

Abstract
Bone fracture is accompanied by trauma, mechanical stresses, and inflammation – conditions known to induce the mitochondrial permeability transition. This phenomenon occurs due to opening of the mitochondrial permeability transition pore (MPTP) promoted by cyclophilin D (CypD). MPTP opening leads to more inflammation, cell death and potentially to disruption of fracture repair. Here we performed a proof-of-concept study and tested a hypothesis that protecting mitochondria from MPTP opening via inhibition of CypD improves fracture repair. First, our in vitro experiments indicated pro-osteogenic and anti-inflammatory effects in osteoprogenitors upon CypD knock-out or pharmacological inhibition. Using a bone fracture model in mice, we observed that bone formation and biomechanical properties of repaired bones were significantly increased in CypD knock-out mice or wild type mice treated with a CypD inhibitor, NIM811, when compared to controls. These effects were evident in young male but not female mice, however in older (13 month-old) female mice bone formation was also increased during fracture repair. In contrast to global CypD knock-out, mesenchymal lineage-specific (Prx1-Cre driven) CypD deletion did not result in improved fracture repair. Our findings implicate MPTP in bone fracture and suggest systemic CypD inhibition as a modality to promote fracture repair.
Keywords: bone fracture, osteoprogenitors, cyclophilin D, mitochondria, permeability transition
Introduction
The incidence of bone fractures is increasing worldwide as a consequence of the growth of the aged population with osteoporosis and increased bone fragility (1). Currently, 5-10 % of fractures result in fracture non-unions, a condition for which few bone-anabolic treatment modalities have proven effective (2). Fracture healing is a multistage process involving cells of various lineages including immune cells, chondro- and osteo- progenitors, endothelial cells, etc. Among them, bone marrow stromal (a.k.a. mesenchymal stem) cells (BMSCs) and periosteal osteoprogenitors are essential to the fracture healing process as these cells can differentiate indirectly into bone via a cartilage intermediate or directly into bone; processes known as endochondral ossification and intramembranous ossification respectively (3). BMSCs are characterized by their tri-lineage (osteo-, adipo-, and chondro-genic) differentiation potential, surface markers (e.g. cd31-,cd45-, cd29+, cd105+), and adherence to plastic (4). BMSCs together with periosteal cells are needed for bone maintenance during normal remodeling and for repair in response to fracture (5). Differentiation of BMSCs into OBs is an energy demanding process for which mitochondria are the most suitable sources of energy due to their highly efficient ATP production. Mitochondria also serve as major biosynthetic and signaling hubs and can supply substrates for acetylation and methylation reactions required for both post-translational protein regulation and for epigenetic reactions (6).
To this effect we and others have shown that differentiation of osteoprogenitors into osteoblasts (OB) involves activation of mitochondrial oxidative phosphorylation (OxPhos) (7–9). It has also been well established that in various pathological settings including tissue trauma, mitochondrial dysfunction is a pronounced phenotype. Mitochondrial dysfunction results in impaired OB differentiation and maturation (10) often mediated through the sudden increase in permeability of the inner mitochondrial membrane to solutes of up to 1500 Da, i.e. mitochondrial permeability transition (MPT). The MPT phenomenon is a result of prolonged opening of the mitochondrial permeability transition pore (MPTP) (11) leading to a loss of the inner mitochondrial membrane integrity, mitochondrial swelling, and initiation of cell death. Such a prolonged opening can be induced by increases in mitochondrial ROS, calcium, misfolded proteins, and various other excessive stresses. Mitochondrial dysfunction is even more pronounced in aging. However, the relationship between mitochondrial metabolism and fracture healing has not been studied (12).
To date, a mitochondrial peptidyl-prolyl isomerase, cyclophilin D (CypD), encoded by the nuclear Ppif gene is the only genetically proven positive regulator of MPTP opening (13) and its genetic deletion desensitizes MPTP to opening, thereby maintaining mitochondrial membrane integrity and increasing the threshold of activation for the MPT phenomenon (14). The earliest known inhibitor of CypD and, thus of MPTP opening is the calcineurin binding immunosuppressor, cyclosporine A (CsA). (15). CypD genetic deletion or pharmacological inhibition are extensively used in studies of the heart and brain pathologies, where loss of CypD function has been shown to protect against injury, e.g. after ischemia-reperfusion. We first demonstrated that CypD knockout (KO) mice are protected against aging-related bone loss (16) however, the role of CypD and MPTP in bone fracture repair remains poorly defined (11, 13, 17–19). It is known that fracture is associated with inflammation and oxidative stress which can induce MPTP opening (20) however a functional inflammatory response is a critical step in fracture repair. Therefore non-immunosuppressive derivatives of CsA such as NIM811, Debio025, and JW47 may generate more specific insights into the pathophysiologic function of CypD in fracture (15,21,22).
In this work, we test the hypothesis that protecting mitochondria from MPT through inhibition of CypD will upregulate osteogenesis and promote fracture repair. We examine the effect of inhibition of MPT via CypD KO or treatment with MM811 in primary mouse BMSCs and using a mouse tibial bone fracture model. Overall, our findings described below suggest systemic CypD/MPTP inhibition as a new avenue for bone anabolic strategies in fracture repair.
Results
Genetic or pharmacological inhibition of CypD leads to increased resistance of mitochondria to MPTP opening
Bone marrow- and periosteum-derived osteoprogenitors are responsible for osteoblastogenesis and new bone formation during fracture repair. To our knowledge, the effect of MPTP inhibition on osteoprogenitors has not been studied. First, using the calcium retention capacity (CRC) assay, we confirmed that genetic deletion or pharmacological inhibition of CypD increases resistance of mitochondria in osteoprogenitors to MPTP opening. BMSCs were isolated from long bones of CypD KO and WT control littermate mice (Fig. 1A). To verify CypD deletion, we performed immunoblotting, which showed complete absence of CypD protein in CypD KO BMSCs (Supplementary Fig. S1A). Of note, BMSC numbers were similar in CypD KO and WT bone marrow as measured with immunolabeling and flow cytometry and CFU-F assay (Supplementary Fig. S1 B & C). These cells were then permeabilized to expose mitochondria and subjected to CRC assay. Figure 1B shows typical calcium retention/release traces from these cells. Our data show that when compared to WT controls, CypD KO BMSCs show higher mitochondrial CRC, indicative of increased resistance to MPTP opening. Mitochondria in CypD KO BMSCs retain higher amounts of calcium (7 vs 3 nmoles Ca2+/106 cells) before undergoing permeability transition (Fig. 1B & C). To confirm that this higher CRC was not due to higher number of mitochondria in CypD KO cells, we measured mitochondrial mass in WT and CypD KO BMSCs with nonyl acridine orange (NAO) and flow cytometry and found no significant difference (Supplementary Fig. S1D). We also observed no difference in mtDNA content between WT and CypD KO cells measured with qPCR (Supplementary Fig. S1E). Pharmacologic inhibition of CypD using NIM811 was performed in cells isolated from C57BL/6J mice and subjected to CRC assay in the presence or absence of NIM811. Figure 1D shows that NIM811 significantly increased mitochondrial CRC in BMSCs to the same extent as genetic ablation of CypD. NIM811 treatment is known to inhibit MPTP-gating activity of CypD without significant effect on its expression which was verified with real-time RT-PCR (Supplementary Fig. S1F). These experiments show that targeting CypD is effective in inhibiting MPTP in BMSCs without affecting BMSC numbers or mitochondrial content.
Figure 1. Genetic or pharmacological inhibition of CypD inhibits MPTP opening in BMSCs.
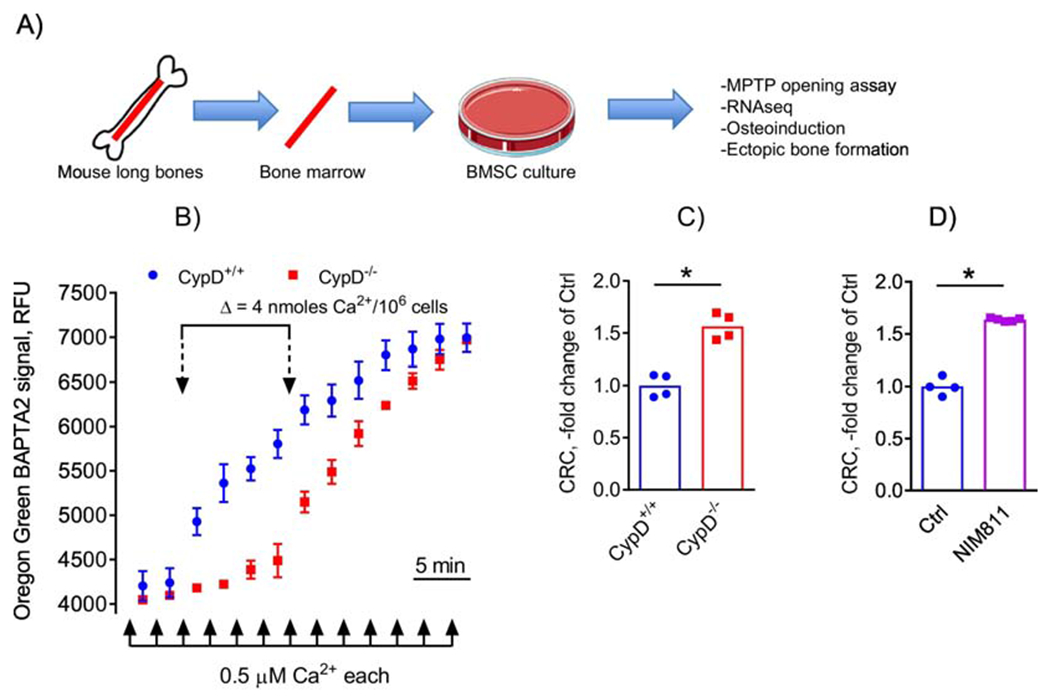
A) BMSCs were isolated from mouse long bones, cultured and subjected to various assays; B) BMSCs from CypD+/+ and CypD−/− littermates were subjected to calcium retention capacity (CRC) assay which measures sensitivity to MPTP opening after excessive calcium uptake by mitochondria. CypD−/− BMSC mitochondria accumulate more calcium before opening the pore and, thus show higher CRC; C) CRC was calculated and plotted as a fold change of control CypD+/+. CypD−/− BMSCs show significantly higher CRC and, thus lower MPTP opening when compared to CypD+/+ controls; D) C57BL/6J BMSCs treated with the CypD inhibitor, NIM811, show significantly higher CRC and, thus lower MPTP opening when compared to vehicle-treated controls. Data in (B) are Means ± SD (n = 4). Plots in (C) and (D) show actual data points and calculated means. *, p < 0.05, determined by unpaired t-test. See also Supplementary Figure S1.
Genetic deletion of CypD imposes distinct pro-osteogenic and anti-inflammatory molecular signatures in BMSCs
As CypD KO BMSCs have never been characterized before, we performed RNAseq transcriptome analysis in CypD KO and WT control BMSCs. RNAseq data bulk analysis and hierarchical clustering indicated very distinct molecular signatures in CypD KO cells and WT controls (Fig. 2A). Further examination of these signatures with Reactome pathway analysis revealed significant upregulation of genes involved in collagen formation, organization and maturation (Fig. 2B) that are crucial for bone formation. Consistent with Reactome, KEGG analysis showed significant upregulation of pathways relevant to extracellular matrix and downregulation of pathways involved in bone catabolism, such as TNF signaling and rheumatoid arthritis (Fig. 2C). Looking at individual genes involved in bone catabolism, we detected a general trend towards downregulation with expression of catabolic genes, such as MMP’s, significantly decreased in CypD KO cells (Fig. 2D). In concert with these findings, Ingenuity Pathway Analysis (IPA) showed significant downregulation of various pro-inflammatory factors, such as NF-κB and TLR4 (Supplementary Figure S2A), and of TNF Receptor and IL2 pathways (Supplementary Figure S2B & C). Taken together these findings indicate very distinct pro-osteogenic and anti-inflammatory gene signatures in CypD KO BMSCs when compared to WT controls.
Figure 2. CypD KO BMSCs show a distinct pro-osteogenic molecular signature.
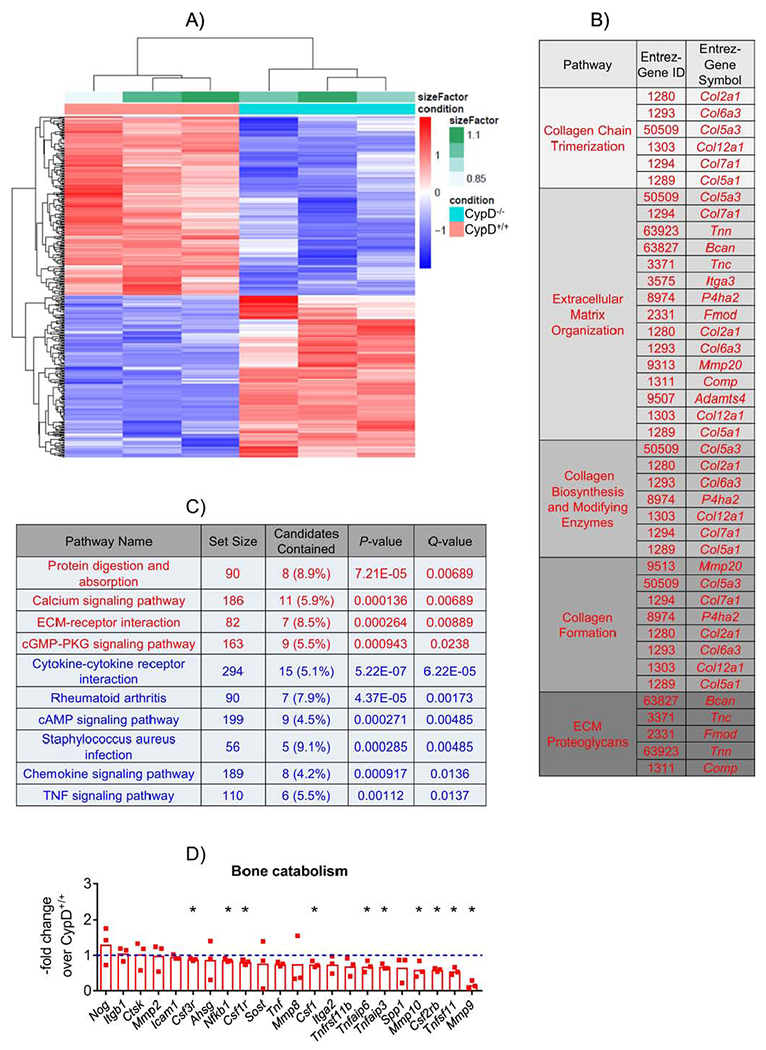
BMSCs from CypD−/− and control CypD+/+ littermates (n=3) were cultured in growth media and analyzed by RNAseq. A) CypD−/− BMSCs show distinct opposite trends in upregulated and downregulated genes compared to control BMSCs in the heatmap; B) Reactome pathway analysis shows upregulation of genes related to bone formation in CypD−/− vs CypD+/+ BMSCs; C) KEGG pathway analysis of upregulated (red) and downregulated (blue) genes in CypD−/− vs CypD+/+ BMSCs; D) Genes related to bone catabolism are mostly downregulated in CypD−/− vs CypD+/+ BMSCs. Plot shows actual data points and calculated means. *, p < 0.05 determined by unpaired t-test. See also Supplementary Figure S2.
Genetic deletion or pharmacological inhibition of CypD enhances osteogenic function of BMSCs
We next investigated whether the pro-osteogenic transcriptomic signature of CypD KO BMSCs translates into a pro-osteogenic effect in vitro. We performed osteoinduction in CypD KO and WT BMSC cultures and observed a more pronounced increase in osteogenic phenotypic markers, alkaline phosphatase (ALP, Fig. 3A) detected via specific staining and mineralization measured with alizarin red staining (Fig. 3B) in CypD KO BMSCs when compared to WT controls. We also detected significantly higher expression of an osteogenic marker gene, osteocalcin (Bglap), in CypD KO BMSCs when compared to the WT cells as measured with real-time RT-PCR at both Day 0 and Day 14 of osteoinduction (Fig. 3C). We also tested whether pharmacological inhibition of CypD with NIM811 would exert similar pro-osteogenic effects in BMSCs. Figure 3D shows that osteoinduction of BMSCs in the presence of NIM811 for 14 days increased osteogenic markers Alpl and Bglap when compared to vehicle-treated controls. In order to demonstrate the increased osteogenic potential of CypD KO BMSC in vivo, we performed BMP2-mediated ectopic bone (ossicle) formation assay in which grafted BMSCs create bone and support both development of vascularized bone marrow of recipient origin (23). At 6 weeks following implantation of both WT (left flank) and CypD KO BMSCs (right flank), we were able to confirm ossicle formation in grafts from both genotypes (Fig. 3E). CypD KO BMSCs grafts led to significantly larger ossicles as assessed with histomorphometry (Fig. 3F). Importantly, both CypD KO and WT BMSCs grafts led to formation of ectopic bone, marrow,and vascularized stroma (Fig. 3G).Altogether, these data confirm that genetic or pharmacological inhibition of CypD has a pronounced pro-osteogenic effect in osteoprogenitors.
Figure 3. CypD KO BMSCs show increased osteogenic potential.
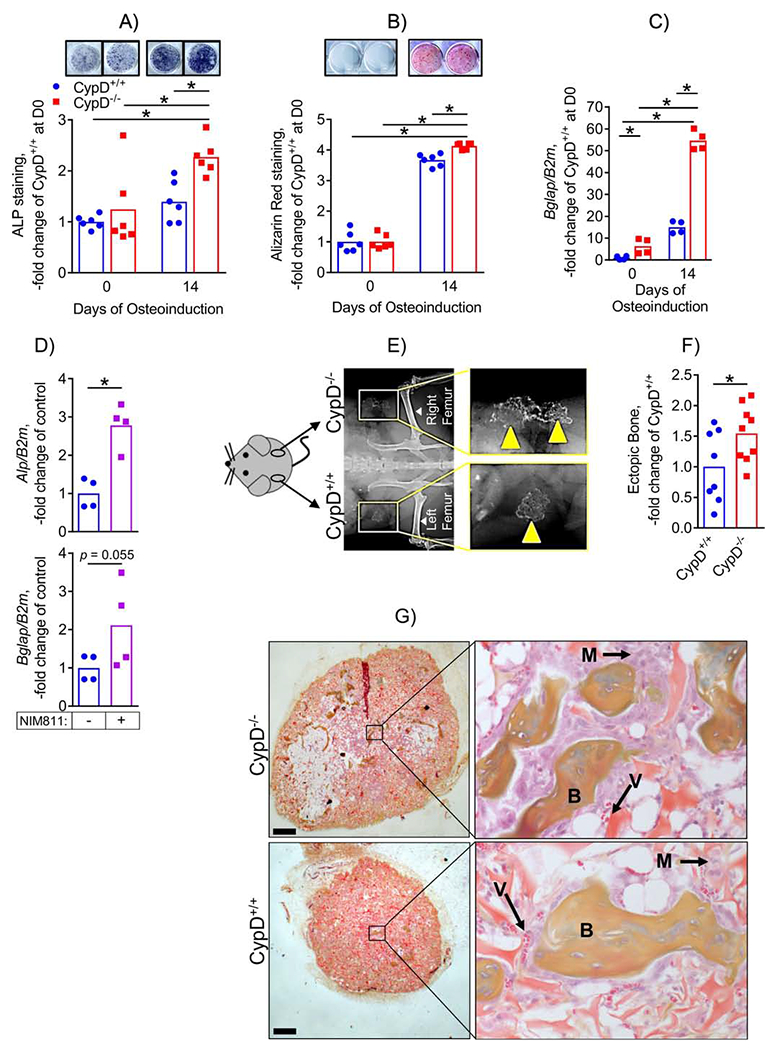
BMSCs from CypD−/− and control CypD+/+ littermates were osteoinduced for 0 or 14 days and analyzed by staining for alkaline phosphatase (ALP, A) and for mineralization with alizarin red (ARed, B) and by real-time RT-PCR (C). CypD−/− BMSCs show more intense ALP and ARed staining (A & B, respectively) and higher expression of osteogenic gene marker, Bglap (C). D) BMSCs isolated from C57BL/6J mice were osteoinduced for 14 days in the presence of NIM811 at 0.5 mM or vehicle control; and osteogenic marker gene expression was measured with real-time RT-PCR. NIM811-treated cells had significantly higher expression of Alp. BMSCs were also implanted into backs of immunocompromised nude mice for an ectopic bone formation assay (E – G). Ossicle formation was analyzed after 6 weeks; E) X-ray showing ectopic bone formation; F) Ectopic bone was measured with histomorphometry. CypD−/− BMSCs show a significant increase in ectopic bone formation; G) Histology sections of ectopic bones with boxed areas magnified showing bone (B), marrow (M), and blood vessels (V). Data are presented as fold changes over undifferentiated control CypD+/+ BMSCs. Plots show actual data points and calculated means. *, p < 0.05, determined by ANOVA followed by post-hoc analysis (A μ C) or unpaired t-test (D, F).
Genetic deletion of CypD leads to more efficient bone fracture repair
Given our observation of a strong pro-osteogenic effect upon inhibition of CypD both in vitro and in vivo, we hypothesized that genetic or pharmacological inhibition of CypD would stimulate bone formation during fracture healing in mice. To test this, CypD KO and WT control littermates were subjected to unilateral tibial fractures; and fractured and contralateral unfractured bones were collected at various time-points to access fracture healing as outlined in the diagram in Figure 4A. Representative X-ray images of fractured bones at post-fracture day (PFD) 0 and 35 shown in Figure 4B demonstrate both genotypes were capable of radiographically complete healing in our time frame (stabilized tibial fracture healing in mice takes on average 35 days (3) after which the fracture is considered biomechanically healed). We next assessed the quality of repaired bone via the biomechanical torsion test of both fractured and unfractured bones at PFD 35 in both male and female littermates. CypD KO mice showed significantly increased torsional biomechanical properties (torsional rigidity and maximum torque) of repaired tibiae when compared to WT controls (Fig. 4C, top panels) while torsional biomechanical properties in unfractured CypD KO and WT bones showed no significant differences (Supplementary Fig. S3A&B). This is in agreement with our previous observations showing that young CypD KO and WT mice have a similar skeletal phenotype at baseline (4) and that protective effects of CypD KO ensue mostly under a pathological stress, e.g. aging or trauma. To our knowledge, this is the first report of a role of CypD/MPTP in fracture repair. Interestingly this difference was only observed in males, with female fractures showing no change in biomechanical properties (Fig. 4C, bottom panels). We hypothesized that this is due to the well-known inhibitory effect of estrogen on MPTP opening (24,25) which would mask any additional effect of CypD KO. To test this, we studied the effect of estradiol (E2) on MPTP opening in BMSCs using CRC assay. E2 significantly increased CRC and, thus inhibited MPTP opening to the same extent as NIM811 or CypD KO while estrogen depletion from the media significantly reduced CRC (Supplementary Fig. S4A). These results indicate that estrogen by itself is protective against MPTP opening, which can explain why we do not see an improved fracture repair phenotype in female CypD KO mice. As aging may unmask the effect of CypD inhibition according to our previous study, we obtained 13 mo old female mice, performed fractures and treated mice with either NIM811 or vehicle (Supplementary Figure S4B). Even though 13 month-old mice are not considered aged, they show a pronounced bone loss and delayed fracture repair (16, 26). While further studies of this cohort are needed, our initial data on serum bone formation marker, P1NP, indicate a significant increase in NIM811-treated group at PFD35 (Supplementary Figure S4C). To our knowledge, this is the first report of a role of CypD/MPTP in fracture repair. Given our observation of sexual dimorphism of the effect, our further efforts included only male mice in order to better characterize the role of CypD/MPTP in fracture repair.
Figure 4. Genetic deletion or pharmacological inhibition of CypD results in stronger repaired bone in male mice at post fracture day 35.
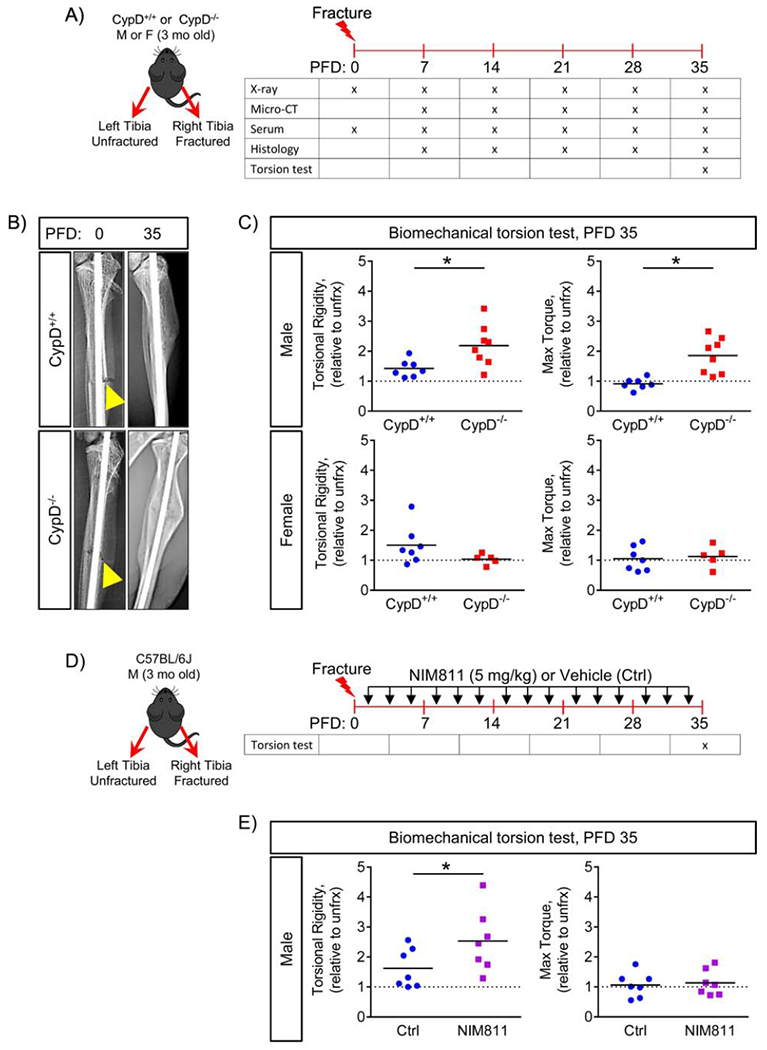
A) Tibial fractures were performed on 3-month-old male and female mice; and fractured and contralateral unfractured bones were collected for various analyses; B) Representative X-rays at post fracture day (PFD) 0 and 35. Arrowheads indicate the fracture site; C) Biomechanical properties of repaired bone were measured at PFD 35 by torsion test and reported as torsional rigidity indicating bone toughness and maximum torque indicating bone strength. Data are expressed as relative to the contralateral unfractured limb to account for differences in bone phenotype between animals. CypD−/− male mice show increased torsional properties of repaired bone; whereas, CypD−/− female mice show no difference when compared to control CypD+/+ littermates; D) Tibial fracture was induced similarly to the CypD KO mice and underwent biomechanical torsion test at PFD 35. NIM811 at 5 mg/kg or vehicle was injected intraperitoneally 3 times per week throughout the healing process; E) NIM811-treated mice show an increase in torsional rigidity and no difference in maximum torque when compared to vehicle-treated controls. Plots show actual data points and calculated means. *, p < 0.05 vs CypD+/+ controls as determined by an unpaired t-test. See also Supplementary Figure S3 & S4.
Pharmacologic inhibition of CypD leads to more efficient bone fracture repair in mice
A more clinically relevant approach for CypD inhibition is using a pharmacological inhibitor. We, therefore used NIM811 and observed that mice treated with NIM811 three times per week throughout the duration of fracture repair (Fig. 4D) showed an increase in torsional rigidity when compared to vehicle-treated mice (Fig. 4E, left panel), but no difference in maximum torque (Fig. 4E, right panel). Again, the unfractured tibia showed no baseline difference in biomechanical parameters (Supplementary Fig. S4D). This indicates that a clinically relevant pharmacological inhibition of CypD improves bone fracture repair. Further dose and regimen optimization as well as developing bone-targeted variants may enhance the inhibitor efficacy.
CypD KO mice show increased mineralization and collagen matrix maturation in fracture calluses
Since the torsional strength of the repaired bone is dependent on the mineralization of the callus bridging to reestablish continuity of the cortical ends of the fractured bone, we performed transverse section-by-section analysis of μCT scans and heat-mapping of BMD in sections flanking the bridge. This enabled quantification of mineralization at the bony callus bridge (Fig. 5A). As evident from this analysis, mineralization of the bony bridge was significantly higher in CypD KO vs WT control mice (Fig. 5A, right panel). The strength and rigidity of the repaired bone is also influenced by the organic component of bone matrix, e.g. collagen I. We therefore utilized an optical method based on measuring the ratio of forward-to-backward (F/B) scattering of second harmonic generation (SHG) signal from collagen (27). Changes in F/B ratio reflect differences in the diameter of the fibrils bundled into collagen fibers, as well as their spacing within the fiber, and their packing disorder (28). More mature collagen fibers have increased forward propagating SHG signal and ultimately have a higher F/B ratio compared to less mature collagen (29). F/B ratio analyses of H&E-stained sections of the calluses at PFD 35 showed higher collagen F/B ratios in repaired bone of CypD KO mice when compared to WT controls (Fig. 5B). Thus, CypD KO mice have increased bone maturation at the fracture site.
Figure 5. Genetic deletion of CypD results in better repaired bone quality at post fracture day 35.
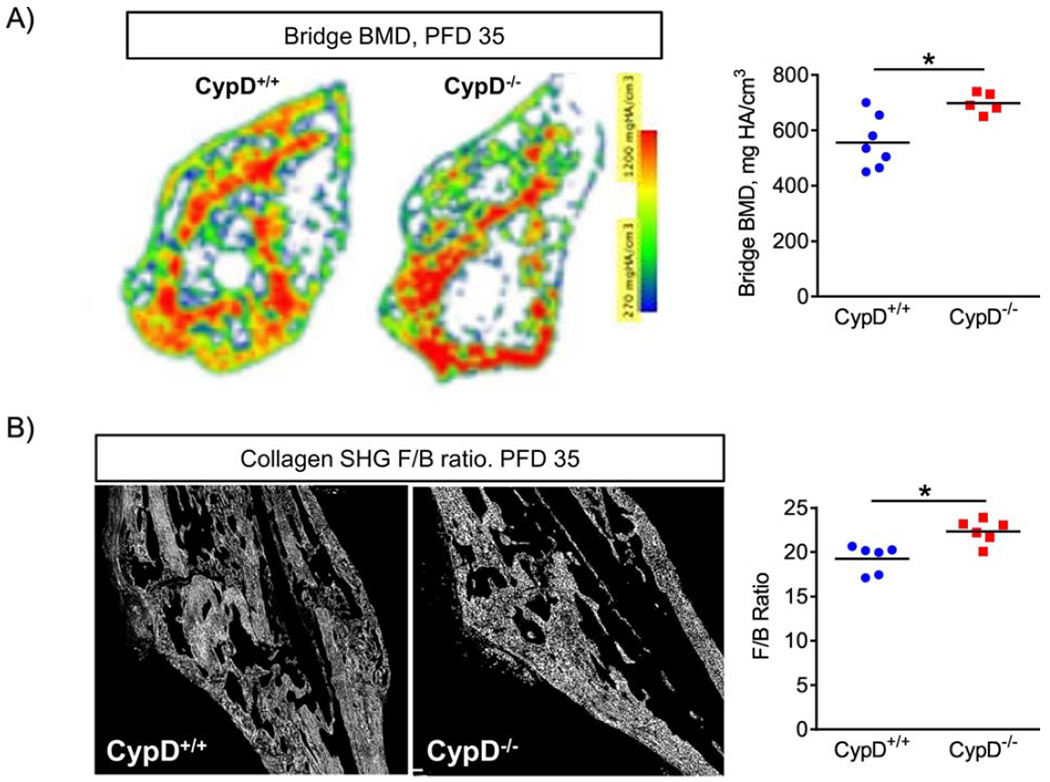
A) Representative heat map of BMD of the bridge region in a transverse section. At PFD 35, CypD−/− mice show an increase in BMD at the bridge; B) Representative second harmonic generation (SHG) images of collagen in sagittal sections through the callus where forward to backward (F/B) scattering was measured using multiphoton microscopy at PFD 35 as described in Methods. CypD−/− mice have higher F/B ratios indicating better aligned and more mature collagen fibers in the callus. Plots show actual data points and calculated means. *, p < 0.05 vs CypD+/+ controls as determined by an unpaired t-test. See also Supplementary Figure S5.
In addition, we performed whole callus μCT analysis at various time-points, which demonstrated with few exceptions (e.g. higher callus BMD in CypD KO vs WT mice at PFD 28) no significant differences between the CypD KO mice and WT controls in the bony callus volume fraction (BV/TV), cortical thickness, and the whole callus BMD (Supplementary Fig. S5). These data indicate that there was increased formation and higher mineralization of the bony bridge along with presence of more mature unidirectional collagen fibers in fracture calluses of CypD KO mice, which together most likely accounted for improved biomechanical properties of the repaired bone.
Genetic deletion of CypD results in accelerated bone formation during fracture healing
As our data suggest that CypD KO improves maturation and quality of the newly formed bone, we asked whether this was due to increased bone formation during fracture healing. Histologic analyses and automated histomorphometry of ABH/OG stained sections of the calluses of CypD KO mice at PFD 7 and 14 detected less cartilage and more bone, suggesting a truncated cartilaginous callus phase and accelerated bone formation (Fig. 6). Immunostaining for osteoblastic marker, osteocalcin, in CypD KO mice revealed an increase in osteocalcin within the callus at PFD 7 and again at PFD 28 when compared to WT controls (Fig. 7A), supporting the notion of accelerated bone formation in CypD KO fracture calluses. To further confirm the effect on bone formation, we measured serum P1NP, a procollagen peptide synthesized by OBs during bone formation process (30). When compared to WT control littermates, CypD KO mice show an increase in serum P1NP levels at PFD 7 which did not reach statistical significance (p = 0.2873) and statistically significant increases at PFD 28 and 35 (Fig. 7B). Together, the increased osteogenic markers at PFD 7 likely indicate accelerated new bone formation while at PFD 28 and 35 accelerated bony callus remodeling during fracture repair in CypD KO mice.
Figure 6. Accelerated callus ossification in CypD knock-out mice.
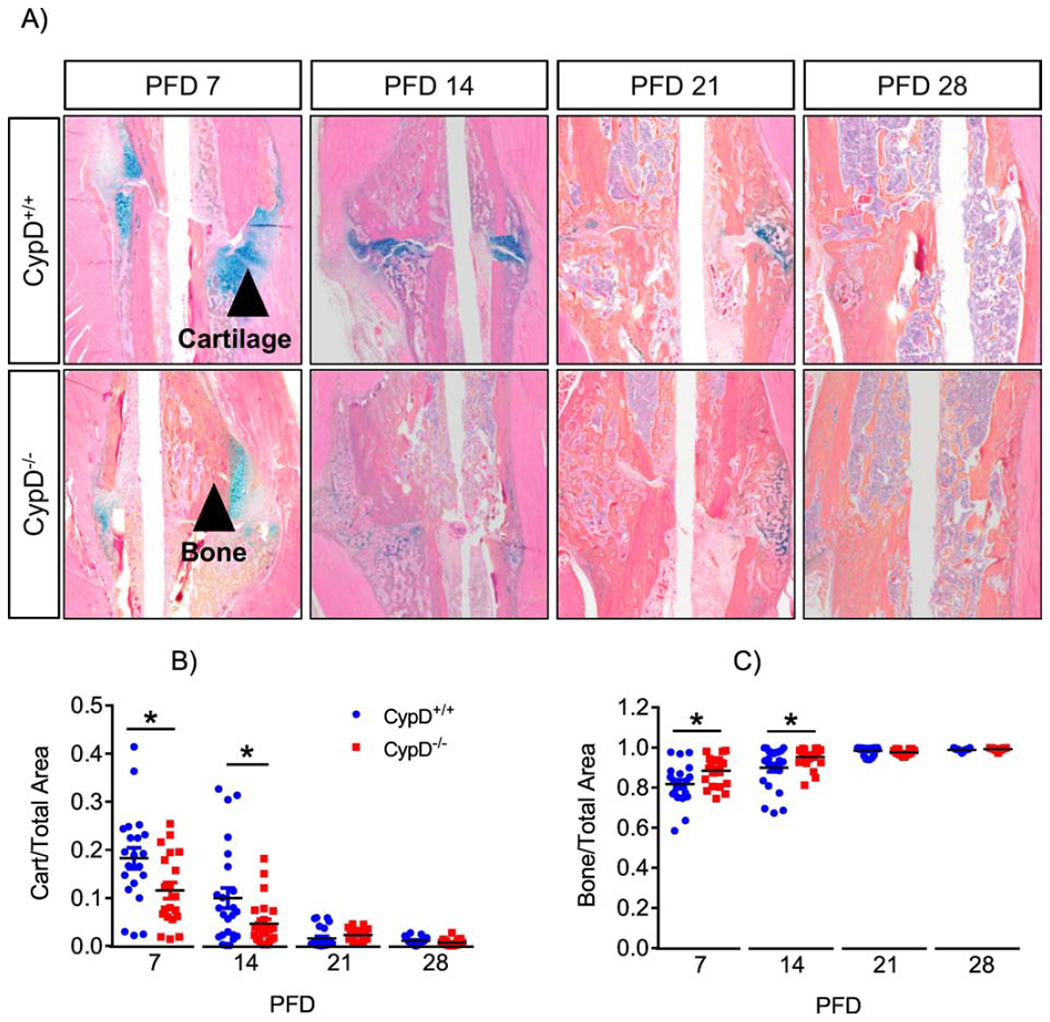
Histological staining using ABH/OG stain followed by histomorphometry of fractured bones was performed at various time points (A). CypD−/− mice show accelerated callus ossification and a truncated cartilaginous phase at PFD 7 and 14 (B & C). Plots show actual data points and calculated means. *, p < 0.05, determined by unpaired t-test of each time point, t-test was used because samples at each time point were collected and analyzed independently of other time points. See also supplementary Figure S6.
Figure 7. Genetic deletion of CypD results in increased bone formation during fracture healing.
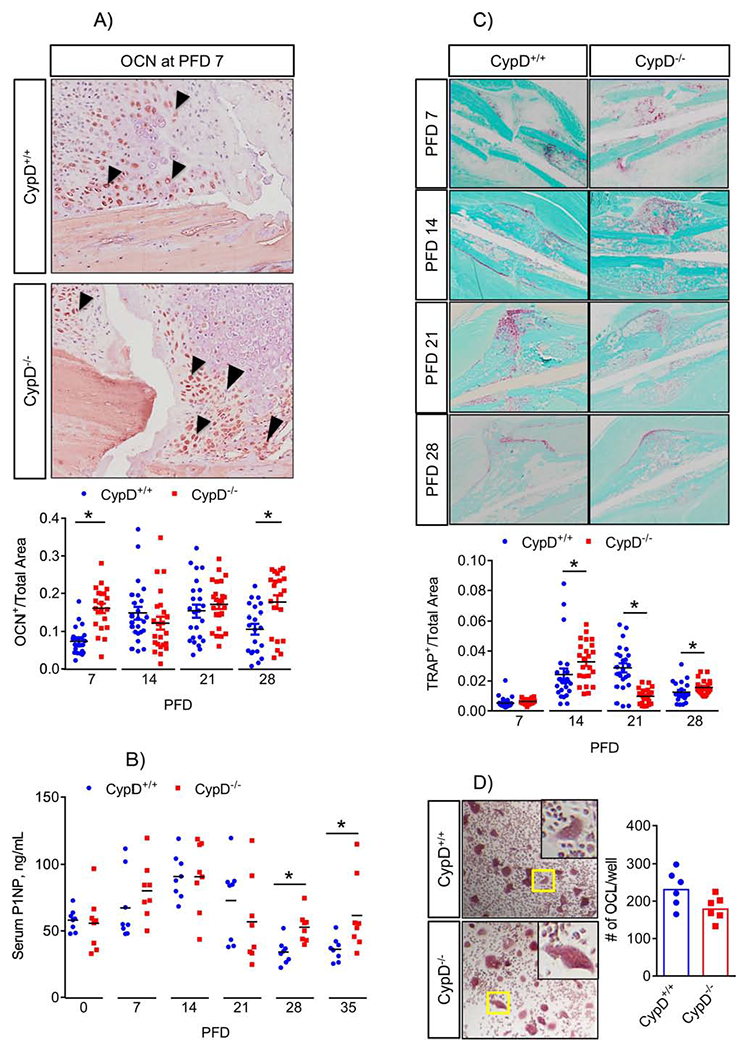
A) Sections of calluses were probed with antibodies against osteocalcin (OCN) at various time-points. CypD−/− mice show increased OCN immunostaining at PFD 7 and 28; B) Serum was collected at various time-points and analyzed for P1NP. CypD−/− mice show increased serum P1NP at PFD 28 and 35 when compared to CypD+/+ control littermates, indicating an increase in bone formation: C) Tissue sections were stained with TRAP-specific stain to detect osteoclast activity at various time-points; D) Osteoclast (OCL) precursors were isolated from bone marrow and OCL differentiation assay was performed as described in Methods. Number of OCL per well was not significantly different between CypD−/− and CypD+/+ control littermates. Boxed regions in (D) are magnified in the inserts to show details of multinucleated TRAP+ cells. Plots show actual data points and calculated means. *, p < 0.05, determined by unpaired t-test of each time point. In (A – C), t-test was used because samples at each time point were collected and analyzed independently of other time points. See also Supplementary Figures S6 & S7.
As this fracture model is known to heal via endochondral ossification, which involves chondrocyte hypertrophy and replacement by and/or transdifferentiation into osteoblasts, we checked whether chondrocyte hypertrophy was affected. Our in vivo data show that the number of hypertrophic chondrocytes in fracture calluses of CypD KO mice was not significantly different from WT controls (Supplementary Fig. S6A). In addition, we compared the rate of chondrocyte maturation/hypertrophy in CypD KO and WT cells in vitro and again found no difference (Supplementary Fig. S6B). We, therefore, conclude that the observed accelerated fracture callus ossification is unlikely caused by changes in chondrocytes.
Next, we assessed fracture callus remodeling and osteoclast activity. Osteoclast-specific TRAP staining revealed mixed results as CypD KO mice remodeled callus more actively than control littermates at PFD 14 and 28 and less actively at PFD 21 (Fig. 7C). To find out whether CypD KO had a direct effect on osteoclasts (OCL), we performed OCL differentiation assay, which showed that OCL differentiation was not significantly different between CypD KO and control (Fig. 7D). OCLs are regulated by BMSC- and OB-derived RANKL and Opg and, in particular, by RANKL/Opg ratio. We, therefore examined expression of their corresponding genes, Tnfsf11 and Tnfrsf11b using our RNAseq data. Our data showed that although the expression of both genes was downregulated in CypD KO BMSCs when compared to controls, their expression ratio was unchanged (Supplementary Fig. S6C). These data indicate that CypD KO effect on bone resorption is not as strong as its effect on bone formation.
As fracture callus ossification may be influenced by changes in callus vascularization or recruitment of immune cells, we counted the number of blood vessels and macrophages in calluses at PFD 7 and 14 using histomorphometry and immune cell composition using flow cytometry. There were no significant differences in the number of vessels in fracture calluses, immune cell composition in the bone marrow or number of macrophages in fracture calluses between CypD KO and WT control littermates (Supplementary Fig. S7A–C). In addition, we assessed cytokine levels in the serum of the WT and CypD KO mice using a cytokine array at PFD 0 and 7 and did not detect any differences (Supplementary Fig. S7D). Neutrophil infiltration within the fracture calluses was also not different between CypD KO and WT mice at PFD7 (Supplementary Fig. S7E). Together, these histological and serological analyses indicate that CypD KO mice have an accelerated callus ossification with truncated cartilaginous phase during fracture healing and these effects are not accompanied by changes in callus vascularization or immune cells.
Specific deletion of CypD in mesenchymal lineage is not sufficient to improve bone fracture repair
Since we observed cell-autonomous pro-osteogenic effect of CypD KO in BMSCs, we asked whether BMSC- and BMSC progeny-specific CypD deletion would recapitulate the stimulatory effect of global CypD KO on fracture repair. We used a mesenchymal lineage-specific, Prx1-Cre-driven deletion of CypD in CypDf/f mice (Supplementary Fig. S8A). First, we confirmed Prx1-Cre specificity by crossing the Prx1-Cre mice to Ai9 (tdTomato) reporter mice and analyzing Ai9 signal in both unfractured and fractured tibiae tissue sections and observed the presence of Tomato signal in chondrocytes (white arrowheads), BMSCs (green arrowheads), OBs (yellow arrowheads) and osteocytes (grey arrowheads) of Cre+ samples. (Supplementary Figure S8B). Importantly no signal was detected in Cre− samples. These data indicate that Prx1-Cre is specific to mesenchymal lineage. Next, we confirmed CypD knock-down in Prx1-Cre; CypDf/f but not in Cre- control mice (Supplementary Fig. S8C). Then, we performed tibial fractures in Prx1-Cre; CypDf/f and control Cre- CypDf/f male littermates. We found that Prx1-Cre-driven deletion of CypD resulted in no difference in maximum torque and torsional rigidity at PFD 35 in fractured tibiae (Supplementary Fig. S8D). These data indicate that mesenchymal lineage specific CypD deletion is not sufficient to improve fracture healing and provides support for using the global CypD KO and systemic pharmacological inhibitors.
In summary, we showed that genetic and pharmacological inhibition of MPTP opening is beneficial for fracture repair in mice and that this effect is not restricted to mesenchymal lineage but is systemic.
Discussion
Cyclophilin D is an exclusively mitochondrially localized prolyl-peptidyl cis-trans isomerase. While the exact role for the enzymatic activity for CypD in mitochondria is not yet well understood, its position as a key regulator of the mitochondrial permeability transition pore has been repeatedly demonstrated. Both pharmacological (i.e. Cyclosporin A, NIM811, Debio025 and JW47) and genetic (CypD KO) inhibition of CypD result in mitochondria with decreased sensitivity to calcium- and reactive oxygen species-induced permeability transition. The impact of CypD KO on cellular processes is widespread including cardiac development (31), ischemia reperfusion injury (27), and now bone formation. Manipulation of CypD as a regulator of the MPTP, and thus mitochondrial integrity and bioenergetics is a useful mechanism for investigating cellular reliance on mitochondria-derived ATP and metabolites.
While the molecular identity of the MPTP remains under debate, its physiologic and pathologic roles have been intensively studied (32–34). The MPTP is characterized as a mega-conductance channel; however in patch clamp experiments it has been observed to have sub-conductances as low as 30 pS, and reaching up to 1.3 nS (35,36). These sub-conductance states of the MPTP are hypothesized to have a non-pathologic or less pathologic or even physiologic role in the mitochondrial membrane. Sub-conductance or brief “flickering” activity of the pore has been reported in striated muscle (37) and neuronal synapses (38) and has a potential role in mitochondrial calcium homeostasis (39) and modulating mitochondrial membrane potential (40) and, thus OxPhos function.
The role of mitochondrial dysfunction in bone pathologies has not been studied extensively. In our previous report, we uncovered evidence for the MPTP-mediated mitochondrial dysfunction in aging mouse bone as a cause of aging-associated bone loss (16). Building on our previous work and existing knowledge of MPTP involvement in the response to trauma and inflammation, we investigated the role of MPTP in osteogenic function in BMSCs and in bone fracture healing. Healing of bone fracture progresses in distinct phases: an inflammation phase that occurs acutely post fracture, soft cartilaginous callus phase, mineralization phase, and a remodeling phase. As acute inflammation involves processes linked to mitochondrially generated reactive oxygen species, and tissue repair is an energy intensive process, we hypothesized that the CypD/MPTP inhibition model would show improved fracture healing.
We first investigated whether MPTP loss-of-function via CypD genetic deletion or pharmacological inhibition exerts any cell-autonomous effect on BMSCs. RNAseq analysis revealed distinct pro-osteogenic and anti-inflammatory molecular profiles in CypD KO BMSCs when compared to WT controls. Osteoinduction studies in vitro as well as ectopic bone formation assay in vivo (23) showed an increase in osteogenic function in BMSC cultures along with greater ectopic bone formation in mice. NIM811-mediated inhibition of CypD/MPTP also produced pro-osteogenic effect in BMSCs. In vivo we observed that CypD KO mice had a significant increase in biomechanical properties of repaired bone. The increased torsional strength and rigidity of the repaired bone was associated with increased mineralization of the bony bridge, and presence of more mature collagen, all indicative of higher OB bone forming activity during fracture healing in CypD KO mice. Histological, immunohistochemical, and serological analyses all indicated that male CypD KO mice show an accelerated bone fracture callus ossification. An important question is why CypD KO did not show improved biomechanical properties of repaired bone in young female mice. One possible explanation, partially confirmed by in vitro data shown here, may come from the known fact that estrogen is a strong inhibitor of MPTP opening; although the mechanism of this protective effect is not yet well understood (24,25). Therefore, because of presence of high estrogen, female mice may be well protected from MPTP opening even under the conditions of trauma such as fracture. Sexual dimorphism of MPTP activity has been reported before in various pathologies (41). These results suggest avenues for future research, such as testing the effect of CypD KO in aged female mice. In this regard, our data showing higher P1NP levels in 13 month-old female mice undergoing fracture and treated with NIM811 suggest that the effect of CypD inhibition will be unmasked in aged female mice. We also used a pharmacological inhibitor of CypD, NIM811, and observed a positive effect on torsional rigidity of the repaired bone. These data suggest that further optimization of the inhibitor dose and regimen as well as developing bone-targeted variants of the inhibitor has a strong potential to enhance the inhibitor efficacy for fracture repair.
Despite the fact that the fracture model used here heals very efficiently in mice and it is, therefore, hard to detect positive effects of treatments, we were able to show significant improvement in our CypD KO and NIM811-treatment models. Thus, a challenged fracture model, e.g. in aged mice, can potentially better define the role of CypD and MPTP in bone healing. The global CypD KO animal model used in our study more closely mimics the expected action of pharmacologic CypD inhibitors lacking cell specificity and provides compelling evidence that CypD is an important therapeutic target for improving outcomes in fracture repair. Another important notion is that we did not detect any significant effects of CypD KO on immune cells or inflammatory cytokines, likely indicating that MPTP opening does not contribute to inflammation in the fracture model used here. This may be because MPTP opens here as a less pathologic, sub-conductance channel described above. Aging is known to induce highly disruptive MPTP opening. This suggests a possibility of a different effect of MPTP inhibition in an aged mouse fracture model, justifying further studies of the effect of aging.
To see if MPTP loss-of-function specifically in osteogenic lineage is sufficient to promote bone fracture repair, we used mesenchymal lineage-specific CypD deletion in Prx1-Cre;CypDf/f mice. Our data show a lack of effect in these mice. This observation is not entirely surprising given the involvement of various other cell types in fracture repair, such as immune cell, endothelial progenitors, etc. However, in our global knockout model we did not detect any changes in chondrocyte hypertrophy, immune cells, or vascularization during fracture repair. The cellular milieu in a fracture callus is very dynamic and all components are involved in cross-talk through various signaling pathways. Although we do not detect changes in the above mentioned cell types, global CypD KO could be involved in the dampening or activation of various signaling pathways. This could affect the cross-talk between OBs and other involved cell types, which would not be shown in quantitative studies. The most pronounced effect was seen in osteogenic lineage, i.e. increased callus ossification, collagen I maturation, as well as osteogenic markers: P1NP and osteocalcin, all indicative of increased OB function. OBs play a key role in hard callus formation in fracture healing, resulting in recruitment of OCLs to initiate remodeling and ultimately healing. This is not surprising as mature OB rely on mitochondria OxPhos for energy. In sum, cell-autonomous effect of CypD deletion on osteoprogenitors is not sufficient to promote bone fracture repair; and systemic loss-of-function of CypD is required to fully unveil the pro-osteogenic effect.
In conclusion, herein we report a novel impact of CypD/MPTP inhibition on bone fracture repair in vivo and osteogenic function of BMSCs in vitro. This work, along with previous work in our lab, provides support for the role of mitochondria, as key regulators of the osteogenic program and bone formation and repair. We posit that CypD inhibition using pharmacological inhibitors of CypD, such as NIM811, Debio025, and JW47 (15,21), as potential candidates to induce bone formation and promote bone repair especially in aging and other pathologies.
Methods
Some methods have been previously reported (8,42) but are briefly described again here with modifications.
MOUSE STRAINS
CypD KO (Ppif−/−) mouse strain (originated in the Molkentin lab at Cincinnati Children’s Hospital and available from the Jackson Laboratory, RRID: IMSR_JAX:009071) was obtained from the Lab of Dr. George Porter at the University of Rochester and backcrossed 5 times onto the C57BL/6J background prior to experiments. CypD KO and control wild-type (WT) littermates were generated by breeding mice heterozygous for CypD allele. CypD floxed (CypDf/f) mice were bought from Jackson Laboratories (originated in the Korsmeyer lab at Dana Farber Cancer Institute, RRID: IMSR_JAX:005737) and bred in house. Prx1-Cre mice were a kind gift from Dr. Ackert-Bicknell (University of Rochester, originated in the Tabin Lab at Harvard Medical School). Prx1-Cre;CypDf/+ were bred from the cross of CypDf/f mice to Prx1-Cre mice. Prx1-Cre;CypDf/+ were then backcrossed with CypDf/f mice to obtain the Prx1-Cre;CypDf/f and CypDf/f littermate mice. Ai9 (tdTomato) mice were bought from Jackson Laboratories (RRID: IMSR_JAX: 007909). C57BL/6J mouse strain was obtained from the Jackson Laboratory (RRID: IMSR_JAX:000664) and bred in house. Thirteen-month-old C57BL/6J mice were obtained from the National Institute of Aging rodent colony and acclimatized in our vivarium for at least one week prior to experiments. All mice were housed at 23°C on a 12-hr light/dark cycle with free access to water and PicoLab Rodent Diet 20 (LabDiet #5053, St. Louis, MO). Mice were in group housing when applicable based on weaning. Test naive mice with an average weight of 28g were used for experiments. The assessments of all animal studies were performed in a blinded and coded manner.
FRACTURE HEALING ASSAYS
Tibia fracture model
Mice were subjected to unilateral tibial fractures following anesthesia as described before (43). Briefly, right tibia was exposed and mid-diaphyseal cut was made with a scalpel to induce fracture. A 27 Ga stainless steel pin was inserted intramedullary for stabilization. Nylon sutures closed the skin. Left tibia was sham operated and served as unfractured control. CypD KO and control WT littermate mice that underwent tibial fracture were collected at post fracture day (PFD) 7, 14, 21, 28, and 35 for analyses. Fractured and unfractured tibiae of Prx1-Cre;Ai9 mice and Cre-negative control Ai9 mice were collected at PFD 7 and processed for frozen sectioning and imaging for lineage tracing. Fractured and unfractured tibiae of Prx1-Cre;CypDf/f mice and Cre-negative control CypDf/f mice were collected at PFD 35 for biomechanical testing. C57BL/6J mice that underwent fracture were randomly divided into two groups and treated with either NIM811 (Labnetwork Inc, Cambridge, MA) or vehicle control. Freshly diluted NIM811 was injected intraperitoneally three times per week for the duration of the fracture healing (35 days) at a dose of 5 mg/kg, while control mice were injected with vehicle. Vehicle consisted of 7 parts PBS, 2 parts Kolliphor oil, and 1 part 100% EtOH. NIM811 dosage and regimen was based on previous rodent and human studies (18,43,44).
Biomechanical torsion testing
Following euthanasia, the fractured and unfractured tibiae were isolated and cleaned of excess soft tissue. Tibiae were stored at −80°C; and pins were removed prior to biomechanical testing. Tibiae were subjected to torsional testing. The ends of the tibias were cemented (Bosworth Company) in aluminum tube holders and tested using an EnduraTec TestBench™ system (Bose Corporation, Eden Prairie, MN). The tibiae were tested in torsion until failure at a rate of 1°/sec. The torque data were plotted against rotational deformation to determine maximum torque and torsional rigidity.
Serum P1NP and cytokine analysis
Blood was collected and serum was prepared by centrifugation and stored at −80°C until assayed. The bone formation marker P1NP was assessed in serum samples using the Rat/Mouse P1NP EIA kit (Immuno Diagnostic Systems, Kit #AC-33F1) according to the manufacturer’s instructions. Absorbance was measured at 450 nm. Data were analyzed with a P1NP curve to determine concentrations in ng/mL. Cytokines were assessed in serum samples from PFD 0 and 7 using the Proteome Profiler Mouse Cytokine Array Kit, Panel A (R & D Systems, Catalog #ARY006) according to the manufacturer’s instructions and were developed using a ChemiDoc XRS+ (BioRad). Intensities of each set of spots were measured and normalized to control spots on the membrane. Eight mice per collection time point were analyzed.
Bone micro-computed tomography
Following euthanasia, the fractured and unfractured tibiae were isolated and cleaned of excess soft tissue. Tibiae were fixed in 10 % neutral buffered formalin (NBF) for 72 hrs and pins were removed prior to micro-computed tomography (micro-CT). Tibiae were imaged using VivaCT 40 tomograph (Scanco Medical). Scanco analysis software was utilized for volume quantification. Trabecular bone vs total volume (BV/TV), bone mineral density (BMD), and cortical thickness were determined for unfractured tibias, while callus total volume, callus BV/TV, and BMD were determined for fractured tibiae.
Histology
NBF-fixed tibiae were processed for histology via decalcification in EDTA for two weeks followed by paraffin embedding. For frozen sectioning, undecalcified tibiae were fixed for three days in 10% NBF. Thereafter, samples were placed in 30% sucrose solution made in PBS followed by Cryomatrix embedding. Sections were cut to 5 μm in three levels of each sample, and then stained with either Hematoxylin/Eosin H&E, ABH/OG, with TRAP counter-stained with FastGreen, or DAPI (frozen sections).
Histomorphometry
ABH/OG-, TRAP- or immunohistochemistry (IHC)-stained slides were scanned in an Olympus VSL20 whole slide imager at 40x magnification and evaluated with VisioPharm automated histomorphometry software. ABH/OG-stained slides were analyzed to measure the bone and cartilage area relative to total non-marrow area of the callus in the fractured tibia. Vessel cross-sections with morphologically defined wall and lumen containing red blood cells were counted in serial ABH/OG-stained sections of the callus at PFD 7 and 14 and expressed as a mean number of vessels per section. TRAP-stained slides were analyzed to measure the TRAP positive area relative to total area of the callus in the fractured tibia. Neutrophils infiltrating fracture calluses were determined morphologically using expertise of our histopathology core and counted. IHC-stained slides probed for osteocalcin and F4/80 were analyzed to measure the osteocalcin or F4/80 positive area relative to total area of the callus in the fracture tibia. Three different levels were counted per mouse and averaged. There were eight mice per collection time point.
Collagen imaging using second harmonic generation
H&E-stained slides were imaged using multiphoton microscopy. An excitation light of 810 nm was directed and focused through a water immersion objective (20X, NA 0.95, Olympus) to the sample. Backward scattered SHG was passed through the objective, filtered via a 405 ± 15 nm bandpass filter and collected in a photomultiplier tube (Hamamatsu HC125-02). The forward propagating signal from the sample was passed through a condenser lens (NA 0.9) and filtered from excess excitation light via a 670 nm short pass dichroic mirror. SHG signal was further filtered with a bandpass filter (405 ± 15 nm) and collected in a photomultiplier tube (Hamamatsu HC125-02). Images were generated using Olympus FluoView software. Forward-to-backward (F/B) scatter ratios were calculated from the gathered images using an in-house written ImageJ macro (45).
Immunohistochemistry
NBF-fixed tibiae were processed as above in the Histology section. IHC was carried out using a primary anti-osteocalcin antibody (RRID:AB_10540992) diluted 1:750 or anti-F4/80 antibody (RRID: AB_323806) diluted 1:500 followed by incubation with a biotinylated anti-rabbit IgG secondary antibody (RRID:AB_2313606) diluted 1:750 and developing using a streptavidin-HRP conjugate diluted 1:1500. The primary antibody solution was composed of PBS, 3% Triton X-100 and 5% Goat Serum, while the secondary and streptavidin-HRP was made in PBS. Antigen retrieval was performed using a pressure cooker and Bull’s Eye Decloaker for osteocalcin and F4/80 and a pressure cooker. Detection was visualized with NovaRED Peroxidase Substrate Kit and counterstained with Hematoxylin QS for osteocalcin or with TrueBlue Chromagen Substrate counterstained with Nuclear Fast Red for F4/80.
BMSCs
Primary bone marrow cells were isolated from CypD−/− and control CypD+/+ littermates or from C57BL/6J mice by dissection and cleaning of femurs and tibiae followed by bone marrow flushing. Cells were plated at 20 × 106 per 10 cm dish in low glucose DMEM (LG-DMEM) supplied with 1 mM glutamine, 10% fetal bovine serum (FBS), and 1% penicillin-streptomycin. After 24h, non-adherent cells were removed and used for osteoclastogenesis assay. Adherent cell population containing bone marrow stromal (a.k.a. mesenchymal stem) cells (BMSCs) were used for BMSC experiments.
BMSC culture and differentiation
BMSCs were cultured in LG-DMEM described above at 37°C, 5% CO2, and 5% O2. Cells were cultured in 5% O2 to mimic their in vivo microenvironment. We used LG-DMEM containing physiological levels of glucose (5 mM) to avoid any artifacts caused by supraphysiological levels of glucose. Media was changed every day for 3 days following BMSC isolation to rid the culture of non-adherent hematopoietic cells. Media was then changed weekly until cells reached 80% confluency, which is when the cells were plated for the appropriate experiments. Osteogenic differentiation was induced by adding β-glycerol phosphate at 2 mM and ascorbate at 50 μg/mL to LG-DMEM. Osteogenic media was changed weekly until collection. To confirm osteogenic differentiation, cells were stained for osteoblast-specific alkaline phosphate (ALP) and for mineralization using alizarin red (ARed) and analyzed for expression of osteoblast-specific genes. Chondrocyte hypertrophy was induced in media containing β-glycerol phosphate at 10 mM, ascorbate at 50 μg/mL, and Insulin-Transferrin-Selenium mix. Alcian blue staining was used to mark chondrogenic differentiation and Alizarin Red to mark maturation and hypertrophy.
Flow cytometry
Freshly isolated bone marrow cells were resuspended in ice-cold FACS buffer (PBS added with 2% FBS) containing a mix of surface marker-specific antibodies carrying different fluorescent tags. For BMSC detection, the following BD Biosciences antibodies were used: anti-cd45 PerCP, anti-cd31 PerCP-eFluor 710, anti-cd105 eFluor450, and anti-cd29 APC, all diluted 1:500. For hematopoietic cell analysis, the following BD Biosciences antibodies were used: anti-cd45 APC Cy7, anti-cdllb AF700, anti-Ly6C BV605, anti-Ly6G BV786, and anti-F4/80 PE-Cy5, all diluted 1:100.
Cells were incubated at +4°C with agitation for 30 min with the above antibodies, centrifuged in a table-top centrifuge at 7000 rpm for 1 minute, washed with 500 μL ice-cold FACS buffer, centrifuged again, and finally resuspended in 250 μL ice-cold FACS buffer for analysis. Flow cytometry was performed at the University of Rochester Core Facility using BD Biosciences LSRII flow cytometer.
CFU assay
Bone marrow cells were isolated as described above and plated at 25,000 cells/cm2. LG-DMEM media change was performed in a similar manner in that media was changed daily for 3 days following isolation and then every week thereafter. Cells were stained with Crystal Violet 14 days after initial plating to assess total fibroblastic colonies (CFU-F). Clusters of greater than 50 cells were considered a CFU.
RNAseq
Total RNA was isolated and processed in the University of Rochester Genomic Core. The TruSeq RNA Sample Preparation Kit V2 (Illumina) was used for library construction. The amplified libraries were hybridized to the Illumina single end flow cell and amplified using cBot (Illumina). Reads were generated and aligned to the organism-specific reference genome. An adjusted p < 0.05 was used to determine significance.
Real-time RT-PCR
Total RNA was isolated using the RNeasy kit and reverse transcribed into cDNA using the qScript cDNA synthesis kit cDNA was subjected to real time reverse transcription-PCR (RT-PCR). The primer pairs used for genes of interest are outlined in Table S1. Real time RT-PCR was performed in the RotorGene system (Qiagen) using SYBR Green. The expression of genes of interest was normalized to expression of B2m (β-2 microglobulin).
Western blot
Cells were lysed with previously described lysis buffer (8) containing protease inhibitors and subjected to 4-12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) followed by transfer to polyvinylidene difluoride (PVDF) membranes and blocking in 5% dry milk reconstituted in PBST (PBS supplemented with Tween 20 at 0.05%), as previously described (8). All antibodies were diluted in 2.5% dry milk in PBST. For CypD detection, blots were probed with monoclonal CypD antibody (RRID: AB 478283) diluted 1:1000 and HRP conjugated goat anti-mouse antibody diluted 1:3000. To verify equal loading, blots were probed with β-actin antibody (RRID: AB 476697) diluted 1:2000 and HRP conjugated goat anti-mouse antibody diluted 1:5000. CypD and β-actin signals were developed with West Pico Substrate. Bands were measured with densitometry using ImageJ software. Signal was normalized to β-actin.
Ectopic bone formation assay
Cells were loaded onto a collagen Gelfoam sponge carrier at a concentration of 2 × 106 per 50 μL of LG-DMEM supplemented with 100 ng/mL of mouse recombinant BMP-2 and subcutaneously implanted into the backs of immunocompromised nude mice. Bone formation was assessed at 6 weeks after implantation by X-ray, histology, and histomorphometry.
OSTEOCLASTOGENESIS
Non-adherent bone marrow cells were collected from total bone marrow isolates cultured for 24 hrs and plated at 5 × 104 cells/well in 96-well plates in α-MEM supplemented with 15% FBS, M-CSF (10 ng/ml, R&D Systems), 1% penicillin-streptomycin (Gibco), 1% non-essential amino acids (Gibco). Cells were cultured for 3 days. Thereafter, to generate osteoclasts (OCL), media was changed; M-CSF (10 ng/ml) and RANKL (10 ng/ml, R&D Systems) were added; and then media was replaced every 2 days with fresh media supplemented with fresh M-CSF and RANKL. After 5 days of RANKL exposure, cells were fixed and stained for tartrate-resistant acid phosphatase (TRAP, Sigma-Aldrich Co, 387A). OCLs were identified as multinucleated (more than 3 nuclei) TRAP-positive cells.
MITOCHONDRIAL ASSAYS
Calcium Retention Capacity
Calcium Retention Capacity (CRC) assay was performed to measure MPTP opening in permeabilized cells exposed to calcium pulses as described before (46), with several modifications. In particular, BMSCs were permeabilized with 0.01% digitonin for 5 min on ice in a KCl-based buffer (140 mM KCl, 2 mM KH2PO4, 6 mM HEPES, pH 7.4) containing EGTA at 1 mM. Digitonin and EGTA were then washed away. Cells remained permeabilized even after digitonin washout as evident from positive staining of cells with Trypan Blue (not shown). One hundred thousand permeabilized cells were placed in 0.1 mL of KCl-based buffer containing 5 mM Succinate, 1 μM cytochrome c, and 1 μM Oregon Green BAPTA2, a fluorescent probe that measures extramitochondrial Ca2+. Of note, we used Oregon Green instead of Calcium Green 5N conventionally used in CRC assay, because of more appropriate Kd value for Ca2+ (0.5 μM vs 14 μM). Permeabilized cells contain significantly smaller number of mitochondria when compared to highly concentrated suspensions of isolated mitochondria usually used in the CRC assay. Therefore, we had to use smaller pulses of Ca2+ (0.5 μM instead of 20 μM) and higher affinity indicator, Oregon Green. Permeabilized cells were exposed to Ca2+ pulses, and Oregon Green fluorescence was monitored using a BioTek plate reader. Because of our plate reader specifications, we could not monitor the spikes of Ca2+ followed by a decrease due to uptake by mitochondria observed in previous publications (46), but could only detect a steady-state signal achieved after the pulse. MPTP opening was detected as an irreversible increase in Oregon Green fluorescence after the pulse. CRC values were expressed in nmoles of accumulated Ca2+ per 106 cells. Some cells were treated with 10 nM estradiol (E2) or 0.5 μM NIM811 or incubated in estrogen-depleted media. As cell culture media estrogen comes from the added serum, estrogen depletion of the media was done by charcoal stripping fetal bovine serum.
Mitochondrial mass assay
Cells were stained with nonyl acridine orange (NAO) at 100 nM, a fluorescent probe that labels cardiolipin present primarily in mitochondrial membranes (47), for 15 min at 37°C. Stained cells were then lifted from plates with cell scraper, washed, and resuspended in PBS. NAO signal was detected in BD Biosciences LSRII flow cytometer.
MtDNA assay
Cells were collected and total DNA isolated using the Wizard SV DNA purification kit (Promega). Quantitative real-time PCR was used to detect the mtDNA-encoded mt-Col using the following primer pair (5’-GCC CCA GAT ATA GCA TTC CC-3’ and 5’-GTT CAT CCT GTT 186 CCT GCT CC-3’) and nuclear-encoded 18s using the following primer pair (5’-TAG AGG GAC AAG TGG CGT TC-3’ and 5’-CGC TGA GCC AGT CAG TGT-3’). MtDNA was normalized to gDNA.
STATISTICS
Three to eight independent experiments were done to derive each panel of the paper, depending on the results of power analysis. Power analysis was carried out using the formula: , where n = sample size, α = type I error, β = type II error, Δ = effect size, σ = standard deviation, and Z is a constant (48). An example of power analysis carried out for biomechanical testing is shown below in Table S2. Data were analyzed using Prism 8 (GraphPad Software). Mean values and standard errors were calculated and the statistical significance (p <0.05) was established using either Student’s t-test when two variables were compared or one-way analysis of variance (ANOVA) when more than two variables were compared based on normal spread of our data.
STUDY APPROVAL
Animal husbandry and experiments were performed in accordance with the Division of Laboratory Animal Medicine, University of Rochester, state and federal law, and National Institutes of Health policy. University of Rochester Institutional Animal Care and Use Committee (IACUC) specifically approved this study.
Supplementary Material
Highlights.
MPT inhibition using CypD knock-out or NIM811 exerts pro-osteogenic effect in BMSCs
Protecting mitochondria from the MPT stimulates bone fracture repair
CypD knock-out promotes osteoblast activity and bone formation in fracture repair
Acknowledgements
We would like to thank the Center for Musculoskeletal Research and the Mitochondrial Interest Group for their fruitful discussions; Frances Fuks, Alexander Chirokikh, and Julian Joseph for contributing to histomorphometry analysis; Katherine Escalera-Rivera, Alex Hollenberg, and Sarah Murphy for assistance with mouse management and prep assistance; Drs. George Porter, Jennifer Jonason and Cheryl Ackert-Bicknell for their contributions to experimental design and data interpretation; Dr. Paul Brookes for the use of Seahorse apparatus; the histology core within the Center for Musculoskeletal Research (Kathy Maltby, Sarah Mack, and Jeffery Fox), the micro-CT core within the Center for Musculoskeletal Research (Michael Thullen), Mary Georger of the Wilmot Cancer Institute Histopathology Core, and the University of Rochester Genomics and Flow Cytometry Cores. Financial support was provided by NIH (R01 AR072601 and R21 AR07928 to R.A.E.; R01 AR070613 to H.A.; and P30 AR069655 to the Center for Musculoskeletal Research) and by a grant 17-004 to R.A.E. from the Orthopaedic Research and Education Foundation with additional funding provided by Musculoskeletal Transplant Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
This document contains two supplemental tables and eight supplemental figures.
The authors have declared that no conflict of interest exists.
References
- 1.Phetfong J, Sanvoranart T, Nartprayut K, Nimsanor N, Seenprachawong K, Prachayasittikul V, Supokawej A. Osteoporosis: the current status of mesenchymal stem cell-based therapy. Cellular & Molecular Biology Letters. 2016:1–20. doi: 10.1186/s11658-016-0013-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ghiasi MS, Chen J, Vaziri A, Rodriguez EK, Nazarian A. Bone fracture healing in mechanobiological modeling: A review of principles and methods. Bom Reports. 2017;6:87–100. doi: 10.1016/j.bonr.2017.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carlier A, Geris L, van Gastel N, Carmeliet G, Van Oosterwyck H. Oxygen as a critical determinant of bone fracture healing—A multiscale model. Journal of Theoretical Biology. 2015;365(C):247–264. doi: 10.1016/j.jtbi.2014.10.012. [DOI] [PubMed] [Google Scholar]
- 4.Friedenstein AJ, Chailakhjan RK, Lalykina KS. The development of fibroblast colonies in monolayer cultures of guinea-pig bone marrow and spleen cells. Cell Prolif. 1970;3(4):393–403. doi: 10.1111/j.1365-2184.1970.tb00347.x. [DOI] [PubMed] [Google Scholar]
- 5.Colnot C Skeletal Cell Fate Decisions Within Periosteum and Bone Marrow During Bone Regeneration. J Bone Miner Res. 2009;24(2):274–282. doi: 10.1359/jbmr.081003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cherry C, Thompson B, Saptarshi N, Wu J, Hoh J. 2016: A “Mitochondria” Odyssey. Trends in Molecular Medicine. 2016;22(5):391–403. doi: 10.1016/j.molmed.2016.03.009. [DOI] [PubMed] [Google Scholar]
- 7.Chen C-T, Shih Y-RV, Kuo TK, Lee OK, Wei Y-H. Coordinated changes of mitochondrial biogenesis and antioxidant enzymes during osteogenic differentiation of human mesenchymal stem cells. Stem Cells. 2008;26(4):960–968. doi: 10.1634/stemcells.2007-0509. [DOI] [PubMed] [Google Scholar]
- 8.Shum LC, White NS, Mills BN, de Mesy Bentley KL, Eliseev RA. Energy Metabolism in Mesenchymal Stem Cells During Osteogenic Differentiation. Stem Cells and Development. 2016;25(2): 114–122. doi: 10.1089/scd.2015.0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fomi MF, Peloggia J, Trudeau K, Shirihai O, Kowaltowski AJ. Murine Mesenchymal Stem Cell Commitment to Differentiation Is Regulated by Mitochondrial Dynamics. Stem Cells. 2015;34(3):743–755. doi: 10.1002/stem.2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baker N, Boyette LB, Tuan RS. Characterization of bone marrow-derived mesenchymal stem cells in aging. Bone. 2015;70:37–47. doi: 10.1016/j.bone.2014.10.014. [DOI] [PubMed] [Google Scholar]
- 11.Bemardi P, Rasola A, Forte M, Lippe G. The Mitochondrial Permeability Transition Pore: Channel Formation by F-ATP Synthase, Integration in Signal Transduction, and Role in Pathophysiology. Physiol Rev. 2015;95(4): 1111–1155. doi: 10.1152/physrev.00001.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Toman J, Fiskum G. Influence of aging on membrane permeability transition in brain mitochondria. J Bioenerg Biomembr. 2011. ;43( 1 ):3–10. doi: 10.1007/s10863-011-9337-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakagawa T, shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434(7033):648–652. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 14.Gutiérrez-Aguilar M, Baines CP. Structural mechanisms of cyclophilin D-dependent control of the mitochondrial permeability transition pore. BBA - General Subjects. 2015;1850(10):2041–2047. doi: 10.1016/j.bbagen.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Javadov S, Jang S, Parodi-Rullán R, Khuchua Z, Kuznetsov AV. Mitochondrial permeability transition in cardiac ischemia-reperfusion: whether cyclophilin D is a viable target for cardioprotection? Cell Mol Life Sci. 2017;74(15):2795–2813. doi: 10.1007/s00018-017-2502-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shum LC, White NS, Nadtochiy SM, de Mesy Bentley KL, Brookes PS, Jonason JH, Eliseev RA. Cyclophilin D knock-out mice show enhanced resistance to osteoporosis and to metabolic changes observed in aging bone. PLoS ONE. 2016. doi: 10.1371/journal.pone.0155709.s009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Du H, Guo L, Fang F, Chen D, A Sosunov A, M McKhann G, Yan Y, Wang C, Zhang H, Molkentin JD, Gunn-Moore FJ, Vonsattel JP, Arancio O, Chen JX, Yan SD. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nature Medicine. 2008; 14(10): 1097–1105. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mbye LH, Singh IN, Sullivan PG, Springer JE, Hall ED. Attenuation of acute mitochondrial dysfunction after traumatic brain injury in mice by NIM811, a non-immunosuppressive cyclosporin A analog. Experimental Neurology. 2008;209(l):243–253. doi: 10.1016/j.expneurol.2007.09.025. [DOI] [PubMed] [Google Scholar]
- 19.Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci USA. 2005; 102(34): 12005–12010. doi: 10.1073/pnas.0505294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rottenberg H, Hoek JB. The path from mitochondrial ROS to aging runs through the mitochondrial permeability transition pore. Aging Cell. 2017;111:10580–13. doi: 10.1111/acel.12650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Briston T, Lewis S, Koglin M, Mistry K, Shen Y, Hartopp N, Katsumata R, Fukumoto H, Duchen MR, Szabadkai G, Staddon JM, Roberts M, Powney B. Identification of ER-000444793, a Cyclophilin D-independent inhibitor of mitochondrial permeability transition, using a high-throughput screen in cryopreserved mitochondria. Sci Rep. 2016;6:37798. doi: 10.1038/srep37798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hansson MJ, Mattiasson G, Månsson R, Karlsson J, Keep MF, Waldmeier P, Ruegg UT, Dumont J-M, Besseghir K, Elmer E. The Nonimmunosuppressive Cyclosporin Analogs NIM811 and UNIL025 Display Nanomolar Potencies on Permeability Transition in Brain-Derived Mitochondria. J Bioenerg Biomembr. 2004;36(4):407–413. doi: 10.1023/B:JOBB.0000041776.31885.45. [DOI] [PubMed] [Google Scholar]
- 23.Robey PG, Kuznetsov SA, Riminucci M, Bianco P. Bone Marrow Stromal Cell Assays: In Vitro and In Vivo. In: Skeletal Development and Repair Nol 1130 Methods in Molecular Biology. Totowa NJ: Humana Press, Totowa, NJ; 2014:279–293. doi: 10.1007/978-1-62703-989-5_21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.La Colla A, Vasconsuelo A, Boland R. Estradiol exerts antiapoptotic effects in skeletal myoblasts via mitochondrial PTP and MnSOD. J Endocrinol. 2013;216(3):331–341. doi: 10.1530/JOE-12-0486. [DOI] [PubMed] [Google Scholar]
- 25.Bopassa JC, Eghbali M, Toro L, Stefani E. A novel estrogen receptor GPER inhibits mitochondria permeability transition pore opening and protects the heart against ischemia-reperfusion injury. American Journal of Physiology-Heart and Circidatory Physiology. 2010;298(1):H16. doi: 10.1152/ajpheart.00588.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Naik AA, Xie C, Zuscik MJ, Kingsley P, Schwarz EM, Awad H, Guldberg R, Drissi H, Puzas JE, Boyce B, Zhang X, O’Keefe RJ. Reduced COX-2 expression in aged mice is associated with impaired fracture healing. J Bone Miner Res. 2008;24(2):251–264. doi: 10.1359/jbmr.081002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen X, Nadiarynkh O, Plotnikov S, Campagnola PJ. Second harmonic generation microscopy for quantitative analysis of collagen fibrillar structure. Nat Protoc. 2012;7(4):654–669. doi: 10.1038/nprot.2012.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Han X, Burke RM, Zettel ML, Tang P, Brown EB. Second harmonic properties of tumor collagen: determining the structural relationship between reactive stroma and healthy stroma. Opt Express. 2008;16(3):1846–1859. [DOI] [PubMed] [Google Scholar]
- 29.Williams RM, Zipfel WR, Webb WW. Interpreting second-harmonic generation images of collagen I fibrils. Biophysical Journal. 2004;88(2): 1377–1386. doi: 10.1529/biophysj.104.047308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Valenti M, Dalle Carbonare L, Mottes M. Osteogenic Differentiation in Healthy and Pathological Conditions. IJMS. 2017;18(1):41. doi: 10.3390/ijms18010041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Horn JR, Quintanilla RA, Hoffman DL, de Mesy Bentley KL, Molkentin JD, Sheu S-S, Porter GA Jr. The Permeability Transition Pore Controls Cardiac Mitochondrial Maturation and Myocyte Differentiation. Developmental Cell. 2011;21(3):469–478. doi: 10.1016/j.devcel.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jonas EA, Porter GA, Beutner G, Mnatsakanyan N, Alavian KN. Cell death disguised: The mitochondrial permeability transition pore as the c-subunit of the F(1)F(0) ATP synthase. Pharmacol Res. 2015;99:3 82–392. doi: 10.1016/j.phrs.2015.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kwong JQ, Molkentin JD. Physiological and pathological roles of the mitochondrial permeability transition pore in the heart. Cell Metabolism. 2015;21(2):206–214. doi: 10.1016/j.cmet.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mnatsakanyan N, Llaguno MC, Yang Y, Yan Y, Weber J, Sigworth FJ, Jonas EA. A mitochondrial megachannel resides in monomeric FIFO ATP synthase. Nature Communications. 2019;10(1):5823. doi: 10.1038/s41467-019-13766-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pavlov EV, Priault M, Pietkiewicz D, Cheng EH, Antonsson B, Manon S, Korsmeyer SJ, Mannella CA, Kinnally KW. A novel, high conductance channel of mitochondria linked to apoptosis in mammalian cells and Bax expression in yeast. J Cell Biol. 2001;155(5):725–73E doi: 10.1083/jcb.200107057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pavlov E, Grigoriev SM, Dejean LM, Zweihorn CL, Mannella CA, Kinnally KW. The mitochondrial channel VDAC has a cation-selective open state. Biochim Biophys Acta. 2005;1710(2–3):96–102. doi: 10.1016/j.bbabio.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 37.Wang W, Fang H, Groom L, Cheng A, Zhang W, Liu J, Wang X, Li K, Han P, Zheng M, Yin J, Wang W, Mattson MP, Kao JPY, Lakatta EG, Sheu S-S, Ouyang K, Chen J, Dirksen RT, Cheng H. Superoxide flashes in single mitochondria. Cell. 2008;134(2):279–290. doi: 10.1016/j.cell.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jonas EA, Buchanan J, Kaczmarek LK. Prolonged activation of mitochondrial conductances during synaptic transmission. Science. 1999;286(5443): 1347–1350. doi: 10.1126/science.286.5443.1347. [DOI] [PubMed] [Google Scholar]
- 39.Elrod JW, Wong R, Mishra S, Vagnozzi RJ, Sakthievel B, Goonasekera SA, Karch J, Gabel S, Farber J, Force T, Heller Brown J, Murphy E, Molkentin JD. Cyclophilin D controls mitochondrial pore–dependent Ca2+ exchange, metabolic flexibility, and propensity for heart failure in mice. J Clin Invest. 2010;120(10):3680–3687. doi: 10.1172/JCI43171DS1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hunter DR, Haworth RA. The Ca2+-induced membrane transition in mitochondria. I. The protective mechanisms. Archives of Biochemistry and Biophysics. 1979;195(2):453–459. doi: 10.1016/0003-9861(79)90371-0. [DOI] [PubMed] [Google Scholar]
- 41.Fels JA, Manfredi G. Sex Differences in Ischemia/Reperfusion Injury: The Role of Mitochondrial Permeability Transition. Neurochem Res. 2019. doi: 10.1007/s11064-019-02769-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shares BH, Busch M, White N, Shum L, Eliseev RA. Active mitochondria support osteogenic differentiation by stimulating β-catenin acetylation. J Biol Chem. 2018;293(41): 16019–16027. doi: 10.1074/jbc.RA118.004102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lawitz E, Godofsky E, Rouzier R, Marbury T, Nguyen T, Ke J, Huang M, Praestgaard J, Serra D, Evans TG. Safety, pharmacokinetics, and antiviral activity of the cyclophilin inhibitor NIM811 alone or in combination with pegylated interferon in HCV-infected patients receiving 14 days of therapy. Antiviral Res. 2011. ;89(3):238–245. doi: 10.1016/j.antiviral.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 44.Garbaisz D, Turoczi Z, Aranyi P, Fulop A, Rosero O, Hermesz E, Ferencz A, Lotz G, Harsanyi L, Szijarto A. Attenuation of skeletal muscle and renal injury to the lower limb following ischemia-reperfusion using mPTP inhibitor NIM-811. PLoS ONE. 2014;9(6):el01067. doi: 10.1371/journal.pone.0101067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Burke K, Tang P, Brown E. Second harmonic generation reveals matrix alterations during breast tumor progression. J Biomed Opt. 2012; 18(3):31106. doi: 10.1117/1.JBO.18.3.031106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Giorgio V, Stockum von S, Antoniel M, Fabbro A, Fogolari F, Forte M, Glick GD, Petronilli V, Zoratti M, Szabo I, Lippe G, Bemardi P. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci USA. 2013; 110(15): 5887–5892. doi: 10.1073/pnas.1217823110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Beutner G, Eliseev RA, Porter GA. Initiation of Electron Transport Chain Activity in the Embryonic Heart Coincides with the Activation of Mitochondrial Complex 1 and the Formation of Supercomplexes. Santos J, ed. PLoS ONE. 2014;9(ll):e113330–25. doi: 10.1371/journal.pone.0113330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bhalerao S, Kadam P. Sample size calculation. Int J Ayurveda Res. 2010; 1(1):55–6. doi: 10.4103/0974-7788.59946. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.