

Abstract
Bioorthogonal chemistry has enabled the development of bioconjugates in physiological environments while averting interference from endogenous biomolecules. Reactions between carbonyl-containing molecules and alkoxyamines or hydrazines have experienced resurgence in popularity in bioorthogonal chemistry owing to advances that allow the reactions to occur under physiological conditions. In particular, ortho-carbonyl substituted phenylboronic acids (CO-PBAs) exhibit greatly accelerated rates of hydrazone and oxime formation via intramolecular Lewis acid catalysis. Unfortunately, the rate of the reverse reaction is also increased, yielding a kinetically less stable bioconjugate. When the substrate is a hydrazine derivative, an intramolecular reaction between the boronic acid and the hydrazone can lead to formation of heterocycle containing a boron-nitrogen bond. We have shown previously that α-amino hydrazides undergo rapid reaction with CO-PBAs to form highly stable, tricyclic products, and that this reaction is orthogonal to the popular azide-alkyne and tetrazine-alkene reactions. In this work, we explore a series of heteroatom-substituted hydrazides for their ability to form tricyclic products with two CO-PBAs, 2-formylphenylboronic acid (2fPBA) and 2-acetylphenylboronic acid (AcPBA). In particular, highly stable products were formed using β-hydroxy hydrazides and 2fPBA. C-terminal β-hydroxy hydrazide proteins are available using conventional biochemical methods, which alleviates one of the difficulties with applications of bioorthogonal chemical reactions: site-specific incorporation of a reactive group into the biomolecular target. Using sortase mediated ligation (SML), C-terminal threonine and serine hydrazides were appended to a model eGFP protein in high yield. Subsequent labeling with 2fPBA functionalized probes could be performed quickly and quantitatively at neutral pH using micromolar concentrations of reactants. The SML process was applied directly to an expressed protein in cellular extract, and the C-terminal modified target protein was selectively immobilized using 2fPBA-agarose. Elution from the agarose yielded a highly pure protein that retained the hydrazide functionality. This strategy should be generally applicable for rapid, efficient site-specific protein labeling, protein immobilization and preparation of highly pure functionalized proteins.
Graphical Abstract

INTRODUCTION
With an abundance of functional groups and reactive residues present in both simple and complicated systems, precise site-specific modification is a challenge for both biologists and chemists seeking to functionalize biomolecules of interest. The need to discover suitable methods for site-specific modification of proteins promotes the integration of multidisciplinary strategies. Bioorthogonal chemistry has developed into one of the most effective solutions to circumvent this challenge.1–3
Bioorthogonal reactions that proceed through pericyclic mechanisms have been successfully applied to a wide range of molecular systems and in multiple disciplines.4–9 The desire to perform multiple bioorthogonal reactions in a single system has promoted the development of orthogonal pairs of reactants that follow cycloaddition pathways.10,11 Bioorthogonal reactions that proceed through other mechanisms are less developed, but the potential to deploy these chemistries simultaneously with cycloaddition-based processes provides impetus to improve existing reactions and discover new ones.
Condensation reactions using hydrazine or alkoxyamine derivatives have been used for bioconjugation for decades, and are more recently used in the context of bioorthogonal ligations.12–14 The utility of these reactions had been restricted by the strong dependence of the reaction rate on pH, which typically peaks near pH 4.15,16 Successful approaches to circumvent the unfavorable kinetics in neutral aqueous conditions include the use of intermolecular nucleophilic catalysis or intramolecular Bronsted acid catalysis.12,17,18 Modifications to the electrophile, such as incorporating electron withdrawing groups into an aromatic aldehyde, can also alleviate the need for an external catalyst.19,20
Intramolecular Lewis acid catalysis of these reactions increases the rate of the condensation reaction at neutral pH by several orders of magnitude, which was reported by three different groups in 2015. Gillingham and co-workers first published that the boronic acid of 2-formylphenylboronic acid (2fPBA, Scheme 1a) is responsible for the exceptionally fast rate of oxime formation between an alkoxyamine and the aromatic aldehyde.21 Gao and co-workers showed that 2-acetylphenylboronic acid (AcPBA) also undergoes rapid reaction with alkoxyamines, but the resulting product is less stable and more reversible than that formed with 2fPBA.22 In this same work, the investigators analyzed hydrazide and hydrazine products with AcPBA. Finally, our group reported that when an aromatic hydrazine serves as the nucleophile, a transient hydrazone undergoes an intramolecular dehydration reaction to yield a boron-nitrogen heterocyclic compound - a substituted 2, 3, 1 - benzodiazaborine (DAB),23 an unusal, stable aromatic heterocycle first reported in the 1960s.24
Scheme. 1.
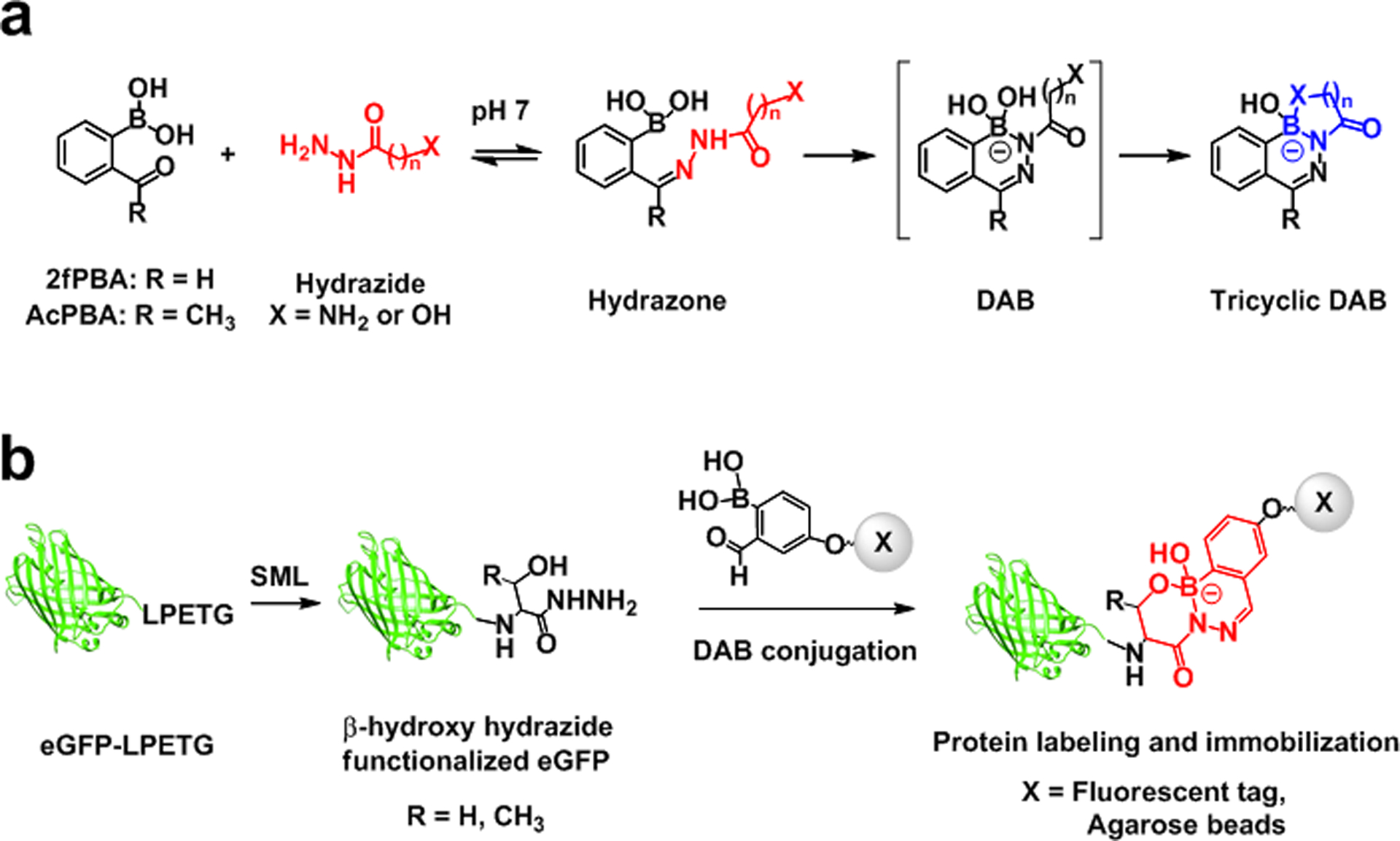
(a) Stepwise representation of tricyclic DAB formation between ortho-carbonyl substituted PBAs and hydrazide derivatives. (b) Fluorescent labeling and immobilization of eGFP through 2fPBA-β-hydroxy hydrazide linkage.
The rate of product formation between an ortho-carbonyl substituted phenylboronic acid (CO-PBA) and a hydrazine derivative has been consistently observed to be orders of magnitude faster than that of a corresponding reaction without the boronic acid. The final product(s) of such reactions, however, is affected greatly by the hydrazine derivative used25 and, to a lesser extent, the nature of the carbonyl.22 For example, aliphatic hydrazides undergo reaction with 2fPBA at neutral pH to yield a heterogenous mixture of the corresponding hydrazone and DAB in solution, the composition of which varies with pH.25 This is unfortunate for the use of CO-PBAs in bioorthogonal chemistry since there are many established methods for incorporating a hydrazide into a biomolecule but few for integrating a hydrazine.
We discovered that when an aliphatic hydrazide possesses an amine at the α-carbon, it will undergo rapid reaction with 2fPBA to form a single, stable product that is indefinitely stable in acidic (pH 4) and basic (pH 9) aqueous solution (Scheme 1a).25 The tetrasubstituted boron in the hydrazide DAB possesses two B-N bonds in a tricyclic structure, neither of which appear to exchange with water in aqueous solution. We recently applied this coupling chemistry to the creation of antibody-payload conjugates that are stable for days in human serum. The potential utility of this chemistry for partnering with established bioorthogonal reactions was also demonstrated by performing the DAB conjugation simultaneously with the well-established strain-promoted azide-alkyne cycloaddition reaction (SPAAC) and an inverse electron demand Diels-Alder reaction (IEDDA).26
Thus, the DAB reaction, properly executed, can be implemented in applications requiring multiple bioorthogonal labeling reactions. A hindrance to the addition of the DAB conjugation to the bioorthogonal repertoire is the need to site-specifically introduce one of the bioorthogonal reactants into the target. Ideally, the process would be easy to accomplish using methods and tools widely used by biologists. One possible route is through carboxy-terminal protein hydrazides, which have been widely used for applications such as native chemical ligation and site-specific labeling, and multiple methods for creating such biomolecules exist.27–32 We reasoned that a protein with a C-terminal hydrazide should form a stable conjugate with a CO-PBA if the side chain of the terminal amino acid could stabilize a tetrahedral boron in the DAB in a manner similar to the amino group of an α-amino hydrazide.
We selected the sortase mediated ligation (SML) to assess the feasibility of this approach. The SML is a versatile chemoenzymatic technique that has been exploited to incorporate a variety of functional moieties on the C-terminus of proteins.33,34 Sortase A (SrtA) is the first and most well-known transpeptidase used for this purpose, although multiple homologues and mutants of sortases have been subsequently uncovered and examined.35–38 SrtA39 recognizes the LPXTG (X= any amino acid) sorting motif on the C-terminus of the target protein and the enzyme’s catalytic cysteine residue cleaves the sorting sequence between threonine and glycine. The thioacyl enzyme intermediate can undergo reaction with an amine-containing nucleophile, forming a covalent linkage between the target protein and functional substrates. When used as a tool to incorporate a probe or bioorthogonal handle on the C-terminus of the protein, SML is robust and efficient and can be performed in biologically complex environments such as a cell lysate.40 Importantly for this application, SrtA has been shown to accept hydrazine as a substrate, resulting in a product that possesses a C-terminal threonine hydrazide.41 We hypothesized that the alcohol on the β-carbon might stabilize a DAB-containing product by virtue of a boron-oxygen bond using the side chain of the threonine. This idea is bolstered by a recent report from Gois and co-workers, who stabilized iminoboronate constructs through the formation of a B-O bond that locked the boron center in a tetrahedral geometry.42
In this work, we survey a series of L-amino acid hydrazides and related molecules to determine the structure(s) of the products formed with 2fPBA and AcPBA and their relative stabilities. These results inform us of the potential of the C-terminal protein labeling methodology using SML and of other possible biomolecule labeling strategies. Next, we use a model eGFP protein43 containing a sortase recognition site to generate C-terminal β-hydroxy hydrazides using the established hydrazine approach and with simple, accessible C-terminal hydrazide peptides. The C-terminal protein hydrazides prepared in this manner maintain their rapid reaction rate with 2fPBA derivatives and form highly stable conjugation products.
The DAB reaction between CO-PBAs and C-terminal β-hydroxy hydrazide-containing proteins has multiple applications, a few of which are illustrated here (Scheme 1b). Site-specific protein labeling is shown to occur in a complex environment through the formation of the β-hydroxy stabilized DAB. The C-terminal β-hydroxy hydrazide modification is further utilized to serve as a handle for protein purification. The SML followed by the DAB reaction can be performed on an expressed protein in bacterial lysate in one pot. Remarkably, immobilization of protein in these mixtures using 2fPBA-functionalized agarose beads is complete in minutes, recapitulating the rapid kinetics of the small molecule model compounds. An overnight elution process performed at neutral pH yields the desired protein in a highly pure form.
RESULTS AND DISCUSSION
Formation of DAB products with functionalized hydrazides.
We previously demonstrated that the sole product of the reaction of 2fPBA with glycine hydrazide (1a) in aqueous solution is a highly stable tricyclic DAB conjugate (5a),25 and that the DAB formed with an α-amino hydrazide motif results in highly stable bioconjugates.26 Unfortunately, the α-amino hydrazide motif has not yet been incorporated directly into a protein using straightforward molecular biology techniques. There are, however, a number of well-established protocols for creating C-terminal protein and peptide hydrazides, which could be harnessed to create a suitably modified carboxy terminus.27–32 Therefore, the first objective was to evaluate whether hydrazides derived from or related to canonical amino acids can form acceptably stable products with 2fPBA or AcPBA. Selected hydrazides bearing various functional groups, including α-amino hydrazides 1b-c, β-amino hydrazide 2, α-hydroxy hydrazide 3, and β-hydroxy hydrazides 4a-c (Figure 1a), were allowed to react with an equimolar concentration of 2fPBA or AcPBA in aqueous solution at neutral pH. The resulting solutions were examined by 1H NMR. Next, the pH of the solution was decreased to pH 4 by the addition of small amounts of concentrated acid, and these solutions were again subjected to NMR analysis.
Figure. 1.
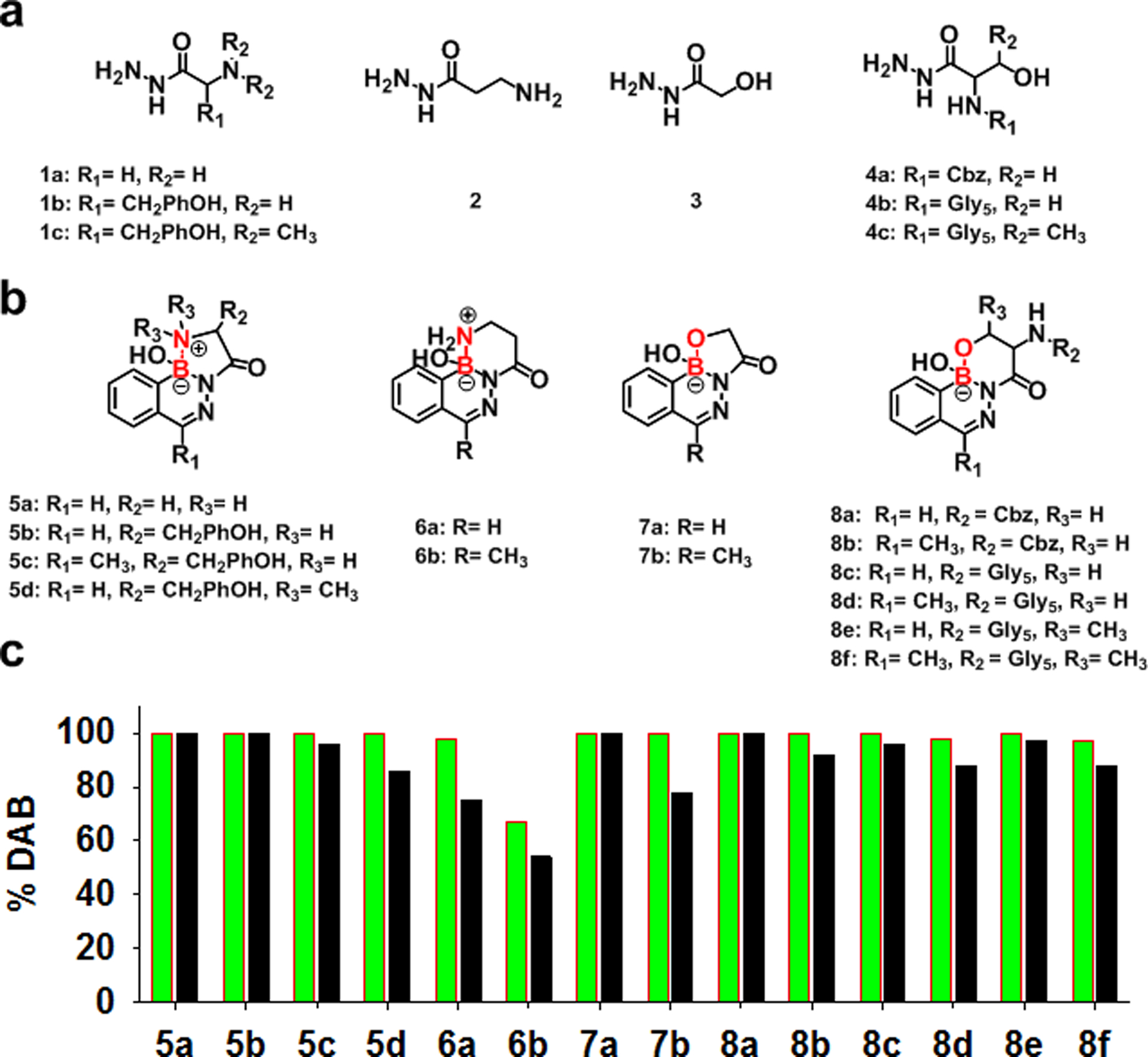
(a) Selected hydrazides bearing various functional groups. (b) Tricyclic DABs generated by reaction between CO-PBAs and 1a - 4c; the boron-heteroatom secondary interactions are labeled in red. (c) Stability of 5a - 8f defined by the percentage of DAB in pH 7 (green) and pH 4 (black) solutions.
The NMR data were used to determine the relative concentration of DAB product (Figure 1b) in solution at pH 7 and pH 4, as described in Supporting Information (SI, section 6). The presence of CO-PBAs, hydrazones and DAB products are clearly distinguishable in 1H NMR spectra. Stability is judged by the relative concentration of the intact DAB product in each solution. These data are shown in Figure 1c.
Some general trends can be extracted from the data. First, consider the products formed with the aldehyde, 2fPBA. As expected, α-amino hydrazides (1a-c) capable of forming a B-N five membered ring in a DAB product form very stable products with the 2fPBA (5a, b & d). The bulky side chain of tyrosine hydrazide had no effect on the stability of the DAB (5b). In fact, addition of two methyl groups to the coordinating nitrogen of tyrosine hydrazide still yielded a reasonably stable product (5d; 86% DAB at pH 4). Crystal structures of 5b and 5d were determined and analyzed. (SI, section 1.2) The side chain of tyrosine had little effect on the overall structure of the 5-membered ring. For example, the bond angles in the 5-membered rings of 5b are similar to those of the previously reported α-amino hydrazide DAB 5a.25 In contrast, the bond angles in the 5-membered ring in 5d are somewhat smaller, particularly the interior bond angle for the dimethylated nitrogen (Table S1). Thus, although substituents on the hydrazide are not completely benign, it seems likely that most α-amino acid hydrazides may serve as partners with 2fPBA to form stable conjugates.
The effect of ring size on the stability of tricyclic DAB in this series was inspected using a model compound, γ-aminobutyric acid hydrazide (2). Interestingly, the tricyclic DAB 6a, which contains a second 6-membered ring, was notably less stable than the corresponding DAB tricycle containing a 5-membered ring (5a). Only 75% of the 6a solution is in the DAB form at pH 4, compared to 100% for 5a.
Next, the effect of the hydroxyl substituent on product stability was assessed. Glycolic acid hydrazide (3) was selected as a model α-hydroxy hydrazide. The resulting product is quite stable. The expected tricyclic structure (7a), which contains a second 5-membered B-O ring, is formed in quantitative yield at pH 7, and no dissociation is observed at pH 4.
The β-hydroxy hydrazides from serine and threonine (4a-c) also produced stable conjugates with 2fPBA (8a, 8c and 8e). At pH 4, more than 97% of the solution was in DAB form. These data show that the methyl group on side chain of threonine had no influence on the stability of the DAB (8e).
In summary, all of the heteroatom-substituted hydrazides examined form tricyclic DABs with 2fPBA at pH 7 in quantitative or nearly quantitative yield. Most of these are also very stable at pH 4. Surprisingly, the least stable in this series was 6a, the tricyclic DAB containing a B-N bond within a 6-membered ring.
Now consider the products formed with the methyl ketone analogue of 2fPBA, AcPBA. This CO-PBA has been used by several investigators for forming iminoboronates and DABs.44,45 AcPBA has a much lower propensity to form thiazolidines with L-cysteine than 2fPBA, which may be useful in certain biological applications.46 Gillingham previously concluded that hydrazone and oxime products of AcPBA are less stable than those with 2fPBA.47 Thus, it is not surprising that the data collected here show that DABs formed with AcPBA and hydrazides also tend to be less stable than those formed with 2fPBA and the corresponding hydrazide. Note, however, that when the product can form a tricyclic DAB containing a 5-membered B-N ring (5b vs. 5c) or a 6-membered B-O ring (8a vs. 8b; 8c vs. 8d; 8e vs. 8f), there is little difference in the stability of the AcPBA- and 2fPBA-derived DAB. In fact, the only DAB that was not formed in essentially quantitative yield at pH 7 was the 6-membered B-N ring product 6b. It should also be noted that there is a larger difference in stability of the DAB with 5-membered B-O ring formed with AcPBA vs. 2fPBA – about 78% of the DAB 7b is intact at pH 4, while no dissociation is observed for 7a under these conditions.
Finally, to confirm that the side chain of the amino acid hydrazide is responsible for the stability of the DAB, the NMR experiments were repeated with two forms of glutamic acid α-amino acid hydrazide (SI, section 6). As expected, the stability profile of the products with 2fPBA are nearly the same as the previously studied propanoic acid hydrazide-2fPBA DAB.25
We therefore conclude that proteins that contain an α- or β-functionalized hydrazide will likely form stable complexes with CO-PBAs provided that the added functional group can also form a stable ring containing the boron atom of the DAB. In the subsequent protein studies, we chose 2fPBA rather than AcPBA for DAB conjugation. AcPBA forms more stable iminoboronates than 2fPBA,48 which is undesirable in this application, and 2fPBA forms more stable DABs than AcPBA with β-hydroxy hydrazides (vide infra).
Kinetics and stability of 2fPBA-β-hydroxy hydrazide conjugate.
One of the attractive features for using 2fPBA and AcPBA to form bioconjugates is the fast rate of product formation normally observed for these systems. It was therefore important to assess the reaction kinetics for formation of β-hydroxy stabilized DABs (8c-f). Reactions were performed under pseudo-first order conditions using a small peptide with a C-terminal serine or threonine hydrazide (4b, 4c) and monitored by absorption difference spectroscopy. Absorption data at a single wavelength as a function of time for the reaction of 4b with 2fPBA and with AcPBA are illustrated in Figure 2a. It is seen that the overall reaction with 2fPBA is complete in less than 5 min, whereas the reaction with AcPBA requires about 30 min to reach completion.
Figure. 2.
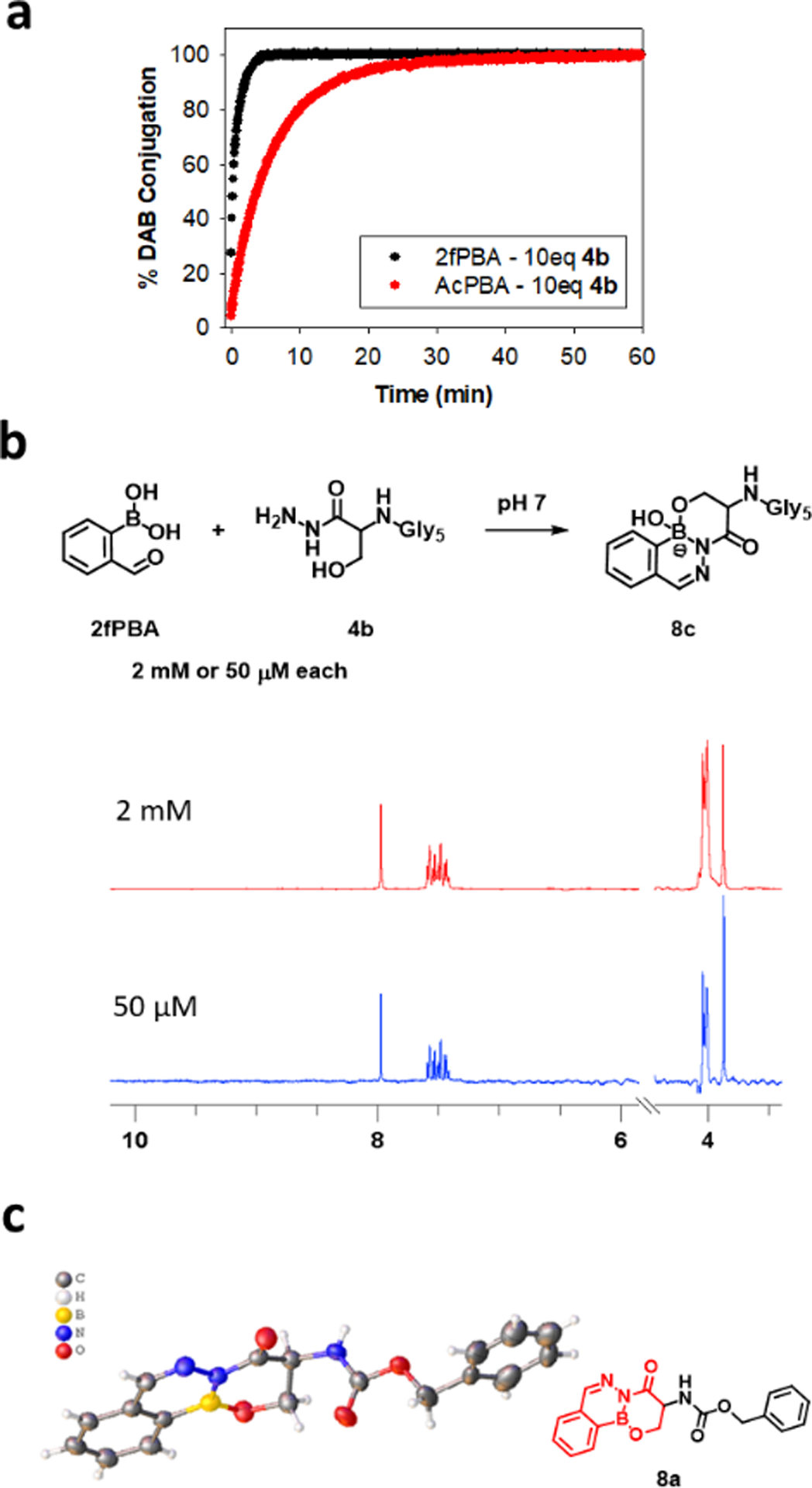
(a) Kinetics of the reactions between 100 μM 4b and 10 μM 2fPBA (black) or AcPBA (red) in 0.1 M phosphate buffer, pH 7. The reaction progress was monitored by the change in absorbance at 302 nm. (b) Equimolar concentrations of 2fPBA and 4b were mixed in pH 7 phosphate buffer to a final concentration of 2 mM or 50 μM. 1H NMR spectra of the resulting solutions were collected. (c) Crystal structure of 8a.
The full spectral data as a function of time for the reaction of 4b with 2fPBA were analyzed using Olis SpectralWorks software (Figure S3). Good fits were obtained with a three-species sequential fit model (A→B→C), where A represents 2fPBA or AcPBA, B is the hydrazone intermediate and C is the final DAB. For the 2fPBA reaction, an apparent second order rate constant for the hydrazone formation step was calculated from these experiments to be 955 ± 15 M−1 s−1, and the combined ring closure event was 0.014 ± 0.0006 s−1. The rate constants are consistent with those obtained with glycine hydrazide and 2fPBA.26 Kinetic analysis for the threonine hydrazide peptide 4c yielded similar results (Figure S5), leading to the conclusion that the side chains of serine and threonine are not adversely affecting the speed of the DAB-forming reaction.
Kinetic data were also collected for AcPBA and 4b (Figure S4). Interestingly, analysis of the data by three-species sequential fit model yielded an apparent rate constant for first step of the reaction (hydrazone formation) of 120 ± 5 M−1 s−1, which is about 8-times slower than hydrazone formation in the 2fPBA reaction. The difference in rate constants is reasonable, as aldehydes are generally more reactive then ketones toward hydrazines and other nucleophiles.12,16 The rate constant for the ring closing process was about the same for AcPBA and 2fPBA (0.012 ± 0.0002 s−1).
Another important consideration for a bioconjugate is the stability of the complex at the low concentrations that would be encountered in biological applications. Therefore, the stability of 2fPBA-β-hydroxy hydrazide conjugate was assessed in dilute solution using NMR and absorption spectroscopy methods. Equimolar volumes of the serine hydrazide peptide 4b and 2fPBA in pH 7 buffer were mixed to a final concentration of 50 μM for each component. Figure 2b demonstrates that conversion to 8c was quantitative as observed by 1H NMR. To assess dissociation at a lower final concentration, the 2 mM solution of 8c was diluted to 10 μM in pH 7 in a spectrophotometer. The absorption intensity at 302 nm decreased by less than 2% over a 24 hours period. (Figure S7–8). If the 2% change is reflective of hydrolysis of the DAB, a dissociation constant for 8c is estimated to be <10 nM (SI, section 2.6). Overall, these data support the idea that a bioconjugate formed using this chemistry will be very stable even at low concentrations.
Finally, direct evidence for a bond between β-hydroxy side chain of the amino acid hydrazide 4a and the boron atom in 2fPBA was obtained by X-ray crystallography (Figure 2c). It is important to note that the dehydrated form of the product is found in the solid state, in which the boron adopts a trigonal geometry. It is known that molecules capable of forming boron-heteroatom dative bonds may form different structures about the boron atom in solid state and solution state.49,50
C-terminal protein labeling through β-hydroxy stabilized DAB conjugation.
Having established that a peptide with a C-terminal serine or threonine hydrazide will form a stable tricyclic structure with 2fPBA or AcPBA, the next task was to determine the applicability of this reaction for site-specific labeling of the C-terminus of a protein. A model protein, eGFP, that contained a sortase sorting sequence LPETG followed by a His tag (designated eGFP-LPETG) was expressed in E. coli and purified by affinity chromatography for this purpose.
The Liu lab has developed a method by which the thioester intermediate of a sortase-bound substrate is trapped by hydrazine rather than the usual polyglycine substrate, yielding threonine hydrazide directly on the C-terminus of protein. This hydrazinolysis is irreversible, since the resulting C-terminal hydrazide is no longer a substrate for SML.41 This methodology has since been used for chemical protein synthesis of phosphorylated p62 protein.51 We applied this method to create a C-terminal threonine hydrazide model protein. eGFP-LPETG was treated with a large excess of hydrazine in the presence of SrtA to generate eGFP-1a (Figure 3a). The purified protein could be labeled by a 2fPBA-functionalized Texas Red fluorophore (2fPBA-TxR) in buffer solution and in bacterial lysate, a chemically complex environment. A single fluorescently labeled protein resulted in each case (Figure 3b), illustrating the bioorthogonality of the reaction.
Figure. 3.
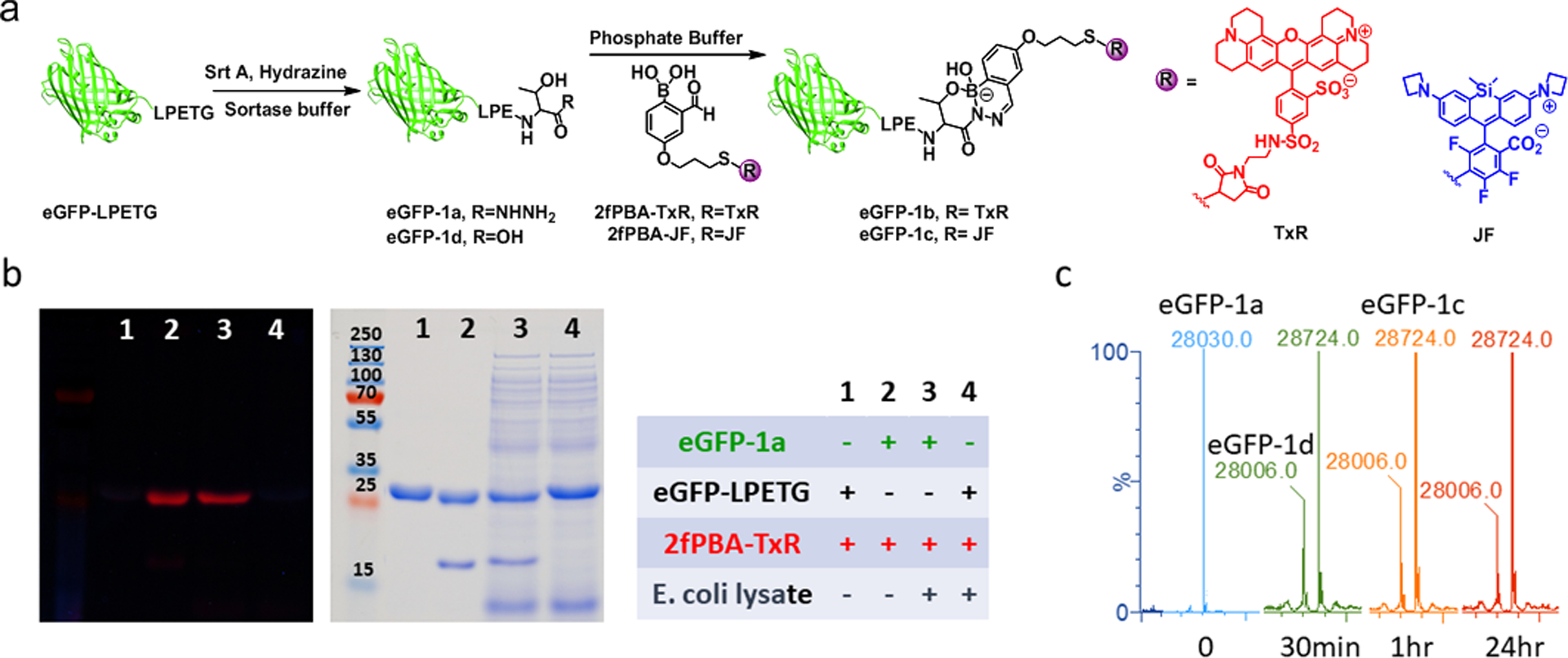
(a) Schematic illustration of sortase mediated hydrazinolysis of eGFP-LPETG to yield eGFP-1a which reacted with 2fPBA-TxR or 2fPBA-JF subsequently; hydrolyzed product eGFP-1d was formed in the hydrazinolysis step. (b) Fluorescence (left) and Coomassie blue staining (right) images of a gel which checked the reaction of eGFP-1a and 2fPBA-TxR in pH 7 phosphate buffer or E. coli lysate. The band at ~18 kDa is SrtA. (c) the reactions of eGFP-1a and 2fPBA-JF were monitored by LCMS after 30 minutes (green), 1 hour (orange) and 24 hours (red).
Quantitative analysis of the sortase-mediated hydrazinolysis reaction was performed using LC-MS. Aliquots of the sortase reaction were desalted and then allowed to react with 10 eq. of 2fPBA-Janelia Fluor 669 (2fPBA-JF). Aliquots were removed at various times for analysis. After 30 min, eGFP-1c was observed in ~77% yield (Figure 3c; full spectra are shown in Figure S14). Longer incubation time (up to 24 hours) did not change the amount of labeled product. Through experiments detailed in the SI, we determined that the 28006 Da species is eGFP-1d, with a C-terminal carboxylate, rather than the slightly larger hydrazide eGFP-1a. (Figure S14)
After multiple attempts to improve the yield of the hydrazide eGFP-1a, we concluded that the high concentration of sortase required for the hydrazinolysis reaction for this protein substrate was also favoring a competing reaction, hydrolysis of the eGFP-LPETG to the carboxylate. Hydrolysis is a known competition reaction in SML when a weak substrate is used to displace the thioester intermediate.52–57 The modified eGFP used by the Liu lab possesses a longer C-terminal sequence than the eGFP used here (LPETGGLE-His6 vs. LPETG-His6), which may be a better substrate for sortase-mediated hydrazinolysis than the construct used in this work.
We therefore turned to the classic nucleophile for sortase ligation, a pentaglycine peptide (Figure 4a). These peptides are very straightforward to prepare using a peptide synthesizer; in fact, preloaded serine and threonine hydrazide resins are commercially available. Using a low concentration of SrtA (2 μM) and a high concentration of 4b or 4c (5 mM), quantitative conversion of eGFP-LPETG to the desired product, C-terminal hydrazide eGFP-2a or eGFP-3a, was observed after 3 hours (Figure 4b and Figure S15).
Figure. 4.
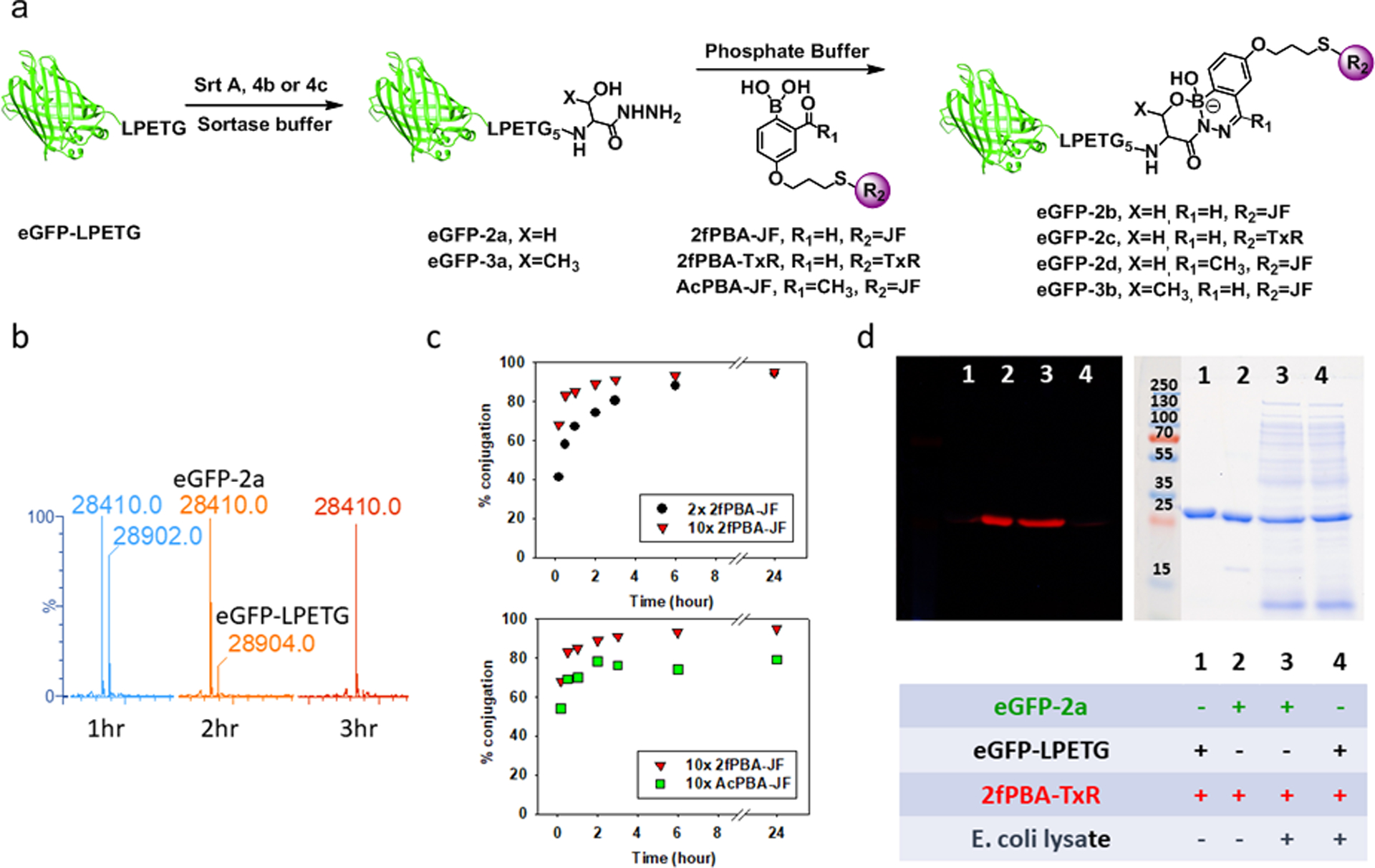
(a) Schematic illustration of sortase mediated ligation of eGFP-LPETG and 4b/4c to yield eGFP-2a/eGFP-3a which reacted with 2fPBA-JF or 2fPBA-TxR subsequently. (b) LC-MS analysis of sortase mediated ligation of eGFP-LPETG and 4b at 1 hour, 2 hours and 3 hours. (c) Kinetics of conjugation of β-hydroxy hydrazide functionalized eGFP (eGFP-2a) with 2fPBA-JF or AcPBA-JF in pH 7 phosphate buffer. Top panel: eGFP-2a (20 μM) reacted with 2 eq. or 10 eq. of 2fPBA-JF. Bottom panel: kinetics comparison of conjugation of GFP-2a (20 μM) with 10 eq. of 2fPBA-JF or AcPBA-JF. Full LC-MS spectra are shown in Figure S16–18. (d) Fluorescence (left) and Coomassie blue stained (right) images of a gel which demonstrates the labeling of eGFP-2a with 2fPBA-TxR in pH 7 phosphate buffer or E. coli lysate, showing covalent attachment and the orthogonality of the β-hydroxy stabilized DAB conjugation.
The rates by which small molecule model compounds undergo reaction with one another are not necessarily the same as the rates at which a desired bioconjugate is formed.58 It is therefore important to assess directly the speed of the bioconjugation reaction on the biomolecule of interest. Each β-hydroxy hydrazide functionalized protein, eGFP-2a or eGFP-3a (20 μM), was incubated with 2fPBA-JF at room temperature, and aliquots were removed at various times for LC-MS. The results obtained with eGFP-2a are illustrated in Figure 4c. With 10-fold excess of the fluorophore, 85% of β-hydroxy hydrazide functionalized eGFP was conjugated with 2fPBA-JF in the first hour and the reaction reached 95% completion in 3 hours (Figure 4c, top panel). When the conjugation was performed with low excess of 2fPBA-JF (2 eq.), the reaction rate was slower, but the conjugation reaction was still 90% complete after 6 hours. Prolonged incubation yielded the same level of conjugation for both conditions.
The kinetic analysis was repeated using AcPBA as the reactive functional group and 10-fold excess of the AcPBA-Janelia Fluor 669 (AcPBA-JF) was applied. These data are compared to the corresponding 2fPBA reaction in Figure 4c (bottom panel). As expected from the model compound reactions (Figure 2a), the rate of product formation was slower for the AcPBA fluorophore, although the difference was not as pronounced as observed for the small molecule hydrazides. Notably, no more than 80% conversion of the conjugate was observed in AcPBA reaction, even after 24 hours. This result reflects the trend of DAB stability observed in the small molecule studies (Figure 1c) and a correspondingly larger Kd for AcPBA derived DAB formation. Therefore, the 2fPBA moiety was confirmed to be more suitable for generating stable bioconjugates.
Protein immobilization and purification based on 2fPBA-β-hydroxy hydrazide conjugation.
Having demonstrated the highly efficient and orthogonal nature of the 2fPBA-β-hydroxy hydrazide conjugation, we next explored immobilization of the C-terminal hydrazide protein on a 2fPBA-functionalized solid support. Ideally, the bond between the desired protein and the solid support would be stable until its release is triggered (Figure 5a). We reasoned that dissociation of the resin-bound protein could be promoted by disturbing the equilibrium between the protein hydrazide and 2fPBA by irreversibly trapping one of the components of the dissociation reaction.
Figure. 5.
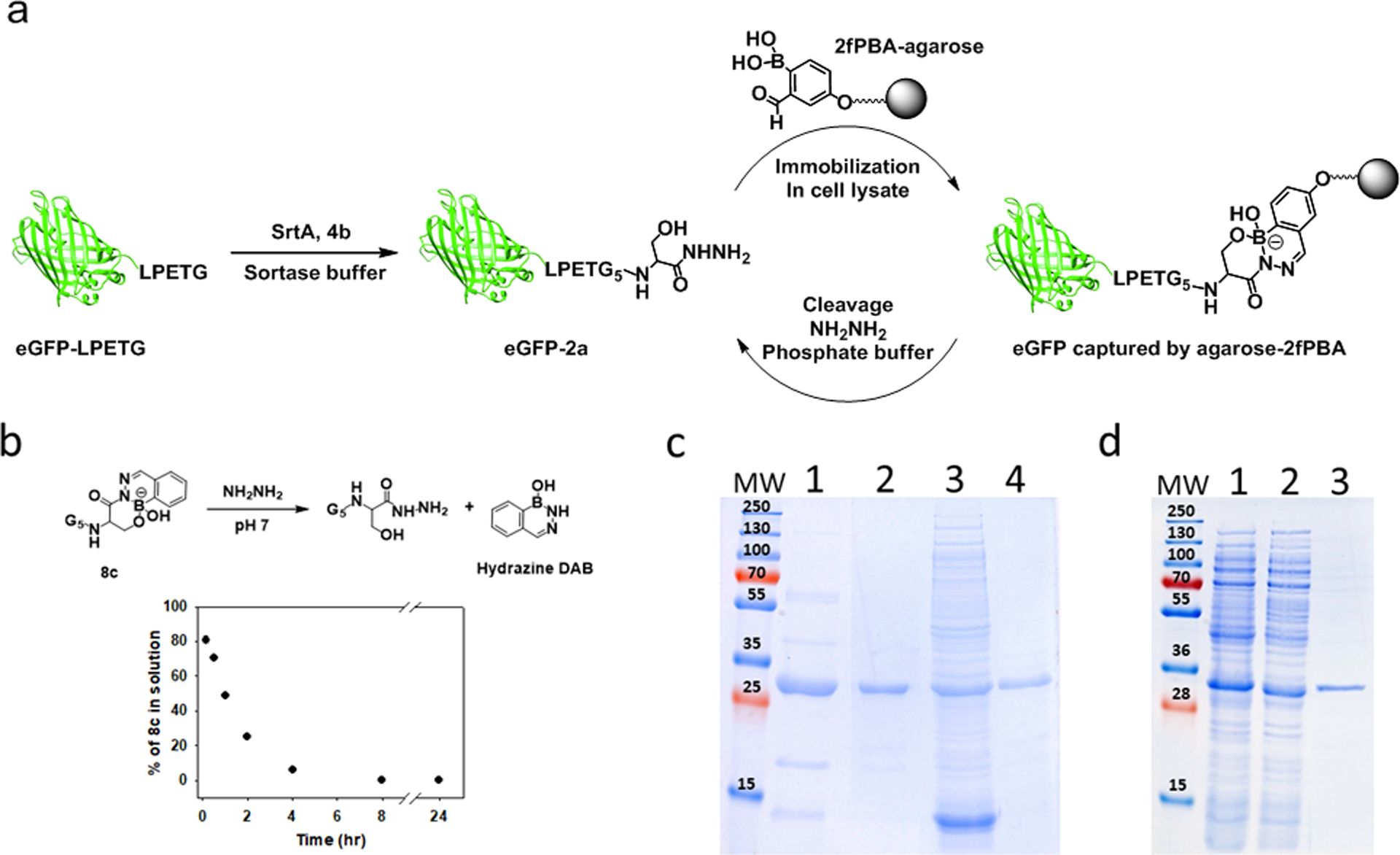
(a) Schematic illustration of protein purification from E. coli cellular lysate. (b) 2fPBA-β-hydroxy hydrazide linkage (8c) reversed by 200 eq. hydrazine in pH 7 buffer. (c) Coomassie blue stained gel which showed successful immobilization of eGFP-2a from impure samples. Lane 1: impure eGFP-2a. Lane 2: eGFP-2a purified by 2fPBA functionalized agarose. Lane 3: cell lysate spiked with eGFP-2a. Lane 4: eGFP-2a purified from cell lysate. (d) Coomassie blue stained gel which showed successful one-pot purification of eGFP from E. coli cellular lysate. Lane 1: E. coli cellular lysate containing eGFP-LPETG. Lane 2: eGFP-2a generated in E. coli cellular lysate. Lane 3: eGFP-2a purified from E. coli cellular lysate.
Hydrazine forms an exceptionally stable DAB with 2fPBA.24 Its ready availability and lack of reactivity with proteins under mild conditions suggested its use for this purpose. Its suitability for the dissociation reaction was first tested using small molecules. Conjugate 8c (2 mM) was treated with 100-fold excess of hydrazine hydrate (200 mM) in pH 7 phosphate buffer. The composition of solution was monitored by 1H NMR over time, which showed a decrease in 8c concentration concomitant with formation of the hydrazine DAB. (Figure 5b; see Figure S9a for full spectra) Increasing the hydrazine concentration to 200-fold excess increased the rate of the reaction, indicating that hydrazine can also participate in the DAB dissociation process (Figure S9b).
An important consideration for applications involving immobilized 2fPBA is non-specific protein immobilization via iminoboronates formed between 2fPBA and lysine residues.48 These bonds form readily at neutral pH; however, the dissociation constants for iminoboronates are low mM at neutral pH44 vs. the apparent nM Kds estimated for the 2fPBA-β-hydroxy hydrazide conjugates created here. Moreover, iminoboronate stability in aqueous solution appears to be mediated by electrostatic interaction in the “solvent inserted” iminoboronate, similar to behavior observed in o-aminomethyl arylboronic acids.44,59,60 Lowering the pH below the pKa of the boronate anion removes this stabilizing interaction and leads to the dissociation of the imine,61 which is shown experimentally with a model iminoboronate (Figure S11). Addition of 1 eq of hydrazine hydrate to the model iminoboronate solution at pH 7 immediately yielded the hydrazine DAB, further illustrating the reversibility of the imine (Figure S12).
To demonstrate this strategy for protein immobilization, we created 2fPBA-functionalized agarose beads and added purified eGFP-2a (43 μg) to the slurry (90 μL; 33% (v/v) beads in PBS). Within 5 minutes, the resin turned green and settled to the bottom of the column, leaving a solution that was colorless and transparent. Absorption spectroscopy showed that the liquid contained less than 5% of the initial mass of eGFP-2a. The resin-bound eGFP-2a was released from the resin by overnight incubation with 200 mM of hydrazine in pH 6.8 phosphate buffer. Figure 5c, lanes 1 and 2, show that the process yielded a very pure protein: even the minor impurities that are frequently observed in His-tagged proteins purified using nickel affinity chromatography have been successfully eliminated.
A more stringent test of the affinity column was performed by adding pure eGFP-2a to an E. coli lysate prior to submitting the protein to the resin. The resin was washed 2x with 0.1 M sodium acetate buffer pH 5, to remove non-specifically bound proteins. The protein was eluted from the resin in the manner described above. Figure 5c, lanes 3 and 4, show that eGFP-2a was obtained in highly pure form from the lysate.
Finally, we applied this procedure to isolate the expressed eGFP-2a protein without prior purification. Since native proteins in E. coli do not contain the sortase sequence, we reasoned that this method could be used to directly isolate a sortase-site containing expressed protein.62 To demonstrate this strategy, eGFP-LPETG was expressed in E. coli. The cells were lysed, and the resulting lysate was subjected to SML by adding SrtA and 4b to the protein mixture. Small molecules were removed, and the lysate was applied to the 2fPBA-agarose resin. After a one-hour incubation, the resin was washed as described to remove loosely associated proteins, followed by overnight incubation with the hydrazine-containing buffer. SDS-PAGE analysis of the eluent showed only eGFP-2a. Thus, successful one-pot protein purification from a protein expressed in E. coli was accomplished. (Figure 5d)
SUMMARY
Examination of a series of α- and β-functionalized hydrazides showed that both hydroxyl and amine functional groups can form stable tricyclic DAB derivatives, allowing rational design of reactive biomolecules for CO-PBA bioorthogonal ligations. Both the aldehyde and the methyl ketone phenylboronic acids yielded tricyclic DAB structures with these hydrazides at neutral pH. The 2fPBA-derived DABs exhibited robust stability in acidic pH, while the AcPBA-derived DABs were less tolerant. Overall, the aldehyde 2fPBA promoted faster reactions and more stable DAB products than the ketone AcPBA, suggesting its preferred use for generating stable bioconjugates.
Widely used methods in molecular biology followed by a sortase mediated ligation enabled the installation of a β-hydroxy hydrazide handle on a model eGFP protein via hydrazine or a polyglycine peptide hydrazide. The peptide hydrazide is conveniently prepared by solid phase peptide synthesis through a well-known protocol and commercially available preloaded resin. Thus, one component of the DAB reaction can be appended to a protein of interest using methods and reagents available in or accessible to most biochemistry laboratories.
The C-terminal serine hydrazide-functionalized protein may be created directly from a precursor protein expressed in E. coli and purified from other components in the cellular milieu using 2fPBA-functionalized resin. The high affinity yet reversible DAB linkage allows for the isolation of highly pure protein from the heterogenous mixture. Cleavage from the resin preserves the hydrazide functionality on the protein, which can then be reused for applications in research and medicine that require homogeneous protein conjugates. Alternative placement of a suitably substituted hydrazide functional group should be accessible using more involved methods of protein modification such as non-canonical amino acid mutagenesis, which would further expand the utility of this coupling reaction.
EXPERIMENTAL SECTION
Synthesis of bifunctional reagents.
Syntheses of the following molecules are described in the Supporting Information: 2fPBA-JF, 2fPBA-TxR, AcPBA-JF and hydrazide derivatives 1c, 4b-c.
Enzymatic modification of eGFP-LPETG with SrtA and hydrazine.
The substrate eGFP-LPETG (30 μM) was allowed to react with the enzyme SrtA (20 μM) and hydrazine (0.2 M) in sortase buffer (50 mM Tris, 150 mM NaCl, 10 mM CaCl2, pH 7.4) at room temperature. After 3 hours, the reaction mixture was subjected to diafiltration using an Amicon Ultra Centrifugal Filter (3K MWCO) to eliminate excess hydrazine, followed by nickel affinity chromatography to remove SrtA. The concentration of the purified product was determined by absorbance of eGFP at 488 nm (ε=55000 M−1 cm−1).
eGFP-1a labeling with 2fPBA-fluorophore.
eGFP-1a (30 μM) was allowed to react with 2fPBA-TxR or 2fPBA-JF (300 μM) in 0.1 M phosphate buffer pH 7 (2% DMSO (v/v)). The 2fPBA-TxR reaction was performed in buffer or in E. coli cellular lysate at room temperature for 3 hours. eGFP-LPETG was treated using the same conditions as a control. The solutions were then subjected to SDS-PAGE. The gel was treated with de-staining solution (50% methanol, 10% acetic acid) overnight to remove unbound fluorophore and photographed under UV irradiation to detect fluorescence of labeled protein. The gel was then stained with Coomassie Brilliant Blue solution (0.1% (w/v)) followed by overnight treatment with de-staining solution and photographed under studio lights. The 2fPBA-JF reaction was performed in buffer and the progress of the reaction was monitored using LC-MS. The stability of the conjugate eGFP-1c was evaluated by storing the labeled sample overnight at room temperature. After 24 hours, LC-MS analysis showed no decomposition of the DAB conjugate. (Figure S14)
Enzymatic modification of eGFP-LPETG with SrtA and pentaglycine peptide (4b, 4c).
The substrate eGFP-LPETG (20 μM) was allowed to react with the SrtA (2 μM) and the nucleophile 4b or 4c (5 mM) in sortase buffer at room temperature. The reaction was complete in 3 hours as assessed by LC-MS. (Figure S15) Unreacted peptide was removed using diafiltration to yield the functionalized eGFP-2a or eGFP-3a.
Labeling reaction of eGFP-2a or eGFP-3a with 2fPBA-JF or 2fPBA-TxR.
eGFP-2a or eGFP-3a (20 μM) was allowed to react with the 2fPBA-fluorophore (200 μM) described for eGFP-1a. Analysis of the conjugate was performed using SDS-PAGE or LC-MS.
Kinetics of labeling of eGFP-2a with 2fPBA-JF or AcPBA-JF.
eGFP-2a (20 μM) was allowed to react with 40 μM (2 eq.) or 200 μM (10 eq.) of 2fPBA-JF or with 200 μM (10 eq.) of AcPBA-JF in 0.1 M phosphate buffer, pH 7 (2% DMSO (v/v)) at room temperature. An aliquot of 20 μL was removed from the reaction at various times (10 minutes, 30 minutes, 1 hour, 2 hours, 3 hours, 6 hours and 24 hours), and analyzed by LC-MS. (Figure 4c, Figure S16–18)
Immobilization of eGFP-2a onto 2fPBA-agarose and elution of immobilized protein.
Functionalized 2fPBA-agarose slurry (90 μL slurry with 30 μL of resin) was loaded onto a spin column and washed three times with 200 μL of 0.1 M phosphate buffer, pH 7. eGFP-2a (15 μM, 100 μL) in 0.1 M phosphate buffer pH 7 or E. coli cellular lysate was introduced to the spin column. eGFP-LPETG was treated using the same conditions as a control. The solution was loaded onto the resin and the column was rotated end-over-end for 10 minutes, at which time no absorbance at 488 nm was detected, indicating the completion of the immobilization of eGFP-2a. The resin turned green after the completion of the reaction. The spin column was centrifuged at 1000xg for 1 minute to remove excess buffer. The resin was then washed three times with 200 μL of 0.1 M sodium acetate buffer pH 5. The immobilized protein was eluted by incubating the resin with 200 μL of 200 mM of hydrazine solution in 0.1 M PBS, pH 6.8. After mixing end-over-end overnight, the column was centrifuged at 1000xg for 2 minutes to collect the eluant. The successful capture of the target protein and its elution were demonstrated by SDS-PAGE. (Figure 5c)
One-pot purification of eGFP-LPETG from E. coli cellular lysate using 2fPBA functionalized solid support.
Cells expressing eGFP-LPETG were lysed according to protocol described in SI followed by buffer exchange into sortase buffer. Cellular lysate containing eGFP-LPETG (15 μM, 100 μL) was allowed to react with 4b (3.75 mM) in the presence of SrtA (1.5 μM) at room temperature for 3 hours. Following diafiltration to remove excess 4b, the solution (100 μL) was loaded onto a spin column containing functionalized 2fPBA-agarose slurry (90 μL slurry with 30 μL of resin) and the column was rotated end-over-end for 1 hour. The completion of the immobilization was indicated by the UV absorbance of the supernatant at 488 nm reading zero (negligible). The spin column was centrifuged at 1000xg for 1 minute to remove unbound proteins. The resin was then washed three times with 200 μL 0.1 M phosphate buffer pH 7, containing 50 mM hydrazine and 1 M KCl. The immobilized protein was eluted from the resin as described above and analyzed by SDS-PAGE and LC-MS.
Supplementary Material
ACKNOWLEDGMENTS
Funding was provided by NIH Grant 1R15CA227747 and the SUNY Research Foundation Accelerator Fund. The authors acknowledge Professor David R. Liu for the generous donation of the plasmids used in this study. The Regional NMR Facility (600 MHz instrument) at Binghamton University is supported by NSF (CHE-0922815). The Dr. G. Clifford & Florence B. Decker Foundation is acknowledged for the generous donation of some of the equipment used in this work. The authors would like to thank Professor L. Nathan Tumey for instrumental access and helpful discussions regarding mass spectrometry, Professor Brian Callahan for instrumental access and helpful discussions regarding protein expression and purification, Tak Ian Chio for helpful discussions, Dr. Juergen Schulte for help with NMR experiments and Mr. David Tuttle for photography. We thank Austin Ablicki, Joshua Shaw and Stella Ebner for their assistance in protein expression and purification and Kendrick Choi for the synthesis of AcPBA-JF.
Footnotes
The authors declare the following competing financial interest(s): S.L.B is an inventor on a patent pertaining to this chemistry. The other authors declare no competing financial interest.
Supplementary methods, including protein expression, synthesis of reagents and additional details of experimental procedures; supplementary figures; and spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
CCDC 1942735–1942737 contains the supplementary crystallographic data for this paper. The data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/structures.
REFERENCES
- (1).Sletten EM; Bertozzi CR Bioorthogonal Chemistry: Fishing for Selectivity in a Sea of Functionality. Angew. Chemie Int. Ed 2009, 48, 6974–6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Lang K; Chin JW Bioorthogonal Reactions for Labeling Proteins. ACS Chem. Biol 2014, 9, 16–20. [DOI] [PubMed] [Google Scholar]
- (3).Devaraj NK The Future of Bioorthogonal Chemistry. ACS Cent. Sci 2018, 4, 952–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Debets MF; Van Berkel SS; Dommerholt J; Dirks AJ; Rutjes FPJT; Van Delft FL Bioconjugation with Strained Alkenes and Alkynes. Acc. Chem. Res 2011, 44, 805–815. [DOI] [PubMed] [Google Scholar]
- (5).Knall A-C; Slugovc C Inverse Electron Demand Diels–Alder (IEDDA)-Initiated Conjugation: A (High) Potential Click Chemistry Scheme. Chem. Soc. Rev 2013, 42, 5131–5142. [DOI] [PubMed] [Google Scholar]
- (6).Braun AC; Gutmann M; Lühmann T; Meinel L Bioorthogonal Strategies for Site-Directed Decoration of Biomaterials with Therapeutic Proteins. J. Controlled Release 2018, 273, 68–85. [DOI] [PubMed] [Google Scholar]
- (7).Meyer JP; Adumeau P; Lewis JS; Zeglis BM Click Chemistry and Radiochemistry: The First 10 Years. Bioconjugate Chem. 2016, 27, 2791–2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Jiang Y; Chen J; Deng C; Suuronen EJ; Zhong Z Click Hydrogels, Microgels and Nanogels: Emerging Platforms for Drug Delivery and Tissue Engineering. Biomaterials. 2014, 35, 4969–4985. [DOI] [PubMed] [Google Scholar]
- (9).Siegrist MS; Swarts BM; Fox DM; Lim SA; Bertozzi CR Illumination of Growth, Division and Secretion by Metabolic Labeling of the Bacterial Cell Surface. FEMS Microbiol. Rev 2015, 39, 184–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Narayanam MK; Liang Y; Houk KN; Murphy JM Discovery of New Mutually Orthogonal Bioorthogonal Cycloaddition Pairs through Computational Screening. Chem. Sci 2016, 7,1257–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Row RD; Prescher JA Constructing New Bioorthogonal Reagents and Reactions. Acc. Chem. Res 2018, 51, 1073–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Kölmel DK; Kool ET Oximes and Hydrazones in Bioconjugation: Mechanism and Catalysis. Chem. Rev 2017, 117, 10358–10376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Shao J; Tam JP Unprotected Peptides as Building Blocks for the Synthesis of Peptide Dendrimers with Oxime, Hydrazone, and Thiazolidine Linkages. J. Am. Chem. Soc 1995, 117, 3893–3899. [Google Scholar]
- (14).Agten SM; Dawson PE; Hackeng TM Oxime Conjugation in Protein Chemistry: From Carbonyl Incorporation to Nucleophilic Catalysis. J. Pept. Sci 2016, 22, 271–279. [DOI] [PubMed] [Google Scholar]
- (15).Anderson BM; Jencks WP The Effect of Structure on Reactivity in Semicarbazone Formation. J. Am. Chem. Soc 1960, 82, 1773–1777. [Google Scholar]
- (16).Cordes EH; Jencks WP General Acid Catalysis of Semicarbazone Formation. J. Am. Chem. Soc 1962, 84, 4319–4328. [Google Scholar]
- (17).Kool ET; Crisalli P; Chan KM Fast Alpha Nucleophiles: Structures That Undergo Rapid Hydrazone/Oxime Formation at Neutral pH. Org. Lett 2014, 16, 1454–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Kool ET; Park D; Crisalli P Fast Hydrazone Reactants: Electronic and Acid/Base Effects Strongly Influence Rate at Biological pH. J. Am. Chem. Soc 2013, 135, 17663–17666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Xu Y; Xu L; Xia Y; Guan CJ; Guo QX; Fu Y; Wang C; Li YM Fast and Catalyst-Free Hydrazone Ligation via Ortho-Halo-Substituted Benzaldehydes for Protein C-Terminal Labeling at Neutral PH. Chem. Commun 2015, 51, 13189–13192. [DOI] [PubMed] [Google Scholar]
- (20).Xu Y; Wang Y; Liu P; Chu GC; Xu H; Li YM; Wang J; Shi J Catalyst Free Hydrazone Ligation for Protein Labeling and Modification Using Electron-Deficient Benzaldehyde Reagents. Org. Biomol. Chem 2018, 16, 7036–7040. [DOI] [PubMed] [Google Scholar]
- (21).Schmidt P; Stress C; Gillingham D Boronic Acids Facilitate Rapid Oxime Condensations at Neutral pH. Chem. Sci 2015, 6, 3329–3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Bandyopadhyay A; Gao J Iminoboronate Formation Leads to Fast and Reversible Conjugation Chemistry of α-Nucleophiles at Neutral pH. Chem. - A Eur. J 2015, 21, 14748–14752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Dilek O; Lei Z; Mukherjee K; Bane S Rapid Formation of a Stable Boron-Nitrogen Heterocycle in Dilute, Neutral Aqueous Solution for Bioorthogonal Coupling Reactions. Chem. Commun 2015, 51, 16992–16995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Dewar MJS; Dougherty RC Boron-Containing Analogs of Isoquinoline. J. Am. Chem. Soc 1962, 84, 2648–2649. [Google Scholar]
- (25).Gu H; Chio TI; Lei Z; Staples RJ; Hirschi JS; Bane S Formation of Hydrazones and Stabilized Boron-Nitrogen Heterocycles in Aqueous Solution from Carbohydrazides and Ortho-Formylphenylboronic Acids. Org. Biomol. Chem 2017, 15, 7543–7548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Chio TI; Gu H; Mukherjee K; Tumey LN; Bane SL Site-Specific Bioconjugation and Multi-Bioorthogonal Labeling via Rapid Formation of a Boron-Nitrogen Heterocycle. Bioconjugate Chem. 2019, 30, 1554–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Thom J; Anderson D; McGregor J; Cotton G Recombinant Protein Hydrazides: Application to Site-Specific Protein PEGylation. Bioconjugate Chem. 2011, 22, 1017–1020. [DOI] [PubMed] [Google Scholar]
- (28).Zheng J-S; Tang S; Qi Y-K; Wang Z-P; Liu L Chemical Synthesis of Proteins Using Peptide Hydrazides as Thioester Surrogates. Nat. Protoc 2013, 8, 2483–2495. [DOI] [PubMed] [Google Scholar]
- (29).Li YM; Yang MY; Huang YC; Li YT; Chen PR; Liu L Ligation of Expressed Protein α-Hydrazides via Genetic Incorporation of an α-Hydroxy Acid. ACS Chem. Biol 2012, 7, 1015–1022. [DOI] [PubMed] [Google Scholar]
- (30).Huang YC; Fang GM; Liu L Chemical Synthesis of Proteins Using Hydrazide Intermediates. Natl. Sci. Rev 2016, 3, 107–116. [Google Scholar]
- (31).Fang GM; Li YM; Shen F; Huang YC; Li J. Bin; Lin Y; Cui HK; Liu L Protein Chemical Synthesis by Ligation of Peptide Hydrazides. Angew. Chemie - Int. Ed 2011, 50, 7645–7649. [DOI] [PubMed] [Google Scholar]
- (32).Zheng JS; Tang S; Huang YC; Liu L Development of New Thioester Equivalents for Protein Chemical Synthesis. Acc. Chem. Res 2013, 46, 2475–2484. [DOI] [PubMed] [Google Scholar]
- (33).Tsukiji S; Nagamune T Sortase-Mediated Ligation: A Gift from Gram-Positive Bacteria to Protein Engineering. ChemBioChem 2009, 10, 787–798. [DOI] [PubMed] [Google Scholar]
- (34).Ritzefeld M Sortagging: A Robust and Efficient Chemoenzymatic Ligation Strategy. Chem. - A Eur. J 2014, 20, 8516–8529. [DOI] [PubMed] [Google Scholar]
- (35).Chen L; Cohen J; Song X; Zhao A; Ye Z; Feulner CJ; Doonan P; Somers W; Lin L; Chen PR Improved Variants of SrtA for Site-Specific Conjugation on Antibodies and Proteins with High Efficiency. Sci. Rep 2016, 6, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Schmohl L; Bierlmeier J; von Kügelgen N; Kurz L; Reis P; Barthels F; Mach P; Schutkowski M; Freund C; Schwarzer D Identification of Sortase Substrates by Specificity Profiling. Bioorganic Med. Chem 2017, 25, 5002–5007. [DOI] [PubMed] [Google Scholar]
- (37).Nikghalb KD; Horvath NM; Prelesnik JL; Banks OGB; Filipov PA; Row RD; Roark TJ; Antos JM Expanding the Scope of Sortase-Mediated Ligations by Using Sortase Homologues. ChemBioChem 2018, 19, 185–195. [DOI] [PubMed] [Google Scholar]
- (38).Mao H; Hart SA; Schink A; Pollok BA Sortase-Mediated Protein Ligation: A New Method for Protein Engineering. J. Am. Chem. Soc 2004, 126, 2670–2671. [DOI] [PubMed] [Google Scholar]
- (39).Chen I; Dorr BM; Liu DR A General Strategy for the Evolution of Bond-Forming Enzymes Using Yeast Display. Proc. Natl. Acad. Sci 2011, 108, 11399–11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Ham HO; Qu Z; Haller CA; Dorr BM; Dai E; Kim W; Liu DR; Chaikof EL In Situ Regeneration of Bioactive Coatings Enabled by an Evolved Staphylococcus Aureus Sortase A. Nat. Commun 2016, 7:11140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Li YM; Li YT; Pan M; Kong XQ; Huang YC; Hong ZY; Liu L Irreversible Site-Specific Hydrazinolysis of Proteins by Use of Sortase. Angew. Chemie - Int. Ed 2014, 53, 2198–2202. [DOI] [PubMed] [Google Scholar]
- (42).Lopes RMRM; Ventura AE; Silva LC; Faustino H; Gois PMPN,O-Iminoboronates: Reversible Iminoboronates with Improved Stability for Cancer Cells Targeted Delivery. Chem. - A Eur. J 2018, 24, 12495–12499. [DOI] [PubMed] [Google Scholar]
- (43).Li M; Tao Y; Shu Y; LaRochelle JR; Steinauer A; Thompson D; Schepartz A; Chen ZY; Liu DR Discovery and Characterization of a Peptide That Enhances Endosomal Escape of Delivered Proteins in Vitro and in Vivo. J. Am. Chem. Soc 2015, 137, 14084–14093. [DOI] [PubMed] [Google Scholar]
- (44).Cambray S; Gao J Versatile Bioconjugation Chemistries of Ortho-Boronyl Aryl Ketones and Aldehydes. Acc. Chem. Res 2018, 51, 2198–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).António JPM; Russo R; Carvalho CP; Cal PMSD; Gois PMP Boronic Acids as Building Blocks for the Construction of Therapeutically Useful Bioconjugates. Chem. Soc. Rev 2019, 48, 3513–3536. [DOI] [PubMed] [Google Scholar]
- (46).Bandyopadhyay A; Cambray S; Gao J Fast Diazaborine Formation of Semicarbazide Enables Facile Labeling of Bacterial Pathogens. J. Am. Chem. Soc 2017, 139, 871–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Gillingham D The Role of Boronic Acids in Accelerating Condensation Reactions of α-Effect Amines with Carbonyls. Org. Biomol. Chem 2016, 14, 7606–7609. [DOI] [PubMed] [Google Scholar]
- (48).Cal PMSD; Vicente JB; Pires E; Coelho AV; Veiros LF; Cordeiro C; Gois PMP Iminoboronates: A New Strategy for Reversible Protein Modification. J. Am. Chem. Soc 2012, 134, 10299–10305. [DOI] [PubMed] [Google Scholar]
- (49).Zhu L; Shabbir SH; Gray M; Lynch VM; Sorey S; Anslyn EV A Structural Investigation of the N-B Interaction in an o-(N,N-Dialkylaminomethyl)Arylboronate System. J. Am. Chem. Soc 2006, 128, 1222–1232. [DOI] [PubMed] [Google Scholar]
- (50).Kanichar D; Roppiyakuda L; Kosmowska E; Faust MA; Tran KP; Chow F; Buglo E; Groziak MP; Sarina EA; Olmstead MM; et al. Synthesis, Characterization, and Antibacterial Activity of Structurally Complex 2-Acylated 2,3,1-Benzodiazaborines and Related Compounds. Chem. Biodivers 2014, 11, 1381–1397. [DOI] [PubMed] [Google Scholar]
- (51).Tan XL; Pan M; Zheng Y; Gao S; Liang LJ; Li YM Sortase-Mediated Chemical Protein Synthesis Reveals the Bidentate Binding of Bisphosphorylated P62 with K63 Diubiquitin. Chem. Sci 2017, 8, 6881–6887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Williamson DJ; Fascione MA; Webb ME; Turnbull WB Efficient N-Terminal Labeling of Proteins by Use of Sortase. Angew. Chemie - Int. Ed 2012, 51, 9377–9380. [DOI] [PubMed] [Google Scholar]
- (53).Möhlmann S; Mahlert C; Greven S; Scholz P; Harrenga A In Vitro Sortagging of an Antibody Fab Fragment: Overcoming Unproductive Reactions of Sortase with Water and Lysine Side Chains. ChemBioChem 2011, 12, 1774–1780. [DOI] [PubMed] [Google Scholar]
- (54).Guimaraes CP; Witte MD; Theile CS; Bozkurt G; Kundrat L; Blom AEM; Ploegh HL Site-Specific C-Terminal and Internal Loop Labeling of Proteins Using Sortase-Mediated Reactions. Nat. Protoc 2013, 8, 1787–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Theile CS; Theile CS; Witte MD; Blom AEM; Kundrat L; Ploegh HL; Guimaraes CP Site-Specific N-Terminal Labeling of Proteins Using Sortase-Mediated Reactions. Nat. Protoc 2013, 8, 1800–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Frankel BA; Kruger RG; Robinson DE; Kelleher NL; McCafferty DG Staphylococcus Aureus Sortase Transpeptidase SrtA: Insight into the Kinetic Mechanism and Evidence for a Reverse Protonation Catalytic Mechanism. Biochemistry 2005, 44, 11188–11200. [DOI] [PubMed] [Google Scholar]
- (57).Huang X; Aulabaugh A; Ding W; Kapoor B; Alksne L; Tabei K; Ellestad G Kinetic Mechanism of Staphylococcus Aureus Sortase SrtA. Biochemistry 2003, 42, 11307–11315. [DOI] [PubMed] [Google Scholar]
- (58).Oliveira BL; Guo Z; Bernardes GJL Inverse Electron Demand Diels-Alder Reactions in Chemical Biology. Chem. Soc. Rev 2017, 46, 4895–4950. [DOI] [PubMed] [Google Scholar]
- (59).Ni W; Kaur G; Springsteen G; Wang B; Franzen S Regulating the Fluorescence Intensity of an Anthracene Boronic Acid System: A B-N Bond or a Hydrolysis Mechanism? Bioorg. Chem 2004, 32, 571–581. [DOI] [PubMed] [Google Scholar]
- (60).Collins BE; Sorey S; Hargrove AE; Shabbir SH; Lynch VM; Anslyn EV Probing Intramolecular B-N Interactions in Ortho-Aminomethyl Arylboronic Acids. J. Org. Chem 2009, 74, 4055–4060. [DOI] [PubMed] [Google Scholar]
- (61).Gutiérrez-Moreno NJ; Medrano F; Yatsimirsky AK Schiff Base Formation and Recognition of Amino Sugars, Aminoglycosides and Biological Polyamines by 2-Formyl Phenylboronic Acid in Aqueous Solution. Org. Biomol. Chem 2012, 10, 6960–6972. [DOI] [PubMed] [Google Scholar]
- (62).Parthasarathy R; Subramanian S; Boder ET Sortase A as a Novel Molecular “Stapler” for Sequence-Specific Protein Conjugation. Bioconjugate Chem. 2007, 18, 469–476. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.