

Abstract
To identify candidate key genes and miRNAs associated with esophageal squamous cell carcinoma (ESCC) development and prognosis, the gene expression profiles and miRNA microarray data including GSE20347, GSE38129, GSE23400, and GSE55856 were downloaded from the Gene Expression Omnibus (GEO) database. Clinical and survival data were retrieved from The Cancer Genome Atlas (TCGA). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of differentially expressed genes (DEGs) was analyzed via DAVID, while the DEG-associated protein-protein interaction network (PPI) was constructed using the STRING database. Additionally, the miRNA target gene regulatory network and miRNA coregulatory network were constructed, using the Cytoscape software. Survival analysis and prognostic model construction were performed via the survival (version 2.42-6) and rbsurv R packages, respectively. The results showed a total of 2575, 2111, and 1205 DEGs, and 226 differentially expressed miRNAs (DEMs) were identified. Pathway enrichment analyses revealed that DEGs were mainly enriched in 36 pathways, such as the proteasome, p53, and beta-alanine metabolism pathways. Furthermore, 448 nodes and 1144 interactions were identified in the PPI network, with MYC having the highest random walk score. In addition, 7 DEMs in the microarray data, including miR-196a, miR-21, miR-205, miR-194, miR-103, miR-223, and miR-375, were found in the regulatory network. Moreover, several reported disease-related miRNAs, including miR-198a, miR-103, miR-223, miR-21, miR-194, and miR-375, were found to have common target genes with other DEMs. Survival analysis revealed that 85 DEMs were related to prognosis, among which hsa-miR-1248, hsa-miR-1291, hsa-miR-421, and hsa-miR-7-5p were used for a prognostic survival model. Taken together, this study revealed the important roles of DEGs and DEMs in ESCC development, as well as DEMs in the prognosis of ESCC. This will provide potential therapeutic targets and prognostic predictors for ESCC.
1. Introduction
Esophageal carcinoma (EC) remains the sixth leading cause of cancer-associated mortality worldwide, with approximately 442,000 new cases and 440,000 mortalities globally in 2014 [1, 2]. As the predominant subtype of EC, esophageal squamous cell carcinoma (ESCC) is frequently diagnosed in Eastern Asian countries, including China, where it accounts for 95% of all EC cases [3, 4]. A series of risk factors, such as tobacco smoking and consumption of alcohol and salted vegetables, are reported to be associated with the high incidence of ESCC [5]. Currently, despite advances in diagnosis and treatment of ESCC, its prognosis remains poor, with a 5-year overall survival rate of less than 40% [6, 7]. Therefore, identification of the molecular mechanisms underlying the progression and prognosis of ESCC is of utmost importance.
As a gene detection technique, gene profiling or gene chips have been widely used during the last decade for the comprehensive screening of differentially expressed genes (DEGs) [8]. With the development and application of gene chips, more and more data have been generated and stored in public databases, which will provide valuable clues for new research. Currently, an increasing number of studies have reported the identification of DEGs in ESCC. For example, Yuan et al. [9] analyzed the gene expression profile in the GSE20347 dataset, identifying a total of 538 DEGs. Likewise, Xing and Liu [10] identified 1204 DEGs based on the GSE23400 dataset generated from ESCC and matched mucosa tissues. Furthermore, Hu et al. [11] focus on biallelic loss and its relation to mRNA expression based on the GSE38129 array data. Recently, differentially expressed miRNAs (DEMs) has been reported to be associated with differentiation, invasion, and metastasis of patients with ESCC [12]. Moreover, miRNA expression profiling analysis was also recently performed [13]. Jang et al. [14] identified prognostic markers for postoperative recurrence of ESCC by building an easy-to-use prognostic model with three small noncoding RNAs (sncRNAs) based on the GSE55856 dataset. However, the majority of these reports were based on a single dataset, which leads to the results being limited or inconsistent. Hence, the combination of bioinformatics methods and expression profiling techniques using different datasets may provide valuable information for the development of therapeutic strategies for patients with this disease.
In the present study, we obtained the original microarray data of the GSE20347, GSE38129, and GSE23400 datasets, as well as the miRNA microarray data of the GSE55856 dataset from the Gene Expression Omnibus (GEO). Clinical data and survival data were retrieved from The Cancer Genome Atlas (TCGA). Subsequently, the DEGs or DEMs were screened. Pathway enrichment analyses were performed, and protein-protein interaction (PPI) networks were created for the DEGs, in order to identify key genes and their biological function in ESCC. Additionally, the miRNA target gene regulatory network and miRNA coregulatory network were constructed to investigate the underlying functions of these miRNAs. As such, based on survival analysis of DEMs and univariate Cox analysis, a prognostic survival model based on the expression of different miRNAs was constructed.
2. Materials and Method
2.1. Microarray Data Collection and Preprocessing
Gene expression profiles from the GSE20347 (34 samples), GSE38129 (60 samples), and GSE23400 (106 samples) datasets between ESCC samples and matched normal controls were obtained from the GEO (http://www.ncbi.nlm.nih.gov/geo/) database using the Affymetrix Human Genome U133A 2.0 Array platform. In addition, the miRNA microarray GSE55856 dataset, which is composed of 216 samples (108 ESCC samples and 108 normal controls), was obtained using the Affymetrix Multispecies miRNA-2_0 Array.
For the preprocessing of the gene expression profile chip, the raw data of the GSE20347, GSE38129, and GSE23400 datasets were preprocessed using the R Affy package (version: 1.46.1) with a standardized RMA method [15]. The processing included background corrections, normalization, and calculation of gene expression. Afterwards, the probe ID was converted to a gene symbol, with probes that had no corresponding gene symbols being removed. As for the case where multiple probes correspond to the same gene symbol, we selected the mean value of the probes as the final gene expression value. The preprocessing of miRNA microarray data was done in a similar manner using the miRNA chip platform of Affy.
2.2. Identification of DEGs and DEMs
The limma package of R (version: 3.30.2) [16] was used to identify genes or miRNAs that were significantly differentially expressed between the tumor and normal tissues. P < 0.05 and log2 fold change (FC) ≥ 0.58 were selected as the cutoff values for statistically significant DEGs or miRNAs. Subsequently, we selected 3 groups of DEGs and then analyzed whether the genes were also significantly differentially expressed in the 3 datasets.
2.3. Prediction of DEGs Based on a Meta-analysis
By screening DEGs based on a meta-analysis, more reliable DEGs can be obtained due to the collection of multiple experimental datasets and enhancement of statistical ability. In order to integrate the DEGs that were combined in the three datasets, the MetaDE package of R (version: 1.0.5) was used [17]. Gene expression values were examined for heterogeneity with statistic parameters including tau2, Q value, and QP value. Criteria standards of tau2 = 0 and QPval > 0.05 were selected as the homogeneity test parameter. A P value of < 0.05 was the threshold for a significant difference in gene expression. Moreover, a heatmap was generated with the pheatmap [18] R package (version: 3.25).
2.4. Pathway Enrichment Analysis for DEGs
To investigate the biofunctions of DEGs, Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis was performed using DAVID (version: 6.8) [19]. P < 0.05 was selected as the cutoff criterion.
2.5. Identification of PPI Network and Key Genes
To better understand the interactions of the DEGs, the Search Tool for the Retrieval of Interacting Genes (STRING) database (version: 10) was employed to develop a DEG-encoded PPI [20], with a reliability threshold of >0.9. The prediction methods were derived from the neighborhood, gene fusion, cooccurrence, coexpression, experiments, databases, and text mining. The Cytoscape software (version: 3.2.1) was utilized to construct the PPI.
Next, the random walk algorithm was used to analyze important nodes in the PPI network. Briefly, the random walk was started at the seed node and the importance of each node was expressed by calculating the frequency of each node after the random walk between nodes in the network. The corresponding higher frequency genes may be candidate genes that have important physiological regulatory functions. The RWOAG package of R [21] was used to calculate the network node score.
2.6. Construction of the miRNA-Target Gene Regulatory Network
Mirwalk2 (http://zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk2) [22] was used for the prediction of target genes regulated by miRNAs, and differentially expressed target genes were filtered by using the “validated target” module. Based on the data of differentially expressed miRNAs and DEGs, the opposite relationship pairs (upregulated miRNA-downregulated gene or downregulated miRNA-upregulated gene) were selected from the miRNA-target gene data. The regulatory network of miRNA-target genes was constructed using the Cytoscape software [23]. Meanwhile, we screened several miRNAs from the miR2Disease database (http://www.mir2disease.org/) [24] which were reported to be related to ESCC.
2.7. Functional Analysis of miRNAs
In order to obtain information regarding the pathways associated with the identified miRNAs, we performed KEGG pathway analysis for differentially expressed target genes using the clusterProfiler package of R (version: 3.3.1). The enrichment significance P value was corrected with the BH method and a P value of less than 0.05 was considered to be significant.
2.8. Construction of miRNA Coregulatory Network
Based on the regulatory network of miRNAs and their target genes, miRNA pairs that regulate the same target genes were screened to construct the coregulatory network between miRNAs.
2.9. Survival Analysis of DEMs
Clinical and survival data from 251 patients with ESCC were retrieved from TCGA, which were downloaded from the database of University of California Santa Cruz (UCSC) Xena (https://xenabrowser.net). Moreover, miRNA-seq data was downloaded from http://gdac.broadinstitute.org/runs/stddata__2016_01_28/data/ESCA/20160128/.
In general, TCGA data directly downloaded cannot be directly analyzed. Therefore, in order to link different data, we need to match, select, and delete different data by screening samples. In this study, the DEMs obtained from the integration analysis of different GEO databases intersected with the miRNA-seq data filenames downloaded by TCGA using an R package [25]. Additionally, miRNAs with value = 0 in more than half of the total samples were removed from the existing intersection data.
The optimal miRNA cutoff was determined via the surv_cutpoint of survminer (version 0.4.3) of R package, with >optimal cutoff being considered high expression and <optimal cutoff being considered low expression. Survival analysis was conducted with the survival (version 2.42-6) R package, and P values < 0.05 were taken as the threshold. miRNAs with significant correlation to prognosis were selected and survival curves were made.
2.10. Univariate Cox Analysis and Prognostic Model Construction
Univariate survival Cox analysis was continued for miRNAs significantly correlated with survival, and miRNAs with P values < 0.05 were used for the construction of the prognostic model.
After univariate analysis, there were still many significant univariate factors, which were not conducive to inclusion in the prognostic model. Therefore, some dimensionality reduction methods were adopted to select the most important univariate factors to be included in the prognostic model for downstream analysis. In this study, the rbsurv R package was used to investigate the robustness of univariate survival analysis. Briefly, 3/4 samples were randomly selected as training data and the remaining 1/4 samples as validation data. Multivariate Cox analysis was carried out on the obtained models in the test training set and verification set, and risk scores for survival verification of the model were obtained. Finally, the overall evaluation effect of the model on prognosis was checked.
3. Results
3.1. Identification of DEGs and DEMs
After data preprocessing, a total of 2575, 2111, and 1205 DEGs between ESCC and normal tissues were identified in the gene expression profile of the GSE20347, GSE38129, and GSE23400 datasets, respectively, based on the cutoff criteria. Moreover, 226 DEMs were identified in GSE55856, of which 190 were upregulated and 36 were downregulated.
3.2. Meta-analysis of DEGs and Hierarchical Clustering
Based on the meta-analysis, 1001 DEGs, including 700 upregulated genes and 301 downregulated genes were obtained. As shown in Figure 1, hierarchical clustering revealed that the DEGs obtained from the meta-analysis and DEMs could effectively cluster the samples from the GSE20347, GSE38129, GSE23400, and GSE55856 datasets, which suggests that the ESCC samples could easily be distinguished from the normal controls by analyzing the DEGs or miRNAs.
Figure 1.
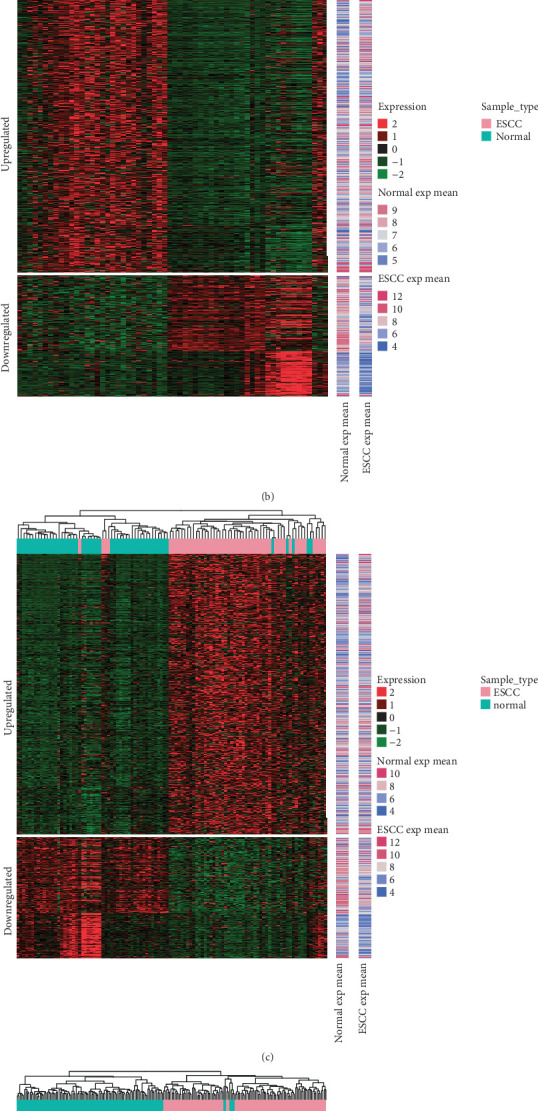
Heatmap clustering of the differentially expressed genes (DEGs) and miRNAs (DEMs) between ESCC and normal tissues samples in the GSE20347 (a), GSE38129 (b), GSE23400 (c), and GSE55856 (d) datasets. “Red” represents high relative expression and “green” represents a low relative expression.
3.3. Functional Enrichment Analysis of DEGs Screened from the Meta-analysis
As illustrated in Figure 2, DEGs were classified into four functional categories, including pathways, biological process, cellular components, and molecular function. KEGG pathway analysis revealed that the upregulated DEGs were mainly enriched in 18 pathways, including DNA replication, cell cycle, proteasome, base excision repair, and the spliceosome signaling pathway (all, P < 0.05; Table 1). Similarly, 18 KEGG pathways were significantly enriched in downregulated DEGs, such as regulation of actin cytoskeleton, glycine, serine, and threonine metabolism, cGMP-PKG signaling pathway, valine, leucine, and isoleucine degradation and the arginine and proline metabolism signaling pathway (all, P < 0.05).
Figure 2.
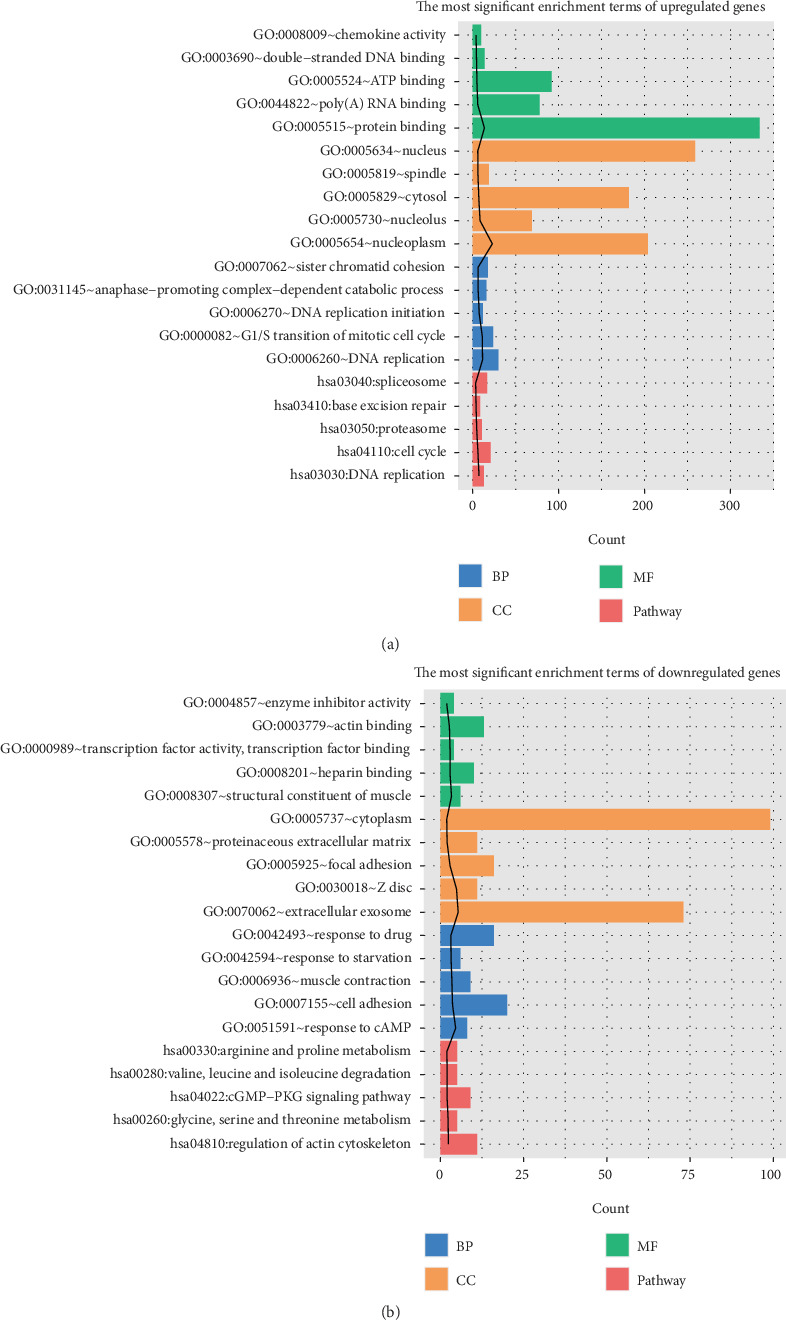
DEG functional enrichment analysis in ESCC. Upregulated (a) and downregulated (b) DEGs were enriched in four functional categories, including pathways, biological processes, cellular components, and molecular functions.
Table 1.
Signaling pathway enrichment analysis of DEGs in ESCC.
| Term | Count | P value | |
|---|---|---|---|
| Up | hsa03030:DNA replication | 13 | 3.51E-08 |
| hsa04110:cell cycle | 21 | 8.02E-07 | |
| hsa03050:proteasome | 11 | 2.33E-05 | |
| hsa03410:base excision repair | 9 | 9.52E-05 | |
| hsa03040:spliceosome | 17 | 3.83E-04 | |
| hsa03430:mismatch repair | 7 | 4.78E-04 | |
| hsa00240:pyrimidine metabolism | 14 | 9.29E-04 | |
| hsa03420:nucleotide excision repair | 8 | 5.35E-03 | |
| hsa05222:small-cell lung cancer | 11 | 5.50E-03 | |
| hsa00480:glutathione metabolism | 8 | 8.42E-03 | |
| hsa05230:central carbon metabolism in cancer | 9 | 8.77E-03 | |
| hsa04062:chemokine signaling pathway | 17 | 1.18E-02 | |
| hsa03013:RNA transport | 16 | 1.29E-02 | |
| hsa04510:focal adhesion | 17 | 2.83E-02 | |
| hsa00230:purine metabolism | 15 | 3.25E-02 | |
| hsa04115:p53 signaling pathway | 8 | 3.39E-02 | |
| hsa04512:ECM-receptor interaction | 9 | 4.68E-02 | |
| hsa04145:phagosome | 13 | 5.00E-02 | |
|
| |||
| Down | hsa04810:regulation of actin cytoskeleton | 11 | 4.23E-03 |
| hsa00260:glycine, serine, and threonine metabolism | 5 | 4.86E-03 | |
| hsa04022:cGMP-PKG signaling pathway | 9 | 9.30E-03 | |
| hsa00280:valine, leucine and isoleucine degradation | 5 | 9.45E-03 | |
| hsa00330:arginine and proline metabolism | 5 | 1.17E-02 | |
| hsa00072:synthesis and degradation of ketone bodies | 3 | 1.29E-02 | |
| hsa00410:beta-alanine metabolism | 4 | 1.72E-02 | |
| hsa04710:circadian rhythm | 4 | 1.72E-02 | |
| hsa04270:vascular smooth muscle contraction | 7 | 1.91E-02 | |
| hsa04924:renin secretion | 5 | 2.68E-02 | |
| hsa04713:circadian entrainment | 6 | 2.68E-02 | |
| hsa04510:focal adhesion | 9 | 3.02E-02 | |
| hsa00982:drug metabolism-cytochrome P450 | 5 | 3.25E-02 | |
| hsa04610:complement and coagulation cascades | 5 | 3.40E-02 | |
| hsa00360:phenylalanine metabolism | 3 | 3.59E-02 | |
| hsa04020:calcium signaling pathway | 8 | 4.01E-02 | |
| hsa05146:amoebiasis | 6 | 4.02E-02 | |
| hsa00071:fatty acid degradation | 4 | 4.30E-02 | |
3.4. Identification of Key Candidate Genes and Pathways by DEGs PPI Network Analysis
As shown in Figure 3, a series of DEGs were filtered into the PPI network, which contained 448 nodes and 1144 interaction pairs. Among the nodes, the key candidate node genes were identified by filtering the random walk score. The top 20 nodes, including 17 upregulated and 3 downregulated genes were summarized in Table 2. Among these DEGs, the MYC protooncogene (MYC) had the highest score.
Figure 3.
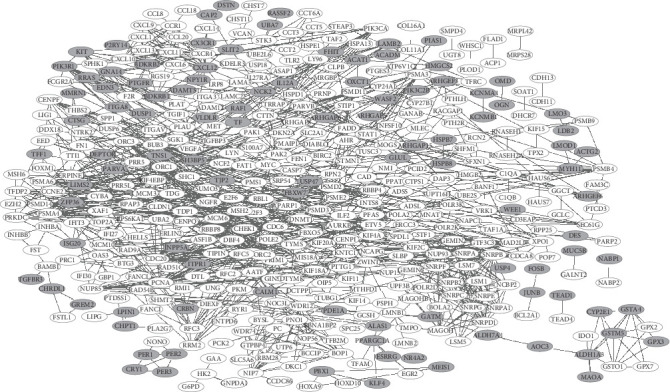
DEGs protein-protein interaction (PPI) network in ESCC. There were 448 nodes and 1144 interactions identified in the network. White represents upregulated genes and gray represents downregulated genes.
Table 2.
The key genes (top 20) in the PPI network in ESCC.
| Node | Direction | Random walk score |
|---|---|---|
| MYC | Up | 0.00656632 |
| PCNA | Up | 0.005105377 |
| AURKB | Up | 0.005066962 |
| STAT1 | Up | 0.004433487 |
| CXCL12 | Down | 0.003986094 |
| POLR2K | Up | 0.003855735 |
| CDC20 | Up | 0.003688161 |
| PIK3CA | Up | 0.003613145 |
| CDC6 | Up | 0.003570703 |
| PRKDC | Up | 0.00355442 |
| SHMT2 | Up | 0.003539976 |
| CHEK1 | Up | 0.003533405 |
| PPARGC1A | Down | 0.003496619 |
| MAGOH | Up | 0.003355163 |
| SERPINE1 | Up | 0.003290157 |
| CCR1 | Up | 0.003277042 |
| LYN | Up | 0.003267606 |
| HK2 | Up | 0.003262363 |
| WDR12 | Up | 0.003251239 |
| PIK3R1 | Down | 0.003241252 |
3.5. Regulatory Network of miRNA Target Genes
According to the screening principles of an upregulated miRNA-downregulated gene or downregulated miRNA-upregulated gene, we constructed the miRNA-target gene regulatory network. As shown in Figure 4, a total of 72 upregulated miRNAs which targeted 130 downregulated genes, as well as 19 downregulated miRNAs which targeted 133 upregulated genes were filtered in the network. Based on the miRNAs associated with ESCC obtained from miR2Disease (http://www.mir2disease.org/), 8 miRNAs, including miR-196a, miR-21, miR-205, miR-194, miR-103, miR-223, miR-203, and miR-375, were found to be significantly differentially expressed in miRNA microarray datasets. Among these miRNAs, 7 miRNAs excluding miR-203 were found in the network. Moreover, the gene with the highest score in the PPI network, MYC, was coregulated by miR-125a-3p, miR-940, and miR-375, among which miR-375 has been reported to be related to ESCC.
Figure 4.

MiRNA-target gene regulatory network. There were 72 upregulated miRNAs and 130 downregulated target genes, as well as 19 downregulated miRNAs and 133 upregulated target genes in the network. Gray represents a downregulated expression, while white represents an upregulated expression. Ovals represent the differentially expressed target gene, and diamonds represents the DEMs (blue edge represents miRNAs reported to be disease related).
3.6. Coregulatory Network between miRNAs
In order to construct the coregulatory network, the miRNAs that regulated the same target gene were identified via the miRNA-target gene regulatory network. As illustrated in Figure 5, several miRNAs which are reported to be disease related, such as hsa-miR-198a, hsa-miR-103, hsa-miR-223, hsa-miR-21, hsa-miR-194, and hsa-miR-375, had common target genes with other DEMs. These miRNAs have played an essential in proliferation, invasion, and metastasis of malignant disease, which is closely related to pathogenesis and prognosis.
Figure 5.
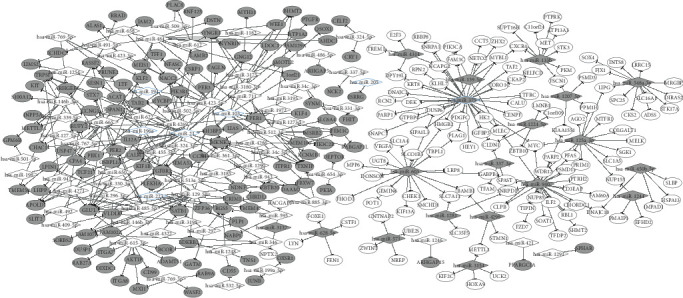
The coregulatory network between miRNAs. Several reported disease-related miRNAs shared common target genes with other DEMs. Gray represents a downregulated expression and white represents an upregulated expression. The blue thickened edge represents miRNAs reported to be disease related.
3.7. Survival Analysis of DEMs
After processing of the TCGA data described above, a total of 184 cancer samples and 174 differentially matched miRNAs were obtained. Survival analysis revealed that there were 91 DEMs significantly correlated with the outcome of ESCC patients (Supplementary Table 1).
3.8. Univariate Cox Analysis and Prognostic Model Construction
A total of 13 DEMs were obtained after the univariate Cox analysis. According to the model analysis, a prognostic survival model with 4 DEMs, including hsa-miR-1248, hsa-miR-1291, hsa-miR-421, and hsa-miR-7-5p was obtained. Among these miRNAs, hsa-mir-1248, hsa-miR-1291, and hsa-miR-421 were the DEMs in the GEO data. Moreover, hsa-miR-7-5p was concentrated as the precursor of hsa-miR-7 in the GEO data.
Multivariate Cox analysis was carried out for the following 4 DEMs, hsa-miR-1248, hsa-miR-1291, hsa-miR-421, and hsa-miR-7-5p, in the training set and validation set, respectively, and the regression coefficients were obtained (Table 3). Furthermore, the corresponding risk score was calculated for survival analysis and survival test. The threshold determination of the prognostic model was performed. The threshold of the cutoff point in the training set is 1.48 (Figure 6(a)) and in the validation set is 1.56 (Figure 6(b)), respectively. As illustrated in Figures 6(c) and 6(d), the survival analysis results of the risk score obtained by the prognostic model composed of the 4 miRNAs were appropriate in both the training and validation sets (both, P < 0.01).
Table 3.
The regression coefficients of 4 miRNAs.
| Training set | Validation set | ||
|---|---|---|---|
| hsa-miR-1248 | 0.62396 | PANTR1 | 0.37498 |
| hsa-miR-1291 | 0.40231 | LINC01266 | 0.09693 |
| hsa-miR-421 | -0.02532 | FGF13-AS1 | 0.10180 |
| hsa-miR-7-5p | 0.19866 | TMEM132D-AS1 | 0.34272 |
Figure 6.
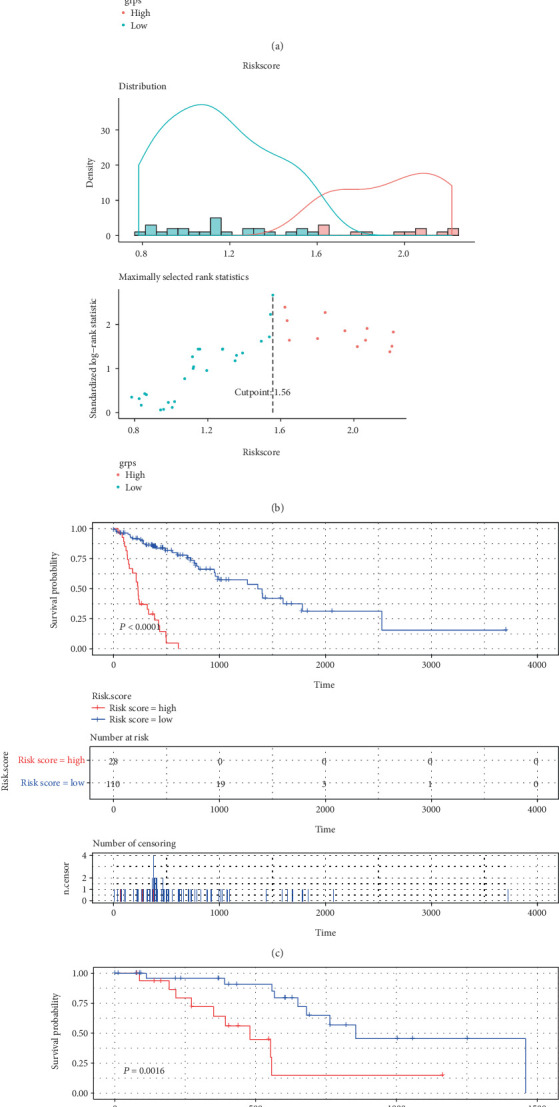
The threshold determination and survival test of the risk score of the prognostic model. The threshold of the cutoff point training set (a) and validation set (b). Survival results of the risk score obtained in the training set (c) and validation set (d) of the prognostic model composed of 4 miRNAs.
4. Discussion
During the past few decades, an increasing number of studies have investigated the causes and potential mechanisms of ESCC tumorigenesis. However, the high incidence and mortality of ESCC worldwide still pose a challenge, as most studies only focus on a single genetic event [9, 26]. Gene profiling or microarray technologies have been widely used to predict potential targets for the treatment of different tumors. In our study, we downloaded three original microarray data and identified 2575, 2111, and 1205 DEGs, as well as 226 DEMs, between ESCC and normal tissues. Moreover, the meta-analysis was used to further obtain more reliable DEGs, and 1001 genes that could be used for sample clustering of each dataset were identified. Furthermore, based on the clinical, survival, and miRNA-seq data downloaded from TCGA, a total of 85 DEMs were found to be significantly associated with the outcome of ESCC patients. As such, a prognostic survival model composed of 4 DEMs, including hsa-miR-1248, hsa-miR-1291, hsa-miR-421, and hsa-miR-7-5p was constructed.
It has been known that tumorigenesis is a complex process that involves the interaction of various genes and signaling pathways [26, 27]. In ESCC, an increasing number of signaling pathways have been reported to play important roles in the progression of the disease [28–30]. Therefore, the analysis of pathways related to neoplasia could provide information regarding tumor development. In the present study, the KEGG pathway analysis revealed that both upregulated and downregulated DEGs were mainly enriched in 18 pathways, such as the DNA replication, cell cycle, p53, proteasome, BcGMP-PKG signaling pathway, valine, leucine, and isoleucine degradation, beta-alanine metabolism, and arginine and proline metabolism signaling pathways. Among these pathways, recent studies have shown a correlation between the proteasome pathway and ESCC carcinogenesis [31]. Zhou et al. [32] revealed that the cell growth and apoptosis of ESCC could be regulated via activation of the p53 signaling pathway. Moreover, the beta-alanine metabolism pathway was reported as a novel subpathway related to ESCC, by the cooperative use of different genes in different pathways [33]. Taken together, the detection of molecules related to these pathways may help predict the occurrence and progression of ESCC.
Furthermore, the PPI network of proteins encoded by the identified DEGs was constructed. It contained 448 nodes and 1144 interactions. MYC, with the highest random walk score, could be regarded as the key gene in the PPI network. Indeed, accumulating evidence has demonstrated that MYC is an important factor in biological development and tumorigenesis. For instance, Kwon et al. [34] showed that MYC was overexpressed in ESCC patients and that its expression could predict better overall survival (OS) for patients. Likewise, Zhong et al. [35] revealed that MYC is involved in the tumorigenicity of ESCC by regulating the expression of the hydroxymethylglutaryl coenzyme A reductase (HMGCR). Therefore, further research on MYC will provide a basis for targeted therapy against ESCC.
miRNAs, which are endogenous, noncoding single-stranded RNAs, have been reported to play critical roles in various biological processes via binding to their target mRNAs [36, 37]. In the present study, a miRNA-target gene regulatory network using the identified DEMs was constructed, and seven miRNAs associated with ESCC, including miR-196a, miR-21, miR-205, miR-194, miR-103, miR-223, and miR-375 were identified in both the microarray data and the miRNA-target gene regulatory network. Among these miRNAs, miR-196a, miR-21, and miR-205 were previously found to be abnormally expressed in ESCC [38–40]. Notably, miR-125a-3p, miR-940, and miR-375 collectively regulated the expression of MYC, suggesting that these miRNAs might play a role in ESCC by regulating the MYC expression. Additionally, the miRNAs that regulated the same target gene were identified using the coregulatory network, and miR-198a, miR-103, miR-223, miR-21, miR-194, and miR-375 were reported to be miRNAs related to ESCC [41–44]. Survival analysis showed that a total of 85 DEMs were related to prognosis, among which hsa-miR-1248, hsa-miR-1291, hsa-miR-421, and hsa-miR-7-5p were used in a prognostic survival model. Indeed, miR-1248 has been reported to be involved in the microRNA signature model for the prediction of prognosis in patients with the Wilms tumors [45]. Moreover, miR-1291 and miR-421 are associated with OS in patients with lung adenocarcinoma [46, 47]. However, a prognostic survival model using these miRNAs has not been constructed for ESCC. The results of our study revealed that the tumorigenesis of ESCC may be the result of the coregulation of multiple miRNAs. Moreover, several miRNAs, such as hsa-miR-1248, hsa-miR-1291, hsa-miR-421, and hsa-miR-7-5p could be used to construct a prognostic survival model for the prediction of ESCC patient outcomes.
In conclusion, in the present study, we conducted a comprehensive bioinformatics analysis of DEGs, DEMs, and pathways, based on different datasets. As such, we identified DEGs such as MYC, miRNAs such as miR-125a-3p, miR-940, miR-375 miR-196a, miR-21, miR-205, miR-194, miR-103, miR-223, and miR-198a, and pathways such as the proteasome, p53, and beta-alanine metabolism pathways, which may be involved in ESCC development. Notably, survival analysis showed that 85 DEMs were related to prognosis, among which hsa-miR-1248, hsa-miR-1291, hsa-miR-421, and hsa-miR-7-5p were used to construct a prognostic survival model. Taken together, these findings have important clinical significance, as they can improve our understanding of the pathogenesis and molecular mechanisms of ESCC. Moreover, our results provide potential biomarkers for the prediction of ESCC prognosis. However, further studies are still needed to confirm the function of the identified genes.
Acknowledgments
This study was supported by grants from the Hunan Provincial Key Research and Development Program for Social Development (2016SK2006), National Natural Science Foundation of Hunan Province (2019JJ50353), Natural Science Foundation of Hunan Province National Health Commission (B2019091), Natural Science Foundation of Changsha Science and Technology Bureau (kp1901084), Wu Jieping Medical Foundation (320.6750.18368), and Cancer Foundation of China (NCC2018B58).
Data Availability
The data used to support the findings of this study are included within the article.
Conflicts of Interest
The authors declare that they have no competing interests.
Authors' Contributions
Le-meng Zhang and Jianhua Chen contributed to the clinical data collection. Tianli Cheng, Hua Yang, Changqie Pan, Haitao Li, and Yongzhong Luo contributed to the data statistics analysis. Le-meng Zhang contributed to the study design, manuscript writing, and data analysis. All authors read and approved the final manuscript.
Supplementary Materials
Supplementary Table 1: 85 miRNAs significantly correlated with prognosis.
References
- 1.Zeng H., Zheng R., Zhang S., et al. Esophageal cancer statistics in China, 2011: estimates based on 177 cancer registries. Thoracic Cancer. 2016;7(2):232–237. doi: 10.1111/1759-7714.12322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lagergren J. Oesophageal cancer in 2014: advances in curatively intended treatment. Nature Reviews Gastroenterology & Hepatology. 2015;12(2):74–75. doi: 10.1038/nrgastro.2014.213. [DOI] [PubMed] [Google Scholar]
- 3.Zhang P., Li X. M., Zhao X. K., et al. Novel genetic locus at MHC region for esophageal squamous cell carcinoma in Chinese populations. PLoS One. 2017;12(5, article e0177494) doi: 10.1371/journal.pone.0177494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen W., Zheng R., Baade P. D., et al. Cancer statistics in China, 2015. Ca: A Cancer Journal for Clinicians. 2016;66(2):115–132. doi: 10.3322/caac.21338. [DOI] [PubMed] [Google Scholar]
- 5.Tran G. D., Sun X. D., Abnet C. C., et al. Prospective study of risk factors for esophageal and gastric cancers in the Linxian general population trial cohort in China. International Journal of Cancer. 2005;113(3):456–463. doi: 10.1002/ijc.20616. [DOI] [PubMed] [Google Scholar]
- 6.Best L. M., Mughal M., Gurusamy K. S. Non-surgical versus surgical treatment for oesophageal cancer. Cochrane Database of Systematic Reviews. 2016;3(3, article CD011498) doi: 10.1002/14651858.cd011498.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Hagen P., Hulshof M., van Lanschot, et al. Preoperative chemoradiotherapy for esophageal or junctional cancer. New England Journal of Medicine. 2012;366(22):2074–2087. doi: 10.1056/nejmoa1112088. [DOI] [PubMed] [Google Scholar]
- 8.Vogelstein B., Papadopoulos N., Velculescu V. E., Zhou S., Jr D. L., Kinzler K. W. Cancer genome landscapes. Science. 2013;339(6127):1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yuan Y., Xue L., Fan H. Screening of differentially expressed genes related to esophageal squamous cell carcinoma and functional analysis with DNA microarrays. International Journal of Oncology. 2014;44(4):1163–1170. doi: 10.3892/ijo.2014.2262. [DOI] [PubMed] [Google Scholar]
- 10.Xing J., Liu C. Identification of genes associated with histologic tumor grade of esophageal squamous cell carcinoma. FEBS Open Bio. 2017;7(9):1246–1257. doi: 10.1002/2211-5463.12228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu N., Wang C., Clifford R. J., et al. Integrative genomics analysis of genes with biallelic loss and its relation to the expression of mRNA and micro-RNA in esophageal squamous cell carcinoma. BMC Genomics. 2015;16:1–11. doi: 10.1186/s12864-015-1919-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fu H. L., Wu D. P., Wang X. F., et al. Altered miRNA expression is associated with differentiation, invasion, and metastasis of esophageal squamous cell carcinoma (ESCC) in patients from Huaian, China. Cell Biochemistry & Biophysics. 2013;67(2):657–668. doi: 10.1007/s12013-013-9554-3. [DOI] [PubMed] [Google Scholar]
- 13.Slotta-Huspenina J., Drecoll E., Feith M., et al. MicroRNA expression profiling for the prediction of resistance to neoadjuvant radiochemotherapy in squamous cell carcinoma of the esophagus. Journal of Translational Medicine. 2018;16(1):p. 109. doi: 10.1186/s12967-018-1492-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jang H. J., Lee H. S., Burt B. M., et al. Integrated genomic analysis of recurrence-associated small non-coding RNAs in oesophageal cancer. Gut. 2017;66(2):215–225. doi: 10.1136/gutjnl-2015-311238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gautier L., Cope L., Bolstad B. M., Irizarry R. A. Affy—analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20(3):307–315. doi: 10.1093/bioinformatics/btg405. [DOI] [PubMed] [Google Scholar]
- 16.Smyth G., Smyth G. K. Limma: linear models for microarray data. In: Gentleman R., Carey V. J., Huber W., Irizarry R. A., Dudoit S., editors. Bioinformatics and Computational Biology Solution Using R and Bioconductor. New York, NY: Springer; 2013. (Statistics for Biology and Health). [DOI] [Google Scholar]
- 17.Qi C., Hong L., Cheng Z., Yin Q. Identification of metastasis-associated genes in colorectal cancer using metaDE and survival analysis. Oncology Letters. 2016;11(1):568–574. doi: 10.3892/ol.2015.3956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kolde R. pheatmap: pretty heatmaps. 2015.
- 19.Huang D. W., Sherman B. T., Lempicki R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 20.Szklarczyk D., Franceschini A., Wyder S., et al. STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Research. 2015;43(D1):D447–D452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Köhler S., Bauer S., Horn D., Robinson P. N. Walking the interactome for prioritization of candidate disease genes. American Journal of Human Geneticss. 2008;82(4):949–958. doi: 10.1016/j.ajhg.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dweep H., Gretz N. miRWalk2.0: a comprehensive atlas of microRNA-target interactions. Nature Methods. 2015;12(8):p. 697. doi: 10.1038/nmeth.3485. [DOI] [PubMed] [Google Scholar]
- 23.Jiang Q., Wang Y., Hao Y., et al. miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Research. 2009;37(Supplement 1):D98–D104. doi: 10.1093/nar/gkn714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu G., Wang L. G., Han Y., He Q. Y. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics-a Journal of Integrative Biology. 2012;16(5):284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Team R. C. R: A Language and Environment for Statistical Computing. R foundation for statistical computing; 2013. [Google Scholar]
- 26.Wang X., Li G., Luo Q., Gan C. Identification of crucial genes associated with esophageal squamous cell carcinoma by gene expression profile analysis. Oncology Letters. 2018;15:8983–8990. doi: 10.3892/ol.2018.8464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Long J., Liu Z., Xingda W. U., Yuanhong X. U., Chunlin G. E. Gene expression profile analysis of pancreatic cancer based on microarray data. Molecular Medicine Reports. 2016;13(5):3913–3919. doi: 10.3892/mmr.2016.5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xiaoyu H., Yiru Y., Shuisheng S., et al. The mTOR pathway regulates PKM2 to affect glycolysis in esophageal squamous cell carcinoma. Technology in Cancer Research & Treatment. 2018;17 doi: 10.1177/1533033818780063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tu Y., Tan F., Zhou J., Pan J. Pristimerin targeting NF-κB pathway inhibits proliferation, migration, and invasion in esophageal squamous cell carcinoma cells. Cell Biochemistry & Function. 2018;36(4):228–240. doi: 10.1002/cbf.3335. [DOI] [PubMed] [Google Scholar]
- 30.Li X., Chen D., Li M., Gao X., Shi G., Zhao H. The CADM2/Akt pathway is involved in the inhibitory effect of miR-21-5p downregulation on proliferation and apoptosis in esophageal squamous cell carcinoma cells. Chemico-Biological Interactions. 2018;288:76–82. doi: 10.1016/j.cbi.2018.04.021. [DOI] [PubMed] [Google Scholar]
- 31.Yang Q., Lin W., Liu Z., et al. RAP80 is an independent prognosis biomarker for the outcome of patients with esophageal squamous cell carcinoma. Cell Death & Disease. 2018;9(2):p. 146. doi: 10.1038/s41419-017-0177-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou P., Zhang R., Wang Y., et al. Cepharanthine hydrochloride reverses the mdr1 (P-glycoprotein)-mediated esophageal squamous cell carcinoma cell cisplatin resistance through JNK and p 53 signals. Oncotarget. 2017;8(67):111144–111160. doi: 10.18632/oncotarget.22676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li C., Wang Q., Ma J., et al. Integrative pathway analysis of genes and metabolites reveals metabolism abnormal subpathway regions and modules in esophageal squamous cell carcinoma. Molecules. 2017;22(10):p. 1599. doi: 10.3390/molecules22101599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kwon D., Ji Y. Y., Keam B., Kim Y. T., Jeon Y. K. Prognostic implications ofFGFR1andMYCstatus in esophageal squamous cell carcinoma. World Journal of Gastroenterology. 2016;22(44):9803–9812. doi: 10.3748/wjg.v22.i44.9803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhong C., Fan L., Yao F., Shi J., Fang W., Zhao H. HMGCR is necessary for the tumorigenecity of esophageal squamous cell carcinoma and is regulated by Myc. Tumour Biology. 2014;35(5):4123–4129. doi: 10.1007/s13277-013-1539-8. [DOI] [PubMed] [Google Scholar]
- 36.Shukla G. C., Singh J., Barik S. MicroRNAs: processing, maturation, target recognition and regulatory functions. Molecular & Cellular Pharmacology. 2011;3:p. 83. [PMC free article] [PubMed] [Google Scholar]
- 37.Bartel D. P. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fendereski M., Zia M. F., Shafiee M., Safari F., Saneie M. H., Tavassoli M. MicroRNA-196a as a potential diagnostic biomarker for esophageal squamous cell carcinoma. Cancer Investigation. 2017;35(2):78–84. doi: 10.1080/07357907.2016.1254228. [DOI] [PubMed] [Google Scholar]
- 39.Wang K., Chen D., Meng Y., Xu J., Zhang Q. Clinical evaluation of 4 types of microRNA in serum as biomarkers of esophageal squamous cell carcinoma. Oncology Letters. 2018;16:1196–1204. doi: 10.3892/ol.2018.8720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu H., Yao Y., Meng F., et al. Predictive value of serum miR-10b, miR-29c, and miR-205 as promising biomarkers in esophageal squamous cell carcinoma screening. Medicine. 2015;94(44, article e1558) doi: 10.1097/MD.0000000000001558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang Z., Zhang L., Zhu D., Shan X., Zhou X. A novel serum microRNA signature to screen esophageal squamous cell carcinoma. Cancer Medicine. 2017;6(1):109–119. doi: 10.1002/cam4.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li F., Lv J.-H., Liang L., et al. Downregulation of microRNA-21 inhibited radiation-resistance of esophageal squamous cell carcinoma. Cancer Cell International. 2018;18(1):p. 39. doi: 10.1186/s12935-018-0502-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu C., Wang C., Guan X., et al. Diagnostic and prognostic implications of a serum miRNA panel in oesophageal squamous cell carcinoma. PLoS One. 2014;9(3, article e92292) doi: 10.1371/journal.pone.0092292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yi J., Jin L., Chen J., et al. MiR-375 suppresses invasion and metastasis by direct targeting of SHOX2 in esophageal squamous cell carcinoma. Acta biochimica et biophysica Sinica. 2017;49(2):159–169. doi: 10.1093/abbs/gmw131. [DOI] [PubMed] [Google Scholar]
- 45.Gong Y., Zou B., Chen J., et al. Potential five-microRNA signature model for the prediction of prognosis in patients with Wilms tumor. Medical Science Monitor. 2019;25:5435–5444. doi: 10.12659/MSM.916230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sathipati S. Y., Ho S.-Y. Identifying the miRNA signature associated with survival time in patients with lung adenocarcinoma using miRNA expression profiles. Scientific Reports. 2017;7(1):p. 7507. doi: 10.1038/s41598-017-07739-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li Y., Cui X., Li Y., Zhang T., Li S. Upregulated expression of miR-421 is associated with poor prognosis in non-small-cell lung cancer. Cancer Management and Research. 2018;Volume 10:2627–2633. doi: 10.2147/CMAR.S167432. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1: 85 miRNAs significantly correlated with prognosis.
Data Availability Statement
The data used to support the findings of this study are included within the article.