

Abstract
Background
The bleeding risks for nonsyndromic platelet function disorders (PFDs) that impair aggregation responses and/or cause dense granule deficiency (DGD) are uncertain.
Objectives
Our goal was to quantify bleeding risks for a cohort of consecutive cases with uncharacterized PFD.
Methods
Sequential cases with uncharacterized PFDs that had reduced maximal aggregation (MA) with multiple agonists and/or nonsyndromic DGD were invited to participate along with additional family members to reduce bias. Index cases were further evaluated by exome sequencing, with analysis of RUNX1‐dependent genes for cases with RUNX1 sequence variants. Bleeding assessment tools were used to estimate bleeding scores, with bleeding risks estimated as odds ratios (ORs) relative to general population controls. Relationships between symptoms and laboratory findings were also explored.
Results
Participants with uncharacterized PFD (n = 37; 23 index cases) had impaired aggregation function (70%), nonsyndromic DGD (19%) or both (11%), unlike unaffected relatives. Probable pathogenic RUNX1 variants were found in 2 (9%) index cases/families, whereas others had PFD of unknown cause. Participants with PFD had increased bleeding scores compared to unaffected family members and general population controls, and increased risks for mucocutaneous (OR, 4‐207) and challenge‐related bleeding (OR, 12‐43), and for receiving transfusions for bleeding (OR, 100). Reduced MA with collagen was associated with wound healing problems and bruising, and more severe DGD was associated with surgical bleeding (P < .04).
Conclusions
PFDs that impair MA and/or cause nonsyndromic DGD have significantly increased bleeding risks, and some symptoms are more common in those with more severe DGD or impaired collagen aggregation.
Keywords: blood platelet disorders, hemorrhage, hemostasis, odds ratio, platelet storage pool deficiency, wound healing
Essentials.
Bleeding risks for nonsyndromic platelet function disorders (PFDs) are unknown.
We examined bleeding in PFDs and its relationship to certain laboratory findings.
Nonsyndromic PFD had 4‐ to 207‐fold increased risk of bleeding.
Bleeding symptom differed between severe dense granule deficiency and impaired collagen aggregation.
1. INTRODUCTION
Platelet function disorders (PFDs) are important causes of bleeding. 1 The molecular causes of many rare, severe, and syndromic PFDs and thrombocytopenic disorders are now defined. 2 , 3 PFDs that present with impaired maximal aggregation (MA) responses to multiple agonists by light transmission aggregometry (LTA) 4 , 5 , 6 and/or dense granule (DG) deficiency (DGD) by whole mount electron microscopy (EM) 7 are largely uncharacterized disorders, with a minority caused by mutations in transcription factors (TF), such as RUNX1 and FLI1. 8 , 9
Uncharacterized PFDs that impair LTA and/or cause nonsyndromic DGD are known to increase bleeding scores, assessed by bleeding history assessment tools (BAT), such as the ISTH‐BAT. 10 , 11 , 12 , 13 Recently, we illustrated a proof of principle: that odds ratio (OR) analyses could be used to estimate the bleeding risks for such PFD, using data for a family with 6 affected individuals and a cohort of general population controls. 12 We now extend these bleeding risk analyses to a larger, consecutive‐case cohort with uncharacterized PFDs, with exploration for potential relationships between symptoms and laboratory findings.
2. MATERIALS AND METHODS
The study was conducted in accordance with the recently revised Declaration of Helsinki, with approval of the Hamilton Integrated Research Ethics Board (HiREB) and written informed consent of participants.
Index cases were recruited from a historic, consecutive case cohort, evaluated at Hamilton Health Sciences between September 2012 and March 2018, and diagnosed with an uncharacterized, nonsyndromic PFD after testing by the Hamilton Regional Laboratory Medicine Program. None had prior molecular testing. Inclusion criteria were:
Confirmed LTA abnormalities, with reduced MA responses with ≥2 agonists (based on validated reference intervals for normal and low‐platelet‐count samples 5 , 6 , 14 ) not due to well‐characterized PFD such as Glanzmann’s thrombasthenia or Bernard Soulier syndrome, and/or
Confirmed DGD with <4.9 DG/platelet, based on whole mount EM. 7
Cases with abnormal platelet ATP release without DGD or LTA abnormalities were excluded. 13
To minimize bias, relatives (affected and unaffected) of index cases were also recruited, which required assessments at Hamilton. Participants were asked to indicate on their consent if their physician should be notified if potential disease‐causing mutations were discovered. In consideration of ethical concerns, 15 a revised HiREB‐approved consent was used for further recruitment when families were found to have a probable disease‐causing PFD mutation with other health implications (eg, leukemia).
The cohort included a previously reported family with a RUNX1 frameshift mutation. 12 General population controls with no history of bleeding (n = 60) were recruited.
2.1. Clinical laboratory data
Medical record data collected included hemoglobin; platelet counts; mean platelet volume (MPV); prothrombin times/International Normalized Ratio; activated partial thromboplastin times; thrombin times; Clauss fibrinogen; VWD screen findings, with “low” von Willebrand factor (VWF) defined as 0.30‐0.49 U/mL VWF antigen and a similar reduction in VWF activity 16 ; MA responses (assessed as described 5 , 6 ) to 2.5 and 5.0 µmol/L ADP, 1.25 and 5.0 µg/mL Horm collagen, 6.0 µmol/L epinephrine, 1.6 mmol/L arachidonic acid (AA), 1.0 µmol/L thromboxane analogue U46619 (U46619), and 0.5 and 1.25 mg/mL ristocetin; mean DG numbers per platelet (assessed as described 6 , 7 , 17 , 18 ); ABO blood group; ferritin; and ATP release by lumiaggregometry, 13 if available.
2.2. Bleeding history assessment
Bleeding histories were evaluated using ISTH‐BAT 10 and Clinical History Assessment Tool – Platelets (CHAT‐P), 12 with comparison to general population controls (whose BAT data was previously published 7 , 12 , 13 ) and review by 1‐2 hematologists, who cross‐checked information against medical histories. Participants were contacted if details needed clarification. Discrepancies in BAT scores and affection status (obtained by medical record review) were resolved by consensus. 12 , 13
2.3. Analysis of platelet biomarkers
Samples from participants that provided additional sample donations were used to analyze platelet MYH10, and for cases with RUNX1 sequence variants, platelet transcript levels for several RUNX1‐regulated genes. MYH10 was assessed by immunoblotting, as described 12 , 19 using 30 μg total platelet protein/lane (60‐90 μg on ≥2 samples if suspected to be falsely negative) and known normal and abnormal controls. 12 Platelet MYH10, PF4, MYL9, and ALOX12 transcript levels (cDNA prepared as described 20 ) were assessed at the Centre for Applied Genomics, using the QX200 Droplet Digital PCR (dPCR) system with QuantaSoft v1.7.4 (Bio‐Rad Laboratories, Hercules, CA, USA) and duplex reaction mixes of 10 μL of 2× dPCR SuperMix for Probes (Bio‐Rad Laboratories), 1 μL of the target assay (FAM labeled Taqman probes: MYH10 Hs00413181_m1, MYL9 Hs00382913_m1, ALOX12 Hs00167524_m1, PF4 HS00236998_m1; Life Technologies, Rockville, MD, USA), 1 μL of the endogenous control assay (PPIA probe Hs99999904_m1, VIC label; Life Technologies), 5.5 μL of nuclease‐free water, and 2.5 μL of platelet cDNA (in parallel with no template, no reverse transcriptase and Human Universal RNA controls), with cycling conditions of: 95°C for 10 minutes, 45 cycles of 94°C for 30 seconds, 58°C for 1 minute, 98°C for 10 minutes, and finally a 10°C hold on a Veriti thermal cycler (Life Technologies).
2.4. Genomic DNA isolation, whole exome and Sanger sequencing, and variant annotation
Peripheral blood genomic DNA (300 ng) was prepared for whole exome sequencing (WES) of index cases at the Genetic Molecular Epidemiology Laboratory, using the Illumina Hiseq1500 (2 × 100 bp reads) and TruSeq Exome Capture kit (Catalog # FC‐930‐1012) as described 12 (F1 cases only), or Ion Torrent sequencing technology (1 × 125 bp reads) and the Ampliseq Exome Enrichment kit (Catalog A38262). Raw sequence reads were aligned to the human genome reference sequence (version hg19) using the Torrent Mapping Alignment Program, variant calling was performed using the Torrent Variant Calling (version 5.0.3) pipeline, and poor‐quality variants were filtered out according to the schema described by Damiati et al. 21
“Annovar” (release date: February 1, 2016) was used to annotate variant sequence change effects based on RefGene transcript boundaries. 22 Only rare nonsynonymous sequence variants within bleeding‐associated genes were examined. Rare sequence variants were defined as those having a minor allele frequency (MAF) of <0.01 within both external databases (including the National Heart, Lung, and Blood Institute GO Exome Sequencing Project, the 1000 Genomes project phase 3, and the genome Aggregation Database r2.0.2) and internal databases (from ~1000 samples sequenced from both Ion Torrent and Illumina systems). A MAF threshold of 0.01 was applied within each major ethnic strata of external databases (Europeans, Africans, Latin Americans, East Asians, South Asians) such that if a variant was common (MAF ≥ 0.01) in even a single ethnic group, it was excluded. Nonsynonymous sequence variants included missense, stopgain, stoploss, splicing, and insertion/deletion mutations. Sequencing data was examined for mutations in 63 genes associated with inherited PFD (genes listed in Megy et al 2 Lentaigne et al, 3 and Heremans and Freson 23 including IZKF5), and for these genes, average read depth was 117× (standard deviation [SD] = 58×) across all target exons and 89.2% (SD = 14%) had high sequencing depth (>20×), with RUNX1 and FLI1 sequenced at average depths of 170× and 142×, respectively. Variants were reviewed by multiple individuals to adjudicate variant pathogenicity, with rare protein‐altering sequence variants within the relevant genes assessed using the Mendelian Clinically Applicable Pathogenicity scheme. 24 Probable pathogenic sequence variants and variants of uncertain significance (VUS) were further evaluated by PCR and Sanger sequencing by the MOBIX Laboratory 12 (Appendix S1 lists primers). Copy number variation calling was performed for all samples using the eXome Hidden Markov Model method. 25
2.5. Statistical analyses
The most recent laboratory results and presenting ferritins were analyzed. Two‐tailed Mann Whitney tests were used for 2 group comparisons, 1‐way analysis of variance was used for multiple group comparisons, chi‐squared tests were used to assess proportional differences, and Fisher’s exact test was used when cell counts were <5. CHAT‐P data were used to estimate bleeding risks as ORs with 95% confidence intervals (CIs), after adding 0.5 to all contingency table cells with 0 values, as described, 12 with separate analysis of (i) each sex for symptoms showing sex differences among affected individuals and (ii) families with ≥5 affected individuals. Relationships between bleeding symptoms and LTA findings and/or DG counts were evaluated using Mann‐Whitney tests, chi‐squared tests, and multiple logistic regressions. P values < .05 were considered statistically significant.
3. RESULTS
Most (23/24) eligible index cases consented to participate. After recruiting relatives, the cohort included 37 persons with PFD (median 2/family, range: 1‐6/family; 11 sole participants; 11% children; laboratory finding details in Table S1) and 23 unaffected relatives (median 2/family, range: 0‐6/family; 17% children; none with multiple aggregation defects; n = 1 classified as unaffected with a borderline DG count [4.6/platelet], suggestive of a false positive, 7 declined further testing). Their ages and sex distribution resembled controls (Appendix S2).
3.1. Clinical laboratory findings
Most PFD participants (30/37, 81%) had normal platelet counts (Figure 1A, Table S1), MPV similar to unaffected relatives (Appendix S2), and reduced MA with multiple agonists (29/36, 81%; details in Table S1), commonly with 1.0 μmol/L U46619 (74%), 1.6 mmol/L AA (62%), and/or 1.25 μg/mL collagen (60%) (Figure 1B). Among the 11 with confirmed DGD (Figure 1A, Table S1), 36% had abnormal LTA, and 88% (n = 3 not tested) had impaired ATP release with ≥2 agonists. Appendix S2 summarizes additional laboratory findings.
FIGURE 1.

Platelet findings for the cohort of participants with an uncharacterized platelet function disorder. Blue shading indicates the range of normal results, dashed lines denote the lower limit of the reference interval (RI), closed symbols denote index cases, and open symbols denote nonindex cases. (A) Platelet counts (RI, 150‐450 × 109/L) and average numbers of dense granules/platelet (lower limit of RI, 4.9). (B) Light transmittance platelet aggregometry findings, shown as the percent maximal aggregation (MA) in responses to 2.5 and 5.0 µmol/L ADP, 1.25 and 5.0 µg/mL Horm collagen, 6.0 µmol/L epinephrine, 1.6 mmol/L arachidonic acid (AA), 1.0 µmol/L U46619, and 1.25 mg/mL ristocetin. The percentage of the cohort with impaired MA is shown for each agonist.
3.2. Findings for whole exome sequencing and biomarker analysis
Whole exome sequencing (summarized in Tables S2 and S3) indicated the index cases for 2 families (Caucasian) had probable pathogenic RUNX1 sequence variants predictive of haploinsufficiency, including (i) family 1, with a RUNX1 frameshift mutation shared by 6 affected individuals 12 (1 who since developed leukemia); and (ii) family 8 with a different, novel RUNX1 frameshift mutation (chr21:g. 36231795_36231796delTG) (protein: RUNX1: p.Thr196 SerfsTer16) shared by mother and son; her other son had a PFD with thrombocytopenia (deceased, unrelated cause; family history otherwise negative). Platelets from affected members of these 2 families showed reduced expression of the RUNX1 target genes PF4, MYL9, and ALOX12 (Figure S1) and increased MYH10 expression, but the findings for some overlapped controls (Figure S1). Although 5 of 6 cases with RUNX1 haploinsufficiency mutations had increased platelet MYH10 protein, one was consistently negative (Figure S2). The family 21 index case (sole participant, negative family history of blood problems) had increased MYH10 protein (Figure S2) but was classified as having a RUNX1 VUS (Table S2) based on normal platelet transcript levels for PF4, MYL9, ALOX12, and MYH10 (Figure S1). Platelet MYH10 was increased in 2 of 4 cases from family 2 (Figure S2), with an uncharacterized PFD, but no identified RUNX1 or FLI1 sequence changes. The family 5 index case had DGD, a FLI VUS (Table S2) without detectable MYH10 (Figure S2), and deletion of 1 copy of AP3D1 (Table S3) without other features of Hermansky Pudlak syndrome associated with AP3D1 mutations. 26 , 27 Other index cases did not have identified mutations in PFD genes.
3.3. Bleeding scores
PFD participants had higher ISTH‐BAT scores than unaffected relatives and general population controls (respective medians: 9 vs 1 vs 0; P < .001) (Figure 2A,B). ISTH‐BAT scores showed no association to age (R 2 = .28, P = .23); however, only four affected individuals were children. Affected females had higher ISTH‐BAT scores than affected males (respective medians: 11.5 vs 4.0; ranges: 1‐20 vs 0‐9; P = .002), even after excluding female‐specific items (respective medians: 8.5 vs 4; ranges: 1‐15 vs 0‐9; P = .002), reflecting more cutaneous bleeding (respective medians: 3 vs 0; all ranges: 0‐3; P = .039). ISTH‐BAT scores were higher for index compared to nonindex PFD cases (respective medians, ranges: 9 vs 6.5, 3‐15 vs 0‐9, P = .001; females only: 12.5 vs 7.5, 6‐20 vs 6‐11; P = .02; males only: 5.5 vs 3; ranges 0‐9 vs 3‐4; P = .22) and for PFD participants with prior surgery (medians: 9.5 vs 4; ranges: 0‐20 vs 1‐9; P = .024) and/or wound healing problems (medians: 10.5 vs 8; ranges: 4‐18 vs 0‐15; P = .022).
FIGURE 2.
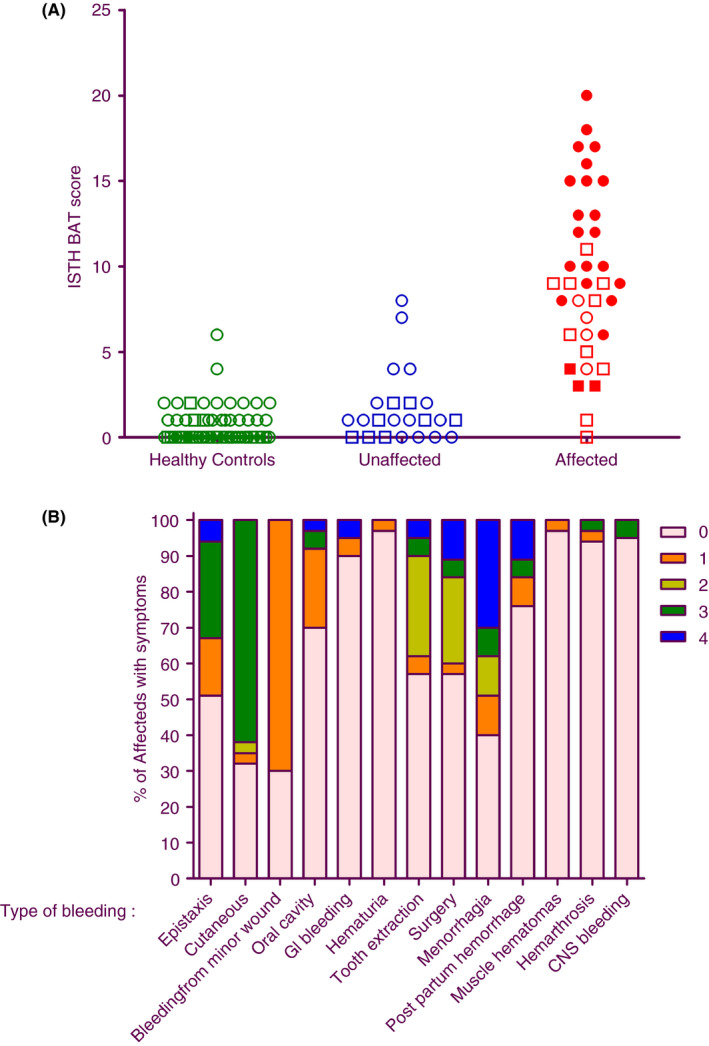
ISTH‐BAT data for the cohort with an uncharacterized platelet function disorder. (A) Bleeding scores are compared for general population controls (n = 60), and unaffected (n = 23) and affected (n = 37) family members. Red closed and open symbols, respectively, indicate index and nonindex PFD cases. Circles and squares, respectively, indicate females and males. (B) Details of the ISTH‐BAT scores for PFD participants, shown as the percentages that reported bleeding symptoms by category and severity (4 indicates the most severe, 1 indicates the least severe). None reported “Other bleedings.” BAT, bleeding history assessment tool; CNS, central nervous system; GI, gastrointestinal; PFD, platelet function disorder
3.4. Bleeding risk estimates
Figure 3 summarizes the estimated risks for important mucocutaneous and challenge‐related bleeding symptoms (full details in Table S4; analyses by sex in Tables S5 and S6; family 2 estimates in Table S7 [Figure S3 shows pedigree]). Compared to general population controls, participants with PFD had increased risks for receiving transfusions for bleeding (OR, 100) and for prolonged nosebleeds (OR, 28); bleeding from minor wounds lasting >1 hour (OR, 44); wound healing problems (OR, 15); excessive oral or dental bleeding (OR, 43); bleeding from operation(s) (OR, 20); hematuria with urinary tract infections (OR, 33); and gastrointestinal bleeding (OR, 4.1) (Figure 3A) but not for muscle, joint, or intracranial bleeding (Table S4). Females with PFD had increased risks for excessive bleeding with childbirth/miscarriage (OR, 17) and prolonged (>7 days) menses (OR, 8.8) (Figure 3B, Table S4). They also had higher risks than affected males for experiencing excessive bleeding or bruising (OR, 207 vs 87), bruising without trauma (OR, 35 vs 14) and bruising disproportionate to trauma (OR, 26 vs 14) (Figure 3B, Tables S5 and S6). The increased likelihood of a family history of leukemia/bone marrow problems was not evident when families with RUNX1 mutations were excluded (Table S4).
FIGURE 3.
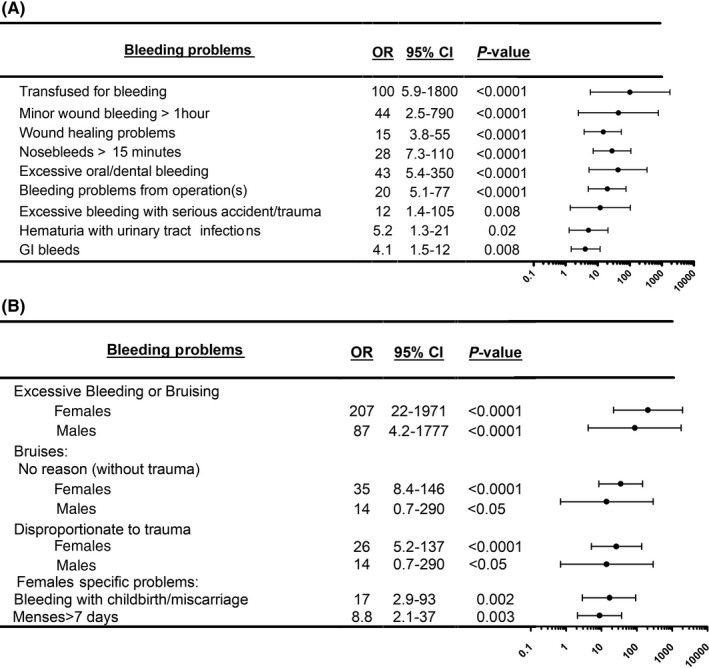
Bleeding risk estimates for important mucocutaneous and challenge‐related bleeding symptoms for participants with an uncharacterized platelet function disorder compared to general population controls. Bleeding risks are summarized as odds ratios (ORs) with 95% confidence intervals (CIs) for findings without (A) and with sex differences (B) (additional details in Tables S4‐S7). GI, gastrointestinal
3.5. Relationships between laboratory findings and bleeding symptoms
Analyses for relationships between symptoms and laboratory findings for participants with PFD revealed that reduced MA with collagen was associated with more bruises (median numbers for those with or without reduced MA: 5 vs 2; P < .01) and wound healing problems (OR, 9.3; 95% CI 1.1‐110; P < .04). Among participants with low DG counts, DG counts were significantly lower for those reporting surgical bleeding (1.2 vs 2.2; P < .04).
4. DISCUSSION
Our primary goals were to evaluate bleeding risks for uncharacterized PFD that impair aggregation responses to multiple agonists and/or cause nonsyndromic DGD, and explore possible relationships between bleeding and MA or DG abnormalities. We excluded well‐characterized and syndromic PFDs, as these PFDs are less common and better understood. We found probable pathogenic RUNX1 sequence variants in ~9% (2/23) of index cases, verifying that these disorders represent an important PFD subgroup. 8 , 9 WES failed to find a probable cause of the PFD in most index cases, which is in keeping with other studies. 9 , 28 , 29 , 30 , 31 We purposely evaluated both index cases and additional family members to minimize potential bias in clinical and laboratory data, noting that index cases had more bleeding but similar laboratory findings to other affected individuals. Aggregation abnormalities were present in the majority of our affected participants (81%), and DGD was present in 30%. Many with DGD (64%) had normal aggregation findings, and 1 had normal ATP release, verifying that these tests do not detect all cases with DGD. 7 Unlike unaffected family members and general population controls, our PFD cases had elevated ISTH‐BAT scores, like others with PFD. 11 , 32 Our PFD cases reported challenge‐related bleeding about as often as reported for severe PFD. 33 ISTH‐BAT scores were higher for PFD participants who had prior surgery or wound healing problems, which is interesting, as platelets release mediators that contribute to wound healing. 34 Our PFD cohort did not report some bleeding problems (eg, joint and muscle bleeds, spontaneous hematuria) that occur in Quebec platelet disorder, 35 suggesting that CHAT‐P might be useful to phenotype PFD. Unlike ISTH‐BAT scores, likelihood estimates allowed us to quantify bleeding while censoring data for those without exposure to certain challenges. Our bleeding risk estimates clarify that uncharacterized PFDs have significantly increased bleeding risks compared to the general population (Figure 3). Like other studies, females were seen more often than males for bleeding assessment, 5 , 6 , 7 , 13 , 36 possibly because females with PFD experience more spontaneous and disproportionate bruises, in addition to prolonged menses and childbirth‐related bleeding (Figure 3). We noted that participants appreciated learning about their estimated bleeding risks, which aided treatment discussions. As many PFD participants reported surgical and dental bleeding only when prophylactic treatment was not given, earlier diagnosis and treatment could improve outcomes, including the need for transfusions.
Our study purposely focused on the commonest type of PFD encountered in hematology practice. While 9% of our index cases (representing 2 families) had probable pathogenic TF mutations affecting RUNX1, 1 had no family history of leukemia or myelodysplasia. RUNX1 mutations alter gene expression, 19 , 37 , 38 , 39 , 40 , 41 , 42 , 43 and we found reduced expression of PF4, MYL9, and ALOX12 in platelets from PFD participants with RUNX1 haploinsufficiency mutations, with some but not all showing increased MYH10 transcript and protein levels compared to controls. Accordingly, we caution against analyzing MYH10 for diagnostic purposes. At present, there is no consensus on when to evaluate PFD cases for TF mutations and ethical concerns have been raised about testing for mutations with other health implications. 15 , 19 , 42 In consideration of these issues, we gave participants the choice of having their physician informed if a potential disease‐causing mutation was found (with the recommendation for further testing by a clinical genetics laboratory), and if the testing of a family uncovered a PFD mutation with other health implications, we used a modified consent form that disclosed this information to recruit additional family members.
Our study provides important evidence that uncharacterized PFD, presenting with impaired aggregation responses and/or nonsyndromic DGD, without a likely a priori cause, are associated with clinically important increased bleeding risks that are relevant to evidence‐based care. Uniquely, we identified that impaired collagen aggregation is associated with more bruising and wound healing problems, and that more severe DGD is associated with surgical bleeding. These interesting associations suggest that platelet‐collagen interactions reduce bruising and promote wound healing, and that platelet DGs (which store polyphosphate and ADP for stimulus‐induced release) help limit surgical bleeding. 34 , 44
RELATIONSHIP DISCLOSURE
The authors declare nothing to report.
AUTHOR CONTRIBUTION
JB, MB, JI, ST, LG, MC, and CH performed the research and analyzed data. CH, AP, and GP designed the research study. MC analyzed and wrote the WES findings. CH and GR developed the CHAT‐P tool. CH, JB, MC, and ST led the writing of the paper with contributions from all authors.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the subjects for their participation in the study.
Brunet J, Badin M, Chong M, et al. Bleeding risks for uncharacterized platelet function disorders. Res Pract Thromb Haemost. 2020;4:799–806. 10.1002/rth2.12374
Handling Editor: Yotis Senis
Funding information
This work was supported by the Canadian Hemophilia Society (CPMH, ADP, GP) and Canada Research Chairs in Molecular Hemostasis (CPMH), the Genetics of Complex Diseases (ADP), and Genetic and Molecular Epidemiology (GP).
REFERENCES
- 1. Hayward CP. Diagnostic evaluation of platelet function disorders. Blood Rev. 2011;25:169–73. [DOI] [PubMed] [Google Scholar]
- 2. Megy K, Downes K, Simeoni I, Bury L, Morales J, Mapeta R, et al. Curated disease‐causing genes for bleeding, thrombotic, and platelet disorders: communication from the SSC of the ISTH. J Thromb Haemost. 2019;17:1253–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lentaigne C, Greene D, Sivapalaratnam S, Favier R, Seyres D, Thys C, et al. Germline mutations in the transcription factor IKZF5 cause thrombocytopenia. Blood. 2019;134:2070–81. [DOI] [PubMed] [Google Scholar]
- 4. Quiroga T, Goycoolea M, Matus V, Zuniga P, Martinez C, Garrido M, et al. Diagnosis of mild platelet function disorders. Reliability and usefulness of light transmission platelet aggregation and serotonin secretion assays. Br J Haematol. 2009;147:729–36. [DOI] [PubMed] [Google Scholar]
- 5. Castilloux JF, Moffat KA, Liu Y, Seecharan J, Pai M, Hayward CP. A prospective cohort study of light transmission platelet aggregometry for bleeding disorders: is testing native platelet‐rich plasma non‐inferior to testing platelet count adjusted samples? Thromb Haemost. 2011;106:675–82. [DOI] [PubMed] [Google Scholar]
- 6. Hayward CP, Pai M, Liu Y, Moffat KA, Seecharan J, Webert KE, et al. Diagnostic utility of light transmission platelet aggregometry: results from a prospective study of individuals referred for bleeding disorder assessments. J Thromb Haemost. 2009;7:676–84. [DOI] [PubMed] [Google Scholar]
- 7. Brunet JG, Iyer JK, Badin MS, Graf L, Moffat KA, Timleck M, et al. Electron microscopy examination of platelet whole mount preparations to quantitate platelet dense granule numbers: Implications for diagnosing suspected platelet function disorders due to dense granule deficiency. Int J Lab Hematol. 2018;40:400–7. [DOI] [PubMed] [Google Scholar]
- 8. Daly ME. Transcription factor defects causing platelet disorders. Blood Rev. 2017;31:1–10. [DOI] [PubMed] [Google Scholar]
- 9. Stockley J, Morgan NV, Bem D, Lowe GC, Lordkipanidze M, Dawood B, et al. Enrichment of FLI1 and RUNX1 mutations in families with excessive bleeding and platelet dense granule secretion defects. Blood. 2013;122:4090–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rodeghiero F, Tosetto A, Abshire T, Arnold DM, Coller B, James P, et al. ISTH/SSC bleeding assessment tool: a standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J Thromb Haemost. 2010;8:2063–5. [DOI] [PubMed] [Google Scholar]
- 11. Lowe GC, Lordkipanidze M, Watson SP, Group UGs . Utility of the ISTH bleeding assessment tool in predicting platelet defects in participants with suspected inherited platelet function disorders. J Thromb Haemost. 2013;11:1663–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Badin MS, Iyer JK, Chong M, Graf L, Rivard GE, Waye JS, et al. Molecular phenotype and bleeding risks of an inherited platelet disorder in a family with a RUNX1 frameshift mutation. Haemophilia. 2017;23:e204–13. [DOI] [PubMed] [Google Scholar]
- 13. Badin MS, Graf L, Iyer JK, Moffat KA, Seecharan JL, Hayward CP. Variability in platelet dense granule adenosine triphosphate release findings amongst patients tested multiple times as part of an assessment for a bleeding disorder. Int J Lab Hematol. 2016;38:648–57. [DOI] [PubMed] [Google Scholar]
- 14. Hayward CP, Moffat KA, Pai M, Liu Y, Seecharan J, McKay H, et al. An evaluation of methods for determining reference intervals for light transmission platelet aggregation tests on samples with normal or reduced platelet counts. Thromb Haemost. 2008;100:134–45. [PubMed] [Google Scholar]
- 15. Greinacher A, Eekels JJM. Simplifying the diagnosis of inherited platelet disorders? The new tools do not make it any easier. Blood. 2019;133:2478–83. [DOI] [PubMed] [Google Scholar]
- 16. Graf L, Moffat KA, Carlino SA, Chan AK, Iorio A, Giulivi A, et al. Evaluation of an automated method for measuring von Willebrand factor activity in clinical samples without ristocetin. Int J Lab Hematol. 2014;36:341–51. [DOI] [PubMed] [Google Scholar]
- 17. Hayward CP, Moffat KA, Plumhoff E, Timleck M, Hoffman S, Spitzer E, et al. External quality assessment of platelet disorder investigations: results of international surveys on diagnostic tests for dense granule deficiency and platelet aggregometry interpretation. Semin Thromb Hemost. 2012;38:622–31. [DOI] [PubMed] [Google Scholar]
- 18. Hayward CP, Moffat KA, Spitzer E, Timleck M, Plumhoff E, Israels SJ, et al. Results of an external proficiency testing exercise on platelet dense‐granule deficiency testing by whole mount electron microscopy. Am J Clin Pathol. 2009;131:671–5. [DOI] [PubMed] [Google Scholar]
- 19. Antony‐Debre I, Bluteau D, Itzykson R, Baccini V, Renneville A, Boehlen F, et al. MYH10 protein expression in platelets as a biomarker of RUNX1 and FLI1 alterations. Blood. 2012;120:2719–22. [DOI] [PubMed] [Google Scholar]
- 20. Hayward CP, Liang M, Tasneem S, Soomro A, Waye JS, Paterson AD, et al. The duplication mutation of Quebec platelet disorder dysregulates PLAU, but not C10orf55, selectively increasing production of normal PLAU transcripts by megakaryocytes but not granulocytes. PLoS One. 2017;12:e0173991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Damiati E, Borsani G, Giacopuzzi E. Amplicon‐based semiconductor sequencing of human exomes: performance evaluation and optimization strategies. Hum Genet. 2016;135:499–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res. 2010;38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Heremans J, Freson K. High‐throughput sequencing for diagnosing platelet disorders: lessons learned from exploring the causes of bleeding disorders. Int J Lab Hematol. 2018;40(Suppl 1):89–96. [DOI] [PubMed] [Google Scholar]
- 24. Jagadeesh KA, Wenger AM, Berger MJ, Guturu H, Stenson PD, Cooper DN, et al. M‐CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat Genet. 2016;48:1581–6. [DOI] [PubMed] [Google Scholar]
- 25. Fromer M, Moran JL, Chambert K, Banks E, Bergen SE, Ruderfer DM, et al. Discovery and statistical genotyping of copy‐number variation from whole‐exome sequencing depth. Am J Hum Genet. 2012;91:597–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mohammed M, Al‐Hashmi N, Al‐Rashdi S, Al‐Sukaiti N, Al‐Adawi K, Al‐Riyami M, et al. Biallelic mutations in AP3D1 cause Hermansky‐Pudlak syndrome type 10 associated with immunodeficiency and seizure disorder. Eur J Med Genet. 2019;62:103583. [DOI] [PubMed] [Google Scholar]
- 27. Ammann S, Schulz A, Krageloh‐Mann I, Dieckmann NM, Niethammer K, Fuchs S, et al. Mutations in AP3D1 associated with immunodeficiency and seizures define a new type of Hermansky‐Pudlak syndrome. Blood. 2016;127:997–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Johnson B, Lowe GC, Futterer J, Lordkipanidze M, MacDonald D, Simpson MA, et al. Whole exome sequencing identifies genetic variants in inherited thrombocytopenia with secondary qualitative function defects. Haematologica. 2016;101:1170–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bastida JM, Lozano ML, Benito R, Janusz K, Palma‐Barqueros V, Del Rey M, et al. Introducing high‐throughput sequencing into mainstream genetic diagnosis practice in inherited platelet disorders. Haematologica. 2018;103:148–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Leinoe E, Zetterberg E, Kinalis S, Ostrup O, Kampmann P, Norstrom E, et al. Application of whole‐exome sequencing to direct the specific functional testing and diagnosis of rare inherited bleeding disorders in patients from the Oresund Region, Scandinavia. Br J Haematol. 2017;179:308–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Downes K, Megy K, Duarte D, Vries M, Gebhart J, Hofer S, et al. Diagnostic high‐throughput sequencing of 2396 patients with bleeding, thrombotic, and platelet disorders. Blood. 2019;134:2082–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gresele P, Orsini S, Noris P, Falcinelli E, Alessi MC, Bury L, et al. Validation of the ISTH/SSC bleeding assessment tool for inherited platelet disorders: a communication from the Platelet Physiology SSC. J Thromb Haemost. 2019;18:732–9. [DOI] [PubMed] [Google Scholar]
- 33. Orsini S, Noris P, Bury L, Heller PG, Santoro C, Kadir RA, et al. Bleeding risk of surgery and its prevention in patients with inherited platelet disorders. Haematologica. 2017;102:1192–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Golebiewska EM, Poole AW. Platelet secretion: from haemostasis to wound healing and beyond. Blood Rev. 2015;29:153–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McKay H, Derome F, Haq MA, Whittaker S, Arnold E, Adam F, et al. Bleeding risks associated with inheritance of the Quebec platelet disorder. Blood. 2004;104:159–65. [DOI] [PubMed] [Google Scholar]
- 36. Hayward CP, Moffat KA. Laboratory testing for bleeding disorders: strategic uses of high and low‐yield tests. Int J Lab Hematol. 2013;35:322–33. [DOI] [PubMed] [Google Scholar]
- 37. Rao AK, Poncz M. Defective acid hydrolase secretion in RUNX1 haplodeficiency: Evidence for a global platelet secretory defect. Haemophilia. 2017;23:784–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kaur G, Jalagadugula G, Mao G, Rao AK. RUNX1/core binding factor A2 regulates platelet 12‐lipoxygenase gene (ALOX12): studies in human RUNX1 haplodeficiency. Blood. 2010;115:3128–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jalagadugula G, Mao G, Kaur G, Goldfinger LE, Dhanasekaran DN, Rao AK. Regulation of platelet myosin light chain (MYL9) by RUNX1: implications for thrombocytopenia and platelet dysfunction in RUNX1 haplodeficiency. Blood. 2010;116:6037–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sun L, Gorospe JR, Hoffman EP, Rao AK. Decreased platelet expression of myosin regulatory light chain polypeptide (MYL9) and other genes with platelet dysfunction and CBFA2/RUNX1 mutation: insights from platelet expression profiling. J Thromb Haemost. 2007;5:146–54. [DOI] [PubMed] [Google Scholar]
- 41. Glembotsky AC, Bluteau D, Espasandin YR, Goette NP, Marta RF, Marin Oyarzun CP, et al. Mechanisms underlying platelet function defect in a pedigree with familial platelet disorder with a predisposition to acute myelogenous leukemia: potential role for candidate RUNX1 targets. J Thromb Haemost. 2014;12:761–72. [DOI] [PubMed] [Google Scholar]
- 42. Bluteau D, Glembotsky AC, Raimbault A, Balayn N, Gilles L, Rameau P, et al. Dysmegakaryopoiesis of FPD/AML pedigrees with constitutional RUNX1 mutations is linked to myosin II deregulated expression. Blood. 2012;120:2708–18. [DOI] [PubMed] [Google Scholar]
- 43. Lordier L, Bluteau D, Jalil A, Legrand C, Pan J, Rameau P, et al. RUNX1‐induced silencing of non‐muscle myosin heavy chain IIB contributes to megakaryocyte polyploidization. Nat Commun. 2012;3:717. [DOI] [PubMed] [Google Scholar]
- 44. Kligman MD, Zyromski NJ, McCullough DG, Gunning WT. Platelet‐dense granule deficiency causes postoperative hemorrhage in patients receiving enoxaparin: a novel observation with dramatic clinical implications. Am J Surg. 2009;197:365–70. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material