

Abstract
Automated genotyping of drug-resistant Mycobacterium tuberculosis (MTB) directly from sputum is challenging for three primary reasons. First, the sample matrix – sputum – is highly viscous and heterogeneous, posing a challenge for sample processing. Second, acid-fast MTB bacilli are difficult to lyse. And third, there are hundreds of MTB mutations that confer drug resistance. An additional constraint is that MTB is most prevalent where test affordability is paramount. We address the challenge of sample homogenization and cell lysis using magnetic rotation of an external magnet – at high (5000) rpm – to induce rotation of a disposable stir disc that causes chaotic mixing of glass beads (“MagVor”). Nucleic acid is purified using a pipette tip with an embedded matrix that isolates nucleic acid (“TruTip”). We address the challenge of cost and genotyping multiple mutations using 203 porous three-dimensional gel elements printed on a film substrate and enclosed in a microfluidic laminate assembly (“Lab-on-a-Film”). This Lab-on-a-Film Assembly (LFA) serves as a platform for amplification, hybridization, washing and fluorescent imaging, while maintaining a closed format to prevent amplicon contamination of the workspace. We integrated and automated MagVor homogenization, TruTip purification, and LFA amplification in a multi-sample, sputum-to-genotype system. Using this system, we report detection down to 43 cfu/mL of MTB bacilli from raw sputum.
Keywords: Microfluidic, tuberculosis, drug-resistance, molecular diagnostic, sample-to-answer
Graphical Abstract
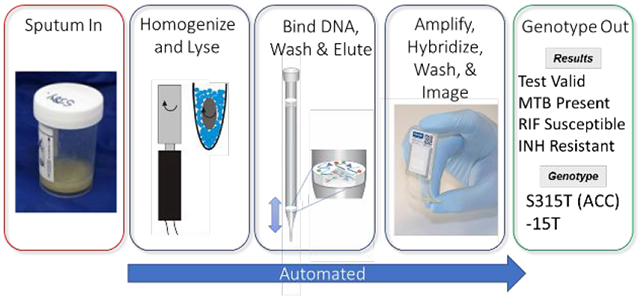
Evidence of tuberculosis (TB) dates back 5000 years; mummies were discovered to have lesions caused by Mycobacterium tuberculosis (MTB).1 At the beginning of the 19th century in Europe, as many as 1 in 333 people died from TB.1 Today, TB still ranks in the top ten worldwide causes of death and now ranks ahead of HIV.2 Perhaps, most alarming is the global prevalence of drug-resistant TB (DR-TB): each year there is more than half a million new cases of multi drug-resistant TB (MDR-TB) – defined as MTB resistance to isoniazid (INH) and rifampicin (RIF).2
The differences between treating drug-susceptible vs drug-resistant TB is dramatic. In the United States, treatment costs $17k for patients with drug-susceptible TB, and for patients with MDR-TB, treatment costs $134k – almost ten times more.3 The difference in treatment duration is also dramatic: 6 months for patients infected with drug-susceptible TB and 18 months for patients infected with MDR-TB.4 Furthermore, consider the following difference in treatment duration for those infected with INH-resistant (RIF-susceptible) MTB as compared with those infected with RIF-resistant (INH-susceptible) MTB. The World Health Organization (WHO) guidelines for treating the former patients include a short-course (6 month) regimen of rifampin, ethambutol, pyrazinamide, and levofloxacin. The short-course is not recommended for treating patients infected with RIF-resistant (INH-susceptible) MTB or MDR-TB. Rather, the guidelines for these patients are to use a more complex, longer course (18 month) regimen. This drastic difference in treatment courses emphasizes the rationale for WHO’s encouragement “towards universal testing of both isoniazid and rifampicin resistance.”4
Immediate INH and RIF resistance detection is also reported to be more cost effective than either immediate MTB diagnosis followed by delayed INH and RIF resistance testing or immediate RIF-resistance detection followed by delayed INH-resistance testing – even in regions of low levels of drug resistance (MDR-TB > 1%).5
The prevalence of RIF susceptible, INH resistant MTB is notable. WHO reports this prevalence as 7.1% in new TB cases and 7.9% in previously-treated cases.2 One study in Kuwait reports it at 163/264 MTB isolates (67%).6 Immediately detecting MTB resistance to RIF – and INH – is therefore important. Identifying the mutations that cause resistance – particularly for INH – has important clinical implications. For example, mutations in the inhA gene are associated with low levels of INH resistance, and mutations in the katG gene are associated with high levels of INH resistance.7 Lempens et al. report that for strains with the katG Ser315Thr mutation, the minimum inhibitory concentration increased three-fold when inhA c-15t is also present, further suggesting that the levels of INH resistance are dependent on the genotype.8
Developing molecular tests for MDR-TB is inherently complex because: (1) the sample type (typically sputum) is highly viscous and heterogenous, and thus extracting and purifying microbial DNA presents significant challenges, (2) the thick, waxy, lipid-rich cell wall of the MTB bacilli is difficult to lyse, (3) there are hundreds of genetic mutations that confer TB-drug resistance, and (4) the market is cost sensitive because those infected are often economically disadvantaged.
The dissemination of over 20 million Cepheid Xpert MTB/RIF tests has certainly contributed to the steady decline in deaths caused by TB.9 This test, however, is limited to detection of MTB and RIF resistance, and it uses a method of “sloppy molecular beacons”, which does not report specific mutations.10,11 Another XDR-TB test is in development, which includes markers for INH but does not include markers for RIF, thus it is intended as a reflex test to their MTB/RIF test.12,13 Ao et al. also describes an MTB/RIF test, which reports mutations, but it does not include INH resistance.14 There are a number of molecular MDR-TB tests that are high complexity (do not include integrated sample preparation) available from: Hain (LPA15 and Fluorotype16), Capital Bio17, InSilixa18, Veredus19, Abbott20, and Roche.21 The number of MDR-TB tests that include integrated sample preparation are more limited. These include: Becton Dickinson MAX MDR-TB assay22 and published works such as that described in the review by Mani et al.23 A limitation with real-time PCR systems such as the Xpert and the BD Max is that they detect but do not report mutations because of the limited number of optical channels (i.e, a mismatch between the target and the reporter probe results in a mutant call without identifying the specific mutation).
Sequencing methods hold promise as being able to achieve this goal. Traditionally, whole genome sequencing requires removal of contaminating or human DNA, which is often accomplished by culturing the MTB bacilli.24 Nimmo et al use a SureSelect protocol, which requires molecular biologists to perform numerous protocol steps, to isolate the MTB bacilli prior to sequencing.25 Votintseva et al. developed a modified MolYsis Basic5 kit (Molzyme, Germany) that digests human DNA and then lyses the bacilli and extracts the MTB DNA for library preparation and sequencing using either Illumina’s MiSeq/MiniSeq or Oxford Nanopore Technology’s Minion.26 The protocol requires a number of manual steps such as centrifugation, pelleting, and removing the supernatant, which limit the applicability to high complexity laboratories with skilled technicians. Targeted next generation sequencing can possibly avoid the requirement for MTB DNA enrichment by using a multiplex PCR reaction. However, after amplification the PCR tube is opened for the subsequent library preparation steps, introducing a contamination risk of the laboratory and other adjacent samples.
Despite the tremendous progress in MTB detection and identification, an affordable automated sputum-to-genotype system for detection and reporting MDR-TB mutations, deletions, and insertion elements remains an unmet clinical need. 2,4
We previously reported an automated nucleic acid extraction method,27 MDR-TB detection using a manually-operated microfluidic consumable,28 and the translation of this device to a film substrate.29 Here we report the integration of these approaches to address this unmet need.
MATERIALS AND METHODS
System Description
The analyzer, shown in Figure 1A, consists of sub-systems for: sample preparation, thermal cycling, and imaging. The consumables are loaded together on a moveable stage (carriage) to allow them to travel to the appropriate location and establish the interfaces shown in Figure 1B. The automated protocol initiates with MagVor for improved lysis and homogenization (beyond an initial liquefaction and heat kill step); the effect of this improvement is reported in Thakore et al.27 The subsequent steps include: purification with TruTip, which also serve as the liquid handling pipette tips; transfer sample to heater tubes for the heated DNA binding step; wash, air dry, and elute from the DNA binding matrix in the TruTips using reagents in a deep-well plate; mix elution with PCR master mix; transfer mixture to the LFA; thermal cycle; wash the gel element array; image and analyze; and eject the TruTips.
Figure 1.
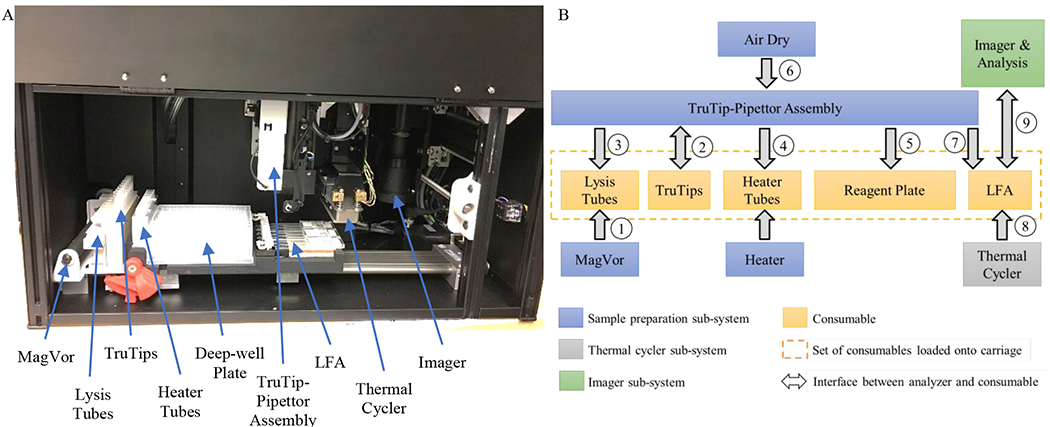
A. Photograph of the interior of the analyzer depicting the MagVor for lysis and homogenization, the TruTip for purification, the deep-well plate so that all liquids are part of the consumable, pipetting station, the Lab-on-a-Film Assembly (LFA), the thermal cycler, and the imager. B. Illustration of the interfaces of the analyzer sub-systems with the consumables; the order of the analyzer operations is numbered. (Note, Step 6 - Air Drying occurs at Column 7 of the deep-well plate). The carriage moves the consumable to each interface on the analyzer.
As illustrated, the sample preparation sub-system includes sub-assemblies for rotating the MagVor magnet, aspirating and dispensing from and into the TruTips, heating to improve binding to the porous matrix embedded in the TruTip, and air drying the TruTips in order to remove residual organics. We previously reported this sample preparation sub-system in the form factor of a bench-top workstation intended for nucleic acid isolation for multiple down-stream analysis methods such as sequencing, real-time PCR and microarrays.30 The details of the sub-system can be found in that publication. Briefly, it consists of a method of Magnetically-induced Vortexing (“MagVor”) by rotating a diametrically polarized cylindrical neodymium magnet. The rotation of this magnet induces a changing magnetic field inside of a tube (lysis tube) that includes a magnetic disc. The changing magnetic field causes the disc to rotate and stirs the glass beads, which serve to homogenize the sample and lyse the MTB bacilli.27 We custom designed the lysis tube for this application to allow for: a fill volume up to 600 μL, a pierceable cap that is hinged (connected) to the tube, and conformance to 9 mm spacing to correspond with the deep-well reagent plate. An injection-molded (Icomold, Holland, OH) interconnected strip of heater tubes with a square top slides into an aluminum block that has a “U-shaped” profile, allowing intimate contact with two side walls and the bottom of the tubes. The aluminum block includes an integrated 50W 24 V DC cartridge heater (NextThermal, Battle Creek, MI) that has a 6.35 mm diameter and a 108 mm length and an NTC 10kΩ thermistor (#235-1399-ND, Digi-key, Thief River Falls, Minnesota) for feedback control and another one that serves as a high limit cutoff. The thermistors are connected to an embedded controller (#sbRIO-9627, National Instruments, Austin, TX), which provides Proportional, Integral, Differential (PID) control through a LabVIEW (National Instruments) software interface. The setup is then calibrated to the liquid temperature in the heater tubes. Following the heated DNA binding step, the matrix in the TruTip is washed and air dried with an air pump that connects to a custom designed linear selection valve to allow air flow through one TruTip at a time to ensure the same flow across each matrix. As described previously,27,30 the system also includes a pipetting system for aspirating and dispensing liquids into and out of the TruTip. The TruTip matrix is recessed 26.5 mm from the distal end of the pipette tip allowing liquid transfer up to 180 μL without contacting the matrix. Thus, the TruTip also serves as a standard liquid transferring pipette tip. A stage translates a carriage in the X direction while the TruTips move in a Z direction and the imaging system translates in a Y direction across each of the chambers.
The analyzer also includes a thermal cycler which consists of a heat spreader with embedded Peltier thermal electric coolers (#1MC06-126-05AN, TEC Microsystems, Berlin, DE), and three integrated thermistors at the edges and the center of the spreader. The spreader dimensions (29 mm x 86 mm x 2.8 mm) span the entire volume for all reaction chambers. A linear actuator (#L12-50-210-12-P, Actuonix, British Columbia, CA) translates the thermal cycler assembly down onto the LFA. A custom circuit board serves as the connector interface to the thermal cycler assembly, which also includes a 5 cfm 40 mm height x 40 mm width x 9.9 mm depth fan (#1939K43, McMaster-Carr, Elmhurst, IL) and a custom heat sink. Control of the thermal cycler is accomplished with a Thermal Cycler Module (#TCM-M207, Easy Shining, Chengdu, China).
The imager sub-system, previously a stand-alone unit described in Linger et al., consists of an excitation and an emission optical train.28 The emission optical train illuminates the array from an “oblique” angle using a mirror to direct the collimated 530 nm LED light at approximately a 60° incident angle. The emission optical train is perpendicular to the gel element array with a 29 mm focal length (distance from the objective lens to the object plane) to allow clearance of the carriage and consumables without interference. The emission optical train includes a macro-fluorescence lens, collimating lenses, and a CMOS camera.
A segregated electrical control box supplies power and controls to the analyzer. A laptop computer serves as the Graphical User Interface for the user. The Labview program provides control and measurement of all sub-systems, allows user-defined scripts, and automates imaging and analysis so that once the user “executes all,” the system operates automously without interuption.
Consumables
Figure 2 shows the consumables, which includes lysis tubes (#402-00100, Akonni Biosystems, Frederick, MD), a heater strip (#402-002, Akonni Biosystems, Frederick, MD), 1.2 mL SPT TruTips (#302-80021, Akonni Biosystems, Frederick, MD), a deep well plate (#1896-2110, USA Scientific, Orlando, FL) filled with reagents, and an LFA. All of these components were described in Thakore et al. except for the LFA.27 The LFA, shown in the Figure 3 inset, consists of a Lab-on-a-Film consumable for six tests, described in Kukhtin et al., and a housing of inlet ports.29 Briefly, the LFA consists of an array of 203 gel elements31 printed on polyester substrates, a spacer that circumscribes the array and forms the amplification and hybridization chamber, and a hydrophilic cover film, described in Cooney et al.32 Additionally, a waste chamber is attached to the LFA. The inlet port housing consists of pierceable septa (#AC052, MiniValve, Netherlands). The LFA design has a capacity for up to 8 chambers, but only 6 chambers were used for this study because our current printing infrastructure (platens, UV curing system, and wash bath) are configured for 25 mm x 75 mm film substrates. Thus, two arrays were printed on each of three film substrates. We used the PCR master mix and the array map, which includes probes for 37 mutations, deletions, or insertion elements, reported in Linger et al.28 A polyvinylchloride insulation foam (#9318K64, McMaster-Carr) with (black) tape (#T743, Thorlabs, Newton, NJ) (for reducing the background during image capture) was inserted between the LFA and its receptacle on the carriage.
Figure 2.
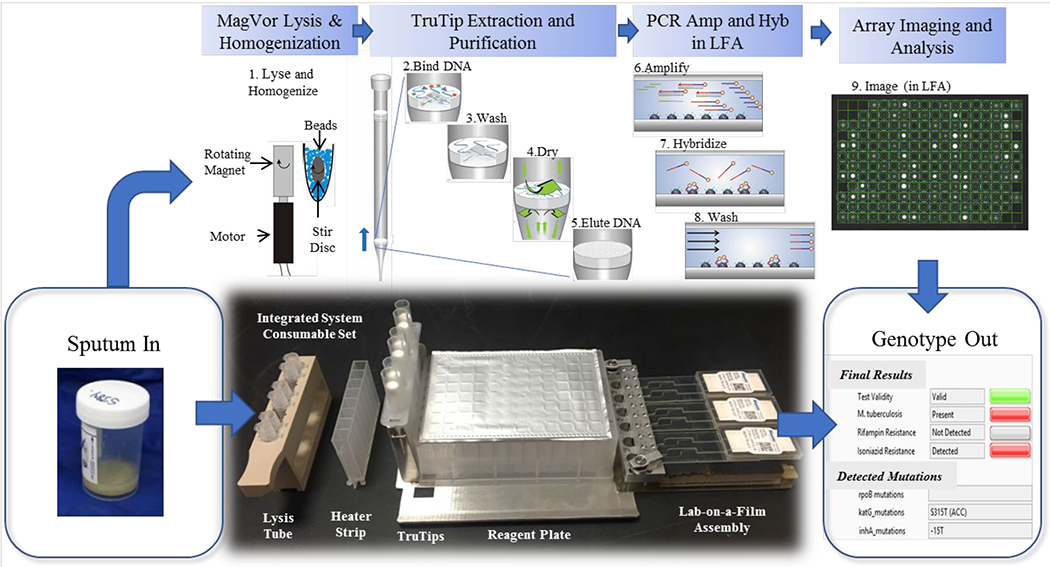
Layout of the consumable for 6 samples in which the following steps occur. (1) Lysis and homogenization occur in lysis tubes, which include a magnetized stir disc and glass beads. (2) The TruTip aspirates the sample mixed with a binding buffer so that it flows through the pores of the matrix in the tip resulting in DNA bound to the matrix. (3) The porous matrix is washed to remove the impurities. (4) The matrix is dried with air. (5) The bound DNA is eluted into an elution buffer. (6) The purified DNA is amplified with an asymmetric PCR reaction. (7) The product (with fluorescent labels) hybridize to the gel elements. (8) The gel elements are washed to remove unbound product. And, (9) an image of the array is captured and analyzed.
Figure 3.
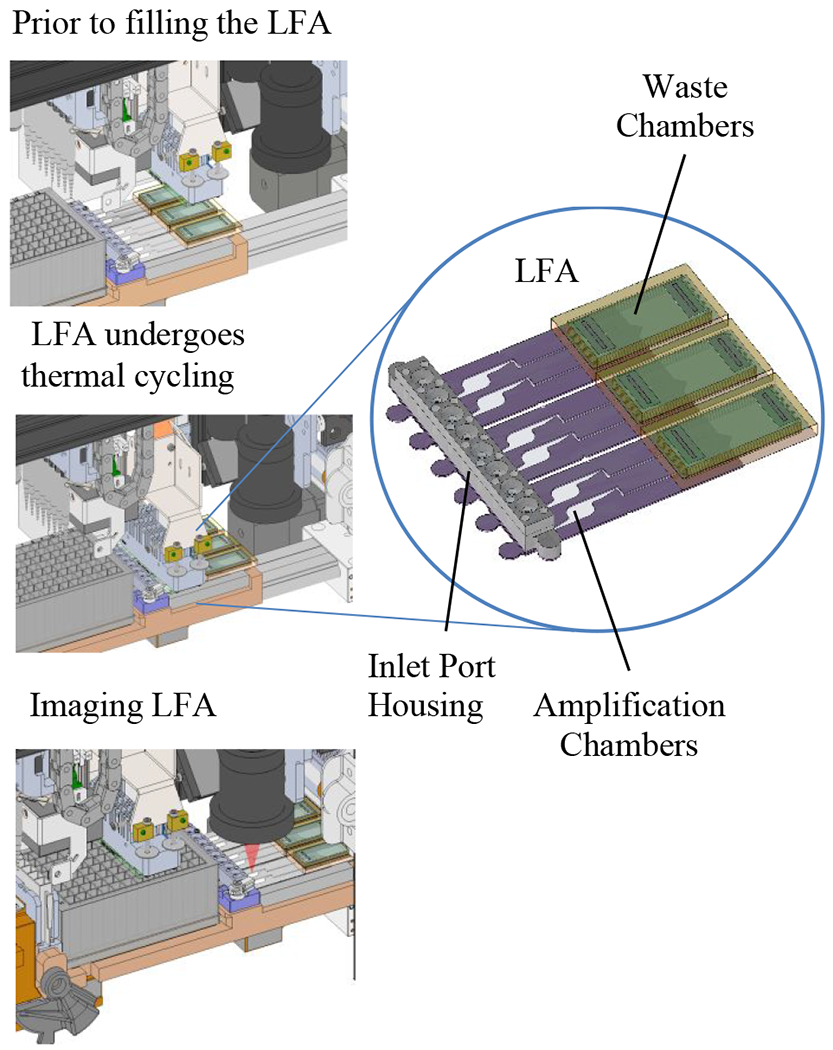
Illustration of the LFA on the system with the thermal cycler in the (1) “up” (disengaged) position, (2) in the “down” (engaged) position, and (3) with the imager capable of individually imaging and analyzing each array of gel elements.
Sample Collection and Preparation
Two sample types were used in these studies: purified MTB DNA and F137Ra bacilli with the latter spiked in MTB-negative sputa. The DNA was either purchased from ATCC (#25177D-5, Manassas, VA), or it was extracted from heat-killed MTB bacilli, provided by the Special Programme for Research and Training in Tropical Diseases (TDR) Tuberculosis Strain Bank (now integrated under BCCM/TIM).33 The extraction procedure consisted of lysis using a BD Geneohm Lysis Kit (#441243 Becton Dickinson Sparks, MD) followed by purification with a DNA Mini Kit (#51304 Qiagen, Hilden, DE).
De-identified sputum samples were purchased from BioIVT (Westbury, NY) and pooled and tested to ensure it was MTB-negative using our real time IS6110 MTB PCR assay described in Thakore et al.27
H37Ra bacilli (#25177, ATCC) were grown in culture and enumerated with plate counting as described previously.27 As described in Thakore et al., liquefaction buffer was manually pipetted at a ratio of 8 parts liquefaction buffer to 100 parts sputum.27 The spiked samples were prepared by manually pipetting 972 μL (9 parts) of the pre-liquefied sputa with 108 μL (1 part) of heat-killed (5 min at 90°C) H37Ra cells. The final titer concentrations were: 4000 cfu/mL, 1200 cfu/mL, 400 cfu/mL, 130 cfu/mL, and 43 cfu/mL in 1.08 mL aliquots of sputa.
The subsequent automated “on system” protocols are shown in Table 1. The sputum-to-genotype protocol requires about 8 hours for the full protocol shown in Table 1 and additional initialization, stage movement and wait steps (not shown). The automated analysis algorithm was the same as that described previously.28
Table 1.
Automated extraction protocol on the system for sputum-to-genotype testing.
| Operation | Programmed Parameter(s) |
|---|---|
| Sample homogenization | Rotate MagVor at 4950 rpm. 10 min * |
| Add sample to lysis buffer | Add 250 μL of sample (in two steps; 125 μL per transfer) to the 380 μL of lysis buffer in the heater strip. ~2.5 min |
| Heat incubation | 10 min at 56°C |
| Add 95% ethanol | 500 μL. ~1 min |
| Binding Step | 20 pipetting cyclest†; 20 min |
| TruTip Wash 1 | 10 pipetting cycles; 7 min |
| TruTip Wash 2 | 5 pipetting cycles; ~2.5 min |
| TruTip Wash 3 | 5 pipetting cycles; ~2.5 min |
| Air dry TruTip matrix | 90 sec forced air per TruTip; 10 min |
| Elute purified DNA | 100 μL, 10 pipetting cycles; ~2 min |
| Add DNA to Master mix | Transfer 40 μL of DNA solution from one column on deep-well reagent plate to the next column with 92 μL of PCR mix‡; ~1 min |
| DNA-to-Genotype protocol starts here. | |
| Add DNA to Master mix | Transfer 40 μL of DNA solution from one column on the deep-well reagent plate to the next column with 92 μL of PCR mix. ~1 mm |
| Mix | 100 μL pipetting volume; 20 pipetting cycles; ~1.5 min |
| Transfer mix to LFA | Transfer 60 μL from deep-well reagent plate and introduce 52 μL of liquid through inlet ports of housing in order to fill LFA; ~1 mm |
| PCR | Lower thermal cycler onto LFA. PCR Protocol: initial denaturation for 5 min at 89°C; 30 cycles of 87°C for 50 s, 60 to 55°C (touchdown) for 65 s, and 65°C for 35 s; 20 cycles of 87°C for 50 s, 55°C for 65 s, and 65°C for 35 s; final extension at 65 °C for 3 min. Total time 3h 10 min. |
| Hybridization | 54oC;3h |
| Wash | 125 μL 3 times; ~4.5 min |
| Image and Analyze | Acquire image and analyze for each array. Move camera to each chamber, acquire image, analyze; ~5 min |
TruTip was left off of lane with DNA sample (external positive control).
All pipetting operations were programmed at 135 μL sec−1 flow rate.
Tip was added to nozzel of lane with DNA sample (external positive control) to serve as a means of evaluating the DNA-to-genotype testing.
RESULTS AND DISCUSSION
Approach to Integration
Integrating numerous microfluidic processes on a single cartridge or chip can be expensive. Yet, complex protocols are often required for sensitive molecular diagnostic tests in order to satisfy stringent clinical requirements.34 Our strategy for addressing this challenge was to simplify the microfluidics by using a sequence of short and simple steps that can easily be automated. For sample preparation, we simplified the fluidics by integrating a nucleic acid binding matrix inside of a pipette tip,35,36 and we simplified amplification and hybridization using an integrated Lab-on-a-Film assembly.
Fluidic Mechanisms
The fluidic handling steps on the system can be categorized into three different mechanisms: pipetting, air flow from the air pump, and capillary action. The first of these steps (pipetting) was implemented in order to simplify the microfluidics. To achieve liquid transport between consumables (lysis tube, heater strip, deep-well plate, and LFA), we used the TruTip. The second of these mechanisms, the air pump, served as a “blow out” of residual liquid droplets in the pipette tip and as a mechanism for drying the matrix. And, the third of these mechanisms, capillary action, occurred within the LFA as a means of uniformly filling the LFA due to the hydrophilic properties of the cover film and the absorbent in the waste chamber.
The LFA has four distinct microfluidic features that control fluidic behavior. The first of these features is the hydrophilic film that serves as the LFA cover, which is particularly important for filling along the walls of the spacer. The radius of curvature at the intersection of the cover with the spacer approaches zero, thus the capillary pressure theoretically approaches infinity. Practically, the radius of curvature, and thus capillary pressure, is limited by the roughness of the laser-cut wall of the spacer tape. The high capillary pressure at the walls of the microfluidic chamber result in liquid preferentially filling these regions, minimizing the air gaps along the walls.
A second feature is a “staircase” that separated the microarray amplification chamber from the waste chamber. The staircase consists of a series of abrupt 90 degree turns that serves to pin the contact line resulting in a positive (convex) curvature. When the advancing contact line (meniscus) changes from negative (concave) to positive curvature (convex), the liquid does not advance. This feature prevents liquid from entering the waste chamber during thermal cycling.
The third LFA feature is the absorbent in the waste chamber. After the wash buffer is introduced into the LFA, the pressure in the liquid rises higher than the positive curvature of the convex mensicus. When the wash buffer advances into the small pores of the absorbent, the capillary pressure is very high (225 mm/30 min flow rate), higher than the capillary pressure in the chamber. The liquid then imbibes into the absorbent and evacuates the amplification/hybridization chamber.
The fourth feature is the housing of inlet ports, which is the interface between the TruTip and the LFA. The core component of this housing is a pierceable check valve. This valve opens and makes a tight seal with the TruTip such that liquids dispensed through the TruTip flow into the LFA. When the TruTip is retracted from the septum, the lips of the valve re-seal and prevent liquid from escaping, creating a sealed system. The characteristic of the valve – as a check valve – allows for imbibition into the waste chamber absorbent, which includes but prevents liquid flow in the reverse direction (back out of the valve). The valve reseals once the TruTip retracts. To evaluate the efficacy of the sealing, we attempted to flow air in the reverse direction by submerging the assembled housing under water and depressing a P1000 pipettor to the second (blow out) stop. No air bubbles were observed in the bath of water indicating an effective seal (n=62 different valves). We additionally confirmed that the liquid remains in the chamber after performing the thermal cycling protocol in Table 1 (n=36). And, we confirmed that the liquid does not drain from the reaction chamber to the waste chamber as a result of hydrostatic pressure (by holding the LFA upright), indicating that the valve is properly sealed after being pierced (n=52).
Interface Between Thermal Cycler and LFA
There are five aspects of the thermal cycler – LFA interface that facilitate efficient heating and cooling of the chamber: (1) heating is through a thin cover film, (2) the back side of the LFA is insulated to minimize thermal losses, (3) force from the thermal cycler is applied at the interface to enforce uniform contact between the LFA and the thermal cycler, (4) multiple thermistors are integrated throughout the thermal cycler heat spreader to allow for discrete heating zones for more uniform control across the interface, and (5) the thermal electric cooler consists of high-density Peltier junctions for rapid heating and cooling and thus finer feedback control.
Lowering the thermal cycler down onto the LFA and heating “from above” has the following advantages. First, the cover (top) of the LFA is a 0.1 mm thick film compared to the substrate (bottom), which is 0.175 mm. Our gel element printing process required a thicker substrate in order to secure the film to the platen during printing so that it doesn’t bow; however, the cover film was available in a thinner size. Thus, the thermal transfer is more efficient through the cover. Second, we insulated the opposite side of the LFA to improve thermal efficiency and temperature uniformity across and within chambers, which was facilitated by the use of a rigid disposable foam. And third, we used a black background (optical grade masking tape) to control the background of the chamber during the imaging step. This black tape reduced the background fluorescent intensity and non-uniformity, due to artifacts such as dust particles and scratches to the background surface. By heating from “above” (the same side as imaging), we transferred the background uniformity requirement from the instrument (e.g., thermal cycler surface) to the consumable, which can be more easily controlled through the use of removable protective masks to prevent particulates and scratches to the substrates. Whereas another approach is to print the gel elements on an obaque (black) film, we prefer to use a transpartent substrate film to allow better visualization for quality checking the morphology and placement of the gel elements on transparent substrates.29
DNA-to-Genotype Testing
To characterize the behavior of the integrated system we performed separate DNA-to-genotype and liquefied sputum-to-genotype studies. The DNA-to-genotype studies were performed with MTB DNA from strains that are wild-type, INH mono-resistant, and RIF mono-resistant. For these studies, we used 1 pg of MTB DNA, which is equivalent to ~200 genomic copies (gc) determined by the calculation of length of base pairs of the MTB genome (4 million) and average molecular weight per base pair (660 g/mol). The integrated system reported correct detection for all three strains in duplicate. We selected 1pg because, based on the TruTip extraction efficiency and using a standard curve to correlate DNA concentration to Cp value, 1pg of MTB DNA corresponds to approximately 400 cfu/mL, which is equivalent to the reported detection limit of the GeneXpert in Cepheid’s 510(k) application.10 Table 3 shows ratios for the three relevant markers for three strains. The full list of ratios for each marker can be found in Table S1. The integrated system and companion Lab-on-a-Film MDR-TB test reported “wild-type” for all unique markers for the wild-type sample (i.e., the signal intensities from the 16 wild-type markers were higher than their corresponding 36 mutant markers and 1 deletion marker [507-del for rpoB]). The system also correctly detected and reported the S315T (ACC) mutation in the katG gene and −15T in the inhA gene for the INH mono-resistant sample (TDR-0015); all other markers were correctly reported as wild-type. And, the system correctly reported only the S531W mutation for the mono-resistant RIF sample (TDR-0018). This data illustrates the feasibility of the system to discriminate ratios of mutant to wild-type probes as a means of conferring drug resistance to INH and RIF.
Table 3.
Hybridization ratios for select set of probes after processing from purified DNA to genotype. The algorithm reports the correct genotype; positive values are wild-type and negative values are mutants.
| H37Rvc | H37Rvc | TDR-0015d | TDR-0015d | TDR-0018e | TDR-0018e | |
|---|---|---|---|---|---|---|
| rpoBa S531W | 0.74 | 0.85 | 0.65 | 0.77 | −0.96 | −0.92 |
| katGb S315T(ACC) | 0.94 | 0.91 | −0.79 | −0.48 | 0.93 | 0.90 |
| inhAb −15T | 0.94 | 0.92 | −0.58 | −0.49 | 0.97 | 0.95 |
Gene target for RIF resistance.
Gene targets for INH resistance.
Wild-type sample.
INH mono-resistant sample.
RIF mono-resistant sample.
Sputum-to-Genotype Testing
We also performed 8 sputum-to-genotype runs on the integrated system with titers ranging from 4000 cfu/mL to 43 cfu/mL of MTB spiked in raw sputum. Table 2 shows an example of the results for one run. The first four runs consisted of challenging the integrated system with four replicates of titers of 4000, 400, 130, and 43 cfu/mL using a single titer for each run. These runs included two external 1pg H37Ra DNA positive controls in the outermost lanes. The remaining 4 runs were performed with 4 different titers (1200, 400, 130, and 43 cfu/mL) within each run. For these runs, we included the same DNA positive control as used in the first three runs as well as a negative sputum sample. Figure 4 shows the intensities from the universal markers for the set of 8 runs. These “universal markers” target highly conserved regions in each of the respective genes to serve as indicators of PCR amplification efficiency, where low signal intensities typically correspond to poor amplification yield. The signals are a summation of background-subtracted pixel intensities within the image spot of the corresponding universal marker averaged across three replicate spots. The brightness of the spot correlates to the fluorescence of the gel element, or more specifically to the quantity of Cy3-labeled amplicons hybridized to the complementary probes immobilized in the gel element. The ratios of all wild-type markers with respect to their corresponding mutant probes were positive and therefore were correctly reported as wild type except for a 531 rpoB mutation for 1 sample at 43 cfu/mL. Table 4 summarizes the results of these 8 runs, which consists of 36 samples and 12 external DNA controls.
Table 2.
Example of results from a sputum-to-genotype run on the system. The first sample is a DNA-to-genotype external positive control.
| Sample | H37Ra DNA, 1 pg | 1200 CFU/ml | 400 CFU/ml | 130 CFU/ml | 43 CFU/ml | Negative sputum |
|---|---|---|---|---|---|---|
| MTB present wild type | MTB present wild type | MTB present wild type | MTB present wild type | MTB present wild type | MTB Absent | |
| Test Validity | VALID | VALID | VALID | VALID | VALID | VALID |
| M. tuberculosis Complex | PRESENT | PRESENT | PRESENT | PRESENT | PRESENT | Absent |
| Rifampin Resistance | Not Detected | Not Detected | Not Detected | Not Detected | Not Detected | N/A |
| Isoniazid Resistance | Not Detected | Not Detected | Not Detected | Not Detected | Not Detected | N/A |
Figure 4.
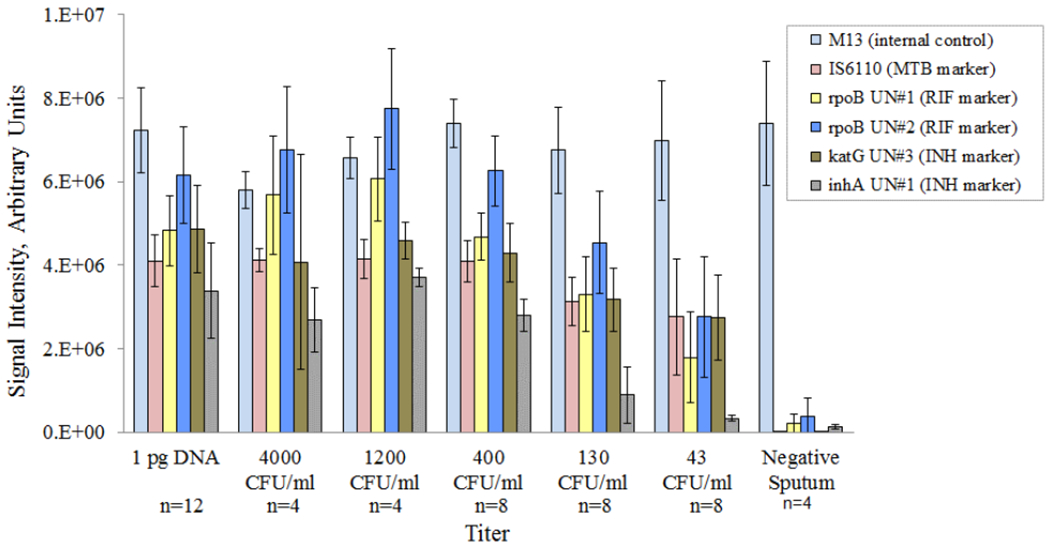
Sputum-to-genotype amplification signals across 8 runs with correct detection (genotyping) down to 43 cfu/mL. Also shown are the signals for the external control (1pg H37Ra DNA).
Table 4.
Summary of results reported by sputum-to genotype tests from 8 runs of sputum with MTB.
| MTB (IS6110) | RIF (rpoB) | INH (katG) | INH (inhA) | ||
|---|---|---|---|---|---|
| Titer | 4000 cfu/mL | 4/4 | 4/4 | 4/4 | 4/4 |
| 1200 cfu/mL | 4/4 | 4/4 | 4/4 | 4/4 | |
| 400 cfu/mL | 8/8 | 8/8 | 8/8 | 8/8 | |
| 130 cfu/mL | 8/8 | 8/8 | 8/8 | 8/8 | |
| 43 cfu/mL | 8/8 | 7/8 | 8/8 | 8/8 | |
| Negative | 4/4 | 4/4 | 4/4 | 4/4 | |
Future Work
Although the processing time is within the 3.5 to 10.5 hour range of four MDR-TB systems evaluated by the WHO21, we have not evaluated the effects of reducing the total analysis time. Our future work, therefore, includes: (1) studies that will investigate the total analysis time versus sensitivity, (2) an increase in the number of tests per run from six to eight , and additional studies to include an increased number of strains, clinical samples, and markers for detection of resistance to additional drugs.
CONCLUSIONS
This work shows the integration of TruTip and a Lab-on-a-Film consumable applied to the detection of drug-resistant MTB from raw sputum. We developed an instrument that automates the TruTip protocol and transfers liquid to a “closed” LFA to prevent amplicon contamination for potential use in clinics and hospital laboratories. We were able to demonstrate that the system effectively processed liquefied sputum, lysed MTB bacilli, isolated MTB DNA, amplified MTB DNA and detected drug-resistant mutations at very sensitive detection levels.
Supplementary Material
ACKNOWLEDGMENTS
The authors wish to thank the National Institutes of Health (U19 AI109755, R44 EB011274, R01 AI111435, R44 AI138903, R44 AI085650, HHSN272201700001C). The authors also wish to thank MRI Global for allowing us to set up the integrated system in their laboratory and for providing access to the laboratory to do the sample-to-answer experiments.
Crude extracts from the TDR Tuberculosis Strain Bank were provided by the United Nations Children’s Fund/United Nations Development Programme/World Bank/World Health Organization Special Programme for Research and Training in Tropical Diseases (TDR), Geneva, Switzerland.
Footnotes
CONFLICT OF INTEREST DISCLOSURE
AVK, RN, AB, PQ, DPC, RCH and CGC were employed by Akonni when this work was conducted. AVK, DPC, RCH and CGC are also shareholders at Akonni.
REFERENCES
- (1).Kato-Maeda M; Bifani PJ; Kreiswirth BN; Small PM The Journal of Clinical Investigation 2001, 107, 533–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Global tuberculosis report 2018; World Health Organization: Geneva, 2018. [Google Scholar]
- (3).Marks SM; Flood J; Seaworth B; Hirsch-Moverman Y; Armstrong L; Mase S; Salcedo K; Oh P; Graviss EA; Colson PW Emerging infectious diseases 2014, 20, 812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).WHO consolidated guidelines on drug-resistant tuberculosis treatment; World Health Organization: Geneva, 2019. [PubMed] [Google Scholar]
- (5).Oxlade O; Falzon D; Menzies D European Respiratory Journal 2012, 39, 626–634. [DOI] [PubMed] [Google Scholar]
- (6).Al-Mutairi NM; Ahmad S; Mokaddas E; Eldeen HS; Joseph S BMC Infect Dis 2019, 19, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Bollela VR; Namburete EI; Feliciano CS; Macheque D; Harrison LH; Caminero JA The international journal of tuberculosis and lung disease 2016, 20, 1099–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Lempens P; Meehan CJ; Vandelannoote K; Fissette K; de Rijk P; Van Deun A; Rigouts L; de Jong BC Scientific reports 2018, 8, 3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Cazabon D; Suresh A; Oghor C; Qin ZZ; Kik SV; Denkinger CM; Pai M Eur Respir J 2017, 50. [DOI] [PubMed] [Google Scholar]
- (10).K131706 Cepheid Xpert MTB/RIF 510(k) FDA Decision Summary: Silver Spring, MD, 2013. [Google Scholar]
- (11).K143302 Cepheid Xpert MTB/RIF 510(k) FDA Decision Summary: Silver Spring, MD, 2015. [Google Scholar]
- (12).Chakravorty S; Roh SS; Glass J; Smith LE; Simmons AM; Lund K; Lokhov S; Liu X; Xu P; Zhang G; Via LE; Shen Q; Ruan X; Yuan X; Zhu HZ; Viazovkina E; Shenai S; Rowneki M; Lee JS; Barry CE 3rd, et al. J Clin Microbiol 2017, 55, 183–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Xie YL; Chakravorty S; Armstrong DT; Hall SL; Via LE; Song T; Yuan X; Mo X; Zhu H; Xu P; Gao Q; Lee M; Lee J; Smith LE; Chen RY; Joh JS; Cho Y; Liu X; Ruan X; Liang L, et al. N Engl J Med 2017, 377, 1043–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Ao W; Aldous S; Woodruff E; Hicke B; Rea L; Kreiswirth B; Jenison R J Clin Microbiol 2012, 50, 2433–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Ranganath R; Kumar GV; Javali V; Ranganath R Annual Research & Review in Biology 2014, 246–257. [Google Scholar]
- (16).de Vos M; Derendinger B; Dolby T; Simpson J; van Helden PD; Rice JE; Wangh LJ; Theron G; Warren RM J Clin Microbiol 2018, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Fang H; Shangguan Y; Wang H; Ji Z; Shao J; Zhao R; Wang S; Zheng L; Jin X; Huang S; Xu K; Sheng J Int J Infect Dis 2019, 81, 46–51. [DOI] [PubMed] [Google Scholar]
- (18).Hassibi A In 2014 IEEE Hot Chips 26 Symposium (HCS); IEEE, 2014, pp 1–32. [Google Scholar]
- (19).Cabibbe AM; Miotto P; Moure R; Alcaide F; Feuerriegel S; Pozzi G; Nikolayevskyy V; Drobniewski F; Niemann S; Reither K Journal of clinical microbiology 2015, 53, 3876–3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Scott L; David A; Noble L; Nduna M; Da Silva P; Black A; Venter F; Stevens W Journal of clinical microbiology 2017, 55, 2491–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).WHO Meeting Report of a Technical Expert Consultation: Accuracy of centralized assays for TB detection and detection of resistance to rifampicin and isoniazid.; World Health Organization: Geneva, 2019. [Google Scholar]
- (22).Shah M; Paradis S; Betz J; Beylis N; Bharadwaj R; Caceres T; Gotuzzo E; Joloba M; Mave V; Nakiyingi L Clinical infectious diseases: an official publication of the Infectious Diseases Society of America 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Mani V; Wang S; Inci F; De Libero G; Singhal A; Demirci U Adv Drug Deliv Rev 2014, 78, 105–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).McNerney R; Clark TG; Campino S; Rodrigues C; Dolinger D; Smith L; Cabibbe AM; Dheda K; Schito M Int J Infect Dis 2017, 56, 130–135. [DOI] [PubMed] [Google Scholar]
- (25).Nimmo C; Shaw LP; Doyle R; Williams R; Brien K; Burgess C; Breuer J; Balloux F; Pym AS BMC genomics 2019, 20, 389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Votintseva AA; Bradley P; Pankhurst L; del Ojo Elias C; Loose M; Nilgiriwala K; Chatterjee A; Smith EG; Sanderson N; Walker TM J Clin Microbiol 2017, 55, 1285–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Thakore N; Norville R; Franke M; Calderon R; Lecca L; Villanueva M; Murray MB; Cooney CG; Chandler DP; Holmberg RC PLoS One 2018, 13, e0199869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Linger Y; Knickerbocker C; Sipes D; Golova J; Franke M; Calderon R; Lecca L; Thakore N; Holmberg R; Qu P; Kukhtin A; Murray MB; Cooney CG; Chandler DP J Clin Microbiol 2018, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Kukhtin AC; Sebastian T; Golova J; Perov A; Knickerbocker C; Linger Y; Bueno A; Qu P; Villanueva M; Holmberg RC; Chandler DP; Cooney CG Lab on a Chip 2019, 19, 1217–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Thakore N; Garber S; Bueno A; Qu P; Norville R; Villanueva M; Chandler DP; Holmberg R; Cooney CG J Microbiol Methods 2018, 148, 174–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Golova JB; Chernov BK; Perov AN; Reynolds J; Linger YL; Kukhtin A; Chandler DP Anal Biochem 2012, 421, 526–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Cooney CG; Sipes D; Thakore N; Holmberg R; Belgrader P Biomed Microdevices 2012, 14, 45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Vincent V; Rigouts L; Nduwamahoro E; Holmes B; Cunningham J; Guillerm M; Nathanson CM; Moussy F; De Jong B; Portaels F; Ramsay A The International Journal of Tuberculosis and Lung Disease 2012, 16, 24–31. [DOI] [PubMed] [Google Scholar]
- (34).High-priority target product profiles for new tuberculosis diagnostics:report of a consensus meeting; World Health Organization: Geneva, 2014. [Google Scholar]
- (35).Chandler DP; Griesemer SB; Cooney CG; Holmberg R; Thakore N; Mokhiber B; Belgrader P; Knickerbocker C; Schied J; St George K J Virol Methods 2012, 183, 8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Griesemer SB; Holmberg R; Cooney CG; Thakore N; Gindlesperger A; Knickerbocker C; Chandler DP; St George K J Clin Virol 2013, 58, 138–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.