

Summary
Background:
The initiating events of chronic gastrointestinal (GI) inflammation in Crohn’s disease (CD) and ulcerative colitis (UC) are not well-defined, but GI infections are implicated.
Aims:
We conducted a systematic review with the primary aim of defining the role of GI infections in risk of incident inflammatory bowel disease (IBD), and secondarily, of synthesizing the current body of relevant translational data to provide biological context for associations between GI infections and IBD risk.
Methods:
We systematically reviewed electronic databases through July 2019. Clinical studies that provided risk estimates of the association between GI infections and incident IBD were included. Inclusion criteria were broader for translational studies aiming to define mechanisms of GI infections and predisposition to or protection from IBD.
Results:
Of the studies identified, 63 met full inclusion criteria. Among studies of clinical gastroenteritis, bacteria—specifically, Salmonella species, Campylobacter species, and Clostridioides difficile—demonstrated consistent positive associations with risk of incident IBD. Of viruses, norovirus was associated with increased risk of incident CD. Regarding inverse associations with incident IBD, Helicobacter pylori and helminth infections were associated with a generally consistent reduced risk of IBD. Based on a qualitative analysis of the translational data, putative mechanisms involve multiple microbial and immunologic pathways.
Conclusions:
Based on this systematic review, certain enteric pathogens are associated with an increased risk of incident IBD, while others are potentially protective. Prospective studies are required to clarify the clinical implications of these enteric pathogens on the risk and course of IBD, not to mention possible therapeutic or preventative benefit.
Keywords: Enteric infection, epidemiology, pathogenesis, microbiome, mucosal immunology
Introduction
Inflammatory bowel disease (IBD), comprising Crohn’s disease (CD) and ulcerative colitis (UC), develops as a result of environmental and microbial factors that elicit a deleterious inflammatory response in genetically susceptibility persons.1,2 An altered gut microbiota, or dysbiosis, impacts mucosal immunity through several pathways, such as by disrupting the integration between environmental, genetic, and immune determinants that are important in metabolism, immunity, and host responses to commensal and pathogenic organisms.3 Any disturbance to this complex network may result in disease, particularly immune-mediated diseases.4 The immune response to gastrointestinal (GI) dysbiosis exemplifies the relationship between the environment and genetics that underlies the cellular and phenotypic range of IBD.4,5 Cross-sectional studies demonstrate unique microbial signatures and T-cell populations for IBD subtypes.6,7 GI infection is a common cause of gut dysbiosis, and several studies have reported an association between enteric infection, functionally altered commensal bacteria, and IBD.5,8,9 By contrast, some microbes such as Helicobacter pylori (H pylori) and helminths, are inversely associated with IBD, particularly CD.
The initiating events and determinants of recurrent, persistent immune dysregulation that ultimately result in chronic GI inflammation are not known, but may precede disease development in the preclinical phase of IBD.10 Given the increasing burden of IBD worldwide, focusing on modifiable risk and protective factors implicated in new-onset IBD is imperative, particularly GI infections. We therefore conducted a comprehensive systematic review with the primary aim of delineating the role of GI infections as environmental determinants of new-onset IBD. We primarily focused on the role of specific enteric infections in the pathogenesis of incident IBD rather than pathobionts or observational changes in the gut microbiome. Accordingly, we did not evaluate association studies of GI pathogens and IBD among patients with prevalent IBD. We contend that a common theme throughout this review is the inability to establish causality and the possibility that certain pathogens may be present at higher frequencies in individuals without contributing to disease pathogenesis. Due to the inability to fully account for measured and unmeasured confounders and establish causality, we secondarily aimed to qualitatively analyze and synthesize the translational data that inform our understanding of mechanisms via which GI infections may predispose to or protect the host from incident IBD.
Methods
Search strategy
We searched the following electronically available databases through July 2019: PUBMED, Ovid, Scopus, ScienceDirect, IMBIOMED, Scielo, IngentaConnect, Nature Publishing Group, and Cochrane database. Studies assessing IBD and GI infections were identified. MeSH terms and/or text words included the broad terms IBD; GI infection; enteric infection. We also searched specific pathogens including: bacteria, viruses, parasites, and fungi. The pathogen search was combined (AND) with “inflammatory bowel disease OR Crohn* disease OR colitis, ulcerative OR indeterminate colitis.” We also manually searched the references of all studies meeting inclusion criteria and any pertinent review articles in order to identify additional studies.
Study selection
Clinical trials and observational studies (e.g. cohort studies) were eligible for inclusion. Cross-sectional studies in which temporality of GI infections and IBD onset could not be determined, case-series, review articles, and conference abstracts were excluded. Studies were included if they met the following criteria: 1) diagnosis of IBD made according to standard diagnostic criteria; 2) diagnosis of GI infection as defined by positive testing on any of the following modalities: histology, culture, serology, polymerase chain reaction (PCR) and/or other molecular technique; 3) ability to confirm that no individuals had IBD at the study inception (i.e. prevalent IBD) 4) sufficient information provided to interpret or calculate comparative effect estimates; 5) full-text available in English. Inclusion criteria were broader for translational or experimental studies and included studies aiming to define mechanisms underlying the association between specific GI infections and predisposition to or protection from experimental or human IBD.
Data abstraction and quality assessment
A standardized data abstraction form was generated and the following data were abstracted from all included articles: authors, publication year, title, study design, study country, pathogen type, pathogen testing modality, IBD type and diagnostic methods, risk estimates, and study follow up, among other study-specific details. We additionally documented the time course of GI infection relative to incident IBD, where available, such as in cohort studies. While meta-analysis was considered a priori, significant heterogeneity among the studies, many lacking in comparator groups, and the dearth of literature for each pathogen separately precluded this. Quality assessment was performed using the Newcastle–Ottawa Scale for nonrandomized studies and the Cochrane Risk of Bias tool for randomized controlled trials.
Results
Systematic review results
Of the 1894 unique studies identified, 423 studies were reviewed in full text, of which 18 ultimately met the predefined strict inclusion criteria for clinical studies of pathogens and incident IBD; 45 met criteria for translational/experimental data. No additional studies were identified based on review of references (Figure 1).
Figure 1.
Diagram of study selection.
Gastrointestinal infections and increased risk of IBD
In general, population-based studies of gastroenteritis and the subsequent development of IBD demonstrate positive associations, most often with Campylobacter and Salmonella species (Table 1).11–18
Table 1.
Clinical gastroenteritis and risk of inflammatory bowel disease.
| Study | Study location and Period | Study Design | Study population | Patient-years or years of follow up | Number of exposed individuals | GE Diagnosis | Number of IBD diagnoses | Risk Assessment (95% CI) |
|---|---|---|---|---|---|---|---|---|
| Garcia Rodriguez, 200616 | UK, 1992–2001 | Cohort | General Practice Research Database in UK 94,013 |
325,743 | 43,013 (All GE) | ICD coding | 64 (UC) | UC: HR 2.3 (1.5–3.6; All GE) |
| Porter 200814 | USA, 1999–2006 | Case-control | US military 14,665 |
Max 7 years per patient | 828 (All GE) | ICD coding | 115 (UC) | UC: OR 1.4 (1.1–1.7; All GE) |
| Gradel, 200911 | Denmark, 1991–2003 | Cohort | Nationwide 39,264 |
Max 15 years per patient | 6,685 (Campylobacter) | ICD coding | 79 (UC) | UC: HR 2.8 (1.7–4.6; Salmonella) |
| Jess, 201117 | Denmark, 1992–2008 | Cohort | Nationwide NR |
94,264,447 | 49,420 (Campylobacter) | Stool culture | 487 (UC) | UC: IRR 3.0 (2.6–3.4; Salmonella) |
| Porter, 201715 | USA, 2001–2009 | Cohort | US military 82,107 |
470,060 | 18 (All GE, in IBD only) | ICD coding | 49 (UC) | UC: HR 2.9 (1.4–6.0; All GE) |
| Castaño-Rodríguez, 201713 | Pooled | Meta-analysis | Pooled, 1652 | - | - | - | 519 (IBD) | IBD: P-OR 2.97 (1.33 to 6.63, |
| Axelrad, 201918 | Sweden, 2002–2014 | Case-control | Nationwide 480,721 |
Max 12 years per patient | 20,790 (All GE) | ICD coding | 1,672 (UC) | UC: OR 1.6 (1.5–1.7; All GE) |
IBD: Inflammatory bowel disease; UC: ulcerative colitis; CD: Crohn’s disease; IBD-U: Inflammatory bowel disease- Unclassified; GE: gastroenteritis; HR: hazard ratio; OR: odds ratio; IRR: incidence rate ratio; NR: not reported; CI: confidence interval
In a nationwide cohort study of 94,013 patients from the United Kingdom, a diagnosis of any gastroenteritis was associated with a greater than 3-fold increased risk of CD adjusted for age, sex, and calendar year (adjusted hazard ratio [aHR] 3.1, 95% confidence interval [CI] 1.7–5.7) and 2-fold increased risk of UC (aHR 2.3, 95%CI 1.5–3.6).16 In a nationwide registry-based cohort study from Denmark evaluating 49,420 stool cultures positive for Campylobacter jejuni and 41,628 for Salmonella species, both UC and CD were more common following an episode of gastroenteritis with a stool culture positive for Salmonella (CD IRR [incidence rate ratio] 2.2, 95%CI 1.7–2.7; UC IRR 3.0, 95%CI 2.6–3.4) or Campylobacter (CD IRR 2.2, 95%CI 1.8–2.7; UC IRR 2.6, 95%CI 2.3–3.0) compared to patients without gastroenteritis. However, CD and UC effect estimates were significantly higher after a negative stool test compared to a positive test, suggesting detection bias.17 In this study, patients without stool cultures were excluded; additionally, viral gastroenteritis, recurrent episodes of gastroenteritis, and antimicrobial therapies were not analyzed.17
A recent nationwide case-control study of 480,721 patients in Sweden reported that—after adjusting for sex, age, birth year, place of residence, previous GI surgery, autoimmune disease, and family history of IBD—a diagnosis of any gastroenteritis was associated with an increased odds of incident IBD (CD aOR [adjusted odds ratio] 1.7, 95%CI 1.6–1.8; UC aOR 1.6, 95%CI 1.5–1.7). An increased number of episodes was associated with higher odds of IBD. For at least 10 years following an episode of gastroenteritis, there remained a statistically significant 30% higher likelihood of developing IBD (aOR 1.3, 95%CI 1.2–1.3).18 In addition, receiving antimicrobial therapy for an episode was associated with a 4-fold higher odds of IBD (aOR 4.2, 95%CI 3.3–5.3).18 This study also reported odds of IBD-subtype according to specific pathogens. Detection of Salmonella, Campylobacter, Yersinia, C. difficile, Amoeba, and norovirus predicted higher odds of CD. Salmonella, Escherichia coli, Campylobacter, and C. difficile predicted a higher aOR of UC.18
Gastrointestinal infections and attenuated risk of IBD
In general, a diverse body of literature comprising population-based, observational, cross-sectional, and retrospective studies consistently demonstrates an inverse relationship between H. pylori (Table 2) and intestinal helminths and IBD.
Table 2.
Helicobacter pylori meta-analyses and risk of inflammatory bowel disease.
| Study | Study population | Number of IBD diagnoses | Risk Assessment (95% CI) |
|---|---|---|---|
| Luther, 201024 | Pooled, 5903 | 1108 (UC) | UC: RR 0.75 (0.62–0.90) |
| 1654 (CD) | CD: RR 0.60 (0.49–0.72) | ||
| Wu, 201523 | Pooled, 3116 | 548 (UC) | UC: RR 0.55 (0.48–0.64) |
| 751 (CD) | CD: RR 0.43 (0.37–0.50) | ||
| Rokkas, 201525 | Pooled, 9163 | 1874 (UC) | UC: RR 0.53 (0.42–0.67) |
| 2526 (CD) | CD: RR 0.38 (0.31–0.47) | ||
| Castaño-Rodríguez, 201713 | Pooled, 80,789 | 2520 (UC) | UC: OR 0.53 (0.44 to 0.65) |
| 2938 (CD) | CD: OR 0.38 (0.31 to 0.47) | ||
| 386 (IBDU) | IBDU: OR 0.43 (0.23 to 0.80) | ||
| Tepler, 201922 | Pooled, 1748 | 272 (UC) | CagA seropositive |
| 688 (CD) | UC: OR 0.68 (0.35–1.32) | ||
| CD: OR 0.25 (0.17–0.38) | |||
| CagA seronegative | |||
| UC: OR 0.97 (0.76–1.24) | |||
| CD: OR 0.97 (0.82–1.14) | |||
| Piovani, 201926 | Pooled, 83,798 | 6284 (UC) | UC: OR 0.53 (0.44–0.65) |
| 7869 (CD) | CD: OR 0.38 (0.31–0.47) |
IBD: Inflammatory bowel disease; UC: ulcerative colitis; CD: Crohn’s disease; IBD-U: Inflammatory bowel disease- Unclassified; OR: odds ratio; RR: relative risk; CI: confidence interval; CagA: cytotoxin-associated gene A
To date, five meta-analyses on H. pylori exposure and IBD have been published.13,22–25 All were based on a variety of study designs, including association studies and lacking comparators. A meta-analysis reported that, among 40 case-control studies with 6130 IBD cases and 74,659 controls, H. pylori exposure was associated with 57% lower odds of IBD (pooled OR [pOR] 0.43, 95%CI: 0.36–0.50); the effect was more pronounced for CD (pOR 0.38, 95%CI: 0.31–0.47) compared to UC (pOR 0.53, 95%CI: 0.44–0.65), but similar to IBD-U (pOR 0.43, 95%CI: 0.23–0.80).26 The inverse association was maintained irrespective of age and geography, but was notably stronger for pediatric versus adult populations and Asian versus Caucasian populations. Other meta-analyses, one of which only included Asian studies, reported consistent findings, demonstrating a significant inverse association between H. pylori and IBD, more pronounced for CD versus UC.23 We identified only one study analyzing the association between H. pylori-strain specific constituents and likelihood of IBD, but no studies analyzing the association with incident, new-onset IBD. Based on meta-analysis of three studies with 1748 people (688 with CD, 272 with UC; 788 ‘controls’ without IBD), compared to CagA seronegativity, CagA seropositivity was associated with a significantly reduced odds of IBD overall (0.35, 95% CI: 0.21–0.44), that was statistically significant for CD (OR 0.25, 95% CI: 0.17–0.38) but not UC (OR 0.68, 95% CI: 0.35–1.32). There was no difference in odds of IBD, CD or UC between H. pylori exposed, CagA seronegative individuals and H. pylori non-exposed individuals.
Recently, a population-based study of nearly 80,000 patients from Taiwan demonstrated that patients with peptic ulcer disease (PUD) who were treated for H. pylori were at the highest risk for autoimmunity or IBD (aHR, 2.36, 95%CI 2.14–2.59), particularly CD, compared to patients with PUD who did not receive anti H. pylori treatment (aHR, 1.91, 95%CI 1.73–2.11).27 It should be noted, though, that H. pylori eradication was not confirmed in the study.
Few population-based studies have aimed to delineate the association between helminths, such as Trichuris suis and IBD,28,29 and no studies analyzed the association between parasites and incident IBD specifically.
Gastrointestinal infections and IBD: Associations between bacteria, viruses, and fungi and IBD
As our primary objective focused on new-onset IBD, we excluded studies reporting associations between pathogens and established IBD. We identified several studies that did not meet our strict inclusion criteria, but were informative. Although no singular, causative organisms and associated time courses have been identified, association studies implicate numerous pathogens in IBD (Table 3).
Table 3.
Specific pathogens associated with risk of IBD.
| Increased Risk | Decreased risk | |
|---|---|---|
| Bacteria | Salmonella species | Helicobacter pylori |
| Escherichia coli | ||
| Yersinia species | ||
| Campylobacter species | ||
| Enterohepatic Helicobacter species | ||
| Mycobacterium avium paratuberculosis | ||
| Clostridioides difficile | ||
| Listeria monocytogenes | ||
| Proteus mirabilis | ||
| Klebsiella pneumoniae | ||
| Citrobacter species | ||
| Viruses | Norovirus | |
| Cytomegalovirus | ||
| Epstein–Barr virus | ||
| Human herpes virus 3 | ||
| Human herpes virus 6 | ||
| Human herpes virus 8 | ||
| Measles virus | ||
| Mumps virus | ||
| Rubella virus | ||
| Rotavirus | ||
| Adenovirus | ||
| Fungi | Candida species | |
| Aspergillus species | ||
| Cryptococcus neoformans | ||
| Parasites | Amoeba/Entamoeba histolytica | Trichuris suis |
| Toxoplasma gondii | Hymenolepis diminuta | |
| Schistosoma species | ||
| Nector americanus |
Translational data on gastrointestinal infections and IBD: Pathogenic mechanisms
Translational data, primarily generated using animal models, demonstrate an association between enteric infection and GI microbial dysbiosis, with downstream effects on intestinal inflammation.5,32–42 Animal models provide unique opportunities to examine causality and the relative contributions of specific pathogens, commensal communities, and host genotype in acute and chronic intestinal inflammation. That said, there are no experimental models that perfectly recapitulate human IBD and findings must therefore be interpreted in context.
Bacteria
Several studies have examined bacterial pathogens as causal in chronic intestinal inflammation in susceptible hosts. The biological plausibility of Mycobacterium avium paratuberculosis (MAP) in CD pathogenesis has been demonstrated in several studies. In one small study of T-cell lines from intestinal biopsies of 11 patients with CD, 13 with UC, and 10 controls, 71% of patients with active CD had a strong Th1/Th17 response to MAP.43 Moreover, MAP reactive CD4 T-cell clones produced IL-17 and/or interferon (IFN)-gamma suggesting a role of mycobacteria in CD.43 Similarly, levels of tumor necrosis factor-alpha (TNF-a) correlated with the presence of MAP.44 Of 202 patients with IBD, peripheral blood from patients with active CD showed the highest MAP DNA prevalence (68%), with infliximab treatment associated with a decrease in MAP DNA and corresponding reduction in inflammatory burden.45 Despite a plethora of studies, there is still no conclusive evidence that MAP is directly etiopathogenic in CD onset.
In a mouse model of dysregulated type 1 immunity, the presence of Proteus mirabilis and Klebsiella pneumoniae correlated with experimental colitis in mice deficient in the transcription factor T-bet. Enterobacteriaeceae species from these mice triggered colitis in the wild-type mice, but not in germ-free mice, suggesting that microbial communities function in conjunction with specific colitis-promoting microbes.46 In a study of Citrobacter rodentium-induced acute gastroenteritis, which represents a mouse model for human enteropathogenic E. coli (EPEC) and enterohemorrhagic E. coli (EHEC), mice colonized with adherent-invasive E. coli (AIEC), a bacterial pathobiont of CD, experienced a post-infectious recovery period characterized by increased AIEC epithelial expansion and delayed improvement of colitis.47 Additionally, mutations in the CD risk genes NOD2 and ATG16L1 increased susceptibility to and enhanced the inflammatory responses to C. rodentium.48–50 The effect of Citrobacter varies among different strains of mice, further suggesting a role for genetic factors. The linking of a GI pathogen with an increase in immunopathology, barrier defects, and delays in mucosal restitution following pathogen clearance is consistent with epidemiological data presented above.
In mice, oral infection with Yersinia pseudotuberculosis remodeled mesenteric adipose tissue and lymph nodes, resembling chronic lymphadenopathy. Y. pseudotuberculosis- induced scarring of the mesentery interfered with the ability of the host to develop tolerance and adaptive immunity to antigens, demonstrating that that acute infections may have cumulative and long-term immunologic consequences.51 Mesenteric adipose and lymphatic changes occur in CD, including preclinical CD. Several studies report an association between Y. pseudotuberculosis and CD, although none specifically report risk of incident IBD. One association study demonstrated that NOD2 mutation frequencies were significantly higher in areas of past exposure to plague.52 In a study of knock-in mice for a variant of ATG16L1, there was defective clearance of Y. enterocolitica with ileitis resembling CD.53
Inoculation with Salmonella enterica Typhimurium caused intestinal inflammation in mice. Interestingly, the inflammation persisted after pathogen clearance and irreversibly escalated in severity with repeated infections.5 Repeated S. enterica Typhimurium infection progressively disabled a host mechanism of protection that resulted in the accumulation of lipopolysaccharide-phosphate (LPS-P).5 LPS activated Toll-like receptor (TLR)4-dependent inflammation with increases of the pro-inflammatory TLR4 ligand, LPS-P. This manifested primarily in the colon with pathology similar to UC.5 It is also not clear whether these effects are unique to Salmonella.
Viruses
Several studies have aimed to define the role of viruses in IBD pathogenesis, especially CD.34 Murine norovirus (MNV) caused epithelial barrier disruption in IL-10 deficient mice representing a potent enterocolitogenic stimulus dependent on the presence of the enteric microbiota.54 Filamentous bacteria colonization abolished MNV-induced colitis, suggesting that colitis depends on the microbial context.55 In another mouse model, norovirus infection in the setting of a polymorphism in the CD susceptibility autophagy gene ATG16L1 produced Paneth cell abnormalities and intestinal pathology resembling CD.33,34 The ability of norovirus infection to induce Th1-immune responses provides one plausible explanation for the selective association with CD, which is typically associated with a more pronounced Th1 signature compared to UC.33,42 Norovirus tropism for the small bowel might also be relevant.56
In a study of monozygotic twins, CMV discordant monozygotic twins showed reductions in many immune cell types, including effector CD8+ and gamma-delta T-cells, as well as attenuated cell signaling responses, most notably in response to IL-10 and IL-6 stimulation, compared to CMV negative monozygotic twins. These data demonstrate how a single viral exposure can significantly modulate the overall immune profile of an individual and might be relevant to IBD pathogenesis in susceptible individuals.57
Parasites
Oral route of infection with Toxoplasma gondii in mice resulted in loss of tolerance to commensals and differentiation of microbiota-specific T-cells into inflammatory effector and memory cells, which were phenotypically and functionally consistent with pathogen-specific T-cells. These data suggest that during an acute GI infection, the immune response to commensals may parallel the response to pathogens.35,58
Translational data on gastrointestinal infections and IBD: Protective Mechanisms
There are several mechanisms by which H. pylori is hypothesized to protect against IBD.13,22,59 One recent systematic review that included experimental studies of H. pylori and IBD, identified five studies meeting inclusion criteria, all of which used mouse models of experimental colitis.60–64 Different protocols were used with respect to timing, duration, and strain of H. pylori (or H. pylori DNA extract or other constituents). Irrespective of protocol, all studies demonstrated that exposure attenuated experimental colitis manifested as either complete prevention or a less severe phenotype of experimental colitis compared to nonexposure. H. pylori exposure was associated with a blunted Th17 response, enhanced T-reg and Th-2 response, or higher immunoregulatory to immunostimulatory ratio. Only one of the included studies analyzed CagA-dependency of this protective effect and demonstrated that infection with H. pylori strain SS1, which has a functional cag pathogenicity island, was associated with less severe colitis. Supportive evidence also comes from H pylori-induced protection against other non-IBD immune-mediated diseases, such as asthma and allergy.65–67
In animal models, helminth infection attenuates colitis as well as modulates the gut microbiota.68–71 In mice, secreted products of T. suis reduced barrier function through altered expression of tight junction proteins in intestinal epithelial cells, resulting in increased passage of soluble compounds and downstream effects on dendritic cell function.72 Dendritic cells mediate the balance between immunity and tolerance, and altered dendritic cell function is implicated in autoimmunity. Secreted products of T. suis also suppressed LPS-induced pro-inflammatory cytokine production in intestinal epithelial cells. These data provide evidence that T. suis promotes immune tolerance and protects against immune-mediated diseases, partly mediated through dendritic cell modulation.72 In another study, mice infected with Hymenolepis diminuta were protected from chemical colitis, while in those previously infected mice, H. diminuta antigen triggered an immunological memory response with increased IL-4 and −10 that reduced the severity of chemical colitis.73
Schistosomes also modulate experimental colitis. In a study of rat immunization with a recombinant schistosome enzymatic protein, chemical colitis was attenuated through eosinophil-dependent modulation of the immune system toward a Th2 profile, with local and systemic increases of IL-13 and IL-5.74,75
In addition to regulatory responses, helminth infection may also attenuate the risk of IBD via direct alterations to the gut microbiota. Infection with T. muris and Heligmosomoides polygyrus protected mice deficient in NOD2 from intestinal abnormalities by inhibiting inflammatory Bacteroides species colonization and instead promoting a protective microbiota enriched in Clostridiales.30 Helminth-colonized individuals among an indigenous Malaysian population, the Orang Asli, harbored a similar protective microbiota; however, deworming therapy reduced levels of Clostridiales and increased Bacteroidales .30 Similar findings have been described in macaques where exposure to T. trichiura ameliorated colitis by restoring mucosal barrier functions and reducing overall bacterial attachment, as well as by altering the communities of adherent bacteria.76
These experimental data motivated several trials of helminths as IBD therapy.77 In a trial of T. suis ova versus placebo in 252 patients with active ileocolonic CD, patients randomized to T. suis ova demonstrated a clinically relevant, dose-dependent immunological response.78 However, no dose showed a clinically relevant effect compared to placebo for induction of clinical remission or response in mildly-to-moderately active, ileocolonic CD.
Discussion
We conducted a systematic review …. and identified significant and relevant association between GI infections (gastroenteritis) and new-onset IBD, along with corroborating biologically plausible mechanisms of action. Based on these data, specific enteric infections and their downstream effects (e.g. metabolomic profiles), might plausibly: 1) trigger gut dysbiosis; 2) function as surrogate markers to identify patients with existing dysbiosis; 3) exacerbate already present dysbiosis; 4) induce immunological scarring, even to commensals, that alters subsequent immune responses; or 5) directly injury the intestinal barrier and activate mucosal immune responses; all contributing to preclinical and clinical IBD pathogenesis or IBD relapse in genetically susceptible individuals. Alternatively, other pathogens such as Helicobacter and certain helminths, might prevent or mitigate underlying dysbiosis and or suppress pro-inflammatory products and pathways (Figure 2).
Figure 2.
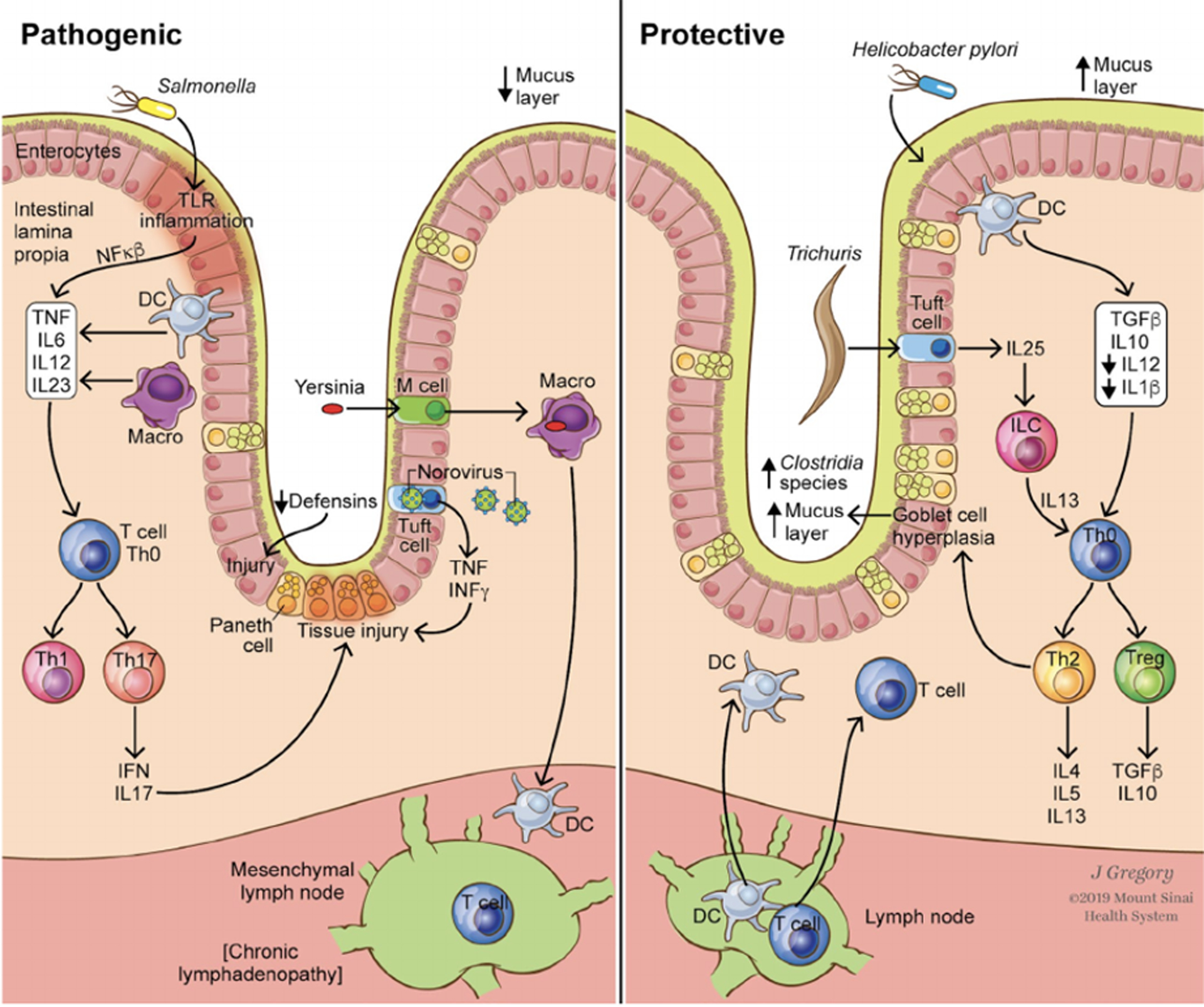
Model of pathogenic and protective mechanisms of bacterial, viral, and parasitic pathogens on onset and flare of IBD.
Several methodological challenges need to be considered when interpreting the association between gastroenteritis and IBD, namely misclassification and detection bias. Studies often analyze subset species as a composite genus, and there is the possibility of diluted effect for species-specific or strain level differences. This is particularly relevant for Campylobacter species. Several Campylobacter species are recognized as human pathogens and implicated in IBD pathogenesis, including Campylobacter jejuni (C. jejuni), C. coli, C. ureolyticus, C. showae and C. concisus.13 In addition, heterogeneity may confound interpretation of Campylobacter, particularly with respect to the diagnostic tools used to detect Enterohepatic Helicobacter species (EHS) as there is molecular overlap with H. pylori-like (HPL)-EHS and Campylobacter. H. pylori itself is inversely associated with IBD, as further detailed below, although this might also be strain-specific instead of species-specific.
It is also likely that most people are universally exposed to various pathogens and experience several episodes of gastroenteritis through different phases in their lifetime, with varying levels of susceptibility related to both non-genetic and plausibly genetic factors as well. Despite a growing body of literature on this topic, there is an inherent limitation that studies are typically restricted to common pathogens detected and managed in clinical practice. While not yet completely defined, there are likely gut microbial factors and/or the coinfection with protective pathogens that may mitigate the development of IBD following certain GI infections. Relatedly, in developing countries where there is generally also a higher burden of enteric infections, the observed incidence of IBD is also lower, although there are most certainly other underlying contributory factors. Another consideration is that only severely symptomatic patients, those with non-resolving symptoms, or also those with high healthcare utilization may present to medical care; whether or not patients undergo diagnostic stool testing at their presentation also varies. With respect to the latter, because the clinical symptoms of gastroenteritis and IBD overlap, there is inconsistent utilization of diagnostic testing and the data are subject to bias, heterogeneity, and unmeasured confounders, such as non-genetic and genetic factors underlying differential susceptibility. In one study of 319 patients with known UC, 6 IBD-risk loci were associated with an increased risk of Clostridioides difficile infection (CDI) while 2 risk loci were protective.19 Genetic risk alleles explained a larger proportion of the variance in CDI risk than clinical factors, suggesting that host immunity is also a fundamental player in CDI susceptibility.19 These data support the hypothesis that CDI might be relevant in IBD pathogenesis.
H. pylori is the most common chronic bacterial infection worldwide, with over 4.4 billion people estimated to be infected.20 There is marked global variation in H. pylori prevalence, however, with colonization significantly more common in developing or recently developed countries, including Asian-Pacific and Central/South American countries, with a much lower overall prevalence in Western industrialized countries.20 From a population standpoint, the decline in H. pylori corresponds to an increase in immune-mediated diseases, particularly IBD. Early-in life H. pylori exposure is associated with important immunomodulatory effects and alterations in the gut microbiome.21,22 IBD is now increasingly diagnosed in populations where the decline in H. pylori has been most striking, especially the Asia-Pacific region. Epidemiologic studies demonstrate an inverse association between H. pylori exposure and IBD, particularly CD. It is hypothesized that H. pylori strain-specific constituents, such as CagA, are important mediators of this protective response, although this has not been well-studied. A recent small meta-analysis did provide some supportive evidence for this hypothesis, however, and demonstrated significantly lower odds of IBD associated with CagA seropositivity versus CagA seronegativity that was driven primarily by the reduced odds in CD as opposed to UC. Notably, there was no significant protective benefit of H. pylori exposure with respect to odds of IBD, CD or UC in the absence of CagA seropositivity, suggesting that CagA seropositivity itself or as a surrogate of other unmeasured effects accounted in large part for the protective benefit observed.22 The consistency in H. pylori and risk of IBD across geographic regions, albeit with a stronger association in Asian-Pacific countries, should be highlighted, as other environmental exposures connected with incident IBD, such as childhood antibiotics, appendicitis, and Campylobacter, have not demonstrated this same broad geographic consistency. It should be emphasized though, that not all Helicobacter species are inversely associated with IBD, particularly EHS.
In addition to H. pylori, we also identified that helminths are generally consistently associated with reduced risk of new-onset IBD. Broadly, helminths play an important immunoregulatory role in the intestinal microbiome, and the lack of identifiable exposure to intestinal helminths has been associated with the development of IBD.30 These clinical data are supported by a platform of biological plausibility, as demonstrated by our qualitative analysis results. The notion that infectious agents may confer protection from a variety of immune-related disorders is generally referred to as the ‘hygiene hypothesis’. This concept posits that lack of childhood exposure to infectious antigenic stimuli may decrease gut microbial diversity, favoring a T helper [Th] 2 proinflammatory immune response, in contrast to a Th1 response in areas with lower hygiene.31 Mechanisms of H. pylori protection against IBD depend on host-microbial interactions and their timing, as well as environmental influences. Supporting mechanisms include systemic H. pylori-induced immunomodulation, molecular mimicry between H. pylori and pathogenic non-HPL EHS and Campylobacter species, and H. pylori production of antibacterial peptides, among others. Early H. pylori infection is also associated with increased diversity of the gut microbiome with greater compositions of bacteria that contribute to intestinal mucosal integrity and immunoregulation, the compromise of which is implicated in IBD pathogenesis.44 Despite multiple plausible mechanisms, there are several other considerations in addition to timing of exposure and strain- and host-specific non-genetic and genetic determinants, such as hygiene, rural versus urban dwelling, and antibiotic exposures, all of which might also be associated with H. pylori colonization and modulation of mechanisms.
Less is known regarding the timeline of events in the preclinical phase that herald overt clinical symptoms and new presentation of IBD. Indeed, anti‐microbial antibodies to gut commensals, such as ASCA‐IgA/IgG, are present years before a diagnosis of IBD, suggesting that the microbiota and immune response are critical in the pathogenesis of disease.10 Based on the above research, gastrointestinal pathogenic insults may initiate these events. Deep sequencing technologies including metabolomics and proteomics will continue to shed light on the intricate relationship between gut pathogens and IBD in the preclinical phase. The metabolic byproducts of these organisms may partly explain the variability in IBD development, phenotype, and activity. Another area where clarity is needed relates to the concept of cumulative infections over time. We are currently unable to accurately identify prior infectious events, as PCR and culture techniques do not capture the total enteric infectious burden of a single individual. It is perhaps the collective enteric infectious history in the context of one’s gut microbiota, as well as non-genetic/genetic susceptibility, rather than an individual microbe that is more pertinent to IBD.
Conclusions
In summary, specific gastrointestinal infections might modify the risk of subsequent IBD development. Although many of the clinical studies meeting inclusion criteria were subject to methodological limitations and biases that we have noted, and causality cannot be definitively established, these data are nevertheless informative for our understanding of factors driving the preclinical and clinical phases of IBD. A vast amount of translational data defining mechanisms by which microbes trigger pathogenic inflammatory pathways in the intestine provide complementary plausible and convincing evidence. Ideally, a better understanding of these mechanisms and modulating factors can be leveraged for preventative/risk attenuating and possibly even therapeutic interventions in IBD. Well-designed prospective studies, while certainly subject to logistic limitations, will be invaluable for investigating the clinical implications of various enteric infections on the risk and course of IBD.
We look forward to the outcomes of currently ongoing collaborative study efforts that are focused on the preclinical stage of IBD and will enable the use of patient biospecimens to explore disease pathogenesis and facilitate the development of personalized solutions, such as microbial therapeutics. Collectively, better defining these complex environmental factors and gene-environment interactions will further promote our efforts toward IBD prevention and a more personalized approach so that we can improve disease-related outcomes.
Acknowledgments:
The authors would like to acknowledge Jill Gregory for her assistance with figure design. This is the peer reviewed version of the following article: Axelrad JE, Cadwell KH, Colombel JF, Shah SC. Systematic review: gastrointestinal infection and incident inflammatory bowel disease. Aliment Pharmacol Ther. 2020 May 5. which has been published in final form at DOI: 10.1111/apt.15770. This article may be used for non-commercial purposes in accordance with Wiley Terms and Conditions for Use of Self-Archived Versions.
Funding: None
Footnotes
Conflict of Interest: Authors report no relevant personal or financial conflicts of interest.
References
- 1.Ungaro R, Mehandru S, Allen PB, et al. Ulcerative colitis. Lancet 2017;389:1756–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Torres J, Mehandru S, Colombel J-F, et al. Crohn’s disease. Lancet 2017;389:1741–1755. [DOI] [PubMed] [Google Scholar]
- 3.Thaiss CA, Zmora N, Levy M, et al. The microbiome and innate immunity. Nature 2016;535:65–74. [DOI] [PubMed] [Google Scholar]
- 4.Joeris T, Müller-Luda K, Agace WW, et al. Diversity and functions of intestinal mononuclear phagocytes. Mucosal Immunol. 2017;10:845–864. [DOI] [PubMed] [Google Scholar]
- 5.Yang WH, Heithoff DM, Aziz PV, et al. Recurrent infection progressively disables host protection against intestinal inflammation. Science 2017;358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gui X, Li J, Ueno A, et al. Histopathologic Features of Inflammatory Bowel Disease are Associated with Different CD4+ T Cell Subsets in Colonic Mucosal Lamina Propria. J. Crohns Colitis 2018. [DOI] [PubMed] [Google Scholar]
- 7.Leung JM, Davenport M, Wolff MJ, et al. IL-22-producing CD4+ cells are depleted in actively inflamed colitis tissue. Mucosal Immunol. 2014;7:124–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Axelrad JE, Joelson A, Green PHR, et al. Enteric Infections Are Common in Patients with Flares of Inflammatory Bowel Disease. Am. J. Gastroenterol 2018;113:1530–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Axelrad JE, Joelson A, Nobel YR, et al. Enteric infection in relapse of inflammatory bowel disease: the utility of stool microbial PCR testing. Inflamm. Bowel Dis 2017;23:1034–1039. [DOI] [PubMed] [Google Scholar]
- 10.Torres J, Burisch J, Riddle M, et al. Preclinical disease and preventive strategies in IBD: perspectives, challenges and opportunities. Gut 2016;65:1061–1069. [DOI] [PubMed] [Google Scholar]
- 11.Gradel KO, Nielsen HL, Schønheyder HC, et al. Increased short- and long-term risk of inflammatory bowel disease after salmonella or campylobacter gastroenteritis. Gastroenterology 2009;137:495–501. [DOI] [PubMed] [Google Scholar]
- 12.Keithlin J, Sargeant J, Thomas MK, et al. Systematic review and meta-analysis of the proportion of Campylobacter cases that develop chronic sequelae. BMC Public Health 2014;14:1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Castaño-Rodríguez N, Kaakoush NO, Lee WS, et al. Dual role of Helicobacter and Campylobacter species in IBD: a systematic review and meta-analysis. Gut 2017;66:235–249. [DOI] [PubMed] [Google Scholar]
- 14.Porter CK, Tribble DR, Aliaga PA, et al. Infectious gastroenteritis and risk of developing inflammatory bowel disease. Gastroenterology 2008;135:781–786. [DOI] [PubMed] [Google Scholar]
- 15.Porter CK, Welsh M, Riddle MS, et al. Epidemiology of inflammatory bowel disease among participants of the Millennium Cohort: incidence, deployment-related risk factors, and antecedent episodes of infectious gastroenteritis. Aliment. Pharmacol. Ther 2017;45:1115–1127. [DOI] [PubMed] [Google Scholar]
- 16.García Rodríguez LA, Ruigómez A, Panés J. Acute gastroenteritis is followed by an increased risk of inflammatory bowel disease. Gastroenterology 2006;130:1588–1594. [DOI] [PubMed] [Google Scholar]
- 17.Jess T, Simonsen J, Nielsen NM, et al. Enteric Salmonella or Campylobacter infections and the risk of inflammatory bowel disease. Gut 2011;60:318–324. [DOI] [PubMed] [Google Scholar]
- 18.Axelrad JE, Olén O, Askling J, et al. Gastrointestinal Infection Increases Odds of Inflammatory Bowel Disease in a Nationwide Case-Control Study. Clin. Gastroenterol. Hepatol 2019;17:1311–1322.e7. [DOI] [PubMed] [Google Scholar]
- 19.Ananthakrishnan AN, Oxford EC, Nguyen DD, et al. Genetic risk factors for Clostridium difficile infection in ulcerative colitis. Aliment. Pharmacol. Ther 2013;38:522–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hooi JKY, Lai WY, Ng WK, et al. Global Prevalence of Helicobacter pylori Infection: Systematic Review and Meta-Analysis. Gastroenterology 2017;153:420–429. [DOI] [PubMed] [Google Scholar]
- 21.Wang C, Nishiyama T, Kikuchi S, et al. Changing trends in the prevalence of H. pylori infection in Japan (1908–2003): a systematic review and meta-regression analysis of 170,752 individuals. Sci. Rep 2017;7:15491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tepler A, Narula N, Peek RM, et al. Systematic review with meta-analysis: association between Helicobacter pylori CagA seropositivity and odds of inflammatory bowel disease. Aliment. Pharmacol. Ther 2019;50:121–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu X-W, Ji H-Z, Yang M-F, et al. Helicobacter pylori infection and inflammatory bowel disease in Asians: a meta-analysis. World J. Gastroenterol 2015;21:4750–4756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luther J, Dave M, Higgins PDR, et al. Association between Helicobacter pylori infection and inflammatory bowel disease: a meta-analysis and systematic review of the literature. Inflamm. Bowel Dis 2010;16:1077–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rokkas T, Gisbert JP, Niv Y, et al. The association between Helicobacter pylori infection and inflammatory bowel disease based on meta-analysis. United European Gastroenterol. J 2015;3:539–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Piovani D, Danese S, Peyrin-Biroulet L, et al. Environmental Risk Factors for Inflammatory Bowel Diseases: An Umbrella Review of Meta-analyses. Gastroenterology 2019;157:647–659.e4. [DOI] [PubMed] [Google Scholar]
- 27.Lin K-D, Chiu G-F, Waljee AK, et al. Effects of Anti-Helicobacter pylori Therapy on Incidence of Autoimmune Diseases, Including Inflammatory Bowel Diseases. Clin. Gastroenterol. Hepatol 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wammes LJ, Mpairwe H, Elliott AM, et al. Helminth therapy or elimination: epidemiological, immunological, and clinical considerations. Lancet Infect. Dis 2014;14:1150–1162. [DOI] [PubMed] [Google Scholar]
- 29.Chu KM, Watermeyer G, Shelly L, et al. Childhood helminth exposure is protective against inflammatory bowel disease: a case control study in South Africa. Inflamm. Bowel Dis 2013;19:614–620. [DOI] [PubMed] [Google Scholar]
- 30.Ramanan D, Bowcutt R, Lee SC, et al. Helminth infection promotes colonization resistance via type 2 immunity. Science 2016;352:608–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Okada H, Kuhn C, Feillet H, et al. The “hygiene hypothesis” for autoimmune and allergic diseases: an update. Clin. Exp. Immunol 2010;160:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jostins L, Ripke S, Weersma RK, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012;491:119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matsuzawa-Ishimoto Y, Shono Y, Gomez LE, et al. Autophagy protein ATG16L1 prevents necroptosis in the intestinal epithelium. J. Exp. Med 2017;214:3687–3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cadwell K, Patel KK, Maloney NS, et al. Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell 2010;141:1135–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hand TW, Dos Santos LM, Bouladoux N, et al. Acute gastrointestinal infection induces long-lived microbiota-specific T cell responses. Science 2012;337:1553–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Negroni A, Colantoni E, Vitali R, et al. NOD2 induces autophagy to control AIEC bacteria infectiveness in intestinal epithelial cells. Inflamm Res 2016;65:803–813. [DOI] [PubMed] [Google Scholar]
- 37.Stevens C, Henderson P, Nimmo ER, et al. The intermediate filament protein, vimentin, is a regulator of NOD2 activity. Gut 2013;62:695–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Henderson P, Wilson DC, Satsangi J, et al. A role for vimentin in Crohn disease. Autophagy 2012;8:1695–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuballa P, Huett A, Rioux JD, et al. Impaired autophagy of an intracellular pathogen induced by a Crohn’s disease associated ATG16L1 variant. PLoS One 2008;3:e3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cadwell K, Liu JY, Brown SL, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 2008;456:259–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Small C-LN, Reid-Yu SA, McPhee JB, et al. Persistent infection with Crohn’s disease-associated adherent-invasive Escherichia coli leads to chronic inflammation and intestinal fibrosis. Nat. Commun 2013;4:1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kernbauer E, Ding Y, Cadwell K. An enteric virus can replace the beneficial function of commensal bacteria. Nature 2014;516:94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Olsen I, Tollefsen S, Aagaard C, et al. Isolation of Mycobacterium avium subspecies paratuberculosis reactive CD4 T cells from intestinal biopsies of Crohn’s disease patients. PLoS One 2009;4:e5641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Clancy R, Ren Z, Turton J, et al. Molecular evidence for Mycobacterium avium subspecies paratuberculosis (MAP) in Crohn’s disease correlates with enhanced TNF-alpha secretion. Dig. Liver Dis 2007;39:445–451. [DOI] [PubMed] [Google Scholar]
- 45.Nazareth N, Magro F, Machado E, et al. Prevalence of Mycobacterium avium subsp. paratuberculosis and Escherichia coli in blood samples from patients with inflammatory bowel disease. Med Microbiol Immunol 2015;204:681–692. [DOI] [PubMed] [Google Scholar]
- 46.Garrett WS, Gallini CA, Yatsunenko T, et al. Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe 2010;8:292–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang H, Yu HB, Bhinder G, et al. TLR9 limits enteric antimicrobial responses and promotes microbiota-based colonisation resistance during Citrobacter rodentium infection. Cell Microbiol. 2019;21:e13026. [DOI] [PubMed] [Google Scholar]
- 48.Martin PK, Marchiando A, Xu R, et al. Autophagy proteins suppress protective type I interferon signalling in response to the murine gut microbiota. Nat. Microbiol 2018;3:1131–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marchiando AM, Ramanan D, Ding Y, et al. A deficiency in the autophagy gene Atg16L1 enhances resistance to enteric bacterial infection. Cell Host Microbe 2013;14:216–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim Y-G, Kamada N, Shaw MH, et al. The Nod2 sensor promotes intestinal pathogen eradication via the chemokine CCL2-dependent recruitment of inflammatory monocytes. Immunity 2011;34:769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fonseca DM da, Hand TW, Han S-J, et al. Microbiota-Dependent Sequelae of Acute Infection Compromise Tissue-Specific Immunity. Cell 2015;163:354–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dumay A, Gergaud O, Roy M, et al. Is Crohn Disease the price to pay today for having survived to the Black Death? J. Crohns Colitis 2019. [DOI] [PubMed] [Google Scholar]
- 53.Murthy A, Li Y, Peng I, et al. A Crohn’s disease variant in Atg16l1 enhances its degradation by caspase 3. Nature 2014;506:456–462. [DOI] [PubMed] [Google Scholar]
- 54.Basic M, Keubler LM, Buettner M, et al. Norovirus triggered microbiota-driven mucosal inflammation in interleukin 10-deficient mice. Inflamm. Bowel Dis 2014;20:431–443. [DOI] [PubMed] [Google Scholar]
- 55.Bolsega S, Basic M, Smoczek A, et al. Composition of the Intestinal Microbiota Determines the Outcome of Virus-Triggered Colitis in Mice. Front. Immunol 2019;10:1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Karst SM, Wobus CE. A working model of how noroviruses infect the intestine. PLoS Pathog. 2015;11:e1004626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brodin P, Jojic V, Gao T, et al. Variation in the human immune system is largely driven by non-heritable influences. Cell 2015;160:37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Burger E, Araujo A, López-Yglesias A, et al. Loss of Paneth Cell Autophagy Causes Acute Susceptibility to Toxoplasma gondii-Mediated Inflammation. Cell Host Microbe 2018;23:177–190.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shah SC, Tepler A, Peek RM, et al. Association Between Helicobacter pylori Exposure and Decreased Odds of Eosinophilic Esophagitis-A Systematic Review and Meta-analysis. Clin. Gastroenterol. Hepatol 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Engler DB, Leonardi I, Hartung ML, et al. Helicobacter pylori-specific protection against inflammatory bowel disease requires the NLRP3 inflammasome and IL-18. Inflamm. Bowel Dis. 2015;21:854–861. [DOI] [PubMed] [Google Scholar]
- 61.Wu Y-Z, Tan G, Wu F, et al. H. pylori attenuates TNBS-induced colitis via increasing mucosal Th2 cells in mice. Oncotarget 2017;8:73810–73816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang H, Dai Y, Liu Y, et al. Helicobacter pylori Colonization Protects Against Chronic Experimental Colitis by Regulating Th17/Treg Balance. Inflamm. Bowel Dis 2018;24:1481–1492. [DOI] [PubMed] [Google Scholar]
- 63.Luther J, Owyang SY, Takeuchi T, et al. Helicobacter pylori DNA decreases pro-inflammatory cytokine production by dendritic cells and attenuates dextran sodium sulphate-induced colitis. Gut 2011;60:1479–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Higgins PDR, Johnson LA, Luther J, et al. Prior Helicobacter pylori infection ameliorates Salmonella typhimurium-induced colitis: mucosal crosstalk between stomach and distal intestine. Inflamm. Bowel Dis 2011;17:1398–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Oertli M, Sundquist M, Hitzler I, et al. DC-derived IL-18 drives Treg differentiation, murine Helicobacter pylori-specific immune tolerance, and asthma protection. J. Clin. Invest 2012;122:1082–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Arnold IC, Dehzad N, Reuter S, et al. Helicobacter pylori infection prevents allergic asthma in mouse models through the induction of regulatory T cells. J. Clin. Invest 2011;121:3088–3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kyburz A, Urban S, Altobelli A, et al. Helicobacter pylori and its secreted immunomodulator VacA protect against anaphylaxis in experimental models of food allergy. Clin. Exp. Allergy 2017;47:1331–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Walk ST, Blum AM, Ewing SA-S, et al. Alteration of the murine gut microbiota during infection with the parasitic helminth Heligmosomoides polygyrus. Inflamm. Bowel Dis 2010;16:1841–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cançado GGL, Fiuza JA, Paiva NCN de, et al. Hookworm products ameliorate dextran sodium sulfate-induced colitis in BALB/c mice. Inflamm. Bowel Dis. 2011;17:2275–2286. [DOI] [PubMed] [Google Scholar]
- 70.Hunter MM, Wang A, Hirota CL, et al. Neutralizing anti-IL-10 antibody blocks the protective effect of tapeworm infection in a murine model of chemically induced colitis. J. Immunol 2005;174:7368–7375. [DOI] [PubMed] [Google Scholar]
- 71.Elliott DE, Setiawan T, Metwali A, et al. Heligmosomoides polygyrus inhibits established colitis in IL-10-deficient mice. Eur. J. Immunol 2004;34:2690–2698. [DOI] [PubMed] [Google Scholar]
- 72.Hiemstra IH, Klaver EJ, Vrijland K, et al. Excreted/secreted Trichuris suis products reduce barrier function and suppress inflammatory cytokine production of intestinal epithelial cells. Mol. Immunol 2014;60:1–7. [DOI] [PubMed] [Google Scholar]
- 73.Arai T, Lopes F, Shute A, et al. Young mice expel the tapeworm Hymenolepis diminuta and are protected from colitis by triggering a memory response with worm antigen. Am. J. Physiol. Gastrointest. Liver Physiol 2018;314:G461–G470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Driss V, El Nady M, Delbeke M, et al. The schistosome glutathione S-transferase P28GST, a unique helminth protein, prevents intestinal inflammation in experimental colitis through a Th2-type response with mucosal eosinophils. Mucosal Immunol. 2016;9:322–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Capron M, Béghin L, Leclercq C, et al. Safety of P28GST, a Protein Derived from a Schistosome Helminth Parasite, in Patients with Crohn’s Disease: A Pilot Study (ACROHNEM). J Clin Med 2019;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Broadhurst MJ, Ardeshir A, Kanwar B, et al. Therapeutic helminth infection of macaques with idiopathic chronic diarrhea alters the inflammatory signature and mucosal microbiota of the colon. PLoS Pathog. 2012;8:e1003000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sandborn WJ, Elliott DE, Weinstock J, et al. Randomised clinical trial: the safety and tolerability of Trichuris suis ova in patients with Crohn’s disease. Aliment. Pharmacol. Ther 2013;38:255–263. [DOI] [PubMed] [Google Scholar]
- 78.Schölmerich J, Fellermann K, Seibold FW, et al. A Randomised, Double-blind, Placebo-controlled Trial of Trichuris suis ova in Active Crohn’s Disease. J. Crohns Colitis 2017;11:390–399. [DOI] [PMC free article] [PubMed] [Google Scholar]