are likely anti-CRISPRs that inhibit CRISPR-Cas systems in their respective hosts. Interestingly, there are instances where core bacterial genomes encode AcrIIA1NTD orthologues that are short ~70–80 amino acid proteins possessing only the HTH domain. One example is in Lactobacillus delbrueckii, where strains contain an AcrIIA1NTD homolog (35% identical, 62% similar to AcrIIA1ΦA006) with key residues conserved (e.g. L10 and T16). Given that AcrIIA1NTD represses anti-CRISPR transcription, we wondered whether bacteria could co-opt this regulator and exploit its activity in trans, preventing a phage from deploying its anti-CRISPR arsenal. Remarkably, we observed that the L. delbrueckii AcrIIA1NTD homolog is always a genomic neighbor of either the Type I-E, I-C, or II-A CRISPR-Cas systems in this species (Figure 4A), and these CRISPR-associated AcrIIA1NTD proteins are highly conserved (>95% sequence identity). This association is supportive of an “anti-anti-CRISPR” role that aids CRISPR-Cas function by repressing the deployment of phage inhibitors against each system. Although there are no specific anti-CRISPR proteins identified in Lactobacillus phages and prophages that express anti-CRISPRs, we reasoned that phages with their own acrIIA1 homolog might have acr loci that would be vulnerable to repression by the host protein. Fluorescent reporters were built, driven by seven different Lactobacillus phage or prophage promoters that possess an acrIIA1 homolog in their downstream operon (Figure S3C). This enabled the identification of one promoter, from phage Lrm1, that was robustly repressed by L. delbrueckii host AcrIIA1NTD. This confirms that a bona fide acr locus in a Lactobacillus phage can be repressed by a host version of a hijacked acr repressor (Figure 4B).
Figure 4. AcrIIA1NTD Encoded from a Bacterial Host Displays “anti-anti-CRISPR” Activity.

(A) Schematic of host-AcrIIA1NTD homologs encoded in core bacterial genomes next to Type II-A, I-C, and I-E CRISPR-Cas loci in Lactobacillus delbrueckii strains. (B) Seven promoters from the indicated phages and prophages were placed upstream of RFP, in the presence or absence of host-encoded AcrIIA1NTD, and fluorescence readout as in Figure 3. (C) Left panels: Plaquing assays where the indicated L. monocytogenes phages are titrated in ten-fold dilutions (black spots) on lawns of L. monocytogenes (gray background) expressing anti-CRISPRs from plasmids, LmoCas9 from a strong promoter (pHyper-cas9) or lacking Cas9 (Δcas), and the natural CRISPR array containing spacers with complete or partial matches to the DNA of each phage. (†) Denotes the absence of a spacer targeting the ΦJ0161a phage. Representative pictures of at least 3 biological replicates are shown. Right panel: Schematic of bacterial “anti-anti-CRISPR” activity where host-encoded AcrIIA1NTD (hA1NTD) blocks the expression of anti-CRISPRs from an infecting phage. (D) Nanoluciferase (NLuc) expression from the anti-CRISPR locus promoter or a (E) late viral promoter during lytic infection (Meile et al., 2020). L. monocytogenes 10403S strains expressing AcrIIA1 or AcrIIA1NTD from a plasmid were infected with reporter phages ΦA006acr::nluc or ΦA006 ΔLCR ply::nluc. Data are shown as the mean fold change in RLU (relative luminescence units) of three biological replicates ± SD (error bars).
To interrogate the anti-anti-CRISPR prediction in a native phage assay, we expressed AcrIIA1NTD from a plasmid (Figure 4B and S4B) or from an integrated single-copy acrIIA1NTD driven by its cognate phage promoter (Figure S4B) in L. monocytogenes. A panel of distinct anti-CRISPR-encoding phages became vulnerable to Cas9 targeting when AcrIIA1NTD was expressed by the host (Figures 4C and S4B), whereas expression of full-length AcrIIA1, AcrIIA1CTD, or AcrIIA4 had the expected anti-CRISPR phenotype (Figures 4C and S4A). Each of these phages possesses complete or partial spacer matches to the Lmo10403s CRISPR array. In contrast, replication of the non-targeted phages, ΦJ0161a (Figure 4C) and ΦP35 (Figure S4B), was unperturbed. Additionally, the acr::nluc reporter phage was used in a similar experiment, confirming that acr expression rapidly occurs during infection and can be silenced by expression of AcrIIA1 or AcrIIA1NTD (Figure 4D), while a model late promoter (ply::nluc) was not silenced (Figure 4E). These data demonstrate that hosts can use the anti-CRISPR repressor to block anti-CRISPR synthesis, rendering a phage unable to express its Acr proteins.
DISCUSSION
The Listeria phage anti-CRISPR AcrIIA1 was first described as a Cas9 inhibitor, and here we demonstrate that it is also a transcriptional autorepressor of the acr locus required for optimal lytic growth and prophage induction. Notably, this bi-functional regulatory anti-CRISPR has the ability to tune acr transcription in accordance with Cas9 levels.
Transcriptional autorepression is seemingly the predominant regulatory mechanism in bacteria and phages, as 40% of transcription factors in E. coli exert autogenous negative control (Thieffry et al., 1998). Due to their short response times, negative autoregulatory circuits are thought to be particularly advantageous in dynamic environments where rapid responses improve fitness. A strong promoter initially produces a rapid rise in transcript levels and after some time, repressor concentration reaches a threshold, shutting off its promoter to maintain steady-state protein levels (Madar et al., 2011; Rosenfeld et al., 2002). During infection, phages must rapidly produce anti-CRISPR proteins to neutralize the preexisting CRISPR-Cas complexes in their bacterial host. Consistent with the rapid response times exhibited by negatively autoregulated promoters, we observed a burst of anti-CRISPR locus expression within ten minutes post infection using a reporter phage (Figures 4C and S4C). During lysogeny, autorepression by AcrIIA1 presumably tempers anti-CRISPR locus expression, generating steady-state anti-CRISPR levels to maintain Cas9 inactivation.
Negative autoregulation maintains precise levels of the proteins encoded by the operon to prevent toxic effects caused by their overexpression (Thieffry et al., 1998), as classically observed with the λ phage genes cII and N (Shimatake and Rosenberg, 1981). In this study, the engineered ΦA006-IIA1CTD phage, which only contains the AcrIIA1CTD and lacks the AcrIIA1NTD autorepressor, displayed a pronounced lytic growth defect, even stronger than the defect of the ΦA006Δacr phage that completely lacks anti-CRISPRs (Figure 1B). This suggests that the AcrIIA1NTD autoregulatory domain is fused to AcrIIA1CTD in nature to limit the expression of an anti-CRISPR domain that can be toxic to the phage. Phages expressing only AcrIIA4 or AcrIIA12 were only mildly affected by the absence of AcrIIA1NTD (Figure 1B). However, other Listeria phage anti-CRISPRs (such as AcrIIA3) have been shown to exert toxic effects (Rauch et al., 2017), underscoring the need for an autoregulatory mechanism that tempers anti-CRISPR levels. The ΦJ0161a phage displays a remarkably strong growth defect when AcrIIA1 is absent (ΦJ0161aΔacrIIA1–2, Figure 1A), which is suppressed by promoter mutations or deletion of orfA (Figure 1C), suggesting that misregulation of a gene within the acr locus may be deleterious. Constitutively strong promoter activity may also have other deleterious effects. A recent study demonstrated that neighboring phage genes can be temporally misregulated in the absence of an anti-CRISPR locus autorepressor, Aca1 (Stanley et al., 2019).
Beyond cis regulatory auto-repression, prophages may also use AcrIIA1NTD to combat phage superinfection, benefitting both the prophage and host cell. The phage lambda cI protein, for example, represses prophage lytic genes and prevents superinfection by related phages during lysogeny (Johnson et al., 1981). Similarly, a lysogen could use AcrIIA1NTD to bolster the activity of a second CRISPR-Cas system in its host (such as the Type I-B system that is common in Listeria) by preventing incoming phages from expressing their Type I-B anti-CRISPRs. Host expressed AcrIIA1NTD does manifest as an anti-anti-CRISPR, blocking anti-CRISPR expression from infecting or integrated phages (Figures 4B and S4B). We also demonstrate that AcrIIA1NTD orthologues that reside in non-mobile regions of bacterial genomes can perform as a bona fide anti-CRISPR repressor. Thus, the importance of the conserved anti-CRISPR locus repression mechanism may represent a weakness in the phage, which can be exploited by the host through the co-opting of this anti-CRISPR regulator.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Please direct any requests for further information or reagents to the lead contact, Joseph Bondy-Denomy (joseph.bondy-denomy@ucsf.edu).
Materials Availability
Listeria strains, plasmids, and phages constructed and used in this study are disclosed in Table S2 (Excel spreadsheet).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Microbe Strains
Listeria monocytogenes strains (10403s) were cultured in brain-heart infusion (BHI) medium at 30°C. To ensure plasmid maintenance in Listeria strains, BHI was supplemented with tetracycline (2 μg/mL) for pPL2oexL integrated constructs or erythromycin (7.5 μg/mL) for pLEB579-derived constructs. Escherichia coli (DH5α, XL1Blue, NEB 10-beta, or NEB Turbo for plasmid maintenance and SM10 for conjugation into Listeria) and Pseudomonas aeruginosa (PAO1) were cultured in LB medium at 37°C. To maintain plasmids, LB was supplemented with chloramphenicol (25 μg/mL) for pPL2oexL in E. coli, erythromycin (250 μg/mL) for pLEB579 in E. coli, gentamicin (30 μg/mL) for pHERD30T in E. coli and P. aeruginosa, or carbenicillin (250 μg/mL for P. aeruginosa, 100 μg/mL for E. coli) for pMMB67HE. For maintaining pHERD30T and pMMB67HE in the same P. aeruginosa strain, media was supplemented with 30 μg/mL gentamicin and 100 μg/mL carbenicillin. The Listeria strains, plasmids, and phages constructed and used in this study are listed in Table S2.
Phages
Listeria phages A006, A118, A502, A620, J0161a, P35, and their derivatives were all propagated at 30°C on acrIIA1NTD-expressing L. monocytogenes 10403sɸcure (Δcas9, ΔtRNAArg::pPL2oexL-acrIIA1NTD) to allow optimal lytic growth of phages lacking their own acrIIA1NTD. The Pseudomonas DMS3m-like phage (JBD30) was propagated on PAO1 at 37°C. All phages were stored in SM buffer (100 mM NaCl, 8 mM MgSO4•7H2O, 50 mM Tris-HCl pH 7.5, 0.01% (w/v) gelatin), supplemented with 10 mM CaCl2 for Listeria phages, at 4°C.
METHOD DETAILS
Listeria and Pseudomonas strain construction
DNA fragments were PCR-amplified from genomic, plasmid, or synthesized DNA and cloned by Gibson Assembly into Listeria plasmids: episomal pLEB579 (Beasley et al., 2004) or the pPL2oexL single-copy integrating plasmid derived from pPL2 (Lauer et al., 2002) or P. aeruginosa plasmids: pMMB67HE or pHERD30T. To generate all Listeria monocytogenes strains, pPL2oexL plasmids were conjugated (Lauer et al., 2002; Simon et al., 1983) and pLEB579 plasmids were electroporated (Hupfeld et al., 2018; Park and Stewart, 1990) into Lmo10403s. For all Pseudomonas strains, plasmids were electroporated into PAO1 (Choi et al., 2006).
Isogenic ϕA006 anti-CRISPR phage engineering
Isogenic ϕA006 phages encoding distinct anti-CRISPRs from the native anti-CRISPR locus were engineered by in vitro-assemby of synthetic bacteriophage DNA as subsequent genome activation in L. monocytogenes L-form cells (EGDe strain variant Rev2) as previously described (Kilcher et al., 2018). Denoted acr genes (*) contain the strong ribosomal binding site (RBS) naturally associated with the first gene in the natural ϕA006 anti-CRISPR locus (orfA) whereas unmarked genes contain their native RBS. Note: the acrIIA1 RBS is weaker than the orfA RBS. The reporter phage ϕA006_acr::nluc was constructed by inserting a codon-optimized [optimized for L. monocytogenes using JCat (Grote et al., 2005)] nanoluciferase (nluc) gene sequence upstream of acrIIA1 using the endogenous acrIIA1 RBS (gene synthesis: ThermoFisher). DNA sequence of codon-optimized nanoluciferase (5’–3’):
ATGGTTTTCACTTTAGAAGATTTCGTTGGTGATTGGCGTCAAACTGCTGGTTACAACTTAGATCAAGTTTTAGAACAAGGTGGTGTTTCTTCTTTATTCCAAAACTTAGGTGTTTCTGTTACTCCAATCCAACGTATCGTTTTATCTGGTGAAAACGGTTTAAAAATCGATATCCATGTTATCATCCCATACGAAGGTTTATCTGGTGATCAAATGGGTCAAATCGAAAAAATCTTCAAAGTTGTTTACCCAGTTGATGATCATCATTTCAAAGTTATCTTACATTACGGTACTTTAGTTATCGATGGTGTTACTCCAAACATGATCGATTACTTCGGTCGTCCATACGAAGGTATCGCTGTTTTCGATGGTAAAAAAATCACTGTTACTGGTACTTTATGGAACGGTAACAAAATCATCGATGAACGTTTAATCAACCCAGATGGTTCTTTATTATTCCGTGTTACTATCAACGGTGTTACTGGTTGGCGTTTATGTGAACGTATCTTAGCTTAA
Listeria phage titering
A mixture of 150 μl stationary Listeria culture and 3 mL molten LC top agar (10 g/L tryptone, 5 g/L yeast extract, 10 g/L glucose, 7.5 g/L NaCl, 10 mM CaCl2, 10 mM MgSO4, 0.5% agar) was poured onto a BHI plate (1.5% agar) to generate a bacterial lawn, 3 μL of phage ten-fold serial dilutions were spotted on top, and after 24 hr incubation at 30°C, plate images were collected using the Gel Doc EZ Documentation system (BioRad) and Image Lab (BioRad) software.
Quantification of phage plaque forming units
Listeria phage infections were conducted using the soft agar overlay method: 10 μL phage dilution was mixed with 150 μL stationary Listeria culture in 3 mL molten LC top agar supplemented with 300 μg/mL Tetrazolium Violet (TCI Chemicals) to generate contrast for plaque visualization (Hurst et al., 1994) and poured onto a BHI-agar plate. After 24 hr incubation at 30°C, phage plaque-forming units (PFU) were quantified.
Isolation of J0161Δacr suppressor phages
A high titer lysate of the J0161ΔacrIIA1–2 was plated on Δcas9 strains that do not express acrIIA1. This caused a reduction in apparent titer by ~5 orders of magnitude but low frequency plaques were picked and propagated through three rounds of plaque purification. After plaque purification, the acr locus was PCR amplified from phage DNA and amplicons were Sanger sequenced to identify mutations.
Construction of Listeria lysogens
Lysogens were isolated from plaques that emerged after titering phages (ϕJ0161a, ϕA006, or their derivatives) on a lawn of Lmo10403sɸcureΔcas9 or LmoEGD-e (see “Listeria phage titering”). Lysogeny was confirmed by prophage induction with mitomycin C (0.5 μg/mL) treatment as previously described (Estela et al., 1992) and by PCR amplification and Sanger sequencing of the phage anti-CRISPR locus. All Lmo10403s strains containing prophages were lysogenized and verified prior to introducing additional constructs (integrated pPL2oexL or episomal pLEB579).
Listeria reporter phage assays
To quantify acr-locus expression during lytic infection, over-night cultures of the indicated host cells were diluted to an OD600=0.01 and infected with ϕA006 acr::nluc at an MOI=1. Time-course infection assays were performed at 30°C. At indicated time-points, 20 μL was removed from the infection, mixed with 20 μL Nano-GLO substrate, and bioluminescence quantified on a Glo-Max NAVIGATOR device (Promega, integration time = 5 s). Relative luminescence units (RLUs) were background corrected (luminescence of a phage-only control) and divided by values of a control infection with wild-type ϕA006. ϕA006 acr::nluc lysogens were produced as described in “construction of Listeria lysogens” and confirmed by PCR (Primer1: TAATTTGCTTAACTGATACC; Primer2: TGACTACTACGTATATTCG), by measuring bioluminescence, and by assessing homo-immunity. To quantify acr-locus expression from ϕA006 acr::nluc lysogens, log-phase cultures were diluted to an OD600=0.05 and bioluminescence quantified and divided by background values obtained from non-lysogenized parental strains.
Prophage induction efficiency quantification
Prophages were induced from Lmo10403s::ΦJ0161 lysogens expressing cis-acrIIA1 from the prophage Acr locus or trans-acrIIA1 from the bacterial host genome by treating with 0.5 μg/mL mitomycin C as previously described (Estela et al., 1992). After overnight incubation with continuous shaking at 30°C, cells were pelleted by centrifugation at 8000 g for 10 min and phage-containing supernatants were harvested. To quantify the amount of phage induced from each lysogen, phage-containing supernatants were used to infect Lmo10403sΦcure lacking cas9 and expressing AcrIIA1NTD (Δcas9;IIA1NTD, to bypass the lytic growth defect of ΦJ0161ΔacrIIA1–2) as described in “plaque forming unit (PFU) quantification of Listeria phages” and the resulting PFUs were quantified. Data are displayed as the mean PFU/mL after prophage induction of four biological replicates ± SD (error bars).
acr promoter transcriptional repression
To generate acr promoter transcriptional reporters, the nucleotide sequences (~100–350 base pairs) upstream of putative acr loci encoding acrIIA1 homologs were synthesized (Twist Bioscience) and cloned upstream of an mRFP gene into the pHERD30T vector. Promoter sequences are listed in Table S1. Transcriptional reporters were electroporated into P. aeruginosa PAO1 strains containing pMMB67HE-AcrIIA1-variants. Saturated overnight cultures of Pseudomonas were diluted 1:10 in LB supplemented with 30 μg/mL gentamicin, 100 μg/mL carbenicillin, and 1 mM IPTG to induce AcrIIA1 expression in a 96-well special optics microplate (Corning). Cells were incubated at 37°C with continuous double-orbital rotation for 24 hr in the Synergy H1 Hybrid Multi-Mode Reader (BioTeK) and measurements of OD600 and RFP (excitation 555 nm, emission 610 nm) relative fluorescence units (RFU) recorded every 5 min with the Gen5 (BioTek) software. Background fluorescence of growth media was subtracted and the resulting RFU values were normalized to OD600 (). Data are displayed as the mean normalized fluorescence of three biological replicates ± SD. Data are shown as the mean percentage RFP repression (RFU values at 960 min for AcrIIA1 mutants and 1170 min for homologs, normalized to OD600) in the presence of AcrIIA1 relative to controls lacking AcrIIA1 of at least three biological replicates ± SD (error bars).
Acr protein expression and purification
N-terminally 6xHis-tagged Acr proteins were expressed from the pET28 vector. Recombinant protein expression was induced with 0.25 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) at 18 °C overnight. Cells were harvested by centrifugation and lysed by sonication in buffer A (50 mM Tris-HCl pH 7.5, 500 mM NaCl, 0.5 mM DTT, 20 mM imidazole, 5% glycerol) supplemented with 1 mM PMSF and 0.25 mg/mL lysozyme (Sigma). Cell debris was removed by centrifugation at 20000 g for 40 min at 4 °C and the lysate incubated with Ni-NTA Agarose Beads (Qiagen). After washing, bound proteins were eluted with Buffer A containing 300 mM imidazole and dialyzed overnight into storage buffer (20 mM HEPES-NaOH pH 7.4, 150mM KCl, 10% glycerol, 2mM DTT).
in vitro AcrIIA1–anti-CRISPR promoter binding
The affinities of AcrIIA1 and individual domains for DNA were measured in triplicate using microscale thermophoresis (MST) on the Monolith NT.115 instrument (NanoTemper Technologies GmbH, Munich, Germany). Single-stranded complementary oligonucleotides were annealed to generate 40 bp acr promoter fragments harboring WT or mutated palindrome. The DNA substrate at 0.15 nM to 5 μM concentrations was incubated with 12.5 nM RED-tris-NTA-labeled AcrIIA1/domains at room temperature for 10 min in 1x buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 10 mM MgCl2, 0.05 % Tween-20). Samples were loaded into Monolith NT.115 Capillaries and measurements were performed at 25 °C using 40% LED power and medium microscale thermophoresis power. Data analyses were carried out using NanoTemper analysis software. DNA substrate sequences used are as follows:
5’-AACTATTGACTACTACGTATATTCGTAGTATAATGTGAAT-3’ (Wild-type)
5’-AACTATTGACAACTACGTATATTCGTAGTTTAATGTGAAT-3’ (Terminal Mutations)
5’-AACTATTGACAACAACCTATATTGGTTGTTTAATGTGAAT-3’ (Six Mutations)
Listeria protein samples for immunoblotting
Saturated overnight cultures of Lmo10403s strains overexpressing FLAG-tagged Cas9 (Δcas9, ΔtRNAArg::pPL2oexL-LmoCas9–6xHis-FLAG) were diluted 1:10 in BHI with appropriate antibiotic selection (see “microbes”), grown to log phase (OD600 0.2–0.6), 1.6 OD600 units of cells were harvested by centrifugation at 8000 g for 5 min at 4°C. Cells were lysed with lysozyme treatment: cell pellets were resuspended in 200 μL of TE buffer supplemented with 2.5 mg/mL lysozyme and 1x cOmplete mini EDTA-free protease inhibitor cocktail (Roche), samples were incubated at 37°C for 30 min, quenched with one-third volume of 4X Laemmli Sample Buffer (Bio-Rad), and boiled for 5 min at 95°C.
Immunoblotting
Protein samples were separated by SDS-PAGE using 4–20% Mini-PROTEAN TGX gels (BioRad) and transferred in 1X Tris/Glycine Buffer onto 0.22 micron PVDF membrane (Bio-Rad). Blots were probed with the following antibodies diluted 1:5000 in 1X TBS-T containing 5% nonfat dry milk: rabbit anti-FLAG (Sigma-Aldrich Cat# F7425, RRID:AB_439687), mouse anti-FLAG (Sigma-Aldrich Cat# F1804, RRID:AB_262044), HRP-conjugated goat anti-Rabbit IgG (Bio-Rad Cat# 170–6515, RRID:AB_11125142), and HRP-conjugated goat anti-mouse IgG (Santa Cruz Biotechnology Cat# sc-2005, RRID:AB_631736). Blots were developed using Clarity ECL Western Blotting Substrate (Bio-Rad) and chemiluminescence was detected on an Azure c600 Imager (Azure Biosystems).
QUANTIFICATION AND STATISTICAL ANALYSIS
All numerical data, with the exception of the microscale thermophoresis (MST) data, were analyzed and plotted using GraphPad Prism 6.0 software. The MST data were analyzed using the NanoTemper analysis software (NanoTemper Technologies GmbH) and plotted using GraphPad Prism 6.0 software. Statistical parameters are reported in the Figure Legends.
Data and Code Availability
The AcrIIA1 homolog protein accession numbers and associated promoter sequences are disclosed in Table S1.
Supplementary Material
Table S2. Listeria strains, plasmids, and phages constructed and used in this study, Related to STAR Methods
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rabbit anti-FLAG | Sigma-Aldrich | Cat# F7425; RRID: AB_439687 |
| mouse anti-FLAG | Sigma-Aldrich | Cat# F1804; RRID: AB_262044 |
| HRP-conjugated goat anti-Rabbit IgG | Bio-Rad | Cat# 170-6515; RRID: AB_11125142 |
| HRP-conjugated goat anti-mouse IgG | Santa Cruz Biotechnology | Cat# sc-2005; RRID: AB_631736 |
| Bacterial and Virus Strains | ||
| Listeria monocytogenes 10403s | Rauch et al., 2017 | RefSeq: NC_017544.1 |
| Listeria monocytogenes 10403s derivatives | This paper | See Table S2 |
| Pseudomonas aeruginosa strain PAO1 | Laboratory of Alan Davidson | RefSeq: NC_002516.2 |
| Pseudomonas aeruginosa strain PAO1 derivatives | This paper | N/A |
| Escherichia coli DH5α | New England Biolabs | Cat #C2982I |
| Escherichia coli SM10 | Laboratory of Daniel Portnoy | N/A |
| Listeria phage A006 | This paper | RefSeq: NC_009815.1 |
| Listeria phage A006 derivatives | This paper | See Table S2 |
| Listeria phage A118 | This paper | RefSeq: NC_003216.1 |
| Listeria phage A502 | This paper | RefSeq: MDRA00000000 |
| Listeria phage A620 | This paper | N/A |
| Listeria phage J0161a | Rauch et al., 2017 | RefSeq: NC_017545.1 |
| Listeria phage J0161a derivatives | This paper | N/A |
| Listeria phages P35 | This paper | RefSeq: NC_009814.1 |
| Pseudomonas phage JBD30 | Laboratory of Alan Davidson | RefSeq: NC_020198.1 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| AcrIIA1 protein homologs tested for promoter repression | This paper | See Table S1 |
| Purified protein: AcrIIA1 | This paper | N/A |
| Monolith His-Tag Labeling Kit RED-tris-NTA | Nanotemper Technologies | Cat #MO-L018 |
| Tetrazolium Violet | TCI Chemicals | Cat #T0174 |
| Critical Commercial Assays | ||
| Gibson Assembly Master Mix | New England Biolabs | Cat #E2611 L |
| Phusion Hot Start Flex DNA Polymerase | New England Biolabs | Cat #M0535S |
| Oligonucleotides | ||
| Listeria reporter phage lysogen confirmation Primer1: TAATTTGCTTAACTGATACC | This paper | N/A |
| Listeria reporter phage lysogen confirmation Primer2: TGACTACTACGTATATTCG | This paper | N/A |
| Wild-type Acr promoter for in vitro binding assay: AACTATTGACTACTACGTATATTCGTAGTATAATGTGAAT | This paper | N/A |
| Terminal Mutations Acr promoter for in vitro binding assay: AACTATTGACAACTACGTATATTCGTAGTTTAATGTGAAT | This paper | N/A |
| Six Mutations Acr promoter for in vitro binding assay: AACTATTGACAACAACCTATATTGGTTGTTTAATGTGAAT | This paper | N/A |
| Recombinant DNA | ||
| AcrIIA1-associated promoter sequences | Twist Bioscience | See Table S1 |
| pKSV7 | Rauch et al., 2017 | addgene.org/26686/ |
| pKSV7-derivative plasmids | This paper | See Table S2 |
| pPL2oexL | Rauch et al., 2017 | https://doi.org/10.1016/j.cell.2016.12.009 |
| pPL2oexL-derivative plasmids | This paper | See Table S2 |
| pLEB579 | Beasley et al., 2004 | https://doi.org/10.1093/ps/83.1.45 |
| pLEB579-derivative plasmids | This paper | See Table S2 |
| pHERD30T | Laboratory of Alan Davidson | GenBank: EU603326.1 |
| pHERD30T-derivative plasmids | This paper | N/A |
| pMMB67HE | ATCC | http://www.snapgene.com/resources/plasmid_files/basic_cloning_vectors/pMMB67HE/ |
| pMMB67HE-derivative plasmids | This paper | N/A |
| pET28 protein expression plasmid | Laboratory of David Morgan | N/A |
| pET28-6xHis-AcrIIA1 protein expression plasmid | This paper | N/A |
| Software and Algorithms | ||
| Prism 6.0 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Gen 5 | BioTek | https://www.biotek.com/products/software-robotics-software/gen5-microplate-reader-and-imager-software/ |
| Image Lab 5.2.1 | BioRad | http://bio-rad.com/en-cn/product/image-lab-software |
| NanoTemper Analysis Software | NanoTemper Technologies | https://nanotempertech.com/monolith/ |
| Other | ||
| Synergy H1 Microplate Reader | BioTek | https://www.biotek.com/products/detection-hybrid-technology-multi-mode-microplate-readers/synergy-h1-hybrid-multi-mode-reader/ |
| Azure c600 Imager | Azure Biosystems | https://www.azurebiosystems.com/imaging-systems/azure-600/ |
| Monolith NT.115 | NanoTemper Technologies | https://nanotempertech.com/monolith/ |
Highlights.
Listeria anti-CRISPR protein AcrIIA1 serves as an anti-CRISPR and a vital autorepressor
The rapid and strong acr promoter must be repressed for maximal phage fitness
AcrIIA1 allows prophages to tune Acr expression to Cas9 levels
AcrIIA1 homologs have been co-opted by the host as a “anti-anti-CRISPRs”
ACKNOWLEDGEMENTS
We would like to thank Daniel A. Portnoy (UC Berkeley) for providing the pLMB3C-pRhamnose plasmid, and Jonathan Asfaha (David Morgan Lab, UCSF) and Ujjwal Rathore (Alex Marson Lab, UCSF) for experimental advice and reagents. The J.B.-D lab was supported by the UCSF Program for Breakthrough Biomedical Research funded in part by the Sandler Foundation, the Searle Fellowship, the Vallee Foundation, the Innovative Genomics Institute, an NIH Director’s Early Independence Award DP5-OD021344, and NIH R01GM127489. The S.Ki. lab was supported by an Ambizione Fellowship (Swiss National Science Foundation, PZ00P3_174108).
Footnotes
DECLARATION OF INTERESTS
J.B.-D. is a scientific advisory board member of SNIPR Biome and Excision Biotherapeutics and a scientific advisory board member and co-founder of Acrigen Biosciences, and is an inventor on patents relating to anti-CRISPR proteins.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Beasley SS, Takala TM, Reunanen J, Apajalahti J, and Saris PEJ (2004). Characterization and Electrotransformation of Lactobacillus Crispatus Isolated from Chicken Crop and Intestine. Poult. Sci. 83, 45–48. [DOI] [PubMed] [Google Scholar]
- Birkholz N, Fagerlund RD, Smith LM, Jackson SA, and Fineran PC (2019). The autoregulator Aca2 mediates anti-CRISPR repression. Nucleic Acids Res. 47, 9658–9665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges AL, Davidson AR, and Bondy-Denomy J (2017). The Discovery, Mechanisms, and Evolutionary Impact of Anti-CRISPRs. Annu. Rev. Virol. 4, null. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouns SJJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJH, Snijders APL, Dickman MJ, Makarova KS, Koonin EV, and van der Oost J (2008). Small CRISPR RNAs Guide Antiviral Defense in Prokaryotes. Science 321, 960–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi K-H, Kumar A, and Schweizer HP (2006). A 10-min method for preparation of highly electrocompetent Pseudomonas aeruginosa cells: Application for DNA fragment transfer between chromosomes and plasmid transformation. J. Microbiol. Methods 64, 391–397. [DOI] [PubMed] [Google Scholar]
- Estela LA, Sofos JN, and Flores BB (1992). Bacteriophage Typing of Listeria monocytogenes Cultures Isolated From Seafoods. J. Food Prot. 55, 13–17. [DOI] [PubMed] [Google Scholar]
- Garneau JE, Dupuis M-È, Villion M, Romero DA, Barrangou R, Boyaval P, Fremaux C, Horvath P, Magadán AH, and Moineau S (2010). The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468, 67–71. [DOI] [PubMed] [Google Scholar]
- Grote A, Hiller K, Scheer M, Münch R, Nörtemann B, Hempel DC, and Jahn D (2005). JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 33, W526–W531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hupfeld M, Trasanidou D, Ramazzini L, Klumpp J, Loessner MJ, and Kilcher S (2018). A functional type II-A CRISPR–Cas system from Listeria enables efficient genome editing of large non-integrating bacteriophage. Nucleic Acids Res. 46, 6920–6933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst CJ, Blannon JC, Hardaway RL, and Jackson WC (1994). Differential Effect of Tetrazolium Dyes upon Bacteriophage Plaque Assay Titers. Appl Env. Microbiol 60, 3462–3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang S, and Maxwell KL (2019). Meet the Anti-CRISPRs: Widespread Protein Inhibitors of CRISPR-Cas Systems. CRISPR J. 2, 23–30. [DOI] [PubMed] [Google Scholar]
- Johnson AD, Poteete AR, Lauer G, Sauer RT, Ackers GK, and Ptashne M (1981). λ Repressor and cro—components of an efficient molecular switch. Nature 294, 217–223. [DOI] [PubMed] [Google Scholar]
- Ka D, An SY, Suh J-Y, and Bae E (2018). Crystal structure of an anti-CRISPR protein, AcrIIA1. Nucleic Acids Res. 46, 485–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauer P, Chow MYN, Loessner MJ, Portnoy DA, and Calendar R (2002). Construction, Characterization, and Use of Two Listeria monocytogenes Site-Specific Phage Integration Vectors. J BACTERIOL 184, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loessner MJ (1991). Improved procedure for bacteriophage typing of Listeria strains and evaluation of new phages. Appl. Environ. Microbiol. 57, 882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loessner MJ, Goeppl S, and Busse M (1991). Comparative inducibility of bacteriophage in naturally lysogenic and lysogenized strains of Listeria spp. by u.v. light and Mitomycin C. Lett. Appl. Microbiol. 12, 196–199. [Google Scholar]
- Madar D, Dekel E, Bren A, and Alon U (2011). Negative auto-regulation increases the input dynamic-range of the arabinose system of Escherichia coli. BMC Syst. Biol. 5, 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meile S, Sarbach A, Du J, Schuppler M, Saez C, Loessner MJ, Kilcher S (2020). Engineered reporter phages for rapid bioluminescence-based detection and differentiation of viable Listeria cells. Appl Environ Microbiol. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mojica FJM, Díez-Villaseñor C, García-Martínez J, and Soria E (2005). Intervening Sequences of Regularly Spaced Prokaryotic Repeats Derive from Foreign Genetic Elements. J. Mol. Evol. 60, 174–182. [DOI] [PubMed] [Google Scholar]
- Osuna BA, Karambelkar S, Mahendra C, Christie KA, Garcia B, Davidson AR, Kleinstiver BP, Kilcher S, and Bondy-Denomy J (2020a). Listeria phages induce Cas9 degradation to protect lysogenic genomes. co-submitted companion manuscript [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SF, and Stewart GSAB (1990). High-efficiency transformation of Listeria monocytogenes by electroporation of penicillin-treated cells. Gene 94, 129–132. [DOI] [PubMed] [Google Scholar]
- Rauch BJ, Silvis MR, Hultquist JF, Waters CS, McGregor MJ, Krogan NJ, and Bondy-Denomy J (2017). Inhibition of CRISPR-Cas9 with Bacteriophage Proteins. Cell 168, 150–158.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld N, Elowitz MB, and Alon U (2002). Negative Autoregulation Speeds the Response Times of Transcription Networks. J. Mol. Biol. 323, 785–793. [DOI] [PubMed] [Google Scholar]
- Shimatake H, and Rosenberg M (1981). Purified λ regulatory protein c II positively activates promoters for lysogenic development. Nature 292, 128–132. [DOI] [PubMed] [Google Scholar]
- Simon R, Priefer U, and Pühler A (1983). A Broad Host Range Mobilization System for In Vivo Genetic Engineering: Transposon Mutagenesis in Gram Negative Bacteria. Bio/Technology 1, 784–791. [Google Scholar]
- Stanley SY, Borges AL, Chen K-H, Swaney DL, Krogan NJ, Bondy-Denomy J, and Davidson AR (2019). Anti-CRISPR-Associated Proteins Are Crucial Repressors of AntiCRISPR Transcription. Cell 178, 1452–1464.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern A, and Sorek R (2011). The phage-host arms race: Shaping the evolution of microbes. BioEssays 33, 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thieffry D, Huerta AM, Pérez-Rueda E, and Collado-Vides J (1998). From specific gene regulation to genomic networks: a global analysis of transcriptional regulation in Escherichia coli. BioEssays 20, 433–440. [DOI] [PubMed] [Google Scholar]
- Trasanidou D, Gerós AS, Mohanraju P, Nieuwenweg AC, Nobrega FL, and Staals RHJ (2019). Keeping crispr in check: diverse mechanisms of phage-encoded anti-crisprs. FEMS Microbiol. Lett. 366, fnz098. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2. Listeria strains, plasmids, and phages constructed and used in this study, Related to STAR Methods
Data Availability Statement
The AcrIIA1 homolog protein accession numbers and associated promoter sequences are disclosed in Table S1.