

Abstract
Tilimycin is an enterotoxin produced by the opportunistic pathogen Klebsiella oxytoca that causes antibiotic-associated hemorrhagic colitis (AAHC). This pyrrolobenzodiazepine (PBD) natural product is synthesized by a bimodular nonribosomal peptide synthetase (NRPS) pathway comprised of three proteins: NpsA, ThdA and NpsB. We describe the functional and structural characterization of the fully reconstituted NRPS system and report the steady-state kinetic analysis of all natural substrates and cofactors as well as the structural characterization of both NpsA and ThdA. The mechanism of action of tilimycin was confirmed using DNA adductomics techniques through the detection of putative N-2 guanine alkylation after tilimycin exposure to eukaryotic cells, providing the first structural characterization of a PBD-DNA adduct formed in cells. Finally, we report the rational design of small-molecule inhibitors that block tilimycin biosynthesis in whole cell K. oxytoca (IC50 = 29 ± 4 μM) through the inhibition of NpsA (KD = 29 ± 4 nM).
Keywords: Nonribosomal peptide synthetase, adenylation, microbiome, tilimycin, pyrrolobenzodiazepine, Klebsiella oxytoca
Graphical Abstract
INTRODUCTION
The gastrointestinal (GI) tract of every human houses an extraordinarily complex ecosystem of microorganisms, collectively known as the GI microbiota, with representation of over one thousand different species from all domains of life.1 Advances in metagenomic screening over the past decade have begun to reveal the profound influence of the GI microbiota on human health and disease.2–7 Normally, these commensal microorganisms work harmoniously with the human host to maintain a symbiotic relationship,8,9 but disturbance of the gut microbiota leads to dysbiosis, which can have far reaching effects.10,11 While many chronic diseases including inflammatory bowel disease, obesity, diabetes, and neurodegenerative disorders have been correlated to dysbiosis of gut microbial communities,12–22 the causative links between microbiota and disease are often shrouded by the complexity of the host-microbiota interactions.
Klebsiella oxytoca and its role in antibiotic-associated hemorrhagic colitis (AAHC) serves as a model for understanding the molecular basis of a disease caused by an imbalance of the gut microbiota.23–25 K. oxytoca is an opportunistic gram-negative bacterial species that resides at low abundance in the colon of 2–10 percent of healthy individuals.23,26 Following β-lactam antibiotic treatment, K. oxytoca utilizes its constitutively expressed β-lactamase to survive while commensal bacteria are eliminated. In the absence of an otherwise competitive microbial community, K. oxytoca thrives, triggering severe dysbiosis. Subsequent expansion of K. oxytoca in the colon leads to overproduction of tilivalline and related small molecule enterotoxins that cause apoptosis of colonic epithelium cells.27–30 Originally, the cytotoxicity and corresponding AAHC was attributed solely to tilivalline,23,31,32 but further analysis led to the identification of tilimycin,27–29 which is a more potent cytotoxin and likely plays a greater role in the pathogenicity of K. oxytoca,30,33
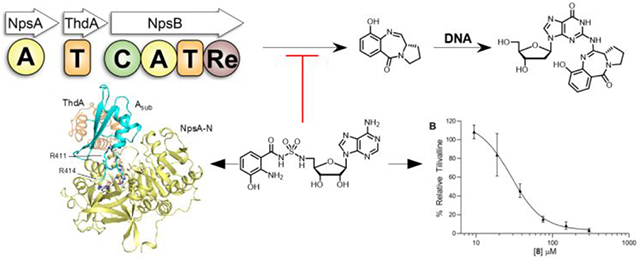
Tilimycin is a tricyclic pyrrolobenzodiazepine synthesized by K. oxytoca via a nonribosomal peptide synthetase (NRPS) pathway (Figure 1).24,28,29 NRPSs are large multi-modular proteins responsible for the synthesis of short peptides in an assembly line like fashion, independent of mRNA ribosomal machinery. The formation of a single amide bond in a NRPS module requires three unique domains. First, an adenylation domain (A) is responsible for amino acid selection, activation and transfer onto the downstream thiolation domain. The thiolation domain (T) contains a flexible and mobile phosphopantetheinyl cofactor arm which carries the amino acid that is connected via a thioester linkage. Finally, a condensation domain (C) catalyzes amide bond formation between two loaded amino acids.34 Tilimycin biosynthesis is performed by a bimodular NRPS comprised of NpsA, ThdA and NpsB. NpsA and ThdA are stand-alone proteins for adenylation and thiolation respectively while NpsB contains a complete C-A-T module along with a C-terminal reductase (Re) domain for release of tilimycin. Bioinformatic analyses of signature sequences within the adenylation domains suggest 3-hydroxyanthranilic acid is the substrate for NpsA while NpsB confers specificity for l-proline.24 Thus, NpsA is predicted to activate and load the stand alone thiolation protein ThdA with 3-hydroxyanthranilic acid, while NpsB activates and loads l-proline on to its corresponding thiolation domain. The condensation domain is predicted to catalyze amide bond formation between the 3-hydroxyanthranilate thioester and the free amine of l-proline to afford the l-N-(3-hydroxyanthraniloyl)prolyl-S~TNpsB acyl-peptide intermediate followed by reductive release of the thioester to l-N-(3-hydroxyanthraniloyl)prolinal mediated by the reductase domain.24 Ring-chain tautomerization of the amino aldehyde to the close-chain hemiaminals yields tilimycin. which can undergo non-enzymatic reaction with indole, produced in large quantities endogenously in K. oxytoca, to afford tilivalline.33
Figure 1.
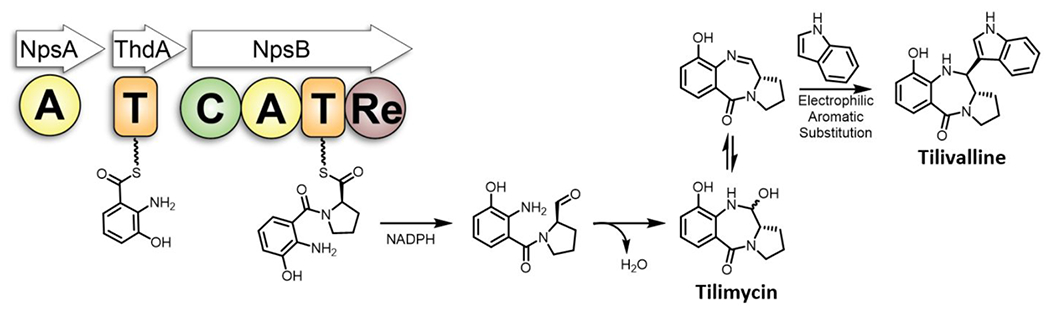
The NRPS pathway for the biosynthesis of tilimycin. Reductive release of the dipeptide affords l-N-(3-hydroxyanthraniloyl)prolinal that spontaneously cyclizes to a mixture of diastereomeric aminals known as tilimycin that are in equilibrium with the imine species. Non-enzymatic condensation with indole yields tilivalline. A = adenylation domain, T = thiolation domain, C = condensation domain, Re = thioester-reductase domain.
Herein we report the detailed functional and structural characterization of the tilimycin biosynthetic pathway that extends prior metabolic profiling and mutasynthesis studies to define the overall pathway and substrate tolerance.28 We then describe the synthesis of two classes of rationally designed bisubstrate inhibitors which enabled structural characterization of NpsA and a NpsA-ThdA fusion protein in the adenylation and thioester conformations, respectively. One of the bisubstrate inhibitors was shown to selectively inhibit tilimycin synthesis in cell culture without impairing bacterial replication. Finally, to investigate the molecular mechanism of action of the pyrrolobenzodiazepine class of natural products, we utilized DNA adductomic techniques to successfully detect and structurally characterize alkylated DNA from eukaryotic cells exposed to tilimycin, helping to further clarify the causative link between a microbiota derived natural product and human disease.
RESULTS
Cloning, Expression and Purification of NpsA, holo-ThdA and holo-NpsB.
The thdA and npsB genes were PCR amplified from K. oxytoca strain MIT-5243 genomic DNA and cloned into a pET15b vector containing a TEV cleavage site and a 5-His affinity tag at the N-terminus. The npsA gene was codon optimized for expression in E. coli, synthesized and cloned into a pET15b vector containing an identical 5-His TEV cleavage site. The plasmids were transformed and overexpressed in E. coli BL21(DE3) cells. Two step purification was performed starting with immobilized metal affinity chromatography (IMAC). The partially purified apo-ThdA and apo-NpsB were incubated with the promiscuous phosphopantetheinyl transferase Sfp from Bacillus subtilis to ensure conversion to the holo protein form. Subsequent size exclusion chromatography afforded 20 mg/L of NpsA, 27 mg/L of holo-ThdA and 3 mg/L of holo-NpsB. The molecular mass of the proteins determined by SDS-PAGE were consistent with their calculated molecular weight: ~56 kDa (55,772 kDa calculated) for NpsA, ~10 kDa (9,486 kDa calculated) for holo-ThdA, and ~150 kDa (164,015 kDa calculated) for holo-NpsB (Figure S1).
Biochemical Characterization of NpsA and ThdA.
NpsA catalyzes a two-step ATP-dependent acylation of the phosphopantetheine prosthetic group of holo-ThdA to yield 3-hydroxyanthraniloyl-ThdA (acyl-ThdA) that proceeds through a 3-hydroxyanthraniloyl adenosine monophosphate (3HA-AMP) intermediate (Figure 2A). Catalysis of these two steps requires dynamic movement of the C-terminal hinge region of NpsA as observed in other adenylating enzymes.35–37 Initially, the “closed” adenylation conformation allows for binding and ligation of aryl-acid and ATP substrates. The “open” thiolation conformation enables holo-ThdA recruitment and subsequent acylation. While prior in vitro mutasynthesis studies have revealed that NpsA possesses relaxed substrate specificity for anthranilic acid derivatives27–29, steady-state kinetic characterization of NpsA has not been reported. To assess the steady-state kinetics of the complete adenylation-acylation reaction, we used a continuous coupled assay wherein the pyrophosphate (PPi) byproduct generated in the initial adenylation reaction is converted to phosphate by inorganic pyrophosphatase and then purine nucleoside phosphorylase catalyzes the phosphorolysis of 7-methylthioguanosine to 7-methylthioguanine (MesG), whose formation can be monitored at 360 nm.38,39 We confirmed the reaction velocity was linearly dependent on NpsA concentration for this coupled assay. ThdA was also required for full activity and the reaction rate decreased 140-fold in the absence of ThdA. Initial velocity experiments were performed by varying one substrate at fixed saturating concentrations of the other two substrates and provided apparent KM values of 63, 15 and 3.4 μM for 3-hydroxyanthranilic acid, ATP and holo-ThdA, respectively, with an average apparent kcat equal to 0.89 s−1 (Figure 2A, Table 1). Because of reduced rates at high concentrations of ThdA, the kinetic data were fit to a substrate-inhibited model providing a substrate inhibition constant, Ki of 15 μM. Interestingly, substrate inhibition by thiolation domains has not been previously observed. We next explored the substrate specificity of 3-hydroxyanthranilic acid with 3-hydroxybenzoic acid and anthranilic acid to probe the importance of the 2-amino or 3-hydroxyl substituents. Removal of the 3-hydroxy group in anthranilic acid led to a 52-fold decrease in the specificity constant (kcat/KM), while deletion of the 2-amino group in 3-hydroxybenzoic acid provided a more pronounced 200-fold decrease in specificity constant indicating the 2-amino group is more critical for catalytic efficiency than the 3-hydroxy group.
Figure 2:
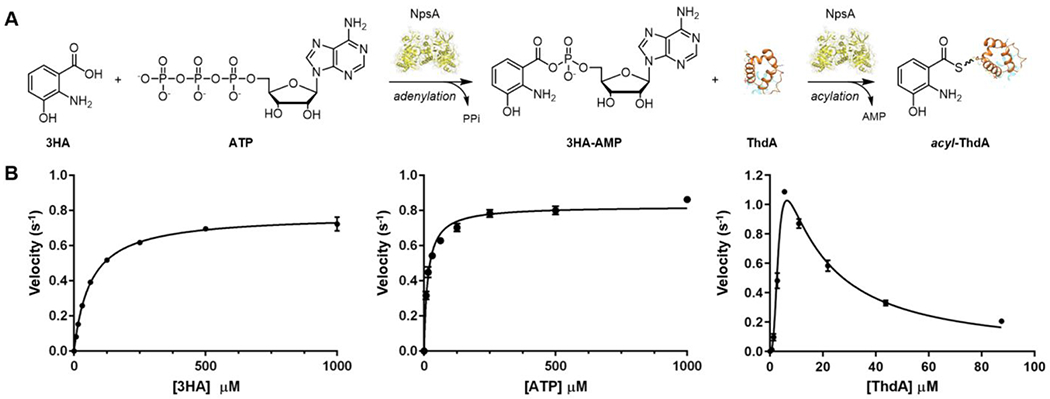
A) Adenylation-ligation enzymatic reaction catalyzed by NpsA. B) Initial velocity vs substrate concentration [3HA], [ATP] and [ThdA]. Data were fit to the Michaelis-Menten equation or substrate inhibition model. Each reaction contained 5 mM ATP and 20 μM ThdA (for varying 3HA), 500 μM 3-HA and 20 μM ThdA (for varying 3-ATP), 500 μM 3HA and 5 mM ATP (for varying ThdA) in 50 mM Tris, 150 mM NaCl, 10 mM MgCl2 pH 8 buffer with 20 nM NpsA. Data are average (±SD) of triplicate experiments.
Table 1.
Apparent Kinetic parameters for NpsA.
| Substrate | KM (μM) | kcat (s−1) | kcat/KM (M−1·s−1) |
|---|---|---|---|
| 3-Hydroxyanthranilic acid | (6.3 ± 0.2) × 101 | 0.78 ± 0.01 | (1.8 ± 0.1) × 104 |
| 3-Hydroxybenzoic acid | (4.0 ± 0.9) × 103 | 0.35 ± 0.07 | (8.8 ± 2.6) × 101 |
| Anthranilic acid | (8.5 ± 0.7) × 102 | 0.29 ± 0.01 | (3.4 ± 0.3) × 102 |
| ATP | (1.5 ± 0.1) × 101 | 0.82 ± 0.01 | (5.6 ± 0.4) × 104 |
| holo-ThdA | 3.4 ± 0.2, (Ki = 15 ± 2) | 1.08 ± 0.02 | (3.2 ± 0.2) × 105 |
Characterization of NpsB and Fully Reconstituted Assay.
The next downstream enzyme in the NRPS pathway is NpsB with a C-A-T-Re multidomain architecture. Unlike the standalone proteins NpsA and ThdA, the enzymatic activities of NpsB cannot be easily decoupled. We initially attempted to interrogate the activity of the embedded adenylation domain of NpsB employing hydroxylamine to facilitate enzyme turnover by reaction with the acyl-adenylate intermediate using the coupled assay employed for NpsA.40,41 Addition of hydroxylamine to the NpsB assay containing saturating l-proline and ATP did not stimulate enzyme turnover as measured by PPi release suggesting acyl transfer to the prolyl-loaded thiolation domain (l-Pro-S~TNpsB) was rapid or the initial l-Pro-adenylate was inaccessible for reaction with hydroxylamine (Figure S2). The observed steady-state rate for PPi formation of 0.01 sec−1 (that is dependent on both l-proline and ATP) is consistent with the leak of the acyl-adenylate from the adenylation domain, which enables slow enzyme turnover. We next explored whether addition of NADPH to the NpsB assay containing saturating l-proline and ATP could induce turnover of l-prolyl-S~TNpsB through reduction of the proline thioester by the reductase (Re) domain to a prolinal shunt product (Figure 3A, red arrow). However, addition of NADPH did not stimulate enzyme turnover as measured by PPi release or NADPH consumption which suggests the Re domain is only operative on the native l-A-(3-hydroxyanthraniloyl)prolyl-S~TNpsB substrate. To test this hypothesis, we developed a fully reconstituted assay with stoichiometric 3-hydroxyanthraniloyl-S~ThdA as substrate, which could be generated by preincubating stoichiometric ThdA with catalytic NpsA and 3-hydroxyanthranilic acid. Incubation of the substrates 3-hydroxyanthraniloyl-S~ThdA, l-proline, ATP and NADPH with catalytic NpsB afforded tilimycin as confirmed with an authentic synthetic standard (Figure 3A, blue arrow). To investigate the apparent steady-state enzyme kinetic parameters of NpsB, we measured NADPH consumption at 340 nm and varied the concentration of one substrate (i.e. l-proline) at fixed saturating concentration of the other two substrates (i.e. ATP and NADPH) along with 20 μM 3-hydroxyanthraniloyl-S~ThdA. Initial velocity experiments provided apparent KM values of 1.8 × 103, 21 and 57 μM for of l-proline, ATP and NADPH, respectively with an average apparent kcat equal to 0.050 s−1 (Figure 3B, Table 2)
Figure 3.
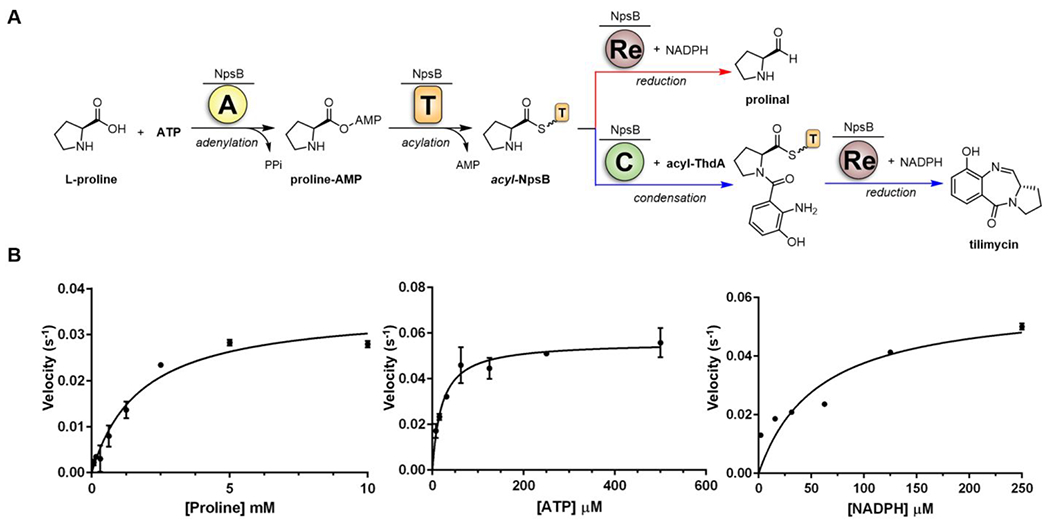
A) Overall enzymatic reaction catalyzed by NpsB. The A-domain of NpsB activates l-proline to the prolyl-adenylate species and transfers it to the downstream T-domain of NpsB. The intermediate l-Pro-S~T can undergo two competitive reactions: direct reduction by the reductase domain (red arrow) or condensation with acyl-ThdA (blue arrow), which can subsequently be reduced by the Re domain to afford tilimycin. B) Initial velocity vs substrate concentration [l-Proline], [ATP] and [NADPH], Data were fit to the Michaelis-Menten equation. Each reaction contained 5 mM ATP and 0.5 mM NADPH (for varying l-Proline), 5 mM l-Proline and 0.5 mM NADPH (for varying ATP), 5 mM l-Proline and 5 mM ATP (for varying NADPH) in 50 mM Tris, 150 mM NaCl, 10 mM MgCl2 pH 8 buffer with 250 nM NpsB and 20 μM 3-hydroxyanthraniloyl-S~ThdA. Data are average (±SD) of triplicate experiments.
Table 2.
Apparent Kinetic parameters for NpsB.
| Substrate | KM (μM) | kcat (s−1) | kcat/KM (M−1·s−1) |
|---|---|---|---|
| l-Proline | (1.8 ± 0.3) × 103 | 0.037 ± 0.002 | (2.1 ± 0.4) × 101 |
| ATP | (2.1 ± 0.3) × 101 | 0.056 ±0.002 | (2.7 ± 0.4) × 103 |
| NADPH | (5.7 ± 1.7) × 101 | 0.058 ± 0.007 | (1.0 ± 0.3) × 103 |
Rational Design and Synthesis of NpsA Inhibitors.
Adenylation enzyme inhibitors that mimic the acyl-AMP intermediate (Figure 4A and 4B) can be developed by replacing the labile phosphate moiety of the acyl-AMP with a stabile non-cleavable bioisostere such as sulfamide to afford an acyl adenosine monosulfamide (acyl-AMS) inhibitor.42 The sulfamide moiety closely mimics the tetrahedral geometry and charge distribution of the ionized phosphate group, but is chemically and enzymatically stabile. Acyl-AMS inhibitors derive their high affinity by simultaneously interacting with the carboxylic acid and ATP binding pockets of the adenylation domain while their specificity arises from the acyl group. The NRPS assembly line responsible for tilimycin biosynthesis contains two potential adenylation domain targets, NpsA and NpsB. The stand-alone adenylating enzyme NpsA was selected as a target because it uses the esoteric substrate, 3-hydroxyanthranilic acid, ensuring higher selectivity than NpsB, whose adenylation domain is functionally related to prolyl tRNA synthetases found in all domains of life.43
Figure 4.
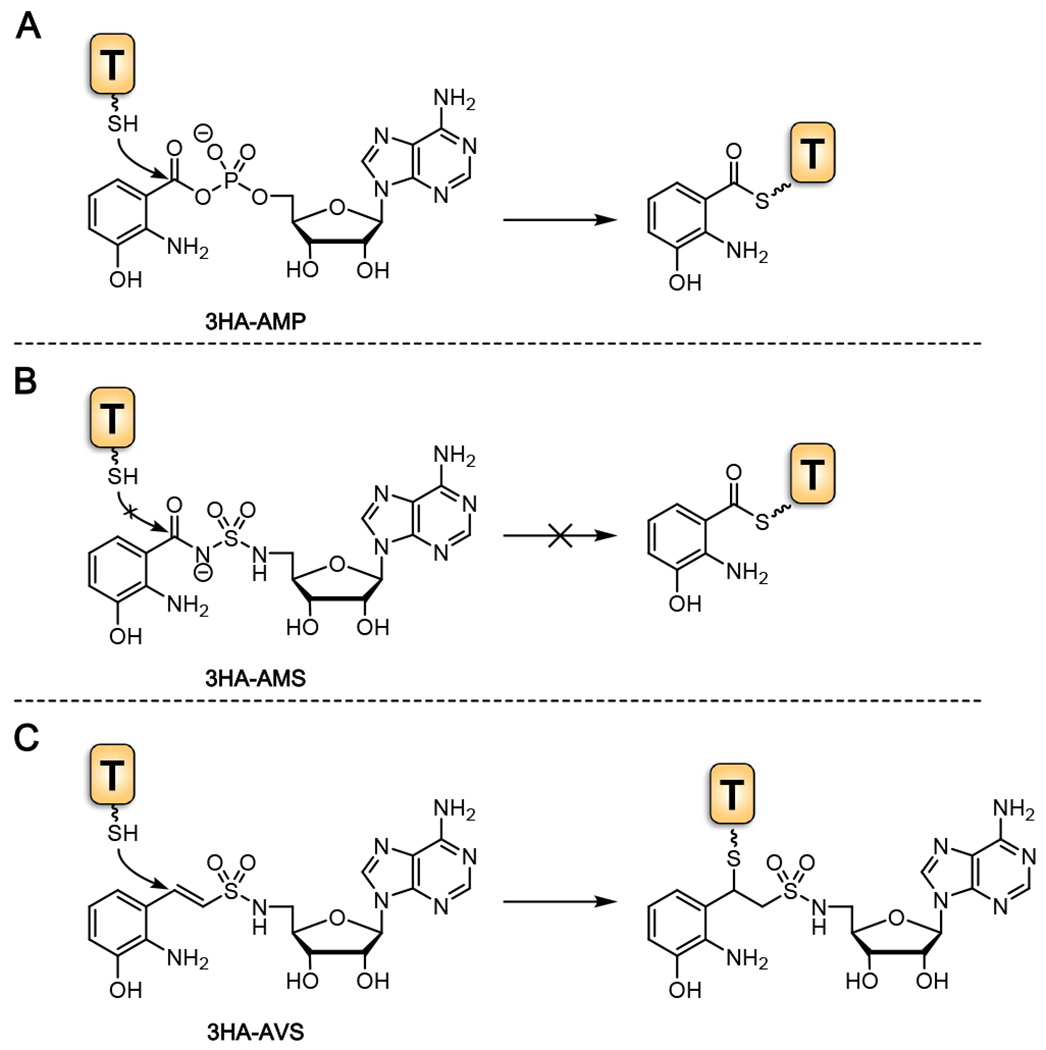
A) The mechanism in which 3-hydroxyanthraniloyl-adenylate is loaded to the holo-ThdA phosphopantetheinyl arm yielding acyl-ThdA. B) Non-cleavable bioisostere 3-hydroxyanthraniloyl-AMS mimicking the adenylate intermediate, inhibiting acylation of the downstream ThdA. C.) Vinyl-sulfonamide 3-hydroxyanthraniloyl-AVS participates in a hetero-Michael addition with the terminal phosphopantetheinyl thiol of holo-ThdA resulting in covalent modification and irreversible inhibition.
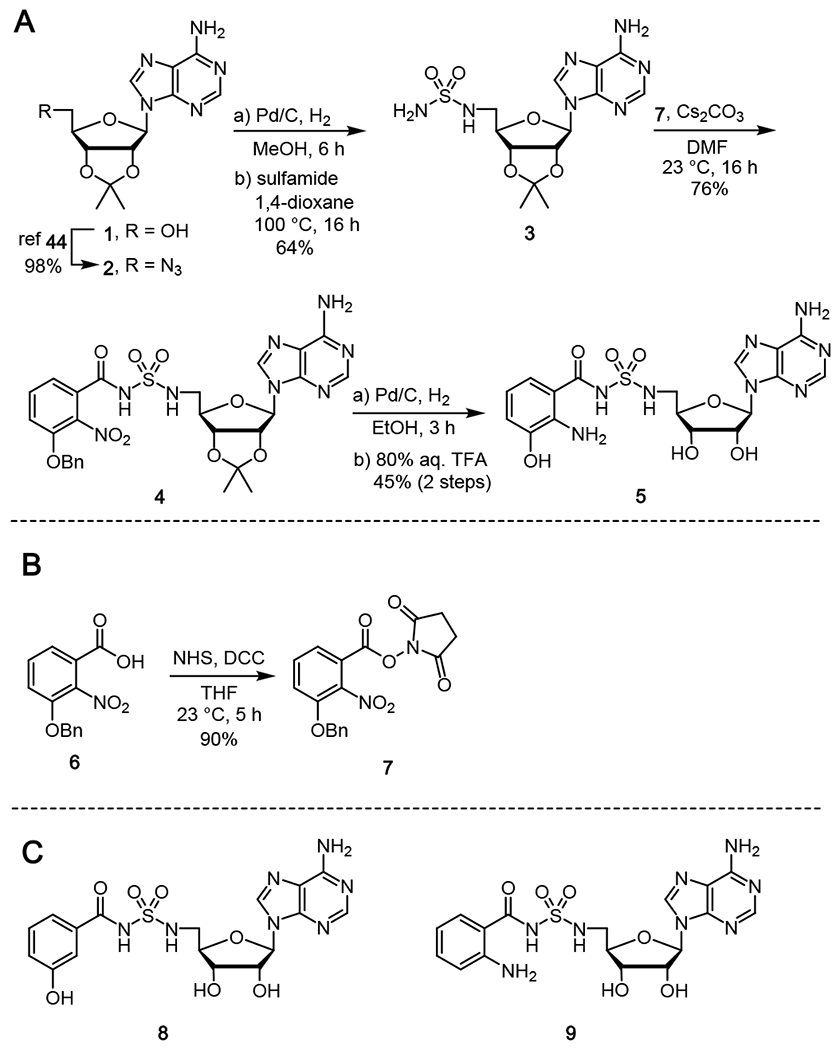
Based on the above considerations, we designed the inhibitor 3-hydroxyanthraniloyl-AMS 5. The synthesis commenced from 2′,3′-isopropylideneadenosine 1 that was converted to the 5′-azido nucleoside 244 followed by catalytic hydrogenation to the intermediate amino derivative, which was directly condensed with sulfamide using our recently developed method by refluxing in 1,4-dioxane to afford 3 (Scheme 1A)45 Acylation of 3 with N-hydroxysuccinimidyl (NHS) ester 7 promoted by Cs2CO3 in DMF furnished acyl-sulfamide 4. Catalytic hydrogenolysis of the 3-benzyloxy ether and concomitant reduction of the 2-nitro group using Pd/C in methanol followed by TFA deprotection of the acetonide afforded the final product 3-hydroxyanthraniloyl-AMS 5 in a 45% overall yield after purification by preparative reverse-phase high-performance liquid chromatography (HPLC). The N-hydroxysuccinimidyl ester 7 was synthesized from the 3-benzyloxy-2-nitrobenzoic acid 646 by DCC-mediated coupling with N-hydroxysuccinimide (Scheme 1B). To explore the SAR of 3-hydroxyanthraniloyl-AMS 5, we also prepared two closely related analogues, 3-hydroxybenzoyl-AMS 8 and anthraniloyl-AMS 9 through deletion of the 2-amino and 3-hydroxy functional groups present in the aryl acid moiety (Scheme 1C). These were prepared analogously to 5 as described in the Supporting Information starting from 2-anthranilic acid and 3-hydroxybenzoic acid.
Scheme 1.
Synthesis of 3-hydroxyanthraniloyl-AMS (5).
We next synthesized a second class of adenylation inhibitor that incorporates a vinylsulfonamide isostere of the acyl-phosphate linkage.47,48 Adenosine vinylsulfonamide (AVS) inhibitors mimic the acyl-adenylate intermediate, but contain a reactive Michael acceptor at the precise position of the incoming phosphopantetheine thiol that irreversibly binds the downstream thiolation domain. AVS inhibitors thus cross-link the adenylation and thiolation domains, stabilizing their transient interaction and are useful for capturing the adenylation domain in the thioester conformation (Figure 4C).49,50
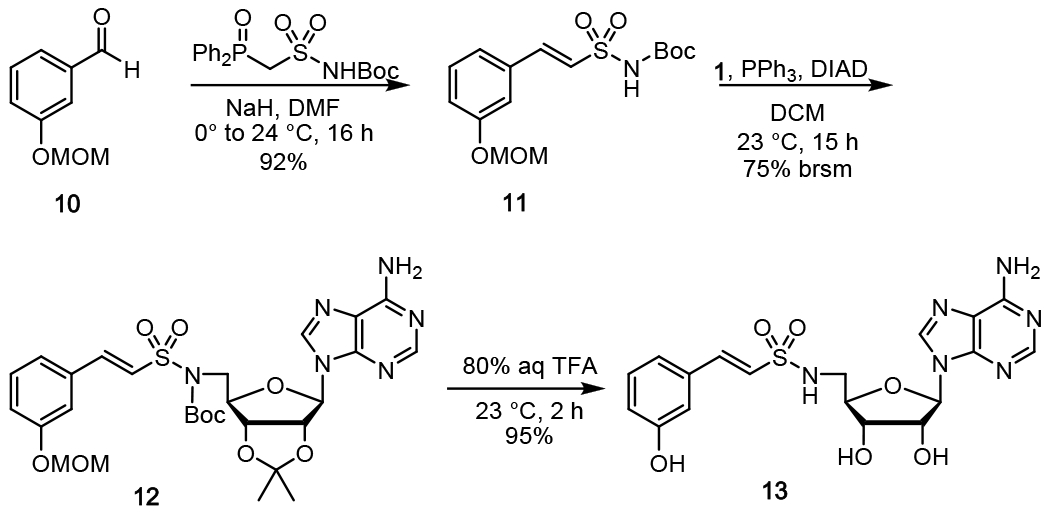
We designed 3-hydroxybenzoyl-AVS 13 for structural characterization of NpsA in complex with ThdA. Notably, 13 lacks a 2-amino group on the aryl ring due to the oxidative instability of the ortho-aminophenol moiety observed under the extended crystallization trials. The 3-hydroxy group was retained since it is more important for NpsA binding (vide infra). Synthesis of ligand 13 was achieved in three steps beginning with Horner-Wittig condensation of 3-hydroxybenzaldehyde 10 and N-Boc-diphenylphosphoryl sulfonamide51 to afford E-vinylsulfonamide 11 (Scheme 2). Subsequent Mitsunobu coupling of 11 with 2′,3′-isopropylideneadenosine 1 gave 12. Global deprotection of the MOM, Boc and acetonide groups using TFA yielded the final 3-hydroxybenzoyl-AVS 13.
Scheme 2.
Synthesis of 3-hydroxybenzoyl-AVS (13).
Biophysical and Biochemical Characterization of AMS inhibitors.
Acyl-adenylate inhibitors 5, 8, and 9 were evaluated for enzyme inhibition of NpsA using our coupled assay with 20 nM NpsA and 20 μM ThdA under initial velocity conditions in the presence of saturating concentrations of 3-hydroxyanthranilic acid (500 μM) and ATP (5 mM). The concentration-response plots were fit to the Morrison equation for tight binding inhibition to determine the inhibition constants (Figure 5A). The lead inhibitor 3-hydroxylanthraniloyl-AMS 5 exhibited an appKi = 54 nM (Table 3). Analog 8 lacking the 2-amino group had a modest 4-fold loss of potency while analog 9 devoid of the 3-hydroxyl group was 22-fold less potent than 5.
Figure 5.
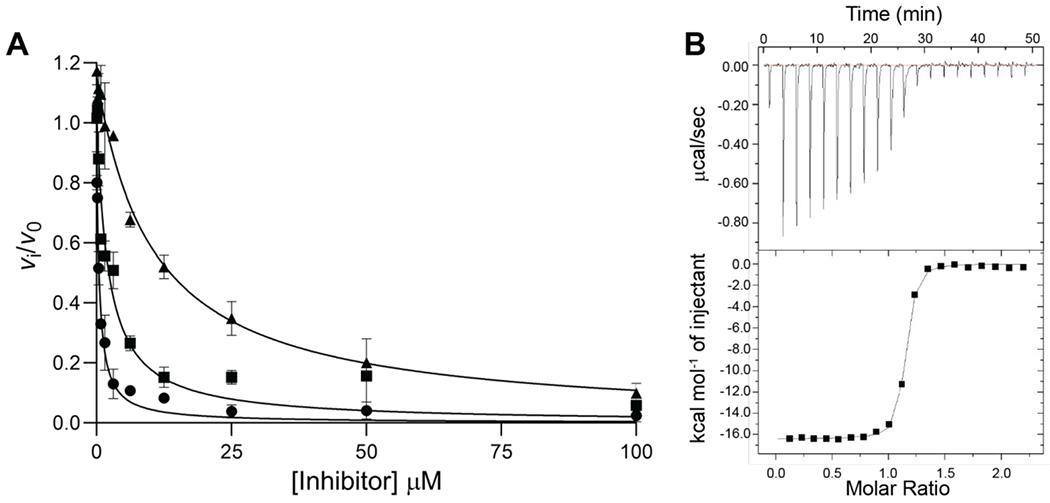
A) Concentration-response plot for NpsA inhibition by 5, 8 and 9: 3-hydroxyanthraniloyl-AMS 5 (circle), 3-hydroxybenzoyl-AMS 8 (square), anthraniloyl-AMS 9 (triangle). The curve represents the best non-linear fit to the Morrison equation. Data are average (±SD) of triplicate experiments. B) Representative ITC binding isotherm of binding 5 to NpsA. (Top) Data obtained by titration of 25 nM NpsA with 0.25 μM 5. (Bottom) The integrated curve showing experimental points and the best fit (−).
Table 3:
Summary of characterization of all three inhibitors against purified NpsA.
| Cmpd | appKi, (nM) | KD (mM) | ΔH (kcal/mol) | TΔS (kcal/mol) | ΔG (kcal/mol) | n |
|---|---|---|---|---|---|---|
| 5 | (5.4 ± 0.8) × 101 | (2.93 ± 0.28) × 101 | (−1.61 ± 0.16) × 101 | −5.67 ± 0.21 | (1.04 ± 0.16) × 101 | 1.06 ± 0.03 |
| 8 | (2.3 ± 0.4) × 102 | (1.12 ± 0.68) × 102 | (−1.64 ± 0.29) × 101 | −6.92 ± 2.91 | 9.45 ± 0.23 | 0.94 ± 0.01 |
| 9 | (1.2 ± 0.1) × 103 | (4.85 ± 2.50) × 102 | (−1.09 ± 0.35) × 101 | −2.31 ± 0.47 | 8.59 ± 0.31 | 1.17 ± 0.02 |
We also determined the binding affinities of 5, 8 and 9 to NpsA by isothermal titration calorimetry to provide the dissociation constant (KD), Gibbs free energy (ΔG), enthalpy (ΔH), and entropy (ΔS) of the protein-ligand interactions (Table 3, Figure 5B). Compound 5 binds tightly with a KD of 29 nM, a value in close agreement to the apparent Ki, value of 54 nM measured using the functional kinetic assay. Binding is enthalpically driven with a ΔH of −16.1 kcal/mol with a small unfavorable entropic component. Deletion of the 2-amino group in compound 8 resulted in a 4-fold decrease in KD relative to 5 due to an increased entropic penalty, while deletion of the 3-hydroxyl in 9 provided a 17-fold decrease in KD compared to 5 caused by a 5.5 kcal/mol loss in enthalpy that was partially compensated by an entropic gain. The relative binding affinities of 8 and 9 compared to 5 determined by ITC closely mirror the relative inhibition constants from the steady-state kinetic assay.
Structures of NpsA and ThdA.
To help gain further insight into molecular interactions underpinning the observed trends in ligand affinities, we obtained an X-ray crystal structure (2.9 Å) of NpsA co-crystallized with 3-hydroxyanthraniloyl-AMS 5 (Figure 6A). The structure features a single chain of NpsA with 3-hydroxyanthraniloyl-AMS 5 occupying the active site and the C-terminal subdomain, Asub, positioned in the adenylate forming conformation identifiable by the contact between Lys492 within the Asub and the pro-R oxygen atom of the acyl-sulfamide.42 The moderate resolution of this initial structure precluded unequivocal determination of the orientation of the aryl ring. Therefore, we obtained X-ray crystal structures of the NpsA N-terminal domain (in which the C-terminal subdomain of NpsA was truncated from the Asn405 hinge residue onward) in complex with compounds 5, 8 and 9 (1.7-2.2 Å). The resolution of the 3-hydroxyanthraniloyl-AMS 5 containing structure unequivocally displays the orientation of the aryl ring suggested by the full-length structure (Figure 6A) in which the 3′-hydroxyl group form a tripartite H-bond with Asn207 and Ser271 (Figure 7B). This pose was used to deduce the orientation of the ligand in the full-length structure (Figure 7A). Each liganded structure features canonical nucleotide A domain interactions including: Asp388 with the ribose 2′,3′-hydroxyls; Thr298 with the pro-k sulfamide oxygen; Asn293 with the adenine 6-amino group. In the aryl ring of the ligands, the 3′-hydroxy group presumably participates as an H-bond acceptor with an Asn207 donor and an H-bond donor with a Ser271 acceptor, while anthraniloyl derivatives contain the additional 2′-amino group that could potentially participate as an H-bond donor with a Ser271 acceptor.
Figure 6.
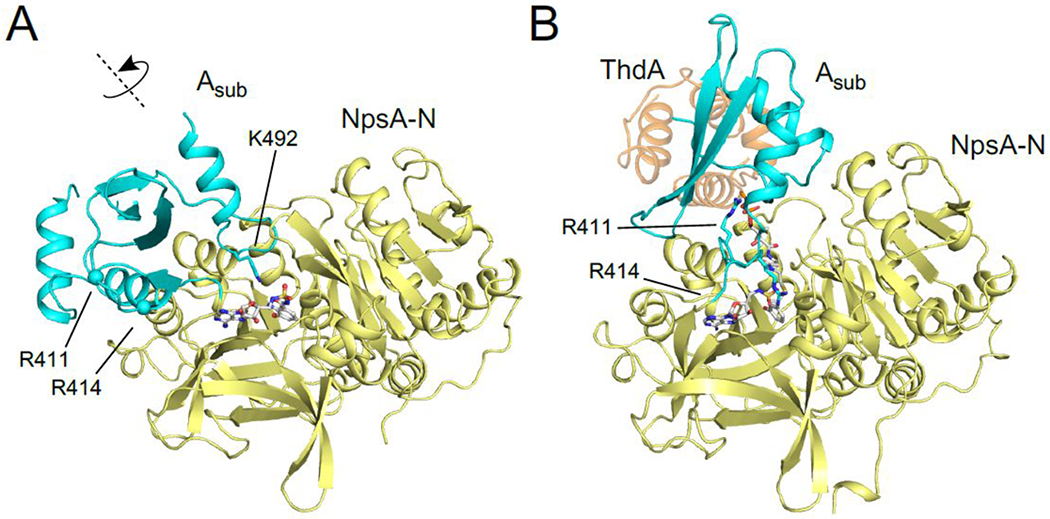
A) 3-hydroxyanthraniloyl-AMS 5 bound to full length NpsA showing Asub domain in the adenylation conformation. B) 3-hydroxybenzoyl-AVS 13 bound to NpsA-ThdA fusion protein showing A sub domain in the acylation configuration binding auxiliary protein ThdA.
Figure 7.
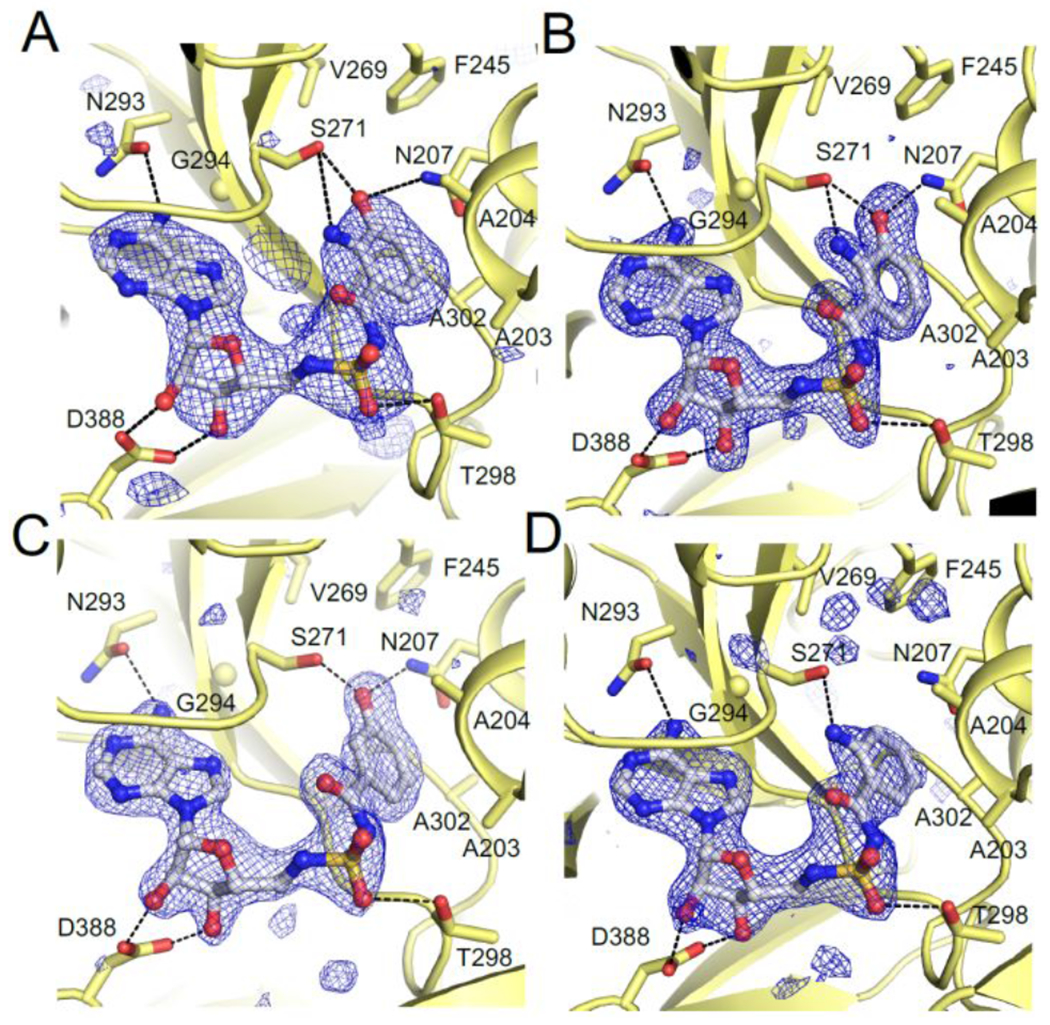
A) Full-length NpsA and 3-hydroxyanthraniloyl-AMS 5, B) NpsA-N and 3-hydroxyanthraniloyl-AMS 5, C) NpsA-N and 3-hydroxybenzoyl-AMS 8, and D) NpsA-N and 2-anthraniloyl-AMS 9. Each panel displays conserved A domain nucleotide binding residues (Asn293, Thr298, Asp388). Simulated annealing electron density is calculated with coefficients of the form mFo-DFc and contoured at 3σ.
We also generated an engineered NpsA-ThdA fusion construct and subsequently determined the 2.9 Å crystal structure of NpsA-ThdA bound to 3-hydroxybenzoyl-AVS 13 (Figure 6B). The final model contains four NpsA adenylation domains and four ThdA thiolation domains. The linker joining the NpsA and ThdA domains are disordered in all four chains however the termini of the respective molecules suggest that the proteins have adopted a domain swapped configuration in which the carrier domain of one protein chain is donated to a neighboring adenylation domain. This configuration was seen previously in the similar fusion construct from enterobactin biosynthesis.48 Despite the disorder in the linker and ThdA, the interface between the adenylation and thiolation domains is consistent and is similar to the interface seen in other structures of trapped adenylation-thiolation structures.48–50 The Asub residues Arg411 and Arg414 have moved from a surface exposed loop in the adenylation conformation (Figure 6A) to form a clamp over the Ser542 phosphodiester and 13, respectively in the thiolation conformation. (Figure 6B). The majority of the NpsA-ThdA contact is mediated by a parallel helix-helix interaction between ThdA helix 2 and a helix on the NpsA N-terminal domain that runs from Gly248 to Val257. The residue interactions between the two appear as Arg411/Arg445/Asn541 that is analogous to the Arg437/Lys473/Asp574 motif, which is conserved among the enterobactin, acinetobactin, bacillibactin and vibriobactin systems.48
Inhibition of Tilimycin Biosynthesis in Klebsiella oxytoca.
3-Hydroxyanthraniloyl-AMS 5 was next evaluated for its ability to inhibit tilimycin production in the clinical isolate K. oxytoca strain MIT 10-5243. Due to the nonenzymatic addition of indole observed in endogenous production of tilimycin, a high-performance liquid chromatography tandem mass spectrometry (LC-MS/MS) method was developed to monitor the MS2 transition of tilivalline (334.4→199.1 m/z) instead. We observed tilivalline production commenced as the K. oxytoca culture transitioned to stationary phase at 6 hours, with peak concentrations being detected after 27 hours before declining due to degradation (Figure S4). We first showed 3-hydroxyanthraniloyl-AMS 5 does not inhibit growth of K. oxytoca at 300 μM highlighting the selectivity and nonmicrobicidal activity of this adenylation inhibitor (Figure S5). Tilivalline levels were subsequently measured at 8-hours following incubation of 300 μM 5, 8, and 9 with K. oxytoca. Unfortunately, inhibition of tilivalline was not detected for compound 5 and more careful examination of the growth curves revealed 5 paradoxically stimulated growth of K. oxytoca in earlier timepoints likely leading to the modest increase of tilivalline production observed (Figure S5). We speculated metabolism or degradation of 5 under the culture conditions may have been responsible for the lack of tilivalline inhibition. We quantified 5 in the culture of K. oxytoca during the full time course and observed 75% degradation of 3-hydroxyanthraniloyl-AMS 5 by the 8-hour timepoint (Figure S4). Deletion of either the 2-amino or 3-hydroxy groups of the ortho-aminophenol in 5 with analogs 8 and 9 was expected to abolish the potential oxidative instability and subsequent degradation. Gratifyingly, 3-hydroxybenzoyl-AMS 8 and 2-anthraniloyl-AMS 9 reduced tilivalline levels with 8 completely ablating tilivalline production under the experimental conditions (Figure 8A). The concentration-response plot for inhibition of tilivalline production of the most promising inhibitor 3-hydroxybenzoyl-AMS 8 yielded an IC50 value of 29 μM (Figure 8B).
Figure 8.
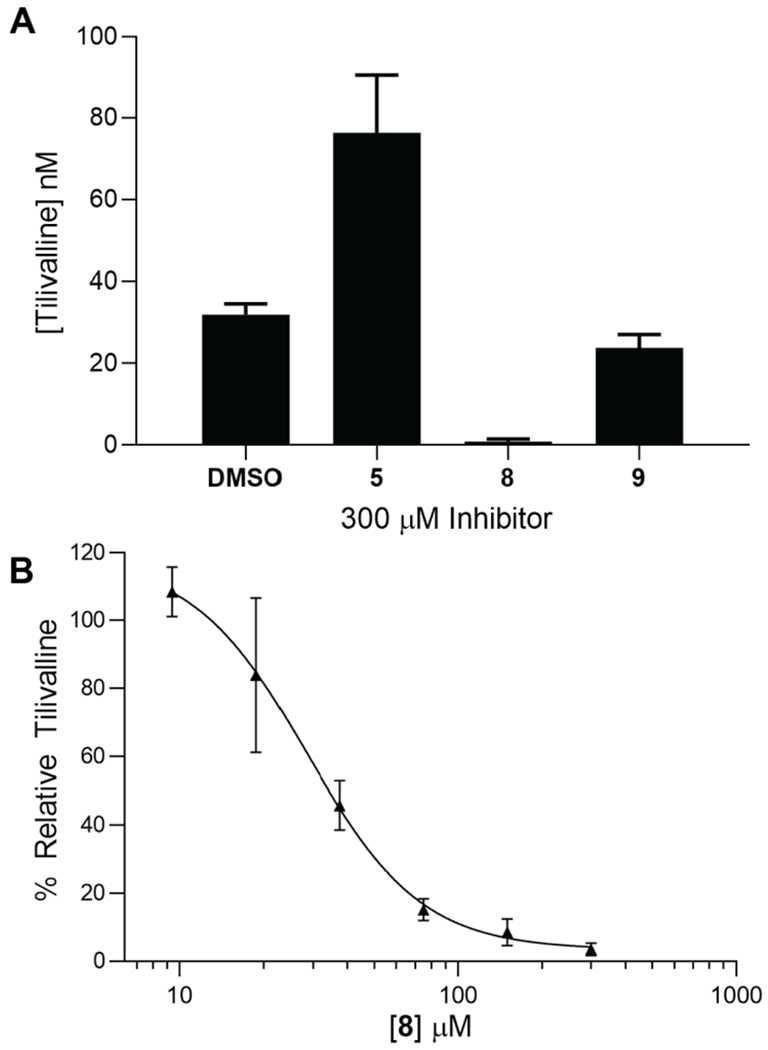
A) Cultures inoculated with K. oxytoca to a starting OD600 = 0.01 with 300 μM of 5, 8, 9 or 0.6% DMSO as a negative control were grown for 8 hours at 37 °C in CASO medium shaking at 250 rpm. Tilivalline production was quantified using LC-MS/MS and the m/z transitions 334.4→199.1 [tilivalline (analyte)] and 424→199.1 [O-benzyltilivalline (internal standard)]. B) Concentration-response plot of the normalized % production of tilivalline at 8 hours under the incubation conditions described in panel A relative to a DMSO-only control versus the concentration 3-hydroxybenzoyl-AMS 8. The curve was fit to a four-parameter Hill equation to provide an IC50 value of 29 ± 4 μM and a Hillslope of −2.1 ± 0.5. The data points represent the mean (±SD) of two independent experiments.
DNA Adductomics.
Tilimycin belongs to a class of NRPS secondary metabolites called pyrrolobenzodiazepines.52,53 These compounds have been observed to react in vitro with DNA via the electrophilic imine at the C-11 position and the nucleophilic N-2 of guanine forming a covalent aminal bond.54–56 Tilimycin has been predicted to react in an analogous fashion and ultimately results in single and double stranded DNA breaks.57 Downstream effects of DNA adduct formation have been observed, however, direct detection of the DNA adducts in cells has not been observed.33 To further verify tilimycin’s mechanism of toxicity we used an LC-MS11 DNA adductomics approach to simultaneously detect both anticipated DNA adducts from direct alkylation of DNA by tilimycin, and any unanticipated DNA adducts which may form.58 Data acquisition consists of MS and MS2 data dependent acquisition with MS3 fragmentation triggered upon neutral loss of 2′-deoxyribose (116.0473 m/z) or one of the four nucleobases (151.0494, 135.0545, 126.0429, 111.0433 m/z).
We began with exposure of either purified calf thymus DNA or 15N bacterial DNA with tilimycin at 37 °C for 24 hours. The DNA was then enzymatically hydrolyzed, and the resulting mixture was purified and enriched by solid phase extraction and fractionated into three portions (10% MeOH/H2O, 100% MeOH, 2% formic acid/MeOH). Each fraction was analyzed to detect and identify DNA adducts which formed due to the exposure. The proposed adduct dG-tilimycin (484.1939 m/z) was observed in the 100% MeOH fraction with two distinct chromatographic peaks (retention times of 14.5 and 15.1 min), likely corresponding to the two diastereomers which could form upon nucleophilic addition of dG to the C-11 imine of tilimycin (Figure 9A, Figure S6). MS2 fragmentation of the parent ion of dG-tilimycin (484.1939 m/z) resulted in the neutral loss of deoxyribose (368.1466 m/z) which triggered MS3 fragmentation that was dominated by a product ion of 269.0789 m/z (Figure 9E). Confirmation of the adduct identity was aided by the 15N bacterial DNA exposure which resulted in an isotopically labeled dG-tilimycin (489.1791 m/z; +4.9852 m/z for 5 15N atom incorporation in the nucleobase) with fragmentation to 373.1323 m/z and 274.0637 m/z product ions which maintain all 5 15N atoms of guanine (Figure 10). Along with the proposed dG-tilimycin adduct, we observed a dA-tilimycin adduct in very low abundance (Figure S7). In parallel, we analyzed calf thymus DNA exposed tilivalline and did not detect any adducts, consistent with the hypothesis of other laboratories that tilivalline neither degrades back into tilimycin nor interacts with DNA.
Figure 9.
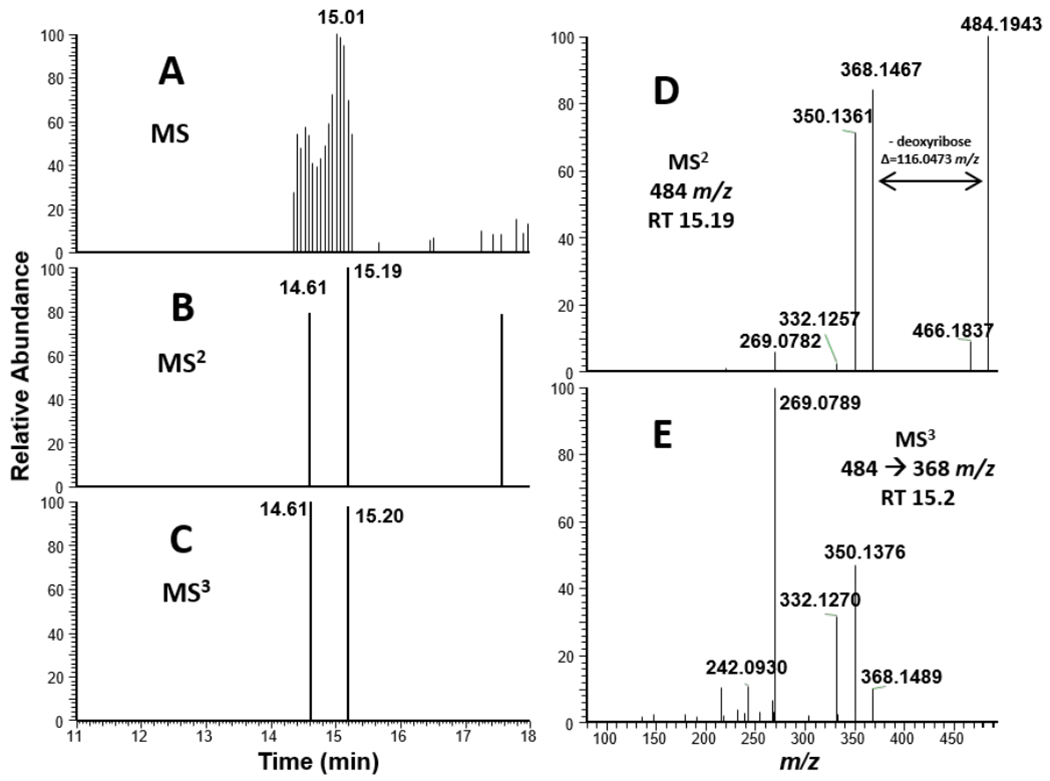
Mass spectral data for ctDNA treated with tilimycin. A.) MS1 EIC of 484.1939 m/z. B.) MS2 data dependent event for 484.1939 m/z. C.) MS3 triggered event for 484.1939→368.1466 m/z (neutral loss of deoxyribose). D.) MS2 mass spectrum from fragmentation of 484 m/z at retention time = 15.19 min. E.) MS3 mass spectrum from fragmentation of 484.1939→368.1466 m/z at retention time = 15.20 min.
Figure 10.
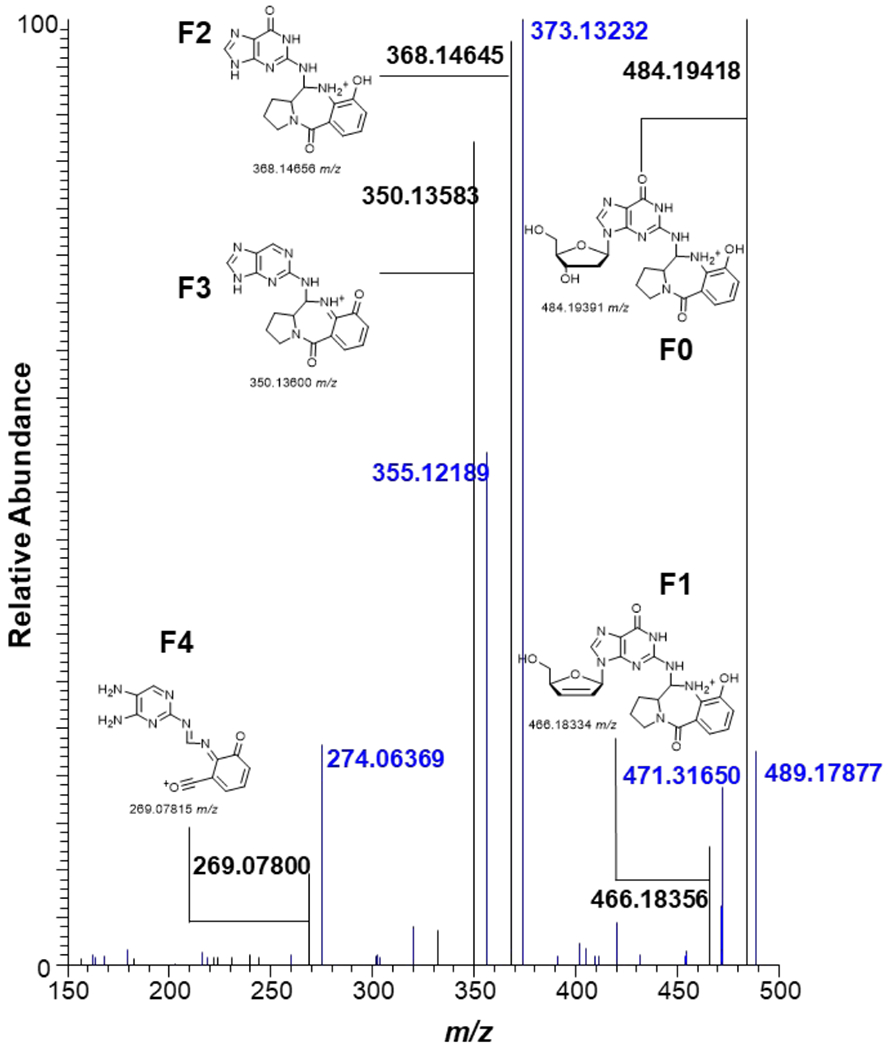
MS2 Fragmentation pattern of dG-tilimycin adduct (possible structure) from tilimycin exposure to ctDNA (black) overlaid with identical exposure to 15N bacterial DNA (blue). Possible product ions F0-F4 shown.
To determine if these metabolites are detectable from whole cell exposure, HEK cells were treated with 1 μM tilimycin (IC25; Figure S13) and incubated at 37 °C for 24 hours. The cells were lysed, and the DNA was extracted, hydrolyzed, purified and enriched identically to the DNA exposure studies as describe previously. Targeted mass spectrometry of the MS2 fragmentation of 484.1939 m/z yielded distinct extracted ion chromatogram (EIC) peaks of 368.1446 m/z (neutral loss of 2′-deoxyribose) and 350.1360 m/z (neutral loss of 2′-deoxyribose and H2O) between 13.2-14.0 min (Figure 11) consistent with the proposed dG-tilimycin adduct observed in the tilimycin-treated ctDNA (Figure 9). The dA-tilimycin adduct was not observed, likely indicating it is below the limit of detection of the assay.
Figure 11.
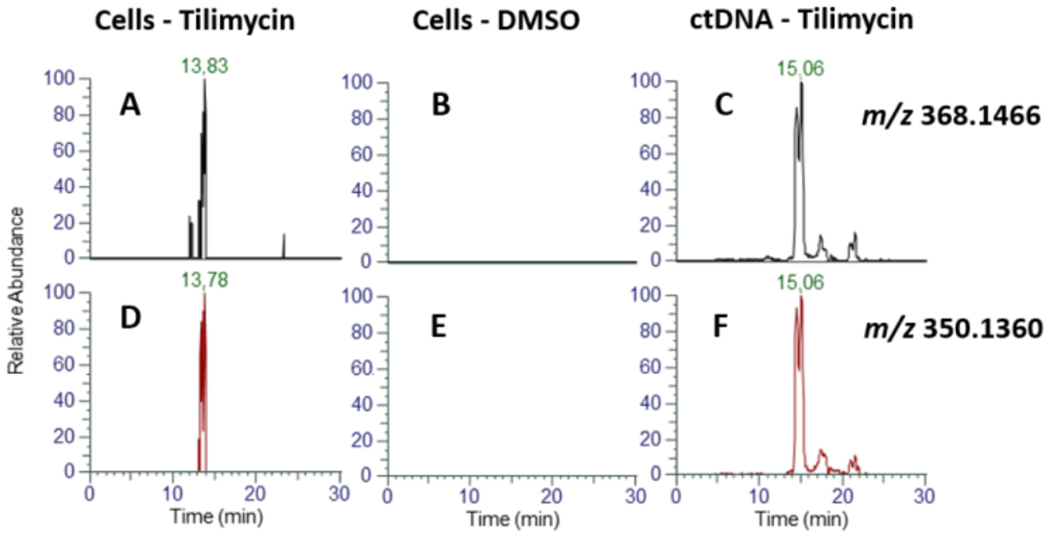
MS2 EIC of 484.1939→368.1466 m/z (neutral loss of deoxyribose) for tilimycin-treated HEK cells (A), DMSO-only HEK cells (B), and tilimycin treated ctDNA (C). MS2 EIC of 484.1939→350.1360 m/z (neutral loss of deoxyribose, H2O) for tilimycin-treated cells (D), DMSO-only HEK cells (E), and tilimycin-treated ctDNA (F).
DISCUSSION
Tilimycin belongs to a large class of NRPS secondary metabolites called pyrrolobenzodiazepines (PBD) that contain a tricyclic ring system formed by condensation of anthranilic acid and l-proline derivatives.59 PBDs are largely produced by the Streptomyces genus of bacteria and their exceptional cytotoxicity52 has been exploited to design synthetic PBD dimers as investigational chemotherapeutic agents.53 Targeted delivery of PBD dimers to tumor cells through antibody-drug conjugate techniques has been investigated in multiple Phase I-III clinical trials.60–62 Thus, the presence of an endogenously produced PBD within the human microbiota is concerning.24
The biosynthesis of prototypical PBDs including anthramycin59, tomaymycin63, and sibiromycin64,65 have been elucidated by functional assignment of the corresponding biosynthetic gene clusters through homology analysis, gene inactivation, and chemical complementation studies. The PBD core is assembled by a bimodular NRPS comprised of a loading module with an adenylation and thiolation domain, either as a single protein or separate stand-alone proteins, and an extension module with a C-A-T-Re architecture. Muller and co-workers recently reported the first attempt to biochemically reconstitute a complete PBD NRPS system using LC-MS to directly detect phosphopantetheine tethered substrates including anthranilic acid and proline derivatives. A dipeptide intermediate attached to the thiolation domain of the extension module was also observed.63 However, only trace quantities of PBD were produced, which the authors speculated may be due to insufficient channeling between the recombinant NRPS proteins under the in vitro assay conditions.63 We successfully reconstituted the complete tilivalline NRPS employing recombinant NspA, ThdA, and NspB proteins and were able to dissect the activities of each module using continuous coupled assays that measured production of pyrophosphate or NADPH consumption.
We first interrogated the activity of the loading module, which was greatly facilitated by the unique organization of the tilimycin NRPS system wherein the adenylation and thiolation domains, NpsA and ThdA, are stand-alone proteins, enabling use of stoichiometric quantities of ThdA to promote turnover. Typically, adenylation domain activity is evaluated using a non-physiologically relevant pyrophosphate exchange assay that only measures the first half-reaction in the reverse direction through incorporation of pyrosphosphate into ATP. Instead, we evaluated the loading of ThdA by NpsA by quantifying formation of the pyrophosphate byproduct using a coupled enzyme system consisting of pyrophosphatase and purine nucleoside phosphorylase.38,40 NpsA catalyzed the efficient loading of ThdA with 3-hydroxyanthranilic acid yielding a kcat of 0.89 s−1 and KM values of 63, 15 and 3.4 μM for 3-hydroxyanthranilic acid, ATP and holo-ThdA, respectively. The KM for holo-ThdA is commensurate with other values reported for thiolation domains with their cognate adenylation domains; however, the apparent KM of ATP and 3-hydroxyanthranilic acid are an order of magnitude lower and higher than related substrates with aryl acid adenylation domains, respectively.36,41,66 Apparent KM values determined for these NRPS adenylation enzymes are strongly affected by the assay utilized (hydroxamate-MesG versus the classical pyrophosphate exchange) as well as substrate concentrations, which could explain the observed discrepancies.
We also observed ThdA possessed strong substrate inhibition with a Ki = 15 ± 2 μM. The three dimensional structure of our NpsA:ThdA fusion construct shows the interface between the ThdA loop 1 and the Asub domain of NpsA are composed of many electrostatic and non-specific hydrophobic interactions. These interactions are also conserved in the homologous natural didomain of the Xenorhabdus species.67 These observations support the hypothesis that the interface observed in the NpsA-ThdA crystal structure is physiologically relevant. This interaction also explains the observed inhibition at high concentrations of ThdA; the substrate binding pocket is occluded by ThdA and Asub when ThdA is bound to NpsA in the thiolation conformation. There may be an equilibrium between adenylation and thiolation conformations that shift in favor of thiolation in the presence of holo-ThdA.
Interrogation of NpsB activity proved much more challenging and initial attempts using our coupled assay systems for pyrophosphate or NADPH consumption failed to show any turnover. However, catalytic turnover could be achieved employing stoichiometric concentrations of pre-loaded ThdA as the substrate for NpsB in the presence of the other co-substrates l-proline and NADPH. The time-dependent formation of tilivalline was confirmed by LC-MS analysis employing an authentic synthetic standard. We were able to demonstrate for the first time the efficient production of a PBD in vitro with an overall kobs of 0.013 s−1 using this product formation assay (Figure S16). The inability of NpsB to turnover l-proline in the presence of ATP and NADPH suggests the reductase domain possesses high specificity for the native l-N-(3-hydroxyanthraniloyl)prolyl-S~TNpsB intermediate over l-prolyl-S~TNpsB, which stands in stark contrast to other reported Re domains that more rapidly reduce the amino thioester monomers over the corresponding dipeptide thioesters.68 The close correspondence between the kobs determined in the product formation and the kcat of 0.05 s−1 independently determined using a NADPH consumption assay confirm reduction is tightly coupled to dipeptide formation. While the overall kcat is low, it represents a composite of the microscopic rate constants for l-proline adenylation and thioesterification onto the thiolation domain of NpsB, condensation of the resulting l-prolyl-S~TNpsB with the upstream 3-hydroxyanthraniloyl-S~ThdA, and thioester reduction of the l-N-(3-hydroxyanthraniloyl)prolyl-S~TNpsB intermediate to a dipeptide aldehyde species that spontaneously cyclizes to tilimycin as a diastereomeric mixtures of cyclic aminals. We hypothesize the rate-limiting step is likely reductive release since previous studies with Re domains68–69 show low turnover numbers (apparent kcat s ranging from 0.03 to 0.003 sec−1). By contrast, the reaction rates of isolated adenylation and condensation domains are typically two-to-three orders of magnitudes greater than our observed kcat.39,50,68,70,71
The major metabolite isolated from K. oxytoca in cell culture as well as the gut microbiome is not tilimycin, but rather tilivalline due to non-enzymatic and irreversible reaction of tilimycin with indole by electrophilic aromatic substitution.24,29,33 The reaction proceeds through an iminium species produced by dehydration of tilimycin. The initial report by Zechner and coworkers identified tilivalline as the major metabolite produced by K. oxytoca, however, subsequent studies have shown tilimycin is also formed in vitro and in vivo,29,33 Tilimycin is an order of magnitude more cytotoxic than tilivalline and is also a genotoxic DNA-damaging agent, whereas tilivalline inhibits microtubule formation.33 Consequently, tilimycin likely plays a more significant role in AAHC and may additionally lead to cellular transformation upon long-term exposure.33
Interestingly, the relationship between the dipeptides tilimycin and tilivalline, which are produced by enzymatic and non-enzymatic processes and mediate different cellular effects, bears similarity to other NRPS systems. Fischbach et al. has shown that NRPS biosynthetic gene clusters harboring terminal reductase domains that afford dipeptide aldehydes are ubiquitous in the gut microbiota 72 Dipeptide aldehydes produced in the gut can potentially modulate host-microbe interactions through inhibition of cysteine proteases.72 Cyclization of typical dipeptide aldehydes lacking an anthranilate moiety occurs slowly over several hours in the gut yielding dihydropyrazinone and pyrazinone metabolites, which are predicted to have important physiological roles in mediating microbe-microbe interactions.68,73
In parallel to our characterization of NpsA we synthesized the bisubstrate inhibitors 3-hydroxyanthraniloyl-AMS 5, 3-hydroxybenzoyl-AMS 8 and anthraniloyl-AMS 9. Biophysical characterization by isothermal titration calorimetry revealed the 3-hydroxyl of the aryl ring is more important than the 2-amino substituent since removal of the 3-hydroxyl group in 8 led to a greater than 20-fold loss of affinity and concordant 4 kcal/mol decrease in binding enthalpy. The ITC result is consistent with the removal of the two hydrogen bonds (Asn207/Ser271) that could contribute ~2 kcal/mol each to binding enthalpy as seen in the co-crystal structures. By contrast, deletion of the 2-amino group in 9 led to no reduction in binding enthalpy and a modest 4-fold reduction in affinity that can be attributed to an entropic penalty from removal of the possible prearrangement via intramolecular hydrogen bonding between the 2-amino and the A-acyl sulfamide. The trend in binding affinities for 3-hydroxybenzoyl ligand 8 (KD = 112 nM) and anthraniloyl ligand 9 (KD = 485 nM) is counterintuitively opposite the trend in KM values for the respective substrates 3-hydroxybenzoic acid (KM = 4,000 μM) and anthranilic acid (KM = 854 nM) with NpsA. These results indicate the 3-hydroxy is more critical for binding, whereas the 2-amino group is important for catalytic efficiency.
The design of inhibitors of specific enzymes in gut microbial pathways mechanistically linked to human diseases represents a novel strategy to develop a new class of pharmacological agents.74–76 The tilimycin pathway has been genetically validated for causing AAHC.24 Moreover, tilimycin and many PBDs are potent genotoxins,33 thus small-molecule inhibitors of tilimycin biosynthesis could have therapeutic utility for not only treating AAHC, but also to prevent transformation of colorectal cells in the 2–10% of people colonized with K. oxytoca. Attempts to ‘drug the microbiome’ are still in the early stages of conception; however, nonantibiotic approaches may have utility, especially in light of the rise of antimicrobial resistance and the revolution of precision medicine.77–78 As a prelude to future studies for treating AAHC, we evaluated our bisubstrate inhibitors for inhibition of tilimycin in K. oxytoca cell cultures. We initially examined our most potent enzyme inhibitor 3-hydroxyanthraniloyl-AMS 5 against K. oxytoca and were dismayed to observe no inhibition of tilimycin production. We speculated the lack of activity may be due to oxidative degradation of the ortho-aminophenol moiety of 3-hydroxyanthraniloyl-AMS 5 under the aerobic culture conditions.79 Indeed, we observed rapid degradation of 5 under the culture conditions, so we examined 3-hydroxybenzoyl-AMS 8 and anthraniloyl-AMS 9 that lack the sensitive o/Vrio-ami nophenol moiety. Consistent with its improved biochemical potency, 3-hydroxybenzoyl-AMS 8 was found to possess superior activity inhibiting 50% tilimycin production at 29 μM, but no observable growth inhibition of K. oxytoca at 300 μM, the highest concentration evaluated. Previous studies of related adenylation inhibitors have shown they possess poor bioavailability and thus are expected to accumulate to high levels in the gastrointestinal tract.45 Consequently, inhibitors such as 8 may have ideal characteristics for selective inhibition of tilimycin inhibition while sparing both the host and the microbiome.
PBDs share a common mechanism of action through the covalent binding to guanosine in double-stranded DNA, which was established by a series of seminal studies in the 1970s.57,80 A single X-ray co-crystal structure of anthramycin bound to the synthetic oligonucleotide 5′-CCAACGTTGG-3′ confirmed anthramycin binds in the minor groove and alkylates N-2 of the guanine nucleobase.81 However, limited binding is available for most PBDs and DNA interaction is inferred using molecular modeling and thermal stabilization assays measuring unfolding of double-strand DNA with short synthetic oligonucleotides.82–86 A few recent studies have quantified overall DNA binding using mass spectrometry, but detection of defined DNA adducts in vivo has never been reported for any PBD.87,88 To this end, we sought to identify for the first time, PBD-like DNA adducts caused by tilimycin exposure to eukaryotic cells. The major metabolite observed in calf thymus DNA and HEK cell exposure was the hypothesized dG-tilimycin adduct with a parent mass of 484.1939 m/z. This molecular weight supports the previous reports of the N-2 regioselective alkylation of guanine. The N-7 of guanine is the most nucleophilic nitrogen on the nucleobase, however, when alkylation of the N-7 occurs, like in the case of nitrogen mustards, the anomeric linkage is weakened and the 2′-deoxyribose is rapidly cleaved. In the case of tilimycin, the parent mass of this N-7 metabolite would be 368.1467 m/z (-ribose), which was not observed. MS3 fragmentation of the dG-tilimycin adduct reveals a unique fragment with a mass of 269.0789 m/z. We hypothesized that this fragment was a result of loss of pyrrolidine from tilimycin which maintained the aminal linkage to guanine. When calf thymus DNA was exposed to high levels of tilimycin we were also able to detect a novel dA-tilimycin adduct. This adduct was not detectable in the exposure of tilimycin to HEK cells, which may indicate a higher sensitivity requirement to monitor the low abundance of the metabolite.
CONCLUSION
Although pyrrolobenzodiazepines were discovered nearly sixty years ago and have been extensively studied, fundamental questions remained concerning their biosynthesis and mechanism of action. In this study we sought to more fully characterize the NRPS biosynthetic machinery and mechanism of action of the microbiota-derived metabolite, tilimycin. We provided the first kinetic characterization of a bimodular NRPS responsible for PBD biosynthesis. First, we established that NspA efficiently loads ThdA with a preference for anthranilate and demonstrated the 2-amino group is more critical than the 3-hydroxyl group for catalytic efficiency. Unexpectedly, we showed ThdA exhibits strong substrate inhibition by occluding the substrate binding pocket of NpsA. We next studied the extension module NpsB and showed it orchestrates the biosynthesis of tilimycin in a tightly controlled manner with reductive release only occurring on the dipeptide chain intermediate to afford a dipeptidyl aldehyde that spontaneously cyclizes to tilimycin. The bisubstrate AMS and AVS inhibitors proved crucial for the structural characterization of NpsA and ThdA and allowed us to observe both the adenylation and thioester conformations of NpsA. As a potential therapeutic strategy for treating antibiotic associated hemorrhagic colitis, we showed 3-hydroxybenzoyl-AMS 8 could completely block biosynthesis of tilimycin in K. oxytoca providing a nonmicrobiocidal approach to drugging the microbiome. Finally, in order to conclusively characterize the mechanism of action of tilimycin, we detected the pyrrolobenzodiazepine-like N-2 deoxyguanine adduct when HEK cells were exposed to tilimycin and detected a novel deoxyadenosine DNA adduct when exposed calf thymus DNA to tilimycin. To the best of our knowledge, this is the first structural characterization of a PBD-DNA adduct formed in cells.
Methods
Cloning:
The K. oxytoca gene encoding for NpsA (NpsA; WP_004103551.1) was commercially synthesized (GENEWIZ, South Plainfield, NJ) containing Ndel (5′) and Xhol (3′) restriction sites appended to the ends of the gene. A single silent mutation was designed at the codon for Ala5 to eliminate a natural Ndel site contained within the gene. The gene was subcloned using Ndel/Xhol) into a pET15b vector containing an N-terminal 5xHis-tag and TEV protease recognition site (pET15b-TEV-NpsA; Genewiz; South Plainfield, NJ, EISA). K. oxytoca strain MIT-5243 was obtained from BEI Resources (Manassas, VA) and grown in small cultures for the generation of genomic DNA. The genes encoding ThdA and NpsB were PCR amplified from genomic DNA and cloned into the pET15b-TEV plasmid using Ndel and Xhol restriction sites.
Protein expression and purification. Wild-type (wt)-NpsA.
The ampicillin-resistant pET15b-wt-NpsA plasmid was transformed into BL21(DE3) cells for expression. A single colony from the transformed cells was used to inoculate a 3 mL starter culture of LB containing ampicillin (100 μg/mL), The starter culture was grown overnight at 37 °C with shaking at 250 rpm. The following day, the 3 mL culture was used to inoculate 1 L of terrific broth (24 g/L yeast extract, 20 g/L tryptone, 4 mL/L glycerol, 0.017 M KH2PO4, 0.072 M K2HPO4) containing ampicillin (100 μg/mL). The culture was shaken at 250 rpm and 37 °C until the OD595 reached 0.8, then the temperature was lowered to 16 °C and the culture induced with IPTG (750 μM) and incubated overnight at 16 °C with shaking at 250 rpm.
Cells were harvested by centrifugation (5,000×g, 20 min, 4 °C), resuspended in lysis buffer (50 mM HEPES, 150 mM NaCl, 20 mM imidazole, 0.2 mM TCEP, pH 6.8) and lysed by French press. The lysed cells were centrifuged (35,000×g, 40 min, 4 °C) and the supernatant was loaded onto Ni-NTA resin (2 mL, Takara; Mountain View, CA. USA). The resin was washed with lysis buffer (10 mL). wt-NpsA was eluted with elution buffer (50 mM HEPES, 150 mM NaCl, 300 mM imidazole, 0.2 mM TCEP, pH 6.8) and subsequently loaded onto a HiLoad 16/600 Superdex 75 μg gel-filtration column (GE Healthcare) using SEC buffer (50 mM HEPES, 150 mM NaCl, 0.2 mM TCEP, 10% glycerol, pH 6.8). The fractions containing NpsA were pooled, exchanged into storage buffer (25 mM HEPES, 75 mM NaCl, 0.2 mM TCEP, 10% glycerol, pH 6.8) and concentrated using a 10 kDa centrifugation. The resulting protein stock was stored at −80 °C and thawed prior to use.
ThdA.
Expression was the same as wt-NpsA with the following exceptions: After IMAC chromatography the partially purified protein was incubated with TEV protease (8% w/w) and dialyzed in 50 mM HEPES, 250 mM NaCl, 0.2 mM TCEP, pH 8.0 buffer for 16 h at 4 °C. Further incubation of ThdA with the promiscuous phosphopantethineyl transferase Sfp (10 nM), 12.5 mM MgCb and 1 mM CoA for 60 minutes at 24 °C ensured full conversion of apo-ThdA to holo-ThdA.49,89 The storage buffer was (20 mM HEPES, 40 mM NaCl, 0.2 mM TCEP, pH 7). holo-ThdA was concentrated using a 10 kDa centrifugation filter and fractionated into 20 μL aliquots frozen at −80 °C until used. The total protein yield was determined by nanodrop to be 26.5 mg/L of culture.
NpsB.
The ampicillin-resistant pET15b-NpsB plasmid was transformed into BL21(DE3) cells for expression. A single colony from the transformed cells were used to inoculate a 3 mL starter culture of LB containing ampicillin (100 μg/mL), The starter culture was grown overnight at 37 °C with shaking at 250 rpm. The following day, the 3 mL culture was used to inoculate 1 L of terrific broth (24 g/L yeast extract, 20 g/L tryptone, 4 mL/L glycerol, 0.017 M KH2PO4, 0.072 M K2HP04) containing ampicillin (100 μg/mL). The culture was shaken at 250 rpm and 37 °C until the OD595 reached 0.6, then the temperature was lowered to 16 °C and the culture induced with IPTG (250 μM) and incubated overnight at 16 °C with shaking at 250 rpm.
Cells were harvested by centrifugation (5,000μg, 20 min, 4 °C), resuspended in lysis buffer (50 mM HEPES, 250 mM NaCl, 20 mM imidazole, 0.2 mM TCEP, 10% glycerol, pH 6.8) and lysed by French press. The lysed cells were centrifuged (35,000×g, 40 min, 4 °C) and the supernatant was loaded onto Ni-NTA resin (2 mL, Takara; Mountain View, CA. EISA). The resin was washed with lysis buffer (10 mL). NpsB was eluted with elution buffer (50 mM HEPES, 250 mM NaCl, 300 mM imidazole, 0.2 mM TCEP, 10% glycerol, pH 6.8) and subsequently loaded onto a HiLoad 16/600 Superdex 200 μg gel-filtration column (GE Healthcare) using SEC buffer buffer (50 mM HEPES, 250 mM NaCl, 0.2 mM TCEP, 10% glycerol, pH 6.8). The fractions containing NpsB were pooled, exchanged into storage buffer (50 mM HEPES, 150 mM NaCl, 0.2 mM TCEP, 10% glycerol, pH 6.8) and concentrated using a 10 kDa centrifugation filter. The resulting protein stock was stored at −80 °C and thawed prior to use. The total protein yield was 2.7 mg/L of culture.
NpsA Assay
Reactions were performed under initial velocity conditions using wt-NpsA (20–200 nM), 0.1 U purine nucleoside phosphorylase (Sigma N8264-100UN), 0.04 U inorganic phosphatase (Sigma I1643-100UN), and 100 μM MesG (Berry & Associates), with varying concentrations of ATP, aryl acid, and ThdA in assay buffer (50 mM Tris, 150 mM NaCl, 10 mM MgCl2, pH 8) in a total volume of 100 μL. The reaction was initiated by addition of wt-NpsA. Reactions were run in triplicate in 96 half-area UV transparent plates (Corning 3679) and cleavage of MesG was continuously monitored at A360 every 10 sec on a Molecular Devices Spectra Max M5e over 5 min.
Determination of Kinetic Parameters and Substrate Specificity of NpsA.
Steady-state kinetic parameters of aryl acids, ATP and holo-ThdA were determined using the wt-NpsA assay described above by varying one substrate concentration while maintaining the other two substrates at fixed saturating levels. The enzyme and substrate concentrations for kinetic characterization were: aryl acid (7.8–1000 μM) with ATP (5 mM) and ThdA (20 μM) held constant employing 200 nM wt-NpsA; ATP (7.8–1000 μM) with aryl acid (500 μM) and ThdA (20 μM) held constant employing 20 nM wt-NpsA; and ThdA (0.31–80 μM) with aryl acid (500 μM) and ATP (5 mM) held constant employing 20 nM wt-NpsA. The normalized initial velocities from triplicate experiments were then plotted against the varied substrate concentration to generate a saturation curve, which was fit by nonlinear regression analysis to the Michaelis-Menten equation (eq 1) or the substrate inhibition model (eq 2) using GraphPad Prism 6.0.
| (1) |
| (2) |
Measurement of Inhibition Constants.
The determination of the apparent inhibition constants (appKi) for adenylation inhibitors 5, 8 and 9 was performed using the NpsA assay described above and performed in triplicate. All inhibitors were tested from 11–1000 nM at a constant 1% DMSO concentration with 20 nM wt-NpsA in the presence of saturating concentrations of 3-hydroxyanthranilic acid (500 μM), ATP (5 mM) and ThdA (20 uM). The inhibitors displayed tight-binding inhibition (Kiapp < 200·[E]), thus the fractional activity (vi/v0), where vi, is the reaction velocity at a given [I] and v0 is the reaction velocity of the DMSO control, were fit by nonlinear regression analysis using GraphPad Prism 6.0 to the Morrison equation (eq 3),90 constraining [E] to 20 nM to obtain the appKi values, which were converted to Ki values using the Cheng-Prushoff equation (eq 4) for a competitive inhibitor, where [S] and KM correspond to the concentration and Michaelis constant of 3-hydroxyanthranilate.
| (3) |
| (4) |
Determination of Kinetic Parameters of NpsB.
Steady-state kinetic parameters of l-proline, ATP and NADPH were determined by monitoring NADPH consumption (ΔA340) with or without the presence of NpsA (20 nM), ThdA (20 μM), and 3-hydroxyanthranilic acid (500 μM) in assay buffer (50 mM Tris, 150 mM NaCl, 10 mM MgCl2, pH 8). The enzyme and substrate concentrations for kinetic characterization were: l-proline (0.078–10 mM) with ATP (5 mM) held constant employing 250 nM NpsB; ATP (7.8–500 μM) with l-proline (5 mM) held constant employing 250 nM NpsB; and NADPH (7.8–250 μM) with l-proline (5 mM) and ATP (5 mM) held constant employing 250 nM NpsB. The initial velocities from triplicate experiments were then plotted against the varied substrate concentration to generate a saturation curve, which was fit by nonlinear regression analysis to the Michaelis-Menten equation (eq 1).
Isothermal Titration Calorimetry.
ITC experiments were performed on an AutoITC200 (Malvern Instruments). The experiments were performed at 25 °C in assay buffer (50 mM HEPES, 150 mM NaCl, 5 mM MgCl2 pH 6.8). Buffer exchange into assay buffer was performed on wt-NpsA prior to experiments using centrifugation filtration (Amicon 10 kDa filter) and the final filtrate was used to prepare a solution of the inhibitors from a 50 mM stock in DMSO. The percentage DMSO in the final inhibitor solution was matched in the wt-NpsA protein solution. The 200 μL sample cell was filled with wt-NpsA (30 μM, concentration determined using the calculated extinction coefficient Protparam tool) and the 40 μL injection syringe was filled with inhibitor (300 μM). Titrations were carried out with a stirring speed of 750 rpm and a 200 s interval between 2 μL injections. The first injection for each sample was excluded from data fitting. The experimental data was fitted to a titration curve using the Origin™ software package (version 7.0) to determine KA, n, and ΔH. The thermodynamic parameters (ΔG and TΔS) were calculated using equation 5:
| (5) |
where ΔG, ΔH, and ΔS are the changes in free energy, enthalpy, and entropy of binding, respectively; R = 1.98 cal· mol−1K−1; T is the absolute temperature (298 K). The affinity of the ligands for the protein is given as the dissociation constant (KD = 1/KA). ITC experiments were run in triplicate and analyzed independently, and the thermodynamic values were averaged.
Inhibition of Tilivalline Biosynthesis in K. oxytoca.
Production of tilivalline by K. oxytocci (BEI Resources, MIT 10-5243) was performed in duplicate in 14 mL snap-cap tubes containing 3-mL cultures in CASO medium (17 g/L casein peptone, 2.5 g/L K2HPO4, 2.5 g/L, D(+)-glucose, 5 g/L NaCl, 3 g/L soy peptone). Cultures were incubated at 37 °C at 250 rpm. Prior to every experiment, K. oxytoca was streaked out on an LB-ampicillin agar plate and one colony was chosen for the subsequent experiments. Cultures were inoculated with an overnight CASO culture from a single colony to a starting OD600 = 0.01 with varying concentrations of 5, 8 and 9 or 0.6% DMSO as a negative control. Aliquots (20 μL) of the cultures were taken at 8 and 24-hour timepoints. The aliquots were diluted with 80 μL acetonitrile, vortexed for 20 sec and stored at −20 °C for 2 h to ensure full precipitation. The solutions were centrifuged at 15,000×g for 5 min and 50 μL of the supernatant was transferred to a conical vial containing 50 nM benzyl-tilivalline as an internal standard. The samples were then subjected to LC-MS/MS analysis.
Tilivalline Quantitation.
Samples were analyzed by LC-MS/MS (Shimadzu UFLC XR-AB SCIEX QTRAP 5500). Reverse phase LC was performed on a Kinetix C18 column (50 mm × 2.1 mm, 2.6 μm particle size; Phenomenex, Torrance, CA). Mobile phase A was 0.1% aqueous formic acid while mobile phase B was 0.1% formic acid in acetonitrile. Initial conditions were 5% B from 0 to 0.5 min, after which the %B was increased to 95% from 0.5 to 3 min. The column was washed in 95% B for 2 min, returned to 5% over 0.2 min, and allowed to re-equilibrate for 2.8 min in 5% B to provide a total run time of 8 min. The flow rate was 0.5 mL/min and the column oven was maintained at 40 °C. The injection volume was 10 μL. Tilivalline was monitored by MS in positive ionization mode by multiple reaction monitoring (MRM). To determine the optimum mass spectrometry settings, an authentic tilivalline standard was infused at a concentration of 1 μM (1:1 water/acetonitrile containing 0.1% formic acid) onto the MS by a syringe pump at a flow of 10 μL/min. The following transitions were utilized: 334.4→199.1 [tilivalline (analyte)] 424→199.1 [benzyl-tilivalline (internal standard)]. Analyte and internal standard peak areas were calculated (MultiQuant, version 2.0.2, AB SCIEX). Analyte peak areas were normalized to internal standard peak areas, and the analyte concentrations were determined with an appropriate standard curve.
Cloning, expression, and purification of NpsA, NpsA-N and NpsA-ThdA fusion protein for X-ray crystallography.
Wt-NpsA proved refractory to initial crystallization attempts with an in-house sparse matrix screen. To enhance the probability of successful crystal nucleation and growth, a surface entropy reduction (SER) mutation, EEQ→AAA, was identified with the SERp server and introduced at positions 312-314 via QuikChange mutagenesis. All subsequent constructs generated for crystallographic analysis contain this EEQ→SER mutation. A construct encoding NpsA with the C-terminal subdomain, Asub (Asn405-Val506), removed was also cloned into pET15b-TEV and designated pET15b-TEV-NpsA-N. The artificial di-domain fusion construct encoding for NpsA-ThdA was cloned by inserting the ThdA gene with a six bp 5′ linker encoding for the insertion of an Ala-Ser dipeptide at the 3′ end of NpsA to generate pET15b-TEV-NpsA-ThdA (Figure S8–S12).
NpsA, NpsA-ThdA, and NpsA-N for crystallization studies were produced in E. coli BL21(DE3) cells. 1 L of LB media (100 μg/mL ampicillin) was inoculated with 1 mL of an LB overnight culture (100 μg/mL ampicillin, 37 °C, 250 rpm). Cultures were grown to an OD600 of approximately 0.6 (37 °C, 250 rpm) then incubated at 4 °C (30 min, 250 rpm) after which IPTG (500 μM) was added followed by incubation at 16 °C (20 h, 250 rpm). Cells were harvested via centrifugation (5000×g, 35 min, 4 °C) and flash frozen in liquid nitrogen. Subsequent purification steps were carried out at 4 °C.
Cell pellets were resuspended in lysis buffer (NpsA, NpsA-N; 50 mM HEPES pH 8.0, 150 mM NaCl, 20 mM imidazole, 0.2 mM TCEP; 10 mL/g) or lysis buffer with 10% v/v glycerol (NpsA-ThdA; 20 mL/g). Cells were lysed via mechanical disruption (Branson sonifier) and the resulting lysate was clarified via ultracentrifugation (40,000xg, 40 min, 4 °C), filtered (0.45 μm) and passaged over a Ni-loaded HisTrap column followed by a 3 column volume (CV) washout with lysis buffer. The column was then washed with 5 CVs of 10% v/v elution buffer (NpsA, NpsA-N; 50 mM HEPES pH 8.0, 150 mM NaCl, 300 mM imidazole, 0.2 mM TCEP) or elution buffer with 10% v/v glycerol (NpsA-ThdA) followed by a linear gradient of 10→100% v/v elution buffer over 5 CVs. For NpsA-N, protein-laden fractions were pooled, TEV protease was added (1:100 w/w, TEV:NpsA/NpsA-N), and then fraction pools were dialyzed against lysis buffer for approximately 16 h. TEV protease and uncleaved NpsA-N were removed by a second passage over the Ni-loaded HisTrap column following the initial day 1 procedure.
NpsA-ThdA was subjected to simultaneous TEV cleavage and phosphopantetheinylation. Following day 1 Ni-IMAC, NpsA-ThdA containing fractions were pooled and combined with TEV protease (1:100 w/w, TEV:NpsA-ThdA), promiscuous phosphopantetheinyl transferase Sfp (400 nM), MgCl2 (1 mM), and coenzyme A (1 mM; ~5 equivalents NpsA-ThdA). The reaction mixture was rocked gently at 23°C for 3 h and then dialyzed against lysis buffer (4 h, 4°C). TEV protease, Sfp, and uncleaved NpsA-ThdA were removed by a second passage over the Ni-loaded HisTrap column following the initial day 1 procedure. Following the final IMAC purification step fraction pools were concentrated (Amicon® Ultra Filter; NpsA/NpsA-N: 30 kDa MWCO, NpsA-ThdA: 50 kDa MWCO) and further purified via gel filtration (Superdex200, 16/60; NpsA, NpsA-N: 50 mM HEPES pH 8.0, 150 mM NaCl, 0.2 mM TCEP; NpsA-ThdA: 50 mM HEPES pH 8.0, 150 mM NaCl, 0.2 mM TCEP, 10% v/v glycerol; Figure S14. Following gel filtration samples were exchanged to a crystallization buffer (NpsA: 10 mM HEPES pH 8.0, 25 mM NaCl, 0.4 mM TCEP; NpsA-ThdA: 10 mM HEPES pH 8.0, 25 mM NaCl, 0.4 mM TCEP, 2.5 mM MgCl2, 5% v/v glycerol) via serial concentration and dilution or concentrated directly in gel filtration buffer (NpsA-N). Final protein concentrations (NpsA-N: 15.0 mg/mL, NpsA: 7.0 mg/mL, holo-NpsA-ThdA: 19.0 mg/mL) were determined using extinction coefficients at 280 nm (NpsA-N: 31860 M−1cm’, NpsA: 38850 M−1cm’, holo-NpsA-ThdA: 44350 M−1cm’). NpsA-N and NpsA-ThdA samples protein samples were flash frozen as pellets in liquid nitrogen and stored at −80°C. NpsA protein samples were stored at 4°C for approximately 2 days prior to crystallization plate setup.
Crystallization of NpsA variants.
NpsA-N.
Unliganded NpsA-N crystals were grown with hanging drop vapor diffusion at 20 °C. Thawed NpsA-N stock solution (15 mg/mL, 1 μL) was mixed with well solution (100 mM HEPES pH 7.5, 150 mM KBr, 30% w/v PEG3350, 1 μL) and the resultant drop was incubated over 500 μL of well solution in a 24-well linbro style plate. Unliganded NpsA-N crystals were cryoprotected by soaking in well solution supplemented with 15%v/v glycerol for approximately 15–30 seconds prior to vitrification in liquid nitrogen. 3-hydroxyanthraniloyl-AMS 5, 3-hydroxybenzoyl-AMS 8 and anthraniloyl-AMS 9 ligands were introduced into unliganded NpsA-N crystals following the same general cryoprotection/soaking procedure. Three separate solutions containing increasing PEG3350 concentration (34, 41, 48% w/v), 100 mM HEPES pH 7.5, and 150 mM KBr, were supplemented with 2 mM 5/8/9. Ligand stock solutions (50 mM) were prepared with either gel filtration buffer (5/8) or DMSO (9). The NpsA-N crystals were harvested and soaked for approximately 1 h in each PEG3350 solution in order of increasing PEG concentration at 23 °C. During each soaking step drops were sealed in a 72 well microbatch plate with paraffin oil. Crystals were vitrified directly in liquid nitrogen after the final soak.
NpsA.
Crystals of full length NpsA were grown in the presence of 5 using the microbatch under paraffin oil technique at 23 °C. NpsA stock solution was concentrated (10 kDa MWCO Amicon® filter) and combined with a 5 stock solution (30 mM in NpsA crystallization buffer) so that the composition of the final solution was 16 mg/mL NpsA, 5 mM 5 in NpsA crystallization buffer. The NpsA/5 solution (2 μL) was combined with well solution (400 mM imidazole pH 6.6, 10% w/v PEG HMW, 2.5% w/v 1,3-dimethylimidazolium dimethyl phosphate; PEG HMW is a 1:1:1:1 mixture of PEG8000/10000/12000/20000; 1 μL) and the resultant drop was overlaid with paraffin oil in an untreated, polystyrene 72 well microbatch plate from Hampton Research (Aliso Viejo, CA, USA). Crystals, which formed concurrently with heavy precipitate, were extracted from the drops and soaked for approximately 5 min in a solution of well solution supplemented with 25% v/v pentaerythritol propoxylate (5/4 PO/OH; PEP426) prior to vitrification in liquid nitrogen.
NpsA-ThdA.
Holo-NpsA-ThdA was crystallized in the presence of a 3-hydroxybenzoyl-AVS 13 inhibitor using the microbatch under paraffin oil technique at 14 °C. A stock solution of 13 (160 mM, DMSO) was added directly to a thawed solution of holo-NpsA-ThdA (19 mg/mL, 300 μM) in approximately 4-fold excess (1.2 mM). The resultant NpsA-ThdA/13 solution was incubated on ice for approximately 30 min and then filtered (0.2 μm) prior to crystallization plate setup. The protein/ligand solution (1.2 μL) was mixed with well solution (0.2 M sodium formate, 20% w/v PEG3350; 1 μL) and overlaid with paraffin oil. Crystals began to form approximately 1-2 h after plate setup. (+/−)-2-Methyl-2,4-pentanediol (neat, ~1 μL) was added directly to the drop and after approximately 5 min crystals were harvested and vitrified in liquid nitrogen.
X-ray diffraction data collection, structure solution, and refinement.
Diffraction data were collected from single crystals on either APS beamline 23-IDD (unliganded NpsA-N, NpsA-N/5, NpsA-N/8, NpsA/9), SSRL beamline 12-2 (NpsA-ThdA/13), or SSRL beamline 9-2 (NpsA-N/5) (Table S1). In each case data were indexed and integrated with XDS followed by merging in the appropriate space group with AIMLESS. Optimal processing parameters were generated with the AutoPROC software package.
NpsA-N.
Two separate datasets were collected for unliganded NpsA-N crystals, which occurred in distinct monoclinic space groups with unit cell constants that corresponded to one (C2) or two (P21) chains in the asymmetric unit. Each structure was solved via molecular replacement. A protein basic local alignment search tool (BLAST) search of the protein data bank (PDB) yielded 53 sequences ranging from 22–33% identity. Due to the lack of a clearly distinguished homology model, the program MRage, as implemented in the PHENIX suite, was used to identify a viable search model from among the BLAST results and to solve the NpsA-N C2 structure. Ultimately, a solution to the C2 structure was found using a search model derived from 1AMU91 (29% identity with NpsA; phenylalanine activating domain of gramicidin synthase 1) processed with phenix.sculptor and with the C-terminal subdomain removed. To improve these initial phases the structure was solved and rebuilt with MR-Rosetta and phenix.autobuild using the sculpted 1AMU N-terminal domain as a search model. Portions of the model were rebuilt manually with iterative real space refinement in Coot and reciprocal space refinement in phenix.refine. Final rounds of refinement employed translation-libration-screw (TLS) parameterization. The P21 unliganded NpsA-N structure was solved with two chains in the asymmetric unit using Phaser and the C2 unliganded NpsA-N structure as a search model. NpsA-N crystals soaked with 5, 8, and 9 each crystallized in space group P21 with unit cell constants that were consistent with those of the unliganded NpsA-N P21 crystal. For both liganded structures a copy of the two chain unit from the unliganded NpsA-N P21 structure was placed in the unit cell with Phaser followed by iterative real space refinement with Coot and reciprocal space refinement with phenix.refine. Ligand restraints for 5, 8, and 9 were generated with AM1 optimization as implemented inphenix.elbow.92 Final rounds of refinement employed translation-libration-screw (TLS) parameterization.
NpsA.
Full-length NpsA was co-crystallized with 5 in space group I222 and with one chain in the asymmetric unit. The structure was solved via molecular replacement using Phaser and a single chain unliganded NpsA-N (P21) search model. Difference density (mFo-DFc) corresponding to the C-terminal subdomain was observed after an initial round of coordinate and atomic displacement parameter (ADP) refinement with phenix.refine. The C-terminal subdomain, Asub, was built in manually using the Cα-baton building mode in Coot. Following manual placement of Asub and several rounds of iterative real space refinement in Coot and reciprocal space refinement in phenix.refine and phenix.rosetta_refine the structure was subjected to molecular dynamics flexible fitting (MDFF) with the ChimeraX plugin ISOLDE93. Map coefficients (2mFo-DFc) for real space MDFF via ISOLDE were generated with phenix.maps by filling in missing Fobs reflections and excluding Rfree flagged reflections. Following MDFF in ISOLDE, ADP values were refined with phenix.refine. Final rounds of refinement employed TLS parameterization; the C-terminal subdomain, Asub, was excluded from TLS refinement.
NpsA-ThdA.
NpsA-ThdA co-crystallized with the mechanism-based inhibitor 13 in space group P1 with four chains in the asymmetric unit. The structure was solved via molecular replacement using Phaser and a single chain unliganded NpsA-N (P21) search model. Difference density (mFo-DFc) corresponding to Asub and ThdA (PCP domain) was observed after an initial round of coordinate and atomic displacement parameter (ADP) refinement with phenix.refine. Asub and ThdA domains were built in manually to varying degrees among the four chains with iterative rounds of real space and reciprocal space refinement using Coot and phenix.refine respectively. Following this initial refinement problematic regions of the model, i.e. rotamer and Ramachandran outliers, were relaxed with ISOLDE into a static 2mFo-DFc map generated with phenix.maps with Rfree flagged reflections excluded and missing Fobs reflections filled. Ligand restraints were generated with phenix.elbow for a single entity comprised of a phosphopantetheine arm covalently tethered to 13. Tetrahedral bond restraints were applied to the phosphopantetheine/Ser542 phosphodiester bond. Final rounds of refinement included bond length and angle outlier correction with phenix .real_space_refine followed by ADP refinement and TLS parameterization in phenix.refne; Asub and ThdA domains were excluded from TLS refinement.
Alamar Blue Cell Viability Assay.
Human embryonic kidney cell (HEK293T, ATCC: CRL-3216) cells were cultured in Dulbecco’s minimum essential medium (MEM) supplemented with 10% (v/v) fetal bovine serum, 100 U/mL penicillin and 100 μg/mL streptomycin under a 5% CO2 atmosphere at 37 °C. Cells were seeded into a 96 well plate (Corning 3595) at a density of 10,000 cells/well. After incubation for 24 h, the media was removed from adherent cells and replaced with FBS free MEM containing tilimycin yielding a final concentration of 0.125, 0.25, 0.5, 1, 2, 4, 8, 16, 32, 64, 128 μM tilimcyin or DMSO (0.2%) and subsequently allowed to incubate for an additional 24 h. All experiments were performed in triplicate for each concentration. Following the 24 h incubation, 10 μL of alamar blue cell viability reagent (Invitrogen A50100) was added. The plate was further incubated for 4 h in the dark at 37 °C after which fluorescence was measured at Ex560/Em590 560:590 on a Molecular Devices Spectra Max M5e plate reader. Cell viability was calculated by normalizing fluorescence signal to the DMSO treated control (Figure S13).
Reagents for DNA Adductomics.
Methanol (LC-MS grade), acetonitrile (LC-MS grade), isopropanol (IPA) and formic acid (FA, 98% v/v) were purchased from Fluka (St. Louis, MO, USA). Distilled water was purified by a Milli-Q system (Milford, MA, USA). DNase I recombinant expressed by Pichia pastoris (R-DNase, 10,000 U/mg), phosphodiesterase-1 extracted from Crotalus adamanteus (PDE-1, 0.4 U/mg), recombinant alkaline phosphatase expressed by Pichia pastoris (R-ALP, 7,000 U/mg), and calf thymus DNA (CtDNA) were purchased from Roche (St. Louis, MO, USA). Single membrane filtration devices Microcon (10 kDa cutoff, 0.5 mL) were purchased from Amicon (Billerica, MA, USA). Silanized vials (0.3 mL, 1.2 mL and 4 mL) were purchased from ChromTech (Apple Valley, MN, USA). Internal standards [15N5-N2-ethyl-deoxyguanosine (dG) and [l5N5]-N6-methyl-deoxyadenosine (dA) were synthesized as described by Wang et al.94,95
15N-DNA Generation.
Escherichia coli (MG1655 strain) was cultured in 15N-labeled minimal medium (5 mL) at 37 °C overnight, for three generations to obtain a uniformly labeled 15N-strain. The OD600 was used to monitor bacterial growth until stationary phase. The bacteria culture was centrifuged at 3000×g for 10 min at 24 °C. The bacterial pellet was resuspended in 50% glycerol in bacterial medium solution, frozen and stored in −80 °C until use. 15N-labeled minimal medium (1 L) was prepared using 200 mL of M9 salts (Na2HPO4•7H2O, 64 g KH2PO4, 15 g, NaCl, 2.5 g, 15NH4C1, 5.0 g, deionized H2O, to 1 L), 20 mL of glucose (20%; Sigma-Aldrich), 2 mL of MgSO4 (1 M; Fisher Scientific), 100 μL of CaCl2 (1 M; Fisher Scientific). A starter culture was done by inoculating 10 μL of 15N and 14N-stock bacteria into 5 mL of medium and grown overnight. 50 μL of cells were then inoculated in 1 L of medium and incubated overnight. Cells were centrifuged and pellet stored at −80 °C until use.
Bacterial 15N-DNA Isolation.
Cells were resuspended in 25 mL of Cell Lysis Solution (Qiagen) and treated with 150 μL of Proteinase-K (24 h, 24 °C) and RNase-A (2 h, 24 °C). Proteins were precipitated by adding 7.5 mL of Protein Precipitation Solution (Qiagen). The pellet was discarded and the supernatant was transferred into a new tube containing an equal amount of cold IPA to precipitate the DNA. The mixture was centrifuged, the supernatant was discarded and the DNA pellet washed with IPA 70 % v/v in water and then with 100% IPA. The pellets were dried, resuspended in DNA buffer (20 mM Tris, 2 mM MgCl2 pH 7.4), and stored at −20°C. The yield and purity of the DNA was assessed using a nanodrop UV/Vis spectrophotometer monitoring the 260 nm and 280 nm wavelengths.
DNA Exposure and Digestion.
CtDNA (100 μg), bacterial DNA (100 μg), and 15N-bacterial DNA (100 μg), were dissolved in Tris buffer (20 mM Tris, 2 mM MgCl2 pH 7.4). Tilimycin and tilivalline were added to the DNA solutions separately (100 μM). The samples total volume was 200 μL and they were incubated at 37 °C overnight. DNA exposure to DMSO (1% v/v) was used as a negative control. DNA isolation was performed by IPA precipitation. Briefly, 1 mL of cold IPA was added to each sample vial. The precipitated DNA was isolated, washed twice with 1 mL 70% IPA and 1 mL 100% IPA and dried under a nitrogen stream. All steps of the protocol were performed using silanized glass vials.
DNA Isolation from HEK Cells.
HEK293T cells in MEM media containing 10% FBS, 100 U/mL penicillin and 100 μg/mL streptomycin under a 5% C02 atmosphere at 37 °C were grown to 90% confluency in T75 flasks (Corning CLS3275; ~6 x 106 cells). The cells were then washed with PBS and incubated with 1 μM tilimycin or DMSO (0.2%) in FBS free MEM media for 24 h at 37 °C. Following treatment, the cells were washed PBS, suspended in a trypsin-containing buffer, pelleted at 500×g for 5 min, and further washed 3 times in PBS. Cells were resuspended in 15 mL of Cell Lysis Solution (Qiagen) and treated with 150 μL of Proteinase-K (24h, RT) and RNase-A (2 h, RT). Proteins were precipitated by adding 7.5 mL of Protein Precipitation Solution (Qiagen). The pellet was discarded and the supernatant was transferred into a new tube containing an equal amount of cold IPA to precipitate the DNA. The mixture was centrifuged and the supernatant was discarded and the DNA pellet washed with IPA 70 % v/v in water and then with 100% IPA. The pellets were dried, resuspended in appropriate buffer, and stored at −20°C. The yield and purity of the DNA was assessed using a nanodrop UV/Vis spectrophotometer monitoring the 260 nm and the 280 nm wavelengths.
DNA Hydrolysis and Deoxyguanosine Quantitation via HPLC.
DNA was enzymatically hydrolyzed. Recombinant-DNase was added (0.5 U/μg DNA) and the samples were incubated overnight at 24 °C. Then, Recombinant-DNase (0.5 U/μg DNA), R-ALP-1 (0.4U U/μg DNA) and PDE-1 (0.02 mU/μg DNA) were added and the samples were incubated at 37 °C for 2 h and additionally, overnight at 24 °C. Enzymes were removed before dG quantitation via filtration (Microcon, 10 kDa cutoff). The recovery of the DNA adducts was determined by spiking the samples with isotopically labeled internal standards ([15N5]-N2-ethyl-dG and [l5N5]-N6-methyl-dA 100 fmol each).
Quantitation of dG was carried out with a Dionex UltiMate 3000 RSLCnano System (Thermo Scientific, Waltham, MA) with a UV detector set at 254 nm. A 300 μm ID × 15 cm C18 column (2 μm, 100 Å) (Thermo Scientific, Waltham, MA) was used with water (A) and CH3OH (B) as mobile phases, an injection volume of 1 μL, and a flow rate of 15 μL/min. The elution gradient was as follows: an initial condition of 5% B for 2 min, a linear gradient from 2% to 25% B in 10 min, and then from 25% to 95% B in 3 min, followed by a 5 min hold, a return to the starting conditions over 2 min and re-equilibration for 3 min (25 min total run time). A calibration curve for dG (0.0625-50 ng/μL) was performed in triplicate. The amount of dG in each sample was calculated using the peak area, the slope of the calibration curve, and the fraction of the total volume injected. The amount of dG was then used to calculate the amount of DNA in each sample using the ratio of 1 mg of DNA per 0.66 μmol of dG.
Sample Enrichment and Purification.
Samples were purified by solid-phase extraction using Strata-X cartridges (30 μm, Phenomenex, Torrance, CA) after washing with 3 mL of CH3OH and equilibrating with 1 mL of H20. The samples were loaded on the cartridges, washed with 1 mL of H2O, and eluted with 1 mL of 10% CH3OH, 100% CH3OH and 2% formic acid in CH3OH sequentially. The last two eluted fractions were evaporated to dryness and reconstituted in 2% CH3OH/H2O to a final volume of 20 μL.
LC Conditions for MS Analysis.
The dried hydrolyzed DNA samples were reconstituted in 20 μL of 2% CH3OH/H2O (LC-MS grade, Fluka) and 1 μL analyzed by LC-MS. The LC was performed using a nanoflow UPLC (Ultimate 3000 RSLCnano UPLC, Thermo Scientific, Waltham, MA). The UPLC was equipped with a 5 μL loop and reversed-phase chromatographic separation was performed using a hand-packed commercially available fused-silica emitter (230 × 0.075 mm ID, 15 μm orifice, New Objective, Woburn MA) with C18 stationary phase (5 μm, 100, Luna Phenomenex, Torrance, CA). The mobile phase consisted of an aqueous solution of 0.05% (v/v) formic acid (A) and CH3CN (B). The elution program included an isocratic step (2% of B for 5.5 min at 1 μL/min), followed by a two steps linear gradient of B (1.3%/min for 37 min and 24%/min for 2 min, both at 0.3 μL/min) and it concluded with a washing isocratic step, performed at 98% of B for 2 min at 0.3 μL/min. At the end of the elution program, the LC-system was equilibrated for 3 min at isocratic conditions (2% of B, 1 μL/min). The injection valve position was switched at 6 min to take the injection loop out of the flow path.
Mass Spectrometry.
All mass spectrometry-based analysis was performed using a hybrid high-field Orbitrap mass spectrometer (Lumos, Thermo Scientific, Waltham, MA). The LC system was interfaced to the mass spectrometer using a Nanoflex ESI ion source (Nanoflex Thermo Scientific, Waltham, MA), which operated in positive ion mode at 2.5 kV. The transfer ion tube temperature was 300 °C, and S-Lens setting was 60.
Untargeted Adductomic Screening Analysis.
The screening method was performed using a data dependent acquisition-constant neutral loss/MS3 (DDA-CNL/MS3) approach, consisting of full scanning with data dependent MS2 and neutral loss MS3 acquisition (NL-MS3). The full scan (150–1,200 m/z) was performed with quadrupole filtering, maximum injection time of 50 ms, automatic gain control target of 2×l05, and a resolution setting of 120,000. Data dependent parameters were: dynamic exclusion of 30 s, mass tolerance of ± 5 ppm, repeat count of 1, minimum intensity of 2×l04, and a cycle time of 3 s. An MS2 exclusion list of 18 masses (Table S2), consisting of unmodified nucleosides and their electrostatically bound dimer ions, was used with a mass tolerance of ± 5 ppm. MS2 scan events were triggered based on an intensity threshold of 2×l04. The MS2 fragmentation was performed with a quadrupole isolation width of 1.5 m/z, stepped HCD collision energy (15, 25, 35%), AGC value of 2×l05, maximum injection time of 54 ms, and Orbitrap detection at a resolution setting of 30,000. For the NL-MS3 data acquisitions, MS2 product ions were isolated in the ion trap with an isolation window of 2 m/z, and MS3 fragmentation triggered upon observation of the neutral loss of the 2′-deoxyribose (dR) or the base moieties (-dR; 116.0474, -G; 151.0494, -A: 135.0545, -T; 126.0429; -C; 111.0433 ± 5 ppm), with HCD fragmentation (40%), Orbitrap detection at a resolution of 15,000, AGC value of 200,000 and a maximum injection time of 200 ms. A set of scan events was included for the targeted fragmentation of hypothesized ions of interest. 6 tilimycin derived-DNA adducts (484.1946 m/z, 468.1997 m/z, 368.1472 m/z, 352.1523 m/z, 459.1881 m/z, 444.1884 m/z), was investigated with quadrupole isolation widths of 1.5 m/z, maximum injection time of 54 ms, AGC of 200,000, and resolution setting of 30,000. Fragmentation was performed using HCD with stepped collision energy (16, 26, 36%). LC and injection conditions are as reported in the LC Conditions for MS Analysis section.
Targeted Adduct Analysis.
The targeted adduct analysis was performed using targeted MS2 analysis of 4 tilimycin derived-DNA adducts (484.1946 m/z, 468.1997 m/z, 368.1472 m/z, 352.1523 m/z), with quadrupole isolation widths of 1.5 m/z, maximum injection time of 246 ms, AGC of 1×106, and resolution setting of 120,000. Fragmentation was performed using HCD with a stepped collision energy (16, 26, 36%). The fragmentation of the DNA adduct precursor ions [MH]+ results in neutral loss of the dR, G, A, T or C moiety to produce the corresponding [MH-dR]+, [MH-G]+, [MH-A]+, [MH-T]+, and [MH-C]+ product ions whose masses are extracted for detection of the adducts. LC and injection conditions are as reported in LC Conditions for MS Analysis section.
Data Analysis.
For DNA adduct screening data analysis, RawConverter (http://fields.scripps.edu/rawconv/) was used to convert the Thermo Scientific raw data files to mzXML files which were imported into a SQL database, where data filtration on the basis of MS3 triggering ion m/z and retention time was performed. MS3 triggering ions present in the exposed samples with both m/z (±5 ppm) and retention times (± 30 s) different than those in the control (non-exposed) samples, were selected and reported as putative DNA adducts. The absence of putative DNA adduct ion signals in the control samples were further confirmed by generating extracted ion chromatograms for all putative DNA adduct precursor masses at 5 ppm mass tolerance using the Qualbrowser component of the Xcalibur 3.0 software package (Thermo Scientific, Waltham, MA). MS2 and MS3 spectra of each putative DNA adduct were subsequently evaluated for structural information and the peak area of the precursor mass was determined. Ions uniquely found in exposed samples were then confirmed by manually searching the the corresponding 15N-version of the hypothesized adduct in the 15N-labelled-DNA sample. MS2 and MS3 spectra of 15N-molecules were also compared with the ones observed in unlabelled or cell-derived DNA.
General Methods for Chemistry.
All commercial reagents (Sigma-Aldrich, Acros Organics, and Alfa-Aesar) were used as provided unless otherwise indicated. Compounds 296, 646, and 1097 were synthesized as described. ACS and HPLC grade solvents were purchased from Fischer Scientific. An anhydrous solvent dispensing system (MBRAUN) using packed columns of neutral alumina was used for drying CH2Cl2 and the solvents were dispensed under nitrogen. All reactions were performed under an inert atmosphere of argon in oven dried (130-150 °C) glassware. Thin layer chromatography (TLC plate, Merck EMD Millipore) was performed on a pre-coated silica gel 60 F254 plates. The detection of compounds was carried out with UV light. Flash chromatography was performed with silica gel P60 (Silicycle) with the indicated solvent system. All NMR spectra were recorded on a Varian 400 MHz spectrometer. 1H NMR spectra were referenced to residual CDCl3 (7.27 ppm) or CD3OD (3.31 ppm); 13C NMR spectra were referenced to CDCl3 (77.23 ppm) or CD3OD (49.0 ppm). Data for 1H NMR are reported as follows: chemical shift (multiplicity, coupling constant in Hertz (Hz), number of hydrogens). Abbreviations are as follows: s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, m = multiplet, br = broad. High-resolution mass spectra (HRMS) were obtained at the Department of Chemistry, University of Minnesota, on a Bruker BioTOF II instrument.
Synthesis of Tilimycin and Tilivalline
Tilimycin, tilivalline and O-benzyltilivalline were synthesized as described and the 1H NMR and HRMS data matched previously reported data.29,98
General Procedure 1: Global Deprotection.
Fully protected adenosine acyl-sulfamide adenylate (4, S3, S4) (0.1 mmol) was dissolved in EtOH (20 mL), then charged with 5% w/w Pd/C (10 wt%). The flask was evacuated and backfilled with H2 three times, then stirred under an atmosphere of H2 for 3 h. The Pd/C was removed by filtering through a pad of celite and the supernatant was concentrated to yield a waxy brown solid. The solid was dissolved in TFA:H20 (4:1) and stirred at room temperature for an additional 3 h. The acidic solution was concentrated in vacuo to yield a crude orange solid. The crude material was redissolved (10 mg/mL) in 1:1 MeCN-50 mM triethylammonium bicarbonate (pH 7.5) and filtered to remove insoluble solids. The resulting solution was purified by preparative reverse phase HPLC with a Phenomenex Gemini C18 (250 mm × 21 mm) column at a flow rate of 21.0 mL/min employing a gradient of 5→30% acetonitrile (Solvent B) in 50 mM aqueous triethylammonium bicarbonate at pH 7.5 for 15 minutes. Fractions containing the product were pooled and lyophilized to afford the final compound as the triethylammonium salt.
5′-[N-Sulfamoyl]amino-2′,3′-O-isopropylidene-5′-deoxyadenosine (3).
Compound 2 (690 mg, 2.08 mmol, 1 equiv) was dissolved in EtOH (15 mL). The flask was charged with 5% w/w Pd/C (70 mg, 10 wt%) evacuated and backfilled with H2 three times, then stirred under an atmosphere of H2 for 2 h. The Pd/C was removed by filtering through a pad of celite and the supernatant was concentrated to yield a yellow oil. The crude material was redissolved in 1,4 dioxanes (25 mL) and refluxed for 2 h. The reaction mixture was concentrated and purified by silica gel flash chromatography (dry loading, 1→5% MeOH/EtOAc) affording the title compound (510 mg, 64%) as a white solid: Rf = 0.24 (5:95 MeOH/EtOAc); 1H NMR (400 MHz, CD3OD) δ 1.36 (s, 3H), 1.60 (s, 3H), 3.36 (dd, 2H, J = 3.90, 5.24 Hz), 4.47 (td, 1H, 7= 2.32, 3.78, 3.78 Hz), 5.09 (dd, 1H, J = 2.45, 6.23 Hz), 5.32 (dd, 1H, J = 4.10, 6.26 Hz), 6.05 (d, 1H, J = 4.07 Hz), 8.22 (s, 1H), 8.26 (s, 1H). MS (ESI+) m/z calcd for C13H19N7O5SNa+ [M+Na]+ 408.1, found 408.6.
5′-[N-(3-Benzyloxy-2-nitrobenzoyl)sulfamoyl]amino-2′,3′-O-isopropylidene-5′-deoxyadenosine (4).
The NHS-ester 7 (74 mg, 0.2 mmol, 1.5 equiv) and adenosine monosulfamide (3) (50 mg, 0.13 mmol, 1 equiv) were dissolved in DMF (5 mL). The reaction mixture was cooled to 0 °C and Cs2CO3 (130 mg, 0.4 mmol, 3 equiv) was added. The reaction was allowed to warm to room temperature and stirred for 18 h. The excess Cs2CO3 was removed by filtration with a sintered glass filter and the supernatant was concentrated to yield a yellow solid, which was purified by silica gel flash chromatography (dry loading, 2.5→15% MeOH/EtOAc) afforded the title compound (53 mg, 65%) as a white solid: Rf = 0.25 (10:90 MeOH/EtOAc); 1H NMR (400 MHz, CD3OD) δ 1.32 (s, 3H), 1.56 (s, 3H), 3.35–3.37 (m, 2H), 4.41–4.42 (m, 1H), 5.08 (dd, J = 6.1, 2.5 Hz, 1H), 5.16 (s, 2H), 5.32 (dd, J = 6.2, 3.8 Hz, 1H), 6.07 (d, J = 3.7 Hz, 1H), 7.25–7.39 (m, 8H), 8.23 (s, 1H), 8.26 (s, 1H); 13C NMR (100 MHz, CD3OD) δ 25.6, 27.6, 46.3, 72.2, 83.4, 84.7, 85.7, 92.4, 115.6, 117.7, 120.6, 122.5, 128.3, 129.1, 129.14, 129.5, 131.8, 133.3, 137.3, 141.7, 150.0, 151.0, 154.0, 157.2, 170.3; HRMS (ESI–) m/z calcd for C27H27N8O9S− [M−H]− 639.1627, found 639.1657 (error 4.5 ppm).
5′-[N-(2-Amino-3-hydroxybenzoyl)sulfamoyl]amino-5′-deoxyadenosine triethylammonium salt (5).
The title compound was prepared from 4 using general procedure 1 to afford a colorless foam (20 mg. 45%): HPLC purity = 93.8%, tR = 8.86 min, k′ = 2.5; 1H NMR (400 MHz, CD3OD) δ 1.29 (t, J = 8 Hz, 27 H, excess Et3N), 3.16 (q, J = 8 Hz, 18 H, excess Et3N), 3.29–3.41 (m, 2H), 4.26 (q, J = 3.4 Hz, 1H), 4.35 (dd, 7= 5.4, 2.5 Hz, 1H), 4.86–4.90 (m, 1H), 5.91 (d, J = 6.8 Hz, 1H), 6.47 (t, J = 7.9, 1H), 6.76 (d, J = 7.7 Hz, 1H), 7.23 (d, 7= 8.1 Hz, 1H), 8.26 (s, 1H), 8.39 (s, 1H), 8.55 (s, 2H); 13C NMR (100 MHz, CD3OD) δ 9.3, 46.5, 47.8, 73.2, 74.6, 85.9, 91.0, 115.1, 115.9, 119.5, 120.2, 138.7, 140.7, 145.2, 148.8, 152.7, 155.7, 171.8; HRMS (ESI-) m/z calcd for C17H19N8O7S− [M−H]− 479.1103, found 479.1088 (error 3.6 ppm).
3-Benzyloxy-2-nitrobenzoic acid N-hydroxysuccinimide ester (7).
Aryl acid 646 (270 mg, 1 mmol, 1 equiv) was dissolved in THF (25 mL). The solution was cooled to 0 °C. N-hydroxysuccimide (250 mg, 2.2 mmol, 2.2 equiv) was added followed by DCC (450 mg, 2.2 mmol, 2.2 equiv). The reaction mixture was allowed to warm to room temperature and stirred for 12 h. The resulting precipitate was filtered with a sintered glass filter and the supernatant was concentrated and purified by silica gel flash chromatography (dry loading, 10→50% EtOAc/hexanes) affording the title compound (315 mg, 85%) as a white solid: Rf = 0.30 (50:50 EtOAc/hexanes); 1H NMR (400 MHz, CDC13) δ 2.88 (s, 4H), 5.24 (s, 2H), 7.33–7.40 (m, 6H), 7.51 (t, J = 8.19 Hz, 1H), 7.76 (d, J = 7.86 Hz, 1H); 13C NMR (100 MHz, CDC13) δ 25.8, 71.7, 118.7 121.0, 122.8, 127.2, 128.7, 129.0, 131.2, 134.8, 150.5, 158.5, 168.6 (missing 1 aryl carbon); HRMS (ESI+) m/z calcd for C18H14N2O7Na+ [M+Na]+ 393.0693, found 393.0692 (error 0.4 ppm).
5′-[N-(3-Hydroxybenzoyl)sulfamoyl]amino-5′-deoxyadenosine triethylammonium salt (8).
The title compound was prepared from S3 using general procedure 1 afford a colorless foam (23 mg, 49%): HPLC purity = 99.8%, tR = 12.10 min, k′ = 3.5; 1H NMR (400 MHz, CD3OD) δ 1.27 (t, J = 8.0 Hz, 9H), 3.15 (q, J = 8.0 Hz, 6H), 3.31–3.35 (m, 2H), 4.24 (q, J = 3.4 Hz, 1H), 4.34–4.36 (dd, J = 5.4, 2.8 Hz, 1H), 4.85–4.86 (m, 1H), 5.92 (d, J = 6.5 Hz, 1H), 6.87 (d, J = 2.6 Hz, 1H), 7.18 (t, J = 7.9 Hz, 1H), 7.38 (s, 1H), 7.43 (d, J = 7.9 Hz, 1H), 8.28 (s, 1H), 8.33 (s, 1H); 13C NMR (100 MHz, CD3OD) δ 9.3, 46.4, 47.8, 73.1, 74.8, 85.9, 90.7, 116.3, 119.3, 120.7, 120.8 129.9, 139.5, 141.9, 150.4, 154.2, 157.4, 158.3 (missing 1 aryl carbon); HRMS (ESI-) m/z calcd for C17H18N7O7S− [M−H]− 464.0994, found 464.0978 (error 3.4 ppm).
5′-[N-(2-Aminobenzoyl)sulfamoyl]amino-5′-deoxyadenosine triethylammonium salt (9).
The title compound was prepared from S4 using general procedure 1 to afford a colorless foam (12 mg, 25%): HPLC purity = 90.5%, tR = 9.98 min, k′ = 2.9; 1H NMR (400 MHz, CD3OD) δ 1.23 (t, J = 8.0 Hz, 9H), 3.05 (q, J = 8.0 Hz, 6H), 3.31–3.34 (m, 2H), 4.24 (q, J = 3.3 Hz, 1H), 4.36 (dd, J = 5.4, 2.9 Hz, 1H), 4.82–4.85 (m, 1H), 5.94 (d,J = 6.3 Hz, 1H), 6.56 (t, J = 7.5, 1H), 6.67 (d, J = 8.2 Hz, 1H), 7.10 (t, J = 8.3 Hz, 1H), 7.80 (d, J = 8.0 Hz, 1H), 8.31 (s, 2H); 13C NMR (100 MHz, CD3OD) δ 9.6, 46.5, 47.7, 73.0, 74.9, 85.9, 90.4, 117.0, 118.0, 120.5, 120.7, 131.8, 132.8, 141.8 150.5, 150.7, 154.1, 157.3, 175.9; HRMS (ESI-) m/z calcd for C17H19N8O6S− [M−H]− 463.1154, found 463.1130 (error 5.0 ppm).
(E)-tert-Butyl {2-[3-(methoxymethoxy)phenyl]ethenyl}sulfonylcarbamate (11)
tert-Butyl[(diphenylphosphoryl)methyl]sulfonyl carbamate49 (150 mg, 0.37 mmol, 1 equiv) was dissolved in DMF (5 mL) under an atmosphere of argon. The solution was cooled to 0 °C and NaH (27 mg, 1.1 mmol, 3 equiv) was added. The reaction mixture was allowed to warm to 24 °C and stirred for an additional 30 min to ensure complete conversion to the ylide. Aldehyde 1097 (63 mg, 0.37 mmol, 1 equiv) was added as a solution in DMF (2 mL) and the reaction was stirred overnight. The reaction mixture was concentrated to a yellow oil and partitioned between H20 (20 mL) and EtOAc (20 mL). The organic layer was collected, dried over Na2S04 and concentrated to a light-yellow oil. Purification by flash chromatography (15→30% EtOAc/Hexanes) afforded the title compound (120 mg, 92%) as a white solid: Rf = 0.45 (33:67 EtOAc/Hexanes); 1H NMR (400 MHz, CDC13) δ 1.85 (s, 9H), 3.50 (s, 3H), 5.21 (s, 2H), 5.30 (s, 1H), 6.99 (d, J = 15.5 Hz, 1H), 7.13–7.18 (m, 2H), 7.21 (s, 1H), 7.35 (t, J = 8.0 Hz, 1H), 7.64 (d, J = 15.5 Hz, 1H); 13C NMR (100 MHz, CDC13) δ 28.1, 56.2, 84.3, 94.6, 116.0, 119.5, 122.5, 124.5, 130.3, 133.6, 144.4, 149.6, 157.9; HRMS (ESI+) m/z calcd for C15H22NO6S+ [M+H]+ 344.1162, found 344.1101 (error 1.6 ppm).
(E)-5′-Deoxy-5′-N-teri-butoxycarbonyl-2′,3′-O-isopropylidene-5′N-({2-[3-(methoxymethoxy)phenyl]ethenyl}sulfonyl)adenosine (12)
To a solution of 2′,3′-O-isopropylideneadenosine 1 (32 mg, 0.1 mmol), 11 (30 mg, 0.09 mmol) and triphenylphosphine (34 mg, 0.13 mmol), in THF (1 mL) at 0 °C was added a solution of DIAD (26 μL, 0.13 mmol) in THF (1 mL) over 10 min. The solution was allowed to warm to 24 °C and stirred for 15 h. The mixture was concentrated and purified by flash chromatography (100% EtOAc) to afford a white foam (10 mg, 75% brsm): Rf = 0.12 (100% EtOAc); 1H NMR (400 MHz, CD3OD) δ 1.38 (s, 3H), 1.41 (s, 9H), 1.58 (s, 3H), 3.46 (s, 3H), 4.03–4.06 (m, 2H), 4.44–4.48 (m, 1H), 5.18 (dd, J = 6.4, 3.3 Hz, 1 H), 5.23 (s, 2H), 5.52 (d, 6.3 Hz, 1H), 6.25 (d, J = 2.0 Hz, 1H), 7.02 (d, J = 15.4 Hz, 1H), 7.12 (t, J = 9.5 Hz, 1H), 7.18 (s, 1H), 7.34 (t, J = 8.0 Hz, 1H), 7.36 (d, J = 15.5 Hz, 1H), 8.23 (s, 1H), 8.27 (s, 1H). MS (ESI+) m/z calcd for C28H37N6O9S+ [M+H]+ 633.2, found 633.3.
(E)-5′-Amino-5′-deoxy-5′N-{[2–3-hydroxyphenyl)ethenyl]sulfonyl}adenosine (13)
To a solution of 12 (7 mg, 0.01 mmol) was added 80% aqueous TFA (5 mL). The solution was stirred at 24 °C for 2 h then concentrated. Purification by flash chromatography (1→10% MeOH/EtOAc) afforded a white solid (5 mg, 95%): HPLC purity = 94.1%, tR = 11.97 min, k′ = 3.6 Rf = 0.30 (1:9 MeOH/EtOAc); 1H NMR (600 MHz, CD3OD) δ 3.41 (d, J = 3.5 Hz, 1H), 4.28 (q, J = 3.2 Hz, 1H), 4.37 (dd, J = 5.4, 2.7 Hz, 1H), 4.87–4.89 (m, 1H), 5.93 (d, J = 6.6 Hz, 1H), 6.85 (dd, J = 8.2, 2.5 Hz), 6.95 (d, J = 15.4 Hz, 1H), 6.97 (s, 1H), 7.02 (d, J = 7.6 Hz, 1H), 7.22 (t, J = 7.9 Hz, 1H), 7.37 (d, J = 15.4 Hz, 1H), 8.24 (s, 1H), 8.26 (s, 1H); 13C NMR (100 MHz, CD3OD) δ 45.8, 73.0, 74.4, 85.9, 91.5, 115.5, 118.9, 120.8, 121.2, 126.7, 131.1, 135.5, 142.2, 142.3, 150.0, 153.7, 157.6, 159.2; HRMS (ESI+) m/z calcd for C18H21N6O6S+ [M+H]+ 449.1237, found 449.1230 (error 1.6 ppm).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants (GM116957 and GM136235 to A.M.G.) from the National Institutes of Health. Mass spectrometry for tilivalline quantitation was carried out in the Center for Mass Spectrometry and Proteomics Mass spectrometry for DNA adductomics was carried out in the Analytical Biochemistry Shared Resource of the Masonic Cancer Center, University of Minnesota and supported in part by the U.S. National Institutes of Health and National Cancer Institute (Cancer Center Support Grant CA-77598). Isothermal titration calorimetry was carried out using an ITC-200 microcalorimeter, funded by the NIH Shared Instrumentation Grant S10-OD017982. GM/CA@APS has been funded in whole or in part with Federal funds from the National Cancer Institute (ACB-12002) and the National Institute of General Medical Sciences (AGM-12006). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (P41GM103393).
ABBREVIATIONS
- 3HA-AMP
3-hydroxyanthraniloyl adenosine monophosphate
- Asub
adenylation domain C-terminal subdomain
- AAHC
antibiotic associated hemorrhagic colitis
- ACP
acyl-carrier protein
- acyl-AMS
acyl adenosine monosulfamide
- ATP
adenosine triphosphate
- AVS
adenosine vinylsulfonamide
- CASO
tryptic soy broth
- ctDNA
calf thymus DNA
- dG
2’-deoxyguanosine
- dA
2-deoxyadenosine
- ΔG
gibbs free energy
- GI
gastrointestinal
- ΔH
enthalpy
- HPLC
high performance liquid chromatography
- IMAC
immobilized metal affinity chromatography
- ITC
isothermal titration calorimetry
- KD
dissociation constant
- LC-MS
liquid chromatography-mass spectrometry
- LC-MS/MS
liquid chromatography-tandem mass spectrometry
- l-Pro-S~TNpsB
prolyl-loaded thiolation domain
- NHS
N-hydroxysuccinimidyl
- NL
neutral loss
- NRPS
nonribosomal peptide synthetase
- MDFF
molecular dynamics flexible fitting
- MEM
Dulbecco’s minimum essential medium
- MesG
7-methylthioguanine
- PBD
pyrrolobenzodiazepine
- PPi
pyrophosphate
- ΔS
entropy
- SEC
size exclusion chromatography
- SER
surface entropy reduction
- sfp
4′-phosphopantetheinyl transferase
- TEV
tobacco etch virus
Footnotes
Supporting Information: The Supporting Information is available free of charge via the Internet at http://pubs.acs.org. A complete list of LC-MS/MS parameters and fragmentation data, X-ray crystallographic data for NpsA and ThdA and 1H and 13C NMR of all compounds.
The authors declare no competing financial interests.
References
- (1).Savage DC (1977) Microbial Ecology of the Gastrointestinal Tract. Annu. Rev. Microbiol 31, 107–133. [DOI] [PubMed] [Google Scholar]
- (2).Eloe-Fadrosh EA, and Rasko DA (2013) The Human Microbiome: From Symbiosis to Pathogenesis. Annu. Rev. Med 64, 145–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Nougayrede J-P (2006) Escherichia coli Induces DNA Double-Strand Breaks in Eukaryotic Cells. Science 313, 848–851. [DOI] [PubMed] [Google Scholar]
- (4).Peery AF, Dellon ES, Lund J, Crockett SD, McGowan CE, Bulsiewicz WJ, Gangarosa LM, Thiny MT, Stizenberg K, Morgan DR, Ringel Y, Kim HP, DiBonaventura MD, Carroll CF, Allen JK, Cook SF, Sandler RS, Kappelman MD, and Shaheen NJ (2012) Burden of Gastrointestinal Disease in the United States: 2012 Update. Gastroenterology 143, 1179–1187.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Belizario JE, and Napolitano M (2015) Human microbiomes and their roles in dysbiosis, common diseases, and novel therapeutic approaches. Front. Microbiol 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Marchesi JR, Adams DH, Fava F, Hermes GDA, Hirschfield GM, Hold G, Quraishi MN, Kinross J, Smidt H, Tuohy KM, Thomas LV, Zoetendal EG, and Hart A (2016) The gut microbiota and host health: a new clinical frontier. Gut 65, 330–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Lynch SV, and Pedersen O (2016) The Human Intestinal Microbiome in Health and Disease. N. Engl. J. Med. (Phimister, E. G., Ed.) 375, 2369–2379. [DOI] [PubMed] [Google Scholar]
- (8).Lloyd-Price J, Abu-Ali G, and Huttenhower C (2016) The healthy human microbiome. Genome Med. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Huttenhower C, Gevers D, Knight R, Abubucker S, Badger JH, Chinwalla AT, Creasy HH, Earl AM, FitzGerald MG, Fulton RS, Giglio MG, Hallsworth-Pepin K, Lobos EA, Madupu R, Magrini V, Martin JC, Mitreva M, Muzny DM, Sodergren EJ, Versalovic J, Wollam AM, Worley KC, Wortman JR, Young SK, Zeng Q, Aagaard KM, Abolude OO, Allen-Vercoe E, Alm EJ, Alvarado L, Andersen GL, Anderson S, Appelbaum E, Arachchi HM, Armitage G, Arze CA, Ayvaz T, Baker CC, Begg L, Belachew T, Bhonagiri V, Bihan M, Blaser MJ, Bloom T, Bonazzi V, Paul Brooks J, Buck GA, Buhay CJ, Busam DA, Campbell JL, Canon SR, Cantarel BL, Chain PSG, Chen I-MA, Chen L, Chhibba S, Chu K, Ciulla DM, Clemente JC, Clifton SW, Conlan S, Crabtree J, Cutting MA, Davidovics NJ, Davis CC, DeSantis TZ, Deal C, Delehaunty KD, Dewhirst FE, Deych E, Ding Y, Dooling DJ, Dugan SP, Michael Dunne W, Scott Durkin A, Edgar RC, Erlich RL, Farmer CN, Farrell RM, Faust K, Feldgarden M, Felix VM, Fisher S, Fodor AA, Forney LJ, Foster L, Di Francesco V, Friedman J, Friedrich DC, Fronick CC, Fulton LL, Gao H, Garcia N, Giannoukos G, Giblin C, Giovanni MY, Goldberg JM, Goll J, Gonzalez A, Griggs A, Gujja S, Kinder Haake S, Haas BJ, Hamilton HA, Harris EL, Hepburn TA, Herter B, Hoffmann DE, Holder ME, Howarth C, Huang KH, Huse SM, Izard J, Jansson JK, Jiang H, Jordan C, Joshi V, Katancik JA, Keitel WA, Kelley ST, Kells C, King NB, Knights D, Kong HH, Koren O, Koren S, Kota KC, Kovar CL, Kyrpides NC, La Rosa PS, Lee SL, Lemon KP, Lennon N, Lewis CM, Lewis L, Ley RE, Li K, Liolios K, Liu B, Liu Y, Lo C-C, Lozupone CA, Dwayne Lunsford R, Madden T, Mahurkar AA, Mannon PJ, Mardis ER, Markowitz VM, Mavromatis K, McCorrison JM, McDonald D, McEwen J, McGuire AL, McInnes P, Mehta T, Mihindukulasuriya KA, Miller JR, Minx PJ, Newsham I, Nusbaum C, O’Laughlin M, Orvis J, Pagani I, Palaniappan K, Patel SM, Pearson M, Peterson J, Podar M, Pohl C, Pollard KS, Pop M, Priest ME, Proctor LM, Qin X, Raes J, Ravel J, Reid JG, Rho M, Rhodes R, Riehle KP, Rivera MC, Rodriguez-Mueller B, Rogers Y-H, Ross MC, Russ C, Sanka RK, Sankar P, Fah Sathirapongsasuti J, Schloss JA, Schloss PD, Schmidt TM, Scholz M, Schriml L, Schubert AM, Segata N, Segre JA, Shannon WD, Sharp RR, Sharpton TJ, Shenoy N, Sheth NU, Simone GA, Singh I, Smillie CS, Sobel JD, Sommer DD, Spicer P, Sutton GG, Sykes SM, Tabbaa DG, Thiagarajan M, Tomlinson CM, Torralba M, Treangen TJ, Truty RM, Vishnivetskaya TA, Walker J, Wang L, Wang Z, Ward DV, Warren W, Watson MA, Wellington C, Wetterstrand KA, White JR, Wilczek-Boney K, Wu Y, Wylie KM, Wylie T, Yandava C, Ye L, Ye Y, Yooseph S, Youmans BP, Zhang L, Zhou Y, Zhu Y, Zoloth L, Zucker JD, Birren BW, Gibbs RA, Highlander SK, Methe BA, Nelson KE, Petrosino JF, Weinstock GM, Wilson RK, and White O (2012) Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Hajishengallis G, and Lamont RJ (2016) Dancing with the Stars: How Choreographed Bacterial Interactions Dictate Nososymbiocity and Give Rise to Keystone Pathogens, Accessory Pathogens, and Pathobionts. Trends Microbiol. 24, 477–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Petersen C, and Round JL (2014) Defining dysbiosis and its influence on host immunity and disease: How changes in microbiota structure influence health. Cell. Microbiol 16, 1024–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, Challis C, Schretter CE, Rocha S, Gradinaru V, Chesselet M-F, Keshavarzian A, Shannon KM, Krajmalnik-Brown R, Wittung-Stafshede P, Knight R, and Mazmanian SK (2016) Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 167, 1469–1480.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Kowalski K, and Mulak A (2019) Brain-Gut-Microbiota Axis in Alzheimer’s Disease. J. Neurogastroenterol. Motil 25, 48–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Haran JP, Bhattarai SK, Foley SE, Dutta P, Ward DV, Bucci V, and McCormick BA (2019) Alzheimer’s Disease Microbiome Is Associated with Dysregulation of the Anti-Inflammatory P-Glycoprotein Pathway. mBio (Pettigrew, M. M., Ed.) 10, e00632–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Pickard JM, Zeng MY, Caruso R, and ^nez G (2017) Gut microbiota: Role in pathogen colonization, immune responses, and inflammatory disease. Immunol. Rev 279, 70–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, and Burcelin R (2008) Changes in Gut Microbiota Control Metabolic Endotoxemia-Induced Inflammation in High-Fat Diet-Induced Obesity and Diabetes in Mice. Diabetes 57, 1470–1481. [DOI] [PubMed] [Google Scholar]
- (17).Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W, Guan Y, Shen D, Peng Y, Zhang D, Jie Z, Wu W, Qin Y, Xue W, Li J, Han L, Lu D, Wu P, Dai Y, Sun X, Li Z, Tang A, Zhong S, Li X, Chen W, Xu R, Wang M, Feng Q, Gong M, Yu J, Zhang Y, Zhang M, Hansen T, Sanchez G, Raes J, Falony G, Okuda S, Almeida M, LeChatelier E, Renault P, Pons N, Batto J-M, Zhang Z, Chen H, Yang R, Zheng W, Li S, Yang H, Wang J, Ehrlich SD, Nielsen R, Pedersen O, Kristiansen K, and Wang J (2012) A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 490, 55–60. [DOI] [PubMed] [Google Scholar]
- (18).Davis-Richardson AG, and Triplett EW (2015) A model for the role of gut bacteria in the development of autoimmunity for type 1 diabetes. Diabetologia 58, 1386–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Xue M, Kim CS, Healy AR, Wernke KM, Wang Z, Frischling MC, Shine EE , Wang W, Herzon SB, and Crawford JM (2019) Structure elucidation of colibactin and its DNA cross-links. Science 365, eaax2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).De Angelis M, Francavilla R, Piccolo M, De Giacomo A, and Gobbetti M (2015) Autism spectrum disorders and intestinal microbiota. Gut Microbes 6, 207–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Louis P, Hold GL, and Flint HJ (2014) The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol 12, 661–672. [DOI] [PubMed] [Google Scholar]
- (22).Vizcaino MI, and Crawford JM (2015) The colibactin warhead crosslinks DNA. Nat. Chem 7, 411–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Hogenauer C, Lippe IT, Krause R, and Hinterleitner TA (2006) Klebsiella oxytoca as a Causative Organism of Antibiotic-Associated Hemorrhagic Colitis. N. Engl. J. Med 9. [DOI] [PubMed] [Google Scholar]
- (24).Schneditz G, Rentner J, Roier S, Pletz J, Herzog KAT, Bucker R, Troeger H, Schild S, Weber H, Breinbauer R, Gorkiewicz G, Hogenauer C, and Zechner EL (2014) Enterotoxicity of a nonribosomal peptide causes antibiotic-associated colitis. Proc. Natl. Acad. Sci 111, 13181–13186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Youn Y, Lee SW, Cho H-H, Park S, Chung H-S, and Seo JW (2018) Antibiotics-Associated Hemorrhagic Colitis Caused by Klebsiella oxytoca: Two Case Reports. Pediatr. Gastroenterol. Hepatol. Nutr 21, 141–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Darby A, Lertpiriyapong K, Sarkar U, Seneviratne U, Park DS, Gamazon ER, Batchelder C, Cheung C, Buckley EM, Taylor NS, Shen Z, Tannenbaum SR, Wishnok JS, and Fox JG (2014) Cytotoxic and Pathogenic Properties of Klebsiella oxytoca Isolated from Laboratory Animals. PLoS ONE (Bereswill, S., Ed.) 9, e100542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Dornisch E, Pletz J, Glabonjat RA, Martin F, Lembacher-Fadum C, Neger M, Hogenauer C, Francesconi K, Kroutil W, Zangger K, Breinbauer R, and Zechner EL (2017) Biosynthesis of the Enterotoxic Pyrrolobenzodiazepine Natural Product Tilivalline. Angew. Chem. Int. Ed 56, 14753–14757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).von Tesmar A, Hoffmann M, Abou Fayad A, Huttel S, Schmitt V, Herrmann J, and Muller R (2018) Biosynthesis of the Klebsiella oxytoca Pathogenicity Factor Tilivalline: Heterologous Expression, in Vitro Biosynthesis, and Inhibitor Development. ACS Chem. Biol 13, 812–819. [DOI] [PubMed] [Google Scholar]
- (29).Tse H, Gu Q, Sze K-H, Chu IK, Kao RY-T, Lee K-C, Lam C-W, Yang D, Tai SS-C, Ke Y, Chan E, Chan W-M, Dai J, Leung S-P, Leung S-Y, and Yuen K-Y (2017) A tricyclic pyrrolobenzodiazepine produced by Klebsiella oxytoca is associated with cytotoxicity in antibiotic-associated hemorrhagic colitis. J. Biol. Chem 292, 19503–19520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Hering NA, Fromm A, Bücker R, Gorkiewicz G, Zechner E, Högenauer C, Fromm M, Schulzke J-D, and Troeger H (2019) Tilivalline- and Tilimycin-Independent Effects of Klebsiella oxytoca on Tight Junction-Mediated Intestinal Barrier Impairment. Int. J. Mol. Sci 20, 5595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Mohr N, and Budzikiewicz H (1982) Tilivalline, a New Pyrrolo[2, 1-c][1, 4]Benzodiazepine Metabolite From Klebsiella. Tetrahedron 38, 147–152. [Google Scholar]
- (32).Minami J, Okabe A, Shiode J, and Hayashi H (1989) Production of a unique cytotoxin by Klebsiella oxytoca. Microb. Pathog 7, 203–211. [DOI] [PubMed] [Google Scholar]
- (33).Unterhauser K, Poltl L, Schneditz G, Kienesberger S, Glabonjat RA, Kitsera M, Pletz J, Josa-Prado F, Dornisch E, Lembacher-Fadum C, Roier S, Gorkiewicz G, Lucena D, Barasoain I, Kroutil W, Wiedner M, Loizou JI, Breinbauer R, Diaz JF , Schild S, Hogenauer C, and Zechner EL (2019) Klebsiella oxytoca enterotoxins tilimycin and tilivalline have distinct host DNA-damaging and microtubule-stabilizing activities. Proc. Natl. Acad. Sci 116, 3774–3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Fischbach MA, and Walsh CT (2006) Assembly-Line Enzymology for Polyketide and Nonribosomal Peptide Antibiotics: Logic, Machinery, and Mechanisms. Chem. Rev 106, 3468–3496. [DOI] [PubMed] [Google Scholar]
- (35).Kuhn ML, Alexander E, Minasov G, Page HJ, Warwrzak Z, Shuvalova L, Flores KJ, Wilson DJ, Shi C, Aldrich CC, and Anderson WF (2016) Structure of the Essential Mtb FadD32 Enzyme: A Promising Drug Target for Treating Tuberculosis. ACS Infect. Dis 2, 579–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Drake EJ, Duckworth BP, Neres J, Aldrich CC, and Gulick AM (2010) Biochemical and Structural Characterization of Bisubstrate Inhibitors of BasE, the Self-Standing Nonribosomal Peptide Synthetase Adenylate-Forming Enzyme of Acinetobactin Synthesis,. Biochemistry 49, 9292–9305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Gulick AM (2009) Conformational Dynamics in the Acyl-CoA Synthetases, Adenylation Domains of Non-ribosomal Peptide Synthetases, and Firefly Luciferase. ACS Chem. Biol 4, 811–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Webb MR (1992) A continuous spectrophotometric assay for inorganic phosphate and for measuring phosphate release kinetics in biological systems. Proc. Natl. Acad. Sci 89, 4884–4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Ehmann DE, Shaw-Reid CA, Losey HC, and Walsh CT (2000) The EntF and EntE adenylation domains of Escherichia coli enterobactin synthetase: Sequestration and selectivity in acyl-AMP transfers to thiolation domain cosubstrates. Proc. Natl. Acad. Sci 97, 2509–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Wilson DJ, and Aldrich CC (2010) A continuous kinetic assay for adenylation enzyme activity and inhibition. Anal. Biochem 404, 56–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Bythrow GV, Mohandas P, Guney T, Standke LC, Germain GA, Lu X, Ji C, Levendosky K, Chavadi SS, Tan DS, and Quadri LEN (2019) Kinetic Analyses of the Siderophore Biosynthesis Inhibitor Salicyl-AMS and Analogues as MbtA Inhibitors and Antimycobacterial Agents. Biochemistry 58, 833–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Lux MC, Standke LC, and Tan DS (2019) Targeting adenylate-forming enzymes with designed sulfonyladenosine inhibitors. J. Antibiot. (Tokyo) 72, 325–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Ueda H, Shoku Y, Hayashi N, Mitsunaga J, In Y, Doi M, Inoue M, and Ishida T (1991) X-ray crystallographic conformational study of 5′O-[N-(l-alanyl)-sulfamoyl]adenosine, a substrate analogue for alanyl-tRNA synthetase. Biochim. Biophys. ActaBBA - Protein Struct. Mol. Enzymol 1080, 126–134. [DOI] [PubMed] [Google Scholar]
- (44).Liu F, and Austin DJ (2001) A General Synthesis of 5’-Azido-5’-deoxy-2’,3’- O - isopropylidene Nucleosides. J. Org. Chem 66, 8643–8645. [DOI] [PubMed] [Google Scholar]
- (45).Dawadi S, Kawamura S, Rubenstein A, Remmel R, and Aldrich CC (2016) Synthesis and Pharmacological Evaluation of Nucleoside Prodrugs Designed to Target Siderophore Biosynthesis in Mycobacterium tuberculosis. Bioorg. Med. Chem 24, 1314–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Tipparaju SK, Joyasawal S, Pieroni M, Kaiser M, Brun R, and Kozikowski AP (2008) In Pursuit of Natural Product Leads: Synthesis and Biological Evaluation of 2-[3-hydroxy-2-[(3-hydroxypyridine-2-carbonyl)amino]phenyl]benzoxazole-4-carboxylic acid (A-33853) and Its Analogues: Discovery of N -(2-Benzoxazol-2-ylphenyl)benzamides as Novel Antileishmanial Chemotypes. J. Med. Chem 51, 7344–7347. [DOI] [PubMed] [Google Scholar]
- (47).Qiao C, Wilson DJ, Bennett EM, and Aldrich CC (2007) A Mechanism-Based Aryl Carrier Protein/Thiolation Domain Affinity Probe. J. Am. Chem. Soc 129, 6350– 6351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Sundlov JA, Shi C, Wilson DJ, Aldrich CC, and Gulick AM (2012) Structural and Functional Investigation of the Intermolecular Interaction between NRPS Adenylation and Carrier Protein Domains. Chem. Biol 19, 188–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Drake EJ, Miller BR, Shi C, Tarrasch JT, Sundlov JA, Leigh Allen C, Skiniotis G, Aldrich CC, and Gulick AM (2016) Structures of two distinct conformations of holo-non-ribosomal peptide synthetases. Nature 529, 235–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Miller BR, Drake EJ, Shi C, Aldrich CC, and Gulick AM (2016) Structures of a Nonribosomal Peptide Synthetase Module Bound to MbtH-like Proteins Support a Highly Dynamic Domain Architecture. J. Biol. Chem 291, 22559–22571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Reuter D, McIntosh J, Guinn A, Madera A (2003) Synthesis of Vinyl Sulfonamides Using the Horner Reaction. Synthesis 15, 2321–2324. [Google Scholar]
- (52).Gerratana B (2012) Biosynthesis, synthesis, and biological activities of pyrrolobenzodiazepines: Activities of Pyrrolobenzodiazepines. Med. Res. Rev 32, 254– 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Mantaj J, Jackson PJM, Rahman KM, and Thurston DE (2017) From Anthramycin to Pyrrolobenzodiazepine (PBD)-Containing Antibody-Drug Conjugates (ADCs). Angew. Chem. Int. Ed 56, 462–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Hertzberg RP, Hecht SM, Reynolds VL, Molineux IJ, and Hurley LH (1986) DNA sequence specificity of the pyrrolo[1,4]benzodiazepine antitumor antibiotics. Methidiumpropyl-EDTA-iron(II) footprinting analysis of DNA binding sites for anthramycin and related drugs. Biochemistry 25, 1249–1258. [DOI] [PubMed] [Google Scholar]
- (55).Hurley LH, Reck T, Thurston DE, Langley DR, Holden KG, Hertzberg RP, Hoover JRE, Gallagher G, and Faucette LF (1988) Pyrrolo[1,4]benzodiazepine antitumor antibiotics: relationship of DNA alkylation and sequence specificity to the biological activity of natural and synthetic compounds. Chem. Res. Toxicol 1, 258–268. [DOI] [PubMed] [Google Scholar]
- (56).Antonow D, Barata T, Jenkins TC, Parkinson GN, Howard PW, Thurston DE, and Zloh M (2008) Solution Structure of a 2:1 C2-(2-Naphthyl) Pyrrolo[2,1- c][1,4]benzodiazepine DNA Adduct: Molecular Basis for Unexpectedly High DNA Helix Stabilization † ‡. Biochemistry 47, 11818–11829. [DOI] [PubMed] [Google Scholar]
- (57).Petrusek R, Uhlenhopp E, Duteau N, and Hurley L (1982) Reaction of Anthramycin with DNA. J. Biol. Chem 257, 6207–6216. [PubMed] [Google Scholar]
- (58).Balbo S, Turesky RJ, and Villalta PW (2014) DNA Adductomics. Chem. Res. Toxicol 27, 356–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Hu Y, Phelan V, Ntai I, Farnet CM, Zazopoulos E, and Bachmann BO (2007) Benzodiazepine Biosynthesis in Streptomyces refuineus. Chem. Biol 14, 691–701. [DOI] [PubMed] [Google Scholar]
- (60).Kung Sutherland MS, Walter RB, Jeffrey SC, Burke PJ, Yu C, Kostner H, Stone I, Ryan MC, Sussman D, Lyon RP, Zeng W, Harrington KH, Klussman K, Westendorf L, Meyer D, Bernstein ID, Senter PD, Benjamin DR, Drachman JG, and McEarchern JA (2013) SGN-CD33A: a novel CD33-targeting antibody-drug conjugate using a pyrrolobenzodiazepine dimer is active in models of drug-resistant AML. Blood 122, 1455–1463. [DOI] [PubMed] [Google Scholar]
- (61).Hartley JA, Spanswick VJ, Brooks N, Clingen PH, McHugh PJ, Hochhauser D , Pedley RB, Kelland LR, Alley MC, Schultz R, Hollingshead MG, Schweikart KM, Tomaszewski JE, Sausville EA, Gregson SJ, Howard PW, and Thurston DE (2004) SJG-136 (NSC 694501), a Novel Rationally Designed DNA Minor Groove Interstrand Cross-Linking Agent with Potent and Broad Spectrum Antitumor Activity. Part 1: Cellular Pharmacology, In vitro and Initial In vivo Antitumor Activity. Cancer Research 64, 6693–6699. [DOI] [PubMed] [Google Scholar]
- (62).Morgensztern D, Besse B, Greillier L, Santana-Davila R, Ready N, Hann CL, Glisson BS, Farago AF, Dowlati A, Rudin CM, Le Moulec S, Lally S, Yalamanchili S, Wolf J, Govindan R, and Carbone DP (2019) Efficacy and Safety of Rovalpituzumab Tesirine in Third-Line and Beyond Patients with DLL3-Expressing, Relapsed/Refractory Small-Cell Lung Cancer: Results From the Phase II TRINITY Study. Clin. Cancer Res 25, 6958–6966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).von Tesmar A, Hoffmann M, Pippel J, Fayad AA, Dausend-Werner S, Bauer A, Blankenfeldt W, and Muller R (2017) Total Biosynthesis of the Pyrrolo[4,2]benzodiazepine Scaffold Tomaymycin on an In Vitro Reconstituted NRPS System. Cell Chem. Biol 24, 1216–1227.e8. [DOI] [PubMed] [Google Scholar]
- (64).Li W, Khullar A, Chou S, Sacramo A, and Gerratana B (2009) Biosynthesis of Sibiromycin, a Potent Antitumor Antibiotic. Appl. Environ. Microbiol 75, 2869–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Giessen TW, Kraas FI, and Marahiel MA (2011) A Four-Enzyme Pathway for 3,5-Dihydroxy-4-methylanthranilic Acid Formation and Incorporation into the Antitumor Antibiotic Sibiromycin. Biochemistry 50, 5680–5692. [DOI] [PubMed] [Google Scholar]
- (66).Sikora AL, Wilson DJ, Aldrich CC, and Blanchard JS (2010) Kinetic and Inhibition Studies of Dihydroxybenzoate-AMP Ligase from Escherichia coli. Biochemistry 49, 3648–3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Wolff H, and Bode HB (2018) The benzodiazepine-like natural product tilivalline is produced by the entomopathogenic bacterium Xenorhabdus eapokensis. PLOS ONE (van Berkel, W. J. H., Ed.) 13, e0194297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Wilson DJ, Shi C, Teitelbaum AM, Gulick AM, and Aldrich CC (2013) Characterization of AusA: A Dimodular Nonribosomal Peptide Synthetase Responsible for the Production of Aureusimine Pyrazinones. Biochemistry 52, 926–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Chhabra A, Haque AS, Pal RK, Goyal A, Rai R, Joshi S, Panjikar S, Pasha S, Sankaranarayanan R, and Gokhale RS (2012) Nonprocessive [2 + 2]e-off-loading reductase domains from mycobacterial nonribosomal peptide synthetases. Proc. Natl. Acad. Sci. U. S. A 109, 5681–5686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Couch R, O’Connor SE, Seidle H, Walsh CT, and Parry R (2004) Characterization of CmaA, an Adenylation-Thiolation Didomain Enzyme Involved in the Biosynthesis of Coronatine. J. Bacteriol 186, 35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Keating TA, Suo Z, Ehmann DE, and Walsh CT (2000) Selectivity of the Yersiniabactin Synthetase Adenylation Domain in the Two-Step Process of Amino Acid Activation and Transfer to a Holo-Carrier Protein Domain †. Biochemistry 39, 2297–2306. [DOI] [PubMed] [Google Scholar]
- (72).Guo C-J, Chang F-Y, Wyche TP, Backus KM, Acker TM, Funabashi M, Taketani M, Donia MS, Nayfach S, Pollard KS, Craik CS, Cravatt BF, Clardy J, Voigt CA, and Fischbach MA (2017) Discovery of Reactive Microbiota-Derived Metabolites that Inhibit Host Proteases. Cell 168, 517–526.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Secor PR, Jennings LK, James GA, Kirker KR, Pulcini, deLancey E, McInnerney K, Gerlach R, Livinghouse T, Hilmer JK, Bothner B, Fleckman P, Olerud JE, and Stewart PS (2012) Phevalin (aureusimine B)Production by Staphylococcus aureus Biofilm and Impacts on Human Keratinocyte Gene Expression. PLoS ONE (Sumby, P., Ed.) 7, e40973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Roberts AB, Gu X, Buffa JA, Hurd AG, Wang Z, Zhu W, Gupta N, Skye SM, Cody DB, Levison BS, Barrington WT, Russell MW, Reed JM, Duzan A, Lang JM, Fu X, Li L, Myers AJ, Rachakonda S, DiDonato JA, Brown JM, Gogonea V, Lusis AJ, Garcia-Garcia JC, and Hazen SL (2018) Development of a gut microbe-targeted nonlethal therapeutic to inhibit thrombosis potential. Nat. Med 24, 1407–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Pellock SJ, Creekmore BC, Walton WG, Mehta N, Biernat KA, Cesmat AP, Ariyarathna Y, Dunn ZD, Li B, Jin J, James LI, and Redinbo MR (2018) Gut Microbial β-Glucuronidase Inhibition via Catalytic Cycle Interception. ACS Cent. Sci 4, 868–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Rooks MG, Veiga P, Reeves AZ, Lavoie S, Yasuda K, Asano Y, Yoshihara K, Michaud M, Wardwell-Scott L, Gallini CA, Glickman JN, Sudo N, Huttenhower C , Lesser CF, and Garrett WS (2017) QseC inhibition as an antivirulence approach for colitis-associated bacteria. Proc. Natl. Acad. Sci 114, 142–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Ashley EA (2016) Towards precision medicine. Nat. Rev. Genet 17, 507–522. [DOI] [PubMed] [Google Scholar]
- (78).Brown ED, and Wright GD (2016) Antibacterial drug discovery in the resistance era. Nature 529, 336–343. [DOI] [PubMed] [Google Scholar]
- (79).Dykens JA, Sullivan SG, and Stern A (1987) Oxidative reactivity of the tryptophan metabolites 3-hydroxyanthranilate, cinnabarinate, quinolinate and picolinate. Biochem. Pharmacol 36, 21 1–217. [DOI] [PubMed] [Google Scholar]
- (80).Glaubiger D, Kohn KW, and Chamey E (1974) The Reaction of Anthramycin with DNA III. Properties of the Complex. Biochim. Biophys. Acta BBA - Protein Struct. Mol. Enzymol 361, 303–311. [DOI] [PubMed] [Google Scholar]
- (81).Kopka ML, Goodsell DS, Baikalov I, Grzeskowiak K, Cascio D, and Dickerson RE Crystal Structure of a Covalent DNA-Drug Adduct: Anthramycin Bound to C-C-A-A-C-G-T-T-G-G and a Molecular Explanation of Specificity. Biochemistry 33, 13593–13610. [DOI] [PubMed] [Google Scholar]
- (82).Puvvada MS, Hartley JA, Jenkins TC, and Thurston DE (1993) A quantitative assay to measure the relative DNA-binding affinity of pyrrolo[2,1-c][l,4]benzodiazepine (PBD) antitumour antibiotics based on the inhibition of restriction endonuclease BamhI. Nucleic Acids Res. 21, 3671–3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Gregson SJ, Howard PW, Hartley JA, Brooks NA, Adams LJ, Jenkins TC, Kelland LR, and Thurston DE (2001) Design, Synthesis, and Evaluation of a Novel Pyrrolobenzodiazepine DNA-Interactive Agent with Highly Efficient Cross-Linking Ability and Potent Cytotoxicity. J. Med. Chem 44, 737–748. [DOI] [PubMed] [Google Scholar]
- (84).Smellie M, Bose DS, Thompson AS, Jenkins TC, Hartley JA, and Thurston DE (2003) Sequence-Selective Recognition of Duplex DNA through Covalent Interstrand Cross-Linking: Kinetic and Molecular Modeling Studies with Pyrrolobenzodiazepine Dimers † Biochemistry 42, 8232–8239. [DOI] [PubMed] [Google Scholar]
- (85).Gregson SJ, Howard PW, Gullick DR, Hamaguchi A, Corcoran KE, Brooks NA, Hartley JA, Jenkins TC, Patel S, Guille MJ, and Thurston DE (2004) Linker Length Modulates DNA Cross-Linking Reactivity and Cytotoxic Potency of C8/C8’ Ether-Linked C2- exo -Unsaturated Pyrrolo[2,l- c ][l,4]benzodiazepine (PBD) Dimers. J. Med. Chem 47, 1161–1174. [DOI] [PubMed] [Google Scholar]
- (86).Wells G, Martin CRH, Howard PW, Sands ZA, Laughton CA, Tiberghien A, Woo CK, Masterson LA, Stephenson MJ, Hartley JA, Jenkins TC, Shnyder SD, Loadman PM, Waring MJ, and Thurston DE (2006) Design, Synthesis, and Biophysical and Biological Evaluation of a Series of Pyrrolobenzodiazepine–Poly( N - methylpyrrole) Conjugates. J. Med. Chem 49, 5442–5461. [DOI] [PubMed] [Google Scholar]
- (87).Rahman KM, James CH, Bui TTT, Drake AF, and Thurston DE (2011) Observation of a Single-Stranded DNA/Pyrrolobenzodiazepine Adduct. J. Am. Chem. Soc 133, 19376–19385. [DOI] [PubMed] [Google Scholar]
- (88).Ma Y, Khojasteh SC, Hop CECA, Erickson ΗK, Poison A, Pillow TH, Yu S-F, Wang H, Dragovich PS, and Zhang D (2016) Antibody Drug Conjugates Differentiate Uptake and DNA Alkylation of Pyrrolobenzodiazepines in Tumors from Organs of Xenograft Mice. Drug Metab. Dispos 44, 1958–1962. [DOI] [PubMed] [Google Scholar]
- (89).Quadri LEN, Weinreb PH, Lei M, Nakano MM, Zuber P, and Walsh CT (1998) Characterization of Sfp, a Bacillus subtilis Phosphopantetheinyl Transferase for Peptidyl Carrier Protein Domains in Peptide Synthetases †. Biochemistry 37, 1585–1595. [DOI] [PubMed] [Google Scholar]
- (90).Copeland RA (2013) Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists. John Wiley & Sons, Inc., Hoboken, NJ, USA. [PubMed] [Google Scholar]
- (91).Conti E (1997) Structural basis for the activation of phenylalanine in the non-ribosomal biosynthesis of gramicidin S. EMBO J. 16, 4174–4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Moriarty NW, Grosse-Kunstleve RW, and Adams PD (2009) Electronic ligand builder and optimization workbench (eLBOW): A tool for ligand coordinate and restraint generation. Acta Crystallogr. D Biol. Crystallogr 65, 1074–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Croll TI (2018) ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr. Sect. Struct. Biol 74, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Wang M, Yu N, Chen L, Villalta PW, Hochalter JB, and Hecht SS (2006) Identification of an Acetaldehyde Adduct in Human Liver DNA and Quantitation as N 2 - Ethyldeoxyguanosine. Chem. Res. Toxicol 19, 319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Wang M, Cheng G, Villalta PW, and Hecht SS (2007) Development of Liquid Chromatography Electrospray Ionization Tandem Mass Spectrometry Methods for Analysis of DNA Adducts of Formaldehyde and Their Application to Rats Treated with N - Nitrosodimethylamine or 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone. Chem. Res. Toxicol 20, 1141–1148. [DOI] [PubMed] [Google Scholar]
- (96).Castro-Pichel J, Garcia-Lopez MT, and De las Heras FG (1987) A facile synthesis of ascamycin and related analogues. Tetrahedron 43, 383–389. [Google Scholar]
- (97).Argiielles AJ, Sun S, Budaitis BG, and Nagorny P (2018) Design, Synthesis, and Application of Chiral C 2 -Symmetric Spiroketal-Containing Ligands in Transition-Metal Catalysis. Angew. Chem. Int. Ed 57, 5325–5329. [DOI] [PubMed] [Google Scholar]
- (98).Mori S, Aoyama T, and Shioiri T (1986) New methods and reagents in organic synthesis. 65. A stereoselective synthesis of tilivalline. Tetrahedron Lett. 27, 6111–6114. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.