

Abstract
Age-related accumulation of postzygotic DNA mutations results in tissue genetic heterogeneity known as somatic mosaicism. While implicated in aging as early as the 1950s, somatic mutations in normal tissues have been difficult to study because of their low allele fractions. With the recent emergence of cost-effective high-throughput sequencing, down to the single-cell level, enormous progress has been made in our capability to quantitatively analyze somatic mutations in human tissues in relation to aging and disease. Here, we first review how recent technological progress has opened up this field, providing the first broad sets of quantitative information on somatic mutations in vivo necessary to gain insight into their possible causal role in human aging and disease. We then propose three major mechanisms that can lead from accumulated de novo mutations across tissues to cell functional loss and human disease.
Keywords: Somatic DNA mutation, Pathogenic effects, Aging, Age-related disease, Genome mosaicism, Transcriptional noise
eTOC blurb – Vijg
Vijg and Dong review recent insights that reveal how common genetic mosaicism is, and propose three mechanisms through which somatic mutations could contribute to aging and age-related pathology.
Introduction
Genome mosaicism of the soma refers to genetic heterogeneity within tissues and organs due to postzygotic mutations, i.e., mutations in the genome of somatic cells not transmitted to the next generation. The concept of genome mosaicism stems from the early 20th century when Boveri, Morgan and others speculated that cancer was a somatic mosaic caused by genetic alterations (Boveri, 1914; Morgan and Bridges, 1919). Mutations were then generally understood to be sources of genetic variation that alter the physical and functional units of heredity, first termed “genes” by Wilhelm Johannsen around that same time (Johannsen, 1911). Later, based on new insights into the mutagenic effects of radiation on mammals, Gioacchino Failla and Leo Szilard proposed in the late 1950’s that both cancer and aging were caused by the accumulation of de novo somatic mutations (Failla, 1958; Szilard, 1959). Almost two decades later, this proved to be correct for cancer, a disease caused by cycles of mutation, selection and new mutations, which can eventually result in a metastatic tumor (Nowell, 1976). However, a causal role for postzygotic mutations and genome mosaicism in aging and age-related diseases other than cancer, proved more difficult to establish and remains unclear. This is because postzygotic mutations are generally unique for each individual cell and could only be detected when they clonally amplify to a large allelic fraction. The latter is rare, which explains why it was not noticed until the era of large-scale genotyping began.
Somatic mutations arise because of errors in the repair or replication of damaged DNA (Fig. 1). In the germ line such errors generate genetic diversity by providing the substrate for natural selection, thereby fueling evolution. Germline mutation rate is itself genetically determined and depends on various factors, including environmental constraints and population size. Mutation rates cannot decrease to zero because this would prevent any subsequent evolutionary change and would have fitness costs for the organism in terms of the advanced repair and replication systems required (Baer et al., 2007; Lynch et al., 2016). In those species that have a soma, i.e., the relatively low abundant multicellular organisms, mutation rate may also vary and there is some evidence for increased somatic genome maintenance in long-lived species (Vijg, 2007). This would suggest that long-lived species have a lower somatic mutation rate than short-lived species, but this remains to be demonstrated. Of course, evolutionary theory does not predict the prevention of somatic mutations for extended time periods after reproduction (Williams, 1957). In addition, somatic mutagenesis also fulfils certain essential physiological roles (see Box 1).
Fig. 1. Causes and consequences of somatic DNA mutations.
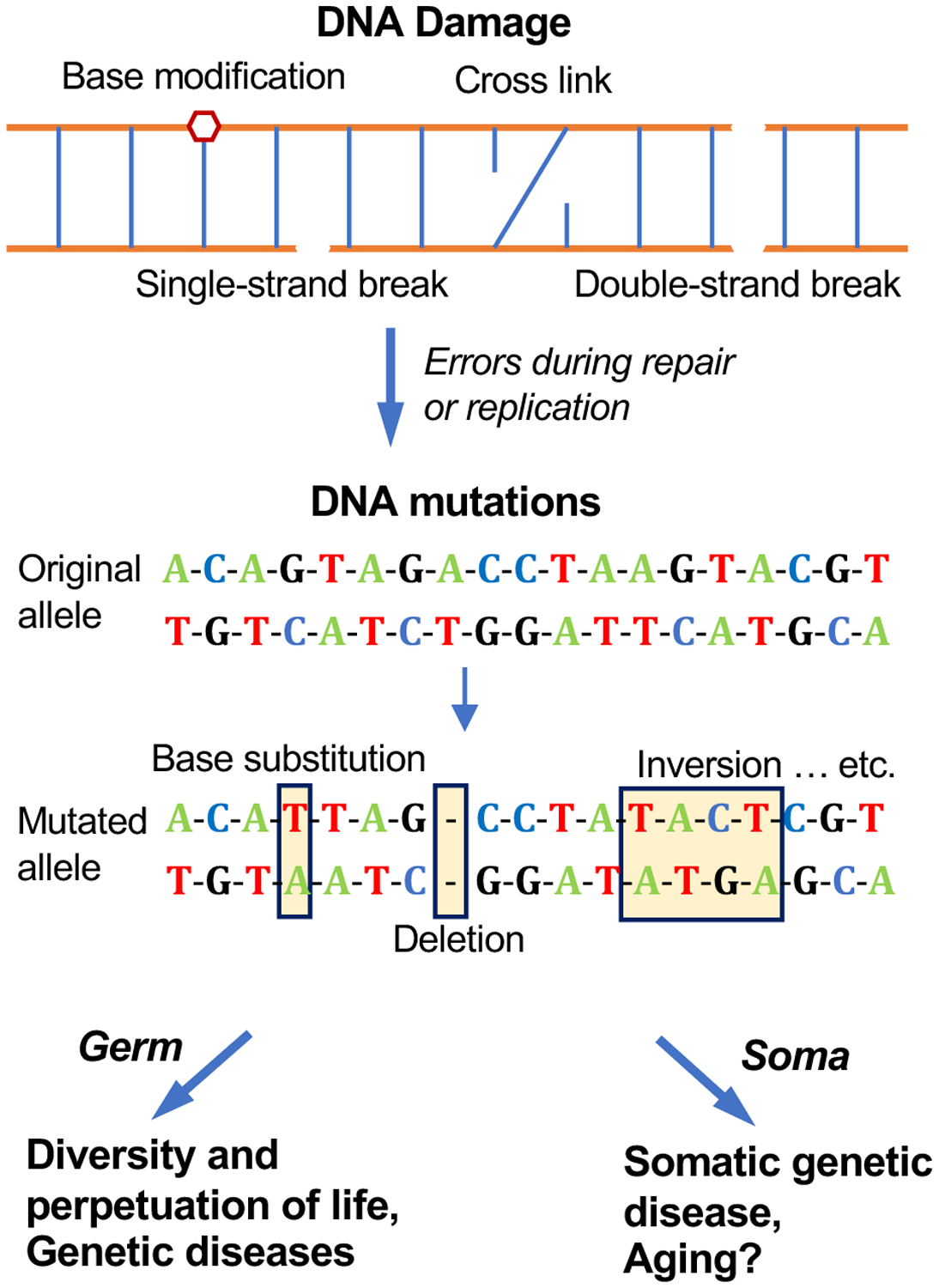
Different types of DNA damage occur daily in every cell of an organism, including strand breaks, base modifications, and cross links. This damage is rapidly repaired through a large variety of pathways. However, errors that occur during DNA repair and replication result in de novo mutations, which range from single nucleotide variations to chromosomal aberrations. In contrast to DNA damage, mutations are irreversible and, therefore, inevitably accumulate over time. When mutations occur in germ cells, they become substrates for evolution, giving rise to diverse life forms and to genetic disease. When they occur in the soma, mutations can result in somatic genetic diseases, age-related diseases and possibly aging.
Box 1. Physiological role of somatic mutations.
Somatic mutagenesis fulfils some important physiological roles. For example, somatic hypermutation generates immunological diversity to produce high-affinity antibodies through the accumulation of point mutations at a very high rate in the V-regions of the immunoglobulin heavy and light chains of B lymphocytes (Odegard and Schatz, 2006). Somatic hypermutation occurs during differentiation in the germinal centers of the lymphoid organs and is mediated by activation-induced cytidine deaminase (AID), which catalyzes targeted deamination of deoxycytidine residues in DNA, turning them into uracil. This leads to mutations at C:G and neighboring A:T base pairs (Di Noia and Neuberger, 2007; Zhang et al., 2019). This process allows vertebrates to produce a vast repertoire of antibody molecules to combat infection.
Somatic mutations might also have important roles in the brain (Paquola et al., 2017) and liver (Duncan et al., 2012). In the brain, most neuronal populations are postmitotic and cannot be renewed, with the exception of the hippocampus and subventricular zone (Gage, 2019). Somatic mutations generated early in development would therefore remain present until old age, generating individual physiological diversity in brain function.
The liver has a very high frequency of aneuploidy, possibly up to 50% of all cells (Duncan et al., 2012) and can regenerate, probably because it is heavily exposed to xenobiotics. This means that it can select specific aneuploid karyotypes to adapt to chronic liver injury. Recently, de novo mutations in specific genes have been found to be enriched for in liver of human cirrhosis patients, including in Pkd1, Kmt2d and Arid1a, that were subsequently demonstrated to promote hepatic clonal fitness and regeneration in mice (Zhu et al., 2019).
Until very recently almost all the available information on de novo mutagenesis in normal cells and tissues had been obtained indirectly, using reporter genes (endogenous genes or transgenes that can be selected for mutations) (Dollé et al., 1997; Grist et al., 1992). In virtually all of these studies, a significant and often tissue-specific increase in somatic mutations with age has been observed (Dollé et al., 1997; Ono et al., 2000). However, while informative, such mutational reporter models have the major limitation that no reporter gene can be considered to represent an entire mammalian genome. Hence, they do not allow obtaining quantitative information about mutation loads across the genome.
Based on the dramatic progress in next-generation sequencing over the last few years, our knowledge about frequency and type of somatic mutations in human tissues during aging has increased enormously. In not much more than half a decade more information on somatic mutations in human and animal tissues and their accumulation as a function of age has been obtained than in the previous half century. Hence, the time has come to discuss strategies to translate the technological breakthroughs that are now leading to a treasure trove of data sets on in vivo somatic mutations into a mechanistic understanding of their possible causal relationship with age-related cell functional loss and chronic disease. Here we will first summarize the status of the field on the hand of the technological advances that enabled this great progress and then discuss the possible mechanisms that can lead from random somatic mutations to age-related cellular degeneration and disease.
How technological advances led to the first accurate account of genome mosaicism
Mutations arise in a stochastic fashion in the different cells of a tissue. In contrast to germline mutations that are copied from the fertilized egg to all somatic cells, postzygotic mutations result in genetic heterogeneity within a tissue or cell population. The far majority of postzygotic mutations are unique to each cell or recurrently present only in very small numbers of cells. This makes their detection a major challenge, which explains the very limited progress in our understanding of somatic mutations, their frequency, how they are generated, their possible tissue-specificity and their relationship to human aging and disease. The exception is cancer because tumors are essentially clones and, therefore, serve as surrogates for the single cells from which they arose. In this section we will discuss the technological advances that made it possible to characterize the somatic mosaicism resulting from postzygotic mutagenesis in some detail in human tissues in relations to aging and disease, including diseases other than cancer.
Genome mosaicism and its increased occurrence in older organism first became apparent with the development of cytogenetic analysis. Using simple Giemsa staining of metaphase spreads, later replaced by Fluorescent In Situ Hybridization (FISH), chromosomal abnormalities were discovered in individual somatic cells, and were later found to increase with age in human blood cells (Jacobs et al., 1964) and regenerating mouse liver (Crowley and Curtis, 1963).
The most widely studied age-related chromosomal abnormality that gives rise to somatic genome mosaicism, reported early in the 1970s from studying metaphases from human blood lymphocytes (Jacobs et al., 1963) and bone marrow (Pierre and Hoagland, 1972), is the mosaic loss of the Y chromosome (LOY) in males during aging, which has now been widely confirmed with more advanced technology. LOY is defined as a lower than expected abundance of DNA from the Y chromosome with a certain threshold of detection, for example, as ≥10% of cells affected (Dumanski et al., 2016). In a recent study of 205,011 men from the UK Biobank LOY was found to affect from 2.5% of men at age 40 to 43.6% at age 70, which makes it the most common de novo somatic mutation over the human lifetime (Thompson et al., 2019). LOY frequency has been associated with shorter life span higher risk of cancer, smoking, Alzheimer’s disease, cardiovascular disease, diabetes, immune deficiencies, and other age-related diseases (Dumanski et al., 2016; Loftfield et al., 2018; Thompson et al., 2019). LOY has a genetic component, and in the aforementioned UK Biobank study, more than 150 autosomal genetic determinants of LOY were identified in the male cohort. LOY is most likely a general biomarker for genome instability in somatic cells. Indeed, the loci found genetically associated with LOY in males were themselves genetically associated, in a female cohort, with female cancers (breast, ovarian and endometrial cancer) and age at natural menopause (Thompson et al., 2019). Of note, early menopause has been genetically associated with DNA damage response (DDR) genes (Day et al., 2015). Based on these results it is tempting to speculate that the association of LOY with a diverse series of age-related pathologies points towards a causal role of somatic mutations in aging and age-related disease.
While being the most common postzygotic mutation, LOY is only one among many. With the emergence of large-scale genotyping technology about a decade ago, genome mosaicism was found to occur at the sub-chromosomal level across the genome in blood. For example, using SNP arrays mosaic copy number variations (CNVs) were found in 0.5% of young individuals and 2–3% of older people (Laurie et al., 2012). With the emergence of high-depth next-generation sequencing of specific genes or whole exomes a general pattern of genome mosaicism was uncovered in blood, called clonal hematopoiesis. For example, in blood genome mosaicism for specific mutations was found at loci frequently mutated in hematologic cancers (Jaiswal et al., 2014). In all these cases the frequency of expanded variants increased with age and was found associated with hematologic cancers, increased mortality, increased risk of type 2 diabetes, atherosclerosis, coronary heart disease and ischemic stroke.
More recent still, ultra-deep sequencing of DNA or RNA from multiple human tissues showed that somatic mutations leading to genome mosaicism does not only occur in blood but is a widespread phenomenon. For example, in numerous small samples taken across a grid of normal esophageal epithelium from nine human organ transplant donors of different ages, ultra-deep (>800-fold), targeted sequencing of known cancer driver genes revealed mutant mosaics that reflect the clonal outgrowth of de novo mutations, with the number of such mutations significantly increasing with age (Martincorena et al., 2018). Similar results were obtained by Yokoyama et al (Yokoyama et al., 2019). Finally, the clonal expansion of somatic mutations has been detected by RNA sequence analysis of normal tissues in the Genotype-Tissue Expression (GTEx) dataset (collected from about 30 non-diseased tissue types across nearly 500 individuals; https://www.gtexportal.org/home/). The results indicate the expected age-related increase in mutations, the association of the number of mutations with tissue-specific proliferation rate, and the enrichment of cancer-driver mutations (Yizhak et al., 2019).
From these findings, it can be concluded that genome mosaicism in blood and in other tissues, ranging from the loss of a Y chromosome in males to large deletions across the genome and point mutations in genes, is common, with the frequency of expanded variants increasing with age. These findings also confirm the aforementioned age-related increase in mutation frequency observed using reporter systems in humans and mice. But they also show that de novo mutations frequently expand into clonal lineages, allowing their detection DNA from bulk tissue.
If aging and age-related diseases occur as a consequence of losing genome sequence integrity, it raises the question of how such a random process can lead to the specific pathophysiology of so many different diseases. This question cannot be easily addressed by studying mutant clones in bulk cell populations. Indeed, the ultra-deep sequencing would uncover only the tip of an iceberg of accumulating mutations that occur independently in every cell. To understand if and how increased mutation loads have a functional impact and predispose to multiple diseases and intrinsic aging, we need to analyse single cells or clones grown out of a single cell.
A number of studies have now used in vitro clonal expansion of single cells, followed by sequencing, to avoid the selective view generated by sequencing bulk DNA for clonal mosaics. This approach has generated complete landscapes of somatic mutations from progenitor cells of the small intestine, liver and colon (Blokzijl et al., 2016), from neuronal stem cells (Bae et al., 2018), and skeletal muscle satellite cells (Franco et al., 2018), and from bronchial epithelial cells (Yoshida et al., 2020), kidney tubular progenitor cells, fat progenitor cells and epithelial progenitor cells (Franco et al., 2019) of human donors as a function of age. This approach has established the total mutation loads of human somatic cells as varying from several hundred base substitutions per genome to well over 3,000, depending on the cell type and donor age. These data have also provided an unselected, comprehensive view of the mutational spectra and signatures of these normal tissues, which in turn provide insights into the mutagen exposure of these tissues. Most importantly, this approach has enabled studying the distribution of mutations across the same genome and their possible interactions, which is critically important in analyzing their potential collective functional impact.
While highly informative, the quantitative analysis of somatic mutation landscapes in single cells clonally expanded in vitro suffers from an important limitation: de novo mutations in postmitotic cells remain out of reach. Moreover, by its very nature, this approach exclusively analyzes mutations in stem or progenitor cells, which generally have a lower mutation rate than differentiated cells. This is best illustrated by the recent, direct comparison of human liver stem cells with fully differentiated hepatocytes, showing an approximately two-fold higher average mutation frequency in the latter (Brazhnik et al., 2020). In addition, fully differentiated cells also displayed a much higher cell-to-cell variation in mutation frequencies, which is probably a reflection of their position in the hierarchy of cellular differentiation.
Direct sequencing of single cells without the selection involved in clonal outgrowth in vitro is the method of choice because it can be applied to every cell type, including postmitotic cell types. However, single-cell methods require whole genome amplification, which is fraud with error. Great progress has been made recently to overcome amplification-induced errors modification of whole genome amplification methods and the introduction of computational ways of detecting artifacts and filter them out (Bohrson et al., 2019; Chen et al., 2017; Dong et al., 2017; Zong et al., 2012). These improved methods showed that the total load of de novo somatic base substitution mutations in different normal human tissues appeared to be between several hundred and well over 5,000, depending on the tissue, the individual cell, and the age of the donor (Brazhnik et al., 2020; Lodato et al., 2018; Zhang et al., 2019), for a review, see (Zhang and Vijg, 2018).
Thus far all sequencing data, from somatic clonal expansion to single-cells yielded information on single nucleotide variants (SNVs), and occasionally small insertion and deletions (INDELs). SNVs are not likely to cause functional changes, except non-synonymous SNVs in protein-coding regions. Hence, these mutations are found in fairly large numbers in normal cells (see above). Small INDELs, most of them only 1 bp insertions or deletions, can be detected using the same principle as used for SNV detection. Their frequency is typically less than tenfold that of SNVs in somatic cells (Franco et al., 2018), which is explained by a much higher likelihood of causing a functional defect. The most impactful mutations are genome structural variations (SVs), i.e., deletions, insertions, inversions and rearrangements, such as interchromosomal and intrachromosomal translocations, mobile element insertions, such as retrotranspositions, and loss of telomere repeats at chromosome ends. SVs, the size of which can vary enormously from dozens of basepairs to whole chromosomes, readily affect cellular functions due to their capacity to rearrange gene regulatory sequences. This is reflected by their much greater role in the diversity and evolution of human genomes, something that was already realized in the 1970s (King and Wilson, 1975).
Because of their high functional impact SVs are now extensively studied. Thus far this has been mainly limited to germline variation and it seems likely that the average individual may differ from another by well over 10,000 SVs (Audano et al., 2019). While that is much less than the 4–5 million SNVs between two individuals, they could collectively impact as many as 20 million bp of sequence (Auton et al., 2015). We know very little about SVs in normal somatic cells. Similar to SVs in the germline genome, the functional effect of SVs in the somatic genome is likely to be much higher than that of SNVs.
Single-cell sequencing data are available for only two types of SVs, CNVs and retrotranspositions; the frequency of both appears to be limited to a few events per cell (Cai et al., 2014; Evrony et al., 2016; McConnell et al., 2013). As yet, there are no single-cell data on other forms of SVs (such as translocations, inversions, large deletions or insertions). It remains technically very difficult to derive accurate SV frequencies from whole genome sequencing data, even in case of bulk DNA (Kosugi et al., 2019). However, large genome rearrangements have been shown to increase with age in tissues such as heart and liver of the mouse using the reporter gene approach (Dollé et al., 1997) and in human and mouse lymphocytes using FISH (see above). Importantly, using a panel of 18 rodent species with diverse lifespans, it has been recently shown that more robust DNA double-strand break (DSB) repair coevolved with longevity (Tian et al., 2019). Since SVs are mostly a consequence of erroneous repair of DNA double-strand breaks it is tempting to suggest that the rate of SV accumulation is an important factor in determining aging rate.
In summary, based on groundbreaking technological advances, we are now able to extensively study somatic mutagenesis and its resulting genome mosaicism, essentially in any human cell type. This is especially true for base substitutions, which can now be accurately called in single cells. Other types of mutations, most notably SVs, still require new and improved analytical approaches. But while this is stil ongoing we can already begin to address the next question: what, if any, is the functional impact of somatic mutations and genome mosaicism? Observations of reduced proliferative capacity of primary cells with increased mutation burden (Franco et al., 2018) and selection against age-related accumulation of mutations in the functional part of the genome (Zhang et al., 2019) suggests that somatic mutations could be causally related to aging. Below we will discuss the mechanisms that could underlie the pathogenic effects of somatic mutations and genome mosaicism.
Pathogenic mechanisms
The main argument against a causal role for random somatic mutations in aging and aging-associated disease has been that spontaneous mutation frequency even at old age is too low to impair cellular function. The exception is cancer where particular driver mutations are selected for a growth advantage. Mutation frequencies in somatic cells were considered low because estimates were based on the mutation frequencies observed in the germline of various species, including human, as deduced from heritable changes in proteins. Indeed, a germline mutation frequency of about 1 ×10−8 per bp per generation was found for humans (Drake et al., 1998), which proved to be almost right when later assessed by the whole genome sequencing of parents and offspring, where all mutations found in the latter and not in the genome of the parents (about 60 new SNVs) were used to calculate the mutation rate per generation (Kong et al., 2012). In this case the mutation rate is almost entirely determined by the males and particularly older males because of the much larger number of germ-line cell divisions in sperm than in oocytes.
However, as discussed above, the new single-cell methods found many more mutations per cell in the soma, i.e., up to several thousands of SNVs, depending on the age of the subject and the cell type. This suggests that somatic mutation rate is higher than germline mutation rate. Indeed, in a direct comparison between germline and somatic mutation rates in humans and mice somatic mutation rate was found to be almost two orders of magnitude more frequent than germline mutation rate (Milholland et al., 2017). To some extent this can be explained by selection against deleterious mutations in the germline. However, most random mutations have no effect. Indeed, mutation frequencies in single sperm were also found to be low (Wang et al., 2012), albeit about 3 times as high as those obtained from genome sequenced pedigree data. This suggests that other factors, including higher germ cell DNA repair rates, could also play a role. Evidence for the latter has come from recent work, which identified a process called transcriptional scanning that maintains DNA sequence integrity specifically in testes (Xia et al., 2020).
That random mutations can have an adverse effect on fitness of cell populations, however, has been shown in an experiment using 3 groups of 75 isogenic strains of E. coli (Elena and Lenski, 1997). Each group harbored one, two or three randomly induced mutations. The results indicated a log-linear decline of average fitness with the increase in the number of mutations. Of course, this result cannot be easily extrapolated to mammalian cells because mammalian genomes are much larger and contain more redundant sequences. As of yet there is very little insight into the mechanism through which random somatic mutations could be pathogenic in aging mammals. Here, we propose that there are essentially three such mechanisms (Fig. 2).
Fig. 2. Pathogenic mechanisms of somatic genome mosaicism.
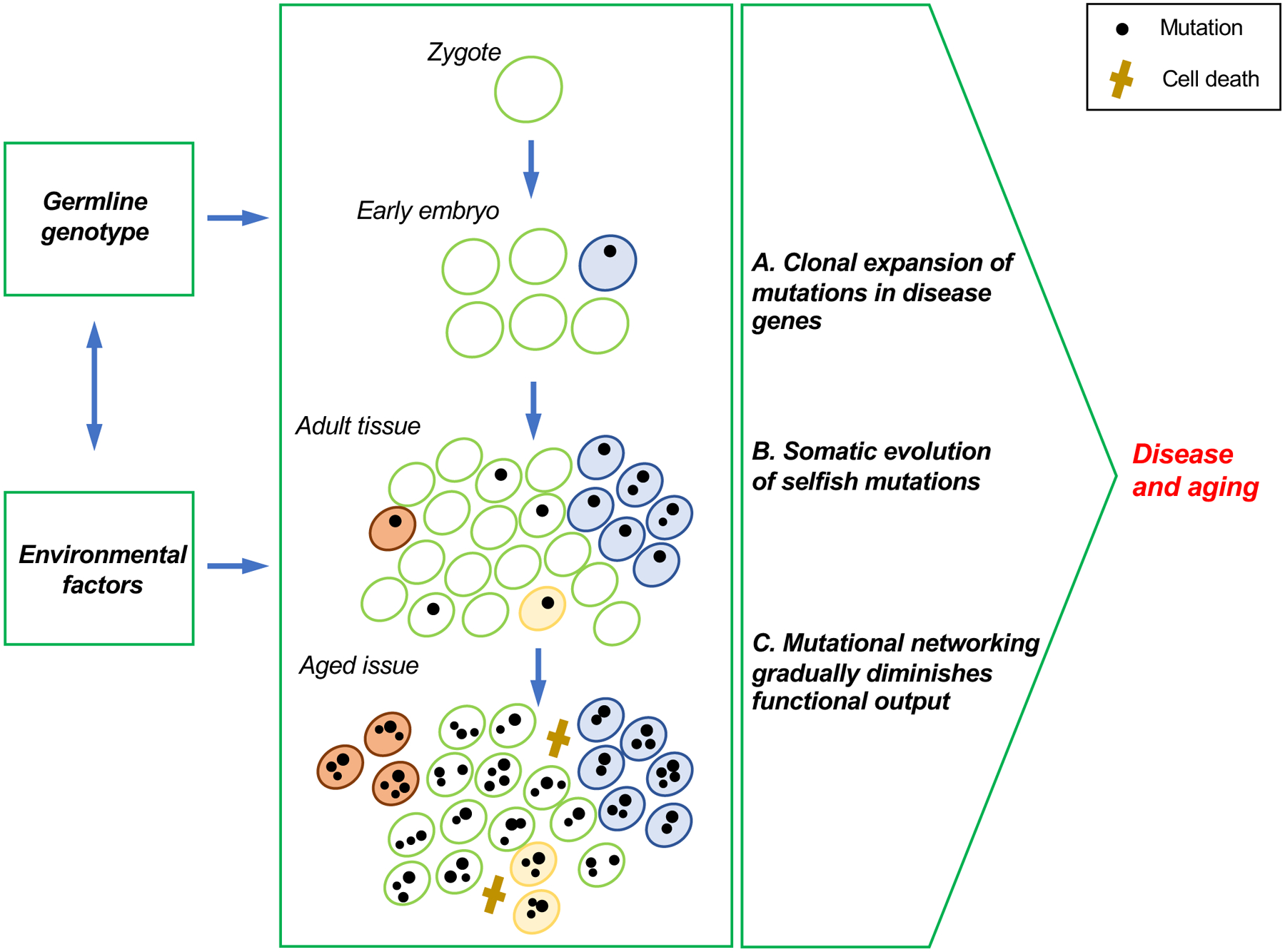
De novo mutations occur and accumulate at all stages during the lifespan of an organism. (A) Mutations that occur early in development are likely to expand clonally, resulting in substantial proportions of cells in a tissue carrying the same mutation, even in the absence of selection. When such mutations disrupt genes associated with Mendelian diseases, their adverse effects may be similar to that of the germline mutations, depending on the proportion of cells affected by the mosaicism. (B) Cells in adult tissues that acquire de novo mutations may clonally expand in a process called somatic evolution. Such selfish mutations can form substantial clonal lineages, which may cause cancer and other age-related diseases that involve loss of proliferative homeostasis, such as atherosclerosis. (C) In each cell, mutations accumulate across its genome, including in those sequences that participate in functional networks. This may result in transcriptional noise, which in turn are likely to affect the signaling networks that are critical for maintaining organismal homeostasis.
Clonal expansion of mutations in human disease genes.
It has been known for some time that many Mendelian genetic diseases have a somatic mutational counterpart. The best examples are overgrowth syndromes in children, such as Proteus syndrome, Neurofibromatosis 1 and 2, McCune-Albright syndrome, and CLOVES disorder (Erickson, 2003, 2010). A fraction of these diseases and sometimes all cases (because the germline variant would be lethal) are caused by somatic mutations that occur early enough in embryonic development to affect a substantial fraction of the tissue in which the disease manifests. Somewhat surprisingly, the fraction of cells in a tissue harboring the disease-causing mutation can be as low as 1% and still show disease, as described for Proteus Syndrome (Lindhurst et al., 2011). In many cases, the somatic mutation confers a growth advantage to mutant cells (hence, the term “overgrowth syndromes”), but often the mutation is simply clonally amplified by chance (Fig. 2A). The most dramatic example of clonal amplification of a human disease gene is a sporadic early-onset patient with Alzheimer’s disease, who showed somatic mosaicism for a presenilin 1 gene mutation. The degree of mosaicism in this patient at the age of presentation was 8% in peripheral lymphocytes and 14% in cerebral cortex with a gene dosage effect (Beck et al., 2004).
In many cases, Mendelian diseases are caused by a germ line and a somatic mutation, each affecting an allelic copy of the same gene, such as in autosomal dominant polycystic kidney disease, in which one allele of the polycystic kidney disease 2 (ADPKD2) gene has been inactivated in the germline and the other by a somatic mutation (Koptides et al., 1999). The cysts in this disease are products of clonal expansion of the second, somatic mutation. Nephron cysts also occur in the absence of disease, during normal aging, in which case their prevalence increases with age, perhaps because of an increase in somatic mutations. This is one of many examples where the difference between disease and aging is blurred, with a possible overlap in mechanism.
Postzygotic mosaic mutation as a cause of disease is not limited to monogenic Mendelian disease. Indeed, in the complex disease autism spectrum disorder (ASD) more than 7% of de novo, causal mutations have been identified as being postzygotic (Lim et al., 2017). These mutations were found in blood with an alternative allele frequency of ≤40%. Interestingly, a large portion of the genes affected were clinically relevant to ASD. The authors hypothesized that the postzygotic mutations arose early in development, which is supported by the observed enrichment of the expression of the affected genes in prenatal brain, most notably amygdala, a part of the brain involved in emotion.
To gain an impression of the potential importance of somatic mutation expansion in human disease genes, we estimated the prevalence of random, postzygotic mosaic mutations affecting any known human disease genes in early development by chance alone. There are 4,200 human genes in which mutations cause Mendelian disorders or complex diseases, as reported in the OMIM database on January 1st 2020 (Amberger et al., 2019). Based on a reported somatic mutation rate of 2.66 × 10−9 per bp per mitosis (Milholland et al., 2017), the number of amino acids encoded per gene (Brocchieri and Karlin, 2005), and the codon usage (Nakamura et al., 2000), the chance of a human embryo to acquire a heterozygous loss-of-function (LOF) mutation at its first zygotic division is 0.058%. This same chance increases when the number of cell increases. For example, at the 7th cell division the chance an embryo is affected has increased to 1.6%. Such a mutation, i.e., at the 7th cell division, could expand to 1% of human somatic cells only by genetic drift, enough to cause disease, as described above (Lindhurst et al., 2011).
Of course, human development and aging cannot be explained by a simple series of cell divisions, like a cell line in culture, but is subject to complex and hierarchically-dictated schemes with some cells dividing much more frequently than others and others becoming subject to apoptosis. Nevertheless, accidental somatic mutation early in development could be a significant mechanism in the etiology of human disease, either alone or in combination with a germline variant. In such cases, the phenotypic effects are straightforward and are associated with the known role of the target gene(s) as a germline defect. However, combinations of low-frequency disease gene mosaics could occur, in which case the phenotypic effects in terms of aging phenotypes in organs and tissues are difficult to predict. Finally, an important question is how a clonally expanded disease gene mutation ending up as a very small fraction of cells can still have pathogenic effects. One possible mechanism could be protein aggregation, which is a hallmark both of normal aging and of some age-related diseases, including Alzheimer’s disease (AD). The age-related deposition of extracellular beta amyloid (Aβ) is associated with AD pathogenesis, and the resulting aggregates have been proposed to seed their own progression and spread (Eisele et al., 2015). In this case the pathogenic effects would be caused by a combination of clonal expansion and prion-like self-propagation of protein misfolding.
Somatic evolution.
Evolution does not only occur in populations of organisms, but also in populations of cells (Fig. 2B), which are genetically heterogeneous due to de novo mutations. For example, variations have been observed in gene copy number and in gene expression and protein levels among 14 HeLa cell strains from 13 international laboratories, cultured under uniform conditions, with consequences for their phenotype, such as responses to Salmonella infection (Liu et al., 2019). Similar findings were reported in an earlier study of 27 strains of the breast cancer cell line MCF7 and other cell lines (Ben-David et al., 2018). Importantly, single-cell derived clones quickly redeveloped genetic heterogeneity. This cell line evolution occurred as a consequence of positive clonal selection that was sensitive to culture conditions (Ben-David et al., 2018).
Not surprisingly, since the landmark paper by Nowell (Nowell, 1976) most attention has been focused on the evolution of somatic cells in relation to the well-documented, age-related increase in cancer incidence and mortality. For example, specific TP53 gene mutations normally found in human skin tumors have also been detected in normal human skin cells, but only after UV irradiation (Nakazawa et al., 1994). Evidence was provided that this could only be a result of selective clonal expansion of TP53 mutant cells. More recently, by performing targeted ultra-deep sequencing of hundreds of sun-exposed normal human skin biopsies, Martincorena et al. discovered somatic mutations in multiple cancer genes, including genes encoding key drivers of cutaneous squamous cell carcinomas (Martincorena et al., 2015). These mutations could be detected because of their somatic amplification into thousands of evolving clones because the mutations gave them a selective advantage over other cells without such mutations. At this stage, the skin cell clones harboring the mutations were still normal skin cells. These results were mostly interpreted in terms of cancer and tumors can be driven by mutations occurring in the still normal cells when exposed to a natural mutagen. However, cancer genes were the only genes analyzed and a more objective way of describing the phenomenon would be as an example of somatic evolution of mutations expanded in a tissue, because they provide the normal cell with a growth and/or survival advantage.
Another example of mutations enriched by somatic evolution in tissues exposed to a mutagen is the observation of cancer driver mutations in normal lung tissue of smokers. Frankling et al observed a single, identical point mutation in TP53 in microdissected bronchial epithelium from multiple sites in both lungs of a subject showing dysplastic changes in the respiratory epithelium, but no cancer (Franklin et al., 1997). The mutation was not found in blood or other organs and must have come from an expanded bronchial epithelial clone in the bronchial mucosa. Indeed, using bronchial epithelial cell clones for detecting somatic mutations across the genome, an approach described above, Yoshida et al (Yoshida et al., 2020) demonstrated that smoking increases overall somatic mutation frequency, with 25% of the cell clones sequenced carrying cancer driver mutations. They found a number of known cancer driver mutations, most notably NOTCH1, TP53 and ARID2, under positive selection in normal bronchial epithelium. Naturally, these somatically expanded mutations in cancer driver genes were assumed to eventually lead to cancer.
However, there is evidence that somatic evolution also causally contributes to age-related diseases other than cancer. It has been proposed that acquired somatic mutations in cancer driver genes that have been frequently found in human bronchial epithelium of smokers, such as TP53, Ras, PTEN, enhance the inflammation and oxidative stress associated with chronic obstructive pulmonary disease (COPD), which is caused mostly by cigarette smoking but without known etiology (Anderson and Bozinovski, 2003).
Somatic evolution has also been considered a potential mechanism for cardiovascular disease, like cancer a major age-related disorder (Andreassi, 2003). The pathogenesis of atherosclerosis has been proposed to involve an inflammatory response to injury and thus cell proliferation. The monoclonal nature of the smooth muscle cells in human atherosclerotic plaques (Benditt and Benditt, 1973) could reflect expansion of mutant cells. Later, evidence for genomic instability in atherosclerotic cells has indeed been reported (Arvanitis et al., 2005; McCaffrey et al., 1997), leading to the hypothesis that expansion of mutant cells could be a major causal factor in cardiovascular disase. However, the nature of the mutant genes(s) remains unknown (Andreassi, 2003). Interestingly, advanced atherosclerotic lesions contain senescent cells, which can further drive pathology through an increase in the expression of atherogenic and inflammatory cytokines and chemokines (Childs et al., 2016). Cellular senescence is the permanent cessation of cellular proliferation, which is often induced by DNA damage and genome instability (Andriani et al., 2019; Campisi, 2013).
More recent data show that the inflammatory end point in cardiovascular disease can also be reached through somatic evolution in blood cells. Indeed, partial bone marrow reconstitution in mice with TET2-deficient cells led to the clonal expansion of these cells and a marked increase in atherosclerotic plaque size (Fuster et al., 2017). While the mechanism of its selective advantage is not entirely clear, all Tet2 deficient cells appear to have enhanced proliferation, which explains their enrichment during aging in hematopoietic cells (Jaiswal et al., 2014).
In addition to a cell growth advantage, mutations can be positively selected by preventing cell death. Using ultra-deep, whole-exome sequencing of post-mortem hippocampus from Alzheimer’s disease patients, clonally expanded somatic mutations were found to increase with age and were significantly enriched for PI3K-AKT, MAPK, and AMPK pathway genes, which are involved in tau phosphorylation (Park et al., 2019). Hyperphosphorylation of tau, which is the main component of neurofibrillary tangles and a hallmark of AD, prevents the apoptosis of neuronal cells in rodents (Li et al., 2007). This mechanism could promote the survival of these mutant cells. As we have seen, even if only a few cells in the hippocampus are affected by these mutations, the protein aggregates could spread from each cell to their surrounding neighbors, as discussed above for beta amyloid.
In summary, in tissues of mammals the adaptive landscape of somatic evolution during aging is similar to the adaptive landscape of evolution (Wright, 1931), but from a different perspective. Indeed, in the aging soma, selection for fitness among individual cells tends to move them away from their optimal peak of functioning in concert with other cells in their host to a more selfish pattern of genetic variation. This pushes the aging process towards loss of functionality and increased risk of disease, most notably loss of proliferative homeostasis, e.g., neoplasia, fibrosis, inflammation, since long recognized as a major aging-related phenomenon (Martin, 2007).
Mutational networking.
As we have discussed, there is now ample evidence that somatic cells carrying mutations in known human heritable disease genes can clonally expand during development and can contribute to diseases other than cancer. In addition, certain acquired gene mutations that are not by themselves disease-causing can confer a selective advantage to the cell, which expands and gradually erode organ and tissue functioning due to increasingly selfish behavior. While the magnitude of the adverse effects of these events in aging still await more extensive studies, there is a third possible mechanism by which randomly accumulating mutations eventually affect cell fitness. This does not require clonal outgrowth and depends on the penetration of such mutations in the DNA sequence components of the gene regulatory networks (GRNs) that provide function to a mammalian organism throughout its life time (Figs. 2C and 3A). Virtually all mutations would accumulate not in the about 1% protein-coding part of the genome but in the gene regulatory regions that make up approximately 11% of all genome sequences (Kellis et al., 2014).
Fig. 3. How can mutation accumulation affect functional output of gene regulatory networks?
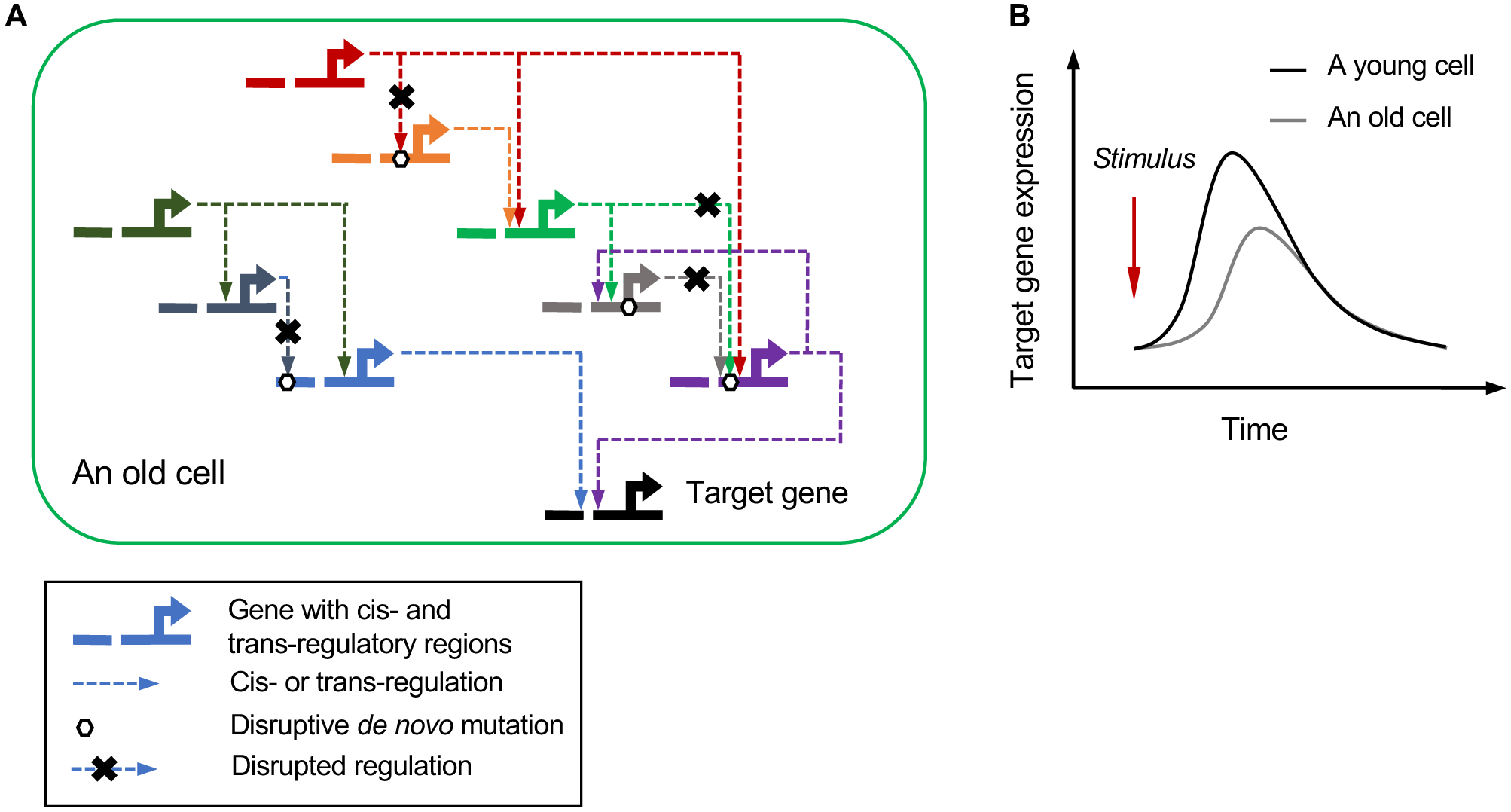
Cellular and organismal function is organized in complex gene regulatory networks (GRNs) involving interactions between large numbers of gene and their regulators, typically genes and their cis- and trans-acting elements. (A) Mutations inevitably accumulate in elements of each functional network in a stochastic manner. While initially insufficient in number to have an effect, eventually these mutations will collectively diminish the functional output of the GRN in an increasingly large number of cells, possibly through increased transcriptional noise (see text). (B) Increased noise in GRNs could explain defects in kinetics of cell signaling during aging, in which the response to external stimuli is blunted.
The topology of GRNs is scale-free, which makes them mutationally robust and minimizes the possibility of a deleterious mutation severely affecting function (Albert et al., 2000). However, GRNs are not immune to mutational effects. Indeed, their very structure - which dampens the effects of mutations - also serves specific biological functions that could be affected by mutations. For example, the most abundant network motif in GRNs, from fruitflies to humans, is the “feed-forward loop” (Boyle et al., 2014). This network motif serves to accelerate or delay the response of a cell to external stimuli (Mangan and Alon, 2003). Even a slightly deleterious mutation could change the kinetics of this process, affecting the time-accurate manner of the response (Fig. 3B). Since GRNs consist of many genes, and many more gene regulatory sequences, the chance that such deleterious mutations might affect network functions is not insignificant. Different sets of random mutations could adversely affect the same GRN through various synergistic interactions. Indeed, a network of 193 noncoding loci has been identified in which different sets of mutations disrupt target gene expression in 88% of tumors in 930 patients across 22 cancer types (Zhang et al., 2018).
Accumulated mutations in GRNs could explain the defects in cell signaling that have been observed with age (Greer and Brunet, 2008). Cells respond to environmental challenges, such as temperature changes, infections and a variety of other stressors through GRNs and their networks of regulatory interactions. While the dynamics of these complex networks in humans are far from understood, their actions are ultimately based on genes and the regulatory sequences that control their expression. Each of the genes in an GRN that is active in a given cell has to work in concert to ensure the specific function, fitness and survival of that cell. All these genes must be expressed at the proper time and in the proper amounts to ensure the appropriate functional output. Of note, normal aging is not the complete abrogation of particular life processes but the gradual erosion of the efficiency of such processes. Examples are thermoregulation, vaccine responses and short-term memory, which are all reduced in elderly but far from completely ablated. Such decline in function could well be explained by the gradual accumulation of weakly deleterious mutations in GRN components (Fig. 3). Naturally, the key question is how mutation accumulation in GRNs would translate in phenotypic deficiencies. One possibility is through transcriptional noise.
Transcriptional noise, increased cell-to-cell transcriptional heterogeneity in a tissue, organ or cell population, is a predicted consequence of mutation accumulation in GRNs. Elevated transcriptional noise was first discovered in both housekeeping and cell-type specific genes in cardiomyocytes of old mice (Bahar et al., 2006). Later, increased transcriptional noise was demonstrated in activated T lymphocytes of aged mice (Martinez-Jimenez et al., 2017) and more recently it was shown that most cell types in the mouse lung show increased transcriptional noise at old age (Angelidis et al., 2019). Transcriptional noise has also been demonstrated in the human pancreas among islet endocrine cells from older donors (Enge et al., 2017). In this study both transcriptional noise and somatic mutation load was shown to be increased in aged human pancreas. Interestingly, cells with high levels of transcriptional noise displayed an increase in non-cell type-specific hormone expression. The authors suggested that such a drift in cell fate could explain the decrease in fitness and organ function associated with aging.
One of the most widely studied molecular changes during aging is telomere attrition, which can cause cellular senescence (Bodnar et al., 1998; Campisi, 2013). The accumulation of senescent cells with age can have adverse effects (Campisi, 2013). Interestingly, telomere attrition can also contribute to transcription noise. Using isogenic pairs of human primary cells in culture with long or short telomeres, expression profiles of genes close to the telomeres were found to be altered with telomere length (Robin et al., 2014). While it is as yet unclear if this finding is relevant in vivo, this mechanism could increase transcriptional noise without the need for telomeres to become critically shortened and cause senescence or cell death.
Apart from non-cell type-specific gene expression and a loss of cell-to-cell coordination in responding to external stimuli, transcriptional noise can also lead to stoichiometric imbalances in protein macromolecular assembly, which is essential for most biological processes. The subsequent, unpartnered components may misfold and accumulate, leading to protein aggregation, which is associated with multiple age-related diseases, especially neuronal degeneration (Lindner and Demarez, 2009). Crude examples of this are the dose imbalances caused by CNVs and by loss-of-function mutations (Veitia et al., 2017). For example, PIN1 loss-of-function somatic mutations in the brains of Alzheimer’s disease patients cause haploinsufficiency, which increases tau phosphorylation and aggregation (Park et al., 2019).
While amply confirmed and likely to explain at least some of the adverse effects of aging, it remains unclear if transcriptional noise is caused by somatic mutations. Indeed, while the somatic mutation rate is at least an order of magnitude greater than originally assumed based on estimated germline mutation frequencies (Milholland et al., 2017), even a few thousand SNVs genome-wide may not affect the expression of a large enough number of genes to cause transcriptional noise. Of course, SNVs are the only mutation type for which we know accurate numbers per cell, with other mutations not yet quantitatively analyzed to the same extent, e.g., SVs, could also accumulate with age and have a much greater functional impact on gene expression patterns.
Alternatively, epimutations, i.e., alterations in the epigenome that control gene expression patterns through chromatin alteration, such as DNA methylation or histone modification, but do not involve changes in the DNA coding sequence, have been proposed to play a more important causal role in aging and disease, possibly through transcriptional noise (Enge et al., 2017). Using single-cell bisulfite sequencing epimutation frequencies of CpG DNA methylation have been observed in mouse liver hepatocytes at least two orders of magnitude higher than somatic mutation frequencies (Gravina et al., 2016). While this suggests that increased epimutations are more likely to play a causal role in the observed increase in transcriptional noise, the effect of a single DNA sequence alteration on average could be much greater than that of a single epimutation. For example, CpG DNA methylation sites may act collectively in controlling gene expression. In other words, many epimutations may be needed to cause the same effect as one sequence alteration.
Nevertheless, epimutations have been identified as bona fide disease-causing events, sometimes directly in somatic cells and sometimes indirectly through genetic mutations in epigenetic modifiers that affect chromatin (Zoghbi and Beaudet, 2016). However, as compared to mutations affecting DNA sequence, well-documented cases of epimutations as a cause of human disease are scarce, possibly because the impact of the average epimutation is not as high as that of a mutation. Naturally, compared to DNA sequence alterations, the field of epimutations as stochastic causes of disease and aging is still in its infancy with little information on epimutational mosaicism. Ultimately, to compare the relative effects of mutations and epimutations in influencing the transcriptome would require the simultaneous sequencing of DNA and RNA from the same cell, as well as computational methods to evaluate the (epi)mutational effect on target-gene transcription and regulatory network kinetics, which are now being developed (Dey et al., 2015; Zhou et al., 2018).
In summary, the genome’s distributed functional organization and high level of integration of the many sequence features that encode specific cellular functions provide robustness and a very high level of redundancy. Initially, therefore, genomes can tolerate fairly high levels of mutation. However, at some point, this type of genome functional organization will begin to amplify the effects of multiple, random mutations. It is possible, therefore, that even with a linear increase in somatic mutations with age, their effects on health and mortality would be exponential, begin to increase rapidly in middle age, following the same Gompertz kinetics as the increase in mortality and disease incidence during the lifetime.
Conclusions
The considerable advances in DNA sequencing technologies over the last decade have provided us with the first accurate picture of the aging-related landscape of somatic mutagenesis. Increasing evidence has revealed that a continuous trickle of de novo mutations in the soma, from early embryogenesis through to adulthood and old age, can cause disease - not only cancer but also a large variety of non-cancer-related disorders. The most surprising finding thus far has been the ubiquity and high frequency of somatic mutations. Whether this is sufficiently high to explain a considerable part of age-related functional decline and disease, alone or in conjunction with epimutations, remains to be tested. Irrespective of a possible causal role of somatic mosaicism in aging, somatic mutations are not the only cause of aging.
Trying to explain aging in terms of a singular process would be in conflict with evolutionary theory. Even if the loss of genome sequence integrity is the most conserved cause of aging and was already active in the first replicators (Vijg, 2007), natural selection would allow a multitude of mutations with late adverse effects to accumulate in the germline, many of which would be positively selected for because of their beneficial effects early in life (Williams, 1957), In this respect, somatic mutation accumulation could be a conserved, inevitable cause of aging, but superposed upon multiple other processes that usually cause the earlier demise of an individual.
Nevertheless, it is clear that previous ideas of a stable genome have now been replaced by a much more dynamic view of considerable cell-to-cell variation in genome structure and function. The purpose of this Perspective was to focus attention on the recent flood of quantitative data on somatic mosaicism, driven by technological advances, that allows for the first time to test if the progressively volatile genome sequence organization could be causally related to aging. The main challenge ahead is to fully characterize mosaic genomes in human tissues in relation to aging and disease. Continuous progress in single-cell sequencing technologies should provide us with such a comprehensive picture in the years ahead, allowing us to make much better estimates of the magnitude of genome mosaicism and its causal relation to human disease and aging. To understand the functional consequences of genome mosaicism, we need to learn how de novo mutations can affect phenotype. This will be greatly facilitated by the current explosion of single-cell, multi-omics technologies (Chappell et al., 2018), which will allow genome alterations to be directly connected to changes at the epigenome, transcriptome and proteome level. Such an integrated analysis of the effect of genome sequence erosion on cell, tissue and organismal phenotypes will eventually allow drawing specific conclusions about the role of somatic mutations as a causal factor in aging and age-related diseases.
Acknowledgements
This work was supported by the NIH (grants P01 AG017242, P01 AG047200, P30 AG038072 and U01 ES029519 to JV; K99 AG056656 to XD), the Glenn Foundation for Medical Research (JV), and the Shanghai Jiao Tong University School of Medicine.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
JV and XD are co-founders and shareholders of SingulOmics Corp.
References
- Albert R, Jeong H, and Barabasi AL (2000). Error and attack tolerance of complex networks. Nature 406, 378–382. [DOI] [PubMed] [Google Scholar]
- Amberger JS, Bocchini CA, Scott AF, and Hamosh A (2019). OMIM.org: leveraging knowledge across phenotype-gene relationships. Nucleic acids research 47, D1038–d1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson GP, and Bozinovski S (2003). Acquired somatic mutations in the molecular pathogenesis of COPD. Trends Pharmacol Sci 24, 71–76. [DOI] [PubMed] [Google Scholar]
- Andreassi MG (2003). Coronary atherosclerosis and somatic mutations: an overview of the contributive factors for oxidative DNA damage. Mutation research 543, 67–86. [DOI] [PubMed] [Google Scholar]
- Andriani GA, Maggi E, Pique D, Zimmerman SE, Lee M, Quispe-Tintaya W, Maslov A, Campisi J, Vijg J, Mar JC, et al. (2019). A direct comparison of interphase FISH versus low-coverage single cell sequencing to detect aneuploidy reveals respective strengths and weaknesses. Sci Rep 9, 10508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelidis I, Simon LM, Fernandez IE, Strunz M, Mayr CH, Greiffo FR, Tsitsiridis G, Ansari M, Graf E, Strom TM, et al. (2019). An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics. Nature communications 10, 963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvanitis DA, Flouris GA, and Spandidos DA (2005). Genomic rearrangements on VCAM1, SELE, APEG1and AIF1 loci in atherosclerosis. J Cell Mol Med 9, 153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audano PA, Sulovari A, Graves-Lindsay TA, Cantsilieris S, Sorensen M, Welch AE, Dougherty ML, Nelson BJ, Shah A, Dutcher SK, et al. (2019). Characterizing the Major Structural Variant Alleles of the Human Genome. Cell 176, 663–675.e619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, and Abecasis GR (2015). A global reference for human genetic variation. Nature 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae T, Tomasini L, Mariani J, Zhou B, Roychowdhury T, Franjic D, Pletikos M, Pattni R, Chen BJ, Venturini E, et al. (2018). Different mutational rates and mechanisms in human cells at pregastrulation and neurogenesis. Science (New York, NY) 359, 550–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baer CF, Miyamoto MM, and Denver DR (2007). Mutation rate variation in multicellular eukaryotes: causes and consequences. Nature reviews Genetics 8, 619–631. [DOI] [PubMed] [Google Scholar]
- Bahar R, Hartmann CH, Rodriguez KA, Denny AD, Busuttil RA, Dolle ME, Calder RB, Chisholm GB, Pollock BH, Klein CA, et al. (2006). Increased cell-to-cell variation in gene expression in ageing mouse heart. Nature 441, 1011–1014. [DOI] [PubMed] [Google Scholar]
- Beck JA, Poulter M, Campbell TA, Uphill JB, Adamson G, Geddes JF, Revesz T, Davis MB, Wood NW, Collinge J, et al. (2004). Somatic and germline mosaicism in sporadic early-onset Alzheimer’s disease. Human molecular genetics 13, 1219–1224. [DOI] [PubMed] [Google Scholar]
- Ben-David U, Siranosian B, Ha G, Tang H, Oren Y, Hinohara K, Strathdee CA, Dempster J, Lyons NJ, Burns R, et al. (2018). Genetic and transcriptional evolution alters cancer cell line drug response. Nature 560, 325–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benditt EP, and Benditt JM (1973). Evidence for a monoclonal origin of human atherosclerotic plaques. Proceedings of the National Academy of Sciences of the United States of America 70, 1753–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blokzijl F, de Ligt J, Jager M, Sasselli V, Roerink S, Sasaki N, Huch M, Boymans S, Kuijk E, Prins P, et al. (2016). Tissue-specific mutation accumulation in human adult stem cells during life. Nature 538, 260–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, and Wright WE (1998). Extension of life-span by introduction of telomerase into normal human cells. Science (New York, NY) 279, 349–352. [DOI] [PubMed] [Google Scholar]
- Bohrson CL, Barton AR, Lodato MA, Rodin RE, Luquette LJ, Viswanadham VV, Gulhan DC, Cortes-Ciriano I, Sherman MA, Kwon M, et al. (2019). Linked-read analysis identifies mutations in single-cell DNA-sequencing data. Nature genetics 51, 749–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boveri T (1914). Zur Frage der Entstehung maligner Tumoren. (Gustav Fischer; ). [Google Scholar]
- Boyle AP, Araya CL, Brdlik C, Cayting P, Cheng C, Cheng Y, Gardner K, Hillier LW, Janette J, Jiang L, et al. (2014). Comparative analysis of regulatory information and circuits across distant species. Nature 512, 453–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazhnik K, Sun S, Alani O, Kinkhabwala M, Wolkoff AW, Maslov AY, Dong X, and Vijg J (2020). Single-cell analysis reveals different age-related somatic mutation profiles between stem and differentiated cells in human liver. Sci Adv 6, eaax2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brocchieri L, and Karlin S (2005). Protein length in eukaryotic and prokaryotic proteomes. Nucleic acids research 33, 3390–3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Evrony GD, Lehmann HS, Elhosary PC, Mehta BK, Poduri A, and Walsh CA (2014). Single-cell, genome-wide sequencing identifies clonal somatic copy-number variation in the human brain. Cell reports 8, 1280–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J (2013). Aging, cellular senescence, and cancer. Annual review of physiology 75, 685–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappell L, Russell AJC, and Voet T (2018). Single-Cell (Multi)omics Technologies. Annual review of genomics and human genetics 19, 15–41. [DOI] [PubMed] [Google Scholar]
- Chen C, Xing D, Tan L, Li H, Zhou G, Huang L, and Xie XS (2017). Single-cell whole-genome analyses by Linear Amplification via Transposon Insertion (LIANTI). Science (New York, NY) 356, 189–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, and van Deursen JM (2016). Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science (New York, NY) 354, 472–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley C, and Curtis HJ (1963). The development of somatic mutations in mice with age. Proceedings of the National Academy of Sciences of the United States of America 49, 626–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day FR, Ruth KS, Thompson DJ, Lunetta KL, Pervjakova N, Chasman DI, Stolk L, Finucane HK, Sulem P, Bulik-Sullivan B, et al. (2015). Large-scale genomic analyses link reproductive aging to hypothalamic signaling, breast cancer susceptibility and BRCA1-mediated DNA repair. Nature genetics 47, 1294–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey SS, Kester L, Spanjaard B, Bienko M, and van Oudenaarden A (2015). Integrated genome and transcriptome sequencing of the same cell. Nature biotechnology 33, 285–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Noia JM, and Neuberger MS (2007). Molecular mechanisms of antibody somatic hypermutation. Annual review of biochemistry 76, 1–22. [DOI] [PubMed] [Google Scholar]
- Dollé ME, Giese H, Hopkins CL, Martus HJ, Hausdorff JM, and Vijg J (1997). Rapid accumulation of genome rearrangements in liver but not in brain of old mice. Nature genetics 17, 431–434. [DOI] [PubMed] [Google Scholar]
- Dong X, Zhang L, Milholland B, Lee M, Maslov AY, Wang T, and Vijg J (2017). Accurate identification of single-nucleotide variants in whole-genome-amplified single cells. Nature methods 14, 491–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake JW, Charlesworth B, Charlesworth D, and Crow JF (1998). Rates of spontaneous mutation. Genetics 148, 1667–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumanski JP, Lambert JC, Rasi C, Giedraitis V, Davies H, Grenier-Boley B, Lindgren CM, Campion D, Dufouil C, Pasquier F, et al. (2016). Mosaic Loss of Chromosome Y in Blood Is Associated with Alzheimer Disease. American journal of human genetics 98, 1208–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan AW, Hanlon Newell AE, Bi W, Finegold MJ, Olson SB, Beaudet AL, and Grompe M (2012). Aneuploidy as a mechanism for stress-induced liver adaptation. J Clin Invest 122, 3307–3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisele YS, Monteiro C, Fearns C, Encalada SE, Wiseman RL, Powers ET, and Kelly JW (2015). Targeting protein aggregation for the treatment of degenerative diseases. Nat Rev Drug Discov 14, 759–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elena SF, and Lenski RE (1997). Test of synergistic interactions among deleterious mutations in bacteria. Nature 390, 395–398. [DOI] [PubMed] [Google Scholar]
- Enge M, Arda HE, Mignardi M, Beausang J, Bottino R, Kim SK, and Quake SR (2017). Single-Cell Analysis of Human Pancreas Reveals Transcriptional Signatures of Aging and Somatic Mutation Patterns. Cell 171, 321–330 e314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson RP (2003). Somatic gene mutation and human disease other than cancer. Mutation research 543, 125–136. [DOI] [PubMed] [Google Scholar]
- Erickson RP (2010). Somatic gene mutation and human disease other than cancer: an update. Mutation research 705, 96–106. [DOI] [PubMed] [Google Scholar]
- Evrony GD, Lee E, Park PJ, and Walsh CA (2016). Resolving rates of mutation in the brain using single-neuron genomics. eLife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Failla G (1958). The aging process and carcinogenesis Annals of the New York Academy of Science 71, 1124–1135. [DOI] [PubMed] [Google Scholar]
- Franco I, Helgadottir HT, Moggio A, Larsson M, Vrtacnik P, Johansson A, Norgren N, Lundin P, Mas-Ponte D, Nordstrom J, et al. (2019). Whole genome DNA sequencing provides an atlas of somatic mutagenesis in healthy human cells and identifies a tumor-prone cell type. Genome biology 20, 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco I, Johansson A, Olsson K, Vrtacnik P, Lundin P, Helgadottir HT, Larsson M, Revechon G, Bosia C, Pagnani A, et al. (2018). Somatic mutagenesis in satellite cells associates with human skeletal muscle aging. Nature communications 9, 800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin WA, Gazdar AF, Haney J, Wistuba II, La Rosa FG, Kennedy T, Ritchey DM, and Miller YE (1997). Widely dispersed p53 mutation in respiratory epithelium. A novel mechanism for field carcinogenesis. J Clin Invest 100, 2133–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, Wu CL, Sano S, Muralidharan S, Rius C, et al. (2017). Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science (New York, NY) 355, 842–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gage FH (2019). Adult neurogenesis in mammals. Science (New York, NY) 364, 827–828. [DOI] [PubMed] [Google Scholar]
- Gravina S, Dong X, Yu B, and Vijg J (2016). Single-cell genome-wide bisulfite sequencing uncovers extensive heterogeneity in the mouse liver methylome. Genome biology 17, 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, and Brunet A (2008). Signaling networks in aging. J Cell Sci 121, 407–412. [DOI] [PubMed] [Google Scholar]
- Grist SA, McCarron M, Kutlaca A, Turner DR, and Morley AA (1992). In vivo human somatic mutation: frequency and spectrum with age. Mutation research 266, 189–196. [DOI] [PubMed] [Google Scholar]
- Jacobs PA, Brunton M, and Brown WM (1964). Cytogenetic studies in leucocytes on the general population: subjects of ages 65 years and more. Annals of human genetics 27, 353–365. [DOI] [PubMed] [Google Scholar]
- Jacobs PA, Brunton M, Court Brown WM, Doll R, and Goldstein H (1963). Change of human chromosome count distribution with age: evidence for a sex differences. Nature 197, 1080–1081. [DOI] [PubMed] [Google Scholar]
- Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, et al. (2014). Age-related clonal hematopoiesis associated with adverse outcomes. The New England journal of medicine 371, 2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannsen W (1911). The genotype conception of heredity. The American Naturalist 45, 129–159. [Google Scholar]
- Kellis M, Wold B, Snyder MP, Bernstein BE, Kundaje A, Marinov GK, Ward LD, Birney E, Crawford GE, Dekker J, et al. (2014). Defining functional DNA elements in the human genome. Proceedings of the National Academy of Sciences of the United States of America 111, 6131–6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King MC, and Wilson AC (1975). Evolution at two levels in humans and chimpanzees. Science (New York, NY) 188, 107–116. [DOI] [PubMed] [Google Scholar]
- Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, Gudjonsson SA, Sigurdsson A, Jonasdottir A, Jonasdottir A, et al. (2012). Rate of de novo mutations and the importance of father’s age to disease risk. Nature 488, 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koptides M, Hadjimichael C, Koupepidou P, Pierides A, and Constantinou Deltas C (1999). Germinal and somatic mutations in the PKD2 gene of renal cysts in autosomal dominant polycystic kidney disease. Human molecular genetics 8, 509–513. [DOI] [PubMed] [Google Scholar]
- Kosugi S, Momozawa Y, Liu X, Terao C, Kubo M, and Kamatani Y (2019). Comprehensive evaluation of structural variation detection algorithms for whole genome sequencing. Genome biology 20, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurie CC, Laurie CA, Rice K, Doheny KF, Zelnick LR, McHugh CP, Ling H, Hetrick KN, Pugh EW, Amos C, et al. (2012). Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nature genetics 44, 642–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HL, Wang HH, Liu SJ, Deng YQ, Zhang YJ, Tian Q, Wang XC, Chen XQ, Yang Y, Zhang JY, et al. (2007). Phosphorylation of tau antagonizes apoptosis by stabilizing beta-catenin, a mechanism involved in Alzheimer’s neurodegeneration. Proceedings of the National Academy of Sciences of the United States of America 104, 3591–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim ET, Uddin M, De Rubeis S, Chan Y, Kamumbu AS, Zhang X, D’Gama AM, Kim SN, Hill RS, Goldberg AP, et al. (2017). Rates, distribution and implications of postzygotic mosaic mutations in autism spectrum disorder. Nature neuroscience 20, 1217–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindhurst MJ, Sapp JC, Teer JK, Johnston JJ, Finn EM, Peters K, Turner J, Cannons JL, Bick D, Blakemore L, et al. (2011). A mosaic activating mutation in AKT1 associated with the Proteus syndrome. The New England journal of medicine 365, 611–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner AB, and Demarez A (2009). Protein aggregation as a paradigm of aging. Biochim Biophys Acta 1790, 980–996. [DOI] [PubMed] [Google Scholar]
- Liu Y, Mi Y, Mueller T, Kreibich S, Williams EG, Van Drogen A, Borel C, Frank M, Germain PL, Bludau I, et al. (2019). Multi-omic measurements of heterogeneity in HeLa cells across laboratories. Nature biotechnology 37, 314–322. [DOI] [PubMed] [Google Scholar]
- Lodato MA, Rodin RE, Bohrson CL, Coulter ME, Barton AR, Kwon M, Sherman MA, Vitzthum CM, Luquette LJ, Yandava CN, et al. (2018). Aging and neurodegeneration are associated with increased mutations in single human neurons. Science (New York, NY) 359, 555–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loftfield E, Zhou W, Graubard BI, Yeager M, Chanock SJ, Freedman ND, and Machiela MJ (2018). Predictors of mosaic chromosome Y loss and associations with mortality in the UK Biobank. Sci Rep 8, 12316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Ackerman MS, Gout JF, Long H, Sung W, Thomas WK, and Foster PL (2016). Genetic drift, selection and the evolution of the mutation rate. Nature reviews Genetics 17, 704–714. [DOI] [PubMed] [Google Scholar]
- Mangan S, and Alon U (2003). Structure and function of the feed-forward loop network motif. Proceedings of the National Academy of Sciences of the United States of America 100, 11980–11985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin GM (2007). The genetics and epigenetics of altered proliferative homeostasis in ageing and cancer. Mechanisms of ageing and development 128, 9–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martincorena I, Fowler JC, Wabik A, Lawson ARJ, Abascal F, Hall MWJ, Cagan A, Murai K, Mahbubani K, Stratton MR, et al. (2018). Somatic mutant clones colonize the human esophagus with age. Science (New York, NY) 362, 911–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, McLaren S, Wedge DC, Fullam A, Alexandrov LB, Tubio JM, et al. (2015). Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science (New York, NY) 348, 880–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Jimenez CP, Eling N, Chen HC, Vallejos CA, Kolodziejczyk AA, Connor F, Stojic L, Rayner TF, Stubbington MJT, Teichmann SA, et al. (2017). Aging increases cell-to-cell transcriptional variability upon immune stimulation. Science (New York, NY) 355, 1433–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaffrey TA, Du B, Consigli S, Szabo P, Bray PJ, Hartner L, Weksler BB, Sanborn TA, Bergman G, and Bush HL Jr. (1997). Genomic instability in the type II TGF-beta1 receptor gene in atherosclerotic and restenotic vascular cells. J Clin Invest 100, 2182–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell MJ, Lindberg MR, Brennand KJ, Piper JC, Voet T, Cowing-Zitron C, Shumilina S, Lasken RS, Vermeesch JR, Hall IM, et al. (2013). Mosaic copy number variation in human neurons. Science (New York, NY) 342, 632–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milholland B, Dong X, Zhang L, Hao X, Suh Y, and Vijg J (2017). Differences between germline and somatic mutation rates in humans and mice. Nature communications 8, 15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan TH, and Bridges CB (1919). The Origin of Gynandromorphs (Carnegie Inst.). [Google Scholar]
- Nakamura Y, Gojobori T, and Ikemura T (2000). Codon usage tabulated from international DNA sequence databases: status for the year 2000. Nucleic acids research 28, 292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa H, English D, Randell PL, Nakazawa K, Martel N, Armstrong BK, and Yamasaki H (1994). UV and skin cancer: specific p53 gene mutation in normal skin as a biologically relevant exposure measurement. Proceedings of the National Academy of Sciences of the United States of America 91, 360–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowell PC (1976). The clonal evolution of tumor cell populations. Science (New York, NY) 194, 23–28. [DOI] [PubMed] [Google Scholar]
- Odegard VH, and Schatz DG (2006). Targeting of somatic hypermutation. Nature reviews Immunology 6, 573–583. [DOI] [PubMed] [Google Scholar]
- Ono T, Ikehata H, Nakamura S, Saito Y, Hosoi Y, Takai Y, Yamada S, Onodera J, and Yamamoto K (2000). Age-associated increase of spontaneous mutant frequency and molecular nature of mutation in newborn and old lacZ-transgenic mouse. Mutation research 447, 165–177. [DOI] [PubMed] [Google Scholar]
- Paquola ACM, Erwin JA, and Gage FH (2017). Insights into the role of somatic mosaicism in the brain. Curr Opin Syst Biol 1, 90–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JS, Lee J, Jung ES, Kim MH, Kim IB, Son H, Kim S, Kim S, Park YM, Mook-Jung I, et al. (2019). Brain somatic mutations observed in Alzheimer’s disease associated with aging and dysregulation of tau phosphorylation. Nature communications 10, 3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierre RV, and Hoagland HC (1972). Age-associated aneuploidy: loss of Y chromosome from human bone marrow cells with aging. Cancer 30, 889–894. [DOI] [PubMed] [Google Scholar]
- Robin JD, Ludlow AT, Batten K, Magdinier F, Stadler G, Wagner KR, Shay JW, and Wright WE (2014). Telomere position effect: regulation of gene expression with progressive telomere shortening over long distances. Genes & development 28, 2464–2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szilard L (1959). On the nature of the aging process. Proceedings of the National Academy of Sciences of the United States of America 45, 30–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson DJ, Genovese G, Halvardson J, Ulirsch JC, Wright DJ, Terao C, Davidsson OB, Day FR, Sulem P, Jiang Y, et al. (2019). Genetic predisposition to mosaic Y chromosome loss in blood. Nature 575, 652–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian X, Firsanov D, Zhang Z, Cheng Y, Luo L, Tombline G, Tan R, Simon M, Henderson S, Steffan J, et al. (2019). SIRT6 Is Responsible for More Efficient DNA Double-Strand Break Repair in Long-Lived Species. Cell 177, 622–638.e622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veitia RA, Govindaraju DR, Bottani S, and Birchler JA (2017). Aging: Somatic Mutations, Epigenetic Drift and Gene Dosage Imbalance. Trends Cell Biol 27, 299–310. [DOI] [PubMed] [Google Scholar]
- Vijg J (2007). Aging of the Genome (Oxford; ). [Google Scholar]
- Wang J, Fan HC, Behr B, and Quake SR (2012). Genome-wide single-cell analysis of recombination activity and de novo mutation rates in human sperm. Cell 150, 402–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GC (1957). Pleiotropy, Natural Selection, and the Evolution of Senescence. Evolution 11, 398–411. [Google Scholar]
- Wright S (1931). Evolution in Mendelian Populations. Genetics 16, 97–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia B, Yan Y, Baron M, Wagner F, Barkley D, Chiodin M, Kim SY, Keefe DL, Alukal JP, Boeke JD, et al. (2020). Widespread Transcriptional Scanning in the Testis Modulates Gene Evolution Rates. Cell 180, 248–262.e221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yizhak K, Aguet F, Kim J, Hess JM, Kubler K, Grimsby J, Frazer R, Zhang H, Haradhvala NJ, Rosebrock D, et al. (2019). RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science (New York, NY) 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama A, Kakiuchi N, Yoshizato T, Nannya Y, Suzuki H, Takeuchi Y, Shiozawa Y, Sato Y, Aoki K, Kim SK, et al. (2019). Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature 565, 312–317. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Gowers KHC, Lee-Six H, Chandrasekharan DP, Coorens T, Maughan EF, Beal K, Menzies A, Millar FR, Anderson E, et al. (2020). Tobacco smoking and somatic mutations in human bronchial epithelium. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Dong X, Lee M, Maslov AY, Wang T, and Vijg J (2019). Single-cell whole-genome sequencing reveals the functional landscape of somatic mutations in B lymphocytes across the human lifespan. Proceedings of the National Academy of Sciences of the United States of America 116, 9014–9019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, and Vijg J (2018). Somatic Mutagenesis in Mammals and Its Implications for Human Disease and Aging. Annual review of genetics 52, 397–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Bojorquez-Gomez A, Velez DO, Xu G, Sanchez KS, Shen JP, Chen K, Licon K, Melton C, Olson KM, et al. (2018). A global transcriptional network connecting noncoding mutations to changes in tumor gene expression. Nature genetics 50, 613–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Theesfeld CL, Yao K, Chen KM, Wong AK, and Troyanskaya OG (2018). Deep learning sequence-based ab initio prediction of variant effects on expression and disease risk. Nature genetics 50, 1171–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu M, Lu T, Jia Y, Luo X, Gopal P, Li L, Odewole M, Renteria V, Singal AG, Jang Y, et al. (2019). Somatic Mutations Increase Hepatic Clonal Fitness and Regeneration in Chronic Liver Disease. Cell 177, 608–621.e612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi HY, and Beaudet AL (2016). Epigenetics and Human Disease. Cold Spring Harb Perspect Biol 8, a019497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong C, Lu S, Chapman AR, and Xie XS (2012). Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science (New York, NY) 338, 1622–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]