

Abstract
Acetylation of histone lysine residues is one of the most well-studied post-translational modifications of chromatin, selectively recognized by bromodomain ‘reader’ modules. Inhibitors of the Bromodomain and Extra Terminal domain (BET) family of bromodomains have shown profound anti-cancer and anti-inflammatory properties, generating much interest in targeting other bromodomain-containing proteins for disease treatment. Herein, we report the discovery of I-BRD9, the first selective cellular chemical probe for Bromodomain-containing protein 9 (BRD9). I-BRD9 was identified through structure-based design, leading to greater than 700-fold selectivity over the BET family and 200-fold over the highly homologous Bromodomain-containing protein 7 (BRD7). I-BRD9 was used to identify genes regulated by BRD9 in Kasumi-1 cells involved in oncology and immune response pathways and to the best of our knowledge, represents the first selective tool compound available to elucidate the cellular phenotype of BRD9 bromodomain inhibition.
Keywords: Bromodomain, BRD9, chemical probe, epigenetics, I-BRD9, inhibitor
Introduction
Chromatin has a highly complex structure composed of nucleosomes, the fundamental packaging elements of genetic information. These structural subunits consist of DNA wrapped around an octameric core of histones, which facilitate the arrangement of DNA in the nucleus of the cell.1 Histone tails are subject to a range of post-translational modifications, aiding the regulation of chromatin accessibility.2 Acetylation of lysine residues is an example of one such modification and is regulated by the dynamic action of histone acetyltransferases (HATs) and histone deacetylases (HDACs), which ‘write’ and ‘erase’ acetylation marks, respectively.3 Bromodomain ‘reader’ modules recognize specific acetyl-lysine (Kac) residues located on histone tails and other proteins. These recognition motifs act together with other chromatin factors to modulate regulation of gene transcription.4 Bromodomains are highly conserved protein modules, comprised of approximately 110 amino acids. The majority are characterized by four alpha-helices, linked by two flexible loop regions, which form the basis of the Kac binding pocket.5
There are at least 56 human bromodomains in 42 bromodomain containing proteins (BCPs) identified to date, neglecting splice variants, each of which has been classified into distinct subgroups based on sequence homology.6,7,8 The Bromodomain and Extra Terminal domain (BET) family of BCPs (BRD2, BRD3, BRD4 and BRDT) contain two bromodomains each, both capable of binding to Kac.3 The most potent and selective bromodomain inhibitors reported to date target all 8 bromodomains of the BET family.4,9,10 Examples include I-BET762 (1),11,12,13 (+)-JQ1 (2),14,15 I-BET151 (3),16,17,18 I-BET726 (4),19 PFI-1 (5)20 and OXF BD 02 (6)21,22 (Figure 1). There are also examples of compounds biased towards binding at either the N-terminal bromodomain (BD1) or C-terminal bromodomain (BD2) of the BET family such as MS436 (7)23 and RVX-208 (8).24,25
Figure 1.

Chemical structures for selected bromodomain inhibitors.
The profound biology associated with the BET family of BCPs suggests there may be therapeutic potential in targeting other members of the bromodomain phylogenetic tree for the treatment of disease. Protein knock-down studies, which block the action of an entire complex, offer a method for probing protein function and linking a BCP to a disease.3 However, as BCPs are multi-domain proteins and often exist as part of multi-body complexes, a chemical probe, which only inhibits the action of the bromodomain may show different effects.26 The therapeutic potential of bromodomain inhibition remains less well explored outside of the BET family,3 in part, due to the limited availability of suitable chemical probes for each member of the human bromodomain phylogenetic tree.27 Therefore, the development of high quality, selective small molecule probes for bromodomains will be a key step in facilitating validation of these reader modules as therapeutic targets. Towards this goal, several groups have generated high quality non-BET bromodomain inhibitors from a diverse set of chemotypes: PFI-3 (9)28 for SMARCA2/4 and PB1(5) domains; I-CBP112 (10),29 SGC-CBP300 (11)30,31 and Ischemin (12)32 for CREBBP and EP300;33 GSK2801 (13)34 for BAZ2B/A and compound 14 for BRPF1 (Figure 1).35 NMR-based screening has been used to identify fragment-sized small molecule inhibitors of PCAF, BAZ2B and ATAD2 providing further evidence for the tractability of non-BET bromodomains.36,37,38 Additionally, a fluorous-tagged multicomponent reaction biased around the 3,5-dimethylisoxazole Kac motif was used to generate the first compounds capable of binding to TAF1.39 There are also several probes available from the Structural Genomics Consortium for the following bromodomains that have not yet had their structures disclosed: BAZ2B/A, BRPF1/2/3, BRPF1.40
Although BRD9 has been reported as a component of the chromatin remodeling SWI/SNF BAF complex,41,42,43 further information regarding its role within disease is somewhat limited.44 The Structural Genomics Consortium have disclosed Bromosporine (15),45 a broad spectrum bromodomain inhibitor, which shows a thermal shift of +3.9 °C at BRD9, suggesting bromodomain binding. In addition, structurally similar compound 16 was reported as the first non-selective inhibitor of BRD9.46,47 This compound shows mixed bromodomain pharmacology, with sub-micromolar activity against BRD9, BRD4, CECR2 and CREBBP. To the best of our knowledge, there are no BRD9 selective chemical probes described in the literature to date.48 Herein, we report the discovery of I-BRD9, the first selective cellular chemical probe for BRD9. The development of I-BRD9 was driven by iterative medicinal chemistry, utilizing structure based design to deliver nanomolar potency at BRD9, > 700 fold selectivity over the BET family and > 70 fold against a panel of 34 bromodomains. I-BRD9 meets and exceeds the chemical probe criteria we defined at the outset of this project, inspired by literature from Bunnage and coworkers,49 who highlighted the importance of high quality chemical probes for target validation: 100 nM or greater potency against the bromodomain of BRD9 as determined by a biochemical assay; 100 fold selectivity over the BET family of bromodomains; 30 fold selectivity over other bromodomain families; cellular activity; and broader selectivity over a range of receptor sites, ion channels, transporters and enzymes.
Results and Discussion
Throughout the course of the investigations to identify a chemical probe for BRD9, selectivity over the BET family of bromodomains was considered of utmost importance. It was felt that the profound biology associated with BET bromodomain inhibition9–11 required at least 100 fold selectivity (as determined using TR-FRET analysis, see Supporting Information), to ensure that any phenotype observed through the use of a BRD9 chemical probe was driven by BRD9 inhibition alone. BRD4 BD1 (N-terminal, bromodomain 1) was used as a representative member of the BET family, driving investigations into selectivity. Analysis of the X-ray crystal structures of apo BRD9 and BRD4 BD1 in complex with I-BET762 (1) revealed a distinct contrast in the architecture of their ZA channels, due to differences in their constituent amino acids. In BRD9, the Tyr106 gate keeper residue blocks access to the lipophilic ‘WPF’ shelf, providing a key difference to the Ile146 gate keeper residue present at the equivalent position of BRD4 BD1 (Figure 2, Table 1). At this stage, it was unclear whether or not selectivity could be gained over the highly homologous BRD7, given its proximity to BRD9 on the phylogenetic tree and sequence similarity within the Kac binding pocket (Figure 3, Table 1).50,51
Figure 2.

X-ray crystal structures of (A) BRD4 BD1 bromodomain (orange) with I-BET762 (1) (cyan); (B) BRD9 bromodomain (blue); (C) BRD4 BD1 surface, and (D) BRD9 surface. In each, the conserved asparagine residue is colored green (BRD4 Asn140 / BRD9 Asn100) and the gatekeeper residue is colored blue (BRD4 Ile146 / BRD9 Tyr106). BRD4 Leu94 and BRD9 Ala54 are colored red
Table 1.
Sequence similarity between the residues of the acetyl-lysine binding sites of BRD4 BD1, BRD9, and BRD7. Colors represent residue properties: green = hydrophobic, red = acidic, blue = basic, orange = polar, purple = proline, white = small. INS = indel.
| BRD4 BD1 | BRD9 | BRD7 | ||
|---|---|---|---|---|
| 1 | TRP81 | GLY43 | ALA154 | WPF motif |
| 2 | PRO82 | PHE44 | PHE155 | WPF motif |
| 3 | PHE83 | PHE45 | PHE156 | WPF motif |
| 4 | GLN84 | ALA46 | SER157 | ZA channel |
| 5 | GLN85 | PHE47 | PHE158 | ZA channel |
| 6 | PRO86 | PRO48 | PRO159 | ZA channel |
| 7 | VAL87 | VAL49 | VAL160 | ZA channel |
| 8 | ASP88 | THR50 | THR161 | ZA loop |
| 9 | ALA89 | ASP51 | ASP162 | ZA loop |
| 10 | LYS91 | INS | INS | ZA loop |
| 11 | LEU92 | INS | INS | ZA loop |
| 12 | ASN93 | ILE53 | ILE164 | ZA loop |
| 13 | LEU94 | ALA54 | ALA165 | ZA loop |
| 14 | TYR97 | TYR57 | TYR168 | Water-binding Tyr |
| 15 | TYR98 | SER58 | SER169 | |
| 16 | ILE101 | ILE61 | ILE172 | |
| 17 | MET132 | MET92 | MET203 | |
| 18 | ASN135 | ASN95 | ASN206 | |
| 19 | CYS136 | ALA96 | ALA207 | Conserved Ala |
| 20 | TYR139 | TYR99 | TYR210 | Conserved Tyr |
| 21 | ASN140 | ASN100 | ASN211 | Conserved Asn |
| 22 | ASP144 | THR104 | THR215 | |
| 23 | ASP145 | VAL105 | ILE216 | |
| 24 | ILE146 | TYR106 | TYR217 | Gatekeeper |
| 25 | VAL147 | TYR107 | TYR218 | |
| 26 | MET149 | LEU109 | ALA220 | |
| 27 | ALA150 | ALA110 | ALA221 |
Figure 3.

Bromodomain phylogenetic tree
Nonetheless, in order to identify inhibitors of the BRD9 bromodomain, a cross-screening strategy of GSK internal compounds was conducted. Thienopyridone 17 was identified from this effort as a potent binder to BRD9 (TR-FRET assay), with pIC50 values of 6.7 and 4.7 against BRD9 and BRD4 BD1, respectively (Figure 4).52
Figure 4.

(A) pIC50 values for compound 17 in the bromodomains of BRD9 and BRD4 BD1 as determined by TR-FRET assays.52 (B) Differences between X-ray crystal structures of 17 in complex with the bromodomain of BRD9 (green) and BRD4 BD1 (blue). (C) Binding mode of 17 in BRD9. Hydrogen-bonds are shown as grey dashed lines and the four conserved water molecules as red spheres. Unfavorable interactions are marked as yellow dashed lines. (D) Binding mode of 17 in BRD4 BD1 (blue) with the position of acetyl-lysine (orange, bromodomains superimposed, from protein data bank entry 2wp2).53 (E,F) Two different orientations of the complex of 17 with BRD9. (G,H) The complex of 17 with BRD4 BD1.
To explain the BRD9 potency of 17 and its selectivity over BRD4, 1.3Å and 1.2Å resolution X-ray crystal structures were solved in BRD9 and BRD4 BD1 respectively. The interactions of the thienopyridone ring are similar in both bromodomains (Figure 4B–D). In BRD9, the carbonyl moiety of the pyridone ring forms a direct hydrogen-bond to the sidechain NH2 group of Asn100, the conserved asparagine residue important for Kac recognition. The pyridone carbonyl also makes a shorter hydrogen-bond to a conserved water molecule bound by Tyr57. The pyridone N-methyl group sits in a small cavity surrounded by four crystallographically conserved water molecules, Phe45 and Val49. The N-methyl pyridone moiety acts as a direct structural analogue of Kac (Figure 4D), whose interactions with BRD9 and BRD4 are typical of those reported for fragments in the corresponding site of BRD4.50,51 The thienopyridone rings fill a tight hydrophobic groove formed by Val49 and Ala54 of the ZA loop on one side, and Tyr106 (the gatekeeper residue) on the other. The 7-dimethoxyphenyl substituent extends outward through the ZA channel, in close contact with Phe44 and Ile53 on either face. The dimethoxy groups participate in a network of hydrogen-bonded water molecules, the 4-methoxy in particular, in a water-mediated interaction with the carbonyl of His42.
At the other end of the molecule, the piperazine amide presents a complex pattern of both desirable and undesirable interactions (Figure 4E). The first unfavorable interaction involves the amide carbonyl oxygen of the linker, which lies 2.7Å from the carbonyl oxygen of Ile53, a close interaction for two electronegative atoms. The second involves the piperidinyl 2-methylene group which lies 2.9Å from the carbonyl oxygen of the conserved asparagine (Asn100) sidechain, preventing its solvation by water. In compensation, the piperazine amide acts as a conformationally restricted linker, directing the terminal methyl sulfonamide to a position where one of its oxygen atoms accepts a hydrogen bond from the backbone NH of Arg101.
The general binding mode of the thienopyridone to BRD4 is the same as to BRD9, but more detailed interactions of this selective compound differ. One difference arises from the double substitution of BRD9 Ala54/Tyr106 for BRD4 Leu94/Ile46 on either side of the thienopyridone ring, leading to a differently shaped pocket illustrated in Figure 2. These result in a tilt of the plane of the thienopyridone ring by approximately 10° away from Leu94 in BRD4 (Figure 4B). The close proximity of Leu94 in BRD4 also forces the thiophene 2-position - carbonyl bond torsion into an almost completely planar conformation, in contrast to the ~30° angle seen in BRD9. This has a knock-on effect on the pendant piperidinyl sulfonamide, which must adopt a different position, and as a result, the sulfonamide is unable to hydrogen-bond to the backbone of Lys141 (the equivalent of Arg101 in BRD9). The electron density for this group is not well defined (Figure S2, Supporting Information), but the best placement of the sulfonamide group is in a region predicted to be electrostatically unfavorable, close to the sidechain oxygen of Tyr139. We believe that this is consistent with the lower potency of 17 for BRD4 compared to BRD9. The hydrogen-bonding potential of Lys141 is satisfied not by the sulfonamide, but instead by a water molecule, which also interacts with the sidechains of Asp144 and Asp140 (Figure 4G, H).
With compound 17 as an encouraging hit, key learnings from the crystallography were taken forward to guide the design of compounds with greater potency and improved selectivity over BRD4. With the hypothesis that the selectivity over BRD4 was driven by the amide carbonyl of compound 17, amide modifications were explored to test our idea.
A synthetic route was developed to access the amide analogues from known bromothienopyridone 1854 (Scheme 1). Halogen-lithium exchange of bromide 18 with n-BuLi at −78 °C, followed by the addition of DMF afforded aldehyde 19. Subsequent electrophilic substitution with NBS, followed by pyridone N-methylation provided key intermediate 21. Amides 17, and 27–29 were synthesized by an initial Pinnick oxidation of aldehyde 21 to give acid 22, followed by amide formation. A subsequent Suzuki-Miyaura coupling with compounds 23–26 was utilized to install the dimethoxyphenyl motif. In contrast, amide 32 was synthesized first by a Suzuki-Miyaura coupling of intermediate 21, followed by a Pinnick oxidation to afford acid 31. Final amide formation using standard peptide coupling conditions furnished compound 32. This alternative route proved more useful, providing access to late stage intermediate 31. Functionalization at the 2-position of 31, through a single amide coupling allowed for the introduction of structural diversity in the final step of the synthesis, providing straightforward access to a range of amides.
Scheme 1.

Synthesis of amides 17 and 27–29 & 32
(a) n-BuLi, THF, −78 °C, then DMF, THF, −60 °C to rt, 65%; (b) NBS, THF, rt, 79%; (c) MeI, Cs2CO3, THF, rt, 71%; (d) NaClO2, NaH2PO4, H2O2, DMSO, H2O, rt, 42–72%; (e) HATU, amine, DIPEA, DMF, rt, 13–79%; (f) (3,4-dimethoxyphenyl)boronic acid, PdCl2(PPh3)2, K2CO3, DME, H2O, microwave, 120 °C, 24–62%.
Analysis of the resulting data relative to compound 17 indicated that the methyl sulfonamide moiety was important for BRD9 potency and, as a result, selectivity over BRD4 BD1 (Table 2). Consistent with the hydrogen-bonding interaction made by the sulfonamide with Arg101 in BRD9 but not Lys141 in BRD4, truncating to piperidine 27 or dimethyl amide 28 reduced BRD9 potency, with little effect on BRD4 activity (compare compounds 17 to 27 and 28). Interestingly, although not selective over BRD4, secondary methyl amide 29 regained the BRD9 potency observed for 17, suggesting that the amide NH makes positive interactions in both BRD4 and BRD9. Finally, cyclic sulfone amide 32 provided a 10 fold improvement in BRD9 activity relative to 29, albeit with little selectivity over BRD4. X-ray crystallography of 32 in the bromodomains of both BRD9 and BRD4 was used to rationalize the observed data (Figure 5).
Table 2. SAR for BRD9 and BRD4 BD1 activity of the thienopyridone amide seriesa.
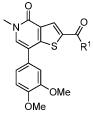 | ||||
|---|---|---|---|---|
| Cmpd | R1= | BRD9 pIC50 | BRD4 BD1 pIC52 | Fold Selectivity |
| 17 | 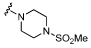 |
6.7 ± 0.16 | 4.7 ± 0.1450 | ×100 |
| 27 | 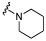 |
5.4 ± 0.14 | 4.8 ± 0.04 | ×4 |
| 28 | 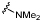 |
5.3 ± 0.10 | 4.9 ± 0.16 | ×3 |
| 29 | 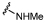 |
6.9 ± 0.12 | 6.7 ± 0.16 | ×2 |
| 32 | 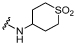 |
7.9 ± 0.09 | 7.3 ± 0.06 | ×4 |
All results are n ≥3 as determined by TR-FRET analysis.
Figure 5.

(A) X-ray crystal structures of BRD9 with 32 (orange) and 17 (green). (B) X-ray crystal structure of 32 in the bromodomains of BRD9 (orange) and BRD4 BD1 (blue). Hydrogen bonds are shown as grey dashed lines and water molecules as red spheres.
The binding mode of the thienopyridone ring of 32 superimposes closely with that seen for 17¸ making identical interactions (Figure 5A). However, the secondary amide of 32 can move closer to the sidechain of Asn100 than is possible for the tertiary amine of 17, forming a direct hydrogen-bond to the asparagine sidechain carbonyl group. This additional interaction may explain the increased potency of 29 relative to 28 and contributes to the greater potency of 32. The electron density for the tetrahydro-2H-thiopyran 1,1-dioxide substituent is quite weak, but interestingly the best fit is with the amide at the axial rather than the equatorial position of the ring (Supporting Information, Figure S3). Another unexpected feature is that when complexed to 32 the ZA loop moves away from the inhibitor compared to its position in the complex with 17 (Figure 5A). This hinge-like motion, involving residues 50-56, results from the tetrahydro-2H-thiopyran ring pushing on the loop through its contacts with Tyr99.
Unexpectedly, the selectivity window for BRD9 over BRD4 is much reduced for 32 than for 17. This can be rationalized by the crystal structures of the compound bound to the two bromodomains (Figure 5B). In contrast to the situation for 17 described above, where different thiophene-amide bond torsion angles in each bromodomain give rise to a selectivity window, the thiophene and amide of 32 are coplanar when bound to either bromodomain. As a result, the sulfone of 32 is able to form a hydrogen bond in each case, to the corresponding backbone NH atoms of Arg101 in BRD9 and Lys141 in BRD4. The similar geometry and interactions in each bromodomain account for the relatively small selectivity window between BRD9 and BRD4.
Despite the low selectivity of 32, the BRD9 potency encouraged further effort to widen the selectivity window. One approach suggested by the X-ray crystal structure of 32 in complex with the bromodomain of BRD9 arose from the observation that the amide carbonyl lies close (within 5 Å) to the backbone carbonyl group of Ile53 (Figure 5A). Although the vectors appeared challenging, it was hypothesized that the replacement of the amide carbonyl group with a hydrogen-bond donor moiety, such as an amidine, could increase activity, either through a direct interaction with the carbonyl of Ile53, or via a through-water hydrogen-bond. Furthermore, it was postulated that selectivity could be gained through exploitation of a key residue difference between BRD9 and BRD4 BD1. Considering the basic nature of the amidine moiety, it was proposed that when charged it would sit more favorably in the less hydrophobic environment alongside Ala54 of BRD9 than beside Leu94 of BRD4 BD1 (Figure 5B). To this end, the amidine analogues of secondary amides 29 and 32 were designed and accessed (Scheme 2).
Scheme 2.

Synthesis of amidines 38–44a
aReagents and conditions: (a) MeI, Cs2CO3, THF, rt, quant.; (b) Zn(CN)2, Pd(PPh3)4, DMF, microwave, 115 °C, 68%; (c) NBS, THF, rt, 86%; (d) NaOMe, MeOH, 75 °C, then amine hydrochloride salt, 87%–quant.; (e) R2B(OH)2, K2CO3, PEPPSI-iPr, IPA, H2O, microwave, 120 °C, 4–52%.
N-Methylation of known thienopyridone 1852 afforded intermediate 33. Following this, a palladium catalysed Negishi cyanation gave nitrile 34. Subsequent bromination at the 7-position of the thienopyridone core with NBS provided 35. Conversion of the nitrile moiety to substituted amidines 36 and 37 was achieved via a Pinner reaction, using NaOMe in MeOH, followed by addition of the appropriate amine.55 With these key intermediates in hand, a Suzuki-Miyaura coupling was utilized to install the different aryl motifs, employing the appropriate boronic acid.
Pleasingly, amidines 38 and 39 retained the BRD9 activity of their direct amide analogues 29 and 32, respectively, with improved levels of selectivity over BRD4 BD1 (Table 3). Transformation of methyl amide 29 to amidine 38 produced a substantial increase in selectivity from 2 to 16 fold. In addition, amidine 39 was 50 fold selective over BRD4, an improvement on the 4 fold seen for its amide analogue, 32.
Table 3. SAR for BRD9 and BRD4 BD1 activity of thienopyridone amidines and their amide analoguesa.
 | ||||
|---|---|---|---|---|
| Cmpd | R = | BRD9 pIC50 | BRD4 BD1 pIC50 | Fold Selectivity |
| 29 | 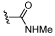 |
6.9 ± 0.12 | 6.7 ± 0.16 | ×2 |
| 32 | 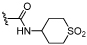 |
7.9 ± 0.09 | 7.3 ± 0.06 | ×4 |
| 38 | 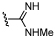 |
7.1 ± 0.13 | 5.9 ± 0.11 | ×16 |
| 39 | 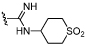 |
8.1 ± 0.19 | 6.4 ± 0.02 | ×50 |
All results are n ≥3 as determined by TR-FRET.
These exciting results provided confidence in our hypothesis that the charged amidine was favored in the less hydrophobic environment of BRD9. With this in mind, further work was conducted in order to gain additional selectivity. The 7-position of the thienopyridone core was selected as an appropriate vector for modification, given its location in the ZA channel. Reaction of advanced intermediate 37 with various boronic acids provided 7-aryl substituted compounds containing both electron-donating and electron-withdrawing substituents (Scheme 2).
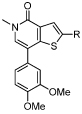
Gratifyingly, all amidines (39-44) delivered ≥ 40 fold selectivity over the first bromodomain of BRD4 (Table 4). These data indicated that both the amidine and phenyl moieties were important for selectivity, with the electronics of the phenyl ring substituents perhaps also playing a role. Unfunctionalized phenyl substituted compound 40, and other electron-donating substituents such as 41 and 42 delivered no significant improvement in selectivity over BRD4 BD1 compared to dimethoxyphenyl derivative 39. However, greater selectivity resulted from replacement of the electron-donating group by electron-withdrawing substituents. Nitrile substituted compound 43 delivered a 60 fold selectivity window, whilst the trifluoromethyl group of compound 44 provided a 160 fold window, satisfying the BET selectivity56 and BRD9 potency probe criteria we defined at the outset of this project. It is of note that the analgous 4-substituted aryl rings to 41-44 demonstrated a similar BRD9 potency profile, however unexpected off target activity prevented their further investigation.
Table 4. SAR for BRD9 and BRD4 BD1 activity of thienopyridone amidinesa.
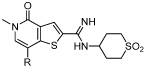 | ||||
|---|---|---|---|---|
| Cmpd | R = | BRD9 pIC50 | BRD4 BD1 pIC50 | Fold Selectivity |
| 39 | 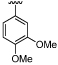 |
8.1 ± 0.19 | 6.4 ± 0.02 | ×50 |
| 40 | 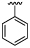 |
7.4 ± 0.13 | 5.8 ± 0.02 | ×40 |
| 41 | 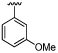 |
7.9 ± 0.12 | 6.1 ± 0.06 | ×60 |
| 42 | 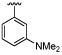 |
8.0 ± 0.23 | 6.3 ± 0.06 | ×50 |
| 43 | 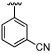 |
7.4 ± 0.12 | 5.6 ± 0.04 | ×60 |
| 44 | 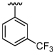 |
7.8 ± 0.21 | 5.6 ± 0.03 | ×160 |
All results are n ≥ 3 as determined by TR-FRET.
X-ray crystal structures of 44 in the bromodomains of BRD9 and BRD4 BD1 provided insights into the interactions responsible for the observed selectivity and supported our hypothesis that led to the synthesis of the amidine. Figure 6A shows the binding mode in BRD9 of 44 compared to that of 32. Altering the substituents of the ZA-channel phenyl ring from 3,4-dimethoxy to 3-trifluoromethyl makes essentially no difference to the observed binding mode of the core of the molecule. The subtle change of the amide of 32 to the amidine of 44 maintains its hydrogen-bond to the conserved Asn100 sidechain (Figure 6A), but leads to more notable changes that suggest significant electrostatic differences between the two molecules. The stronger electrostatics of the amidine NH group allow it to move closer to the backbone carbonyl of Ile53 than was possible with the carbonyl of the amide, resulting in a direct hydrogen-bond between these groups as we had hoped (Figure 6A). One result of this is a ~30° rotation of the thiophene-amidine bond of 44. This allows the tetrahydro-2H-thiopyran 1,1-dioxide amide substituent to resume its equatorial geometry, while maintaining the hydrogen bond between the cyclic sulfone oxygen and the backbone NH of Arg101. The combined effect allows both Tyr99 and residues 50-56 of the ZA loop to relax to a position similar to that seen in the complex with 17, rather than the conformation seen with 32.
Figure 6.

X-ray structure of (A) 32 (orange) and 44 (green) in BRD9, showing the rotation around the thiophene-amidine bond of 44. The significant difference in conformation of the ZA loop that brings the carbonyl group of Ile53 close to the amidine of 44 is also visible. (B) 32 (orange) and 44 (green) in BRD4 BD1 with the 3.6Å distance between the amidine nitrogen and Leu94 shown as a yellow dashed line.
In contrast, the binding mode of 44 in BRD4 BD1 differs little from that of 32, with no movement of the ZA loop (Figure 6B). A very slight movement of the amidine closer to Asn140 probably reflects a tighter charged hydrogen-bonding interaction. The best fit to the density of the tetrahydro-2H-thiopyran 1,1-dioxide amide substituent has equatorial geometry, but this maintains the hydrogen bond between cyclic sulfone and the backbone NH of Lys141. (Figure S2, Supporting Information). An explanation for the reduced BRD4 potency of 44 with respect to 32 is our original hypothesis that a charged amidine group in close proximity to the hydrophobic BRD4 Leu94 sidechain is unfavorable (in the X-ray structure, the distance is 3.6Å). The basic nature of the amidine functionality was confirmed by measurement: the pKa of 44 was determined to be 8.4, so the group would be protonated at neutral pH.
Given the promising activity and selectivity profile displayed by compound 44, this molecule was taken forward for further SAR exploration. It has previously been reported that Kac mimetics containing alkyl groups larger than a methyl can be accommodated in the bromodomain binding pocket.28,57 With this in mind, we sought to investigate the effect of alternative Kac mimetic alkyl groups. To this end, N-ethyl analogue 45 and N-isopropyl analogue 46 were synthesized, via a similar route to that described in Scheme 2.58
Increasing the Kac mimetic alkyl chain length from N-methyl to N-ethyl maintained ≥100 fold selectivity over BRD4 BD1 (Table 5). However, the introduction of the more sterically demanding isopropyl substituent, as in compound 46, reduced BRD9 and BRD4 activities, suggesting that extending beyond N-ethyl is not well tolerated in either of these bromodomains (compare compounds 44 and 46). X-ray crystallography of compound 45 in complex with BRD9 was obtained in order to confirm its binding mode. As expected, 45 adopts a similar conformation to compound 44 (Figure 7A), making identical interactions to Asn100, Ile53 and Arg101. The N-ethyl Kac mimetic is comfortably accommodated in the hydrophobic binding pocket with a slight movement of Phe45, having little effect on the conformation of the remainder of the site.
Table 5. SAR for BRD9 and BRD4 BD1 activity of 3-trifluoromethylphenyl substituted thienopyridone amidinesa.
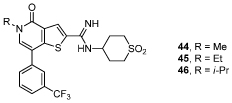 |
|||
|---|---|---|---|
| Cmpd | BRD9 pIC50 | BRD4 BD1 pIC50 | Fold Selectivity |
| 44 | 7.8 ± 0.21 | 5.6 ± 0.03 | ×160 |
| 45 | 7.3 ± 0.18 | 5.3 ± 0.07 | ×100 |
| 46 | 5.5 ± 0.26 | <4.3 | ≥×15 |
All results are n ≥ 3 as determined by TR-FRET analysis.
Figure 7.

(A) Superimposed X-ray crystal structures of the bromodomain of BRD9 complexed with compound 45 (magenta) and 44 (green), showing the subtle movement of Phe45 in response to the N1-methyl/ethyl difference between the two compounds. (B) BROMOscan data for compound 45 (generated at DiscoveRx Corp.)
In order to assess selectivity over non-BET bromodomains, compounds 44 and 45 were profiled in a selection of internal TR-FRET assays, the results of which indicated that compound 45 displayed a superior selectivity profile. Considering this result, compound 45 was progressed for BROMOscan59 profiling against 34 bromodomains to evaluate the effect of the N-ethyl Kac mimetic (Figure 7B). Pleasingly, the results from this broader bromodomain screen indicated nanomolar affinity binding at BRD9, with pKd = 8.7 (for full data, see Supporting Information). Compound 45 showed >700 fold selectivity over the BET family of bromodomains, as well as 200 fold over the highly homologous BRD7 bromodomain (pKd = 6.4). Furthermore, 45 displayed >70 fold selectivity against every other bromodomain tested.
The selectivity of compound 45 over BRD7 is particularly surprising considering the 85% sequence homology between the bromodomains of BRD9 and BRD7 (Table 1). Although an NMR structure of BRD7 has been published,60 there is no crystal structure reported to date. Therefore, it was challenging to rationalize the observed selectivity over this bromodomain. Presumably, selectivity arises from the differences in the constituent amino acids of the WPF shelf (BRD4 nomenclature, see Table 1) and ZA channels of these bromodomains. In BRD9, Gly43 is replaced by Ala154 in the WPF region of BRD7 and Ala46 by Ser157 in the ZA channel. These key changes result in a difference in the architecture of these binding regions, potentially altering the conformation of other residues in the ZA channel.
The observed potency was higher in the BROMOscan format than in house TR-FRET assays. This may be due to use of alternative detection system, protein construct and sample preparation methods. As the rank order is maintained between formats, numerical differences in the reported affinities did not affect decision making.
Based on the high BRD9 affinity and excellent broader bromodomain selectivity results, compound 45 (I-BRD9), was chosen as the chemical probe for BRD9.
As all measurements of binding affinity of I-BRD9 to date had been carried out with truncated bromodomain proteins, we were keen to confirm these findings were consistent with full-length targets in their native context. Using a chemoproteomic competition binding assay in HUT-78 cell lysate, binding of I-BRD9 to endogenous BRD9 displayed >625 fold selectivity against BET family member BRD3 (Figure 8A).56 These data confirms potency at BRD9 and selectivity over the BET family is maintained with endogenous proteins
Figure 8.

(A) Dose-response binding of compound 45 (I-BRD9) for endogenous BRD9 and BRD3 from HuT-78 cell lysates, measured in a chemoproteomic competition binding assay followed by Western blot analysis. (B) BRD9 bromodomain cellular NanoBRET dose-response curve of compound 45 (I-BRD9). (C) qPCR validation of CLEC1, DUSP6, FES and SAMSN1 genes selectively regulated by compound 45 (I-BRD9) (10 μM), but not by I-BET151 (1 μM) mean+/-SD; n=3. Genes were previously identified by full gene transcriptomics in Kasumi-1 cells.
Critically for a bromodomain probe, cellular target engagement of BRD9 and disruption of chromatin binding was demonstrated through a NanoBRET assay measuring displacement of NanoLuc-tagged BRD9 bromodomain from Halo-tagged histone H3.3 (Figure 8B).56
Having established endogenous protein binding, cell and nuclear permeability, excellent selectivity over the BET family and other non-BET bromodomains, further profiling of I-BRD9 was conducted. These experiments aimed to evaluate the selectivity of the compound over a broad range of pharmacological targets including various receptors, transporters, ion channels, kinases and other enzymes. Pleasingly, I-BRD9 showed no activity at less than 5 μM against a panel of 49 targets.56 A summary of the properties of I-BRD9 is given in Table 6.
Table 6.
Summary of Properties of I-BRD9

With a high quality soluble and permeable bromodomain probe in hand, we sought to investigate genes selectively modulated by BRD9 bromodomain inhibition. Accordingly, Kasumi-1 cells were treated with I-BET15115–17 and I-BRD9 for 6 h at pharmacologically matched concentrations. RNA was harvested, reverse transcribed and hybridized on an Illumina expression array. While there was an overlap, the majority of genes were selectively regulated by I-BRD9. Analysis and subsequent validation by qPCR showed 4 genes (CLEC1,62 DUSP6,63 FES64 and SAMSN165) that were strongly down-regulated by I-BRD9 and not by I-BET151 (Figure 8C). These 4 genes are implicated in cancer and immunology pathways, suggesting for the first time a role for the BRD9 bromodomain in their regulation. This down regulation also implies potential utility for synergistic disease modulation by BRD9 inhibition: for example, DUSP6 expression has been implicated in modulating drug sensitivity in cancers which could be important for chemotherapy.61
Conclusion
Through several iterations of structure-based design, starting from tertiary amide 17, we have identified I-BRD9 as the first selective cellular chemical probe for the non-BET bromodomain BRD9. This key contribution provides for the first time the ability to selectively target BRD9 and further enhances the tool set available for targeting human bromodomains. Critical to the success of this endeavor was the selectivity enhancing amidine which drove unfavorable interactions between I-BRD9 and the BET family. This optimized compound is highly potent at BRD9 and displays >700 fold selectivity over the BET family and >70 fold against the panel of 34 bromodomains tested. It is of note that I-BRD9 is selective for a single bromodomain, whereas the vast majority of bromodomain probes bind to ≥2. With proof of bromodomain cellular target engagement, binding to endogenous BRD9 and broader selectivity against a variety of pharmacological targets, we believe this molecule satisfies the requirements necessary for a high quality chemical probe. I-BRD9 was used in the identification a number of BRD9 inhibitor sensitive genes, many of which are involved in immune function and cancer that might indicate the biological role of the BRD9 bromodomain and provides insight and direction for further investigation.
Experimental Section
All solvents were purchased from Sigma Aldrich (Hy-Dry anhydrous solvents) and commercially available reagents were used as received. All reactions were followed by TLC analysis (TLC plates GF254, Merck) or LCMS (liquid chromatography mass spectrometry) using a Waters ZQ instrument. IR spectra were obtained on a Perkin Elmer Spectrum 1 machine. Melting points were measured with a Stuart SMP10, 230 V appartus and were uncorrected.
NMR spectra were recorded at ambient temperature using standard pulse methods on any of the following spectrometers and signal frequencies: Bruker AV-400 (1H = 400 MHz, 13C = 101 MHz), Bruker AV-500 (1H = 500 MHz, 13C = 125 MHz). Chemical shifts are reported in ppm and are referenced to tetramethylsilane (TMS) or the following solvent peaks: CDCl3 (1H = 7.27 ppm, 13C = 77.00 ppm), DMSO-d6 (1H = 2.50 ppm, 13C = 39.51 ppm) and MeOH-d4 (1H = 3.31 ppm, 13C = 49.15 ppm). Coupling constants are quoted to the nearest 0.1 Hz and multiplicities are given by the following abbreviations and combinations thereof: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), br (broad). Column chromatography was performed on pre-packed silica gel columns (30-90 mesh, IST) using a biotage SP4. For detailed LCMS / MDAP / HRMS methodology see Supplementary Methods.
The purity of all compounds tested was determined by LCMS and 1H NMR to be > 95%.
Selected Experimental Procedures
2-Bromo-5-ethylthieno[3,2-c]pyridin-4(5H)-one (47)
A single portion of ethyl iodide (1.40 mL, 17.39 mmol) was added to a solution of bromothieno[3,2-c]pyridin-4(5H)-one (18)52 (2.00 g, 8.69 mmol) and Cs2CO3 (8.50 g, 26.10 mmol) in THF (35 mL) at room temperature. The reaction mixture was heated at 60 °C for 18 h, then allowed to cool to room temperature and diluted with EtOAc (50 mL) and H2O (50 mL). The separated aqueous phase was extracted with EtOAc (3 × 50 mL) and the combined organic layers were passed through a hydrophobic frit and concentrated in vacuo to give 47 (2.30 g, quant.) as a light brown solid. 50 mg of 47 was taken forward for purification by mass directed autopreparation (formic acid), providing suitably pure material (44 mg, 0.17 mmol) for full characterisation; mp 124–125 °C; νmax (solid)/cm–1: 1632, 1577, 1082, 846, 761; 1H NMR (500 MHz, (CD3)2SO) δ ppm 7.62 (s, 1H), 7.56 (d, J = 7.2, 1H), 6.89 (d, J = 7.2, 1H), 3.99 (q, J = 7.2, 2H), 1.24 (t, J = 7.2, 3H); 13C NMR (125 MHz, (CD3)2SO) δ ppm 156.9, 148.9, 134.3, 130.7, 127.5, 112.2, 101.3, 43.8, 15.1; HRMS (M + H)+ calculated for C9H9BrNOS 257.9583; found 257.9585. LCMS (formic acid) (M + H)+ = 258.0, 260.0, Rt = 0.89 min (98%).
5-Ethyl-4-oxo-4,5-dihydrothieno[3,2-c]pyridine-2-carbonitrile (48)
A mixture of 47 (1.10 g, 4.26 mmol), zinc cyanide (1.00 g, 8.52 mmol), and Pd(PPh3)4 (0.44 g, 0.3 mmol) in DMF (10 mL) was heated to 115 °C in a microwave reactor for 3.5 h. This process was repeated to provide a second identical batch. The two batches were allowed to cool to room temperature, diluted with EtOAc (50 mL each) and combined. The solution was filtered through Celite® and concentrated in vacuo. The resulting residue was purified by silica gel chromatography (0–3% MeOH in CH2Cl2). The appropriate fractions were combined and concentrated in vacuo to give 43 (1.70 g, 67%) as a light brown solid. 50 mg of 48 was taken forward for purification by mass directed autopreparation (formic acid), providing suitably pure material (33 mg, 0.16 mmol) for full characterisation; mp 157–158 °C; νmax (solid)/cm–1: 2211, 1637, 1595, 1247, 1078, 879, 780, 769; 1H NMR (400 MHz, (CD3)2SO) δ ppm 8.32 (s, 1H), 7.82 (d, J = 7.2, 1H), 7.03 (d, J = 7.2, 1H), 4.00 (q, J = 7.2, 2H), 1.25 (t, J = 7.2, 3H); 13C NMR (100 MHz, (CD3)2SO) δ ppm 157.6, 151.6, 137.6, 137.2, 129.4, 114.7, 106.9, 101.4, 43.9, 15.0; HRMS (M + H)+ calculated for C10H9N2OS 205.0430; found 205.0429. LCMS (formic acid) (M + H)+ = 205.0, Rt = 0.73 min (98%).
7-Bromo-5-ethyl-4-oxo-4,5-dihydrothieno[3,2-c]pyridine-2-carbonitrile (49)
To a solution of 48 (1.17 g, 5.73 mmol) in THF (20 mL), NBS (1.53 g, 8.59 mmol) was added portion-wise. The reaction mixture was left to stir at room temperature for 48 h. The volatile components were removed in vacuo. The resulting solid was suspended in Et2O (50 mL), filtered under reduced pressure, washed with Et2O (50 mL), collected and dried under vacuum at 40 °C to give 49 (1.69 g, quant.) as an off white solid; mp 197–198 °C; νmax (solid)/cm–1: 3041, 2220, 1654, 1584, 1250, 1090, 766; 1H NMR (500 MHz, (CD3)2SO) δ ppm 8.50 (s, 1H), 8.26 (s, 1H), 4.02 (q, J = 7.2, 2H), 1.26 (t, J = 7.2, 3H); 13C NMR (125 MHz, (CD3)2SO) δ ppm 156.7, 152.9, 138.7, 138.1, 128.8, 114.2, 107.5, 91.0, 44.4, 14.9; HRMS (M + H)+ calculated for C10H8BrN2OS 282.9535; found 282.9534. LCMS (formic acid) (M + H)+ = 283.0, 285.0, Rt = 0.93 min (100%).
7-Bromo-N-(1,1-dioxidotetrahydro-2H-thiopyran-4-yl)-5-ethyl-4-oxo-4,5-dihydrothieno[3,2-c]pyridine-2-carboximidamide (50)
To a suspension of 49 (1.00 g, 3.53 mmol) in MeOH (30 mL) at room temperature, sodium methoxide (25% weight in MeOH, 0.81 mL, 3.53 mmol) was added in a single portion. The reaction mixture was heated at 75 °C for 5 h. 4-Aminotetrahydro-2H-thiopyran-1,1-dioxide hydrochloride (1.98 g, 10.60 mmol) was added and the solution was heated for a further 18 h. The volatile components were removed in vacuo and the resulting solid was suspended in Et2O (50 mL), filtered under reduced pressure, washed with Et2O (50 mL) collected and dried under vacuum at 40 °C to give 50 (1.55 g, quant.) as a white solid. 50 mg of 50 was taken forward for purification by mass directed autopreparation (high pH), providing suitably pure material (25 mg, 0.06 mmol) for full characterisation; mp 284–285 °C; νmax (solid)/cm–1: 1646, 1578, 1284, 1124, 853, 769; 1H NMR (500 MHz, (CD3)2SO) δ ppm 8.18 (s, 1H), 8.01 (s, 1H), 6.80 (br.s, 2H), 4.01 (q, J = 7.2, 2H), 3.68–3.61 (m, 1H), 3.27–3.20 (m, 2H), 3.10–3.03 (m, 2H), 2.09–2.00 (m, 2H), 1.99–1.90 (m, 2H), 1.26 (t, J = 7.2, 3H); 13C NMR (125 MHz, (CD3)2SO) δ ppm 156.9, 150.4, 136.2, 130.2, 122.6, 109.0, 90.6, 48.6, 44.5, 30.0, 15.1; HRMS (M + H)+ calculated for C15H19BrN3O3S2 432.0046; found 432.0046. LCMS (formic acid) (M + H)+ = 432.1, 434.1, Rt = 0.53 min (100%).
N-(1,1-Dioxidotetrahydro-2H-thiopyran-4-yl)-5-ethyl-4-oxo-7-(3-(trifluoromethyl)phenyl)-4,5-dihydrothieno[3,2-c]pyridine-2-carboximidamide (45, I-BRD9)
A mixture of 50 (150 mg, 0.35 mmol), (3-(trifluoromethyl)phenyl)boronic acid (79 mg, 0.42 mmol), PEPPSI-iPr (21 mg, 0.03 mmol) and K2CO3 (115 mg, 0.83 mmol) in H2O (1 mL) and IPA (3 mL) was heated at 120 ºC in a microwave reactor for 0.5 h. The reaction mixture was allowed to cool to room temperature, diluted with EtOAc (40 mL), filtered through Celite® and concentrated in vacuo. The resulting residue was purified by mass directed autopreparation (formic acid). The appropriate fractions were combined and concentrated in vacuo. The resulting residue was dissolved in MeOH (5 mL) and passed through a preconditioned (MeOH, 20 mL) amino propyl column (10 g). The appropriate fractions were combined and concentrated in vacuo to give 45 (50 mg, 29%) as a white solid; mp 228–229 °C; νmax (solid)/cm–1: 1640, 1587, 1340, 1303, 1117; 1H NMR (500 MHz, (CD3)2SO) δ ppm 8.15 (s, 1H), 8.03–7.98 (m, 1H), 7.98–7.94 (m, 1H), 7.91 (s, 1H), 7.82–7.76 (m, 2H), 6.76 (br.s, 2H), 4.10 (q, J = 7.0, 2H), 3.64–3.57 (m, 1H), 3.21–3.12 (m, 2H), 3.10–3.00 (m, 2H), 2.06–1.96 (m, 2H), 1.96–1.85 (m, 2H), 1.32 (t, J = 7.0, 3H); 13C NMR (125 MHz, (CD3)2SO) δ ppm 157.6, 149.7, 147.7, 142.2, 137.5, 134.1, 131.9, 130.7, 130.5, 130.3 (q, JC-F = 31.7), 125.0 (q, JC-F = 3.7), 124.60 (q, JC-F = 3.7), 124.59 (q, JC-F = 272.6), 123.1, 114.4, 49.9, 49.0, 44.0, 30.6, 15.2; 19F NMR (376 MHz, (CD3)2SO) δppm - 61.0; HRMS (M + H)+ calculated for C22H23F3N3O3S2 498.1128; found 498.1110. LCMS (formic acid) (M + H)+ = 498.1, Rt = 0.81 min (100%).
Supplementary Material
Additional text describing all biological methods, chemistry experimental procedures and full BROMOscan data. This material is available free of charge via the Internet http://pubs.acs.org.
Acknowledgment
N. H. T. is grateful to GlaxoSmithKline R&D, Stevenage for PhD. studentship funding. We thank Jacqui Méndez and Danette Daniels of Promega Corporation for facilitating the NanoBRET assay. We also thank Robert Watson and Darren Mitchell for the synthesis of useful building blocks; Matthew Crane for the synthesis of compound 32; Iain Reid for pKa determination; Sean Lynn for assistance with NMR; Marco Smith for high resolution mass spectrometry and Paul White, Keir Sims and Abigail Lucas for crystallization support. We appreciate helpful discussions with Emmanuel Demont during the course of this work.
Abbreviations
- BAF
BRG1/brm-associated factor
- BAZ2A
bromodomain adjacent to zinc finger domain, 2A
- BAZ2B
bromodomain adjacent to zinc finger domain, 2B
- BCP
bromodomain containing protein
- BD
bromodomain
- BET
bromodomain and extra terminal domain
- BRD2
bromodomain containing protein 2
- BRD3
bromodomain containing protein 3
- BRD4
bromodomain containing protein 4
- BRD7
bromodomain containing protein 7
- BRD9
bromodomain containing protein 9
- BRDT
bromodomain containing protein, testis-specific
- CECR2
cat eye syndrome chromosome region, candidate 2
- CREBBP
cAMP response element-binding protein binding protein
- DIPEA
N,N-Diisopropylethylamine
- EP300
E1A binding protein p300
- HAT
histone acetyltransferase
- HATU
O-(7-Azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate
- HDAC
histone deacetylase
- IL-6
interleukin-6
- IPA
isopropanol
- Kac
acetyl lysine
- LCMS
liquid column mass spectrometry
- LPS
lipopolysaccharide
- PB1(5)
PolyBromo 1 (5)
- PCAF
P300/CREBBP-associated factor
- PEPPSI-iPr
pyridine-enhanced precatalyst preparation stabilization and initiation
- PK
pharmacokinetics
- SMARCA2
SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily A, member 2
- SMARCA4
SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily A, member 4
- SWI/SNF
SWItch/sucrose nonfermentable
- TR-FRET
time-resolved Forster (fluorescence) resonance energy transfer
Footnotes
Accession codes
Coordinates have been deposited with the Protein Data Bank (XX) and will be released immediately on publication.
Notes
The authors declare the following competing financial interest(s): All authors except N. H. T., N. C. O. T., H. K. C., A. B., S. C. R. and T. K. were GlaxoSmithKline and Cellzome full-time employees at the time when this study was performed.
References
- 1.Sanchez R, Zhou MM. The role of human bromodomains in chromatin biology and gene transcription. Curr Opin Drug Discovery Dev. 2009;12:659–665. [PMC free article] [PubMed] [Google Scholar]
- 2.Gardner KE, David Allis C, Strahl BD. Operating on chromatin, a colourful language where context matters. J Mol Biol. 2011;409:36–46. doi: 10.1016/j.jmb.2011.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chung CW, Tough DF. Bromodomains: a new target class for small molecule drug discovery. Drug Discov Today: Ther Strategies. 2012;9:111–120. [Google Scholar]
- 4.Chung CW. Small molecule bromodomain inhibitors: extending the druggable genome. Prog Med Chem. 2012;51:1–55. doi: 10.1016/B978-0-12-396493-9.00001-7. [DOI] [PubMed] [Google Scholar]
- 5.Mujtaba S, Zeng L, Zhou MM. Structure and acetyl-lysine recognition of the bromodomain. Oncogene. 2007;26:5521–5527. doi: 10.1038/sj.onc.1210618. [DOI] [PubMed] [Google Scholar]
- 6.Filippakopoulos P, Knapp S. The bromodomain interaction module. FEBS Lett. 2012;586:2682–2704. doi: 10.1016/j.febslet.2012.04.045. [DOI] [PubMed] [Google Scholar]
- 7.Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discovery. 2012;11:384–400. doi: 10.1038/nrd3674. [DOI] [PubMed] [Google Scholar]
- 8.Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, Felletar I, Volkmer R, Muller S, Pawson T, Gingras AC, et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012;149:214–231. doi: 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.For recent reviews of BET bromodomain inhibitors and their therapeutic potential see: Hewings DS, Rooney TPC, Jennings LE, Hay DA, Schofield CJ, Brennan PE, Knapp S, Conway SJ. Progress in the development and application of small molecule inhibitors of bromodomain-acetyl-lysine interactions. J Med Chem. 2012;55:9393–9413. doi: 10.1021/jm300915b. Furdas S, Carlino L, Sippl W, Jung M. Inhibition of bromodomain-mediated protein-protein interactions as a novel therapeutic strategy. Med Chem Commun. 2012;3:123–134. Prinjha RK, Witherington J, Lee K. Place your BETs: the therapeutic potential of bromodomains. Trends Pharmacol Sci. 2012;33:146–153. doi: 10.1016/j.tips.2011.12.002. Knapp S, Weinmann H. Small-molecule modulators for epigenetic targets. ChemMedChem. 2013;3:1885–1891. doi: 10.1002/cmdc.201300344. Sweis RF, Michaelides MR. Recent advances in small-molecule modulation of epigenetic targets: discovery and development of histone methyltransferase and bromodomain inhibitors. Annu Rep Med Chem. 2013;48:185–199. Jennings LE, Measures AR, Wilson BG, Conway SJ. Phenotypic screening and fragment-based approaches to the discovery of small-molecule bromodomain ligands. Future Med Chem. 2014;6:179–204. doi: 10.4155/fmc.13.197. Garnier JM, Sharp PP, Burns CJ. BET bromodomain inhibitors: a patent review. Expert Opin Ther Patents. 2014;24:185–199. doi: 10.1517/13543776.2014.859244.
- 10.For an elegant solution to targeting individual bromodomains of the BET family, see Baud MGJ, Lin-Shiao E, Cardote T, Tallant C, Pschibul A, Chan K-H, Zengerle M, Garcia JR, Kwan TT-L, Ferguson FM, Ciulli A. A bump-and-hole approach to engineer controlled selectivity of BET bromodomain chemical probes. Science. 2014;346:638–641. doi: 10.1126/science.1249830.
- 11.Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, Chandwani R, Marazzi I, Wilson P, Coste H, White J, et al. Suppression of inflammation by a synthetic histone mimic. Nature. 2010;468:1119–1123. doi: 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mirguet O, Gosmini R, Toum J, Clement CA, Barnathan M, Brusq JM, Mordaunt JE, Grimes RM, Crowe M, Pineau O, Ajakane M, et al. Discovery of epigenetic regulator I-BET762: lead optimization to afford a clinical candidate inhibitor of the BET bromodomains. J Med Chem. 2013;56:7501–7515. doi: 10.1021/jm401088k. [DOI] [PubMed] [Google Scholar]
- 13.Chung CW, Coste H, White JH, Mirguet O, Wilde J, Gosmini RL, Delves C, Magny SM, Woodward R, Hughes SA, Boursier EV, et al. Discovery and characterisation of small molecule inhibitors of the BET family bromodomains. J Med Chem. 2011;54:3827–3838. doi: 10.1021/jm200108t. [DOI] [PubMed] [Google Scholar]
- 14.Filippalopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Federov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miyoshi S, Ooike S, Iwata K, Hikawa H, Sugahara K. Int Pat Appl. 2009 WO2009/084693. [Google Scholar]
- 16.Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, Robertson S, Chun CW, Hopf C, Savitski MM, Huthmacher C, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478:529–533. doi: 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mirguet O, Lamotte Y, Donche F, Toum J, Gellibert F, Bouillot A, Gosmini R, Nguyen VL, Delannee D, Seal J, Blandel F, et al. From ApoA1 upregulation to BET family bromodomain inhibition: discovery of I-BET151. Bioorg Med Chem Lett. 2012;22:2963–2967. doi: 10.1016/j.bmcl.2012.01.125. [DOI] [PubMed] [Google Scholar]
- 18.Seal J, Lamotte Y, Donche F, Bouillot A, Mirguet O, Gellibert F, Nicodeme E, Krysa G, Kirilovsky J, Beinke S, McCleary S, et al. Identification of a novel series of BET family bromodomain inhibitors: binding mode and profile of I-BET151 (GSK1210151A) Bioorg Med Chem Lett. 2012;22:2968–2972. doi: 10.1016/j.bmcl.2012.02.041. [DOI] [PubMed] [Google Scholar]
- 19.Gosmini R, Nguyen VL, Toum J, Simon C, Brusq J-MG, Krysa G, Mirguet O, Riou-Eymard AM, Boursier EV, Trottet L, Bamborough P, et al. The discovery of I-BET726 (GSK1324726A), a potent tetrahydroquinoline ApoA1 up-regulator and selective BET bromodomain inhibitor. J Med Chem. 2014;57:8111–8131. doi: 10.1021/jm5010539. [DOI] [PubMed] [Google Scholar]
- 20.Fish PV, Filippakopoulos P, Bish G, Brennan PE, Bunnage ME, Cook AS, Federov O, Gerstenberger BS, Jones H, Knapp S, Marsden B, et al. Identification of a chemical probe for bromo and extra C-terminal bromodomain inhibition through optimization of a fragment-derived hit. J Med Chem. 2012;55(22):9831–9837. doi: 10.1021/jm3010515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hewings DS, Wang M, Philpott M, Fedorov O, Uttarkar S, Filippakopoulos P, Picaud S, Vuppusetty C, Marsden B, Knapp S, Conway SJ, et al. 3,5-dimethylisoxazoles act as acetyl-lysine-mimetic bromodomain ligands. J Med Chem. 2011;54(19):6761–6770. doi: 10.1021/jm200640v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hewings DS, Fedorov O, Filippakopoulos P, Martin S, Picaud S, Tumber A, Wells C, Olcina MM, Freeman K, Gill A, Ritchie AJ, et al. Optimization of 3,5-dimethylisoxazole derivatives as potent bromodomain ligands. J Med Chem. 2013;56(8):3217–3227. doi: 10.1021/jm301588r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang G, Plotnikov AN, Rusinova E, Shen T, Morohasshi K, Joshua J, Zeng L, Mujtaba S, Ohlmeyer M, Zhou M-M. Sturcture-Guided design of potent diazobenzene inhibitors for the BET bromodomains. J Med Chem. 2013;56:9251–9264. doi: 10.1021/jm401334s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McLure KG, Gesner EM, Tsujikawa L, Kharenko OA, Attwell S, Campeau E, Wasiak S, Stein A, White A, Fontano E, Suto RK, et al. RVX-208, an inducer of ApoA-I in humans, is a BET bromodomain antagonist. PLoS One. 2013;8(12):e83190. doi: 10.1371/journal.pone.0083190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Picaud S, Wells C, Felletar I, Brotherton D, Martin S, Savitsky P, Diez-Dacal B, Philpott M, Bountra C, Lingard H, Fedorov O, et al. RVX-208, an inhibitor for BET transcriptional regulators with selectivity for the second bromodomain. Proc Natl Acad Sci USA. 2013;110:19754–19759. doi: 10.1073/pnas.1310658110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muller S, Brown PJ. Epigenetic chemical probes. Clin Pharmacol Ther. 2012;92:689–693. doi: 10.1038/clpt.2012.154. [DOI] [PubMed] [Google Scholar]
- 27.Brand M, Measures AM, Wilson BG, Cortopassi WA, Alexander R, Höss M, Hewings DS, Rooney TPC, Paton RS, Conway SJ. Small molecules inhibitors of boromodomain-acetyl-lysine interactions. ACS Chem Biol. 2015;10:22–39. doi: 10.1021/cb500996u. [DOI] [PubMed] [Google Scholar]
- 28.PFI-3: Selective chemical probe for SMARCA domains. [accessed 06/02/2015]; http://www.thesgc.org/chemical-probes/PFI-3.
- 29.I-CBP112: A CREBBP/EP300-selective chemical probe. [accessed 06/02/2015]; http://www.thesgc.org/chemical-probes/ICBP112.
- 30.SGC-CBP300: A CREBBP/EP300-selective chemical probe. [accessed 06/02/2015]; http://www.thesgc.org/chemical-probes/SGC-CBP30.
- 31.Hay DA, Federov O, Martin S, Singleton DC, Tallant C, Wells C, Picaud S, Philpott M, Monteiro OP, Rogers CM, Conway SJ, et al. Discovery and optimization of small-molecule ligands for the CBP/p300 bromodomains. J Am Chem Soc. 2014;136:9308–9319. doi: 10.1021/ja412434f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Borah JC, Mujtaba S, Karakikes I, Zeng L, Muller M, Patel J, Moshkina N, Morohashi K, Zhang W, Gerona-Navarro G, Hajjar RJ, et al. A small molecule binding to the co-activator CREB-binding protein blocks apoptosis in cardiomyocytes. Chem Biol. 2011;18:531–541. doi: 10.1016/j.chembiol.2010.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.For inhibitors of CREBBP, EP300 and BRD4, see Rooney TPC, Filippakopoulos P, Federov O, Picaud S, Cortopassi WA, Hay DA, Martin S, Tumber A, Rogers CM, Philpott M, Wang M, et al. A series of potent CREBBP bromodomain ligands reveals an induced-fit pocket stabilized by a cation-π interaction. J Angew Chem Int Ed. 2014;53:6126–6130. doi: 10.1002/anie.201402750.
- 34.GSK2801: A selective chemical probe for BAZ2B/A bromodomains. [accessed 06/02/2015]; http://www.thesgc.org/chemical-probes/GSK2801.
- 35.Demont EH, Bamborough P, Chung CW, Craggs PD, Fallon D, Gordon LJ, Grandi P, Hobbs CI, Hussain J, Jones EJ, Le Gall A, et al. 1,3-Dimethyl benzimidazolones are potent, selective inhibitors of the BRPF1 bromodomain. ACS Med Chem Lett. doi: 10.1021/ml5002932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zeng L, Li J, Muller M, Yan S, Mujtaba S, Pan C, Wang Z, Zhou MM. Selective small molecules blocking HIV-1 Tat and coactivator PCAF association. J Am Chem Soc. 2005;127:2376–2377. doi: 10.1021/ja044885g. [DOI] [PubMed] [Google Scholar]
- 37.Ferguson FM, Federov O, Chaikaud A, Philpott M, Muniz JRC, Felletar I, von Delft F, Heightman T, Knapp S, Abell C, Ciulli A. Targeting low-drugability bromodomains: fragment based screening and inhibitor design against the BAZ2B bromodomain. J Med Chem. 2013;56:10183–10187. doi: 10.1021/jm401582c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harner MJ, Chauder BA, Phan J, Fesik SW. Fragment-Based screening of the Bromodomain of ATAD2. J Med Chem. 2014;57:9687–9692. doi: 10.1021/jm501035j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McKeown MR, Shaw DL, Fu H, Liu S, Xu X, Marineau JJ, Huang Y, Zhang X, Buckley DL, Kadam A, Zhang Z, et al. Biased multicomponent reactions to develop novel bromodomain inhibitors. J Med Chem. 2014;57:9019–9027. doi: 10.1021/jm501120z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.BAZ2-ICR: a chemical probe for BAZ2A/B bromdomains; OF-1: A chemical probe for BRPF bromodomains; PFI-4: A chemical probe for BRPF1B; NI-57: A chemical probe for BRPF bromodomains. [accessed 06/02/2015]; http://www.thesgc.org/chemical-probes/epigenetics.
- 41.Middeljans E, Wan X, Jansen PW, Sharma V, Stunnenberg HG, Logie C. SS18 Together with animal-specific factors defines human BAF-type SWI/SNF complexes. PLoS ONE. 2012;7:e33834. doi: 10.1371/journal.pone.0033834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scotto L, Narayan G, Nandula SV, Subramaniyam S, Kaufmann AM, Wright JD, Pothuri B, Mansukhani M, Schneider A, Arias-Pulido H, Murty VV. Integrative genomics analysis of chromosome 5p gain in cervical cancer reveals target over-expressed genes, including Drosha. Mol Cancer. 2008;7:58–67. doi: 10.1186/1476-4598-7-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, Crabtree GR. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet. 2013;45:592–601. doi: 10.1038/ng.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hohmann AF, Vakoc CR. A rationale to target the SWI/SNF complex for cancer therapy. Trends Genet. 2014;30:356–363. doi: 10.1016/j.tig.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bromosporine. [accessed 06/02/2015]; http://www.thesgc.org/chemical-probes/bromosporine.
- 46.Federov O, Lingard H, Wells C, Monteiro OP, Picaud S, Keates T, Yapp C, Philpott M, Martin SJ, Felletar MI, Marsden B, et al. [1, 2, 4]Triazolo[4,3-a]phthalazines: inhibitors of diverse bromodomains. J Med Chem. 2014;57:462–476. doi: 10.1021/jm401568s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.An structurally similar compound with BRD9 activity was also identified, see Guetzoyan L, Ingham RJ, Nikbin N, Rossignol J, Wolling M, Baumert M, Burgess-Brown NA, Strain-Damerell CM, Shrestha L, Brennan PE, Federov O, et al. Machine-assisted synthesis of modulators of the histone reader BRD9 using flow methods of chemistry and frontal affinity chromatography. Med Chem Commun. 2014;5:540–546.
- 48.During manuscript preparation, the Structural Genomics Consortium disclosed the availability of LP99, a chemical probe for BRD9 and BRD7 via their website. [accessed 06/02/2015]; http://www.thesgc.org/node/9361.
- 49.Bunnage ME, Piatnitski Chekler EL, Jones LH. Target validation using chemical probes. Nat Chem Biol. 2013;9:195–199. doi: 10.1038/nchembio.1197. [DOI] [PubMed] [Google Scholar]
- 50.Bamborough P, Diallo H, Goodacre JD, Gordon L, Lewis A, Seal JT, Wilson DM, Woodrow MD, Chung CW. Fragment-based discovery of bromodomain inhibitors part 2: optimization of phenylisoxazole sulfonamides. J Med Chem. 2012;55:587–596. doi: 10.1021/jm201283q. [DOI] [PubMed] [Google Scholar]
- 51.Chung CW, Dean AW, Woolvern JM, Bamborough P. Fragment-based discovery of bromodomain inhibitors part 1: inhibitor binding modes and implications for lead discovery. J Med Chem. 2012;55:576–586. doi: 10.1021/jm201320w. [DOI] [PubMed] [Google Scholar]
- 52.On 2 of 7 test occasions compound 17 showed pIC50 < 4.3 against BRD4 BD1. As such, the mean and standard deviation are calculated from 5 of 7 test occasions
- 53.The acetyl lysine is from a BRDT BD1 structure, see: Moriniere J, Rousseaux S, Steuerwald U, Soler-Lopez M, Curtet S, Vitte AL, Govin J, Gaucher J, Sadoul K, Hart DJ, Krijgsveld J, et al. Cooperative binding by acetylation marks on a histone tail by a single bromodomain. Nature. 2009;461:664–668. doi: 10.1038/nature08397.
- 54.Gentile G, Bernasconi G, Pozzan A, Merlo G, Marzorati P, Bamborough P, Bax B, Bridges A, Brough C, Carter P, Cutler G, et al. Identification of 2-(4-pyridyl)thienopyridones as GSK-3β inhibitors. Bio Org Med Chem Lett. 2011;21:4823–4827. doi: 10.1016/j.bmcl.2011.06.050. [DOI] [PubMed] [Google Scholar]
- 55.Mathews TP, Kennedy AJ, Kharel PC, Kennedy PC, Nicoara O, Sunkara M, Morris AJ, Wamhoff BR, Lynch KR, Macdonald TL. Discovery, biological evaluation, and structure-activity relationship of amidine based sphingosine kinase inhibitors. J Med Chem. 2010;53:2766–2778. doi: 10.1021/jm901860h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Compound 44 showed pIC50 values of 5.1 ± 0.03 against BRD2 BD1; 5.2 ± 0.05 against BRD3 BD1; and 4.9 ± 0.09 against BRDT BD1
- 57.Vollmuth F, Geyer M. Interaction of propionylated and butyrylated histone H3 lysine marks with BRD4 bromodomains. Angew Chem Int Ed. 2010;49:6768–6772. doi: 10.1002/anie.201002724. [DOI] [PubMed] [Google Scholar]
- 58.See Supporting Information for details
- 59.BROMOscan recombinant protein binding assays were carried out at DiscoveRx. http://www.discoverx.com.
- 60.Hongbin S, Jiangxin L, Jiahai Z, Weiqun S, Hongda H, Chao X, Haiming D, Jihui W. Solution structure of BRD7 bromodomain and its interaction with acetylated peptides from histone H3 and H4. Biophys Res Commun. 2007;358:435–441. doi: 10.1016/j.bbrc.2007.04.139. Biochem. [DOI] [PubMed] [Google Scholar]
- 61.Young RJ, Green DVS, Luscombe CN, Hill A. Getting physical in drug discovery II: the impact of chromatographic hydrophobicity measurements and aromaticity. Drug Discov Today. 2011;16:822–830. doi: 10.1016/j.drudis.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 62.C-type lectin-like receptor-1 (CLEC-1); For a role in the immune response, see Sattler S, Reiche D, Sturtzel C, Karas I, Richter S, Kalb ML, Gregor W, Hofer E. The Human C-Type Lectin-Like Receptor CLEC-1 is Upregulated by TGF-ss and Primarily Localized in the Endoplasmic Membrane Compartment. Scand J Immunol. 2012;75:282–292. doi: 10.1111/j.1365-3083.2011.02665.x.
- 63.Dual specificity phosphatase 6 (DUSP6); For a role in cancer, see Bagnyukova TV, Restifo D, Beeharry N, Gabitova L, Li T, Serebriiskii IG, Golemis EA, Astsaturov I. DUSP6 regulates drug sensitivity by modulating DNA damage response. Br J Cancer. 2013;109:1063–1071. doi: 10.1038/bjc.2013.353.
- 64.Feline sarcoma oncogene (FES); For a role in cancer, see Zhang S, Chitu V, Stanley ER, Elliott BE, Greer PA. Fes Tyrosine Kinase Expression in the Tumor Niche Correlates with Enhanced Tumor Growth, Angiogenesis, Circulating Tumor Cells, Metastasis, and Infiltrating Macrophages. Cancer Res. 2011;71:1465–1473. doi: 10.1158/0008-5472.CAN-10-3757.
- 65.SAM domain, SH3 domain and nuclear localization signals 1 (SAMSN1); For a role in cancer, see Yan Y, Zhang L, Xu T, Zhou J, Qin R, Chen C, Zou Y, Fu D, Hu G, Chen J, Lu Y. SAMSN1 is highly expressed and associated with a poor survival in glioblastoma multiforme. PLoS ONE. 2013;8:e81905. doi: 10.1371/journal.pone.0081905.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.